

Abstract
Parkinson’s disease (PD) is the result of dopaminergic (DA) neuronal death in the substantianigra pars compacta (SNc). Current treatments for PD such as L-dopa are limited in effectiveness and fail to address the cause. Targeted anti-inflammatory therapies, particularly directed at nuclear factor kappa B (NF‐κB) activity in alleviating degeneration of DA-neurons is of evolving interest. In the present study, we hypothesised that dexmedetomidine (DEX), an alpha-2 receptor adrenergic agonist, suppress the inflammatory responses associated with PD and restores dopaminergic levels by alleviating substantia nigral degeneration. Male mice (C57Bl/10, 8–11 months old and of 34–40 g of weight) were divided into: the control, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and MPTP + dexmedetomidine (MPTP + DEX) (n = 26 each group). Dex restored dopamine levels in SNpc of MPTP-induced PD mice model. Results of immunohisto staining revealed that Dex treatment post-MPTP induction restored TH-positive cells, with only 12.37% increase (##p < 0.01 vs MPTP) on the third day and a steep 55% increase (###p < 0.001 vs MPTP) following the seventh day of Dex treatment. Moreover, the expressions of proinflammatory markers regulated by NF-κB were diminished in Dex + MPTP group. In addition, cylinder test revealed that Dex treatment improved asymmetric limb usage pattern in MPTP induced mice over the course of 7 days. Hence, in this study, we provided insight on the effect of Dex in the inhibition of NF-κB1 regulated proinflammatory mediators to improve dopamine levels and reduce SNpc dopaminergic neuronal degeneration.
Keywords: Dopaminergic neurons, Degeneration, Dexmedetomidine, Motor activity
1. Introduction
Parkinson’s disease (PD) is the result of dopaminergic (DA) neuronal death in the substantianigra pars compacta (SNc). The most significant pathological characterization of PD, the second most common neurodegenerative disease, is attributed to the dopaminergic (DA) neuronal loss or death in the substantianigra pars compacta (SNpc) region of the brain (Sulzer and Surmeier, 2013). This loss of DA neurons is ascribed to factors that could be both environmental and genetic (Surmeier, 2018). While the precisemolecular mechanisms that eventuallycause PD continue to remainuncertain, the degeneration of neurons producing dopamine leading to PD progression has been recently reported to be the result of chronic activation of inflammatory pathways (Mamelak, 2018). The pro-inflammatory response is predominantlyfacilitated by nuclear factor-kappa B-1 (NF-κB-1), stimulated by other pro-inflammatory signals, thereby regulating the microglial-induced gene expression of the inflammatory mediators (TNF-α, iNOS, IL-1β) (Liu et al., 2017). Since current treatments for PD such as L-dopa are inadequate in their efficacy and work to alleviate only the symptoms without addressing the origin of the disease, developing novel anti-inflammatory therapeutic strategies targeting the action of NF-κBto protect DA-neurons against degeneration are of rising interest to stop and reverse the devastating outcome of PD (Jankovic and Aguilar, 2008).
Dexmedetomidine (DEX) is an alpha-2 receptor adrenergic agonist and is FDA approved, with an elimination half-time of almost 2 h, which is permissible in humans (Giovannitti et al., 2015). Dexmedetomidine has been considered as a vital agent for “functional neurosurgery”, which comprises of craniotomy where the patient needs to be awake for the tumour re-section and the deep brain stimulator (DBS) embedding for PD (Kaur and Singh, 2011). Nevertheless, Dex has been widely reported to display anti-inflammatory activity (Li et al., 2015). Studies on in-vitro and in vivo models have shown that a2-adrenoceptor treatment blocks the release of cytokines, even in cases of endotoxemia (Miksa et al., 2009). Further, Dex with its organ-protective role can prevent cellular death by apoptotis, which is a significant consequence of sepsis pathogenesis (Bao and Tang, 2020). However, the molecular effect of Dex on PD has not been studied.
In this study, we conceptualized that Dex blocks inflammation-mediated progression of PD and restores dopaminergic levels by alleviating substantia nigral degeneration. We measured the expression levels of pro-inflammatory mediators regulated by NFkB1 and monitored the motor activity after Dex treatment in a PD mouse model.
2. Materials and methods
2.1. Animals
Male mice of C57Bl/10 origin, 8–11 months old and of 34–40 g of weight were employed in this study. Animals were maintained in standard conditions with ad-libitum food and water. All the experiments in the present study were permitted by the local ethical committee and carried out according to the guidelines presented in the Declaration of the National Institutes of Health Guide for Care and Use of Laboratory Animals. The mice were divided into three groups (n = 26): the control (administered with 0.9% NaCl), 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) hydrochloride (MPTP-HCl), and dexmedetomidine (MPTP + DEX) groups. The mice were sacrificed on the 3rd and 7th day to test the degeneration of DA neurons in nigrostriatal region. Motor coordination was followed-up for 7 days in all groups. The effect of DEX on expression levels of pro-inflammatory mediators regulated by NFkB1 evaluated post-MPTP treatment by western blot.
2.2. MPTP induction to generate PD mice model
MPTP–HCl procured from Sigma was diluted in 0.9% NaCl (1 mg/ml). It was administered intraperitoneally 4 times at a dose of 10 mg/kg every interval of one hour to finally achieve a complete dose of 40 mg/kg. Similarly, four intraperitonial injections of 0.9% NaCl was given to the mice in the control group.
2.3. Dex treatment
DEX (sigma) was diluted in saline (0.9%). Post-MPTP injections, mice in the MPTP + DEX group were injected intraperitoneally with DEX at a dose of 25 µg/kg per day. Dose of DEX was selected as described in earlier studies (Calhoun et al., 2015). The control and MPTP vehicle groups were given an equivalent volume (200 μL) of saline with identical schedule. The same Dex treatment routine was followed daily up to termination of the experiment. After three and seven days of treatment, six mice in each group were sacrificed to isolate brain tissue for TH immunohistochemistry experiment. At day 7, N = 6 mice in each group were sacrificed to extract protein for western blot analysis of key inflammatory markers.The animals (n = 8 each group) were also assessed formotor function over the course of 7 days post-MPTP injection.
2.4. Tissue processing
Mice were first anesthetized by administering sodium pentobarbital intraperitoneally and sacrificed by exsanguinating ice-cold saline perfusion. Following, brains were quickly removed and sliced into coronal sections of 1 mm with a tissue chopper set. Substantianigra were dissected out from the brain tissue slices and placed on an inverted glass petri dish over ice. The samples were frozen on dry ice, and then stored at −80 °C. For immunohistochemistry samples, the brains were kept in fresh 4% paraformaldehyde solution for 1 h, with the subsequent equilibration in both 20% and 30% sucrose solutions. The samples were kept in 4 °C until they were sliced in cryostat for immunohistochemistry.
2.5. Dopamine concentration evaluation
The dopamine concentration levels were quantified using an ELISA kit (Enzolifesciences) according to the guidelines of the manufacturer. The dissected sections were put in SDS lysis buffer for 30 min on ice for lysis followed by centrifugation at 12000 rpm for 5 min to obtain the supernatant. Then, the lysates were added to the microplates, followed by the sequential addition of assay diluent, mouse dopamine conjugate, substrate solution, as well as stop solution to each well. Microplates were read at 450 nm by an ELISA reader (BioRad). A standard curve was generated to calculate the dopamine concentrations.
2.6. Immunohistochemistry
Mice (n = 6) were sacrificed on the third and seventh day after the MPTP-induction. Horizontal sections of PFA fixed brain (20 mm thickness) were cut to obtain SNpc containing slices using cryostat on gelatin coated slides and kept in −20 °C for storage. The SNpc sections were deparaffinised, washed in PBS for 2.5 min followed by the incubation in H2O2 (0.3%) for 30 min to minimize the endogenous peroxidase action. Further, the sections were rinsed in PBS saline for 2.5 min, followed by 60 min incubation in the blocking solution PBS with 3% horse serum as well as Triton (0.1%). Subsequently, the sections were kept in blocking solution with mouse tyrosine hydroxylase primary antibody (1:20,000, abcam) for 1 h. After rinsing the tissue sections in PBS, they were kept in the blocking solution withbiotin-labelled horse anti-rabbit secondary antibody (1:400). Sections were washed again in PBS and incubated inavidin-peroxidase. The labelled cells were observed using the software NIS-Elements Basic Research software ver 4.00 (Nikon Corporation), and for each group ten microscopic fields at a magnification of 200 X were examined. The number of cells per region were averaged for every mouse.
2.7. Western blot analysis
Striata was dissected immediately after sacrificing miceon seventh day of MPTP-induction (n = 6). The tissue samples were thawed and homogenized with lysis buffer, then centrifuged to collect the supernatant and stored at-80 °C until the day of western blot assay. Total protein (40 µg) from each sample was heated with sodium dodecyl sulphate (SDS) sample buffer at 70 °C for 60 s and the proteins were separated on SDS polyacrylamide gels (10%) and transferred to nitrocellulose membranes by electrophoresis. Primary mouse antibodies for TNF-α, iNOS, IL1β, NFkB1, β-actin (1:1200, Dako) were incubated with the membranes followed by horse anti-rabbit secondary antibody (1:400). The differences in the band density of proteins were detected using ECL kit and measurements were made using ChemiDoc system (Biorad). Relative protein expression levels with respect to β-actin protein levels were calculated.
2.8. Cylinder test
Cylinder test was employed to test the exploratory motor behavioural performance particularly in the usage of forelimb as described previously (Fleming et al., 2013). After 14 days of MPTP-induction, each mouse was placed in a transparent cylinder made of glass of 15 cm diameter and 22 cm height with enough space to move around freely. The observations were recorder by a researcher, who was blinded to the group the mice belonged to. The recordings were made up to a total of twenty contacts using the forepaw of the mice (right and/or left) on cylinder’s wall. Forelimb usage asymmetry of right forelimb was represented as the percent of the total contacts on the wall of the cylinder.
2.9. Statistical analysis
The data are shown as the mean ± SEM. One-way ANOVA was used for comparisons between variable with Bonferroni post-hoc test using GraphPad Prism 8 0.0 (GraphPad Software, Inc.). Statistical significance was defined with the p values of p < 0.01 and p < 0.001. Each experiment was performed in triplicates and repeated thrice.
3. Results
3.1. Dex restores dopamine levels in SNpc of MPTP-induced PD mice model
The low dopamine concentration in SNpcof MPTP induced mice in comparison with the control group was quantified by ELISA (Fig. 1). The dopamine concentration in the SNPc of mice in the control group was measured to be 1.8 µg/mg of tissue. MPTP injected group of mice exhibited a dopamine content of 0.463 µg/mg on third day (***p < 0.001 vs control), whereas 0.36 µg/mg on the seventh day (***p < 0.001 vs control). However, treatment with Dex restored dopamine levels in SNpc of mice injured by MPTP intoxication. MPTP + Dex group showed a dopamine level of 0.72 µg/mg on the third day (p > 0.05, ns). Though the dopamine levels in MPTP + Dex were raised on the third day, the change was found to be non-significant. Interestingly, MPTP + Dex further enhanced dopamine levels to 1.15 µg/mg (###p < 0.001 vs MPTP group), signifying the role of Dex in the restoration of dopamine levels following MPTP-induced Parkinson’s disease model.
Fig. 1.
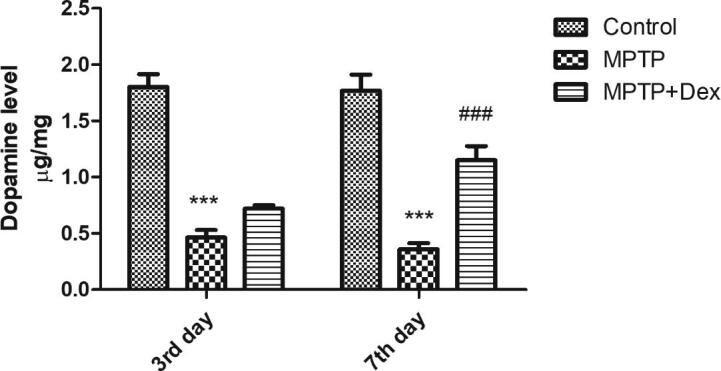
Post-Dex treatment dopamine levels in SNpc of MPTP-induced PD mice model on 3rd and 7th day following MPTP exposure. Data are shown as the mean ± SEM. ###p < 0.001 vs. MPTP; ***P < 0.001 vs control.
3.2. Dex treatment alleviates nigrostriatal degeneration following MPTP treatment
Immunohistochemical staining was performed to test the protein level of TH, an enzyme essential for dopamine synthesis. The number of dopaminergic cells in the SNpc was determined in all three groups (the control, MPTP and MPTP + Dex) at the end of third and the seventh day. Results of immunohistostaining revealed that MPTP injected group of mice displayed lower TH+ neurons in the SNpcas opposed to the control group (Fig. 2 a, b). At the end of the third day, there was a dramatic decreasein the TH-positive cell number by 78.17% (***p < 0.001 vs control) in MPTP against the control group. However, the subsequent dopaminergic neuronal degeneration upto seventh day was gradual with only 6.5% decrease compared with MPTP group on 3rd day, suggesting that the maximum degradation happened within the first three days of MPTP injection. Dex treatment post-MPTP induction restored TH-positive cells, with only 12.37% increase (##p < 0.01 vs MPTP) on the third day and a steep 55% increase (###p < 0.001 vs MPTP) following the seventh day of Dex treatment.
Fig. 2.

Effect of Dex treatment on nigrostriatal degeneration following MPTP treatment (A) Representative immunohistochemical staining of TH showing the effect of Dex treatment on dopaminergic neuronal degeneration (third and seventh day) post-MPTP exposure, Magnification, x200 (B) Quantification of TH + cells in the control, MPTP exposed vehicle treated and Dex treated groups following 3 and 7 days of MPTP exposure. Data are shown as the mean ± SEM. ###p < 0.001 vs. MPTP; ##p < 0.01 vs MPTP; ***P < 0.001 vs. control.
3.3. Dexmedetomidine reduces inflammatory reaction in SNpc of MPTP treated mice
Dex treatment directly suppresses inflammatory reaction following MPTP intoxication as observed by the protein level evaluation of inflammatory regulators NFκB1, TNF-α, IL-1β, and iNOS. TNF-α, IL-1β, and iNOS are generated by microglia and their chronic production causes neuronal tissue destruction. Microglial activation may occur as a result of the DA-neuronal death due to either surge of inflammatory mediatorsor due to the direct effect of neurotoxins, for instance, MPTP. Moreover, NF-κB1 activation is necessary for the microglial-induced generation of these inflammatory mediators. Thus, the expression of these inflammatory markers in SNpc of the brain was determined at day 7 post MPTP injection via western blot analyses. There was a significant rise in the protein levels of TNF-α (5.797 fold increase, ***p < 0.001 vs control), iNOS (3.113 fold increase, **p < 0.01 vs control) IL-1β (3.957 fold increase, ***p < 0.001 vs control) and NFκB1 (6.747 fold increase, ***p < 0.001 vs control) (Fig. 3). Evidently the expressions of these inflammatory markers were diminished in Dex + MPTP group. TNF- α reduced by 5.123 fold, ###p < 0.001 vs MPTP, iNOS exhibited a reduction by 2.217 fold ##p < 0.01 vs MPTP, IL-1β reduced by 2.457 fold, ##p < 0.01 vs MPTP, NFκB1 reduced by 5.647 fold, ###p < 0.001 vs MPTP).
Fig. 3.

Dex treatment at protein level of inflammatory regulators TNF-α, iNOS, IL-1β and NFκB1 following MPTP intoxication. (a) Western blot image showing the band densities of TNF-α, iNOS, IL-1β and NFκB1. (b) Quantification of TNF-α relative expression. (C) Quantification of iNOS relative protein expression. (D) Quantification of relative expression of IL-1βprotein. (e) Quantification of NFκB1 relative expression. Data are presented as mean ± SEM, where ##p < 0.01, ###p < 0.001 vs MPTP; *** p < 0.001vscontrol.
3.4. Dex improves motor behavioural performance post-MPTP induction
To study the outcome of Dex on motor behavioural activity in MPTP-treated mice, cylinder test was performed. It was observed that MPTP mice used right forelimb significantly more frequent by 33.05% (***p < 0.001 vs control) 24 h after MPTP induction with subsequent increase in the right forelimb use asymmetry by 71.62% (***p < 0.001 vs control) on 7th day post-MPTP injection, demonstrating asymmetric limb usage pattern in MPTP induced mice increases over the course of 7 days (Fig. 4). This right forelimb usage frequency in MPTP-treated mice was reduced significantly by 57.47% (###p < 0.001 vs MPTP group) after Dex-treated MPTP-induced mice on 7th day, suggesting that Dex significantly improved the asymmetry of limb usage.
Fig. 4.
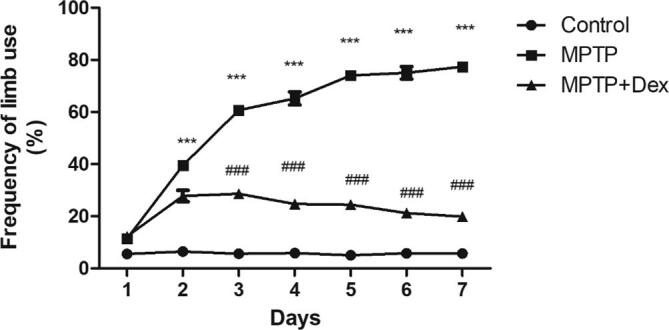
Evaluation of frequency of forelimb usage by cylinder test following Dex treatment in MPTP exposed mice. Data are shown as the mean ± SEM, where ###p < 0.001 vs. MPTP, ***p < 0.001 vs control.
4. Discussion
Here, we demonstrated that Dex treatment renders neuroprotection in Parkinson's disease model of mouse. Dex directly suppresses inflammatory reaction to prevent nigrostriatal degeneration and improve motor activity in mice intoxicated with MPTP, a Parkinson’s disease model. There has been a rising interest in the study of mechanisms involving neuroinflammation affecting PD pathogenesis (Wang et al., 2015, Hirsch et al., 2012). The transcription factor, nuclear factor kappa B (NF‐κB), has been associated with the activation of pro-inflammatory mediators, which in turn leads to the loss of DA neurons in PD (Ghosh et al., 2007, Wang et al., 2020). Blocking the activation of NF‐κB has been deemed as a potential therapeutic drug target for PD (Flood et al., 2011). Reports increasingly demonstrate that inflammatory mediators (TNFα, IL-1β, iNOS), along with other byproducts of active immune cells mightinduce the degeneration of DA neurons in substantianigra in a PD model (Tansey and Goldberg, 2010). The idea of inflammatory activity in PD has been favouredfrom studies of the SN of PD-afflicted post mortem brains from human patients (Hartmann, 2004). Further, it has been shown that levels of these proinflammatory mediators TNFα, IL-1β, IL-6 are higher in both the brainas well as peripheral blood mononuclear cells of individuals affected by PD (Chao et al., 2014). In another light, the presence of nitrite in the cerebrospinal fluid concomitant with the enhanced iNOS expression within the substantianigra was also reported in individuals affected by PD (Jiménez-Jiménez et al., 2014, Percário et al., 2020). A study also reported that the administration of the neurotoxin MPTP that selectively targets dopaminergic neuronsactivates chronic neuroinflammation leading to the progression of neurotoxicity in PD models (Tansey et al., 2007). Hence, in this study, we used MPTP-induced mice as PD model to study the effect of inhibiting the activation of NF-κB1 in improving dopamine levels and reducing SNpc dopaminergic neuronal degeneration.
The existing therapeutics for PD focuses on improving the symptoms rather than addressing the cause of the disease, which makes them less adequate in the context of treatment of PD. The widely used L-dopa for the treatment of PD raises thedopamine levels in the brain and has been utilized for years as an accepted standard for the PD treatment. It controls the PD associated symptoms and is applied in almost every phase of the disease progression yet, largely augments the risk of dyskinesia or involuntary movements (Jankovic and Aguilar, 2008). Besides, the constant usage of the drug for four to five years reduces its effectiveness to combat PD symptoms, making the development of a novel drug essential (Thanvi and Lo, 2004). Therefore, targeting pro-inflammatory mediators may affect the progression of PD, for which the repurposing anti-inflammatory drugs might be a promising solution.
In this context, we tested the efficacy of Dex in PD mice model and how its anti-inflammatory role could be of potential significance in alleviating PD progression. Previous studies have reported the neuroprotective function of Dex against neuronal damage (Endesfelder et al., 2017). Moreover, different studies have shed lighton the anti-inflammatory property of Dex concurrent with the reduction of cell death as well as production of pro-inflammatory mediators both in experimental and clinical studies (Li et al., 2015, Yoon et al., 2016, Ueki et al., 2014). A previous report probed into the mechanistic action of Dex and attributed its anti-infllammatory action partly to the inactivation of TLR-4/NF-κB pathway (Yao et al., 2015), depicting the utility of DEX treatment against focal cerebral I/R injury. Here, we demonstrated that Dex treatment directly suppresses inflammatory reaction following MPTP intoxication as observed by the evaluation of expression of inflammatory regulators TNF-α, IL-1β, iNOS, and NFκB1.
5. Conclusions
The restoration of dopamine levels in SNpc, TH-positive dopaminergic neurons and improved motor activity in mice injured by MPTP intoxication suggested that Dex might be neuroprotective for PD. Thus, the present study provides insight for future research involving direct association of neuro-inflammatory pathways, such as nuclear factor-κB signalling with Dex in PD induced mice models for an effective therapy.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Peer review under responsibility of King Saud University.
References
- Bao N., Tang B. Organ-protective effects and the underlying mechanism of dexmedetomidine. Mediators Inflamm. 2020;2020:6136105. doi: 10.1155/2020/6136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun G., Wang L., Almeida L.E., Kenyon N., Afsar N., Nouraie M., Finkel J.C., Quezado Z.M. Dexmedetomidine ameliorates nocifensivebehavior in humanized sickle cell mice. Eur. J. Pharmacol. 2015;754:125–133. doi: 10.1016/j.ejphar.2015.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao Y., Wong S.C., Tan E.K. Evidence of inflammatory system involvement in Parkinson's disease. Biomed. Res. Int. 2014;2014 doi: 10.1155/2014/308654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endesfelder S., Makki H., von Haefen C., Spies C.D., Bührer C., Sifringer M. Neuroprotective effects of dexmedetomidine against hyperoxia-induced injury in the developing rat brain. PLoS ONE. 2017;12(2) doi: 10.1371/journal.pone.0171498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming S.M., Ekhator O.R., Ghisays V. Assessment of sensorimotor function in mouse models of Parkinson's disease. J. Vis. Exp. 2013;76:50303. doi: 10.3791/50303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood P.M., Qian L., Peterson L.J., Zhang F., Shi J.S., Gao H.M., Hong J.S. Transcriptional factor NF-κB as a Target for therapy in Parkinson's disease. Parkinsons Dis. 2011;2011 doi: 10.4061/2011/216298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A., Roy A., Liu X., Kordower J.H., Mufson E.J., Hartley D.M., Ghosh S., Mosley R.L., Gendelman H.E., Pahan K. Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson's disease. Pro. Nat. Acad. Sci. 2007;104(47):18754–18759. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannitti J.A., Jr, Thoms S.M., Crawford J.J. Alpha-2 adrenergic receptor agonists: a review of current clinical applications. Anesth. Prog. Spring. 2015;62(1):31–39. doi: 10.2344/0003-3006-62.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann A. Postmortem studies in Parkinson's disease. Dialogues Clin. Neurosci. 2004;6(3):281–293. doi: 10.31887/DCNS.2004.6.3/ahartmann. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch E.C., Vyas S., Hunot S. Neuroinflammation in Parkinson's disease. Parkinsonism Relat. Disord. 2012;18(Sl1):S210–S212. doi: 10.1016/S1353-8020(11)70065-7. [DOI] [PubMed] [Google Scholar]
- Jankovic J., Aguilar L.G. Current approaches to the treatment of Parkinson's disease. Neuropsychiatr. Dis. Treat. 2008;4(4):743–757. doi: 10.2147/ndt.s2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Jiménez F.J., Alonso-Navarro H., García-Martín E., Agúndez J.A. Cerebrospinal fluid biochemical studies in patients with Parkinson's disease: toward a potential search for biomarkers for this disease. Front. Cell. Neurosci. 2014;8:369. doi: 10.3389/fncel.2014.00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M., Singh P.M. Current role of dexmedetomidine in clinical anesthesia and intensive care. Anesth. Essays. Res. 2011;5(2):128–133. doi: 10.4103/0259-1162.94750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Li Y., Tian S., Wang H., Wu H., Zhang A., Gao C. Anti-inflammatory effects of perioperative dexmedetomidine administered as an adjunct to general anesthesia: a meta-analysis. Sci. Rep. 2015;5:12342. doi: 10.1038/srep12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Zhang L., Joo D., Sun S.C. NF-κBsignaling in inflammation. Signal Transduct. Target Ther. 2017;2:17023. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamelak M. Parkinson’s disease, the dopaminergic neuron and gammahydroxybutyrate. Neurol. Ther. 2018;7(1):5–11. doi: 10.1007/s40120-018-0091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miksa M., Das P., Zhou M., Wu R., Dong W., Ji Y., Goyert S.M., Ravikumar T.S., Wang P. Pivotal role of the alpha(2A)-adrenoceptor in producing inflammation and organ injury in a rat model of sepsis. PLoS ONE. 2009;4(5) doi: 10.1371/journal.pone.0005504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percário, S., da Silva Barbosa, A., Varela, E.L.P., Gomes, A.R.Q., Ferreira, M.E.S., de Nazaré Araújo Moreira, T., Dolabela, M.F., 2020. Oxidative stress in Parkinson's disease: potential benefits of antioxidant supplementation. Oxid. Med. Cell. Longev. 2020, 2360872. [DOI] [PMC free article] [PubMed]
- Sulzer D., Surmeier D.J. Neuronal vulnerability, pathogenesis, and Parkinson's disease. Mov. Disord. 2013;28:715–724. doi: 10.1002/mds.25187. [DOI] [PubMed] [Google Scholar]
- Surmeier D.J. Determinants of dopaminergic neuron loss in Parkinson's disease. FEBS J. 2018;285(19):3657–3668. doi: 10.1111/febs.14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey M.G., Goldberg M.S. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010;37(3):510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey M.G., McCoy M.K., Frank-Cannon T.C. Neuroinflammatory mechanisms in Parkinson's disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007;208(1):1–25. doi: 10.1016/j.expneurol.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanvi B.R., Lo T.C. Long term motor complications of levodopa: clinical features, mechanisms, and management strategies. Postgrad. Med. J. 2004;80(946):452–458. doi: 10.1136/pgmj.2003.013912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki M., Kawasaki T., Habe K., Hamada K., Kawasaki C., Sata T. The effects of dexmedetomidine on inflammatory mediators after cardiopulmonary bypass. Anaesthesia. 2014;69(7):693–700. doi: 10.1111/anae.12636. [DOI] [PubMed] [Google Scholar]
- Wang Q., Liu Y., Zhou J. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl. Neurodegener. 2015;4:19. doi: 10.1186/s40035-015-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Sun X., Niu M., Zhang X., Wang J., Zhou C., Xie A. RAGE silencing ameliorates neuroinflammation by inhibition of p38-NF-κB signaling pathway in mouse model of Parkinson's disease. Front. Neurosci. 2020;14:353. doi: 10.3389/fnins.2020.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H., Chi X., Jin Y., Wang Y., Huang P., Wu S., Xia Z., Cai J. Dexmedetomidine inhibits TLR4/NF-κB activation and reduces acute kidney injury after orthotopic autologous liver transplantation in rats. Sci. Rep. 2015;5:16849. doi: 10.1038/srep16849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J.Y., Park J.H., Kim E.J., Park B.S., Yoon J.U., Shin S.W., Kim D.W. Dexmedetomidine attenuates H2O2-induced cell death in human osteoblasts. J. Dent. Anesth. Pain. Med. 2016;16(4):295–302. doi: 10.17245/jdapm.2016.16.4.295. [DOI] [PMC free article] [PubMed] [Google Scholar]