

Abstract
Lysosomal free sialic acid storage disorder (FSASD) is an extremely rare, autosomal recessive, neurodegenerative, multisystemic disorder caused by defects in the lysosomal sialic acid membrane exporter SLC17A5 (sialin). SLC17A5 defects cause free sialic acid and some other acidic hexoses to accumulate in lysosomes, resulting in enlarged lysosomes in some cell types and 10–100-fold increased urinary excretion of free sialic acid. Clinical features of FSASD include coarse facial features, organomegaly, and progressive neurodegenerative symptoms with cognitive impairment, cerebellar ataxia and muscular hypotonia. Central hypomyelination with cerebellar atrophy and thinning of the corpus callosum are also prominent disease features. Around 200 FSASD cases are reported worldwide, with the clinical spectrum ranging from a severe infantile onset form, often lethal in early childhood, to a mild, less severe form with subjects living into adulthood, also called Salla disease. The pathobiology of FSASD remains poorly understood and FSASD is likely underdiagnosed. Known patients have experienced a diagnostic delay due to the rarity of the disorder, absence of routine urine sialic acid testing, and non-specific clinical symptoms, including developmental delay, ataxia and infantile hypomyelination. There is no approved therapy for FSASD. We initiated a multidisciplinary collaborative effort involving worldwide academic clinical and scientific FSASD experts, the National Institutes of Health (USA), and the FSASD patient advocacy group (Salla Treatment and Research [S.T.A.R.] Foundation) to overcome the scientific, clinical and financial challenges facing the development of new treatments for FSASD. We aim to collect data that incentivize industry to further develop, obtain approval for, and commercialize FSASD treatments. This review summarizes current aspects of FSASD diagnosis, prevalence, etiology, and disease models, as well as challenges on the path to therapeutic approaches for FSASD.
Keywords: hypomyelination, Infantile Sialic Acid Storage Disorder, lysosomal membrane transporter, N-acetylneuraminic acid, Salla disease, sialic acid, SLC17A5
1. Background
Free sialic acid storage disorder (FSASD; MIM#604369; #269920) is a rare autosomal recessive, progressive, neurodegenerative, multisystem disorder caused by bi-allelic pathogenic variants in the SLC17A5 gene (chromosome 6q13; Gene ID 26503) [1–3]. SLC17A5 encodes the lysosomal membrane transport protein SLC17A5 (also called sialin), a 12-membrane domain lysosomal, proton-coupled carrier that exports sialic acid (N-acetylneuraminic acid, Neu5Ac) and other acidic hexoses from lysosomes [3–8]. Defective SLC17A5 leads to intra-lysosomal free sialic acid accumulation and enlarged ‘vacuolar’ lysosomes, apparent on electron microscopic examination in some cell types (Fig 1A). Individuals with FSASD excrete ~10–100-fold normal amounts of free (i.e., unconjugated) sialic acid in urine (Table 1).
Figure 1: Compilation of FSASD Features.
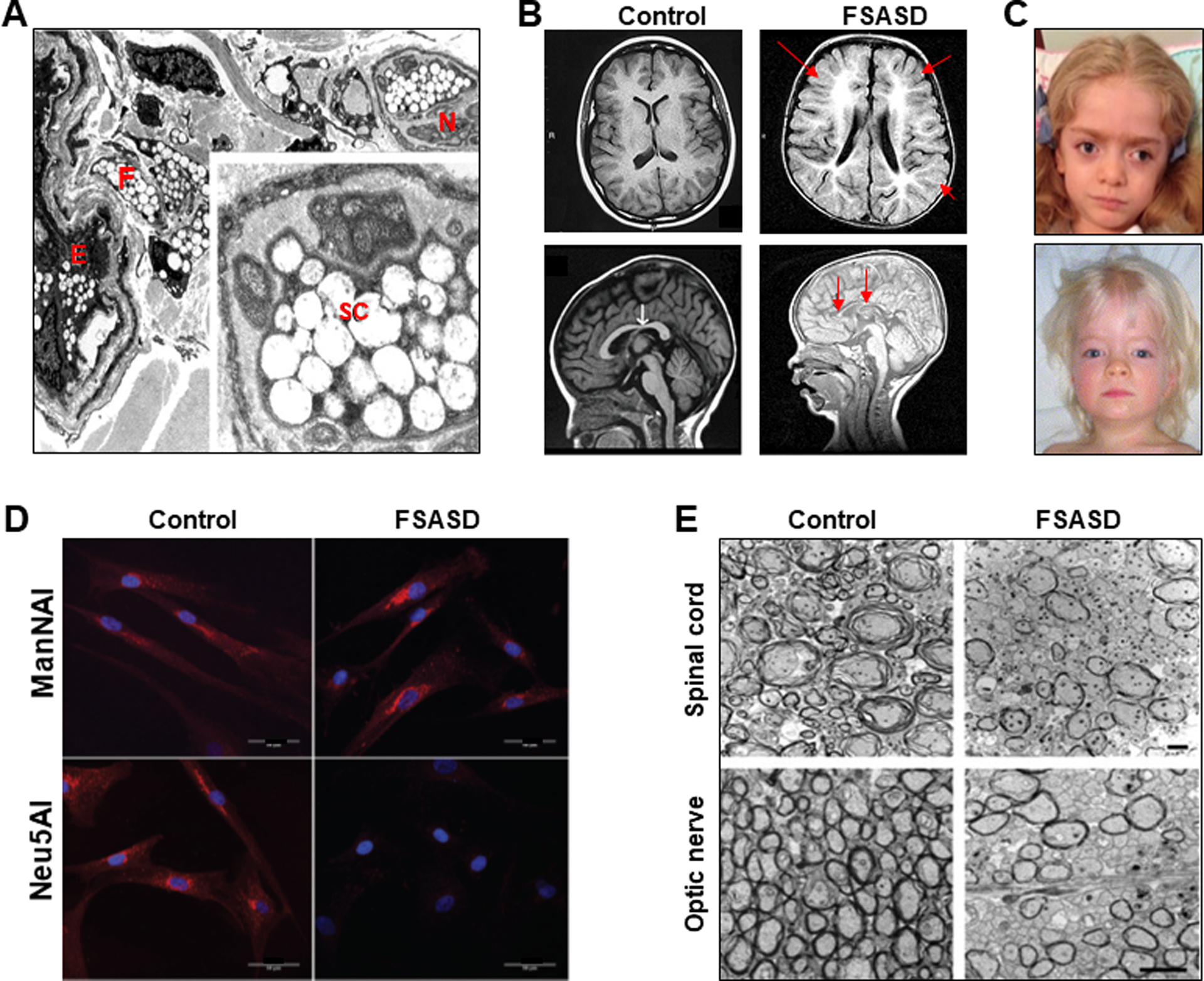
(A) Electron micrograph of a skin biopsy from an intermediate FSASD subject. Dermis revealing blood vessels with endothelial cells (E) and pericytes, a nerve (N) bundle with Schwann cells (SC), and fibroblasts (F). The endothelial cells, fibroblasts, and Schwann cells have numerous enlarged, vacuolar shaped, lysosomes (3860×). Inset: Schwann cell containing enlarged lysosomes, most of which are electron lucent; some contain fine fibrillar material (17,550×). Image derived from [17], with permission from Elsevier Inc.
(B) Brain MRI of the same intermediate FSASD subject as in (A) at 10 months of age (right images) compared to age-matched control images (left). Top: Axial T1-weighted, Bottom: Sagittal midline T1-weighted. Note widespread and profound hypomyelination throughout the cerebral and cerebellar hemispheres and small corpus callosum (red arrows). FSASD images derived from [17], with permission from Elsevier Inc.
(C) Coarse facial features of FSASD include hypertelorism, flat-bridged nose, depressed nasal bridge, broad nasal tip, long philtrum, broad forehead/brachycephaly, depicted in a 4.5 year old girl [10] and a 30-month old girl [17], both presenting with intermediate FSASD. Images with permission from Elsevier Inc.
(D) Ultrastructural images of control and Slc17A5−/− (knock-out, FSASD) mice cervical spinal cord (top) and optic nerve (bottom) cut in cross section demonstrate a decrease in the number of myelinated axons in these tissues in FSASD mice. Scale bars, 2 μm. Image derived from [87] (Copyright 2009 Society for Neuroscience).
(E) Fibroblasts from healthy individuals (Control) and an FSASD patient (FSASD) were metabolically labelled with either ManNAl or Neu5NAl for 8 hours and labeled with AzidoFluor 545 fluorescent probe (red) and the nuclear dye DAPI (blue). Cells were then examined using confocal microscopy (Scale bars: 50 μm). Top images: After incorporation of ManNAl, labeled sialylated glycoconjugates were mainly observed in the perinuclear Golgi-like region of both control and FSASD cells, indicating that FSASD cells have the capacity to transform ManNAl into CMP-Neu5NAl, which was then incorporated into the newly synthesized glycoconjugates. Bottom Images: The FSASD cells labeled with Neu5NAl displayed no staining. These results show the inability of Neu5NAl to reach the cytosol and be converted to CMP-Neu5NAl in FSASD cells, consistent with cellular Neu5Al import through the endocytic pathway [128], thus circumventing the absence of a plasma membrane sialic acid transporter. These results confirm not only the crucial role of SLC17A5 in Neu5NAl metabolism, but also the potential of this metabolic labeling methodology to decipher deficiencies in sialic acid pathways. Images derived from [98], with permission from The Royal Society of Chemistry.
Table 1:
| Disease Form | Urine Free Neu5Ac4
(fold increase) |
Age at onset | Main Clinical Findings | Number of Cases |
|---|---|---|---|---|
|
Mild FSASD Salla disease |
~10-fold | 6–12 mo | Moderate global developmental delay Mild cognitive dysfunction Speech delay Muscle hypotonia, cerebellar ataxia Spasticity Seizures or epilepsy Mostly hypomyelination on brain MRI Motor disability, able to walk With or without coarse facial features Near normal life span |
~ 160 |
|
Intermediate FSASD Intermediate severe SASD |
~15–100-fold | 1–6 mo | Moderate/severe global developmental delay Growth delay or failure to thrive Severe muscle hypotonia Cerebellar ataxia, spasticity Seizures, epilepsy Hypomyelination on brain MRI Mild coarse facial features No or mild organomegaly Nephrosis Shortened life span |
~ 25 |
|
Severe FSASD ISSD |
>100-fold | intrauterine | Intrauterine hydrops, neonatal ascites Failure to thrive Severe global developmental delay Coarse facial features Dysmorphic features Hepatosplenomegaly, cardiomegaly Nephrosis Early death (age < 2 years) |
~ 15 |
| Sialuria | 100–1000-fold | infancy | Coarse facial features Organomegaly Developmental delay |
11 |
| NPL deficiency | 10-fold | childhood | Progressive cardiac myopathy Mild skeletal myopathy |
2 |
Approximately 200 individuals with FSASD have been reported worldwide, of which the majority (> 160 cases) carry the Finnish founder missense variant p.Arg39Cys in SLC17A5 in homozygous or heterozygous form [1, 2, 9, 10]. Clinical features of FSASD include organomegaly, coarse facial features and progressive neurodegenerative symptoms including muscular hypotonia, cerebellar ataxia, and cognitive impairment. Central hypomyelination with thinning of the corpus callosum and cerebellar atrophy are prominent disease features (Fig 1B). FSASD patients manifest a continuous phenotypic spectrum of clinical severity that correlates with the severity of SLC17A5 mutations and the amount of stored free sialic acid in lysosomes [1, 3, 9–13], similar to some other lysosomal storage diseases [14, 15]. FSASD was historically classified in 3 forms [2, 9], ranging from a mild, slowly progressive form with individuals living to adulthood, also called Salla disease [MIM #604369] or Finnish type sialuria and associated with mild (missense) SLC17A5 mutations [1, 16], to an intermediate form [10, 17] and a severe infantile sialic acid storage disorder (ISSD; MIM #269920) form, often lethal in early childhood and associated with severe SLC17A5 mutations [18, 19]. The main aspects of the FSASD clinical spectrum are summarized in Table 1 and detailed in the literature [1–3, 10, 11, 19].
Although sialic acid metabolism, membrane transport, and lysosomal biology have been extensively studied, the pathobiology of FSASD remains poorly understood. Moreover, FSASD is likely underdiagnosed; known patients have experienced a diagnostic delay [2, 11] due to the rarity of the disorder, non-specific clinical symptoms and absence of routine urine sialic acid testing. There is no approved therapy for FSASD, nor are there clinical trials for FSASD listed on clinicaltrials.gov (November 2020). No drug intended to treat FSASD has been granted orphan designation (https://www.accessdata.fda.gov/scripts/opdlisting/oopd/).
The small population of FSASD patients has hindered industry from investing in the pre-clinical and clinical studies necessary to develop therapies [20, 21]. Recently, however, multidisciplinary collaborative efforts involving the National Institutes of Health (NIH), academic clinical scientists, and patient advocacy groups have successfully overcome the scientific, clinical and financial challenges facing the development of new drug treatments for similar rare diseases [20, 22]. Encouraged by these successes, we initiated a collaborative FSASD consortium, including NIH-based and worldwide academic scientists with clinical and basic FSASD research expertise, and the Salla Treatment and Research (S.T.A.R.) Foundation patient advocacy group (https://www.sallaresearch.org/). This consortium will create and study preclinical cell and mouse models, perform basic/translational research, initiate a natural history study to aid in the identification of biomarkers and treatment endpoints, and investigate drug candidates. By generating these data and raising awareness of FSASDs, we hope to incentivize industry to further develop, obtain approval, and commercialize FSASD treatments.
This review addresses the current status, progress, pending requirements and opportunities to advance drug development efforts for this intriguing rare inborn error of sialic acid metabolism.
1. FSASD Disease Nomenclature
When FSASD was first described by Aula et al., 1978 it was named Salla disease after the geographical region in Finnish Lapland where the first known patients resided [16]. Later, individuals outside of Finland with a much more severe clinical course were described as exhibiting infantile sialic acid storage disorder (ISSD) [23]; other reports named the disorder sialic acid storage disorder (SASD) [7, 24] or Finnish type sialuria [25] to distinguish it from the non-lysosomal form of excessive sialic acid production, French type sialuria (MIM#269921) [26, 27]. The term Salla disease is now used in the literature not only for FSASD cases with the Finnish founder variant in SLC17A5, but also for any mild FSASD cases, independent of the mutation or region of origin.
The multiple historic names for this allelic disorder, all caused by defects in the gene SLC17A5, continue to be used in the literature and disease databases. This becomes increasingly confusing for clinicians, patients, researchers, genetic diagnostic laboratories and disease databases and, ultimately, the pharmaceutical rare disease industry. Therefore, we propose to consistently name the disorder ‘Free Sialic Acid Storage Disorder’ (FSASD), referring to the entire spectrum of disease severity and replacing all previous disease definitions. With FSASD referring to the entire spectrum of disorders associated with SLC17A5 deficiency, improvements will follow in worldwide disease awareness, diagnosis, estimations of disease prevalence and, ultimately, support for a path to therapy.
2. Sialic Acid Metabolism
Sialic acids are a diverse family of negatively charged sugars and occupy terminal positions of oligosaccharide chains of most glycans (glycoproteins and gangliosides), on which they mediate a variety of biological functions and play essential roles in disease processes [28, 29]. The most abundant mammalian sialic acid and the precursor of most other sialic acids is N-acetylneuraminic acid (Neu5Ac), generally referred to as sialic acid [29, 30]. Free sialic acid metabolism occurs in different cellular compartments and is divided into three processes, i.e., biosynthesis, salvage and degradation (Fig 2). De novo enzymatic sialic acid biosynthesis occurs mainly in the cytosol but also includes a nuclear step and a negative feedback-inhibition mechanism [31–34]. Free sialic acid salvage from degradation of recycled glycans occurs in lysosomes and free sialic acid exits lysosomes into the cytosol through the SLC17A5 membrane transporter [1, 35, 36]. Catabolic degradation of sialic acid into N-acetylmannosamine (ManNAc) and pyruvate by N-acetyl-neuraminate pyruvate lyase (NPL), also known as sialic acid aldolase, occurs in the cytosol [37, 38].
Figure 2: Intracellular Free Neu5Ac Metabolism and Associated Genetic Disorders.
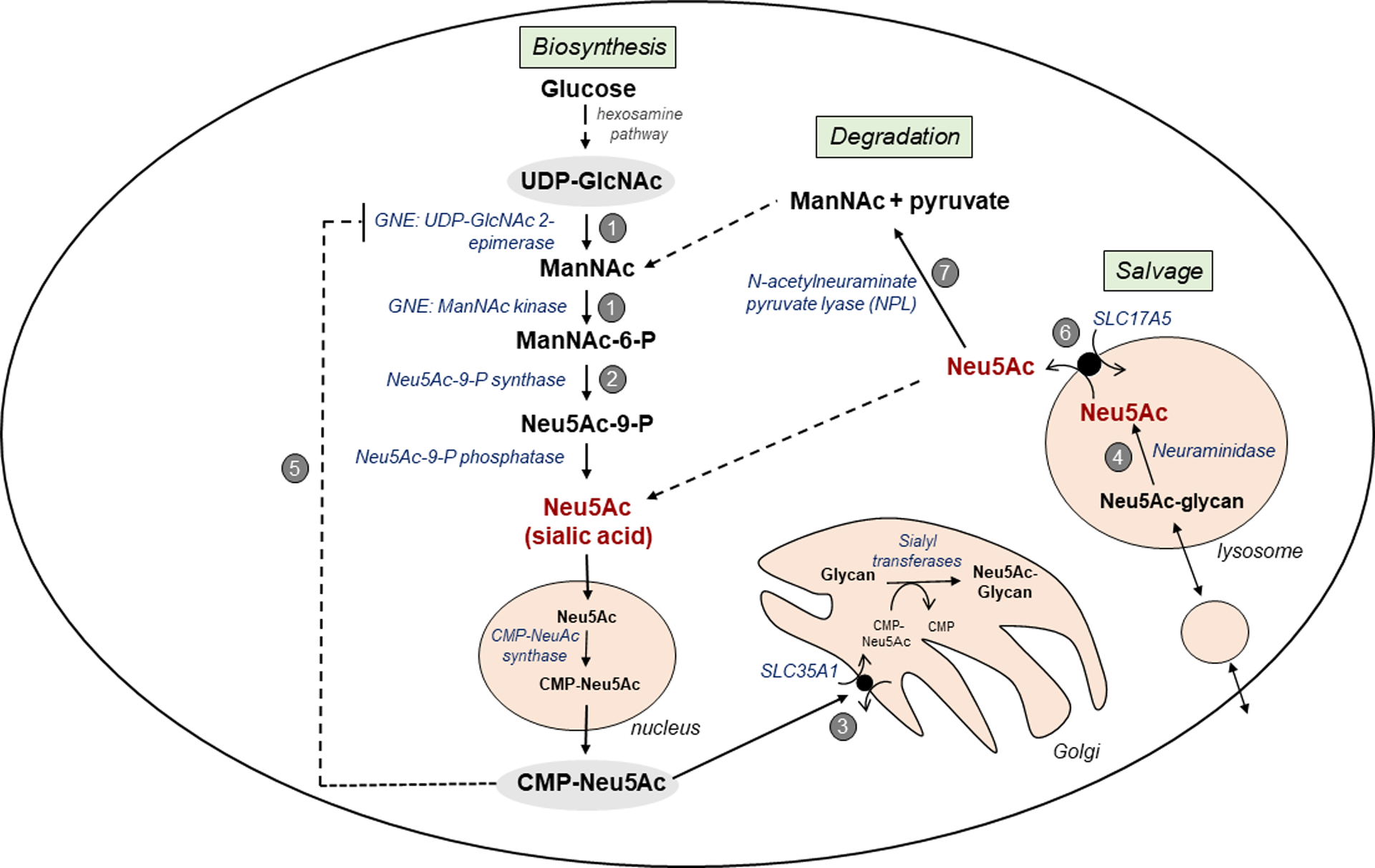
Intracellular free Neu5Ac metabolism comprises three processes:
(A) Cytoplasmic free Neu5Ac biosynthesis is initiated with the conversion of UDP-N-acetyl glucosamine (UDP-GlcNAc) in a few enzymatic steps to Neu5Ac, which is activated in the nucleus to CMP-Neu5Ac and then transported back to the cytosol [31, 32, 40]. Cytosolic CMP-Neu5Ac is transported into the Golgi by SLC35A1 [129] where it serves as a substrate for sialyltransferases that sialylate nascent glycans [130]. Cytosolic CMP-Neu5Ac also strongly feedback-inhibits the first committed enzyme of sialic acid biosynthesis, UDP-GlcNAc 2 epimerase, providing negative feedback regulation of de novo cytoplasmic Neu5Ac synthesis [33, 34].
(B) Intralysosomal free Neu5Ac salvage occurs through recycling of glycans (glycoproteins, gangliosides) through endocytosis by the endo-lysosomal system, where lysosomal enzymes degrade the glycans into their individual building block molecules, including individual monosaccharides. Free Neu5Ac is released from glycans by neuraminidase enzymes [84, 86]. Neu5Ac is then transported from the lysosomal lumen into the cytosol by SLC17A5 [1].
(C) The fate of salvaged free Neu5Ac in the cytoplasm is unclear. A portion may be excreted from the cell, recycled in the Neu5Ac biosynthesis pathway for direct synthesis of CMP-Neu5Ac, or degraded/catabolized by N-acetylneuraminate pyruvate lyase (NPL) [38] into ManNAc and pyruvate. The ManNAc generated in the cytoplasm can either directly re-enter the Neu5Ac biosynthesis pathway or can be converted to N-acetylglycosamine (GlcNAc) for entry in the hexosamine pathway [38].
Several rare genetic disorders are associated with these pathways: (1) GNE myopathy (MIM#605820; ~950 reported cases [131]); (2) N-acetylneuraminic acid phosphate synthase (NANS) deficiency (MIM#605202; ~9 cases [40]); and (3) deficiency of SLC35A1, CDGIIf (MIM#603585; ~3 cases [129]) are characterized by decreased sialylation of glycans; (4) Sialidosis (MIM#256550; >100 cases [132]) is characterized by lysosomal accumulation of sialylated glycans. Three disorders are associated with increased urinary excretion of free Neu5Ac: (5) Sialuria (MIM#269921; ~ 11 cases [33]); (6) FSASD (MIM#269920, #604369; ~200 cases [1]); and (7) NPL deficiency (2 cases [38]).
It remains unclear how free sialic acid biosynthesis, salvage and degradation pathways are regulated and contribute to steady state free sialic acid levels. Studies of inborn errors in free sialic acid metabolism have clarified some aspects (Fig 2) [38–40]. Apart from FSASD, there are two other sialic acid metabolism disorders, sialuria and NPL deficiency, associated with significantly increased urinary free sialic acid (Table 1). The dominant disorder (French type) sialuria (MIM 269921) is due to a monoallelic mutation in the allosteric site of UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE), the initial and rate-limiting enzyme in sialic acid synthesis. The mutation prevents feedback inhibition of GNE by CMP-sialic acid, leading to constitutive production of cytoplasmic free sialic acid and resulting in excessive urinary free sialic acid excretion (100–1000x normal) and increased cytoplasmic free sialic acid in fibroblasts and lymphoblasts (Fig 2, Table 1) [27, 33, 34, 41]. Sialuria has been described in only 11 cases worldwide and presents with relatively mild organomegaly, coarse facial features and varying degrees of developmental delay [33, 41, 42]. NPL deficiency (MIM 611412) is due to biallelic mutations in the NPL gene, leading to decreased cytoplasmic free sialic acid degradation and increased urinary (~ 10x normal) and red blood cell (50–100x normal) free sialic acid levels, but no detectable free sialic acid accumulation in fibroblasts [38]. NPL deficiency, so far described in only 2 siblings, presents with a progressive cardiac myopathy and mild skeletal myopathy. These findings are likely not due to cytosolic accumulation of sialic acid, since they are absent from sialuria subjects with much greater elevations in cytoplasmic free sialic acid compared with NPL deficiency [38].
The apparent rarity of these 3 inborn errors of sialic acid metabolism, all characterized by elevated urinary free sialic acid, can be due to failure to diagnose these diseases because of unfamiliarity with these disorders, the nonspecific nature of the clinical features and, importantly, absence of routine testing for urinary sialic acid. Once increased free sialic acid is detected, these conditions can be easily distinguished by molecular genetic testing of SLC17A5 (for FSASD), GNE (for sialuria) or NPL (for NPL deficiency) and/or determining the cellular localization (cytoplasmic versus lysosomal) of free sialic acid (Table 2). A predominantly lysosomal localization indicates a FSASD; cytoplasmic localization indicates sialuria or NPL deficiency. Of note, other causes of mild elevation in urinary free sialic acid may exist.
Table 2:
Fibroblast Sialic Acid Levels in Disorders of Free Sialic Acid Metabolism1
| Fibroblasts whole cell nmol/mg protein (mean ± SD) |
Fibroblasts % of free Neu5Ac recovered from |
||||||
|---|---|---|---|---|---|---|---|
| N | Free Neu5Ac | Bound Neu5Ac | Lysosomal fraction | Soluble fraction | Microsomal fraction | Nuclear fraction | |
| Controls | 11 | 1.0 ± 0.6 | 14.6 ± 4.6 | 21 % | 54 % | 7 % | 18 % |
| Salla Disease2 | 6 | 10.0 ± 2.9 | 11.9 ± 3.7 | 66 % | 10 % |
54 % |
5.5 % |
| ISSD2 | 5 | 139 ± 92 | 14.1 ± 10 | ||||
| Sialuria | 3 | 143 ± 35 | 8.9 ± 11 | 4 % | 88 % | 2 % | 6 % |
3. FSASD Diagnosis
FSASD should be considered in probands with a clinical presentation of global developmental delay or cognitive impairment, particularly affecting speech development, and regression combined with coarse facies, failure to thrive, organomegaly, truncal muscular hypotonia, ataxia, spasticity, bone anomalies, or short stature [2, 11]. More comprehensive clinical aspects and age of onset of the FSASD spectrum are summarized in Table 1 and detailed in the literature [1–3, 10, 11, 19]. MRI findings (hypomyelination, progressive cerebellar atrophy and small corpus callosum) (Fig 1B) and electron microscopy of skin biopsy (vacuolated cells; Fig 1A) may support the FSASD diagnosis [1, 3, 10, 43]. The non-specific clinical features of FSASD (developmental delay, ataxia, infantile hypomyelination) create an extensive differential diagnosis that contributes to the diagnostic delay [2, 3]. Coarse facial features of FSASD include hypertelorism, flat-bridged nose, depressed nasal bridge, broad nasal tip, long philtrum, broad forehead/brachycephaly (Fig 1C).
A few reported FSASD cases were diagnosed prenatally by biochemical and/or genetic testing of chorionic villi or amniotic fluid cells. These cases had a prior affected sibling or exhibited prenatal features suggestive of FSASD [11, 44–46]. Intrauterine ultrasound examination, fetal autopsy or clinical examinations were reported to show coarse facial features, often with prominent ascites [45, 47, 48] or in some severe cases hydrops fetalis [11, 45, 49]. Importantly, a recent retrospective study of nonimmune hydrops fetalis found that 15–29% of cases were caused by LSDs and 18% (5/28) of those had FSASD, identifying FSASD as one of the most common LSDs associated with nonimmune hydrops fetalis [49].
Detecting elevated free sialic acid in fibroblasts, urine and/or cerebrospinal fluid supports the suspicion of FSASD, although other disorders of free sialic acid excretion are known (Fig 2, Table 1). The FSASD diagnosis was historically confirmed by demonstrating lysosomal (rather than cytoplasmic) localization of elevated free sialic acid in cultured cells [3, 18, 50], but is now mostly confirmed by genetic testing detecting bi-allelic SLC17A5 mutations [1–3, 9, 11].
Although well-established analytic methods to determine free and/or bound sialic acid exist, including colorimetric and fluorometric analysis (thiobarbituric acid assay) [51], 1H-NMR spectroscopy [52], thin-layer chromatography [53, 54], high performance anion-exchange chromatography with pulsed amperometric detection (HPAE-PAD) [55], and liquid chromatography mass spectrometry (LC/MS) [53, 56], there is a lack of routine screening for urinary free sialic acid. This contributes to the considerable diagnostic delay for individuals with FSASD [11]. Identification of additional and reliable FSASD-specific biomarkers would also be clearly of value in diagnoses and therapeutic interventions.
With the current lack of disease-specific (blood-based) biomarkers, we strongly advocate for early genetic testing of suspected cases, since bi-allelic SLC17A5 pathogenic variants ultimately confirm the diagnosis. An early diagnosis is important, to allow for accurate genetic counseling and management of disease symptoms, reduce emotional hardship for families, reduce costs for future diagnostic tests, and make the patient eligible for possible future therapeutic options that may halt progression of this neurodegenerative disease. The lack of blood-based biomarkers also supports the inclusion of SLC17A5 in molecular-based newborn screening, once it is implemented and once FSASD therapies become available. A recent pilot study in Germany showed efficacy of a molecular-based neonatal screening program for cystinosis using the existing national screening framework, leading to neonatal diagnosis and successful treatment of an infant [57].
4. SLC17A5 Molecular Genetics
The SLC17A5 gene, on chromosome 6q13, consists of 11 exons transcribing a main mRNA splice variant 1 (NM_012434) that encodes a 495 amino acid protein (~54 kDa; NP_036566). Recently, 8 additional SLC17A5 mRNA splice variants were added to databases (Gene ID 26503), the biological expression and relevance of which remain to be determined.
As of December 2020, more than 55 pathognomonic SLC17A5 variants were listed in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SLC17A5). Although most reported variants are missense (27 variants), nonsense (6 variants), splicing (7 variants), small deletions (8 variants), gross deletions or insertions (8 variants) have also been reported. Two frequent SLC17A5 missense variants occur, i.e., p.Arg39Cys (c.115C>T; NM_012434), a founder variant originating from the Salla region in Finland, and p.Lys136Glu (c.406A>G; NM_012434), occurring in patients worldwide. The vast majority of reported SLC17A5 variants were identified by direct sequencing in research-based clinical laboratories [1, 2, 9, 11, 45, 58]. Next generation sequencing strategies and inclusion of SLC17A5 gene in commercially available lysosomal storage disease (LSD) gene panels will undoubtedly identify additional cases and SLC17A5 variants in the near future.
SLC17A5 gene variants cause loss of function (transport activity) and/or intracellular mis-localization of the SLC17A5 transporter [4, 59, 60]. Penetrance of FSASD appears complete, although penetrance based on urinary studies alone may be incomplete, since two individuals homozygous for p.Lys136Glu had elevated CSF free sialic acid levels but normal urinary sialic acid levels [58]. Heterozygous carriers of SLC17A5 variants are unaffected, and have urinary free sialic acid levels in the normal range [1, 61]. A genotype-phenotype correlation exists for SLC17A5 variants, apparent in the milder phenotype found in individuals homozygous for the p.Arg39Cys variant [2, 9, 18]. However, phenotypic variation in some individuals with identical SLC17A5 variants suggests involvement of additional genetic or environmental factors [62, 63]. Of note, SLC17A5 variants, including p.Arg39Cys, have been identified as risk factors for Parkinson’s disease [64].
5. Epidemiology
The worldwide prevalence of FSASD is currently estimated at less than 1 per 1,000,000 individuals (https://www.orpha.net/). Higher estimated prevalence rates of 1–9/1,000,000 occur in the Salla region in Finland, where the carrier frequency of the SLC17A5 p.Arg39Cys founder variant is 1 in 100 [9]. There are approximately 200 individuals with FSASD reported worldwide, of which the majority (> 160 cases) carry the p.Arg39Cys variant in homozygous or heterozygous form. A variety of SLC17A5 pathogenic variants are reported in more than 50 individuals worldwide, including Israeli-Bedouin (homozygous for p.Gly328Glu) [63], Canadian-Inuit (homozygous for c.526-2A>G) [65], Old Order Mennonite (homozygous for p.Arg39Cys) [66], Italian, Danish, Spanish, Dominican, Kurdish, and Japanese [11].
For a better understanding of the worldwide FSASD prevalence, we used SLC17A5 gene variants listed in the GnomAD database (https://gnomad.broadinstitute.org/; accessed December 2020). Since FSASD is associated with bi-allelic variants in one autosomal gene locus (SLC17A5), and assuming random mating in an indefinitely large population, we applied the Hardy-Weinberg principle of population genetics (p2 + 2pq + q2 = 1; Table 3, Supp. Table S1) [67–69] to calculate disease prevalence. We aggregated all pathogenic SLC17A5 variants into a single category to use this simple binomial expression (detailed in Supp Table S1).
Table 3:
Estimated Carrier Rates and Prevalence of FSASD1
| General Population2 | Finnish Population (p.Arg39Cys)3 | |
|---|---|---|
| Pathogenic SLC17A5 variants/total alleles (q) | 494/282,862 | 149/25,114 |
| Pathogenic SLC17A5 variant allele frequency | 1/572 | 1/168 |
| Carrier rate (heterozygotes) | 1/286 | 1/84 |
| Predicted FSASD Prevalence | 1/327,865 | 1/28,409 |
| FSASD affected per million | ~ 3 | ~ 35 |
| Estimated number of FSASD cases | ~ 23,000 worldwide4 | ~ 190 in Finland |
Calculated with Hardy-Weinberg principle of population genetics (See Supp Table S1)
Based on pathogenic SLC17A5 variants in GnomAD database
Based on GnomAD data of p.Arg39Cys allele frequency on Finnish alleles
Embryonic lethality of severe FSASD cases and childhood death of intermediate severe cases will reduce the number of living FSASD cases
To avoid over-estimating SLC17A5 variant allele frequencies, we did not include intronic variants more than 2 nucleotides away from exon boundaries, synonymous variants, or any missense variant with a ‘benign’ or ‘likely pathogenic’ pathogenicity score (per Variant Effect Predictor in GnomAD) (Supp. Table S1). We also omitted the number of Finnish alleles with the p.Arg39Cys variant (149 alleles), but we included non-Finnish alleles with this variant (79 alleles). This resulted in a conservative estimate of the prevalence of FSASD to be at least 3 per 1,000,000, with a carrier rate of 1/286 individuals (heterozygotes) (Table 3). Assuming this database represents the worldwide population diversity, these data translate to a prevalence of ~23,000 global FASD cases, with ~13,000 in Asia, ~2,000 in Europe and ~1,700 in North America. However, embryonic lethality of severe cases and childhood death of intermediate severe cases [14, 44, 49] will reduce the number of living FSASD cases significantly. Nevertheless, given that ~200 FSASD cases are reported in the literature, these prevalence values confirm suspicions that many FSASD cases go undiagnosed. Based on GnomAD data of Finnish alleles, we estimate that FSASD due to the homozygous p.Arg39Cys variant has a carrier rate of ~1 per 84 individuals in the Finnish population, translating to a prevalence of ~35 FSASD cases per million (~190 FSASD cases) in Finland (Table 3).
6. FSASD Etiology
The exact pathophysiology of FSASD remains unknown. Effects of SLC17A5 mutations on sialic acid transport activity, SLC17A5 intracellular localization, and amount of stored free sialic acid have been directly correlated with disease severity and survival [19,83,85]. Loss of function of SLC17A5 due to FSASD-associated mutations was demonstrated by utilizing the SLC17A5 N-terminal dileucine lysosomal targeting motif, DRTPLL (Fig 3) [4, 70]. Newly synthesized SLC17A5 traffics to the plasma membrane, from where it is rapidly internalized to the endo-lysosomal system by coat proteins recognizing the dileucine targeting motif [4]. Expression of SLC17A5 with an altered targeting motif results in plasma membrane expression, allowing for the use of whole cell uptake assays to measure transport activity and intracellular localization [4, 59, 71, 72]. While missense variants associated with more severe phenotypes had absent sialic acid transport activity, the variants associated with a milder phenotype (p.Arg39Cys, p.Lys136Glu) had residual transport activity [4, 59, 71]. Some variants also showed partial Golgi retention [72, 73] or endoplasmic reticulum (ER) retention [59]. These findings confirmed an SLC17A5 loss of function disease mechanism and a genotype-phenotype correlation for most tested variants. However, reported clinical heterogeneity in some FSASD siblings with identical mutations also suggests a role for genetic or environmental factors in FSASD clinical variability that might have therapeutic implications [62, 63].
Figure 3: Topology model of SLC17A5.
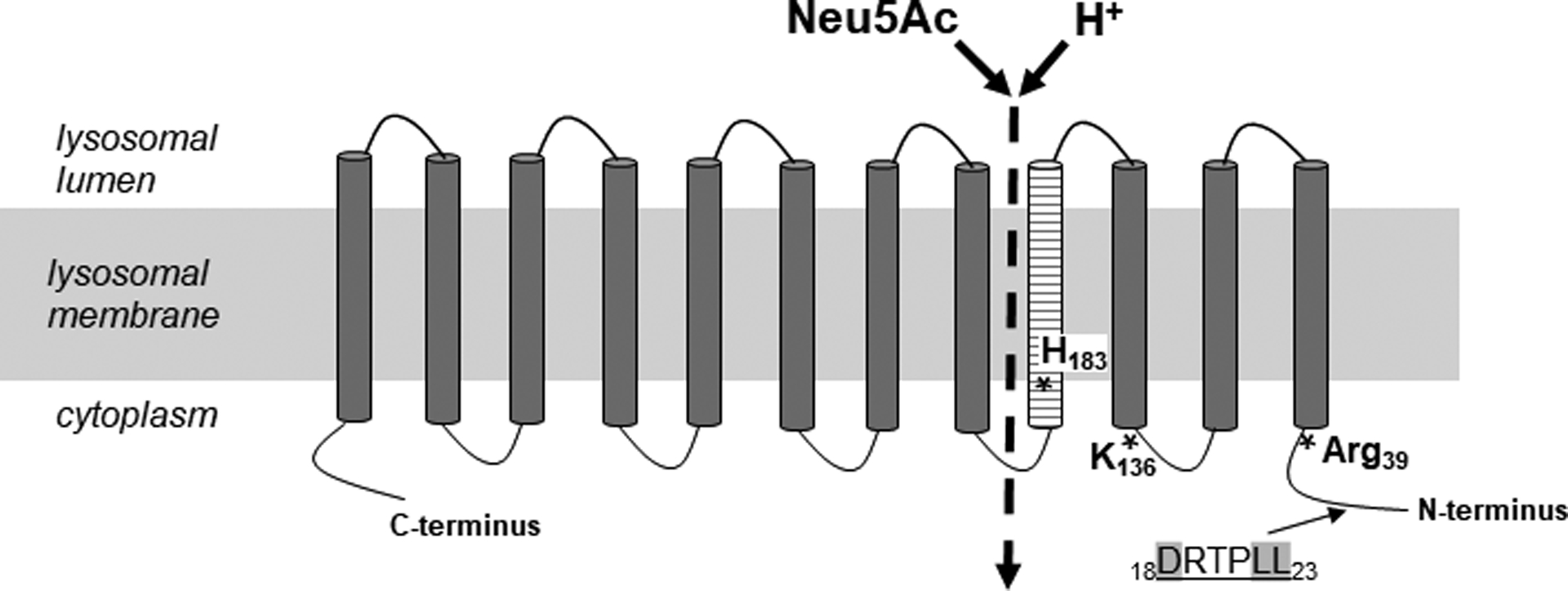
Simplified model of SLC17A5 (not to scale). SLC17A5 consists of 495 amino acids, 12 transmembrane domains and a N-terminal dileucine sorting motif (DRTPLL). Three frequent FSASD mutations are indicated (*). Transmembrane domain 4 (striped) lines a large aqueous cavity that is part of the substrate permeation pathway [4, 5].
It is unknown how accumulated intra-lysosomal free sialic acid or other stored compounds (e.g., glucuronic acid, gluconic acid) contribute to disease pathology [3–6]. Similarly, the clinical effects of alternative transport functions of SLC17A5, i.e., the uptake of glutamate, aspartate or N-aspartyl-glutamate into brain synaptic vesicles [74, 75] and plasma-membrane nitrate transport in salivary gland acinar cells [76], remains enigmatic. Also, the relevance and tissue expression of the 8 recently released human SLC17A5 isoforms (Gene ID 26503) have not been explained. The effects of SLC17A5 deficiency and lysosomal free sialic acid storage on cellular sialic acid metabolism, including protein glycosylation, also remain to be elucidated. These poorly studied features suggest that the function of SLC17A5 may be more complex than simply mediating the efflux of sialic acid from lysosomes.
SLC17A5 might play a role in determining lysosomal pH, since it is a proton-driven transporter [59, 77] and its activity is pH dependent [4, 59]. Changes in the intra-lysosomal milieu due to SLC17A5 deficiency, resulting from reduced trafficking of protons or acidic sugars, may affect other lysosomal functions. Most studies report normal lysosomal enzyme activities in FSASD cultured fibroblasts [53, 78–80], but some studies have reported increased levels and decreased turnover of sialoglycoproteins and gangliosides in lysosomes of FSASD cells [79, 81, 82]. The excessive accumulation of free sialic acid may lead to secondary storage of sialoglycoproteins and gangliosides, since sialic acid is a competitive inhibitor for lysosomal neuraminidases [83, 84]. The accumulation of sialo-glycoconjugates and gangliosides in FSASD tissues may contribute to the development of clinical symptoms, in particular in the central nervous system (CNS) [85–87], similar to other lysosomal storage diseases [88].
The sialylation status of membrane glycoconjugates, in particular brain gangliosides, in FSASD remains to be determined and may contribute to the CNS symptoms and hypomyelination. Reduced ganglioside sialylation is associated with reduced myelination [89], as it affects function of myelin‐associated glycoprotein (MAG), a component of the myelin sheet [90]. Hyposialylation of polysialic acid-neural cell adhesion molecule (PSA-NCAM) also affects CNS myelination [91, 92]. An Slc17a5 knock-out mouse was reported having aberrant expression of PSA-NCAM, possibly underlying the decrease of mature myelinating oligodendrocytes [87].
CNS manifestations in FSASD were also suggested to result from a non-lysosomal brain-specific function of SLC17A5 as a vesicular transporter for glutamate or aspartate [74, 93]. SLC17A5 carrying the p.Arg39Cys variant completely lost aspartate and glutamate transport activity, while it retained residual H+/sialic acid cotransport [74], suggesting that impaired aspartergic and glutamatergic neurotransmission in FSASD may contribute to the CNS dysfunction [74, 94]. This hypothesis supports the fact that neurological symptoms predominate in mild FSASD (p.Arg39Cys mutation), implying that the CNS is more sensitive to SLC17A5 defects than peripheral tissues. However, a role for aspartergic neurotransmission is unlikely as it is not altered in Slc17a5 knock-out mice [95].
7. FSASD Disease Models
FSASD patients’ cells are the most frequently used model for the disorder. FSASD skin fibroblasts and lymphoblasts/leukocytes were historically used for diagnostic purposes and to elucidate parts of the disease mechanism [1, 6, 7, 18, 50, 53, 79, 81, 83]. FSASD cultured fibroblasts were also successfully used for metabolic oligosaccharide engineering (MOE) [96], resulting in a cellular functional assay using chemically modified ManNAc or Neu5Ac that can be traced to newly synthesized sialoglycoconjugates. This assay can be used to screen for therapeutic molecules that restore SLC17A5 function (Fig 1D) [97, 98]. The development of techniques for generating organoids from induced pluripotent stem cells (iPSCs), including brain organoids [99], has created opportunities for new application of patient specific models for FSASD with relevance to the neurodevelopmental and neurodegenerative phenotypes. Therefore, generation of human iPSCs from fibroblasts of FSASD patients should be pursued; they will be a valuable resource to model the disease and screen for therapeutics through differentiation to specialized cell types (such as neurons, oligodendrocytes) or organoids (such as brain) as has been reported for some other lysosomal storage disorders [100–102]. Limited studies on FSASD mouse neuronal cells have been reported so far [87, 93]; such studies would be informative and should be promoted to study FSASD disease mechanisms.
The only reported FSASD model organisms are Slc17a5 knock-out mouse models [87, 103]. These mice experienced growth delays, a severely reduced lifespan, prominent lysosomal vacuolization in central and peripheral tissues, lysosomal accumulation of free sialic acid and glucuronic acid, and a progressive leukoencephalopathy with a postnatal progressive delay of milestone achievement (Fig 1E) [87, 103]. The leukoencephalopathy was characterized by a decreased number of myelinated axons and post-mitotic oligodendrocytes, with the latter associated with an increased percentage of apoptotic cells during later stages of myelinogenesis. Such changes were believed the cause of coordination defects, seizures, and premature death, all of which are consistent with human FSASD. Ultrastructural analysis showed normal migration and proliferation of oligodendrocyte precursor cells (OPCs) but a reduction in mature myelin-producing oligodendrocytes that is likely a consequence of oligodendrocyte lineage apoptosis. A delayed reduction of developmentally regulated PSA-NCAM was proposed as a mechanism for the impaired myelination and reduction in oligodendrocyte number [87]. The short lifespan of the Slc17a5 knockout mice (up to ~ 3 weeks) is restrictive for therapeutic studies, so such studies may benefit from generation of a knock-in FSASD mouse model, preferably mimicking one of the more common FSASD-associated SLC17A5 mutations, i.e., p.Arg39Cys or p.Lys136Glu.
8. FSASD Therapeutic Approaches
There is no approved therapy for FSASD. The medical and psychosocial management of subjects is symptomatic and supportive [2]. The fact that the amount of stored free sialic acid appears to correlate with survival of afflicted individuals [3, 11] suggests reduction of stored material as a therapeutic target. Also, the absent phenotype in heterozygous SLC17A5 carriers (having ~50% transport activity) [7], in combination with the retained SLC17A5 transport activity (~ 10% activity) and milder disease symptoms in cases with certain missense mutations (p.Arg39Cys, p.Lys136Glu, p.Gly409Glu) [4, 59, 72, 104] suggests that therapeutic approaches directed at only partially increasing the expression or stability of mild mutations and/or transport activity of other mutations may prove beneficial. In addition, the majority of reported FSASD cases have at least one p.Arg39Cys mutated allele [9], which makes therapeutic targeting of this variant appealing. In fact, the above-mentioned therapeutic targets were pursued in a recent study, which used a previous three-dimensional (3D) homology model of human SLC17A5 [8] to virtually screen for SLC17A5 chaperones. One compound partially rescued the trafficking defect of the p.Arg39Cys variant, but unfortunately did not rescue SLC17A5 transport activity in mutant cells [105]. This study helps set the stage for future pursuits of effective SLC17A5 therapeutic chaperones.
The increasing interest of cell biologists in lysosomal biology, coupled with rapidly improving experimental and diagnostic tools, new animal models, and increased funding for rare disease research and therapeutics, have recently improved preclinical development of therapies for several other lysosomal membrane transporter disorders; some of these approaches may prove beneficial for FSASD.
Cell-based therapies:
Therapeutic trials of stem cells for FSASD have not occurred. Hematopoietic stem cell transplantations (HSCT) for other disorders of lysosomal membrane transporters are under investigation and, although it may eliminate some symptoms [106], HSCT is not curative for the neurological features [106–110]. Therefore, the risks of HSCT may outweigh those of the disorder itself [106].
Chaperone-based or small molecule therapies:
Such therapies may be effective for certain SLC17A5 point mutations that result in membrane protein misfolding, degradation, trafficking defects, or impaired channel activity. A virtual 3D model-based screening study for SLC17A5 ligands was recently reported [8, 105]. And high-throughput (repurposed) drug screening on FSASD cells should be encouraged, for which drug-based effects might be visualized using metabolic oligosaccharide engineering (MOE) (Fig 1D) [96]. Chaperone-based studies for other membrane transporter disorders, including identification of activating compounds for the mutated transmembrane channels TRPML1 in mucolipidosis type IV [111], HGSNAT in mucopolysaccharidosis IIIC [110], and for the p.Gly551Asp pathogenic variant in the cystic fibrosis transmembrane conductance regulator (CTFR) in cystic fibrosis [112], could inform future chaperone-based studies for SLC17A5 channel activity.
A limitation of most small molecule drugs under preclinical or clinical investigation for other disorders is that while they can reduce disease symptoms or slow disease progression, they do not correct the primary deficiency and are thus not a cure. In addition, these drugs typically require frequent lifelong administration, and, for treatment of neurological symptoms, must repeatedly contend with the difficulty of crossing the blood brain barrier.
Gene Editing/Therapies:
Gene therapy approaches in monogenic diseases like FSASD have the potential to correct underlying genetic defects, offering a cure rather than simply symptom management. Gene therapy may require only a single dose to gain lifelong improvement, and methods that cross the blood brain barrier are evolving [113–115]. While protein-replacement therapies for lysosomal enzymes or other soluble proteins are in clinical development [116], for lysosomal transporter disorders like FSASD this approach is more complex; hence, these disorders may benefit more from investments in gene therapy approaches. So far, gene therapy for other lysosomal membrane transporter disorders has only reached the clinical stage for the lysosomal storage disorders neuronal ceroid-lipofuscinosis 3 (MIM#204200; caused by CLN3 gene defects), for which subjects receive intracranial injections of AAV9-CLN3 (ClinicalTrials.gov Identifier: NCT03770572) [117] and for cystinosis (MIM#606272; caused by CTNS gene defects), for which subjects are transplanted with autologous hematopoietic stem cells ex vivo transduced with a lentiviral vector containing an intact CTNS gene (ClinicalTrials.gov Identifier: NCT03897361) [107, 118]. Gene-based therapy for some other disorders associated with membrane transporter defects, including autosomal dominant osteopetrosis type 2 (OPTA2, MIM#166600; caused by CLCN7 gene defects) [119], are progressing. For FSASD, apart from gene therapy delivering a functional SLC17A5 gene, (CRISPR-based) gene editing approaches, in particular those that correct the common p.Arg39Cys missense variant, may be feasible. Preclinical research in this area should be promoted.
Transcription factor EB (TFEB):
Activation of TFEB has emerged as an exciting therapeutic approach for LSDs [120]. Increasing expression and/or nuclear translocation of TFEB results in upregulated lysosomal biogenesis and function, including exocytosis and autophagy pathways; that helps deplete LSD-lysosomes of their accumulated materials and/or renew lysosomes or cells. The lysosomal membrane-associated mTORC1 kinase complex, which is involved in lysosomal nutrient sensing and TFEB activation [121], is reported to be affected in mucolipidosis type IV (MLIV) [120, 122], but it also appears to be affected in cystinosis [123, 124]. Activation of TFEB with the tyrosine kinase inhibitor genistein rescued lysosomal abnormalities in cystinotic kidney cells [123]. In a mouse model of CLN3, neuropathy and survival improved with either trehalose or MK2206 treatment. Both these drugs prevent TFEB phosphorylation, resulting in its translocation into the nucleus, triggering enhanced clearance of proteolipid aggregates in these CLN3 mice [125]. These findings open new perspectives for clinical application of TFEB-mediated enhancement to FSASD and other lysosomal membrane transporter disorders.
9. Concluding Remarks
While we live in a time of unprecedented opportunities for rare disease research and therapeutics, more than 90% of rare diseases still lack an effective treatment. Long research and development timelines, high development and production costs, and small numbers of patients for each rare disease make industry and academic researchers weigh the cost, time and risks associated with therapy development [20, 21]. Recent multidisciplinary efforts successfully overcame scientific, clinical and financial challenges facing the development of new drug treatments, including an effort for the lysosomal storage disorder Niemann Pick Disease Type C [22].
FSASD is a typical example of one such rare disease, which still lacks therapeutic initiatives two decades after identification of SLC17A5 as causative for FSASD [1]. Our recently initiated multidisciplinary consortium aims to collaboratively accelerate therapeutic development for FSASD. This review summarizes the current status, recent progress and opportunities for FSASD and can be used as a guide to address the substantial number of pending aspects (Table 4) that require our collaborative attention to bring therapeutic options to individuals afflicted with this challenging inborn error of sialic acid metabolism.
Table 4:
Pending Requirements for Collaborative Efforts toward FSASD Therapy
| Requirements | Efforts |
|---|---|
| Disease Awareness | |
| Diagnosis |
|
| Prospective Natural History Study3 |
|
| Disease Models |
|
| Therapeutic Research |
|
| Clinical Trials |
|
Supplementary Material
HIGHLIGHTS:
FSASD is an underdiagnosed neurodegenerative multisystem lysosomal storage disease
FSASD is caused by defects in the lysosomal free sialic acid exporter SLC17A5
FSASD should be considered in individuals with hypomyelination on brain MRI
The SLC17A5 gene should be included in lysosomal storage disease (LSD) gene panels
A research consortium is generating preclinical data for FSASD drug development
Funding:
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
FSASD Consortium members:
David R Adams,a Kostantin Dobrenis,c Jessica Foglio,1 William A Gahl,a Bruno Gasnier,2 Mary Hackbarth,a Marjan Huizing,a Monkol Lek,3 May CV Malicdan,a Liisa E Paavola,4 Marc C Patterson,d Richard Reimer,5 Steven U Walkley,c Melissa Wasserstein,b,c Raymond Y Wang,6 Roberto Zoncu7
1 Salla Treatment and Research (STAR) Foundation, Bronx, NY, 10471, United States
2 Université de Paris, Saints-Pères Paris Institute for the Neurosciences (SPPIN), Centre National de la Recherche Scientifique (CNRS), 75006, Paris, France
3 Department of Genetics, Yale School of Medicine, New Haven, CT, 06519, United States
4 Neural Ltd, Center of Neuropsychology, 90100, Oulu, Finland; Oulu University Hospital, Department of Neurology, 90029, Oulu, Finland
5 Department of Neurology and Neurological Sciences, Stanford University School of Medicine, Stanford, 94305, CA; Palo Alto Veterans Administration Health Care System, Palo Alto, 94304, CA, United States
6 Division of Metabolic Disorders, Children’s Hospital of Orange County, 92868, CA; University of California-Irvine School of Medicine, Irvine, 92868, CA, United States
7 Department of Molecular and Cell Biology, University of California, Berkeley, 94729, CA, United States
Footnotes
Declarations of interest: none
10. References
- [1].Verheijen FW, Verbeek E, Aula N, Beerens CE, Havelaar AC, Joosse M, Peltonen L, Aula P, Galjaard H, van der Spek PJ, Mancini GM, A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases, Nat Genet 23 (1999) 462–465. [DOI] [PubMed] [Google Scholar]
- [2].Adams D, Wasserstein M, Free Sialic Acid Storage Disorders. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (Eds.), GeneReviews, University of Washington, Seattle (WA), 2003. [Updated 2020], p. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1470/. [PubMed] [Google Scholar]
- [3].Aula P, Gahl WA, Disorders of Free Sialic Acid Storage. In: Valle DL, Antonarakis S, Ballabio A, Beaudet AL, G.A. M (Eds.), The Online Metabolic and Molecular Bases of Inherited Disease, McGraw-Hill, 2019, p. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225891389. [Google Scholar]
- [4].Morin P, Sagne C, Gasnier B, Functional characterization of wild-type and mutant human sialin, EMBO J 23 (2004) 4560–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Courville P, Quick M, Reimer RJ, Structure-function studies of the SLC17 transporter sialin identify crucial residues and substrate-induced conformational changes, J Biol Chem 285 (2010) 19316–19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Blom HJ, Andersson HC, Seppala R, Tietze F, Gahl WA, Defective glucuronic acid transport from lysosomes of infantile free sialic acid storage disease fibroblasts, Biochem J 268 (1990) 621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mancini GM, Beerens CE, Aula PP, Verheijen FW, Sialic acid storage diseases. A multiple lysosomal transport defect for acidic monosaccharides, J Clin Invest 87 (1991) 1329–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pietrancosta N, Anne C, Prescher H, Ruivo R, Sagne C, Debacker C, Bertrand HO, Brossmer R, Acher F, Gasnier B, Successful prediction of substrate-binding pocket in SLC17 transporter sialin, J Biol Chem 287 (2012) 11489–11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aula N, Salomaki P, Timonen R, Verheijen F, Mancini G, Mansson JE, Aula P, Peltonen L, The spectrum of SLC17A5-gene mutations resulting in free sialic acid-storage diseases indicates some genotype-phenotype correlation, Am J Hum Genet 67 (2000) 832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Barmherzig R, Bullivant G, Cordeiro D, Sinasac DS, Blaser S, Mercimek-Mahmutoglu S, A New Patient With Intermediate Severe Salla Disease With Hypomyelination: A Literature Review for Salla Disease, Pediatr Neurol 74 (2017) 87–91 e82. [DOI] [PubMed] [Google Scholar]
- [11].Zielonka M, Garbade SF, Kolker S, Hoffmann GF, Ries M, A cross-sectional quantitative analysis of the natural history of free sialic acid storage disease-an ultra-orphan multisystemic lysosomal storage disorder, Genet Med 21 (2019) 347–352. [DOI] [PubMed] [Google Scholar]
- [12].Parazzini C, Arena S, Marchetti L, Menni F, Filocamo M, Verheijen FW, Mancini GM, Triulzi F, Parini R, Infantile sialic acid storage disease: serial ultrasound and magnetic resonance imaging features, AJNR Am J Neuroradiol 24 (2003) 398–400. [PMC free article] [PubMed] [Google Scholar]
- [13].Haataja L, Parkkola R, Sonninen P, Vanhanen SL, Schleutker J, Aarimaa T, Turpeinen U, Renlund M, Aula P, Phenotypic variation and magnetic resonance imaging (MRI) in Salla disease, a free sialic acid storage disorder, Neuropediatrics 25 (1994) 238–244. [DOI] [PubMed] [Google Scholar]
- [14].Zielonka M, Garbade SF, Kolker S, Hoffmann GF, Ries M, A cross-sectional quantitative analysis of the natural history of Farber disease: an ultra-orphan condition with rheumatologic and neurological cardinal disease features, Genet Med 20 (2018) 524–530. [DOI] [PubMed] [Google Scholar]
- [15].Sidransky E, Gaucher disease: complexity in a “simple” disorder, Mol Genet Metab 83 (2004) 6–15. [DOI] [PubMed] [Google Scholar]
- [16].Aula P, Raivio K, Autio S, Thoden CE, Rapola J, Koskela SL, Yamashina I, Four patients with a new lysosomal storage disorder (Salla disease), Monogr Hum Genet 10 (1978) 16–22. [DOI] [PubMed] [Google Scholar]
- [17].Kleta R, Morse RP, Orvisky E, Krasnewich D, Alroy J, Ucci AA, Bernardini I, Wenger DA, Gahl WA, Clinical, biochemical, and molecular diagnosis of a free sialic acid storage disease patient of moderate severity, Mol Genet Metab 82 (2004) 137–143. [DOI] [PubMed] [Google Scholar]
- [18].Kleta R, Aughton DJ, Rivkin MJ, Huizing M, Strovel E, Anikster Y, Orvisky E, Natowicz M, Krasnewich D, Gahl WA, Biochemical and molecular analyses of infantile free sialic acid storage disease in North American children, Am J Med Genet A 120A (2003) 28–33. [DOI] [PubMed] [Google Scholar]
- [19].Lemyre E, Russo P, Melancon SB, Gagne R, Potier M, Lambert M, Clinical spectrum of infantile free sialic acid storage disease, Am J Med Genet 82 (1999) 385–391. [PubMed] [Google Scholar]
- [20].Kaufmann P, Pariser AR, Austin C, From scientific discovery to treatments for rare diseases - the view from the National Center for Advancing Translational Sciences - Office of Rare Diseases Research, Orphanet J Rare Dis 13 (2018) 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Thompson PW, “Developing new treatments in partnership for Primary Mitochondrial Disease: what does industry need from academics, and what do academics need from industry?”, J Inherit Metab Dis Online ahead of print (2020) doi: 10.1002/jimd.12326. [DOI] [PubMed] [Google Scholar]
- [22].Ottinger EA, Kao ML, Carrillo-Carrasco N, Yanjanin N, Shankar RK, Janssen M, Brewster M, Scott I, Xu X, Cradock J, Terse P, Dehdashti SJ, Marugan J, Zheng W, Portilla L, Hubbs A, Pavan WJ, Heiss J, Vite CH, Walkley SU, Ory DS, Silber SA, Porter FD, Austin CP, McKew JC, Collaborative development of 2-hydroxypropyl-beta-cyclodextrin for the treatment of Niemann-Pick type C1 disease, Curr Top Med Chem 14 (2014) 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schleutker J, Leppanen P, Mansson JE, Erikson A, Weissenbach J, Peltonen L, Aula P, Lysosomal free sialic acid storage disorders with different phenotypic presentations--infantile-form sialic acid storage disease and Salla disease--represent allelic disorders on 6q14-15, Am J Hum Genet 57 (1995) 893–901. [PMC free article] [PubMed] [Google Scholar]
- [24].van den Bosch J, Oemardien LF, Srebniak MI, Piraud M, Huijmans JG, Verheijen FW, Ruijter GJ, Prenatal screening of sialic acid storage disease and confirmation in cultured fibroblasts by LC-MS/MS, J Inherit Metab Dis 34 (2011) 1069–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Simila S, Linna SL, Vayrynen M, Autio-Harmainen H, von Wendt L, Ruokonen A, Finnish type of sialic acid storage disease with sialuria (Salla disease): the occurrence and diagnostic significance of cytoplasmic vacuoles in blood lymphocytes, J Ment Defic Res 29 (Pt 2) (1985) 179–186. [DOI] [PubMed] [Google Scholar]
- [26].Montreuil J, Biserte G, Strecker G, Spik G, Fontaine G, Farriaux JP, [Description of a new type of melituria, called sialuria], Clin Chim Acta 21 (1968) 61–69. [DOI] [PubMed] [Google Scholar]
- [27].Leroy JG, Seppala R, Huizing M, Dacremont G, De Simpel H, Van Coster RN, Orvisky E, Krasnewich DM, Gahl WA, Dominant inheritance of sialuria, an inborn error of feedback inhibition, Am J Hum Genet 68 (2001) 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Varki A, Sialic acids in human health and disease, Trends Mol Med 14 (2008) 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schauer R, Kamerling JP, Exploration of the Sialic Acid World, Adv Carbohydr Chem Biochem 75 (2018) 1–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Varki A, Diversity in the sialic acids, Glycobiology 2 (1992) 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hinderlich S, Weidemann W, Yardeni T, Horstkorte R, Huizing M, UDP-GlcNAc 2-Epimerase/ManNAc Kinase (GNE): A Master Regulator of Sialic Acid Synthesis, Top Curr Chem 366 (2015) 97–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kean EL, Munster-Kuhnel AK, Gerardy-Schahn R, CMP-sialic acid synthetase of the nucleus, Biochim Biophys Acta 1673 (2004) 56–65. [DOI] [PubMed] [Google Scholar]
- [33].Seppala R, Lehto VP, Gahl WA, Mutations in the human UDP-N-acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme, Am J Hum Genet 64 (1999) 1563–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kornfeld S, Kornfeld R, Neufeld EF, O’Brien PJ, The Feedback Control of Sugar Nucleotide Biosynthesis in Liver, Proc Natl Acad Sci U S A 52 (1964) 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tettamanti G, Bassi R, Viani P, Riboni L, Salvage pathways in glycosphingolipid metabolism, Biochimie 85 (2003) 423–437. [DOI] [PubMed] [Google Scholar]
- [36].Monti E, Bonten E, D’Azzo A, Bresciani R, Venerando B, Borsani G, Schauer R, Tettamanti G, Sialidases in vertebrates: a family of enzymes tailored for several cell functions, Adv Carbohydr Chem Biochem 64 (2010) 403–479. [DOI] [PubMed] [Google Scholar]
- [37].Schauer R, Sommer U, Kruger D, van Unen H, Traving C, The terminal enzymes of sialic acid metabolism: acylneuraminate pyruvate-lyases, Biosci Rep 19 (1999) 373–383. [DOI] [PubMed] [Google Scholar]
- [38].Wen XY, Tarailo-Graovac M, Brand-Arzamendi K, Willems A, Rakic B, Huijben K, Da Silva A, Pan X, El-Rass S, Ng R, Selby K, Philip AM, Yun J, Ye XC, Ross CJ, Lehman AM, Zijlstra F, Abu Bakar N, Drogemoller B, Moreland J, Wasserman WW, Vallance H, van Scherpenzeel M, Karbassi F, Hoskings M, Engelke U, de Brouwer A, Wevers RA, Pshezhetsky AV, van Karnebeek CD, Lefeber DJ, Sialic acid catabolism by N-acetylneuraminate pyruvate lyase is essential for muscle function, JCI Insight 3 (2018) e122373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Willems AP, van Engelen BG, Lefeber DJ, Genetic defects in the hexosamine and sialic acid biosynthesis pathway, Biochim Biophys Acta 1860 (2016) 1640–1654. [DOI] [PubMed] [Google Scholar]
- [40].van Karnebeek CD, Bonafe L, Wen XY, Tarailo-Graovac M, Balzano S, Royer-Bertrand B, Ashikov A, Garavelli L, Mammi I, Turolla L, Breen C, Donnai D, Cormier-Daire V, Heron D, Nishimura G, Uchikawa S, Campos-Xavier B, Rossi A, Hennet T, Brand-Arzamendi K, Rozmus J, Harshman K, Stevenson BJ, Girardi E, Superti-Furga G, Dewan T, Collingridge A, Halparin J, Ross CJ, Van Allen MI, Rossi A, Engelke UF, Kluijtmans LA, van der Heeft E, Renkema H, de Brouwer A, Huijben K, Zijlstra F, Heise T, Boltje T, Wasserman WW, Rivolta C, Unger S, Lefeber DJ, Wevers RA, Superti-Furga A, NANS-mediated synthesis of sialic acid is required for brain and skeletal development, Nat Genet 48 (2016) 777–784. [DOI] [PubMed] [Google Scholar]
- [41].Enns GM, Seppala R, Musci TJ, Weisiger K, Ferrell LD, Wenger DA, Gahl WA, Packman S, Clinical course and biochemistry of sialuria, J Inherit Metab Dis 24 (2001) 328–336. [DOI] [PubMed] [Google Scholar]
- [42].Ishtiaq H, Siddiqui S, Nawaz R, Jamali KS, Khan AG, Sialuria-Related Intellectual Disability in Children and Adolescent of Pakistan: Tenth Patient Described has a Novel Mutation in the GNE Gene, CNS Neurol Disord Drug Targets 19 (2020) 127–141. [DOI] [PubMed] [Google Scholar]
- [43].Schleutker J, Laine AP, Haataja L, Renlund M, Weissenbach J, Aula P, Peltonen L, Linkage disequilibrium utilized to establish a refined genetic position of the Salla disease locus on 6q14-q15, Genomics 27 (1995) 286–292. [DOI] [PubMed] [Google Scholar]
- [44].Aula N, Aula P, Prenatal diagnosis of free sialic acid storage disorders (SASD), Prenat Diagn 26 (2006) 655–658. [DOI] [PubMed] [Google Scholar]
- [45].Froissart R, Cheillan D, Bouvier R, Tourret S, Bonnet V, Piraud M, Maire I, Clinical, morphological, and molecular aspects of sialic acid storage disease manifesting in utero, J Med Genet 42 (2005) 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Couce ML, Macias-Vidal J, Castineiras DE, Boveda MD, Fraga JM, Fernandez-Marmiesse A, Coll MJ, The early detection of Salla disease through second-tier tests in newborn screening: how to face incidental findings, Eur J Med Genet 57 (2014) 527–531. [DOI] [PubMed] [Google Scholar]
- [47].Carbillon L, Largilliere C, Bucourt M, Scheuer-Niro B, Levaillant JM, Uzan M, Ultrasound assessment in a case of sialic acid storage disease, Ultrasound Obstet Gynecol 18 (2001) 272–274. [DOI] [PubMed] [Google Scholar]
- [48].Gillan JE, Lowden JA, Gaskin K, Cutz E, Congenital ascites as a presenting sign of lysosomal storage disease, J Pediatr 104 (1984) 225–231. [DOI] [PubMed] [Google Scholar]
- [49].Al-Kouatly HB, Felder L, Makhamreh MM, Kass SL, Vora NL, Berghella V, Berger S, Wenger DA, Luzi P, Lysosomal storage disease spectrum in nonimmune hydrops fetalis: a retrospective case control study, Prenat Diagn 40 (2020) 738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Renlund M, Tietze F, Gahl WA, Defective sialic acid egress from isolated fibroblast lysosomes of patients with Salla disease, Science 232 (1986) 759–762. [DOI] [PubMed] [Google Scholar]
- [51].Warren L, The thiobarbituric acid assay of sialic acids, J Biol Chem 234 (1959) 1971–1975. [PubMed] [Google Scholar]
- [52].Haverkamp J, van Halbeek H, Dorland L, Vliegenthart JF, Pfeil R, Schauer R, High-resolution 1H-NMR spectroscopy of free and glycosidically linked O-acetylated sialic acids, Eur J Biochem 122 (1982) 305–311. [DOI] [PubMed] [Google Scholar]
- [53].Renlund M, Chester MA, Lundblad A, Parkkinen J, Krusius T, Free N-acetylneuraminic acid in tissues in Salla disease and the enzymes involved in its metabolism, Eur J Biochem 130 (1983) 39–45. [DOI] [PubMed] [Google Scholar]
- [54].Humbel R, Collart M, Oligosaccharides in urine of patients with glycoprotein storage diseases. I. Rapid detection by thin-layer chromatography, Clin Chim Acta 60 (1975) 143–145. [DOI] [PubMed] [Google Scholar]
- [55].Rohrer JS, Thayer J, Weitzhandler M, Avdalovic N, Analysis of the N-acetylneuraminic acid and N-glycolylneuraminic acid contents of glycoproteins by high-pH anion-exchange chromatography with pulsed amperometric detection, Glycobiology 8 (1998) 35–43. [DOI] [PubMed] [Google Scholar]
- [56].van der Ham M, Prinsen BH, Huijmans JG, Abeling NG, Dorland B, Berger R, de Koning TJ, de MG Sain-van der Velden, Quantification of free and total sialic acid excretion by LC-MS/MS, J Chromatogr B Analyt Technol Biomed Life Sci 848 (2007) 251–257. [DOI] [PubMed] [Google Scholar]
- [57].Hohenfellner K, Bergmann C, Fleige T, Janzen N, Burggraf S, Olgemoller B, Gahl WA, Czibere L, Froschauer S, Roschinger W, Vill K, Harms E, Nennstiel U, Molecular based newborn screening in Germany: Follow-up for cystinosis, Mol Genet Metab Rep 21 (2019) 100514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mochel F, Yang B, Barritault J, Thompson JN, Engelke UF, McNeill NH, Benko WS, Kaneski CR, Adams DR, Tsokos M, Abu-Asab M, Huizing M, Seguin F, Wevers RA, Ding J, Verheijen FW, Schiffmann R, Free sialic acid storage disease without sialuria, Ann Neurol 65 (2009) 753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wreden CC, Wlizla M, Reimer RJ, Varied mechanisms underlie the free sialic acid storage disorders, J Biol Chem 280 (2005) 1408–1416. [DOI] [PubMed] [Google Scholar]
- [60].Sagne C, Gasnier B, Molecular physiology and pathophysiology of lysosomal membrane transporters, J Inherit Metab Dis 31 (2008) 258–266. [DOI] [PubMed] [Google Scholar]
- [61].Seppala R, Tietze F, Krasnewich D, Weiss P, Ashwell G, Barsh G, Thomas GH, Packman S, Gahl WA, Sialic acid metabolism in sialuria fibroblasts, J Biol Chem 266 (1991) 7456–7461. [PubMed] [Google Scholar]
- [62].Varho TT, Alajoki LE, Posti KM, Korhonen TT, Renlund MG, Nyman SR, Sillanpaa ML, Aula PP, Phenotypic spectrum of Salla disease, a free sialic acid storage disorder, Pediatr Neurol 26 (2002) 267–273. [DOI] [PubMed] [Google Scholar]
- [63].Landau D, Cohen D, Shalev H, Pinsk V, Yerushalmi B, Zeigler M, Birk OS, A novel mutation in the SLC17A5 gene causing both severe and mild phenotypes of free sialic acid storage disease in one inbred Bedouin kindred, Mol Genet Metab 82 (2004) 167–172. [DOI] [PubMed] [Google Scholar]
- [64].Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, C. International Parkinson’s Disease Genomics, Heutink P, Shulman JM, Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease, Brain 140 (2017) 3191–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lines MA, Rupar CA, Rip JW, Baskin B, Ray PN, Hegele RA, Grynspan D, Michaud J, Geraghty MT, Infantile Sialic Acid Storage Disease: Two Unrelated Inuit Cases Homozygous for a Common Novel SLC17A5 Mutation, JIMD Rep 12 (2014) 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Strauss KA, Puffenberger EG, Craig DW, Panganiban CB, Lee AM, Hu-Lince D, Stephan DA, Morton DH, Genome-wide SNP arrays as a diagnostic tool: clinical description, genetic mapping, and molecular characterization of Salla disease in an Old Order Mennonite population, Am J Med Genet A 138A (2005) 262–267. [DOI] [PubMed] [Google Scholar]
- [67].Hardy GH, Mendelian Proportions in a Mixed Population, Science 28 (1908) 49–50. [DOI] [PubMed] [Google Scholar]
- [68].Weinberg W, Über den Nachweis der Vererbung beim Menschen, Jahreshefte des Vereins für vaterländische Naturkunde in Württemberg 64 (1908) 368–382. [Google Scholar]
- [69].Mayo O, A century of Hardy-Weinberg equilibrium, Twin Res Hum Genet 11 (2008) 249–256. [DOI] [PubMed] [Google Scholar]
- [70].Bonifacino JS, Traub LM, Signals for sorting of transmembrane proteins to endosomes and lysosomes, Annu Rev Biochem 72 (2003) 395–447. [DOI] [PubMed] [Google Scholar]
- [71].Ruivo R, Anne C, Sagne C, Gasnier B, Molecular and cellular basis of lysosomal transmembrane protein dysfunction, Biochim Biophys Acta 1793 (2009) 636–649. [DOI] [PubMed] [Google Scholar]
- [72].Ruivo R, Sharifi A, Boubekeur S, Morin P, Anne C, Debacker C, Graziano JC, Sagne C, Gasnier B, Molecular pathogenesis of sialic acid storage diseases: insight gained from four missense mutations and a putative polymorphism of human sialin, Biol Cell 100 (2008) 551–559. [DOI] [PubMed] [Google Scholar]
- [73].Aula N, Jalanko A, Aula P, Peltonen L, Unraveling the molecular pathogenesis of free sialic acid storage disorders: altered targeting of mutant sialin, Mol Genet Metab 77 (2002) 99–107. [DOI] [PubMed] [Google Scholar]
- [74].Miyaji T, Echigo N, Hiasa M, Senoh S, Omote H, Moriyama Y, Identification of a vesicular aspartate transporter, Proc Natl Acad Sci U S A 105 (2008) 11720–11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lodder-Gadaczek J, Gieselmann V, Eckhardt M, Vesicular uptake of N-acetylaspartylglutamate is catalysed by sialin (SLC17A5), Biochem J 454 (2013) 31–38. [DOI] [PubMed] [Google Scholar]
- [76].Qin L, Liu X, Sun Q, Fan Z, Xia D, Ding G, Ong HL, Adams D, Gahl WA, Zheng C, Qi S, Jin L, Zhang C, Gu L, He J, Deng D, Ambudkar IS, Wang S, Sialin (SLC17A5) functions as a nitrate transporter in the plasma membrane, Proc Natl Acad Sci U S A 109 (2012) 13434–13439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Mancini GM, de Jonge HR, Galjaard H, Verheijen FW, Characterization of a proton-driven carrier for sialic acid in the lysosomal membrane. Evidence for a group-specific transport system for acidic monosaccharides, J Biol Chem 264 (1989) 15247–15254. [PubMed] [Google Scholar]
- [78].Fois A, Balestri P, Farnetani MA, Mancini GM, Borgogni P, Margollicci MA, Molinelli M, Alessandrini C, Gerli R, Free sialic acid storage disease. A new Italian case, Eur J Pediatr 146 (1987) 195–198. [DOI] [PubMed] [Google Scholar]
- [79].Baumkotter J, Cantz M, Mendla K, Baumann W, Friebolin H, Gehler J, Spranger J, N-Acetylneuraminic acid storage disease, Hum Genet 71 (1985) 155–159. [DOI] [PubMed] [Google Scholar]
- [80].Nakano C, Hirabayashi Y, Ohno K, Yano T, Mito T, Sakurai M, A Japanese case of infantile sialic acid storage disease, Brain Dev 18 (1996) 153–156. [DOI] [PubMed] [Google Scholar]
- [81].Pitto M, Chigorno V, Renlund M, Tettamanti G, Impairment of ganglioside metabolism in cultured fibroblasts from Salla patients, Clin Chim Acta 247 (1996) 143–157. [DOI] [PubMed] [Google Scholar]
- [82].Mendla K, Baumkotter J, Rosenau C, Ulrich-Bott B, Cantz M, Defective lysosomal release of glycoprotein-derived sialic acid in fibroblasts from patients with sialic acid storage disease, Biochem J 250 (1988) 261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Mendla K, Cantz M, Specificity studies on the oligosaccharide neuraminidase of human fibroblasts, Biochem J 218 (1984) 625–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Miyagi T, Yamaguchi K, Mammalian sialidases: physiological and pathological roles in cellular functions, Glycobiology 22 (2012) 880–896. [DOI] [PubMed] [Google Scholar]
- [85].Pshezhetsky AV, Ashmarina M, Keeping it trim: roles of neuraminidases in CNS function, Glycoconj J 35 (2018) 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pan X, De Aragao CBP, Velasco-Martin JP, Priestman DA, Wu HY, Takahashi K, Yamaguchi K, Sturiale L, Garozzo D, Platt FM, Lamarche-Vane N, Morales CR, Miyagi T, Pshezhetsky AV, Neuraminidases 3 and 4 regulate neuronal function by catabolizing brain gangliosides, FASEB J 31 (2017) 3467–3483. [DOI] [PubMed] [Google Scholar]
- [87].Prolo LM, Vogel H, Reimer RJ, The lysosomal sialic acid transporter sialin is required for normal CNS myelination, J Neurosci 29 (2009) 15355–15365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Renaud DL, Lysosomal disorders associated with leukoencephalopathy, Semin Neurol 32 (2012) 51–54. [DOI] [PubMed] [Google Scholar]
- [89].Yoo SW, Motari MG, Susuki K, Prendergast J, Mountney A, Hurtado A, Schnaar RL, Sialylation regulates brain structure and function, FASEB J 29 (2015) 3040–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Yang LJ, Zeller CB, Shaper NL, Kiso M, Hasegawa A, Shapiro RE, Schnaar RL, Gangliosides are neuronal ligands for myelin-associated glycoprotein, Proc Natl Acad Sci U S A 93 (1996) 814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Charles P, Hernandez MP, Stankoff B, Aigrot MS, Colin C, Rougon G, Zalc B, Lubetzki C, Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule, Proc Natl Acad Sci U S A 97 (2000) 7585–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Fewou SN, Ramakrishnan H, Bussow H, Gieselmann V, Eckhardt M, Down-regulation of polysialic acid is required for efficient myelin formation, J Biol Chem 282 (2007) 16700–16711. [DOI] [PubMed] [Google Scholar]
- [93].Aula N, Kopra O, Jalanko A, Peltonen L, Sialin expression in the CNS implicates extralysosomal function in neurons, Neurobiol Dis 15 (2004) 251–261. [DOI] [PubMed] [Google Scholar]
- [94].Miyaji T, Omote H, Moriyama Y, Functional characterization of vesicular excitatory amino acid transport by human sialin, J Neurochem 119 (2011) 1–5. [DOI] [PubMed] [Google Scholar]
- [95].Morland C, Nordengen K, Larsson M, Prolo LM, Farzampour Z, Reimer RJ, Gundersen V, Vesicular uptake and exocytosis of L-aspartate is independent of sialin, FASEB J 27 (2013) 1264–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Mahal LK, Yarema KJ, Bertozzi CR, Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis, Science 276 (1997) 1125–1128. [DOI] [PubMed] [Google Scholar]
- [97].Gilormini PA, Lion C, Vicogne D, Guerardel Y, Foulquier F, Biot C, Chemical glycomics enrichment: imaging the recycling of sialic acid in living cells, J Inherit Metab Dis 41 (2018) 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Gilormini PA, Lion C, Vicogne D, Levade T, Potelle S, Mariller C, Guerardel Y, Biot C, Foulquier F, A sequential bioorthogonal dual strategy: ManNAl and SiaNAl as distinct tools to unravel sialic acid metabolic pathways, Chem Commun (Camb) 52 (2016) 2318–2321. [DOI] [PubMed] [Google Scholar]
- [99].Marton RM, Pasca SP, Organoid and Assembloid Technologies for Investigating Cellular Crosstalk in Human Brain Development and Disease, Trends Cell Biol 30 (2020) 133–143. [DOI] [PubMed] [Google Scholar]
- [100].Luciani M, Gritti A, Meneghini V, Human iPSC-Based Models for the Development of Therapeutics Targeting Neurodegenerative Lysosomal Storage Diseases, Front Mol Biosci 7 (2020) 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kido J, Nakamura K, Era T, Role of induced pluripotent stem cells in lysosomal storage diseases, Mol Cell Neurosci 108 (2020) 103540. [DOI] [PubMed] [Google Scholar]
- [102].Latour YL, Yoon R, Thomas SE, Grant C, Li C, Sena-Esteves M, Allende ML, Proia RL, Tifft CJ, Human GLB1 knockout cerebral organoids: A model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis, Mol Genet Metab Rep 21 (2019) 100513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Stroobants S, Van Acker NG, Verheijen FW, Goris I, Daneels GF, Schot R, Verbeek E, Knaapen MW, De Bondt A, Gohlmann HW, Crauwels ML, Mancini GM, Andries LJ, Moechars DW, D’Hooge R, Progressive leukoencephalopathy impairs neurobehavioral development in sialin-deficient mice, Exp Neurol 291 (2017) 106–119. [DOI] [PubMed] [Google Scholar]
- [104].Myall NJ, Wreden CC, Wlizla M, Reimer RJ, G328E and G409E sialin missense mutations similarly impair transport activity, but differentially affect trafficking, Mol Genet Metab 92 (2007) 371–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Dubois L, Pietrancosta N, Cabaye A, Fanget I, Debacker C, Gilormini PA, Dansette PM, Dairou J, Biot C, Froissart R, Goupil-Lamy A, Bertrand HO, Acher FC, McCort-Tranchepain I, Gasnier B, Anne C, Amino Acids Bearing Aromatic or Heteroaromatic Substituents as a New Class of Ligands for the Lysosomal Sialic Acid Transporter Sialin, J Med Chem 63 (2020) 8231–8249. [DOI] [PubMed] [Google Scholar]
- [106].Teti A, Econs MJ, Osteopetroses, emphasizing potential approaches to treatment, Bone 102 (2017) 50–59. [DOI] [PubMed] [Google Scholar]
- [107].Rocca CJ, Cherqui S, Potential use of stem cells as a therapy for cystinosis, Pediatr Nephrol 34 (2019) 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Nair S, Strohecker AM, Persaud AK, Bissa B, Muruganandan S, McElroy C, Pathak R, Williams M, Raj R, Kaddoumi A, Sparreboom A, Beedle AM, Govindarajan R, Adult stem cell deficits drive Slc29a3 disorders in mice, Nat Commun 10 (2019) 2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Walker MT, Montell C, Suppression of the motor deficit in a mucolipidosis type IV mouse model by bone marrow transplantation, Hum Mol Genet 25 (2016) 2752–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Pshezhetsky AV, Martins C, Ashmarina M, Sanfilippo type C disease: pathogenic mechanism and potential therapeutic applications, Expert Opinion on Orphan Drugs 6 (2018) 635–646. [Google Scholar]
- [111].Chen CC, Keller M, Hess M, Schiffmann R, Urban N, Wolfgardt A, Schaefer M, Bracher F, Biel M, Wahl-Schott C, Grimm C, A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV, Nat Commun 5 (2014) 4681. [DOI] [PubMed] [Google Scholar]
- [112].Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, Moss RB, Pilewski JM, Rubenstein RC, Uluer AZ, Aitken ML, Freedman SD, Rose LM, Mayer-Hamblett N, Dong Q, Zha J, Stone AJ, Olson ER, Ordonez CL, Campbell PW, Ashlock MA, Ramsey BW, Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation, N Engl J Med 363 (2010) 1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Poletti V, Biffi A, Gene-Based Approaches to Inherited Neurometabolic Diseases, Hum Gene Ther 30 (2019) 1222–1235. [DOI] [PubMed] [Google Scholar]
- [114].Gigliobianco MR, Di Martino P, Deng S, Casadidio C, Censi R, New Advanced Strategies for the Treatment of Lysosomal Diseases Affecting the Central Nervous System, Curr Pharm Des 25 (2019) 1933–1950. [DOI] [PubMed] [Google Scholar]
- [115].Ingusci S, Verlengia G, Soukupova M, Zucchini S, Simonato M, Gene Therapy Tools for Brain Diseases, Front Pharmacol 10 (2019) 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Yang Y, Hong Y, Cho E, Kim GB, Kim IS, Extracellular vesicles as a platform for membrane-associated therapeutic protein delivery, J Extracell Vesicles 7 (2018) 1440131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Kohlschutter A, Schulz A, Bartsch U, Storch S, Current and Emerging Treatment Strategies for Neuronal Ceroid Lipofuscinoses, CNS Drugs 33 (2019) 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Harrison F, Yeagy BA, Rocca CJ, Kohn DB, Salomon DR, Cherqui S, Hematopoietic stem cell gene therapy for the multisystemic lysosomal storage disorder cystinosis, Mol Ther 21 (2013) 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Maurizi A, Capulli M, Patel R, Curle A, Rucci N, Teti A, RNA interference therapy for autosomal dominant osteopetrosis type 2. Towards the preclinical development, Bone 110 (2018) 343–354. [DOI] [PubMed] [Google Scholar]
- [120].Ballabio A, The awesome lysosome, EMBO Mol Med 8 (2016) 73–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zoncu R, Efeyan A, Sabatini DM, mTOR: from growth signal integration to cancer, diabetes and ageing, Nat Rev Mol Cell Biol 12 (2011) 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Scotto Rosato A, Montefusco S, Soldati C, Di Paola S, Capuozzo A, Monfregola J, Polishchuk E, Amabile A, Grimm C, Lombardo A, De Matteis MA, Ballabio A, Medina DL, TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway, Nat Commun 10 (2019) 5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Ivanova EA, van den Heuvel LP, Elmonem MA, De Smedt H, Missiaen L, Pastore A, Mekahli D, Bultynck G, Levtchenko EN, Altered mTOR signalling in nephropathic cystinosis, J Inherit Metab Dis 39 (2016) 457–464. [DOI] [PubMed] [Google Scholar]
- [124].Andrzejewska Z, Nevo N, Thomas L, Chhuon C, Bailleux A, Chauvet V, Courtoy PJ, Chol M, Guerrera IC, Antignac C, Cystinosin is a Component of the Vacuolar H+-ATPase-Ragulator-Rag Complex Controlling Mammalian Target of Rapamycin Complex 1 Signaling, J Am Soc Nephrol 27 (2016) 1678–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Palmieri M, Pal R, Nelvagal HR, Lotfi P, Stinnett GR, Seymour ML, Chaudhury A, Bajaj L, Bondar VV, Bremner L, Saleem U, Tse DY, Sanagasetti D, Wu SM, Neilson JR, Pereira FA, Pautler RG, Rodney GG, Cooper JD, Sardiello M, mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases, Nat Commun 8 (2017) 14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Alajoki L, Varho T, Posti K, Aula P, Korhonen T, Neurocognitive profiles in Salla disease, Dev Med Child Neurol 46 (2004) 832–837. [DOI] [PubMed] [Google Scholar]
- [127].Martin RA, Slaugh R, Natowicz M, Pearlman K, Orvisky E, Krasnewich D, Kleta R, Huizing M, Gahl WA, Sialic acid storage disease of the Salla phenotype in American monozygous twin female sibs, Am J Med Genet A 120A (2003) 23–27. [DOI] [PubMed] [Google Scholar]
- [128].Bardor M, Nguyen DH, Diaz S, Varki A, Mechanism of uptake and incorporation of the non-human sialic acid N-glycolylneuraminic acid into human cells, J Biol Chem 280 (2005) 4228–4237. [DOI] [PubMed] [Google Scholar]
- [129].Ng BG, Asteggiano CG, Kircher M, Buckingham KJ, Raymond K, Nickerson DA, Shendure J, Bamshad MJ, University G of Washington Center for Mendelian, M. Ensslen, H.H. Freeze, Encephalopathy caused by novel mutations in the CMP-sialic acid transporter, SLC35A1, Am J Med Genet A 173 (2017) 2906–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Harduin-Lepers A, Vallejo-Ruiz V, Krzewinski-Recchi MA, Samyn-Petit B, Julien S, Delannoy P, The human sialyltransferase family, Biochimie 83 (2001) 727–737. [DOI] [PubMed] [Google Scholar]
- [131].Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, Shmilevich I, Friedmann A, Karpati G, Bradley WG, Baumbach L, Lancet D, Asher EB, Beckmann JS, Argov Z, Mitrani-Rosenbaum S, The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy, Nat Genet 29 (2001) 83–87. [DOI] [PubMed] [Google Scholar]
- [132].d’Azzo A, Machado E, Annunziata I, Pathogenesis, Emerging therapeutic targets and Treatment in Sialidosis, Expert Opin Orphan Drugs 3 (2015) 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.