

Abstract
Top-down mass spectrometry (TD-MS) of peptides and proteins results in product ions that can be correlated to polypeptide sequence. Fragments can either be terminal fragments, which contain either the N- or the C-terminus, or internal fragments that contain neither termini. Normally, only terminal fragments are assigned due to the computational difficulties of assigning internal fragments. Here we describe ClipsMS, an algorithm that can assign both terminal and internal fragments generated by top-down MS fragmentation. Further, ClipsMS can be used to locate various modifications on the protein sequence. Using ClipsMS to assign TD-MS generated product ions, we demonstrate that for apo-myoglobin, the inclusion of internal fragments increases the sequence coverage up to 78%. Interestingly, many internal fragments cover complimentary regions to the terminal fragments that enhance the information that is extracted from a single top-down mass spectrum. Analysis of oxidized apo-myoglobin using terminal and internal fragment matching by ClipsMS confirmed the locations of oxidation sites on the two methionine residues. Internal fragments can be beneficial for top-down protein fragmentation analysis, and ClipsMS can be a valuable tool for assigning both terminal and internal fragments present in a top-down mass spectrum. Data are available via the MassIVE community resource with the identifiers MSV000086788 and MSV000086789.
Keywords: Top-Down Mass Spectrometry (TD-MS), Terminal Fragment, Internal Fragment, Electron Capture Dissociation (ECD)
Graphical Abstract
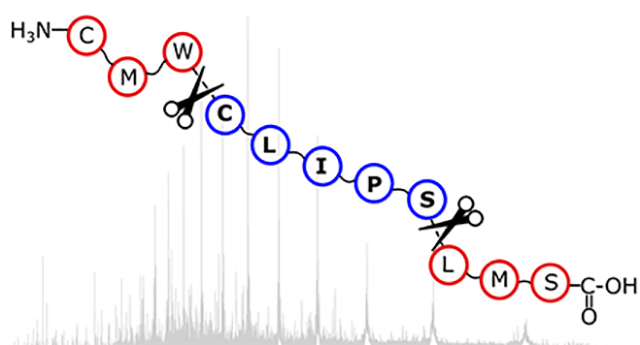
INTRODUCTION
Top-down mass spectrometry (TD-MS) has become a prominent tool for the analysis and characterization of intact proteins and protein complexes.1, 2 TD-MS analysis of proteins and protein complexes has many advantages, including the ability to detect and identify degradation products, sequence variations, post-translational modifications (PTMs), and other proteoforms.3 TD-MS has progressed significantly in the last decade owing to advances in instrumentation and associated technologies.4–6 For example, TD-MS has recently been utilized for the characterization and analysis of heterogeneous samples, large noncovalent protein complexes, and intact monoclonal antibodies.7 Despite these advances, however, the application of TD-MS for profiling PTMs and proteoforms is limited in sensitivity and scope, as data produced by top-down MS methods are not as easily analyzed compared to bottom-up proteomics.
TD-MS analysis of intact proteins typically starts by forming multiply charged gas-phase proteins using electrospray ionization (ESI).8, 9 The protein ions can then be activated and fragmented by collision-,10, 11 photon-,12, 13 or electron-based dissociation methods14–16 to generate product ions that can be assigned to the protein primary sequence.17 Product ions formed by top-down MS can either be i) a terminal fragment ion, which includes the N-terminus (a, b, or c fragment) or the C-terminus (x, y, or z fragment) of the polypeptide sequence,18 or ii) an internal fragment ion that results from multiple cleavage events of the protein backbone to generate ax, ay, az, bx, by, bz, cx, cy, and cz fragment ions, with the first letter designating the cleavage site on the N-terminal side of the fragment and the second letter designating the cleavage on the C-terminal side of the fragment.19–21 The isotopically resolved mass spectral signals22 can be matched to regions of the sequence to return information about the protein’s primary structure. Within a single protein fragmentation mass spectrum, there can be hundreds of product ion signals that could be assignable.
Assignment of mass spectral signals within a mass spectrum can be a long and arduous task that can require manual comparisons of experimentally measured masses to lists of computed theoretical masses to return putative information of the protein sequence. There has been significant development of software tools to aid the deconvolution and automated assignment of TD mass spectral signals. Multiple algorithms have been developed to deconvolute MS spectra including MSDeconv,23 YADA,24 and mMass.25 Other programs such as ProSight PTM 2.026 and ProSightLite27 match deconvoluted mass lists to a theoretical mass list from a given protein sequence. Other assignment algorithms include TopPIC28 and MS-Align+.29 Ge and co-workers have developed MASH Explorer, which allows the user to load raw data from a top-down mass spectrum, deconvolute the peaks present in that spectrum, and match the resulting values to theoretical masses from a given protein sequence.30 Although these and other tools are potentially powerful, they largely consider only the assignment of terminal fragments, which could leave many peaks in a fragmentation mass spectrum, including those representing internal fragments, to be unassigned. These unassigned signals, that we colloquially term as “dark matter” of a fragmentation mass spectrum, could provide valuable information if assigned correctly to the protein sequence.
However, internal fragment ions have been largely ignored due to the difficulty of accurately and efficiently assigning these mass spectral signals. As the size of the protein increases, the number of internal fragments that can be generated increases exponentially, hence increasing the false discovery rates limiting the accuracy of these assignments.31, 32 Due to this, internal fragment ion analysis of top-down mass spectra has been limited to peptides and small proteins. Despite the limitations associated with extending the use of internal fragment ion analysis on larger proteins, the inclusion of accurately assigned internal fragment ions could offer richer sequence and structural information.
Recently, the assignment of internal fragment signals for the analysis of protein ions in TD-MS experiments has been reported. Kelleher and co-workers demonstrated that for collision induced dissociation of ubiquitin (8.6 kDa), the inclusion of internal fragments resulted in a greater fraction of the fragmentation spectrum to be explained.21 Loo and co-workers recently demonstrated that internal fragments can be formed by electron-based fragmentation of ubiquitin and carbonic anhydrase II (29 kDa).32 Although these reports suggest that internal fragments can significantly enhance the information obtained from a TD-MS experiment, which could be beneficial for localizing sites of protein modifications, to date there have been few readily available computational methods that can be utilized to assign internal fragment ions.
Here, we describe an algorithm developed in Python, coined ClipsMS (Comprehensive Localization of Internal Protein Sequences), that can be utilized to assign both terminal and internal fragments resulting from a top-down mass spectrometry experiment. This algorithm generates every possible terminal and internal fragment, compares those fragments against a deconvoluted mass list, and graphically displays the data. We demonstrate the use of ClipsMS for the analysis of top-down mass spectra of wild type (wt) and oxidized apo-myoglobin. Assigning internal fragment masses is shown to increase sequence coverage of the protein sequence and confidence in the location of modified sites present on the protein.
EXPERIMENTAL
Materials:
Apo-myoglobin from equine skeletal muscle was purchased from Sigma-Aldrich (St. Louis, MO, USA) and used without further purification. LC/MS grade water and methanol were obtained from Fisher Chemical (Hampton, NH, USA). Hydrogen peroxide (H2O2) was obtained from Sigma-Aldrich (St. Louis, MO, USA).
Sample Preparation:
The oxidized form of apo-myoglobin was prepared by reaction with hydrogen peroxide at a 1:10 ratio of molar concentration (H2O2/ apo-myoglobin = 1:10) at 37°C for 30 min. Both wild type and oxidized apo-myoglobin were dissolved in water/ methanol/ formic acid (49.5:49.5:1, v/v/v) at a concentration of 20 μM.
Mass Spectrometry:
All experiments were conducted on a 15-T Bruker SolariX Fourier transform ion cyclotron resonance (FTICR)-MS instrument equipped with an infinity ICR cell (Bruker Daltonics, Billerica, MA, USA). Protein solutions were loaded into in-house pulled capillaries coated with gold, and electrosprayed by applying a voltage between 0.8 and 1.2 kV on the ESI capillary. MS1 spectra were collected of wildtype and oxidized myoglobin and the spectra were deconvoluted with UniDec.33 Broadband ECD experiments were conducted without precursor isolation. For ECD fragmentation of wild type apo-myoglobin, the pulse length was set at 0.1s, with a lens voltage at 50 V and an ECD bias voltage at 2V. For ECD fragmentation of the oxidized form, the lens and bias voltage were kept the same, while the pulse length was set at 0.025s to obtain an optimized ECD fragmentation. For each spectrum, 200 scans were obtained. The data was deconvoluted with the SNAP™ 2.0 algorithm from the Bruker Daltonics DataAnalysis software and internally calibrated against theoretical terminal fragments of apo myoglobin.
Algorithm Development and Parameters
The algorithm and GUI were designed in PyCharm 2020.2. The program runs on at least python 3.7 and is available on GitHub (https://github.com/loolab2020/ClipsMS-Version-1.0.0). Because ECD was performed on myoglobin, c, z, and cz fragments were searched for both wildtype and oxidized myoglobin. At the end of every N-terminal fragment a hydrogen atom (1.00783) is added to complete the amino group, and at the end of every C-terminal fragment a hydroxyl group (17.00274) is added to complete the carboxyl group (Figure S1A). The error given to the program was 2 ppm, the smallest internal fragment size is 5, and any fragments containing n-terminal cuts at proline residues were disregarded as false positives. In both instances, the biased version of the algorithm was run. For the oxidized version of the protein, localized modifications corresponding to oxidation (15.99491Da) were added to methionine 55 and methionine 131. An unlocalized modification corresponding to a hydrogen atom (1.00783Da) was also added to each fragment.
Sequence coverage was calculated as number of inter-residue cleavages divided by the total number of inter-residue sites. The number of cleavage site was calculated as the number of unique cleavage sites not already cut by another fragment. The coverage for a PTM site was designated as the number of times that the modified amino acid was covered by a unique fragment.
RESULTS
Graphical user interface of ClipsMS
A graphical user interface (GUI) was designed for ClipsMS so the user can easily compare theoretical fragments of a peptide or protein sequence against a user specified deconvoluted mass list from a top-down mass spectrum (Figure 1A). The algorithm can generate any theoretical terminal and internal fragment from a given amino acid sequence with a user defined minimum sequence length for internal fragments. Users can select the fragment types that can be formed by their experiments and set the mass error tolerance for matching. In addition, modifications can be accounted for: to include localized modifications, where a single amino acid site has been modified, and unlocalized modifications in which modifications can occur on any amino acid site. Once the user inputs the information required, the user can run either a biased version of the algorithm where terminal fragments are weighted higher than internal fragments, or an unbiased version of the algorithm where both terminal and internal fragments are weighted the same.
Figure 1.

A. The graphical user interface (GUI) for ClipsMS. The user can input several key parameters including the error allowed, the smallest internal fragment size, the sequence, the observed fragments, any modifications on the sequence and the type of fragments to search. B. The workflow of the algorithm and how it matches peaks input by the user. The algorithm calculates all theoretical terminal and internal fragments, matches all peaks, makes decisions on which assignments to keep, and automatically generates figures.
Processing and generation of theoretical fragments and fragment matching using ClipsMS
The algorithm calculates a molecular weight based on the amino acid sequence (Table S1) plus a H+ (1.00728 amu) to return the monoisotopic [M+H]+ mass (Figure 1B). Next, all possible a, b, c, x, y, and z terminal fragments of the protein are calculated and stored as a list (Figure 1B, Figure S1A). After terminal fragments are calculated, those fragments are used to calculate the mass of all possible internal fragments: ax, ay, az, bx, by, bz, cx, cy, and cz fragments of a protein (Figure 1B, Figure S1B). All fragment masses are calculated as monoisotopic [M+H]+ masses.
After the base theoretical fragment masses of the amino acid sequence have been generated, these masses are compared against a given deconvoluted mass list (Figure 1B). Each observed mass in the deconvoluted mass list is compared against every theoretical terminal and internal mass for completeness. If modifications have been imported, the shift in mass will be accounted for by the algorithm. The modifications the algorithm accounts for are: (i) localized modifications, (ii) unlocalized modifications, and (iii) terminal modifications. (i) Localized modifications are treated as static modifications that occur on a single amino acid and will not detach from the protein. This would include previously located PTMs, nonstandard mutations (e.g., selenocysteine), the absence of hydrogen atoms from oxidized cysteine residues, and/or user modified proteins. These modifications are added to every terminal and internal fragment that contains that amino acid at a given site before the fragment is compared against the deconvoluted values (Figure 1B). (ii) Unlocalized modifications include modifications that are not attributed to a specific amino acid. This may include addition or subtraction of hydrogen atoms or water molecules, PTMs where the location is not known, or ligands where the location is not known. Unlocalized modifications are added to each mass after the unmodified theoretical masses have been searched (Figure 1B). These modifications were designed to allow for both unmodified and modified peaks to be analyzed. (iii) Terminal modifications are added to the end of every C or N terminal fragment before comparing that fragment against the deconvoluted values (Figure 1B). The terminal modifications do not affect internal fragments matching as they do not contain either terminus. If the measured mass error of an observed fragment compared to a theoretical fragment is within the error tolerance set by the user, the fragment is counted as a match.
After deconvoluted masses are matched with the theoretical masses, matches that cannot occur are automatically deleted from the matched list (Figure 1B). This can include matches that contain c and z fragmentation at proline residues in electron-based fragmentation or matches containing improbable unlocalized modifications. For electron-based fragmentation methods, the α-amino group on proline contains two bonds making it an imino acid, c or z fragmentation at these residues are indicated as improbable34 and removed from the matched list. There is an option to use this feature in the GUI. (Fig. 1A) An example of an improbable unlocalized modification could be a match that contains a mass shift equal to phosphorylation on a fragment not containing a serine, threonine, or tyrosine. If a fragment contains a modification but does not have the amino acids required, the match is designated as a false positive and removed from the list. The masses of unlocalized modifications and the amino acids on which they occur can be input by the user with the GUI (Figure 1A). These safeguards help reduce false positives and generate more accurate results.
After all possible fragments have been matched and improbable fragments have been removed, the algorithm makes decisions on duplicate fragment assignments. For example, a single deconvoluted mass can be assigned to multiple theoretical masses provided these masses fall within the error tolerance set by the user. The algorithm decides which fragment to keep depending on the version run. The unbiased version of the algorithm assigns the fragment with a lower mass error (Figure 1B). If two fragments have exactly the same chemical formula (and as a result the same mass), both fragments are kept and displayed. All matched fragments are output in a table for the user to review. The biased version of the algorithm favors the terminal fragments (Figure 1B). If a terminal fragment and an internal fragment are matched to a single deconvoluted mass, the terminal fragment is chosen as the assignment. If two terminal or two internal fragments are matched to the same deconvoluted mass, the fragment with the lower mass error is assigned. However, two terminal or two internal fragments with exactly the same chemical formula and mass are retained and output in a table for the user to review. These features were incorporated to limit false positive matches.
The output table contains all the information for the observed fragments that were matched with the theoretical fragment list. It includes the fragment type, any localized and unlocalized modifications, any terminal modifications, the observed mass, the theoretical mass calculated by ClipsMS, the starting and ending amino acids, the error of the observed mass compared to the theoretical mass, the sequence of the fragment, the intensity given by the user, and the molecular formula of the fragment (Table S2). This table allows for manual interpretation of the data if desired.
The time it takes for the algorithm to run depends on the length of the protein sequence, the number of fragments uploaded, and the number of modifications on the protein sequence. Peptides and small proteins (< 30 kDa) with a few hundred deconvoluted peaks take less than 1 minute to analyze on a laptop with 12GB of ram with an Intel Core i7–2760QM processor (4 cores @ 2.40 GHz). Larger sequences or sequences with more modifications can take up to a few minutes to complete. At the end of every run, a .csv document with all matched fragments is exported (Table S2) and 3 figures are output representing the fragments that are matched.
Top-down fragmentation data analysis using ClipsMS
To test ClipsMS for top-down fragmentation analysis, apo-myoglobin was prepared in denaturing conditions and electrosprayed on a Bruker 15T FT-ICR MS (Figure S2A). Apo-myoglobin was fragmented with broadband electron capture dissociation (ECD) MS (Figure 2A). In the resulting spectrum, charge reduced precursor ions are present as well as fragment ions (Figure 2A). The data deconvoluted from the SNAP™ algorithm was input to the algorithm along with the intensity values and the biased version of the algorithm was run.
Figure 2.

A. Broadband ECD MS of 20 μM apo-myoglobin formed from acidic denaturing conditions. B. A fragment location map indicating the region of the protein sequence covered by terminal and internal fragments. C. A sequence coverage map for the terminal and internal fragments. Darker regions indicate more coverage. D. A fragment cleavage map indicating the location of inter-amino acid cleavage sites for terminal and internal fragments.
Unlike terminal fragments where one end is fixed, internal fragments contain neither the N nor C terminus. Because neither terminus is fixed, it is difficult to represent internal fragments with the conventional top-down fragmentation map such as the one used by Prosight Lite27 and MASH Explorer.30 To represent both terminal and internal fragments, the algorithm outputs 3 figures: (i) a fragment location map, (ii) a sequence coverage map, and (iii) a fragmentation cleavage site map. Each of these figures displays a key piece of information to describe the data analyzed by the algorithm.
The first figure displays the data in a way so that the number of internal fragments identified is easily determined and the coverage of the protein sequence can be easily shown (Figure 2B). In addition, the size of the dots indicates the relative intensity of the matched fragments. The myoglobin data indicated that 98 c and z terminal fragments were assigned, and 15 cz internal fragments were assigned (Figure 2B). Furthermore, the data indicates that the internal fragments are normally lower in abundance than many of the terminal fragments (Figure 2B). This data also shows that the c and z terminal fragments assigned heavily cover both the N and C terminal regions of the sequence and the cz internal fragments cover the interior of the protein.
The second figure includes the sequence information map of a protein. This map is based off a figure in a paper published by the Kelleher lab.21 The figure represents the areas of the sequence that are covered by the product ions. Darker regions of the sequence indicate regions of the protein that are covered by many fragments while lighter colored regions of the sequence indicate regions of the protein that are covered by fewer fragments. This data indicates regions of the protein that terminal and internal fragments cover. Terminal fragments heavily cover the ends of protein sequences and internal fragments cover the interior of the protein. Internal fragments increase the amount of sequence information of the protein. In the apo-myoglobin data, the sequence coverage is 59% when only terminal fragments are considered; however, by including internal product ions in the search, the sequence coverage was increased to 66% (Figure 2C). Including internal fragments tends to enhance the sequence coverage on proteins by giving information of the amino acid sequence in the center of the protein sequence. This observation has been reported before by our lab for carbonic anhydrase II,32 and an updated fragmentation location map is included (Figure S3).
Figure 2D shows the cleavage sites of terminal and terminal fragments. Analyzing internal fragments can increase the number of fragmentation sites that occur in the protein sequence. The cleavage sites of terminal fragments and the cleavage sites of internal fragments are displayed. In the apo-myoglobin data, it is shown that the terminal fragments identified cleave at 90 of the 152 inter-residue cleavage sites of the protein (Figure 2D). Inclusion of cz internal fragments increased the number of cleavage sites on apo myoglobin to 101 (Figure 2D). Increasing the number of cleavage sites can increase confidence in the sequence identity as more protein information can be extracted.
Unlocalized modification feature of ClipsMS
ClipsMS can also be utilized to investigate unlocalized modifications on protein fragment ions. When unlocalized modifications are considered, ClipsMS will generate a list of fragments with the addition and/or subtraction of a user defined mass after it has searched for the unmodified masses. This list of fragments will then be matched to fragments that the user has input. For wt apo-myoglobin, we investigated the unlocalized modification feature by examining c, z, and cz fragments from wt apo-myoglobin that can be matched with the loss/gain of a hydrogen atom (1.00783Da), which are prevalent in ECD.35 Including the addition of a hydrogen atom increased the number of assignments to 158 (Figure S4B). In addition, the inclusion of internal fragments increased the sequence coverage from 63% to 78% when fragments with an extra hydrogen were searched, (Figure S4C) and the number of cut sites increased from 95 to 118 (Figure S4D). This data indicates that ClipsMS’s unlocalized modification feature can be utilized to increase the number of assignments and the sequence coverage of proteins. In addition to analyzing fragments with neutral mass losses, this feature can also be used to pinpoint PTMs where the location is not specified and/or ligand binding sites of a protein as the inclusion of internal fragments could give more informative data on where PTMs and ligand binding occurs.
Confirmation of localized modification sites using ClipsMS
To test ClipsMS’s localized modification feature, apo-myoglobin was oxidized and fragmented with ECD. An MS1 spectrum of oxidized apo-myoglobin showed a mass difference of 32Da compared to wt apo-myoglobin, suggesting that 2 oxidation sites were present (Figure S2B). Oxidation of intact proteins can occur on methionine residues,36 and apo-myoglobin contains two methionine residues at positions 55 and 131. To confirm that oxidation occurred on these two residues, broadband ECD was performed on the oxidized myoglobin sample (Figure 3A). For oxidized apo-myoglobin, 73 terminal fragments and 20 internal fragments were identified, with 39 of those fragments containing a single oxidation site and 1 fragment containing both oxidation sites (Figure 3B). Modifications on apo-myoglobin were confirmed to be on methionine 55 and 131 and these sites are indicated by a dashed line (Figure 3B). This data indicates that the inclusion of internal fragments enhances the confidence of the location of both oxidation sites.
Figure 3.

A. Broadband ECD MS of 20 μM oxidized apo-myoglobin formed from acidic denaturing conditions. B. A fragment location map indicating the region of the protein sequence covered by terminal and internal fragments. Dashed lines indicate sites of oxidation. C. A sequence coverage map for the terminal and internal fragments assigned indicating terminal and internal fragments cover both oxidation sites. Darker regions indicate more coverage. D. A fragment cleavage map indicating the location of inter-amino acid cleavage sites for terminal and internal fragments. Red amino acids indicate sites of oxidation.
Terminal fragments cover the residues near N and C terminus while the internal fragments cover the residues interior of the protein (Figure 3C). For Met-55, terminal fragments cover the residue 11 times, and 4 additional internal fragments cover Met-55. Similarly, Met-131 was covered by a terminal fragment 23 times, and 3 additional internal fragments cover the modified residue.
When the inter-amino acid cleavage sites of apo-myoglobin are considered, terminal fragments account for 67 inter-amino cleavage sites and inclusion of internal fragments increased the number of inter-amino acid cleavage sites to 84 (Figure 3D). This included a cut site between position 53 and 54, which narrowed down the location of oxidation on Met-55. Increased coverage of residues with PTMs increases confidence that a modification occurs on a particular residue.
DISCUSSION
ClipsMS efficiently assigns both terminal and internal fragments present in top-down mass spectra. Assigning internal fragments can enhance top-down mass spectrometry analysis (Figure 2B). For apo-myoglobin, the sequence coverage obtained was enhanced from 59% to 66%. This data agrees well with previous reports obtained for proteins and peptides.32, 37, 38 Conventionally, internal fragment analysis has been ignored; including internal fragments may help to increase the molecular weight limit of 30 kDa that is often observed for high sequence coverage TD-MS.17, 39, 40 Here, we demonstrate that internal fragments can provide more information of the interior of the protein (Figure 2C). In addition, a plethora of PTMs are located within the interior of protein sequences,41 hence internal fragment assignments can aid in the localization of these PTMs.
Although ClipsMS has been shown to be a powerful tool for top-down fragmentation assignments, there are a few limitations of the algorithm. Duplicated fragments pose a problem for internal fragment analysis. A single deconvoluted mass can be matched to multiple theoretical masses due to those fragments having the same elemental composition. It is possible that a better understanding of top-down fragmentation mechanisms and/or ion mobility analysis of top-down fragments can help overcome this issue. Another current limitation of ClipsMS is that some protein modification types are not considered.35, 42, 43 In the future, the algorithm will include the capability to specifically search for neutral losses and more diverse fragment types such as c+1 fragments, z+1 fragments, and z· fragments. In addition, the ability to search for labile modifications on specific amino acids will be incorporated into the search algorithm. Lastly, larger sequences with more fragments or data containing many modifications can take up to a few minutes to complete. For the myoglobin data, processing times ranged from ~30 seconds to ~11 minutes on the same device (Table S3). A dummy 1023 amino acid sequence (116.3kDa) with 250 fragments took approximately an hour to run on a laptop with 12GB of ram with an Intel Core i7–2760QM processor (4 cores @ 2.40 GHz). Currently however, the architecture of the algorithm is such that it only uses a single core, which limits the processing times. By allowing access to more cores and/or ram the processing time can be significantly reduced. Despite all these limitations, the information obtained from ClipsMS can still be beneficial for top-down protein fragmentation analysis.
As top-down proteomics becomes more mainstream in proteome research, it is becoming increasingly important to efficiently analyze top-down mass spectrometry data. The top-down community for the most part has disregarded internal fragments and opted to only analyze terminal fragments. By analyzing internal fragments, it is possible to gain more insight into the protein sequence. We hope this algorithm will aid researchers to mine some of the previously unknown “dark matter” in top-down mass spectra and will spur research in proteomics and the proteoforms that exist in nature.
Supplementary Material
Figure S1. Types of terminal and internal fragments that can be searched with ClipsMS.
Figure S2. Spectra of wildtype and oxidized myoglobin with their corresponding UniDec spectra.
Figure S3. A fragment location map representing a top-down spectrum of denatured carbonic anhydrase II.
Figure S4. ClipsMS analysis of a top-down mass spectrum of myoglobin including peaks corresponding to fragments with an extra hydrogen atom.
Table S1. Amino acid residue masses used for generating theoretical terminal and internal fragments with ClipsMS.
Table S2. Sample output of ClipsMS analysis for apo myoglobin.
Table S3. Processing times for ClipsMS analysis of myoglobin with different parameters.
ClipsMS (https://github.com/loolab2020/ClipsMS-Version-1.0.0)
ACKNOWLEDGMENTS
The top-down MS data have been deposited to the MassIVE community resource with the data set identifiers MSV000086788 and MSV000086789. Support from the US National Institutes of Health (R01GM103479, S10RR028893), the US National Science Foundation (NSF) (CHE 1808492), and the US Department of Energy (DE-FC02-02ER63421) are gratefully acknowledged. C. L. acknowledges support from the Ruth L. Kirschstein National Research Service Award program (GM007185).
References
- 1.Kelleher NL; Lin HY; Valaskovic GA; Aaserud DJ; Fridriksson EK; Mclafferty FW, Top Down versus Bottom Up Protein Characterization by Tandem High-Resolution Mass Spectrometry. Journal of the American Chemical Society 1999, 121 (4), 806–812. [Google Scholar]
- 2.Lermyte F; Tsybin YO; O’Connor PB; Loo JA, Top or Middle? Up or Down? Toward a Standard Lexicon for Protein Top-Down and Allied Mass Spectrometry Approaches. Journal of the American Society for Mass Spectrometry 2019, 30 (7), 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durbin KR; Fornelli L; Fellers RT; Doubleday PF; Narita M; Kelleher NL, Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa. Journal of Proteome Research 2016, 15 (3), 976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denisov E; Damoc E; Lange O; Makarov A, Orbitrap mass spectrometry with resolving powers above 1,000,000. International Journal of Mass Spectrometry 2012, 325–327, 80–85. [Google Scholar]
- 5.Kelly RT; Tolmachev AV; Page JS; Tang K; Smith RD, The ion funnel: Theory, implementations, and applications. Mass Spectrometry Reviews 2009, 29 (2), 294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw JB; Lin TY; Leach FE 3rd; Tolmachev AV; Tolic N; Robinson EW; Koppenaal DW; Pasa-Tolic L, 21 Tesla Fourier Transform Ion Cyclotron Resonance Mass Spectrometer Greatly Expands Mass Spectrometry Toolbox. J Am Soc Mass Spectrom 2016, 27 (12), 1929–1936. [DOI] [PubMed] [Google Scholar]
- 7.Srzentić K; Fornelli L; Tsybin YO; Loo JA; Seckler H; Agar JN; Anderson LC; Bai DL; Beck A; Brodbelt JS; Van Der Burgt YEM; Chamot-Rooke J; Chatterjee S; Chen Y; Clarke DJ; Danis PO; Diedrich JK; D’Ippolito RA; Dupré M; Gasilova N; Ge Y; Goo YA; Goodlett DR; Greer S; Haselmann KF; He L; Hendrickson CL; Hinkle JD; Holt MV; Hughes S; Hunt DF; Kelleher NL; Kozhinov AN; Lin Z; Malosse C; Marshall AG; Menin L; Millikin RJ; Nagornov KO; Nicolardi S; Paša-Tolić L; Pengelley S; Quebbemann NR; Resemann A; Sandoval W; Sarin R; Schmitt ND; Shabanowitz J; Shaw JB; Shortreed MR; Smith LM; Sobott F; Suckau D; Toby T; Weisbrod CR; Wildburger NC; Yates JR; Yoon SH; Young NL; Zhou M, Interlaboratory Study for Characterizing Monoclonal Antibodies by Top-Down and Middle-Down Mass Spectrometry. Journal of the American Society for Mass Spectrometry 2020, 31 (9), 1783–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fenn J; Mann M; Meng C; Wong S; Whitehouse C, Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246 (4926), 64–71. [DOI] [PubMed] [Google Scholar]
- 9.Donnelly DP; Rawlins CM; Dehart CJ; Fornelli L; Schachner LF; Lin Z; Lippens JL; Aluri KC; Sarin R; Chen B; Lantz C; Jung W; Johnson KR; Koller A; Wolff JJ; Campuzano IDG; Auclair JR; Ivanov AR; Whitelegge JP; Paša-Tolić L; Chamot-Rooke J; Danis PO; Smith LM; Tsybin YO; Loo JA; Ge Y; Kelleher NL; Agar JN, Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nature Methods 2019, 16 (7), 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katta V; Chowdhury SK; Chait BT, Use of a single-quadrupole mass spectrometer for collision-induced dissociation studies of multiply charged peptide ions produced by electrospray ionization. Analytical chemistry 1991, 63 (2), 174–178. [DOI] [PubMed] [Google Scholar]
- 11.McCormack AL; Jones JL; Wysocki VH, Surface-induced dissociation of multiply protonated peptides. Journal of the American Society for Mass Spectrometry 1992, 3 (8), 859–862. [DOI] [PubMed] [Google Scholar]
- 12.Brodbelt JS, Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chemical Society Reviews 2014, 43 (8), 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H; Nguyen HH; Loo RRO; Campuzano ID; Loo JA, An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nature chemistry 2018, 10 (2), 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Syka JE; Coon JJ; Schroeder MJ; Shabanowitz J; Hunt DF, Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proceedings of the National Academy of Sciences 2004, 101 (26), 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zubarev RA; Horn DM; Fridriksson EK; Kelleher NL; Kruger NA; Lewis MA; Carpenter BK; McLafferty FW, Electron capture dissociation for structural characterization of multiply charged protein cations. Analytical chemistry 2000, 72 (3), 563–573. [DOI] [PubMed] [Google Scholar]
- 16.Li H; Sheng Y; McGee W; Cammarata M; Holden D; Loo JA, Structural characterization of native proteins and protein complexes by electron ionization dissociation-mass spectrometry. Analytical chemistry 2017, 89 (5), 2731–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Catherman AD; Skinner OS; Kelleher NL, Top Down proteomics: Facts and perspectives. Biochemical and Biophysical Research Communications 2014, 445 (4), 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zubarev R, Protein primary structure using orthogonal fragmentation techniques in Fourier transform mass spectrometry. Expert Review of Proteomics 2006, 3 (2), 251–261. [DOI] [PubMed] [Google Scholar]
- 19.Zinnel NF; Pai P-J; Russell DH, Ion Mobility-Mass Spectrometry (IM-MS) for Top-Down Proteomics: Increased Dynamic Range Affords Increased Sequence Coverage. Analytical Chemistry 2012, 84 (7), 3390–3397. [DOI] [PubMed] [Google Scholar]
- 20.Michalski A; Neuhauser N; Cox J; Mann M, A Systematic Investigation into the Nature of Tryptic HCD Spectra. Journal of Proteome Research 2012, 11 (11), 5479–5491. [DOI] [PubMed] [Google Scholar]
- 21.Durbin KR; Skinner OS; Fellers RT; Kelleher NL, Analyzing internal fragmentation of electrosprayed ubiquitin ions during beam-type collisional dissociation. Journal of the American Society for Mass Spectrometry 2015, 26 (5), 782–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haverland NA; Skinner OS; Fellers RT; Tariq AA; Early BP; LeDuc RD; Fornelli L; Compton PD; Kelleher NL, Defining Gas-Phase Fragmentation Propensities of Intact Proteins During Native Top-Down Mass Spectrometry. J Am Soc Mass Spectrom 2017, 28 (6), 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X; Inbar Y; Dorrestein PC; Wynne C; Edwards N; Souda P; Whitelegge JP; Bafna V; Pevzner PA, Deconvolution and database search of complex tandem mass spectra of intact proteins: a combinatorial approach. Molecular & Cellular Proteomics 2010, 9 (12), 2772–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carvalho PC; Xu T; Han X; Cociorva D; Barbosa VC; Yates III JR, YADA: a tool for taking the most out of high-resolution spectra. Bioinformatics 2009, 25 (20), 2734–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strohalm M; Hassman M; Košata B; Kodíček M, mMass data miner: an open source alternative for mass spectrometric data analysis. Rapid Communications in Mass Spectrometry: An International Journal Devoted to the Rapid Dissemination of Up-to-the-Minute Research in Mass Spectrometry 2008, 22 (6), 905–908. [DOI] [PubMed] [Google Scholar]
- 26.Zamdborg L; LeDuc RD; Glowacz KJ; Kim Y-B; Viswanathan V; Spaulding IT; Early BP; Bluhm EJ; Babai S; Kelleher NL, ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucleic acids research 2007, 35 (suppl_2), W701–W706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fellers RT; Greer JB; Early BP; Yu X; Leduc RD; Kelleher NL; Thomas PM, ProSight Lite: Graphical software to analyze top-down mass spectrometry data. PROTEOMICS 2015, 15 (7), 1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kou Q; Xun L; Liu X, TopPIC: a software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics 2016, 32 (22), 3495–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X; Sirotkin Y; Shen Y; Anderson G; Tsai YS; Ting YS; Goodlett DR; Smith RD; Bafna V; Pevzner PA, Protein identification using top-down spectra. Molecular & cellular proteomics 2012, 11 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Z; Roberts DS; Melby JA; Wenger K; Wetzel M; Gu Y; Ramanathan SG; Bayne EF; Liu X; Sun R; Ong IM; McIlwain SJ; Ge Y, MASH Explorer: A Universal Software Environment for Top-Down Proteomics. J Proteome Res 2020, 19 (9), 3867–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyon YA; Riggs D; Fornelli L; Compton PD; Julian RR, The Ups and Downs of Repeated Cleavage and Internal Fragment Production in Top-Down Proteomics. J Am Soc Mass Spectrom 2018, 29 (1), 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zenaidee MA; Lantz C; Perkins T; Jung W; Loo RRO; Loo JA, Internal Fragments Generated by Electron Ionization Dissociation Enhance Protein Top-Down Mass Spectrometry. Journal of the American Society for Mass Spectrometry 2020, 31 (9), 1896–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marty MT; Baldwin AJ; Marklund EG; Hochberg GK; Benesch JL; Robinson CV, Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Analytical chemistry 2015, 87 (8), 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Axelsson J; Palmblad M; Håkansson K; Håkansson P, Electron capture dissociation of substance P using a commercially available Fourier transform ion cyclotron resonance mass spectrometer. Rapid communications in mass spectrometry 1999, 13 (6), 474–477. [DOI] [PubMed] [Google Scholar]
- 35.Zhurov KO; Fornelli L; Wodrich MD; Laskay ÜA; Tsybin YO, Principles of electron capture and transfer dissociation mass spectrometry applied to peptide and protein structure analysis. Chemical Society Reviews 2013, 42 (12), 5014–5030. [DOI] [PubMed] [Google Scholar]
- 36.Levine RL; Mosoni L; Berlett BS; Stadtman ER, Methionine residues as endogenous antioxidants in proteins. Proceedings of the National Academy of Sciences 1996, 93 (26), 15036–15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barran PE; Polfer NC; Campopiano DJ; Clarke DJ; Langridge-Smith PRR; Langley RJ; Govan JRW; Maxwell A; Dorin JR; Millar RP; Bowers MT, Is it biologically relevant to measure the structures of small peptides in the gas-phase? International Journal of Mass Spectrometry 2005, 240 (3), 273–284. [Google Scholar]
- 38.Ballard KD; Gaskell SJ, Sequential mass spectrometry applied to the study of the formation of “internal” fragment ions of protonated peptides. International Journal of Mass Spectrometry and Ion Processes 1991, 111, 173–189. [Google Scholar]
- 39.Kelleher NL, Peer Reviewed: Top-Down Proteomics. Analytical Chemistry 2004, 76 (11), 196 A–203 A. [PubMed] [Google Scholar]
- 40.Chen B; Brown KA; Lin Z; Ge Y, Top-Down Proteomics: Ready for Prime Time? Analytical Chemistry 2018, 90 (1), 110–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA, How many human proteoforms are there? Nature chemical biology 2018, 14 (3), 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.TurečEk F, N-Cα Bond Dissociation Energies and Kinetics in Amide and Peptide Radicals. Is the Dissociation a Non-ergodic Process? Journal of the American Chemical Society 2003, 125 (19), 5954–5963. [DOI] [PubMed] [Google Scholar]
- 43.Zubarev RA, Electron-capture dissociation tandem mass spectrometry. Current Opinion in Biotechnology 2004, 15 (1), 12–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Types of terminal and internal fragments that can be searched with ClipsMS.
Figure S2. Spectra of wildtype and oxidized myoglobin with their corresponding UniDec spectra.
Figure S3. A fragment location map representing a top-down spectrum of denatured carbonic anhydrase II.
Figure S4. ClipsMS analysis of a top-down mass spectrum of myoglobin including peaks corresponding to fragments with an extra hydrogen atom.
Table S1. Amino acid residue masses used for generating theoretical terminal and internal fragments with ClipsMS.
Table S2. Sample output of ClipsMS analysis for apo myoglobin.
Table S3. Processing times for ClipsMS analysis of myoglobin with different parameters.
ClipsMS (https://github.com/loolab2020/ClipsMS-Version-1.0.0)