

Abstract
Genome editing technologies, such as CRISPR/Cas9, are promising for treating otherwise incurable genetic diseases. Great progress has been made for ex vivo genome editing; however, major bottlenecks exist in the development of efficient, safe, and targetable in vivo delivery systems, which are needed for the treatment of many diseases. To achieve high efficacy and safety in therapeutic in vivo genome editing, editing activities must be controlled spatially and temporally in the body, which requires novel materials, delivery strategies, and control mechanisms. Thus, there is currently a tremendous opportunity for the biomaterials research community to develop in vivo delivery systems that overcome the problems of low editing efficiency, off-targeting effect, safety, and cell and tissue specificity. In this Review, we summarize delivery approaches and provide perspectives on the challenges and possible solutions, aiming to stimulate further development of engineered materials for in vivo delivery of genome-editing machinery.
Web/TOC summary
In vivo genome editing requires delivery systems that are efficiency, safe, and have tissue specificity. This Review outlines the materials and delivery strategies currently used, and the challenges and potential solutions in in vivo genome editing, aiming to stimulate further development of engineered materials for in vivo delivery of genome-editing machinery.
Introduction
Over the past few decades, the emergence of programmable nucleases has revolutionized the field of genome editing. Programmable nucleases allow for specific and permanent modifications of DNA sequences of choice within a genome. Most of them function by creating a DNA double-strand break (DSB) at the intended target loci in a cell, which is subsequently repaired by non-homologous end joining (NHEJ) or homology-directed repair (HDR) pathways (Box 1)1. Early programmable nuclease platforms include zinc finger nucleases (ZFNs)2,3 and transcription activator-like effector nucleases (TALENs)4 — engineered proteins that are generated through the fusion of a DNA binding domain (zinc finger or Tal effector) with the non-specific FokI nuclease domain1. When a ZFN and TALEN pair binds at two half-sites of the target sequence with the correct orientation and spacing, the FokI domains dimerize, resulting in a DSB. However, the utility of ZFNs and TALENs is restricted by the need to design a new pair of nuclease proteins for each new target site, and difficulties in achieving a high cutting efficiency and multiplexing1,5.
Box 1. CRISPR/Cas9 machinery and genome editing mechanisms.
The CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR associate-protein 9) requires three elements to function: a CRISPR RNA (crRNA), trans-activating CRISPR RNA (tracrRNA), and a Cas9 nuclease protein. The crRNA is homologous to the target sequence and interacts with the auxiliary tracrRNA to form a complex that guides the Cas9 protein to bind to the target DNA sequence1. To simplify the CRISPR system for gene editing, a synthetic single guide RNA (gRNA) is generally engineered by combining the crRNA and tracrRNA into a single RNA transcript109,110, which hybridizes to the target DNA strand (target strand). The gRNA typically comprises a 5’ 17–20 nucleotide sequence complementary to the target DNA sequence and a 3’ sequence that serves as a binding scaffold for Cas9. A protospacer adjacent motif (PAM) of 2–5 nucleotides on the target DNA is required for Cas9 binding and is located directly downstream of the target sequence on the non-target DNA strand. The most-commonly used system SpCas9 recognizes a short 5′-NGG (where N represents any nucleotide and G represents guanine) PAM sequence on the non-target strand and cleaves the DNA target sequence three bases upstream from the PAM.
The Cas9 protein consists of HNH nuclease domain and RuvC nuclease domain. The HNH domain cleaves the DNA strand complementary to the gRNA (target strand) and the RuvC domain cleaves the other strand (non-target strand), forming a double-stand break (DSB). Cas9 is non-specific endonuclease guided by the gRNA to the target site and cleaves the DNA sequence it binds, giving rise to a DSB, which activates DNA repair mechanism(s) in the cell, including the non-homologous end joining (NHEJ) and homology-directed repair (HDR) pathways. The NHEJ pathway may result in mutational insertions and deletions (indels) of short sequences at the repair site, causing disruption of the target gene. Expressing multiple gRNAs targeting different sequences in the same chromosome allows for the precise deletion of large DNA segments. By providing a DNA donor template with sequence homology to the target site, the HDR pathway enables targeted donor insertion at the Cas9 cut site for specific gene correction or addition.
DNA donor templates can be single-stranded oligodeoxyribonucleotide (ssODN)111 or double-stranded DNA (dsDNA)112, which are formed by the desired DNA sequence flanked by sequences homologous to the target site (homology arms). The optimal length of the homology arms and the choice of donor template (ssODN or dsDNA) may depend on the size of precise gene modification being made33. For example, ssODNs with 30–60 bp homology arms may be used to insert or repair small sequences of up to 200 bp113,114. Long dsDNA donors with homology arms of 400–1,000 bp may be used to introduce sequences up to several thousand base pairs at the target site to correct point mutations, small indel mutations, or drive the integration of an entire complementary DNA encoding a gene33,115. The need to deliver a DNA donor template may present an additional challenge for the design of delivery vehicles, especially when long dsDNA donors are used.
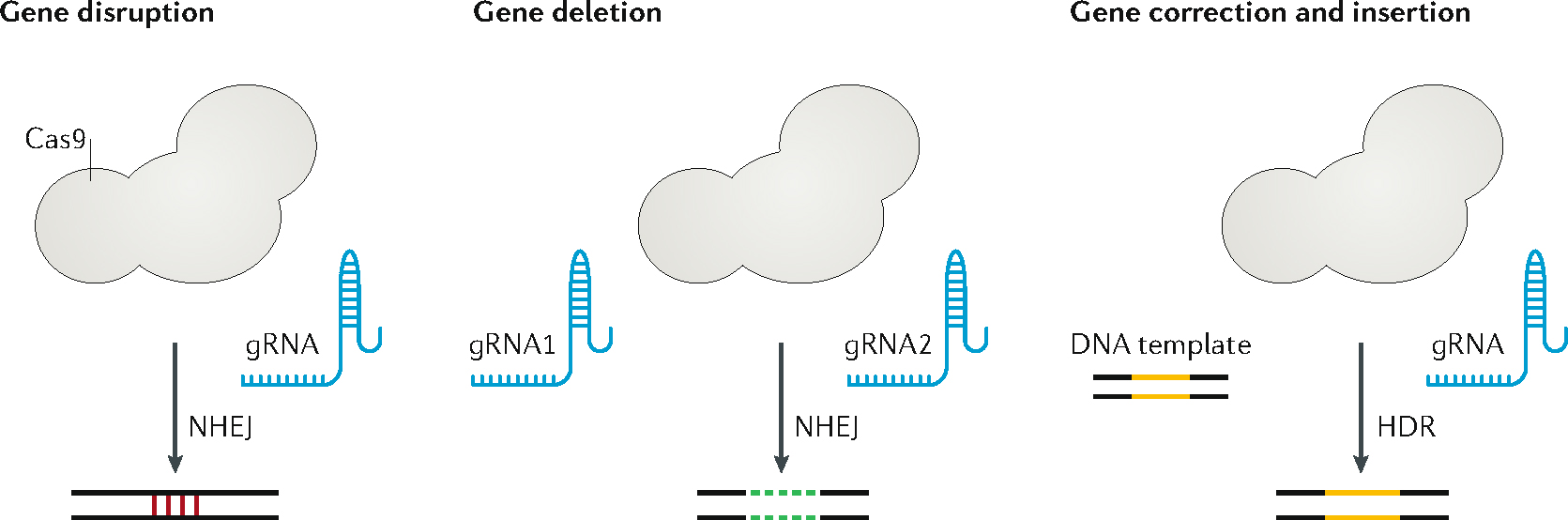
More recently, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) has quickly become the most popular gene editing tool owing to its ease of engineering, versatility, and flexibility6,7. CRISPR was first identified as an adaptive antiviral immune system in bacteria and archaea8–10. Unlike ZFNs and TALENs, the CRISPR/Cas9 system relies on RNA-guided nuclease activity in which target specificity is realized through RNA–DNA Watson–Crick base pairing6,7 and the PAM (protospacer adjacent motif) sequence (Box 2). To date the most widely used system is the wide-type (type II) CRISPR system in Streptococcus pyogenes (SpCas9), which recognizes a short 5′-NGG PAM (where N represents any nucleotide and G represents guanine). CRISPR/Cas9 systems edit the genome using the same Cas9 protein for all target sequences, whereby Cas9 is guided by a single guide RNA (gRNA) via base-pairing to the target sequence. CRISPR/Cas9 systems greatly facilitate genome engineering, for example, for genetic modification of bacteria, plants, and animals; understanding and regulating gene functions; establishing human disease models for basic study and drug discovery; and targeted therapeutic intervention11. In particular, CRISPR/Cas9 can correct or disrupt disease-causing genes, providing potential cures for human genetic diseases11,12.
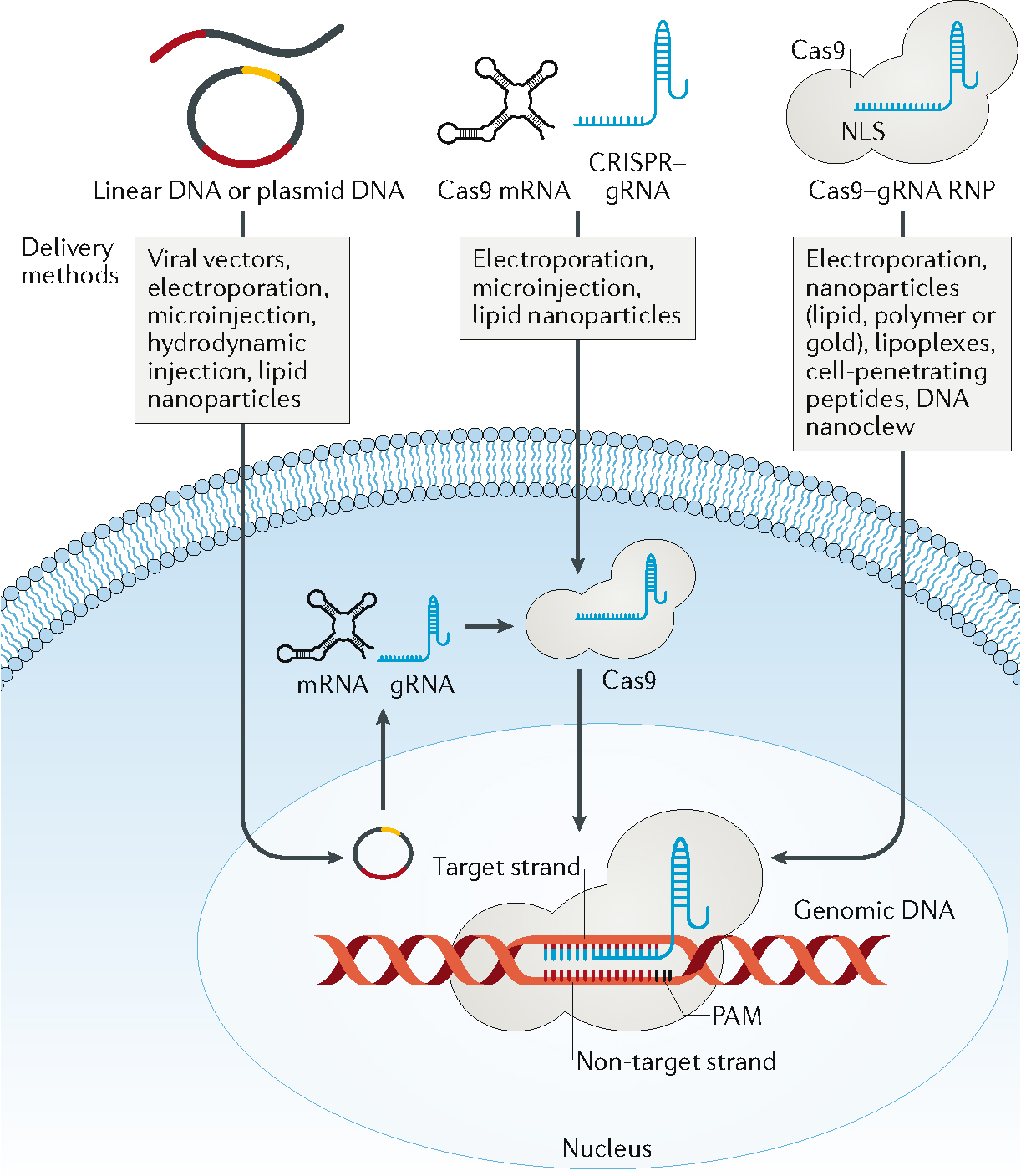
Box 2. Forms of CRISPR/Cas9 cargo and methods for delivery.
The CRISPR/Cas9 system can be delivered as plasmid or linear DNA encoding Cas9 and gRNA; Cas9 mRNA and a separate gRNA; or as Cas9/gRNA ribonucleoprotein (RNP). All forms typically provide one or more nuclear localization signals to promote Cas9 entry into the cell nucleus. When delivered as DNA, the cargo must first enter the cell nucleus and undergo transcription and translation. The resulting Cas9/gRNA complex in the cytoplasm enters the nucleus for gene editing. Plasmid DNA is stable, easy to prepare and results in prolonged transgene expression in cells. In vivo delivery of CRISPR/Cas9 as single-strand or double-strand DNA shares common features with conventional gene delivery; thus, early in vivo genome editing approaches used the gene-transduction mechanisms of viral vectors to deliver genes and induce transgene expression via self-amplification, stable extrachromosomal expression, or even integration into the host genome19,47,58. However, viral vector-based in vivo delivery may lead to unintended mutagenesis, prolonged expression of CRISPR/Cas9, and editing in off-target tissues. High immunogenicity and cost of production are additional challenges for clinical application of viral vectors for genome editing58. As an alternative, nonviral delivery approaches have been developed for in vivo genome editing using organic and inorganic materials, including lipids, peptides, naturally occurring and synthetic polymers, and nanocystals84. However, the editing efficiencies are typically low because of the biological barriers to in vivo transport of delivery vehicles86–89,92,94. When delivered as Cas9 mRNA, the system must enter into the cytoplasm, where it is translated to Cas9 protein, which then forms a complex with gRNA and the resulting RNP translocates into the nucleus. Delivery of Cas9 mRNA co-delivered into cells with gRNA leads to Cas9/gRNA RNP, resulting in faster editing kinetics compared with DNA delivery, which may help reduce off-target effects116. Cas9 mRNA may be delivered into cells in vitro by physical116, chemical117 and viral-based118 methods, and in vivo using nonviral approaches94. The direct delivery of Cas9 protein and gRNA complexes (Cas9/gRNA RNP) into cells using nonviral delivery methods (such as electroporation and nanoparticles) can also result in higher editing efficiency and lower off-target effects than DNA delivery because of faster editing kinetics and the short lifespan of Cas9 protein116.
Although SpCas9 could give rise to >90% indel rates in editing genes in different cell types, it may induce relatively high off-target activity13,14,15. Cleavage at off-target sites may occur in DNA sequences with up to five base mismatches, and DNA and RNA bulges can be tolerated16,17. CRISPR/Cas9 orthologs from different bacterial species and recognizing different PAM sequences have been investigated, including Neisseria meningitidis (NmCas9)18, Streptococcus thermophiles 1 (St1Cas9)6, Staphylococcus aureus (SaCas9)19 and Campylobacter jejuni (CjCas9)20. Cutting by St1Cas9, NmCas9, and SaCas9 requires gRNAs targeting DNA sequences of 21–24 nucleotides near their 5′‐NNAGAAW, 5′‐NNNNGATT, and 5′‐NNGRRT PAM motifs, respectively. CjCas9 recognizes the PAM sequence 5′-NNNNACAC′ or 5′-NNNNRYAC. Cpf1 — a nuclease from Prevotella and Francisella 1, which was later classified as Cas12a — only has a RuvC nuclease domain and does not require a tracrRNA21. It creates a 5′ overhang at the cleavage site producing staggered-end breaks21. Francisella novicida Cpf1 (FnCpf1) recognizes the PAM sequence 5′‐TTN‐3 while both Acidaminococcus spp Cpf1 (AsCpf1) and Lachnospiraceae bacterium Cpf1 (LbCpf1) recognize a 5′ TTTV PAM21.
CRISPR base editors have been developed to alter single DNA bases without the need to generate DSB by linking deactivated Cas9 (dCas9) to DNA deaminases. Two types of base editor have been demonstrated: cytosine base editors that convert G–C to A–T22,23 and adenine base editors that convert A–T to G–C24. Base editors may also be fused to Cas9 nickases22,25. However, substantial off-target effects have recently been reported following base editing with cytosine base editor26,27. Further, owing to its large size, it is very challenging to delivery base editor for in vivo genome editing28. The CRISPR/Cas13a system (previously C2c2), first identified from Leptotrichia shahii, can be used to edit RNA29. This nuclease is guided by a single crRNA and can be directed to cleave single strand RNA targets with complementary protospacers30. Cas13a from Leptotrichia wadei (LwaCas13a) has been shown to knockdown coding and non-coding RNAs in mammalian cells with an efficiency comparable to RNAi but with lower off-target effects31. More recently, the LEAPER (leveraging endogenous ADAR for programmable editing of RNA) system that uses short, engineered RNAs to recruit native ADAR1 or BADAR2 enzymes to change a specific adenosine to inosine has been reported32. This RNA base editing system has the advantage of small size and being deliverable by viral and nonviral vehicles, although repeated in vivo delivery will be required to generate a long-lasting therapeutic effect.
Therapeutic genome editing can be broadly divided into ex vivo and in vivo genome editing approaches12. The former is performed with cells isolated from a patient, where programmable nucleases and donor templates can be delivered into the cell nuclei via biological, chemical, or physical methods, and edited cells can be stored, amplified and, in some cases, sorted ex vivo before delivering back to the patient. Ex vivo genome editing is promising editing hematopoietic stem and progenitor cells for inherited blood disorders (such as sickle cell disease and β-thalassemia) and gene-edited CAR-T cells for cancer33,34. Nearly 20 clinical trials on programmable nuclease-based cell therapies are underway worldwide, most of which are based on CRISPR/Cas9 ex vivo gene-editing (clinicaltrials.gov). However, the treatment of many genetic diseases, including Duchenne muscular dystrophy, spinal muscular atrophy, and hereditary tyrosinemia, requires editing of disease-related genes in the relevant tissue in vivo. Significant challenges exist in specifically and efficiently delivering the CRISPR/Cas9 and donor template to the target cells in vivo.
Herein, we provide an overview of the recent developments in delivering CRISPR/Cas9 and donor template for in vivo genome editing and the associated challenges. We also provide perspectives on possible solutions to the challenges and future development, with the aim to attract more materials scientists to this exciting field.
Delivery challenges for in vivo editing
Efficient in vivo genome editing requires the delivery of the genome-editing machinery (for example, CRISPR/Cas9 and donor template; Box 1) into the nuclei of the target cells. Unintended expression of CRISPR/Cas9 in the non-target tissues and organs should be minimized to avoid off-target mutagenesis. Similarly, after the intended gene editing at the target loci, the persistence of CRISPR/Cas9 expression is undesirable as this may lead to prolonged off-target DNA cleavage. Therefore, effective and safe in vivo genome editing requires stringent spatial and temporal control of the CRISPR/Cas9 activity in the body. Although in vivo delivery of genome-editing machinery shares some features with drug and gene delivery, the complexity of the cargo and its activity present new delivery challenges. There is a range of technological and biological barriers to efficient and specific in vivo genome editing, including the size of the CRISPR/Cas9 system, the limitation of delivery vehicles, the need to deliver genome-editing machinery to cell nucleus, and the control of nuclease activity.
In vivo genome editing can be used to perform gene disruption, gene correction, targeted gene deletion and insertion, and other gene modifications (Box 1). Depending on the applications, the genome-editing machinery may include different variants of Cas9 nuclease, gRNAs, DNA donor template, and other effectors, such as DNA deaminase. As detailed in Box 2, Cas9 nuclease can be delivered as DNA, mRNA, or protein. Cas9 mRNA contains approximately 4500 nucleotides. Cas9-expressing DNA (Cas9 DNA) is larger owing to the additional regulatory elements needed for transcription. The gRNAs and DNA repair templates can be delivered in their original forms, or as part of the DNA cargo including the Cas9 DNA. Successful genome editing requires the presence of all the required components in the cell nucleus in a coordinated manner. Therefore, in vivo genome editing requires proper packaging, dosing, and release of the genome-editing machinery and, if delivered separately, synchronization of individual components in the target cells.
The genome-editing machinery can be delivered either systemically or locally (Figure 1). Systemic delivery, mainly via intravenous injection, takes advantage of the circulatory system, which can distribute blood-borne substances throughout the body. Systemic delivery requires five steps for the gene editing machinery to reach the nucleus of the target cell: distribution in the circulatory system; extravasation from the blood vessels; migration in the interstitial space; cell entry; and intracellular transport into the cell nucleus. Each step presents unique challenges to in vivo genome editing.
Figure 1. Biological barriers to in vivo delivery from systemic circulation to cell nucleus.
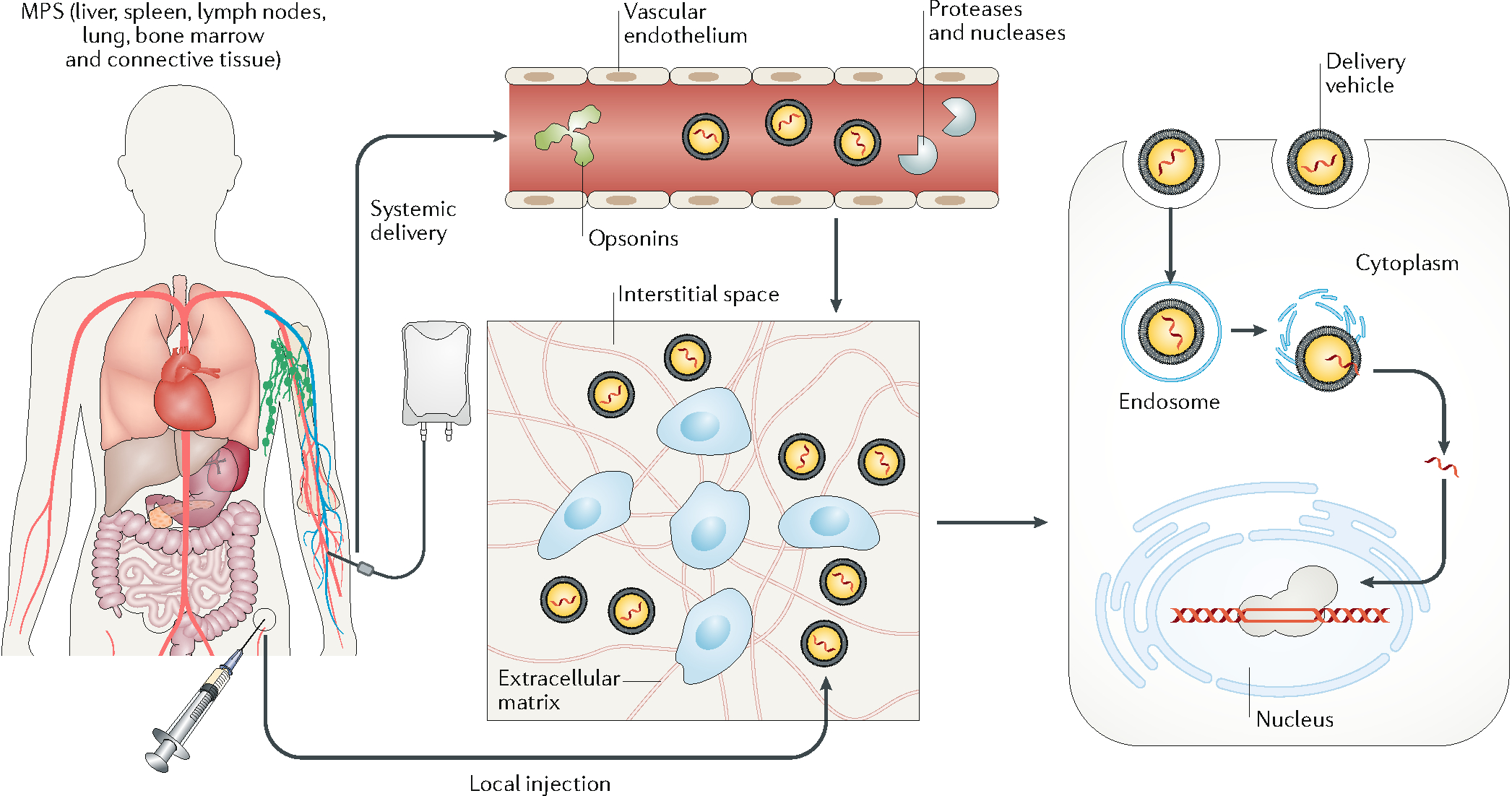
In systemic delivery, the delivery vehicles are distributed throughout the body. To edit target cells, the delivery vehicles need to extravasate, travel across the interstitial space, and pass through the cell membrane into the cell nucleus. With local injection, the delivery vehicles enter the interstitial space directly. In systemic delivery, the delivery vehicles can adsorb opsonins, including antibodies, complement factors, and other proteins in the plasma, which promote their clearance by the mononuclear phagocyte system. In addition, exposure of the genome-editing machinery to the plasma can cause degradation via circulating proteases or nucleases. Another barrier to systemic delivery is the vascular endothelium (unless it is the target tissue). In most tissues, the endothelial cells on the vessel surface are connected to form a continuous layer via cell–cell junctions, which prevents most delivery vehicles from entering the interstitial space. The interstitial transport of delivery vehicles is often hindered by the stroma cells and the extracellular matrix, which may confine systemically delivered vehicles close to the vessel surface and locally injected vehicles to the site of injection. Another rate-limiting step is for the delivery vehicles to pass through the cell membrane via micropinocytosis or endocytosis. The delivery vehicles entering the cells are typically transported from endosomes to lysosomes, where most proteins and nucleic acids are enzymatically digested. Therefore, cargo needs to be released from the delivery vehicle and escape from the endosome to enter the cytosol. Finally, the cargo needs to enter the cell nucleus to perform gene editing at the target locus (except for the case of RNA editing in the cytosol).
After entering the circulatory system, the delivery vehicle mixes with the blood. The components of the editing machinery, such as Cas9 protein, Cas9 mRNA, gRNA, and the DNA donor template, are subject to degradation if exposed to proteases and nucleases in the plasma. In addition, the delivery vehicle can adsorb various plasma proteins, including fibrinogen, albumin, and opsonins. This phenomenon is often described as the formation of protein corona. The composition of the protein corona is determined by the surface charge, hydrophobicity, size, shape, and molecules on the surface of the delivery vehicle35. The protein corona affects the colloidal stability of the delivery vehicle and changes its interactions with the biological system36. In particular, adsorption of the opsonins may induce the recognition and sequestration of the delivery vehicle by the reticuloendothelial system (RES) in the liver, spleen, and lymph nodes as well as the mononuclear phagocytic system (WPS) which mainly consists of the phagocytic cells. Further, a recent study identified preexisting antibodies against SaCas9 and SpCas9 in 78% and 58% of donor samples, respectively, and anti-SaCas9 and anti-SpCas9 T cells in 78% and 67% of samples37. Another study identified SpCas9 specific effector T cells in 96% of donor samples with similar levels of reactive T-cells specific for SaCas9 and Cpf1. Interestingly, SpCas9-reactive regulatory T cells were found to be capable of mitigating SpCas9-reactive effector T cell function in vitro, highlighting a potential solution to overcoming the issue of preexisting immunity38. Typically, nanoparticles smaller than 5 nm will be excreted via the renal system and those larger than 200 nm will be retained in the spleen. In general, when used as a delivery vehicle, more than half of nanoparticles will eventually accumulate in the liver and spleen.
The vascular endothelium is the second barrier to the systemic delivery of genome-editing machinery. In most tissues, the endothelial cells lining the vessel surface are connected via cell–cell junctions, including adherence junctions and tight junctions, which only allow small molecules (<1-nm diameter) to pass through39. Delivery vehicles with diameters greater than 1 nm rely on the less-efficient transcytosis pathways to extravasate from the blood vessel. Vascular permeability is particularly high in the fenestrated vessels in the hepatic sinusoid, and in leaky vessels as a result of inflammation during wound healing or angiogenesis during tumour growth. In such organs and tissues, substantial accumulation of the delivery vehicle often occurs. Conversely, the vascular endothelium in the brain, together with astrocytes and pericytes, forms the blood–brain barrier, which excludes many small polar molecules from entering the brain, as well as large complexes and nanoparticles.
Delivery vehicles extravasated from blood vessels need to travel across the interstitial space to reach the target cells. The transport barriers of the interstitial space are tissue-specific and are controlled by the cell density and the composition and density of the extracellular matrix. Interstitial transport of large molecules and nanoparticles is inefficient owing to their poor diffusivity and the small pores connecting the interstitial space40,41. An additional problem for nonviral delivery is the negatively charged extracellular matrix, which may hinder the transport of positively charged nanoparticles and reduce their cellular internalization. Therefore, nonviral delivery vehicles may only be able to reach the cells close to the exterior of the vessel. In addition, the target tissue may consist of many cell types, including stromal cells (for example, fibroblasts, immune cells, and parenchymal cells). As the (viral or nonviral) delivery vehicle moves through the interstitial space, it may be internalized by any cells it passes. Uptake of delivery vehicles by off-target cells (that is, cells not intended to edit) further reduces the availability of genome editing machinery to modify the target cells.
The cell membrane is formed by a lipid bilayer and various transmembrane proteins. The lipid bilayer is only permeable to small lipophilic molecules42. Large molecules and nanoparticles can enter cells via phagocytosis and endocytosis, or by disrupting the cell membrane43. With both viral and nonviral approaches, the cargo needs to be released from the delivery vehicle after entering the cell. However, genome editing machinery entering cells via endocytosis are transported to lysosomes and broken down by enzymes. The components that escape from the endosome into the cytosol need to further pass the nuclear membrane to reach the cell nucleus. This usually requires the presence of one or more nuclear localization signal peptides on Cas9 protein44. In the cell nucleus, the rate of genome editing is determined by factors including the abundance of individual components of the genome-editing machinery and the accessibility of the target loci in the chromosome. The intracellular transport of the cargo and the genome editing efficiency are dependent on cell cycle and cell type12. For example, the rate of homology-directed repair (HDR) is significantly increased in dividing cells, and gene-editing efficiency is enhanced in highly proliferating cells in which nuclear membrane becomes porous during mitosis leading to elevated nuclear access.
For local delivery via injection, the delivery vehicles are directly introduced into the interstitial space in the target tissue, thus minimizing the dissemination to off-target tissues and organs, and bypassing some barriers in the systemic delivery, such as interactions during circulation and extravasation from the blood vessel. However, the transport of the delivery vehicles can still be restricted by the dense extracellular matrix, which prevents dispersion in the target tissue. Therefore, with local delivery, the genome-editing machinery is often confined in the region around the point of injection, leading to a highly heterogeneous distribution in the target tissue and insufficient editing for the desired outcome.
Viral-based delivery
Viral-based methods remain a popular choice for the delivery of gene editing machinery45,46. Viral vector classes that have been used for in vivo genome editing include adenovirus (AdV), adeno-associated virus (AAV), lentivirus, and retrovirus (Figure 2a), with AAV being the most promising because of its low immunogenicity, good safety profile, and transient transgene expression47–54. To express CRISPR/Cas9 in a target cell, it requires a process involving viral vector uptake, cargo transport and release, transgen transcription and translation (Figure 2b). AAV vectors can be used for targeted delivery based on their serotype, which confers specific tissue tropism55; however, the tissue specificity of AAV vectors is moderate. AAV vectors have already been approved for use in clinical trials for the treatment of diseases, such as α−1 antitrypsin deficiency, haemophilia A and B, and familial hypercholesterolaemia56. Moreover, Luxturna™(voretigene neparvovec-rzyl), an AAV-based therapy for the correction of biallelic RPE65 mutation, which causes inherited retinal disease, has been approved for use in the US. However, AAV vectors still face several limitations, especially the low packaging capacity.
Figure 2. Viral base in vivo delivery of genome editing machinery.
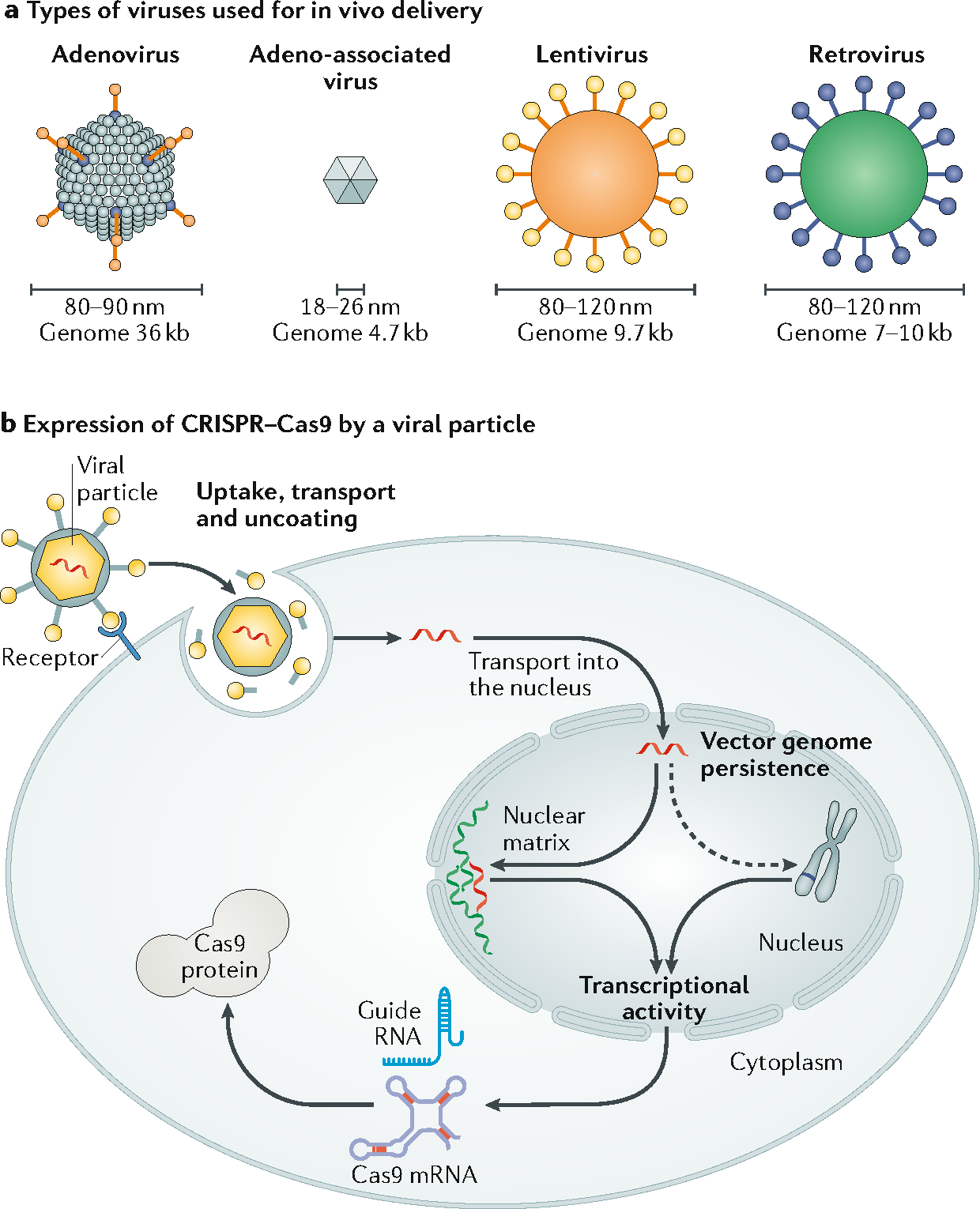
a. Four types of viruses have been used for in vivo delivery of genome editing machinery: adenovirus (AdV), adeno-associated virus (AAV), lentivirus, and retrovirus, with genome sizes of 36kb, 4.7kb, 9.7kb and 7–10kb, respectively. Their physical size as diameters are indicated. b. The process of expressing CRISPR/Cas9 in a target cell by a viral-based delivery vector, including uptake, cargo transport and release; transcriptional activity; and transgene persistence. DNA packaged in the viral vector that encodes Cas9 protein and guide RNA is first being released and transcribed into Cas9 mRNA and gRNA in the cell nucleus, which are then transported to the cytosol where Cas9 protein is produced. Persistent expression of Cas9/gRNA may cause genotoxicity and immune responses.
The large size of the SpCas9 protein (4.3 kb for the coding region) is a challenge for its in vivo delivery with AAV (with a 4.7-kb packing capacity)57. With the addition of regulatory elements, such as promoters and polyadenylation signals, the packaging capacity of AAV is often exceeded for delivering SpCas9-based editing machinery. Thus, it is often necessary to package SpCas9 and gRNA into two separate vectors, which could achieve a delivery efficiency of >70%, as demonstrated in a study to disrupt the Mecp2 gene in the mouse brain47. In contrast, NmCas9, St1Cas9, SaCas9 and CjCas9 require 3.2-, 3.4-, 3.2-, and 2.9-kb coding regions, respectively, allowing for packaging into AAV vectors together with gRNA and regulatory elements58. For example, SaCas9 was packaged into a single AAV with a gRNA to achieve similar editing efficiencies to SpCas919. CjCas9 was packaged into AAV with VEGFA-targeting gRNA, inducing indels of up to 30% in retinal pigment epithelium20. The coding region of Cpf1 is 3.9 kb thus can be readily packaged into AAV59. One strategy to overcome the packaging limit of AAV is to split the transgen into two parts and packaging them into separate AAV vectors, then rejoining the two parts through heterodimerization in the host cell60,61. This strategy has been used to package a base editor system and delivery it to a mouse model for the human disease phenylketonuria to demonstrate the potential in treating this metabolic liver disease28.
Retroviral and lentiviral vectors have a large genome of 7–10kb and 9.7 kb respectively (Figure 2)62,63 and can transduce a large range of cells in vivo64,65. However, these vectors induce the integration of the transgene into the host genome, which can disrupt functional genes and increase off-target gene editing owing to long-term expression of CRISPR/Cas9. To improve their safety profile, integrase-deficient lentiviral vectors (IDLVs) have been developed which harbor integrase mutations that specifically prevent proviral integration, reducing the chances of insertional mutagenesis66. IDLV vectors have been used to deliver CRISPR/Cas9 to develop mouse disease models67, mutate genes in murine primary dendritic cells68, and develop tools to study the immune system69. Recently, a virus-like particle delivery method based on murine leukemia virus, a type of retrovirus, was developed for the in vitro and in vivo delivery of genome editing machinery70. In contrast to packaging DNA encoding CRISPR/Cas9, this method packages Cas9/gRNA RNP, thus having the advantages of limiting the period of Cas9 activity and reducing cost compared with typical viral based delivery.
The packaging capacity of AdV is up to 36 kb, which allows for encapsidation of all the components of a CRISPR/Cas9 gene editing machinery, including the regulatory elements71,72. Recently, SpCas9 with gRNA targeting the mutated form of SERPINA1, which encodes misfolded α1-antitrypsin, was delivered by AdV in a humanized mouse model of α1-antitrypsin73, resulting in a 94% reduction of misfolded α1-antitrypsin in the treated mice compared with the control. Histological analysis of the mouse tissue showed that gene editing reduced liver protein aggregation and fibrosis in the treated mice. Adv has also been used to deliver base editor into the liver of adult mouse to introduce site-specific nonsense mutations, resulting in reduced plasma cholesterol levels74.
Viral-based delivery methods suffer from several additional drawbacks. For example, they induce constitutive expression of Cas9/gRNA (that is, the expression is always ‘on’). This is undesirable because persistent expression of CRISPR/Cas9 may increase off-target effects and cause an anti-Cas9 immune response (Figure 2). To address these issues, a self-deleting AAV system was developed to introduce indels into AAV episomes75. Compared with the standard AAV vector for SaCas9 delivery, the self-deleting AAV vector resulted in a 79% reduction of SaCas9 protein in vivo while maintaining high levels of editing at the on-target sites in multiple genes in the liver75. The safety of viral-mediated therapies may also be lowered by pre-existing immunity to viral capsids76 or CD8+ T cell-mediated response against transduced cells that present viral capsid protein or Cas9 antigens38,77. For clinical applications, this may be overcome through the selection of patients with no or low neutralizing antibodies78, the administration of immunosuppressant drugs prior to treatment79, or decreasing the therapeutic dose administered80. However, the host response to viral vectors remains difficult to predict. In a recent study, piglets and non-human primates treated with high doses of AAV showed signs of severe toxicity and either died or were euthanized 4–14 days after administration81. Moreover, production of viral vectors at a large scale is expensive, and requires specific facilities and expertise. For example, the manufacturing of viral vectors for clinical applications involves expensive single-use culture systems and bioreactors to yield required titers82. A demonstration of this is the AAV-based therapeutic valoctocogene roxaparvovec, which at its Phase 3 clinical trial, and requires a dose of 4 × 1013 viral particles per kilogram (NCT03392974). Therefore, although viral-based in vivo delivery of genome editing machinery has the advantages of being efficient, having good safety profiles (such as that with AAV), and clinical viable, the potential issues of persistent expression, lack of tissue specificity, detrimental transgene integration and limitations in packaging (with AAV) need to be addressed before their widespread applications in in vivo genome editing can be realized.
Synthetic material-mediated delivery
Many types of synthetic material, often in the form of nanoparticles, have been developed as alternatives to viral vectors for in vivo delivery of genome-editing machinery. Synthetic materials have several advantages over viral vectors. For example, they can be tailored for delivering different forms of the CRISPR/Cas9 system, including the Cas9 protein or Cas9/gRNA RNP, Cas9 mRNA with gRNA, and plasmid DNA encoding Cas9 and gRNA. Unlike viral vectors, there is no preexisting immunity against most synthetic materials and their immunocompatibility can be improved by optimizing the size, shape, coating, and surface chemistry43,83. Moreover, synthetic material-mediated delivery does not induce integration of the Cas9 gene into the genome (except random integration). Further, physical formulation or chemical synthesis of nanoparticles is more cost-effective and suitable for large-scale production than viral vectors. However, achieving high delivery efficiency remains a significant challenge for most synthetic material-mediated delivery methods84.
To date, synthetic material-mediated in vivo genome editing has mainly been inspired by conventional gene-delivery methods. The most straightforward approach for in vivo genome editing is to deliver Cas9 RNP (that is, the Cas9 protein complexed with gRNA). Cas9 is a relatively large (160 kDa for SpCas9) and positively charged protein; however, Cas9 RNP has a net negative charge owing to the abundant phosphate groups in the gRNA85. Cas9 RNP can be complexed with delivery vehicles through electrostatic interactions, DNA–gRNA base pairing, nonspecific adsorption, or covalent bonding. Moreover, Cas9 RNP binds to cationic polymers (for example, lipids and peptides), which then bind to the negatively charged cell membrane, thereby enhancing the cellular uptake of RNP. Upon endocytosis, cationic polymers can destabilize the endosomal membrane and release RNP into the cytosol. For example, local injection of Cas9 RNP complexed with a cationic liposomal reagent (Lipofectamine™ 2000) into the cochlea of neonatal Tmc1Bth/+ mice disrupted the dominant deafness-associated allele, increased hair cell survival, and enhanced acoustic startle responses, thus providing a potential strategy for treating autosomal-dominant hearing loss86. In another study, Cas9 and gRNA embedded in crosslinked PEI hydrogel were encapsulated by a cationic lipid membrane, forming liposome-templated hydrogel nanoparticles87. In this study, the lipid membrane was further conjugated with tumour-targeting and cell-penetrating peptides. In a mouse subcutaneous xenograft tumour model, the nanoparticles reached 30% of tumour cells after repeated tail vein injections for three consecutive days87. This study also showed that tail vein injection of nanoparticles inhibited the polo-like kinase 1 (PLK1) expression by more than 60% and reduced the tumour volume by nearly 80% compared to placebo87, demonstrating efficient in vivo gene-editing.
Another promising delivery vehicle is thiolated DNA-coated gold nanoparticles (CRISPR–gold) (Figure 3a)88. Specifically, a DNA donor template was conjugated via hybridization to thiolated DNA and Cas9 RNP was adsorbed via its nonspecific interaction with the DNA molecules on the surface of gold nanoparticles. The nanoparticles were sequentially coated with negatively charged silica and a cationic endosomal disruptive polymer. CRISPR–gold aggregated with an average diameter of approximately 500 nm was administrated in mice via intramuscular injection with cardiotoxin (which activates the proliferation of muscle stem cells by muscle damage) to correct the DNA mutation that causes Duchenne muscular dystrophy88. CRISPR–gold was observed not to up-regulate inflammatory cytokines in the plasma within two weeks, although CD45+ and CD11+ leukocytes increased in treated muscle, which is indicative of local inflammation. Importantly, unlike viral vectors, there was no increase in the plasma level of inflammatory cytokines even after repeated injections of CRISPR–gold, suggesting that it is safe to administer multiple times. A follow-up study showed that intracranial injection of CRISPR–gold rescued exaggerated repetitive behaviours in a mouse model of fragile × syndrome89. This is the first case of behavioural rescue of brain disorder in an animal model using nonviral delivery of gene-editing machinery.
Figure 3. Examples of material systems for in vivo delivery of genome-editing machinery.
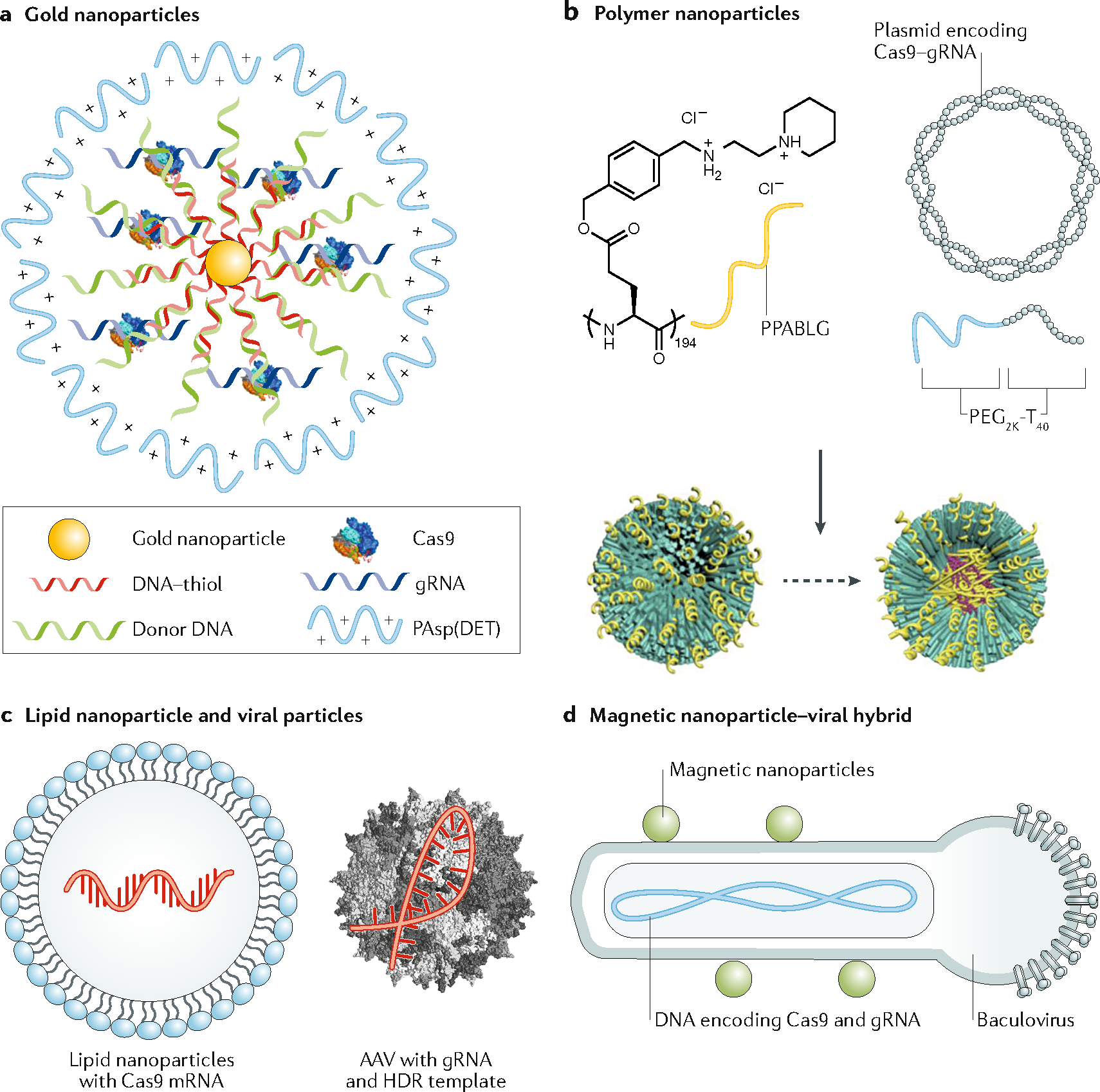
a. Delivery of Cas9/gRNA ribonucleoprotein (RNP) and the template for homology-directed repair (HDR) using gold nanoparticles (CRISPR–gold). CRISPR–gold is composed of gold nanoparticles conjugated with DNA, which are complexed with donor DNA, Cas9 RNP, and the polymer PAsp(DET) for endosomal disruption. Once in the cytoplasm, glutathione releases the DNA from the gold core of CRISPR–gold, which causes the rapid release of Cas9 RNP and donor DNA. b. Nanoparticle-based delivery of Cas9 plasmid and single guide RNA (gRNA). The positively charged α-helical polypeptide PPABLG complexes with Cas9 expression plasmids and gRNAs to form nanoparticles, which are then PEGylated (P-HNPs). P-HNPs can achieve efficient cellular internalization and endosomal escape. c. In vivo delivery of genome-editing machinery by combining lipid nanoparticles carrying Cas9 mRNA with AAV viral particles encoding gRNA and HDR donor template. d. In vivo delivery of CRISPR/Cas9 with spatial control of gene editing. DNA encoding Cas9 and gRNA is packaged into a baculoviral (BV) particle, which is complexed with magnetic nanoparticles (MNP–BV). By applying a magnetic field locally after delivery of MNP–BV, the inactivation of BV by the complement system in the serum can be overcome, leading to spatially controlled genome editing in the target tissue.
Cas9 RNP has also been conjugated to silica-coated upconversion nanocrystals via photocleavable 4-(hydroxymethyl)-3-nitrobenzoic acid (ONA) molecules90. In this example, the surface of the nanoparticles was covered with a layer of Polyethylenimine (PEI) to enhance the transduction efficiency. When the exposed to near-infrared light, the upconversion nanocrystals emit photons in the UV range and release Cas9 RNP from their surface. This allows the in vivo activity of Cas9 to be remotely controlled by optical signals. This study showed that intratumoural injection of nanoparticles suppressed tumour growth by disrupting PLK1.
Because of their strong negative charge, Cas9 mRNA and Cas9 DNA are commonly complexed with cationic polymers for nonviral in vivo delivery. For example, a self-assembled micelle composed of quaternary ammonium-terminated poly(propylene oxide) (PPO-NMe3) and amphiphilic Pluronic F127 was constructed to facilitate DNA binding and enhance cell penetration91. When mixed at an optimal ratio, PPO-NMe3, Pluronic F127, and Cas9 DNA formed micelles ~200 nm in size. In a mouse model of a xenograft HeLa tumour, the micelles with Cas9 DNA were delivered via intratumoural injection, resulting in reduced tumour growth by over 60% compared with the control group91. In another study, cationic α-helical polypeptides and copolymers of poly(ethylene glycol) and polythymine40 were used to bind and condense Cas9 DNA and gRNA into nanoparticles (Figure 3b)92. Polypeptides have high membrane-penetrating ability, which enhances cellular internalization and endosomal escape of the cargo. Intratumoural injection of the nanoparticles targeting PLK1 reduced the PLK1 protein level by 66.7% and partially inhibited the tumour growth in a mouse model of xenograft HeLa tumour. Tail vein injection of positively charged lipid-like nanoparticles carrying Cas9 mRNA and gRNA was able to disrupt hepatitis b virus (HBV) DNA in the liver in a mouse model of HBV infection93.
Current synthetic material-based delivery vehicles have two common features: they are relatively bulky (>100 nm) and have a positively charged surface. The former is due to in part to packaging of the CRISPR/Cas9 system, of which Cas9 RNP, Cas9 mRNA, and DNA encoding Cas9 are relatively large molecules. The latter is necessary for enhancing cell entry and endosomal escape of the cargo. However, these features are not favourable for either systemic or local delivery of genome-editing machinery. In the small number of cases reported using synthetic materials for systemic delivery, the target tissues are limited to the liver and tumour, where the highly permeable vessels allow extravasation of the delivery vehicles into the interstitial space. In most cases, the delivery vehicles are administrated via direct injection into the target tissue. For systemic administration of gene-editing machinery using nanoparticles, repeated injections are often needed to compensate for the poor interstitial dispersion of large nanoparticles and to achieve high delivery efficiency. Although current nonviral delivery methods have yielded exciting results in mouse models where the tissue volume is small, when moving them into clinical studies, it may be challenging to gain comparable therapeutic effects in the human body.
Delivery using viral–nonviral hybrids
Although viral vector-based CRISPR/Cas9 delivery is often associated with increased risk of genotoxicity, the combination of viral and nonviral delivery approaches can take advantage of the efficient transduction machinery of the viral vectors and the controllability of the synthetic materials. One scheme is to deliver the genome-editing machinery with two vehicles: a synthetic material physical method that delivers the Cas9 nuclease and a viral vector that delivers the gRNA(s) and DNA donor template. For example, in an in vitro genome-editing study, electroporation was used to deliver Cas9/gRNA RNP followed by transduction of a DNA donor template packaged in an AAV to efficiently correct sickle mutation in patient-derived hematopoietic stem/progenitor cells33 and induced pluripotent stem cells34. Similarly, in an in vivo study using a mouse model of human hereditary tyrosinemia, lipid nanoparticles carrying Cas9 mRNA and gRNA, and an AAV vector packaging the DNA donor template were administered to mice via tail vein injection, leading to high accumulation in the liver (Figure 3c)94. This treatment resulted in more than 6% gene correction in hepatocytes and alleviated disease symptoms, such as weight loss and liver damage94.
A recent study complexed a baculoviral vector (BV) with magnetic nanoparticles for in vivo delivery (Figure 3d)95. Engineered BVs are derived from an insect virus, Autographa californica multicapsid nucleopolyhedrovirus and can transduce many types of mammalian cells via capsid protein-mediated cell entry, endosomal escape, and transport into the cell nucleus. Unlike most viral vectors used in gene delivery, BV-induced transgene expression lasts for only a few days in mammalian cells owing to the lack of viral replication and viral genome integration. In addition, BVs in the circulatory system can be rapidly inactivated by the complement system, consisting of a set of small plasma proteins that act sequentially to produce a wide range of activities, including cell lysis and opsonization96. When BV is complexed with magnetic nanoparticles (MNP–BV), a locally applied magnetic field can overcome the inactivation, leading to spatially controlled transgene expression in the target tissue95. The interplay between the local magnetic field and the complement system provides an on–off switch for MNP–BV-induced transduction in vivo, which enables local and transient in vivo genome editing when CRISPR/Cas9 is packaged in a BV95.
Future perspective
Although the CRISPR/Cas9 system was only introduced into mammalian cells for genome editing in 20136,7, it has already transformed the field of genome editing and poised to revolutionize many fields of medicine. However, in vivo delivery of genome-editing machinery remains a challenge, and innovative approaches are needed to achieve high delivery efficiency, tissue specificity, and safety for clinical translation. To this end, there are important design features that one could consider (Figure 4). For example, to achieve high efficiency, the delivery vehicle needs to significantly reduce or prevent enzymatic degradation of the CRISPR/Cas9 system during systemic circulation. This can be realized by encapsulation of the delivery vehicle, improving its stability in the blood, and/or chemically modifying the backbones of gRNAs and DNA templates if they are exposed to the blood. The delivery vehicle should have stealth properties to avoid rapid clearance by the mononuclear phagocytic system. This can be achieved by coating with biocompatible polymers such as poly(ethylene glycol) and conjugating with ligands that enable immune evasion. The delivery vehicle should also be able to extravasate from the blood vessel. Studies have shown that extravasation of the delivery vehicles in tissues other than liver and tumour can be realized by targeting tissue-specific ligands that trigger active transcytosis97. Another option is to increase the permeability of local vessels transiently via physical or biological methods98–101. In the interstitial space, the delivery vehicles need to migrate effectively to reach target cells in the tissue. Interstitial transport can be improved by optimizing the size and coating of the nanoparticles and/or modifying the extracellular matrix via enzymatic degradation102. The cellular uptake of nanoparticles can be enhanced by targeting cell surface receptors that activate endocytosis. Upon endocytosis, the delivery vehicles need to release the cargo into the cytosol by destabilizing the endosome membrane with reagents, such as positively charged polymers. In addition to delivering the genome-editing machinery, viral or nonviral systems supplying reagents that improve the accessibility of the target loci in a genome or confer a selective advantage to edited cells (so that they may survive and grow better than non-edited cells) may also improve the overall gene editing efficiency.
Figure 4. Delivery strategies to overcome challenges in the efficiency, specificity, and safety of in vivo delivery of genome-editing machinery.
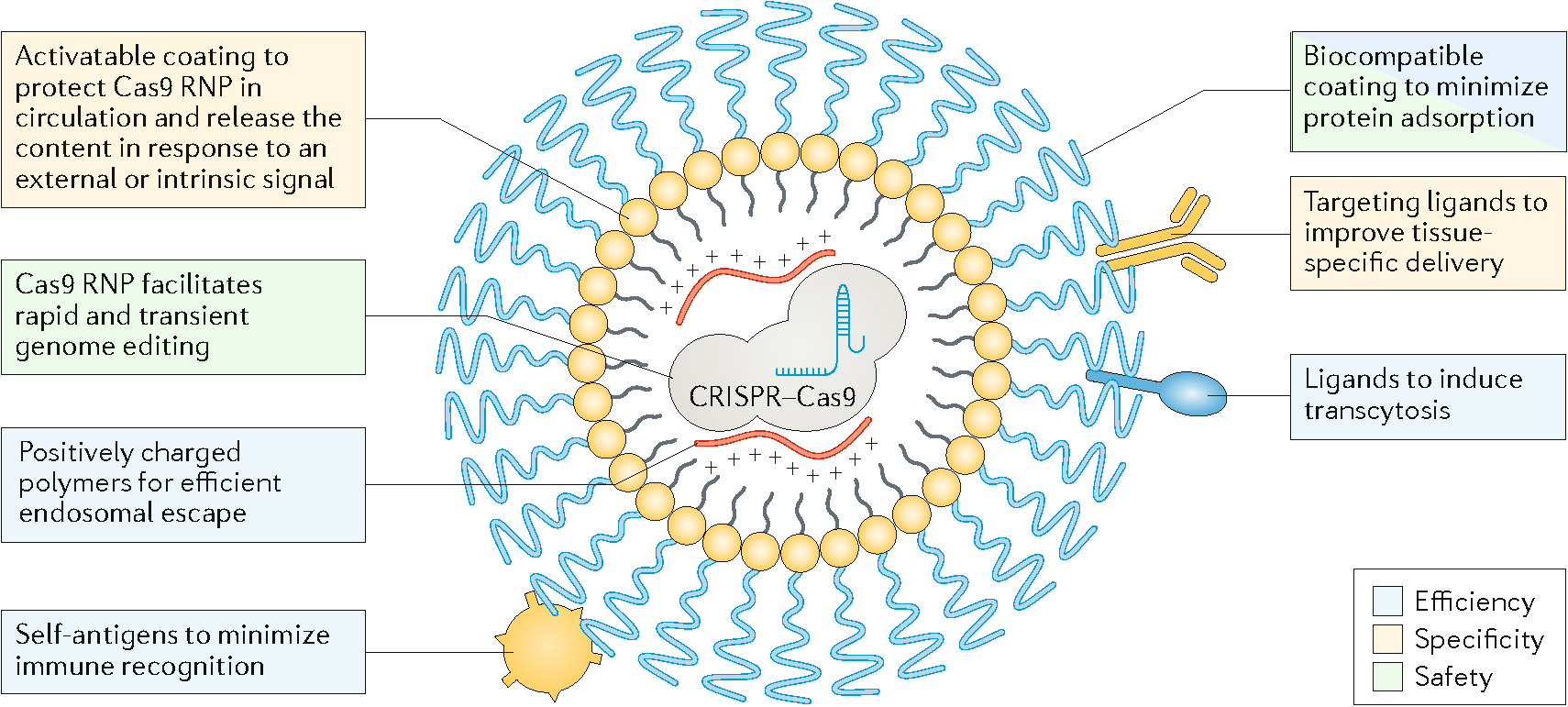
The efficiency of in vivo genome editing can be improved by increasing the bioavailability of CRISPR/Cas9 to the target loci. Approaches to improve efficiency include preventing enzymatic degradation via stable encapsulation; immune evasion via biocompatible coating and self-antigens; increasing extravasation via ligands inducing transcytosis on vascular endothelium; and increasing intracellular and nuclear entry of the CRISPR/Cas9 machinery via cationic polymers. The specificity can be improved by decorating the delivery vehicle with targeting ligands and developing delivery vehicles responding to external signals or tissue-specific cues. Approaches to improve the safety include delivering Cas9 mRNA or protein that only has a short lifespan; implementing mechanisms for local retention or systemic inhibition; and using biocompatible materials to minimize local inflammation.
To improve tissue specificity, the delivery vehicle can be conjugated with cell-specific targeting ligands (Figure 4). Tissue specificity may also be achieved by designing delivery vehicles that can either be activated locally by external optical, thermal or magnetic fields, or that respond to the tissue-specific microenvironment, such as the pH and enzymatic activity. To improve safety, systemic dissemination of CRISPR/Cas9 can be prevented using mechanisms for local retention or systemic inhibition of the delivery vehicle (for example, systemic inhibition of baculoviral vectors by circulating complement factors). In addition, the self-deleting AAV system75, and suicidal or self-deleting strategies that deactivate other types of viral vectors can be used to reduce genotoxicity, thus improving safety while maintaining their intended editing function. Off-target activities of CRISPR/Cas9 can be reduced or minimized by optimizing the dose and/or limiting the duration of Cas9 activity. While temporal control of Cas9 activity can be partially realized by delivering Cas9 mRNA or Cas9/gRNA RNP, other more precise mechanisms may be used, such as fusing a destabilizing domain to Cas9103, using small molecule Cas9 inhibitors104 or light105,106. Further, biocompatible and nonimmunogenic materials are desirable to minimize local inflammatory responses. Finally, in vivo delivery of the genome-editing machinery needs to be designed according to the therapeutic target, the disease state, and the properties of target tissues.
To achieve high efficiency, tissue specificity, and safety in in vivo genome editing using engineered delivery vehicles, modeling, simulation and computational analysis can significantly aid their design, evaluation and optimization107,108. Further, systems approaches can help integrate different functions of a deliver vehicle. For example, an activatable and biocompatible coating could be engineered to protect Cas9 RNP in circulation, minimize protein adsorption, and release the cargo in response to an external or intrinsic signal (Figure 4). Multiple ligands can be conjugated to the surface of nanoparticle carriers to simultaneously improve tissue specific delivery, induce transcytosis, and minimize immune recognition (Figure 4). These and other possibilities make this an exciting time for materials researchers to contribute to the field of in vivo genome editing.
Acknowledgment
This work was supported by the Cancer Prevention and Research Institute of Texas (RR140081 to G.B.), the National Institutes of Health (R01EB026893 to S.T., UG3TR002863 to K.L. and UG3HL151545 to G.B.), and Defense Advanced Research Projects Agency (HR0011-19-2-0009 to K.L.).
Footnotes
Competing Interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gaj T, Gersbach CA & Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller J, McLachlan AD & Klug A Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. 4, 1609–1614 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim YG, Cha J & Chandrasegaran S Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 93, 1156–1160, doi: 10.1073/pnas.93.3.1156 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christian M et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186, 757–761 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin H, Kauffman KJ & Anderson DG Delivery technologies for genome editing. Nat. Rev. Drug Discov 16, 387–399 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolotin A, Quinquis B, Sorokin A & Ehrlich SD Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J & Soria E Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol 60, 174–182 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Pourcel C, Salvignol G & Vergnaud G CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Sander JD & Joung JK CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol 32, 347–355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cox DB, Platt RJ & Zhang F Therapeutic genome editing: prospects and challenges. Nat. Med 21, 121–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee CM, Cradick TJ & Bao G The Neisseria meningitidis CRISPR-Cas9 system enables specific genome editing in mammalian cells. Mol. Ther 24, 645–654 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee CM, Cradick TJ, Fine EJ & Bao G Nuclease target site selection for maximizing on-target activity and minimizing off-target effects in genome editing. Mol Ther 24, 475–487 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cradick TJ, Fine EJ, Antico CJ & Bao G CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 41, 9584–9592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu Y et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol 31, 822–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin Y et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 42, 7473–7485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou Z et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA 110, 15644–15649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ran FA et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim E et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun 8, 14500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zetsche B et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishida K et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353, aaf8729 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Gaudelli NM et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim YB et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol 35, 371–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuo E et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364, 289–292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin S et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 364, 292–295 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Villiger L et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med 24, 1519–1525 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Shmakov S et al. Discovery and functional characterization of diverse Class 2 CRISPR-Cas systems. Mol. Cell 60, 385–397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abudayyeh OO et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abudayyeh OO et al. RNA targeting with CRISPR-Cas13. Nature 550, 280–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu L et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol doi: 10.1038/s41587-019-0178-z (2019). [DOI] [PubMed] [Google Scholar]
- 33.Dever DP et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin RM et al. Highly efficient and marker-free genome editing of human pluripotent stem cells by CRISPR-Cas9 RNP and AAV6 donor-mediated homologous recombination. Cell Stem Cell 24, 821–828 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Walkey CD, Olsen JB, Guo H, Emili A & Chan WC Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J. Am. Chem. Soc 134, 2139–2147 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Walkey CD et al. Protein corona fingerprinting predicts the cellular interaction of gold and silver nanoparticles. ACS Nano 8, 2439–2455 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Charlesworth CT et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med 25, 249–254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner DL et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med 25, 242–248 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Komarova Y & Malik AB Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol 72, 463–493 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Dreher MR et al. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J. Natl. Cancer Inst 98, 335–344 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Yuan F, Krol A & Tong S Available space and extracellular transport of macromolecules: effects of pore size and connectedness. Ann. Biomed. Eng 29, 1150–1158 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Orsi M, Sanderson WE & Essex JW Permeability of small molecules through a lipid bilayer: a multiscale simulation study. J. Phys. Chem. B 113, 12019–12029 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Zhang S, Gao H & Bao G Physical principles of nanoparticle cellular endocytosis. ACS Nano 9, 8655–8671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vakulskas CA et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med 24, 1216–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ginn SL, Amaya AK, Alexander IE, Edelstein M & Abedi MR Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med 20, e3015 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Yanik M et al. In vivo genome editing as a potential treatment strategy for inherited retinal dystrophies. Prog. Retin. Eye Res 56, 1–18 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Swiech L et al. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol 33, 102–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Long C et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson CE et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med 25, 427–432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabebordbar M et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang Y et al. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol 34, 334–338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hakim CH et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight 3, e124297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bak RO & Porteus MH CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Rep. 20, 750–756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maddalena A et al. Triple vectors expand AAV transfer capacity in the retina. Mol Ther 26, 524–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zincarelli C, Soltys S, Rengo G & Rabinowitz JE Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther 16, 1073–1080 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Baruteau J, Waddington SN, Alexander IE & Gissen P Gene therapy for monogenic liver diseases: clinical successes, current challenges and future prospects. J. Inherit. Metab. Dis 40, 497–517 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Z, Yang H & Colosi P Effect of genome size on AAV vector packaging. Mol. Ther 18, 80–86 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen X & Goncalves MA Engineered viruses as genome editing devices. Mol. Ther 24, 447–457 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dai X et al. One-step generation of modular CAR-T cells with AAV-Cpf1. Nat. Methods 16, 247–254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun L, Li J & Xiao X Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat. Med 6, 599–602 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Chew WL et al. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods 13, 868–874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schucht R et al. A new generation of retroviral producer cells: predictable and stable virus production by Flp-mediated site-specific integration of retroviral vectors. Mol. Ther 14, 285–292 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Follenzi A, Sabatino G, Lombardo A, Boccaccio C & Naldini L Efficient gene delivery and targeted expression to hepatocytes in vivo by improved lentiviral vectors. Hum. Gene Ther 13, 243–260 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Blomer U et al. Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J. Virol 71, 6641–6649 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abordo-Adesida E et al. Stability of lentiviral vector-mediated transgene expression in the brain in the presence of systemic antivector immune responses. Hum. Gene Ther 16, 741–751 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wanisch K & Yanez-Munoz RJ Integration-deficient lentiviral vectors: a slow coming of age. Mol. Ther 17, 1316–1332 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heckl D et al. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol 32, 941–946 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Platt RJ et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.LaFleur MW et al. A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nat. Commun 10, 1668 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mangeot PE et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat Commun 10, 45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Candolfi M et al. Effective high-capacity gutless adenoviral vectors mediate transgene expression in human glioma cells. Mol. Ther 14, 371–381 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ding Q et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res 115, 488–492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bjursell M et al. Therapeutic genome editing with CRISPR/Cas9 in a humanized mouse model ameliorates alpha1-antitrypsin deficiency phenotype. EBioMedicine 29, 104–111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chadwick AC, Wang X & Musunuru K In vivo base editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a therapeutic alternative to genome editing. Arterioscler. Thromb. Vasc. Biol 37, 1741–1747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li A et al. A Self-deleting AAV-CRISPR system for in vivo genome editing. Mol. Ther. Methods Clin. Dev 12, 111–122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boutin S et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther 21, 704–712 (2010). [DOI] [PubMed] [Google Scholar]
- 77.Colella P, Ronzitti G & Mingozzi F Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev 8, 87–104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meliani A et al. Determination of anti-adeno-associated virus vector neutralizing antibody titer with an in vitro reporter system. Hum. Gene Ther. Methods 26, 45–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mendell JR et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med 377, 1713–1722 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Vandamme C, Adjali O & Mingozzi F Unraveling the complex story of immune responses to AAV vectors trial after trial. Hum. Gene Ther 28, 1061–1074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hinderer C et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther 29, 285–298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van der Loo JC & Wright JF Progress and challenges in viral vector manufacturing. Hum. Mol. Genet 25, R42–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Getts DR, Shea LD, Miller SD & King NJ Harnessing nanoparticles for immune modulation. Trends Immunol. 36, 419–427 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang HX et al. CRISPR/Cas9-based genome editing for disease modeling and therapy: Challenges and opportunities for nonviral delivery. Chem. Rev 117, 9874–9906 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Zuris JA et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol 33, 73–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gao X et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Z et al. Targeted delivery of CRISPR/Cas9-mediated cancer gene therapy via liposome-templated hydrogel nanoparticles. Adv. Funct. Mater 27, 1703036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee K et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng 1, 889–901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee B et al. Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile × syndrome from exaggerated repetitive behaviours. Nat. Biomed. Eng 2, 497–507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pan Y et al. Near-infrared upconversion-activated CRISPR-Cas9 system: A remote-controlled gene editing platform. Sci. Adv 5, eaav7199 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lao YH et al. HPV oncogene manipulation using nonvirally delivered CRISPR/Cas9 or Natronobacterium gregoryi Argonaute. Adv. Sci 5, 1700540 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang HX et al. Nonviral gene editing via CRISPR/Cas9 delivery by membrane-disruptive and endosomolytic helical polypeptide. Proc. Natl. Acad. Sci. USA 115, 4903–4908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jiang C et al. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res 27, 440–443 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yin H et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol 34, 328–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu H et al. Spatial control of in vivo CRISPR-Cas9 genome editing via nanomagnets. Nat. Biomed. Eng 3, 126–136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nesargikar PN, Spiller B & Chavez R The complement system: history, pathways, cascade and inhibitors. Eur. J. Microbiol. Immunol. (Bp) 2, 103–111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Oh P et al. Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung. Nat. Biotechnol 25, 327–337 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Qiu Y et al. Magnetic forces enable controlled drug delivery by disrupting endothelial cell-cell junctions. Nat. Commun 8, 15594 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rapoport SI Advances in osmotic opening of the blood-brain barrier to enhance CNS chemotherapy. Expert Opin. Investig. Drugs 10, 1809–1818 (2001). [DOI] [PubMed] [Google Scholar]
- 100.Timbie KF, Mead BP & Price RJ Drug and gene delivery across the blood-brain barrier with focused ultrasound. J.Control. Release 219, 61–75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Monsky WL et al. Augmentation of transvascular transport of macromolecules and nanoparticles in tumors using vascular endothelial growth factor. Cancer Res. 59, 4129–4135 (1999). [PubMed] [Google Scholar]
- 102.Wong KM, Horton KJ, Coveler AL, Hingorani SR & Harris WP Targeting the Tumor Stroma: the Biology and Clinical Development of Pegylated Recombinant Human Hyaluronidase (PEGPH20). Curr. Oncol. Rep 19, 47 (2017). [DOI] [PubMed] [Google Scholar]
- 103.Senturk S et al. Rapid and tunable method to temporally control gene editing based on conditional Cas9 stabilization. Nat. Commun 8, 14370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maji B et al. A High-Throughput Platform to Identify Small-Molecule Inhibitors of CRISPR-Cas9. Cell 177, 1067–1079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou XX et al. A Single-Chain Photoswitchable CRISPR-Cas9 Architecture for Light-Inducible Gene Editing and Transcription. ACS Chem. Biol 13, 443–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nihongaki Y, Kawano F, Nakajima T & Sato M Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol 33, 755–760 (2015). [DOI] [PubMed] [Google Scholar]
- 107.Wilhelm S et al. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater 1, 16014 (2016). [Google Scholar]
- 108.Joshi M, Pathak S, Sharma S & Patravale V Design and in vivo pharmacodynamic evaluation of nanostructured lipid carriers for parenteral delivery of artemether: Nanoject. Int. J. Pharm 364, 119–126 (2008). [DOI] [PubMed] [Google Scholar]
- 109.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sternberg SH, Redding S, Jinek M, Greene EC & Doudna JA DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.DeWitt MA et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med 8, 360ra134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Capecchi MR Altering the genome by homologous recombination. Science 244, 1288–1292 (1989). [DOI] [PubMed] [Google Scholar]
- 113.Rivera-Torres N, Banas K, Bialk P, Bloh KM & Kmiec EB Insertional mutagenesis by CRISPR/Cas9 ribonucleoprotein gene editing in cells targeted for point mutation repair directed by short single-stranded DNA oligonucleotides. PLoS One 12, e0169350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park SH et al. Highly efficient editing of the beta-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 47, 7955–7972 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Byrne SM, Ortiz L, Mali P, Aach J & Church GM Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Res. 43, e21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Eoh J & Gu L Biomaterials as vectors for the delivery of CRISPR-Cas9. Biomater Sci 7, 1240–1261 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Yin H et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol 35, 1179–1187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lu B et al. Delivering SaCas9 mRNA by lentivirus-like bionanoparticles for transient expression and efficient genome editing. Nucleic Acids Res. 47, e44 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]