

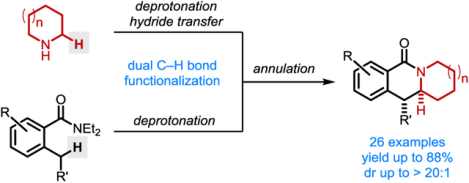
Abstract
Polycyclic lactams are prepared in a single operation from o-toluamides and cyclic amines in a process that involves transient cyclic imines, species that are conveniently obtained in situ from the corresponding lithium amides and simple ketone oxidants. Imines thus generated, such as 1-pyrroline and 1-piperideine, engage lithiated o-toluamides in a facile annulation process. Undesired side reactions such as imine deprotonation and o-toluamide dimerization are suppressed through judicious choice of reaction conditions.
Graphical Abstract
The C–H bond functionalization of amines, in particular alicyclic amines, is an attractive strategy for accessing functionalized amines from their parent heterocycles,1 providing access to valuable pharmacophores.2 However, despite considerable advances, the vast majority of methods developed to date are incompatible with the presence of an amine N–H bond, limiting their utility to tertiary or protected amines.3,4 While notable exceptions exist, such as hydroaminoalkylation and electrochemical α-cyanation,5 these methods are not applicable to direct annulations involving the amine nitrogen atom. Currently, the most general method to achieve the annulation of amines via concomitant N–H and α-C–H bond functionalization appears to be the redox-neutral condensation of amines with aldehydes bearing a covalently linked (pro)nucleophile (e.g., 1 → 2, Figure 1a).3j These transformations are typically facilitated by simple carboxylic acids and have shown to exhibit a rather broad substrate scope.6 An important limitation of redox-annulations is the need for an activated nucleophilic site on the aldehyde reaction partner. While highly attractive from a synthetic point of view, simple aryl groups and benzylic methyl groups are not sufficiently activated to participate in redox-annulations. In addition, redox-annulations of aldehyde substrates on the lower end of the reactivity scale are typically limited to relatively activated amines such as 1,2,3,4-tetrahydroisoquinoline. Here we report a new method for the α-C–H/N–H annulation of alicyclic amines to provide products that are inaccessible via redox-annulation approaches.
Figure 1.
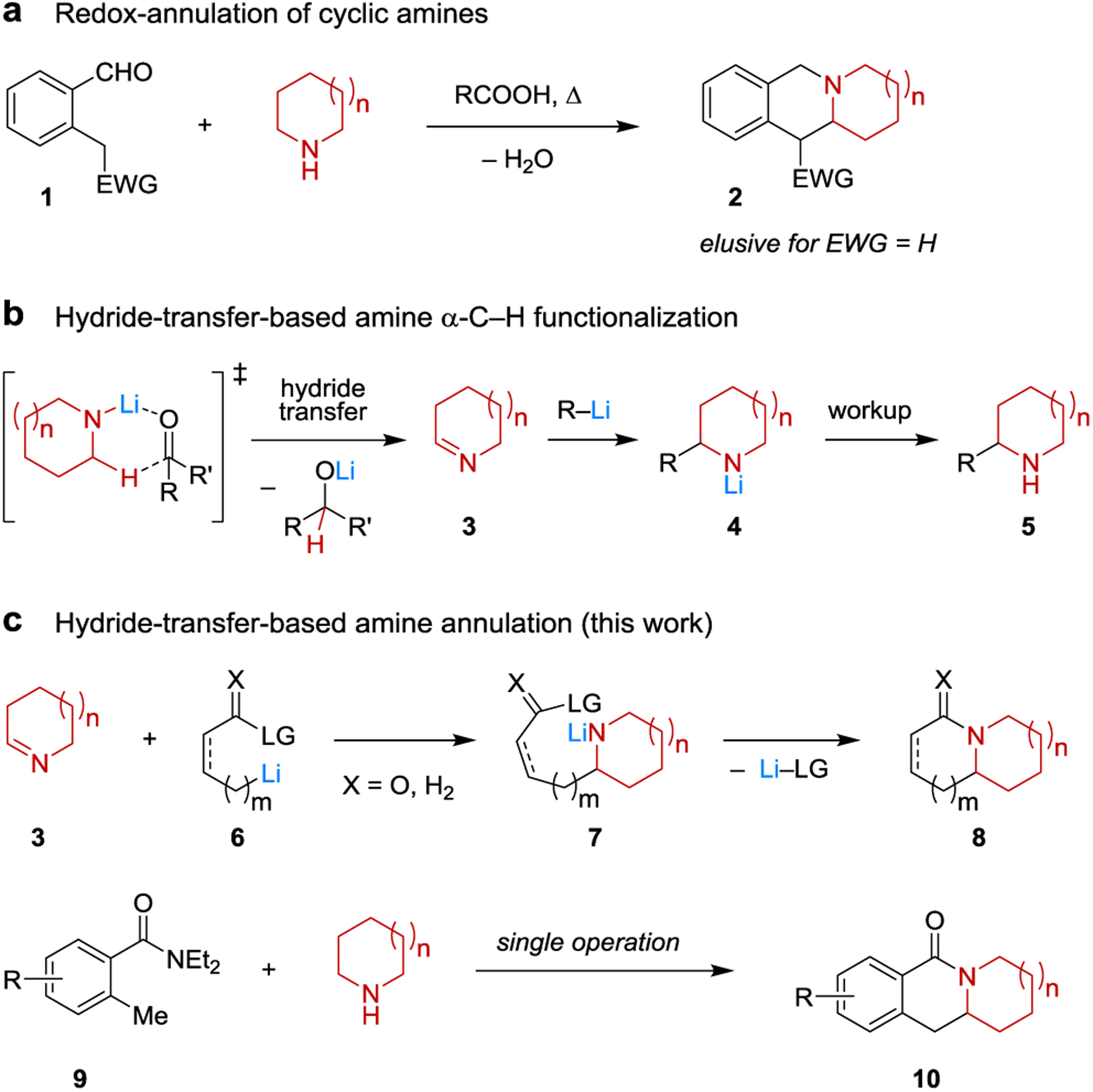
Selected Precedent and New Concept for Amine Annulation.
Inspired by seminal studies by Wittig and coworkers published about half a century ago,7 we recently developed a new method for the α-C–H bond functionalization of secondary amines that utilizes the ability of lithiated amines to act as hydride donors (Figure 1b).8 Readily available ketones such as benzophenone act as hydride acceptors, facilitating the formation of transient cyclic imine 3 and a lithium alkoxide. Imine 3 subsequently engages an organolithium reagent, resulting in lithium amide intermediate 4, providing amine 5 upon workup.8a In the presence of appropriate Lewis acids, other organometallic species such as Grignard reagents and enolates can also be added to 3.8b,8e While some imines of type 3 (e.g., 1-pyrroline and 1-piperideine) are well-known and have been prepared by other means, their propensity to trimerize9 has previously limited their broader use in reactions that involve strong nucleophiles.10 We hypothesized that the hydride transfer strategy to access imine monomers in situ could potentially be applied to an annulation process that further harnesses the reactivity of intermediate 4 without the need for an additional reagent. Specifically, an organolithium nucleophile containing a strategically placed electrophile/leaving group (e.g. 6), upon reacting with imine 3, would form lithium amide intermediate 7 (Figure 1c). The latter could subsequently undergo ring-closure to form annulation product 8. A particularly attractive variant of this strategy would be the synthesis of polycyclic lactams 10 from o-toluamides 9. It should be noted that this processes is significantly more challenging than our previously reported transformations.8 Due to the increased bulk of the nucleophile, deprotonation of the enolizable imine might become competitive with the desired nucleophilic addition. In addition, the reduced nucleophilicity of the annulation partner can likely not be compensated for by the addition of Lewis acid additives, which are expected to be incompatible with the desired transformations.
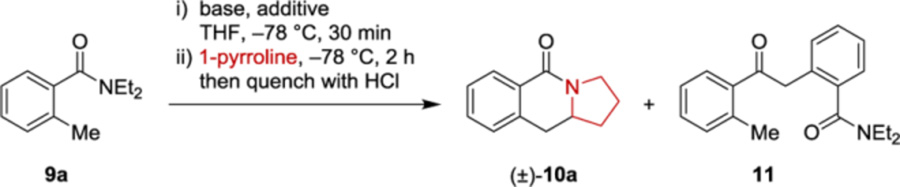
The proposed lactam-forming annulation process was evaluated with pyrrolidine and N,N-diethyl-o-toluamide (9a) as the model substrates (Table 1). Motivation for utilizing this substrate combination was provided by the fact that benzoindolizidinones, benzoquinolizidinones, and other compounds related to structure 10 represent important structural motifs found in a range of natural products and synthetic bioactive materials.11 Methods for the synthesis of such polycyclic compounds remain limited and typically require the multistep construction of the fused rings with a series of functional group interconversions, ultimately limiting the utility of these compounds as a platform for synthesis and drug discovery. While it has been shown that lithiated N,N-diethyl-o-toluamides undergo the corresponding reaction with acyclic imines and stable, non-enolizable dihydroisoquinolines,12 the enolizable nature of alicyclic imines and their propensity to rapidly undergo transformation to unreactive imine trimers represent significant challenges (vide supra). Conditions for the deprotonation of 9a previously developed by Clark and coworkers were tested first.12a Accordingly, 9a was treated with LDA (1.5 equiv) in THF solution at −78 °C for 30 min, followed by addition of an ether solution of 1-pyrroline. The latter was prepared in a separate flask from pyrrolidine, n-BuLi, and benzophenone (2 equiv each). Desired product 10a was obtained in 34% yield (Table 1, entry 1). In addition, side product 11 was isolated in 15% yield, resulting from the undesired dimerization of 9a. A moderate improvement in the yield of 10a was observed with three equiv of imine, with concurrent reduction of the amount of dimer 11 (entry 2). An increase in the amount of LDA used to deprotonate 9a resulted in a further increase in yield (entry 3). Based on precedent, s-butyllithium (s-BuLi) was also tested in the lithiation of o-toluamide. However, a lower yield of product 10a was obtained, despite of no obvious formation of 11 (entry 4). Given their known propensity to impart a strong influence on the aggregation state and the reactivity of organolithium reagents,13 various additives were then evaluated. Strongly coordinating ligands such as TMEDA, HMPA and DMPU all provided improved yields regardless of the base (entries 5–9). In all cases, LDA proved superior to s-BuLi. The sterically more demanding base lithium 2,2,6,6-tetramethylpiperidide (LiTMP) provided results similar to LDA (entry 10). Interestingly, product 10a was obtained in 72% yield upon the addition of LiCl (entry 11).14 A brief evaluation of the hydride acceptor utilized in the preparation of 1-pyrroline revealed that trifluoroacetophenone performed slightly worse than benzophenone (entry 12). Reduced amounts of starting materials (1.5 equiv of base and 2 equiv of 1-pyrroline) were then tested in combination with the two most effective additives (HMPA and LiCl) (entries 13–16). No obvious negative effects on the yield of 10a were observed. Under these more favorable conditions, trifluoroacetophenone was superior to benzophenone as the hydride acceptor. Notably, at most trace amounts of dimer 11 were observed in the presence of any additive tested. Finally, the amount of LiCl was varied. With catalytic amounts of LiCl and no LiCl the yield of the annulation product decreased to 61% and 42%, respectively (entries 17, 18).
Table 1.
Evaluation of Reaction Parameters for the Annulation of N,N-Diethyl-o-Toluamide with 1-Pyrroline.a
 | |||||
|---|---|---|---|---|---|
| entry | base (equiv) | additive (equiv) | hydride acceptor | yield of 10a (%) | yield of 11 (%) |
| 1b | LDA (1.5) | − | Ph2CO | 34 | 15 |
| 2 | LDA (1.5) | − | Ph2CO | 41 | 8 |
| 3 | LDA (2) | − | Ph2CO | 54 | 6 |
| 4 | s-BuLi (2) | − | Ph2CO | 45 | ND |
| 5 | LDA (2) | TMEDA (2) | Ph2CO | 58 | ND |
| 6 | s-BuLi (2) | TMEDA (2) | Ph2CO | 51 | ND |
| 7 | LDA (2) | HMPA (4) | Ph2CO | 68 | ND |
| 8 | s-BuLi (2) | HMPA (4) | Ph2CO | 55 | ND |
| 9 | LDA (2) | DMPU (4) | Ph2CO | 66 | ND |
| 10 | LiTMP (2) | DMPU (4) | Ph2CO | 64 | ND |
| 11 | LDA (2) | LiCl (2) | Ph2CO | 72 | ND |
| 12 | LDA (2) | LiCl (2) | PhCOCF3 | 66 | ND |
| 13b | LDA (1.5) | HMPA (3) | Ph2CO | 59 | ND |
| 14b | LDA (1.5) | HMPA (3) | PhCOCF3 | 65 | ND |
| 15b | LDA (1.5) | LiCl (1.5) | Ph2CO | 56 | ND |
| 16b | LDA (1.5) | LiCl (1.5) | PhCOCF3 | 70 | ND |
| 17b | LDA (1.5) | LiCl (0.3) | PhCOCF3 | 61 | ND |
| 18b | LDA (1.5) | − | PhCOCF3 | 42 | 7 |
Yields correspond to isolated yields. Reactions were performed with 0.5 mmol of 9a. THF (2.5 mL) was used for the lithiation of 9a. 1-Pyrroline was prepared in situ by adding n-BuLi (3 equiv) to a solution of pyrrolidine (3 equiv) in ether (1 mL) at −78 °C, followed by the addition of the hydride acceptor (3 equiv).
2 equiv of pyrrolidine, n-BuLi and the hydride acceptor were used for the preparation of 1-pyrroline.
The scope of the annulation was then examined as summarized in Scheme 1. o-Toluamides bearing various substituents on the phenyl ring readily participated in the reaction. Generally, electron-deficient o-toluamides provided higher yields of the lactam products than those with electron-donating substituents. Multiple factors, acting individually or in concert, could potentially account for this: 1) the increased acidity of protons in the benzylic ortho-position enables a more efficient deprotonation to generate lithiated o-toluamide; 2) better stabilization of the benzylic anion; 3) the cyclization step is facilitated due to the increased electrophilicity of the amide group. Substrates with a substituent in the other ortho position of the amide group required elevated temperatures for the lithiation and provided lower product yields. Most likely, the additional ortho-substituent prevents the amide from being coplanar with the methyl group, a requirement for achieving optimal results in the directed lithiation. The lithiation of the electron-rich o-toluamide required to prepare product 10n was found to be inefficient with LDA. In this case s-BuLi/TMEDA provided superior results. Heteroaromatic amides also participated in this annulation chemistry and provided the corresponding pyridine and furan-containing products 10p and 10q in acceptable yields. The annulation process tolerates a wide scope of cyclic imines. Cyclic imines with expanded ring sizes, bicyclic imines, cyclic imines with remote substituents, and N-alkyl piperazine-derived imines were all viable substrates and produced the corresponding lactams in moderate to good yields and excellent diastereoselectivities. The imine derived from 2-methylpiperidine provided the corresponding product 10v in low yield, presumably due to unfavorable steric interactions in the course of the reaction. The o-ethyl benzamide starting material required for the synthesis of product 10y was a challenging substrate to be lithiated, requiring two equiv of s-BuLi/TMEDA. However, the addition/ring closure steps proceeded smoothly at −78 °C and provided 10y in 56% yield as a 2:1 mixture of diastereomers. An o-benzyl group facilitated the lithiation of the corresponding benzamide. Given the reduced nucleophilicity of the resulting organolithiate, the addition/ring-closure steps required an increase in reaction temperature. Regardless, product 10z was obtained in good yield and excellent diastereoselectivity.
Scheme 1. Scope of the Annulation of N,N-Diethyl-o-Toluamides with Cyclic Imines Generated in Situ and Applications.
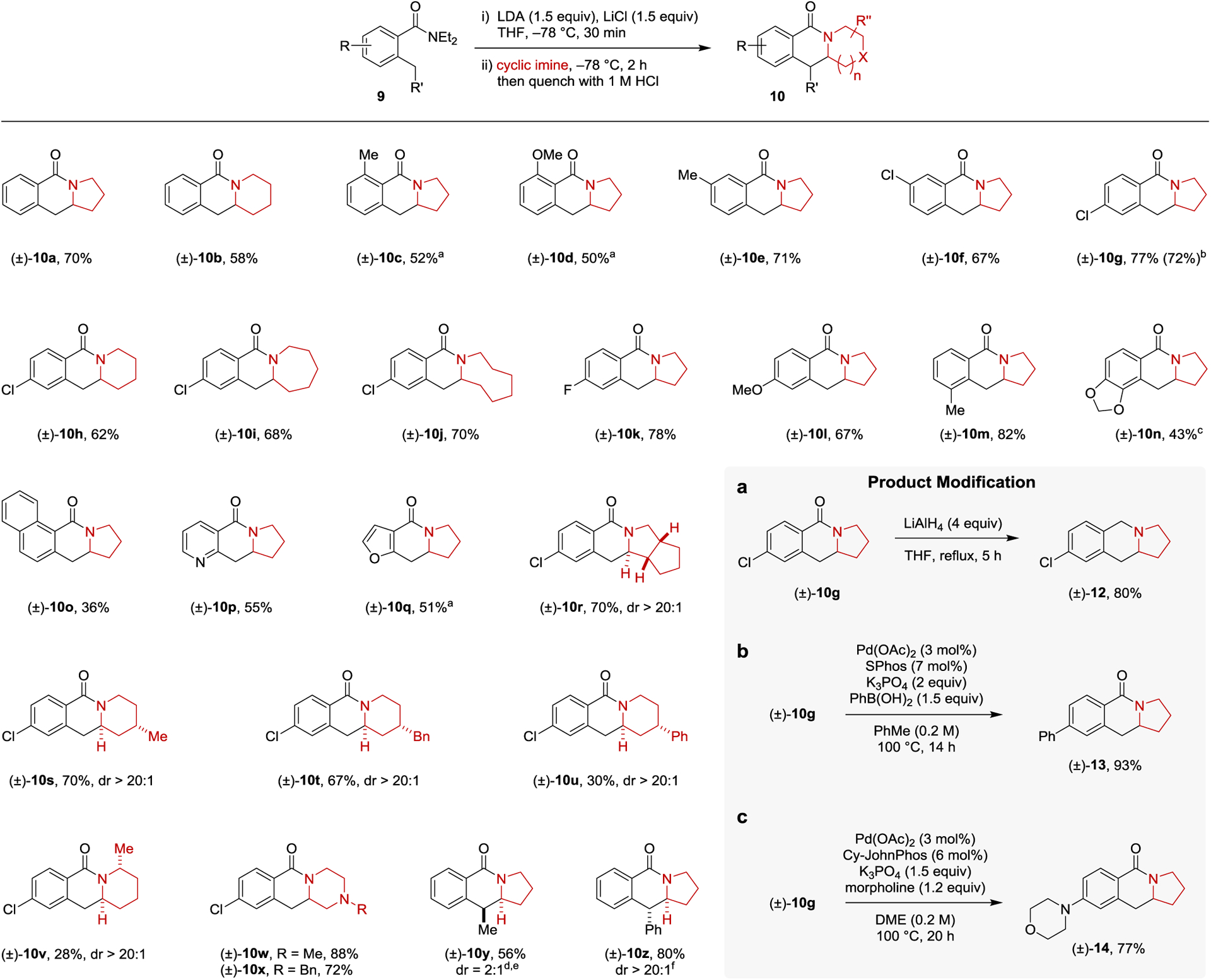
Reactions were performed with 0.5 mmol of 9. Yields correspond to isolated yields. THF (2.5 mL) was used for the lithiation of 9. Cyclic imines were prepared in situ by adding n-BuLi (2 equiv) to a solution of the corresponding cyclic amine (2 equiv) in ether (1 mL) at −78 °C, followed by the addition of trifluoroacetophenone (2 equiv). a o-Toluamide lithiation was performed at −40 °C. b Yield in parenthesis corresponds to reaction conducted on a 2 mmol scale. c o-Toluamide lithiation was performed using s-BuLi/TMEDA (1.5 equiv each) at −78 °C for 30 min. d o-Toluamide lithiation was performed using s-BuLi/TMEDA (2 equiv each) at −78°C for 1 h. e 3 Equiv of 1-pyrroline was used. f Reaction was warmed up to room temperature over 30 min after 1-pyrroline was added. Product Modification a, Reduction of lactam to amine. b, Suzuki-Miyaura coupling. c, Buchwald-Hartwig coupling.
The tricyclic lactams obtained from the annulation of o-toluamides and cyclic imines could be readily utilized to access other structurally diverse compounds. For instance, reduction of compound 10g with lithium aluminum hydride (LiAlH4) furnished benzoindolizidine 12 in 80% yield. Suzuki-Miyaura coupling15 of 10g with phenyl boronic acid resulted in the formation of product 13 in 93% yield. Buchwald-Hartwig coupling16 involving 10g provided 14 in 77% yield. These products would likely be difficult to prepare via direct annulation reactions due to the inaccessibility or unfavorable electronic characteristics of the corresponding starting materials.
In conclusion, we have achieved annulation reactions of lithiated o-toluamides with enolizable cyclic imines, elusive species that were prepared in situ via the intermolecular hydride transfer of the corresponding lithiated amines onto a simple ketone acceptor. This methodology allows for the facile construction of structurally diverse polycyclic lactams in a single operation, dramatically simplifying access to these materials.
Supplementary Material
ACKNOWLEDGMENT
Financial support from the NIH-NIGMS (Grant R01GM101389) is gratefully acknowledged. Mass spectrometry instrumentation was supported by a grant from the NIH (S10 OD021758-1A1).
Footnotes
Supporting Information
Experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Organic synthesis provides opportunities to transform drug discovery. Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Nat. Chem 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- (2).(a) Rings in Drugs. Taylor RD; MacCoss M; Lawson ADG J. Med. Chem 2014, 57, 5845–5859; [DOI] [PubMed] [Google Scholar]; (b) Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. Vitaku E; Smith DT; Njardarson JT J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (3).Selected reviews on amine C–H bond functionalization:; (a) Campos KR, Direct sp3 C-H bond activation adjacent to nitrogen in heterocycles. Chem. Soc. Rev 2007, 36, 1069–1084; [DOI] [PubMed] [Google Scholar]; (b) Jazzar R; Hitce J; Renaudat A; Sofack-Kreutzer J; Baudoin O, Functionalization of Organic Molecules by Transition-Metal-Catalyzed C(sp3)-H Activation. Chem. Eur. J 2010, 16, 2654–2672; [DOI] [PubMed] [Google Scholar]; (c) Yeung CS; Dong VM, Catalytic Dehydrogenative Cross-Coupling: Forming Carbon-Carbon Bonds by Oxidizing Two Carbon-Hydrogen Bonds. Chem. Rev 2011, 111, 1215–1292; [DOI] [PubMed] [Google Scholar]; (d) Mitchell EA; Peschiulli A; Lefevre N; Meerpoel L; Maes BUW, Direct alpha-Functionalization of Saturated Cyclic Amines. Chem. Eur. J 2012, 18, 10092–10142; [DOI] [PubMed] [Google Scholar]; (e) Peng B; Maulide N, The Redox-Neutral Approach to C-H Functionalization. Chem. Eur. J 2013, 19, 13274–13287; [DOI] [PubMed] [Google Scholar]; (f) Girard SA; Knauber T; Li C-J, The Cross-Dehydrogenative Coupling of C sp3-H Bonds: A Versatile Strategy for C-C Bond Formations. Angew. Chem. Int. Ed 2014, 53, 74–100; [DOI] [PubMed] [Google Scholar]; (g) Haibach MC; Seidel D, C-H Bond Functionalization through Intramolecular Hydride Transfer. Angew. Chem. Int. Ed 2014, 53, 5010–5036; [DOI] [PubMed] [Google Scholar]; (h) Vo C-VT; Bode JW, Synthesis of Saturated N-Heterocycles. J. Org. Chem 2014, 79, 2809–2815; [DOI] [PubMed] [Google Scholar]; (i) Qin Y; Lv J; Luo S, Catalytic asymmetric α-C(sp3)–H functionalization of amines. Tetrahedron Lett. 2014, 55, 551–558; [Google Scholar]; (j) Seidel D, The Azomethine Ylide Route to Amine C–H Functionalization: Redox-Versions of Classic Reactions and a Pathway to New Transformations. Acc. Chem. Res 2015, 48, 317–328; [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Beatty JW; Stephenson CRJ, Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res 2015, 48, 1474–1484; [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Mahato S; Jana CK, Classical-Reaction-Driven Stereo- and Regioselective C(sp3)–H Functionalization of Aliphatic Amines. Chem. Rec 2016, 16, 1477–1488; [DOI] [PubMed] [Google Scholar]; (m) Qin Y; Zhu L; Luo S, Organocatalysis in Inert C–H Bond Functionalization. Chem. Rev 2017, 117, 9433–9520; [DOI] [PubMed] [Google Scholar]; (n) Cheng M-X; Yang S-D, Recent Advances in the Enantioselective Oxidative α-C–H Functionalization of Amines. Synlett 2017, 28, 159–174; [Google Scholar]; (o) Chu JCK; Rovis T, Complementary Strategies for Directed C(sp3)–H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem. Int. Ed 2018, 57, 62–101; [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Liu S; Zhao Z; Wang Y, Construction of N-Heterocycles through Cyclization of Tertiary Amines. Chem. Eur. J 2019, 25, 2423–2441; [DOI] [PubMed] [Google Scholar]; (q) Antermite D; Bull JA, Transition Metal-Catalyzed Directed C(sp3)–H Functionalization of Saturated Heterocycles. Synthesis 2019, 51, 3171–3204; [Google Scholar]; (r) Trowbridge A; Walton SM; Gaunt MJ, New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines. Chem. Rev 2020, 120, 2613–2692. [DOI] [PubMed] [Google Scholar]
- (4).Selected recent examples of mechanistically diverse methods for amine C–H bond functionalization:; (a) Jiang H-J; Zhong X-M; Yu J; Zhang Y; Zhang X; Wu Y-D; Gong L-Z, Assembling a Hybrid Pd Catalyst from a Chiral Anionic CoIII Complex and Ligand for Asymmetric C(sp3)–H Functionalization. Angew. Chem. Int. Ed 2019, 58, 1803–1807; [DOI] [PubMed] [Google Scholar]; (b) Ashley MA; Yamauchi C; Chu JCK; Otsuka S; Yorimitsu H; Rovis T, Photoredox-Catalyzed Site-Selective α-C(sp3)–H Alkylation of Primary Amine Derivatives. Angew. Chem. Int. Ed 2019, 58, 4002–4006; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guin S; Dolui P; Zhang X; Paul S; Singh VK; Pradhan S; Chandrashekar HB; Anjana SS; Paton RS; Maiti D, Iterative Arylation of Amino Acids and Aliphatic Amines via δ-C(sp3)–H Activation: Experimental and Computational Exploration. Angew. Chem. Int. Ed 2019, 58, 5633–5638; [DOI] [PubMed] [Google Scholar]; (d) Whitehurst WG; Blackwell JH; Hermann GN; Gaunt MJ, Carboxylate-Assisted Oxidative Addition to Aminoalkyl PdII Complexes: C(sp3)–H Arylation of Alkylamines by Distinct PdII/PdIV Pathway. Angew. Chem. Int. Ed 2019, 58, 9054–9059; [DOI] [PubMed] [Google Scholar]; (e) Ma Y; Yao X; Zhang L; Ni P; Cheng R; Ye J, Direct Arylation of α-Amino C(sp3)-H Bonds by Convergent Paired Electrolysis. Angew. Chem. Int. Ed 2019, 58, 16548–16552; [DOI] [PubMed] [Google Scholar]; (f) Grainger R; Heightman TD; Ley Steven V.; Lima F; Johnson CN, Enabling synthesis in fragment-based drug discovery by reactivity mapping: photoredox-mediated cross-dehydrogenative heteroarylation of cyclic amines. Chem. Sci 2019, 10, 2264–2271; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Vasu D; Fuentes de Arriba AL; Leitch JA; de Gombert A; Dixon DJ, Primary α-tertiary amine synthesis via α-C–H functionalization. Chem. Sci 2019, 10, 3401–3407; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Asako S; Ishihara S; Hirata K; Takai K, Deoxygenative Insertion of Carbonyl Carbon into a C(sp3)–H Bond: Synthesis of Indolines and Indoles. J. Am. Chem. Soc 2019, 141, 9832–9836; [DOI] [PubMed] [Google Scholar]; (i) Lin W; Zhang K-F; Baudoin O, Regiodivergent enantioselective C–H functionalization of Boc-1,3-oxazinanes for the synthesis of β2- and β3-amino acids. Nat. Catal 2019, 2, 882–888; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Chan JZ; Chang Y; Wasa M, B(C6F5)3-Catalyzed C–H Alkylation of N-Alkylamines Using Silicon Enolates without External Oxidant. Org. Lett 2019, 21, 984–988; [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Zhou L; Shen Y-B; An X-D; Li X-J; Li S-S; Liu Q; Xiao J, Redox-Neutral β-C(sp3)–H Functionalization of Cyclic Amines via Intermolecular Hydride Transfer. Org. Lett 2019, 21, 8543–8547; [DOI] [PubMed] [Google Scholar]; (l) Kataoka M; Otawa Y; Ido N; Mori K, Highly Diastereoselective Synthesis of Medium-Sized Carbocycle-Fused Piperidines via Sequential Hydride Shift Triggered Double C(sp3)–H Bond Functionalization. Org. Lett 2019, 21, 9334–9338; [DOI] [PubMed] [Google Scholar]; (m) Lee M; Adams A; Cox PB; Sanford MS, Access to 3D Alicyclic Amine-Containing Fragments through Transannular C–H Arylation. Synlett 2019, 30, 417–422; [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Kapoor M; Chand-Thakuri P; Maxwell JM; Liu D; Zhou H; Young MC, Carbon Dioxide-Driven Palladium-Catalyzed C–H Activation of Amines: A Unified Approach for the Arylation of Aliphatic and Aromatic Primary and Secondary Amines. Synlett 2019, 30, 519–524; [Google Scholar]; (o) Ohmatsu K; Suzuki R; Furukawa Y; Sato M; Ooi T, Zwitterionic 1,2,3-Triazolium Amidate as a Catalyst for Photoinduced Hydrogen-Atom Transfer Radical Alkylation. ACS Catal. 2020, 10, 2627–2632; [Google Scholar]; (p) Roque JB; Kuroda Y; Jurczyk J; Xu L-P; Ham JS; Göttemann LT; Roberts CA; Adpressa D; Saurí J; Joyce LA; Musaev DG; Yeung CS; Sarpong R, C–C Cleavage Approach to C–H Functionalization of Saturated Aza-Cycles. ACS Catal. 2020, 10, 2929–2941; [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Rand AW; Yin H; Xu L; Giacoboni J; Martin-Montero R; Romano C; Montgomery J; Martin R, Dual Catalytic Platform for Enabling sp3 α C–H Arylation and Alkylation of Benzamides. ACS Catal. 2020, 10, 4671–4676; [Google Scholar]; (r) Liu W; Babl T; Röther A; Reiser O; Davies HML, Functionalization of Piperidine Derivatives for the Site-Selective and Stereoselective Synthesis of Positional Analogues of Methylphenidate. Chem. Eur. J 2020, 26, 4236–4241; [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Verma P; Richter JM; Chekshin N; Qiao JX; Yu J-Q, Iridium(I)-Catalyzed α-C(sp3)–H Alkylation of Saturated Azacycles. J. Am. Chem. Soc 2020, 142, 5117–5125; [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Walker MM; Koronkiewicz B; Chen S; Houk KN; Mayer JM; Ellman JA, Highly Diastereoselective Functionalization of Piperidines by Photoredox-Catalyzed α-Amino C–H Arylation and Epimerization. J. Am. Chem. Soc 2020, 142, 8194–8202; [DOI] [PMC free article] [PubMed] [Google Scholar]; (u) Feng K; Quevedo RE; Kohrt JT; Oderinde MS; Reilly U; White MC, Late-stage oxidative C(sp3)–H methylation. Nature 2020, 580, 621–627; [DOI] [PMC free article] [PubMed] [Google Scholar]; (v) Sarver PJ; Bacauanu V; Schultz DM; DiRocco DA; Lam Y.-h.; Sherer E C.; MacMillan DWC, The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)–H trifluoromethylation. Nat. Chem 2020, 12, 459–467; [DOI] [PubMed] [Google Scholar]; (w) McManus JB; Onuska NPR; Jeffreys MS; Goodwin NC; Nicewicz DA, Site-Selective C–H Alkylation of Piperazine Substrates via Organic Photoredox Catalysis. Org. Lett 2020, 22, 679–683; [DOI] [PubMed] [Google Scholar]; (x) Oeschger R; Su B; Yu I; Ehinger C; Romero E; He S; Hartwig J, Diverse functionalization of strong alkyl C–H bonds by undirected borylation. Science 2020, 368, 736–741; [DOI] [PMC free article] [PubMed] [Google Scholar]; (y) Short MA; Blackburn JM; Roizen JL, Modifying Positional Selectivity in C–H Functionalization Reactions with Nitrogen-Centered Radicals: Generalizable Approaches to 1,6-Hydrogen-Atom Transfer Processes. Synlett 2020, 31, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Edwards PM; Schafer LL, Early transition metal-catalyzed C–H alkylation: hydroaminoalkylation for Csp3–Csp3 bond formation in the synthesis of selectively substituted amines. Chem. Commun 2018, 54, 12543–12560; [DOI] [PubMed] [Google Scholar]; (b) Lennox AJJ; Goes SL; Webster MP; Koolman HF; Djuric SW; Stahl SS, Electrochemical Aminoxyl-Mediated α-Cyanation of Secondary Piperidines for Pharmaceutical Building Block Diversification. J. Am. Chem. Soc 2018, 140, 11227–11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Selected examples of redox-annulations:; (a) Zhang C; De CK; Mal R; Seidel D, alpha-Amination of Nitrogen Heterocycles: Ring-Fused Aminals. J. Am. Chem. Soc 2008, 130, 416–417; [DOI] [PubMed] [Google Scholar]; (b) Zheng L; Yang F; Dang Q; Bai X, A Cascade Reaction with Iminium Ion Isomerization as the Key Step Leading to Tetrahydropyrimido[4,5-d]pyrimidines. Org. Lett 2008, 10, 889–892; [DOI] [PubMed] [Google Scholar]; (c) Zhang C; Das D; Seidel D, Azomethine ylide annulations: facile access to polycyclic ring systems. Chem. Sci 2011, 2, 233–236; [Google Scholar]; (d) Dieckmann A; Richers MT; Platonova AY; Zhang C; Seidel D; Houk KN, Metal-Free α-Amination of Secondary Amines: Computational and Experimental Evidence for Azaquinone Methide and Azomethine Ylide Intermediates. J. Org. Chem 2013, 78, 4132–4144; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Richers MT; Breugst M; Platonova AY; Ullrich A; Dieckmann A; Houk KN; Seidel D, Redox-Neutral α-Oxygenation of Amines: Reaction Development and Elucidation of the Mechanism. J. Am. Chem. Soc 2014, 136, 6123–6135; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kang Y; Chen W; Breugst M; Seidel D, Asymmetric Redox-Annulation of Cyclic Amines. J. Org. Chem 2015, 80, 9628–9640; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Chen W; Seidel D, Redox-Annulation of Cyclic Amines and β-Ketoaldehydes. Org. Lett 2016, 18, 1024–1027; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Li J; Fu Y; Qin C; Yu Y; Li H; Wang W, Asymmetric synthesis of isoquinolinonaphthyridines catalyzed by a chiral Bronsted acid. Org. Biomol. Chem 2017, 15, 6474–6477; [DOI] [PubMed] [Google Scholar]; (i) Liu Y; Wu J; Jin Z; Jiang H, Synthesis of 1,2-Fused Bicyclic Imidazolidin-4-ones by Redox-Neutral Cyclization Reaction of Cyclic Amines and α-Ketoamides. Synlett 2018, 29, 1061–1064; [Google Scholar]; (j) Paul A; Chandak HS; Ma L; Seidel D, Redox-Annulations of Cyclic Amines with ortho-Cyanomethylbenzaldehydes. Org. Lett 2020, 22, 976–980; [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Rickertsen DRL; Ma L; Paul A; Abboud KA; Seidel D, Traceless Redox-Annulations of Alicyclic Amines. SynOpen 2020, 04, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Reduction with lithium dialkylamides. Majewski M; Gleave DM J. Organomet. Chem 1994, 470, 1–16; [Google Scholar]; (b) Über Lithium‐diäthylamid als Hydrid‐Donator. Wittig G; Schmidt HJ; Renner H. Chem. Ber 1962, 95, 2377–2383; [Google Scholar]; (c) Hydrid-Übertragung von Lithium-pyrrolidid auf Azomethine. Wittig G; Hesse A. Liebigs Ann. Chem 1971, 746, 174–184. [Google Scholar]
- (8).(a) Direct α-C–H bond functionalization of unprotected cyclic amines. Chen W; Ma L; Paul A; Seidel D Nat. Chem 2018, 10, 165; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) α-Functionalization of Cyclic Secondary Amines: Lewis Acid Promoted Addition of Organometallics to Transient Imines. Paul; Seidel D J. Am. Chem. Soc 2019, 141, 8778–8782; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rapid functionalization of multiple C–H bonds in unprotected alicyclic amines. Chen W; Paul A; Abboud KA; Seidel D. Nat. Chem 2020, 12, 545–550; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Diversification of Unprotected Alicyclic Amines by C-H Bond Functionalization: Decarboxylative Alkylation of Transient Imines. Paul A; Kim JH; Daniel SD; Seidel D, Angew. Chem. Int. Ed 2021, 60, 1625–1628; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) α-C–H Bond Functionalization of Unprotected Alicyclic Amines: Lewis-Acid-Promoted Addition of Enolates to Transient Imines. Kim JH; Paul A; Ghiviriga I; Seidel D, Org. Lett 2021, 23, 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Copper-Catalyzed Asymmetric Propargylation of Cyclic Aldimines. Fandrick DR; Hart CA; Okafor IS; Mercadante MA; Sanyal S; Masters JT; Sarvestani M; Fandrick KR; Stockdill JL; Grinberg N; Gonnella N; Lee H; Senanayake CH. Org. Lett 2016, 18, 6192–6195. [DOI] [PubMed] [Google Scholar]
- (10).Regioselective 2-alkylation and 2-arylation of piperidine and pyrrolidine via organolithiation of cyclic imines. Scully FE. J. Org. Chem 1980, 45, 1515–1517. [Google Scholar]
- (11).Michael JP In The Alkaloids: Chemistry and Biology; Knölker H-J, Ed.; Academic Press: 2016; Vol. Volume 75, p 1–498. [DOI] [PubMed] [Google Scholar]
- (12).(a) Synthesis of 3-substituted and 3,4-disubstituted 3,4-dihydro-1(2H)-isoquinolones by condensation of lithiated N,N-diethyl-o-toluamide with imines. Jahangir Clark, R. J. Org. Chem 1987, 52, 5378–5382; [Google Scholar]; (b) Asymmetric synthesis of protoberberine alkaloids via a tandem nucleophilic addition and intramolecular cyclisation of a chiral o-toluamide anion with 3,4-dihydroisoquinoline. Warrener N, R. Chem. Commun 1997, 2173–2174; [Google Scholar]; (c) Enantioselective Synthesis of Protoberberine Alkaloids via (−)-Sparteine-mediated Asymmetric Condensation-Cyclisation of o-Toluamide Anions with 3,4-Dihydroisoquinolines. Liu L. Synthesis 2003, 2003, 1705–1706. [Google Scholar]
- (13).(a) Lithium Diisopropylamide: Solution Kinetics and Implications for Organic Synthesis. Collum DB; McNeil AJ; Ramirez A Angew. Chem. Int. Ed 2007, 46, 3002–3017; [DOI] [PubMed] [Google Scholar]; (b) Role of Organolithium Aggregates and Mixed Aggregates in Organolithium Mechanisms. Reich H. J. Chem. Rev 2013, 113, 7130–7178. [DOI] [PubMed] [Google Scholar]
- (14).(a) Lithium Diisopropylamide-Mediated Ortholithiations: Lithium Chloride Catalysis. Gupta L; Hoepker AC; Singh KJ; Collum DB J. Org. Chem 2009, 74, 2231–2233; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Regioselective Lithium Diisopropylamide-Mediated Ortholithiation of 1-Chloro-3-(trifluoromethyl)benzene: Role of Autocatalysis, Lithium Chloride Catalysis, and Reversibility. Hoepker AC; Gupta L; Ma Y; Faggin MF; Collum DB J. Am. Chem. Soc 2011, 133, 7135–7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Miyaura N; Suzuki A. Chem. Rev 1995, 95, 2457–2483; [Google Scholar]; (b) A Rationally Designed Universal Catalyst for Suzuki–Miyaura Coupling Processes. Walker SD; Barder TE; Martinelli JR; Buchwald SL. Angew. Chem. Int. Ed 2004, 43, 1871–1876; [DOI] [PubMed] [Google Scholar]; (c) Catalysts for Suzuki-Miyaura Coupling Processes: Scope and Studies of the Effect of Ligand Structure. Barder TE; Walker SD; Martinelli JR; Buchwald SL J. Am. Chem. Soc 2005, 127, 4685–4696. [DOI] [PubMed] [Google Scholar]
- (16).Simple, Efficient Catalyst System for the Palladium-Catalyzed Amination of Aryl Chlorides, Bromides, and Triflates. Wolfe JP; Tomori H; Sadighi JP; Yin J; Buchwald SL J. Org. Chem 2000, 65, 1158–1174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.