

Abstract
Purpose of Review
The goal of this review is to provide an overview of the impact and underlying mechanism of oxidative stress on connexin channel function, and their roles in skeletal aging, estrogen deficiency, and glucocorticoid excess associated bone loss.
Recent Findings
Connexin hemichannel opening is increased under oxidative stress conditions, which confers a cell protective role against oxidative stress-induced cell death. Oxidative stress acts as a key contributor to aging, estrogen deficiency, and glucocorticoid excess-induced osteoporosis and impairs osteocytic network and connexin gap junction communication.
Summary
This paper reviews the current knowledge for the role of oxidative stress and connexin channels in the pathogenesis of osteoporosis and physiological and pathological responses of connexin channels to oxidative stress. Oxidative stress decreases osteocyte viability and impairs the balance of anabolic and catabolic responses. Connexin 43 (Cx43) channels play a critical role in bone remodeling, mechanotransduction, and survival of osteocytes. Under oxidative stress conditions, there is a consistent reduction of Cx43 expression, while the opening of Cx43 hemichannels protects osteocytes against cell injury caused by oxidative stress.
Keywords: Oxidative stress, Connexin channels, Aging, Sex steroids, Glucocorticoids, Osteoporosis
Introduction
Bone is a dynamic organ that undergoes a constant and tightly regulated remodeling process. Osteocytes are the most abundant cell type in bone, communicating with the bone-forming osteoblasts and bone-resorbing osteoclasts, thus maintaining homeostasis and the biomechanical function of the skeleton [1, 2]. The redox balance plays a pivotal role in the regulation of bone remodeling and the generation and survival of bone cells. Oxidative stress, which is induced by excessive reactive oxygen species (ROS) production and/or an impaired antioxidant system, has been revealed as an underlying mechanism for loss of bone mass and quality [3–5]. ROS includes superoxide anion (O2•−), hydroxyl (HO•), singlet oxygen (1O2), and hydrogen peroxide (H2O2). They are highly reactive molecules formed upon incomplete reduction of oxygen during aerobic metabolism [6, 7]. The generation of ROS occurs primarily in mitochondria from the escape of electrons passing through the electron transport chain. ROS act as signaling molecules that affect critical physiological processes, and their accumulation has been shown to cause detrimental effects to DNA, protein, and lipids [8, 9]. Targeting ROS production in bone cells might be a viable therapeutic approach in the prevention of osteoporosis.
Gap junctions are composed of two juxtaposed connexons or hemichannels (HCs). These gap junctions mediate direct cell-cell communication between adjacent osteocytes and osteocytes with other cells residing within the bone surface, such as osteoblasts, osteoclasts, and vasculature cells. Among the different gap junction-forming proteins, connexin 43 (Cx43) is the most abundant connexin present in bone cells [10, 11]. As a key modulator of skeletal homeostasis, Cx43 contributes to bone cell proliferation, survival, and differentiation [12–15]. HCs, unpaired gap junction channels, are extensively involved in the communication between the cytoplasm and the extracellular space and play critical roles in mechanotransduction, cell survival, and autocrine/paracrine signaling [16–19]. These connexin-based gap junctions and HCs allow the passage of small molecules (MW < 1 kDa), such as ions, essential metabolites, and second messengers, such as Ca2+, IP3, NAD+, prostaglandin E2 (PGE2), cAMP, cGMP, ADP, and ATP [20]. Under normal physiological conditions, few HCs are open; their activation (opening) is regulated by multiple factors, including mechanical stimulation, extracellular Ca2+ concentration, pro-inflammatory cytokines, and redox status [21, 22]. In addition, Cxs can be phosphorylated by several kinases, such as protein kinase A, protein kinase C, mitogen-activated protein kinase, and casein kinase 1 at serine/threonine or tyrosine residues [23]. Their phosphorylation plays important roles in the regulation of channel assembly/disassembly, internalization, degradation, trafficking, and gating [24].
Several lines of evidence have indicated that Cx43 gap junctions or HCs regulate susceptibility of H2O2-mediated cell death [25–28]. In this review, we will address the function of Cx43 channels under oxidative stress and summarize the important role of oxidative stress underlying osteoporosis induced by aging, loss of sex steroid, or glucocorticoid treatment. We will particularly focus on the regulations of Cx43 gap junction channels and HCs under the above physiological or pathological conditions. The aim of this review is to provide insights for targeting oxidative stress and Cx43 channels as potential therapeutic means in treating bone loss and osteoporosis.
The Role of Connexin Channels in Response to Oxidative Stress
The study of cellular mechanisms as to how Cx channels respond to oxidative stress in bone is rather limited; however, the modulation of Cx channels by oxidative stress has been reported in several non-skeletal tissues [29–32]. These studies may shed some light on what is taking place in skeletal tissue. In eye lens fiber cells, Cx50 HCs are shown to open in response to H2O2, mediating the transport of reductant glutathione (GSH) [30]. The dominant-negative mutants in Cx50, including Cx50P88S, which inhibits both gap junctions and HCs, and Cx50H156N, which only inhibits HCs, block the protective role of Cx50 in response to H2O2-induced apoptosis, while the Cx50 E48K, which only inhibits gap junctions, does not have such effect. This study highlights a cell protective role of Cx HCs against oxidative damage. In cardiomyocytes, Cx43 can be translocated to the mitochondrial inner membrane, exerting cardioprotection during ischemic preconditioning as well as during hypoxic postconditioning, a process involving ROS as a critical mediator to heart damage [33–35]. The translocation of Cx43 is initiated by heat shock protein 90 as a molecular chaperone and associates with the interaction of a mitochondrial protein, TOM20 [33]. Recent studies show that ATP-sensitive potassium channels in mitochondria (mKATP) also interact with mitochondrial Cx43 HCs and protect cardiomyocytes under mild hypoxic conditions [32, 36, 37]. In astrocytes, Cx43 deficiency or Cx43 channel inhibition results in elevated susceptibility to oxidative stress-induced cell death [38]. H2O2 causes uncoupling of gap junctions, while HC activity is enhanced, which is likely regulated by altered states of protein phosphorylation and intracellular distribution of Cx43. In human retinal pigment epithelial cells, functional Cx43 gap junction channels confer protection from chemical oxidant tert-butyl hydroperoxide (t-BOOH)-induced cell death [26]. In contrast to the cell protective effect, Cx43 HCs have been implicated to reduce astrocyte viability during hypoxia/reoxygenation [39]. Several lines of evidence also indicate the detrimental role of Cx43 channels in response to oxidative stress. Cigarette smoke extract and H2O2 lead to membrane depolarization and opening of HCs, resulting in accelerated cell death of epithelial cells [28]. In another study, cadmium ions (Cd2+) induces higher concentration of ROS and more severe cell injury in Cx43 wild-type fibroblasts than in fibroblasts derived from knockout littermates, potentially caused by the efflux of intracellular GSH and ATP through opened Cx43 channels [31]. A recent study further proves the extracellular loss of the major antioxidant GSH via Cx43 HCs, contributes to Ca2+ depletion-elicited disassembly of cell junctions through regulation of intracellular oxidative status [40]. Moreover, blockade of Cx43 HCs by Cx43 mimetic peptide Gap 19 attenuates the LPS-induced oxidative stress production and cell apoptosis in human umbilical vein endothelial cells [29].
Our previous work has demonstrated a novel cell protective mechanism mediated by Cx43 channels against oxidative stress in osteocytes [27]. We have shown that H2O2 treatment decreases Cx43 expression in a dose-dependent manner and this can be reversed by the antioxidant N-(2-mercaptopropionyl)-glycine (NMPG). In contrast to the reduced gap junction intercellular communication (GJIC), there is an increase of HC function by H2O2, accompanied by increased cell surface expression of Cx43. The impaired GJIC in response to oxidative stress has also been reported in other studies [26, 41], which could be attributed to a reduction of the Cx43 protein available for assembly of functional gap junction channels. While the increased Cx43 expression on cell surface may be caused by either decreased internalization of cell surface Cx43 or increased protein trafficking, resulting in the increased HC activity after H2O2 treatment. The oxidative stress-induced Cx43 migration to the cell surface and HCs opening is inhibited by the depletion of [Ca2+]i with BAPTA-AM, suggesting the [Ca2+]i is critical during this process [42]. Small interfering RNA (siRNA) knockdown of Cx43 renders osteocytes more vulnerable to oxidative stress-induced cell death. This effect is further validated by the Cx43 (E2) antibody, which specifically blocks Cx43 HCs but not gap junctions [43, 44], showing inhibited dye uptake and exacerbation of H2O2-induced osteocytic cell death.
To summarize, whether Cx43 channels play a protective or destructive role in response to oxidative stress is dependent on the type and duration of the insult and may vary among different cell types. In osteocytes, Cx43 HCs contribute to the resistance of oxidative stress; thus, the opening of HCs is beneficial. However, Cx HCs opening can also allow the entry of ROS or release of intracellular GSH, leading to detrimental effects under certain circumstances.
Oxidative Stress and Connexin 43 Channels During Bone Aging
Loss of tissue and organ function has been well documented during the aging process, and oxidative stress contributes to age-related deterioration, including skeletal tissue. With advancing age, both male and female C57BL/6 mice have been shown to exhibit a progressive reduction of cortical bone thickness, trabecular bone volume, bone mineral density, and bone strength [45, 46]. These changes are accompanied by decreased osteoblast and osteoclast numbers, and decreased bone formation rate. In addition, the aged mice show a significant increase of ROS levels, a decrease of GSH reductase activity, and a corresponding increase of the phosphorylation of p53 and p66shc, indicating the activation of signaling transduction pathway leading to apoptosis. Oxidative stress in aged bone tissue may damage mitochondria via the mitochondrial permeability transition pore (MPTP) in bone cells, leading to disruption of tissue homeostasis. Deletion of cyclophilin D (CypD), which is a protein regulator of MPTP, protects mitochondrial function and increases resistance to age-related decline of bone mass and mechanical properties [47]. Kobayashi et al. reports that compared to young mice (12-week-old), the level of cortical bone superoxide is more than twice in 2-year-old mice [48]. Targeted deletion of mitochondrial superoxide dismutase (SOD2) predominantly in osteocytes using dentin matrix protein 1 (DMP1)-cre increases cellular superoxide production and induces remarkable bone loss in an age-dependent manner. SOD2 ablation in osteocytes also results in a disorganized lacunocanalicular network, a decrease in viable osteocytes, and decreased Cx43 expression, indicating the importance of antioxidant proteins in maintaining the osteocytic network.
Osteocytes through their long dendritic processes create an extensively interconnected lacunocanalicular network throughout the mineralized bone matrix. Emerging evidence shows that the extent and connectivity of the lacunocanalicular system degenerates in aged humans as well as in animal models [49–51]. As the major cells responsible for mechanotransduction, osteocytes mediate adaptive responses to mechanical loading by sensing the fluid flow shear stress in the lacunocanalicular system. The impairment of osteocyte connectivity through a combination of dendrite loss and cell death leads to defects in integrating environmental cues, including essential nutrients, survival factors, and mechanical stimuli, and this further increases stress on the remaining osteocytes, forming a vicious cycle [50, 52]. Cx43 gap junctions and HCs are utilized by osteocytes function as key modulators for anabolic and catabolic responses to maintain cell viability. Conditional deletion of Cx43 using DMP1-cre primarily in osteocytes results in increased apoptosis and empty lacunae [14]. Additionally, these mice exhibit an enlarged marrow cavity resulting from enhanced endocortical resorption and compromised mechanical properties, resembling the phenotype of aged mice. Cx43 expression is significantly decreased in osteocytes from old mice as compared to young mice [27]. In vitro studies using the osteocytic MLO-Y4 cell line shows that oxidative stress induces the reduction of Cx43 protein, and this reduction is rescued by the antioxidant NMPG [27]. This observation raises the possibility that the low expression of Cx43 could be due to increased oxidative stress in older animals, considering the direct association of oxidative stress and the aging process. Davis et al. generated a DMP1-8 kb-Cx43/GFP mouse model, which overexpresses Cx43 predominantly in osteocytes, with the percentage of Cx43-positive osteoblasts remaining similar to wild-type littermate controls [53••]. Osteocytic Cx43 overexpression preserves osteocytes viability, increases endocortical bone formation, and decreases resorption in aged mice, along with the increase of circulation marker P1NP and decrease of CTX, leading to improved bone strength. However, the increased osteocytic Cx43 does not prevent age-induced long bone geometrical changes and cancellous bone loss. This could probably be caused by the age of mice (14-month-old) used in this study, which represents a relatively early aging stage in mice, and the time may not be enough for the cellular level changes to translate into bone structural changes. The study of the molecular mechanism underlying Cx43 deficiency and aging-induced osteocyte apoptosis reveals the involvement of miR21 [54]. The reduction of miR21 under both conditions disrupts PTEN/Akt pathway, and the resulting apoptotic osteocytes releases more cytokines RANKL and HMGB1, which induce osteoclast recruitment. Altogether, the above findings point to the contribution of osteocytic Cx43 to the skeletal phenotype of aged mice. The age-related elevation of oxidative stress results in disorganized osteocytic network, osteocyte apoptosis, and decreased Cx43 levels, which is part of the fundamental pathogenic mechanism responsible for the decline of bone mass and strength during aging.
Oxidative Stress and Connexin 43 Channels in Postmenopausal Osteoporosis
Sex steroid deficiency accounts for another critical factor for the development of osteoporosis. Osteoporosis and osteopenia are common among postmenopausal women, which feature a high risk of fragility fracture and microarchitectural deterioration [55]. Like aging, loss of sex steroid promotes an increase of oxidative stress in bone, and a negative correlation between plasma lipid oxidation and bone mineral density values has been reported in osteoporotic postmenopausal women compared with healthy controls [45, 56, 57]. Deficiency of sex steroids compromises the defense against oxidative stress in bone. The levels of the major thiol antioxidants (GSH and thioredoxin) and their reductases fall substantially in rat bone marrow after ovariectomy (OVX), which can be rescued by exogenous 17β-estradiol [58]. The causal link between oxidative stress and OVX-induced bone changes is further confirmed by intraperitoneal injections of the antioxidant N-acetyl cysteine (NAC) and dihydrotestosterone (DHT), which abolish the increase of ROS and OVX-related bone loss, while L-buthionine-(S, R)-sulfoximine (BSO), a specific GSH synthesis inhibitor, causes substantial bone loss [45, 58]. Although oxidative stress is a common feature responsible for the adverse effects of sex steroid deficiency and old age on bone, a recent study indicates that the mechanisms underlying these two processes are distinct [59]. By doing OVX surgery in young (4 months) and old (18 months) mice, the results show that mice remain functional estrogen-sufficient at 19.5-month old, indicating that the decline of bone mass during aging is independent of sex steroid status, which is in agreement with a previous study [45]. Transgenic mouse models expressing the human catalase gene targeted to the mitochondria of myeloid (LysM-Cre) or mesenchymal lineage cells are generated. The attenuation of H2O2 generation by overexpressing catalase in myeloid lineage cells abrogates the loss of cortical bone caused by estrogen deficiency but does not affect age-related cortical or cancellous bone loss.
Estrogen confers regulatory roles on bone remodeling by exerting a pro-apoptotic effect on osteoclasts and an anti-apoptotic effect on osteoblasts and osteocytes [60, 61]. The importance of estrogen on osteogenesis and bone turnover via an antioxidative mechanism has been studied by employing a combined OVX model with bone marrow ablation (BMX) approaches [62]. At 12 and 14 days after BMX, estrogen deficiency increases bone turnover, showing increased osteoblastic and osteoclastic activity. In addition, recent evidence suggests that estrogen regulates the mechanosensitivity of osteocytes [63••, 64]. MLO-Y4 cells are treated with estrogen, estrogen withdrawal from the culture medium, the condition mimicking postmenopausal estrogen deficiency, or with the estrogen receptor inhibitor, fulvestrant. Cells are subjected to oscillatory fluid flow (OFF) after treatment. Osteocytes responded to mechanical loading by a rapid release of NO and PGE2 and elevated intracellular Ca2+ [65–67]. Estrogen treatment enhances OFF-induced intracellular Ca2+ oscillations, NO, and PGE2 release. These effects are diminished in estrogen-withdrawal and fulvestrant-treated groups, along with reduced F-actin fiber formation, and downregulated expression of DMP-1, sclerostin, and other bone-specific genes. Our group has demonstrated that in response to mechanical loading, Cx43 is rapidly redistributed to the cell surface and the opening of Cx43 HCs is increased, which serves as a direct portal for the exit of intracellular PGE2 [18, 68]. The study for the direct regulation of estrogen on Cx43 reveals that 17β-estradiol upregulates Cx43 expression and facilitated gap junction intercellular communication in vitro [64, 69]. Moreover, estrogen withdrawal from culture medium reduces the Cx43 protein level and Cx43 HC function [69••]. Consistently, a significant decrease of osteocytic Cx43 expression is observed in OVX mice compared to sham control. In order to dissect the differential functions of osteocytic Cx43 gap junctions and hemichannels under an estrogen deficiency condition, OVX surgery was performed on two transgenic mouse models driven by the DMP1 promoter that overexpresses Cx43 mutants in osteocytes we previously generated: R76W (dominant-negative mutant blocking only gap junction channels) and Δ130–136 (dominant-negative mutant inhibiting both gap junction channels and HCs). Our study has shown that impairment of Cx43 HCs negatively affects bone remodeling and osteocyte viability [19]. Compared to wild type and R76W groups, Δ130–136 OVX mice have greatly decreased vertebral trabecular bone mass, associated with a significant increase of osteocyte apoptosis, and bone material quality deterioration. Importantly, the ablation of HCs augments the oxidative stress induced by estrogen deficiency, showing increased 4-hydroxynoneanal (4-HNE), a biomarker for lipid peroxidation and SOD2 levels in Δ130–136 OVX mice. In summary, these findings highlight the significance of Cx43 HCs in protecting against catabolic effects due to estrogen deficiency.
Oxidative Stress and Connexin 43 Channels in Glucocorticoid-Induced Bone Loss
Glucocorticoids (GCs), such as dexamethasone (Dex), are widely used in clinical medicine as effective anti-inflammatory and immunosuppressive agents. However, prolonged use of GCs results in bone loss and a high incidence of fracture risk, known as GC-induced osteoporosis (GIO), the most common form of secondary osteoporosis [70]. In addition to exogenous GCs excess, the production of endogenous GCs, as well as sensitivity to GCs, increases with advancing age [71]. GCs act directly on bone cells, which suppress osteoblastogenesis, stimulate osteoblast and osteocyte apoptosis, and prolong the lifespan of osteoclasts [61]. GCs have been reported to induce ROS generation, activating the PKCβ/p66shc/JNK signaling cascade, leading to apoptosis. In response to ROS, the activity of forkhead box O (FoxO) transcription factors is also increased, along with suppressed Akt phosphorylation and attenuated osteoblastogenesis [72]. Recent studies confirm that the oxidative stress can be induced by GC treatment, either via boosting ROS generation or suppressing the antioxidant systems. Dex induces a rapid onset and time-dependent superoxide overproduction in mouse osteoblastic MC3T3-E1 cells in vitro, and decreased GSH levels and SOD activity significantly in femur tissues of treated rats [73, 74]. The natural product propolis from plant and honeybees with antioxidant activities counteract Dex-induced oxidative stress and cytotoxicity. Moreover, our previous novel findings suggest that GC treatment induces the development of autophagy in osteocytes [75, 76]. Interestingly, our recent study reveals that the autophagy level are decreased in H2O2-treated MLO-Y4 cells and osteocytes of superoxide dismutase 1 (SOD1)-deficient mice [77••]. Moderated levels of GCs lead to autophagy, which pre-conditions the osteocytes and conveys a novel cell-protective function against oxidative stress-induced cell death.
Lane et al. have reported the direct effects of GCs on modifying the microenvironment of osteocytes, including an enlargement of the lacunar space and loss of perilacunar mineralized matrix [78]. Furthermore, GCs have been indicated to cause the detachment of osteocytic processes from the lacunar-canalicular walls through the disengagement of integrin from extracellular matrix proteins [79]. In line with previous studies, Gao et al. found that Dex administration induced a robust cytoskeletal rearrangement, coupled with a shortening of the dendritic processes and reduced expression of Cx43 via autophagy-mediated degradation [80••]. Upon the induction of autophagy, Cx43 is internalized into autophagosome/autolysosomes. Lysosomal inhibition using chloroquine, Atg5 knockdown, or activation of Akt-mTOR signaling attenuated the degradation of Cx43. Furthermore, a decreased level of Cx43 RNA and protein by Dex has been described in osteoblastic MC3T3-E1 cells, involving the Akt/mTOR pathway [81]. Dex-induced downregulation of Cx43 and gap junction intercellular communication is further confirmed in human primary osteoblasts. The reduction of Cx43 expression is abolished by the pretreatment with a glucocorticoid receptor blocker, and overexpression of Cx43 reverses the Dex-inhibited osteoblast viability and proliferation, indicating a protective role of Cx43 in the effect of GCs on osteoblastic cells. In addition, previous in vitro study has shown that Cx43 HCs are essential for the transduction of the anti-apoptotic signal of bisphosphonates in response to GCs-induced apoptosis in osteoblasts and osteocytes through the activation of Src/ERKs signal transduction pathway [82]. Interestingly, an in vivo mouse model using osteocalcin promoter-driven Cx43 deletion in mature osteoblasts and osteocytes exhibits no differences with regard to GCs-induced apoptosis and bone loss, compared to littermate controls [13]. The involvement of Cx43 HC function with Dex treatment has recently been shown in a skeletal muscle atrophy model [83]. Activation of Cx43 HCs, increased oxidative stress, and mitochondrial dysfunction are shown upon Dex administration. Antioxidants inhibits Cx HCs and reduces the undesired effects of GCs on skeletal muscles, indicating a deleterious effect of Cx43 on GCs-treated skeletal muscle. However, the mechanism remains elusive for the deleterious effect of Cx43 HCs on GCs-treated skeletal muscle, which is contray to the lack of effect of osteoblastic Cx43 deletion on GCs-induced bone loss. These important questions warrant further investigation.
Conclusions
Emerging evidence has suggested a close relationship between oxidative stress and osteoporotic status. As an important contributor to the pathogenesis of bone loss related to aging, estrogen deficiency, and GC excess, oxidative stress impairs the balance of bone remodeling regulated by osteoblast-mediated bone formation and osteoclast-mediated bone resorption. Importantly, increased oxidative stress levels or lack of antioxidative defenses contribute to osteocyte apoptosis. Osteocytes are key regulators of skeletal homeostasis, orchestrating osteoblasts and osteoclasts via communication through Cx43 gap junction channels and HCs. Cx43 channels play critical roles in bone growth, remodeling, mechanotransduction, and survival of osteocytes. Under elevated oxidative stress conditions, including osteoporosis caused by aging, postmenopausal, or GC treatment, there is a consistent decrease in Cx43 expression, accompanied by reduced gap junction intercellular communication; in contrast, Cx43 HC activation confers a protective role against oxidative stress (Fig. 1). Future studies of cellular mechanisms regarding the modulations of oxidative stress and Cx43 channel function will provide a better understanding of the pathogenesis of osteoporosis and bone loss and will aid in the development of potential new strategies or therapies in treating bone diseases.
Fig. 1.
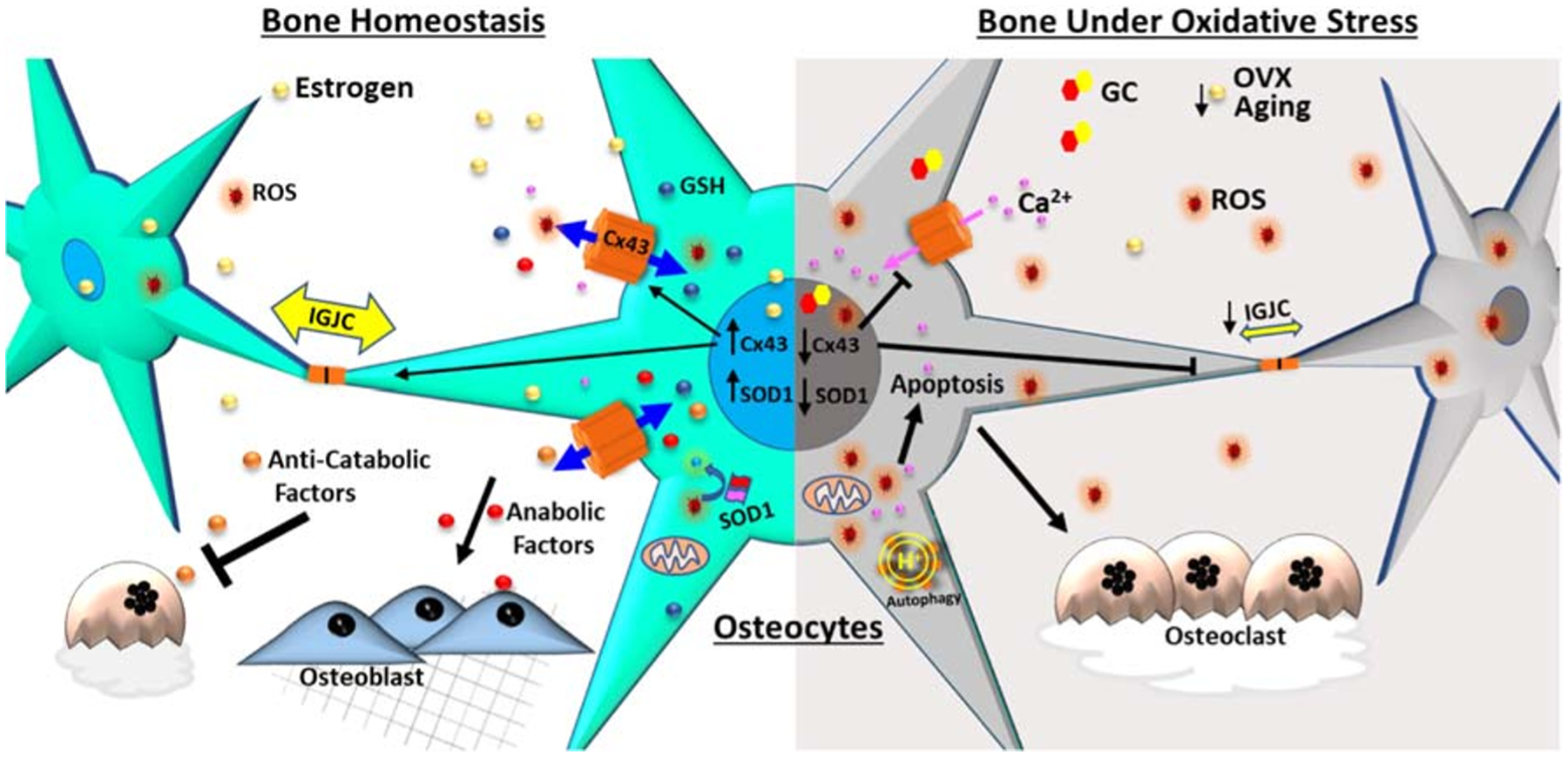
The schematic diagram shows the regulation of connexin 43 (Cx43) channels under oxidative stress in skeletal tissue. Osteocytes are a key regulator of skeletal homeostasis, orchestrating osteoblasts and osteoclasts via communication through Cx43 gap junction channels and hemichannels (HCs). Cx43 channels play critical roles in bone remodeling, mechanotransduction, and survival of osteocytes. Oxidative stress decreases osteocyte viability and impairs the balance of anabolic and catabolic responses. Under oxidative stress (increased ROS level) or lack of antioxidative defense (decreased GSH) conditions, including osteoporosis caused by aging, loss of sex hormones/ovariectomy (OVX), or glucocorticoids (GCs) treatment, Cx43 expression is consistently decreased, accompanied by reduced intercellular gap junction communication (IGJC), while the opening of Cx43 HCs protects osteocytes against cell injury caused by oxidative stress
Acknowledgments
We thank Dr. Eduardo Cardenas at UTHSCSA for critical reading and editing of the paper.
Funding
This work was supported by the National Institutes of Health (NIH) (grant: AR072072) and Welch Foundation (grant: AQ-1507) (to J.X.J.).
Footnotes
Conflict of Interest Rui Hua, Jingruo Zhang, Manuel A. Riquelme, and Jean X. Jiang declare no conflict of interest.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Robling AG, Bonewald LF. The osteocyte: new insights. Annu Rev Physiol. 2020;82:485–506. 10.1146/annurev-physiol-021119-034332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int. 2014;94(1):25–34. 10.1007/s00223-013-9774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altindag O, Erel O, Soran N, Celik H, Selek S. Total oxidative/anti-oxidative status and relation to bone mineral density in osteoporosis. Rheumatol Int. 2008;28(4):317–21. 10.1007/s00296-007-0452-0. [DOI] [PubMed] [Google Scholar]
- 4.Domazetovic V, Marcucci G, Iantomasi T, Brandi ML, Vincenzini MT. Oxidative stress in bone remodeling: role of antioxidants. Clinical cases in mineral and bone metabolism : the official journal of the Italian Society of Osteoporosis, Mineral Metabolism, and Skeletal Diseases. 2017;14(2):209–16. doi: 10.11138/ccmbm/2017.14.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wauquier F, Leotoing L, Coxam V, Guicheux J, Wittrant Y. Oxidative stress in bone remodelling and disease. Trends Mol Med. 2009;15(10):468–77. 10.1016/j.molmed.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Nita M, Grzybowski A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxidative Med Cell Longev. 2016;2016: 3164734–23. 10.1155/2016/3164734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Current biology : CB. 2014;24(10):R453–62. 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163(3):560–9. 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–85. 10.1016/j.redox.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batra N, Kar R, Jiang JX. Gap junctions and hemichannels in signal transmission, function and development of bone. Biochim Biophys Acta. 2012;1818(8):1909–18. 10.1016/j.bbamem.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stains JP, Civitelli R. Connexins in the skeleton. Semin Cell Dev Biol. 2016;50:31–9. 10.1016/j.semcdb.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lecanda F, Warlow PM, Sheikh S, Furlan F, Steinberg TH, Civitelli R. Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J Cell Biol. 2000;151(4): 931–44. 10.1083/jcb.151.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plotkin LI, Lezcano V, Thostenson J, Weinstein RS, Manolagas SC, Bellido T. Connexin 43 is required for the anti-apoptotic effect of bisphosphonates on osteocytes and osteoblasts in vivo. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2008;23(11):1712–21. 10.1359/jbmr.080617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bivi N, Condon KW, Allen MR, Farlow N, Passeri G, Brun LR, et al. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2012;27(2):374–89. 10.1002/jbmr.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watkins M, Grimston SK, Norris JY, Guillotin B, Shaw A, Beniash E, et al. Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol Biol Cell. 2011;22(8):1240–51. 10.1091/mbc.E10-07-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodenough DA, Paul DL. Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol. 2003;4(4):285–94. 10.1038/nrm1072. [DOI] [PubMed] [Google Scholar]
- 17.Plotkin LI. Connexin 43 hemichannels and intracellular signaling in bone cells. Front Physiol. 2014;5:131. 10.3389/fphys.2014.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cherian PP, Siller-Jackson AJ, Gu S, Wang X, Bonewald LF, Sprague E, et al. Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Mol Biol Cell. 2005;16(7):3100–6. 10.1091/mbc.E04-10-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu H, Gu S, Riquelme MA, Burra S, Callaway D, Cheng H, et al. Connexin 43 channels are essential for normal bone structure and osteocyte viability. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2015;30(3):436–48. 10.1002/jbmr.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65: 475–502. 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- 21.Saez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV. Connexin-based gap junction hemichannels: gating mechanisms. Biochim Biophys Acta. 2005;1711(2):215–24. 10.1016/j.bbamem.2005.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orellana JA, Diaz E, Schalper KA, Vargas AA, Bennett MV, Saez JC. Cation permeation through connexin 43 hemichannels is cooperative, competitive and saturable with parameters depending on the permeant species. Biochem Biophys Res Commun. 2011;409(4):603–9. 10.1016/j.bbrc.2011.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36(7):1171–86. 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711(2):172–82. 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Giardina SF, Mikami M, Goubaeva F, Yang J. Connexin 43 confers resistance to hydrogen peroxide-mediated apoptosis. Biochem Biophys Res Commun. 2007;362(3):747–52. 10.1016/j.bbrc.2007.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutnik CM, Pocrnich CE, Liu H, Laird DW, Shao Q. The protective effect of functional connexin43 channels on a human epithelial cell line exposed to oxidative stress. Invest Ophthalmol Vis Sci. 2008;49(2):800–6. 10.1167/iovs.07-0717. [DOI] [PubMed] [Google Scholar]
- 27.Kar R, Riquelme MA, Werner S, Jiang JX. Connexin 43 channels protect osteocytes against oxidative stress-induced cell death. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2013;28(7): 1611–21. 10.1002/jbmr.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramachandran S, Xie LH, John SA, Subramaniam S, Lal R. A novel role for connexin hemichannel in oxidative stress and smoking-induced cell injury. PLoS One. 2007;2(8):e712. 10.1371/journal.pone.0000712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma JW, Ji DD, Li QQ, Zhang T, Luo L. Inhibition of connexin 43 attenuates oxidative stress and apoptosis in human umbilical vein endothelial cells. BMC Pulm Med. 2020;20(1):19. 10.1186/s12890-019-1036-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi W, Riquelme MA, Gu S, Jiang JX. Connexin hemichannels mediate glutathione transport and protect lens fiber cells from oxidative stress. Journal of cell science. 2018;131(6). doi: 10.1242/jcs.212506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang X, Huang T, Zhu Y, Yan Q, Chi Y, Jiang JX, et al. Connexin43 hemichannels contribute to cadmium-induced oxidative stress and cell injury. Antioxid Redox Signal. 2011;14(12): 2427–39. 10.1089/ars.2010.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahmad Waza A, Ahmad Bhat S, Ul Hussain M, Ganai BA. Connexin 43 and ATP-sensitive potassium channels crosstalk: a missing link in hypoxia/ischemia stress. Cell Tissue Res. 2018;371(2):213–22. 10.1007/s00441-017-2736-3. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Sinovas A, Boengler K, Cabestrero A, Gres P, Morente M, Ruiz-Meana M, et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ Res. 2006;99(1):93–101. 10.1161/01.RES.0000230315.56904.de. [DOI] [PubMed] [Google Scholar]
- 34.Boengler K, Konietzka I, Buechert A, Heinen Y, Garcia-Dorado D, Heusch G, et al. Loss of ischemic preconditioning’s cardioprotection in aged mouse hearts is associated with reduced gap junctional and mitochondrial levels of connexin 43. Am J Physiol Heart Circ Physiol. 2007;292(4):H1764–9. 10.1152/ajpheart.01071.2006. [DOI] [PubMed] [Google Scholar]
- 35.Tu RH, Li QJ, Huang Z, He Y, Meng JJ, Zheng HL, et al. Novel functional role of heat shock protein 90 in mitochondrial connexin 43-mediated hypoxic postconditioning. Cell Physiol Biochem. 2017;44(3):982–97. 10.1159/000485399. [DOI] [PubMed] [Google Scholar]
- 36.Ruiz-Meana M, Nunez E, Miro-Casas E, Martinez-Acedo P, Barba I, Rodriguez-Sinovas A, et al. Ischemic preconditioning protects cardiomyocyte mitochondria through mechanisms independent of cytosol. J Mol Cell Cardiol. 2014;68:79–88. 10.1016/j.yjmcc.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Waza AA, Andrabi K, Hussain MU. Protein kinase C (PKC) mediated interaction between conexin43 (Cx43) and K+ (ATP) channel subunit (Kir6. 1) in cardiomyocyte mitochondria: implications in cytoprotection against hypoxia induced cell apoptosis. Cell Signal. 2014;26(9):1909–17. [DOI] [PubMed] [Google Scholar]
- 38.Le HT, Sin WC, Lozinsky S, Bechberger J, Vega JL, Guo XQ, et al. Gap junction intercellular communication mediated by connexin43 in astrocytes is essential for their resistance to oxidative stress. J Biol Chem. 2014;289(3):1345–54. 10.1074/jbc.M113.508390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orellana JA, Hernandez DE, Ezan P, Velarde V, Bennett MV, Giaume C, et al. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia. 2010;58(3):329–43. 10.1002/glia.20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chi Y, Zhang X, Zhang Z, Mitsui T, Kamiyama M, Takeda M, et al. Connexin43 hemichannels contributes to the disassembly of cell junctions through modulation of intracellular oxidative status. Redox Biol. 2016;9:198–209. 10.1016/j.redox.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, et al. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest. 2010;120(1):266–79. 10.1172/JCI39740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riquelme MA, Jiang JX. Elevated intracellular Ca(2+) signals by oxidative stress activate connexin 43 hemichannels in osteocytes. Bone research. 2013;1(4):355–61. 10.4248/BR201304006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riquelme MA, Kar R, Gu S, Jiang JX. Antibodies targeting extracellular domain of connexins for studies of hemichannels. Neuropharmacology. 2013;75:525–32. 10.1016/j.neuropharm.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siller-Jackson AJ, Burra S, Gu S, Xia X, Bonewald LF, Sprague E, et al. Adaptation of connexin 43-hemichannel prostaglandin release to mechanical loading. J Biol Chem. 2008;283(39):26374–82. 10.1074/jbc.M803136200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282(37):27285–97. 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piemontese M, Almeida M, Robling AG, Kim HN, Xiong J, Thostenson JD, et al. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI insight. 2017;2(17). doi: 10.1172/jci.insight.93771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shum LC, White NS, Nadtochiy SM, Bentley KL, Brookes PS, Jonason JH, et al. Cyclophilin D Knock-out mice show enhanced resistance to osteoporosis and to metabolic changes observed in aging bone. PLoS One. 2016;11(5):e0155709. 10.1371/journal.pone.0155709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi K, Nojiri H, Saita Y, Morikawa D, Ozawa Y, Watanabe K, et al. Mitochondrial superoxide in osteocytes perturbs canalicular networks in the setting of age-related osteoporosis. Sci Rep. 2015;5:9148. 10.1038/srep09148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milovanovic P, Zimmermann EA, Hahn M, Djonic D, Puschel K, Djuric M, et al. Osteocytic canalicular networks: morphological implications for altered mechanosensitivity. ACS Nano. 2013;7(9):7542–51. 10.1021/nn401360u. [DOI] [PubMed] [Google Scholar]
- 50.Tiede-Lewis LM, Dallas SL. Changes in the osteocyte lacunocanalicular network with aging. Bone. 2019;122:101–13. 10.1016/j.bone.2019.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tiede-Lewis LM, Xie Y, Hulbert MA, Campos R, Dallas MR, Dusevich V, et al. Degeneration of the osteocyte network in the C57BL/6 mouse model of aging. Aging. 2017;9(10):2190–208. doi: 10.18632/aging.101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riquelme MA, Cardenas ER, Xu H, Jiang JX. The role of connexin channels in the response of mechanical loading and unloading of bone. International journal of molecular sciences. 2020;21(3). doi: 10.3390/ijms21031146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.••.Davis HM, Aref MW, Aguilar-Perez A, Pacheco-Costa R, Allen K, Valdez S, et al. Cx43 overexpression in osteocytes prevents osteocyte apoptosis and preserves cortical bone quality in aging mice. JBMR plus. 2018;2(4):206–16. doi: 10.1002/jbm4.10035. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study indicates that overexpression of Cx43 primarily in osteocytes using DMP1-cre preserves osteocyte viability and cortical bone phenotype, and prevents changes of bone remodeling and circulating markers during aging process.
- 54.Davis HM, Pacheco-Costa R, Atkinson EG, Brun LR, Gortazar AR, Harris J, et al. Disruption of the Cx43/miR21 pathway leads to osteocyte apoptosis and increased osteoclastogenesis with aging. Aging Cell. 2017;16(3):551–63. 10.1111/acel.12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Black DM, Rosen CJ. Clinical practice. Postmenopausal osteoporosis. N Engl J Med. 2016;374(3):254–62. 10.1056/NEJMcp1513724. [DOI] [PubMed] [Google Scholar]
- 56.Sendur OF, Turan Y, Tastaban E, Serter M. Antioxidant status in patients with osteoporosis: a controlled study. Joint bone spine. 2009;76(5):514–8. 10.1016/j.jbspin.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 57.Muthusami S, Ramachandran I, Muthusamy B, Vasudevan G, Prabhu V, Subramaniam V, et al. Ovariectomy induces oxidative stress and impairs bone antioxidant system in adult rats. Clinica chimica acta; international journal of clinical chemistry. 2005;360(1–2):81–6. doi: 10.1016/j.cccn.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 58.Lean JM, Davies JT, Fuller K, Jagger CJ, Kirstein B, Partington GA, et al. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest. 2003;112(6):915–23. 10.1172/JCI18859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ucer S, Iyer S, Kim HN, Han L, Rutlen C, Allison K, et al. The effects of aging and sex steroid deficiency on the murine skeleton are independent and mechanistically distinct. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2017;32(3):560–74. 10.1002/jbmr.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mann V, Huber C, Kogianni G, Collins F, Noble B. The antioxidant effect of estrogen and selective estrogen receptor modulators in the inhibition of osteocyte apoptosis in vitro. Bone. 2007;40(3):674–84. 10.1016/j.bone.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 61.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31(3):266–300. 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi C, Wu J, Yan Q, Wang R, Miao D. Bone marrow ablation demonstrates that estrogen plays an important role in osteogenesis and bone turnover via an antioxidative mechanism. Bone. 2015;79: 94–104. 10.1016/j.bone.2015.05.034. [DOI] [PubMed] [Google Scholar]
- 63.••.Deepak V, Kayastha P, McNamara LM. Estrogen deficiency attenuates fluid flow-induced [Ca(2+)]i oscillations and mechanoresponsiveness of MLO-Y4 osteocytes. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2017;31(7):3027–39. doi: 10.1096/fj.201601280R. [DOI] [PubMed] [Google Scholar]; This study shows that estrogen regulates the mechanosensitivity of osteocytes. Estrogen withdrawl or inhibition of estrogen receptor blunts intracellular Ca2+ oscillations, NO, and PGE2 release induced by oscillatory fluid flow in MLO-Y4 cells.
- 64.Ren J, Wang XH, Wang GC, Wu JH. 17beta estradiol regulation of connexin 43-based gap junction and mechanosensitivity through classical estrogen receptor pathway in osteocyte-like MLO-Y4 cells. Bone. 2013;53(2):587–96. 10.1016/j.bone.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 65.Cheng B, Kato Y, Zhao S, Luo J, Sprague E, Bonewald LF, et al. PGE(2) is essential for gap junction-mediated intercellular communication between osteocyte-like MLO-Y4 cells in response to mechanical strain. Endocrinology. 2001;142(8):3464–73. 10.1210/endo.142.8.8338. [DOI] [PubMed] [Google Scholar]
- 66.Lu XL, Huo B, Chiang V, Guo XE. Osteocytic network is more responsive in calcium signaling than osteoblastic network under fluid flow. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2012;27(3):563–74. 10.1002/jbmr.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McGarry JG, Klein-Nulend J, Prendergast PJ. The effect of cytoskeletal disruption on pulsatile fluid flow-induced nitric oxide and prostaglandin E2 release in osteocytes and osteoblasts. Biochem Biophys Res Commun. 2005;330(1):341–8. 10.1016/j.bbrc.2005.02.175. [DOI] [PubMed] [Google Scholar]
- 68.Cheng B, Zhao S, Luo J, Sprague E, Bonewald LF, Jiang JX. Expression of functional gap junctions and regulation by fluid flow in osteocyte-like MLO-Y4 cells. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2001;16(2):249–59. 10.1359/jbmr.2001.16.2.249. [DOI] [PubMed] [Google Scholar]
- 69.••.Ma L, Hua R, Tian Y, Cheng H, Fajardo RJ, Pearson JJ, et al. Connexin 43 hemichannels protect bone loss during estrogen deficiency. Bone research. 2019;7:11. 10.1038/s41413-019-0050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates the decreased connexin 43 expression and hemichannels activity under estrogen withdrawal condition in MLO-Y4 cells and the protective role of osteocytic connexin 43 hemichannels against ovariectomy induced bone loss and oxidative stress.
- 70.Compston J. Glucocorticoid-induced osteoporosis: an update. Endocrine. 2018;61(1):7–16. doi: 10.1007/s12020-018-1588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O’Brien CA, et al. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in aged mice. Aging Cell. 2010;9(2):147–61. 10.1111/j.1474-9726.2009.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Almeida M, Han L, Ambrogini E, Weinstein RS, Manolagas SC. Glucocorticoids and tumor necrosis factor alpha increase oxidative stress and suppress Wnt protein signaling in osteoblasts. J Biol Chem. 2011;286(52):44326–35. 10.1074/jbc.M111.283481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tolba MF, El-Serafi AT, Omar HA. Caffeic acid phenethyl ester protects against glucocorticoid-induced osteoporosis in vivo: impact on oxidative stress and RANKL/OPG signals. Toxicol Appl Pharmacol. 2017;324:26–35. 10.1016/j.taap.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 74.Lin H, Gao X, Chen G, Sun J, Chu J, Jing K, et al. Indole-3-carbinol as inhibitors of glucocorticoid-induced apoptosis in osteoblastic cells through blocking ROS-mediated Nrf2 pathway. Biochem Biophys Res Commun. 2015;460(2):422–7. 10.1016/j.bbrc.2015.03.049. [DOI] [PubMed] [Google Scholar]
- 75.Xia X, Kar R, Gluhak-Heinrich J, Yao W, Lane NE, Bonewald LF, et al. Glucocorticoid-induced autophagy in osteocytes. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2010;25(11):2479–88. 10.1002/jbmr.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yao W, Dai W, Jiang JX, Lane NE. Glucocorticoids and osteocyte autophagy. Bone. 2013;54(2):279–84. 10.1016/j.bone.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.••.Kar R, Riquelme MA, Hua R, Jiang JX. Glucocorticoid-induced autophagy protects osteocytes against oxidative stress through activation of MAPK/ERK signaling. JBMR plus. 2019;3(4):e10077. doi: 10.1002/jbm4.10077. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals a novel mechanism showing that moderated levels of glucocorticoids leads to autophagy, which pre-conditions the osteocytes and conveys cell-protective function against oxidative stress induced cell death.
- 78.Lane NE, Yao W, Balooch M, Nalla RK, Balooch G, Habelitz S, et al. Glucocorticoid-treated mice have localized changes in trabecular bone material properties and osteocyte lacunar size that are not observed in placebo-treated or estrogen-deficient mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2006;21(3):466–76. 10.1359/JBMR.051103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Plotkin LI, Manolagas SC, Bellido T. Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival. Evidence for inside-out signaling leading to anoikis. J Biol Chem. 2007;282(33):24120–30. 10.1074/jbc.M611435200. [DOI] [PubMed] [Google Scholar]
- 80.••.Gao J, Cheng TS, Qin A, Pavlos NJ, Wang T, Song K, et al. Glucocorticoid impairs cell-cell communication by autophagy-mediated degradation of connexin 43 in osteocytes. Oncotarget. 2016;7(19):26966–78. doi: 10.18632/oncotarget.9034. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study suggests that glucocorticoids treatment reduces osteocytic dendritic processes and connexin 43 expression. The decrease of Cx43 is caused by protein internalization by autophagosome/autolysosomes.
- 81.Shen C, Kim MR, Noh JM, Kim SJ, Ka SO, Kim JH, et al. Glucocorticoid suppresses connexin 43 expression by inhibiting the Akt/mTOR signaling pathway in osteoblasts. Calcif Tissue Int. 2016;99(1):88–97. 10.1007/s00223-016-0121-y. [DOI] [PubMed] [Google Scholar]
- 82.Plotkin LI, Manolagas SC, Bellido T. Transduction of cell survival signals by connexin-43 hemichannels. J Biol Chem. 2002;277(10): 8648–57. 10.1074/jbc.M108625200. [DOI] [PubMed] [Google Scholar]
- 83.Balboa E, Saavedra F, Cea LA, Ramirez V, Escamilla R, Vargas AA, et al. Vitamin E blocks connexin hemichannels and prevents deleterious effects of glucocorticoid treatment on skeletal muscles. International journal of molecular sciences. 2020;21(11). doi: 10.3390/ijms21114094. [DOI] [PMC free article] [PubMed] [Google Scholar]