

Abstract
Insufficient development of new antibiotics and the rising resistance of bacteria to those that we have are putting the world at risk of losing the most widely curative class of medicines currently available. Preventing deaths from antimicrobial resistance (AMR) will require exploiting emerging knowledge not only about genetic AMR conferred by horizontal gene transfer or de novo mutations but also about phenotypic AMR, which lacks a stably heritable basis. This Review summarizes recent advances and continuing limitations in our understanding of AMR and suggests approaches for combating its clinical consequences, including identification of previously unexploited bacterial targets, new antimicrobial compounds, and improved combination drug regimens.
INTRODUCTION
In addition to our immune system, our major defenses against infectious disease are antibiotics, vaccines, sanitation, potable water, sound nutrition, and public health infrastructure. All of these are failing in various parts of the world. One key defense, the use of antibiotics, is beginning to fail worldwide because of the rise of antimicrobial resistance (AMR), which is threatening to undermine the practice of medicine (1–5). AMR is the ability of bacterial pathogens to survive exposure to compounds that were expected to kill them. Although AMR is increasing, we are gaining biological insights and technological advances that can help us to retake lost ground, as we discuss in this Review.
We begin with a discussion of biological insights into bacterial AMR and distinguish its genetic (stable and heritable) and phenotypic forms (reversible and not attributable to a genetic change), also taking stock of the field’s different terminologies. We then summarize current views of antibiotic action as a background for focusing on gaps in our understanding. Although the clinical challenge of genetic AMR is enormous, the clinical recognition and experimental analysis of genetic AMR are relatively straightforward. In contrast, phenotypic AMR is much harder to recognize clinically and analyze experimentally. However, most of the recent advances in understanding AMR mechanisms involve phenotypic AMR and so receive greater emphasis here. Last, we discuss recent technological advances that can leverage efforts to combat AMR through more efficient target identification, compound profiling, and regimen design. Throughout the Review, tuberculosis (TB) caused by the bacterial pathogen Mycobacterium tuberculosis (Mtb) features prominently, because it is the world’s leading cause of infectious death from a single pathogen, probably accounts for the largest number of cases of life-threatening bacterial AMR, and has led to innovations that can serve as examples for combating AMR in other pathogens.
THE BIOLOGY OF AMR
Definitions and conceptual limitations
Genetic AMR arises from mutations in the bacterial genome or receipt of antibiotic resistance genes from other bacteria (Table 1 and Fig. 1). The altered or newly acquired genes confer the capacity for resistance to antibiotics on the bacteria’s progeny. Defined operationally, genetic AMR results in the ability of a bacterial population to grow in the presence of an antibiotic at a much higher concentration, typically at least fourfold higher, than the minimum inhibitory concentration (MIC), where MIC is defined as the lowest concentration of the antibiotic that has routinely prevented visible growth of the same bacterial species in a growth-permissive culture medium. Clinically, genetic AMR leads to primary treatment failure and requires use of alternative antibiotics or other therapeutic approaches. Antibiotics that might be selected as backups may have been passed over in the first place because of their lesser likelihood of efficacy, greater toxicity, or limited availability. For an increasing number of multidrug-resistant bacterial infections, backup antibiotics do not exist. In contrast, phenotypic AMR is the result of changes in a bacterium that arise and subside within individual cells without dependence on a genetic change relative to the sensitive cells in the same population (Table 1 and Fig. 1). Phenotypic AMR to a given antibiotic does not allow growth of the overall bacterial population in the presence of that antibiotic at concentrations at or above the MIC. Stated differently, under conditions that support replication of a given bacterial population, the MIC for a population containing phenotypically resistant cells is the same as that of a population lacking them.
Table 1.
Characteristics of genotypic and phenotypic antimicrobial resistance.
| Characteristic | Genetic AMR | Phenotypic AMR |
|---|---|---|
| Resistance directly attributable to a genetic change (de novo mutation or acquisition of new genes) | Yes | No |
| Resistance manifest in progeny of the resistant bacterial cells | Yes | No or limited |
| MIC, relative to sensitive strain | Increased | Unchanged |
| MBC, relative to sensitive strain | Increased | Increased under some circumstances |
| Clinical outcome | Primary treatment failure | Primary treatment failure |
| Relapse | ||
| Emergence of genetic AMR | ||
| Clinical strategy | Switch antibiotics | Combine antibiotics |
| Include antibiotics that kill bacteria expressing phenotypic AMR to conventional antibiotics | ||
| Increase treatment time |
MIC, minimum inhibitory concentration, the lowest concentration of the antibiotic required to prevent growth of the bacteria over a defined period of time; MBC, minimum bactericidal concentration, the lowest concentration of the antibiotic that kills a given proportion of the population (typically 99%) in a given period of time; AMR, antimicrobial resistance.
Fig. 1. Genetic and phenotypic AMR.
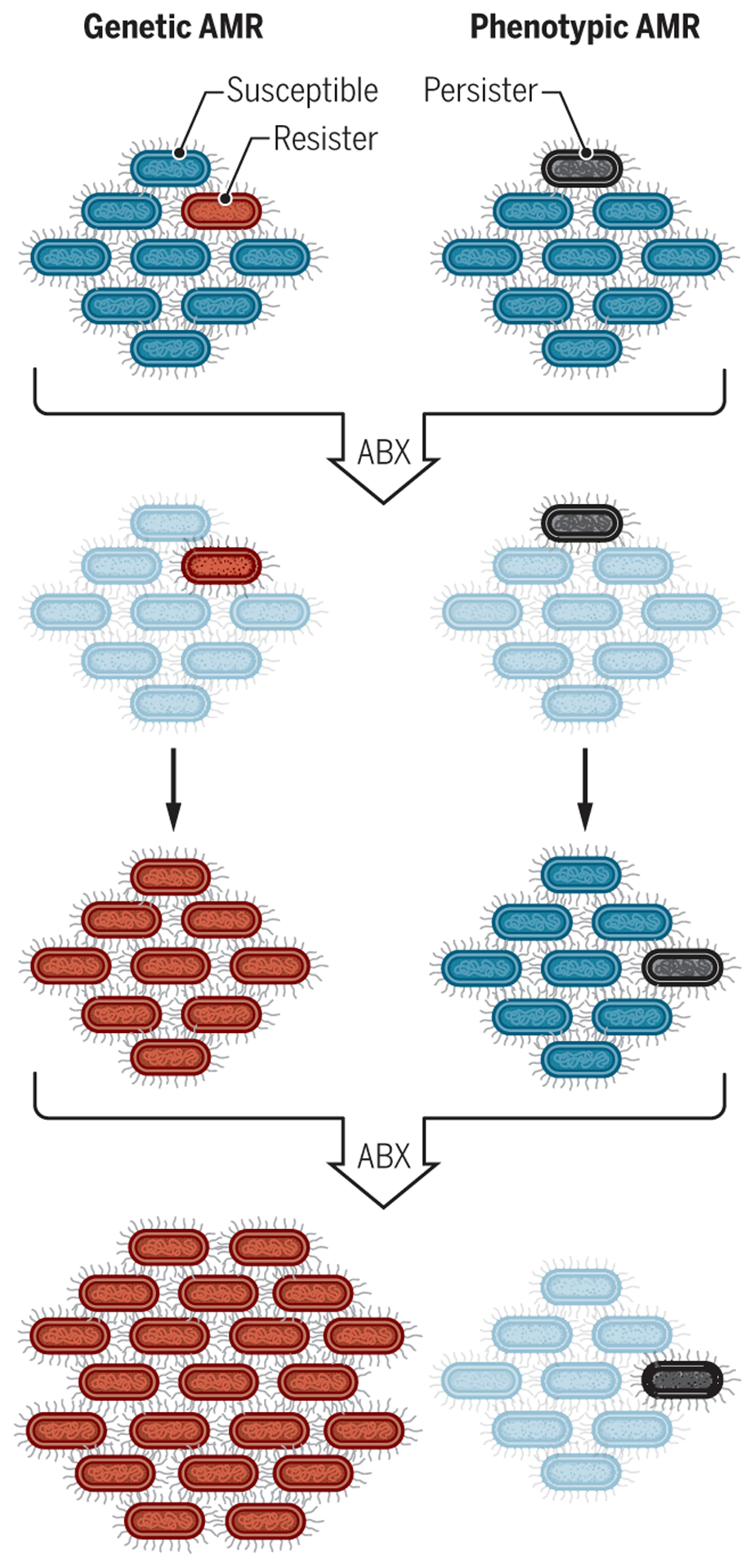
The schematic shows genetic AMR (left) compared to an example of phenotypic AMR (right). Left: In genetic AMR, a bacterium with a mutation that gives it the potential to resist an antibiotic (red cell, first row) survives exposure to that drug (second row) and continues to proliferate, whereas the susceptible majority (blue cells, first row) die (pale blue cells, second row). The resistant bacterium (red) continues to proliferate and pass its mutation on to its progeny even when the antibiotic is removed (third row). Upon a second exposure to antibiotic (fourth row), all the bacterial cells survive and continue to grow during exposure. Right: In contrast, in phenotypic AMR, a bacterial cell is genetically identical to its siblings but happens to be in a metabolic state that is conducive to surviving the first exposure to antibiotic (gray cell, first row). When removed from the antibiotic, this bacterium gives rise to a population resembling that from which it arose so that the second exposure to antibiotic kills the same proportion of the bacterial population as before (fourth row). ABX, antibiotic [adapted from (107)].
Phenotypic AMR can manifest as a slowed rate of killing for the bulk population, a phenomenon sometimes termed “tolerance” (6). Alternatively, phenotypic AMR can be expressed by a subset of bacterial cells that have a much slower rate of killing than most of the population or are not killed at all. This is often called “persistence.” When the survivors are allowed to grow in the absence of the antibiotic, the population to which they give rise has the same MIC as the starting population (Table 1 and Fig. 1).
Besides the MIC, another metric for establishing antibiotic potency is called the MBC, which is the minimum concentration of antibiotic that is bactericidal for a given proportion of the population (typically, 99%) in a given period of time. For replicating bacterial populations, both the MIC and MBC99 are determined routinely; the MBC99 may be the same as the MIC or higher. When antibiotics are tested on bacteria under nonreplicating conditions, an MIC cannot be determined because bacterial numbers have stopped increasing without the antibiotic. Under such conditions, the MBC99 sometimes increases by orders of magnitude or is not reached at all within the range of antibiotic concentrations tested (7), an extreme example of the type of phenotypic AMR that some call tolerance.
Phenotypic AMR can arise stochastically, leading to the descriptors “spontaneous persistence” (6), “stochastic switching” (8), “class 1 persistence” (9), and “type 2 persistence” (10, 11) (Table 2). Phenotypic AMR can also arise upon exposure to altered environments, such as nutrient or oxygen deprivation, acidification, oxidative stress, host immune responses, and exposure to sublethal concentrations of antibiotics. In such cases, the terms applied have included “triggered persistence” (6), “responsive diversification” (8), “class 2 persistence” (9), and “type 1 persistence” (10, 11). Both spontaneous and environmentally induced phenotypic AMR can lead to primary treatment failure or relapse. Clinical strategies for combatting phenotypic AMR include combining antibiotics, using unconventional antibiotics selected for their ability to kill bacteria that are phenotypically resistant to conventional antibiotics, and prolonging antibiotic treatment times (Table 1).
Table 2.
Different terminologies associated with phenotypic AMR.
| Term | Original definition | Redefinition | ||
|---|---|---|---|---|
| Meaning (year) | Meaning (year) | |||
| Phenotypic resistance | Synonym for phenotypic AMR (1944) | (67) | ||
| Bacterial persistence/persisters | Consequence of phenotypic AMR (1944) | (67) | Slow killing of a subpopulation of bacteria in a larger population that is killed rapidly (2019) | (6) |
| Phenotypic tolerance | Synonym for phenotypic AMR (1986) | (72) | Genetically determined delay in onset of replication after transfer to a new culture medium (2014) | (64) |
| Slower killing of an entire population for any reason (2019) | (6) | |||
| Type 1 persistence | Persistence displayed by bacterial cells that remain nonreplicating after transfer from the stationary growth phase (2004) | (10) | Triggered persistence: Persistence in response to a stress stimulus (2019) | (6) |
| Type 2 persistence | Persistence of a slow- growing subpopulation of bacteria whose slow growth is not attributable to having been in stationary growth phase (2004) | (10) | Spontaneous persistence (2019) | (6) |
| Class 1 phenotypic tolerance | Phenotypic AMR of a minority of bacterial cells in a replicating population (2012) | (9) | ||
| Class 2 phenotypic tolerance | Phenotypic AMR of most bacterial cells in a nonreplicating population (2012) | (9) | ||
| Responsive diversification | Phenotypic AMR in response to a stress stimulus (2014) | (8) | ||
| Stochastic switching | Phenotypic AMR manifest before a stress stimulus (2014) | (8) | ||
| Tolerance due to slow growth | Slow killing due to slow growth resulting from genetic impairment or stress, including exposure to antibiotics (2016) | (11) | ||
| Tolerance due to growth lag | Slow killing due to delayed onset of growth resulting from a change in culture conditions (2016) | (11) |
However, the diversity of bacterial adaptive mechanisms frustrates efforts at classification, and definitions of genetic and phenotypic AMR have limitations and exceptions. Some genetic mutations that afford AMR quickly revert, making it hard to distinguish such genetic AMR from phenotypic AMR [e.g., (12, 13)]. This may manifest as “clonal heteroresistance” (14–16), the heritable but short-lived AMR of a subpopulation of bacteria that have acquired single-nucleotide polymorphisms, nucleotide insertions or deletions, or gene amplifications that protect them from the antibiotic. The fitness cost of mutations conferring clonal heteroresistance leads to reversion or compensation by secondary mutations once the bacteria are no longer exposed to the antibiotic (14). Although phenotypic AMR is defined as not arising from a genetic change relative to antibiotic-sensitive members of the same bacterial strain, it is sometimes propagated for several generations, much like clonal heteroresistance. For example, acetylation or methylation of a bacterial histone-like protein led to transiently inherited phenotypic AMR against the antibiotic isoniazid in Mycobacterium smegmatis, a nonpathogenic relative of Mtb (17).
Further blurring the distinction between genetic and phenotypic AMR, the proportion of phenotypically resistant bacteria in a genetically homogeneous (isogenic) population can be increased by mutations in genes not directly related to the antibiotic’s mechanism of action, so-called “high persistence” (hip) mutations.
Rather than striving to define distinct categories of AMR and then listing exceptions, it may be helpful to envision a continuum of resistant states described by three parameters: prevalence of the resistant state in the bacterial population under study, the resistant bacteria’s rate of growth, and the rate at which the antibiotic kills the resistant bacteria (Fig. 2).
Fig. 2. Three characteristics distinguish four types of AMR in vitro.
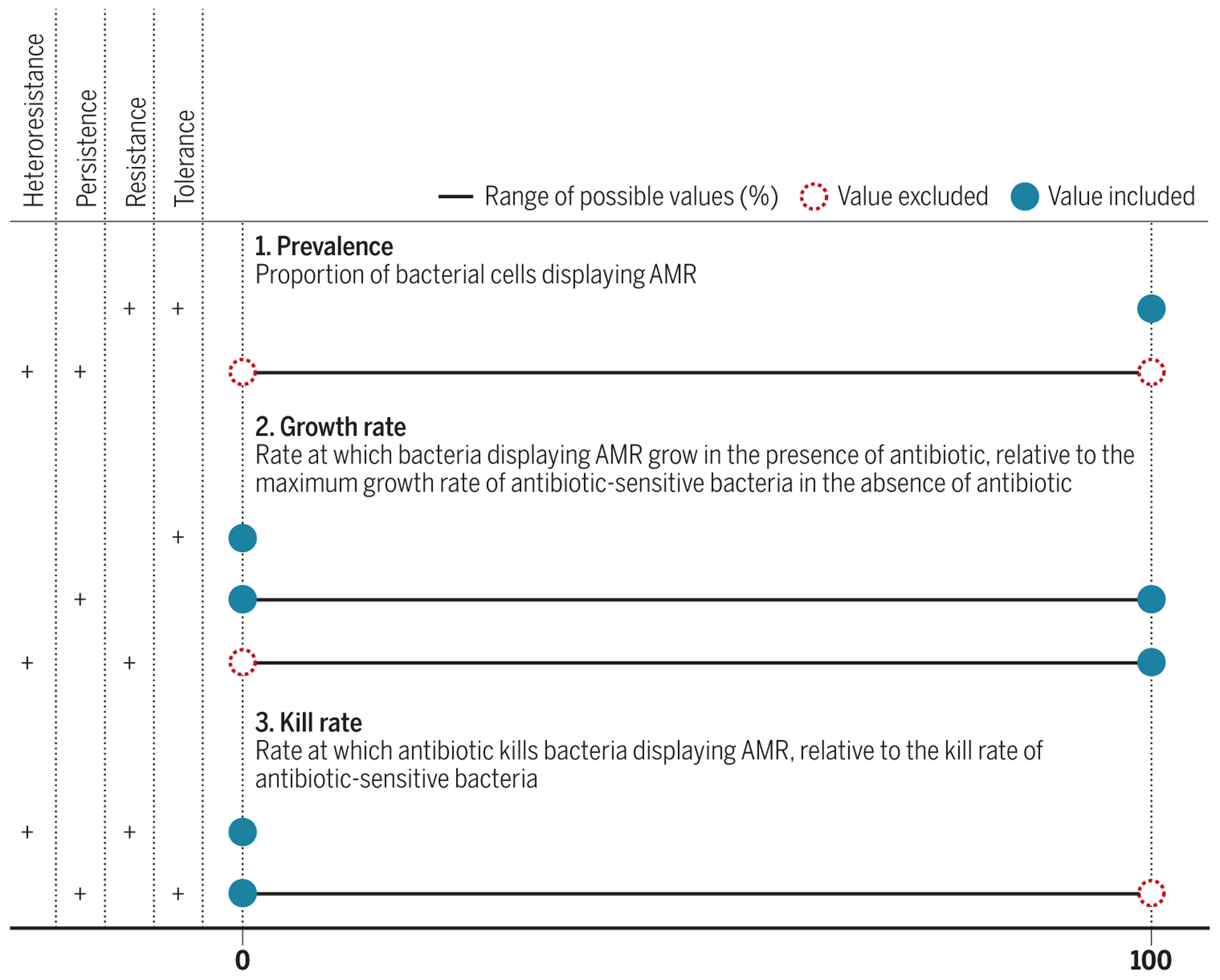
The four types of AMR observed in vitro are as follows: heteroresistance, persistence, resistance, and tolerance. These four types of AMR can be distinguished by a combination of three characteristics: prevalence, growth rate, and kill rate. Different values of these three characteristics enable diverse AMR phenotypes. In clinical settings, different AMR phenotypes may pertain to the disease-causing bacterial pathogen inside or outside host cells, in different anatomical sites, and at different times during the course of the infection and its treatment.
As a further caution to an overemphasis on classification, it is important to acknowledge the shortcomings of the in vitro assays in which such parameters are measured, as discussed next.
Practical limitations to the study of AMR
There are practical limitations to the standard in vitro assays used for studying AMR. To the extent that such assays fail to mirror clinical settings, they may not yield clinically meaningful insights. The assays use culture media that are markedly different from human body fluids in composition and typically expose bacteria to the antibiotic over time periods much shorter than a patient’s course of treatment. Assays to detect AMR rarely model the fluctuations in drug concentrations over time that patients experience, although intermittent antibiotic exposure selects for mutations that increase phenotypic AMR (18). Assays for detecting phenotypic AMR are usually conducted with bacteria in planktonic form (in a liquid medium), whereas most chronic bacterial infections in patients involve biofilms (bacteria embedded in a secreted polymeric matrix) (19). Bacteria in different regions of biofilms can display phenotypic AMR to different antibiotics through different mechanisms (20), and genetic regulators of phenotypic AMR can differ for the same bacterial species in either the planktonic or biofilm state. For example, a comparison of the contributions of more than 500 genetic loci to phenotypic AMR in Pseudomonas aeruginosa cultured in either planktonic or biofilm form identified distinct sets of regulators for each state (21). Standard MIC and MBC assays ignore the so-called inoculum effect, that is, the impact of the size of the initial bacterial inoculum on the antibiotic’s apparent potency. Moreover, a change in MIC of ≤2-fold or ≤4-fold is considered to be within the error of measurement, but recent evidence suggests that minor changes in an antibiotic’s MIC can be clinically consequential (22).
Given these caveats, it is no surprise that insights from in vitro studies of AMR sometimes contrast with those derived in vivo, although few direct comparisons have been reported. For example, a screen conducted in vivo identified mutations in Mtb that either increased or decreased the frequency of phenotypic AMR to isoniazid in mice but had no impact on the proportion of Mtb displaying phenotypic AMR to isoniazid in vitro (23). Most screens for genetic modifiers of phenotypic AMR are conducted in vitro, and seldom are they followed by experiments to test the impact of identified mutations on treatment of an infected host (24).
The limitations of operational definitions based on in vitro assays extend to the concept of bacterial death itself. The “gold standard” for measuring bacterial death during antibiotic exposure involves treating a population of bacterial pathogens with a drug, taking a sample, removing the drug, and placing the cells in conditions that support the replication of their untreated counterparts. The survivors are quantified, usually by counting colony-forming units (CFUs) on a culture medium solidified with agar, and the bacteria that fail to form CFUs are considered dead. Recent studies highlight flaws in this way of thinking (25–31).
Between life and death may lie sickness. As for the patient, so for the pathogen, sickness can lead not just to death but to recovery. Antibiotics can injure bacteria in such a way that the bacteria suspend or slow replication while engaged in molecular repair (25, 26). If survivors are quantified while some bacteria are in repair mode, we may count as dead some bacteria that are on course to regain their ability to replicate and cause disease. Such bacteria often display phenotypic AMR while they are in nonreplicating mode (7, 25, 26).
Evidence is mounting for the microbiological and clinical relevance of this challenge to the operational definition of bacterial death. “Viable but nonculturable” (27, 28), “differentially culturable” (29), and “differentially detectable” (26, 30) are synonymous terms used to describe bacteria that do not form colonies on agar after exposure to various stresses, including antibiotics, but can be shown to be viable by other assays. For example, they can grow in vitro after limiting dilution in liquid medium, which allows the investigator to calculate the original concentration of viable bacteria (26).
Differentially detectable Mtb have been generated in vitro by nutritional stresses with or without exposure to antibiotics (26, 31). Differentially detectable Mtb have been recovered from Mtb-infected mice that were treated with antibiotics until CFUs were undetectable by culture of organ homogenates (32) or were identified in retrospect by the subsequent relapse of remaining mice in the same cohorts (33). Supporting the clinical importance of this phenomenon, differentially detectable Mtb have also been detected in sputum from human participants with TB (29, 30, 34). Two groups reported that 80 to 86% of untreated individuals with TB had orders of magnitude more differentially detectable Mtb than CFUs in their sputum (29, 34), and two studies reported that the proportion of differentially detectable Mtb increased after 2 weeks of antibiotic therapy, even as the total number of viable Mtb declined (30, 34). Moreover, differentially detectable Mtb predominated over CFUs in 10 of 19 specimens from sites of extrapulmonary TB, including lymph nodes, abscesses, pleural fluid, and biopsies of colon and bone (35). In other words, standard measures of bacterial cell death may exaggerate the impact of antibiotics. With these caveats in mind, we can best approach rapidly evolving knowledge about AMR with a contemporary understanding of how antibiotics kill bacteria.
Primary and secondary actions of antibiotics
Recent evidence is strong that antibiotics generally kill bacteria in environments with normal or reduced oxygen by a combination of individual primary actions and common secondary actions (36, 37). When oxygen is absent, the primary actions alone sometimes lead to killing of bacteria (38, 39).
The individual primary actions of most antibiotics involve the corruption of processes essential to the pathogen under the conditions of study and, for clinically effective antibiotics, under the conditions that the bacteria encounter in the host. However, the conditions faced by the pathogen can vary in different niches in a given host at a given time, in a given host at different times, and in different hosts. The essentiality of a bacterial process and its vulnerability to inhibition by antibiotics—that is, the level of inhibition required to stop growth of the bacteria or kill them—may vary with those conditions.
In addition to these primary actions, the engagement of antibiotics with their primary targets frequently triggers a series of secondary actions that can result in increased concentrations of reactive oxygen species (ROS). This can result from either the increased generation or decreased catabolism of ROS. Both of those effects can stem from the altered bacterial metabolism that results from the primary actions of antibiotics (40–45). However, it is unclear exactly how antibiotics acting against their classic bacterial targets—synthesis of nucleic acids, proteins, and cell walls—affect bacterial metabolism. The specific secondary mechanisms that contribute to antibiotic killing likely vary as a function of the antibiotic and bacterial species. One study identified the metabolic responses of Escherichia coli to ampicillin, ciprofloxacin, and gentamicin that were linked to the lethal effects of these drugs as increased flux through central carbon metabolism and an apparent deficiency of adenine (46). The latter led to increased adenine synthesis, resulting in increased synthesis of adenosine triphosphate (ATP) and consumption of oxygen (46), which conceivably could lead to increased ROS generation. Secondary actions such as generation of ROS can continue after no more antibiotic remains in the bacterial cells and the antibiotic’s primary action has ceased (43).
ROS can contribute to antibiotic lethality by damaging bacterial macromolecules and metabolites and impairing the transport of electrons, protons, and other ions. For example, misfolding of proteins can produce a toxic loss or gain of function as well as further ROS generation (43). These toxic effects are countered by bacterial ROS detoxification reactions and molecular repair pathways, including protein refolding and DNA repair, as well as by the degradation or sequestration of macromolecules that are irreparably damaged (47).
Gaps in understanding genetic AMR
Genetic AMR has emerged to every clinically used antibiotic shortly after its introduction into clinical practice (48). Genetic AMR sometimes emerges even sooner, during preregistration clinical trials, precluding deployment of an antibiotic that it may have taken hundreds of millions of dollars to develop (49). Thus, the specter of genetic AMR severely handicaps antibiotics in their competition for pharmaceutical companies’ resources. One remedy for the producer is to sell as much of a given antibiotic as possible before resistance renders it profitless. Agribusiness is an eager customer. More tons of antibiotics are used to promote the growth of healthy food animals than to treat people or animals with infections (50). As a result, antibiotics are prevalent in the urine of healthy people who did not knowingly ingest them (51), likely hastening the selection of drug-resistant bacteria. Additional practices in various parts of the world that select for resistant bacteria include prescription of antibiotics for the treatment of viral illnesses, availability of antibiotics without prescription, and prescription of antibiotics by those who sell them (4).
Most of the known mechanisms of genetic AMR were found decades ago, although new examples continue to be identified. Best known are mutations of the antibiotic’s target that reduce binding of the drug but preserve enough of the target’s function to sustain the pathogen’s ability to replicate, mutations that lead to posttranslational modifications of the target with the same effect, increased expression of the target so that intracellular accumulation of the antibiotic is insufficient to prevent bacterial replication, chemical modification of the drug so it no longer binds to the target, increased catabolism of the drug, increased export of the drug, decreased uptake of the drug, decreased activation of the prodrug, or expression of a pathway that compensates for the inhibition of the target. Recently, another mechanism was found: loss of function of an enzyme whose physiological action reverses the action targeted by the antimicrobial agent (52).
These mechanisms of genetic AMR all pertain to the primary actions of antibiotics. Now that we appreciate the existence of secondary actions stemming from increased ROS, we might predict the existence of genetic AMR linked to increased capacity to prevent or repair oxidative injury. Such examples have been reported (53), but their relative rarity presents a challenge to the theory that these secondary actions are important contributors to antibiotic lethality.
Perhaps mutations that increase resistance to the secondary actions of an antibiotic confer a smaller increase in MIC than that attainable by mechanisms that diminish the antibiotic’s primary action, such that the shift is considered within experimental error and the mutant bacterium is not classed as resistant. Perhaps some such mutations are copy number variations, which are typically not assessed when bacterial genomes are sequenced. Perhaps these types of mutations tend to impose fitness costs that select for revertants to the wild-type sequence during expansion of resistant clones for sequencing (12, 13). Mutations that increase the proportion of bacterial persisters (hip mutations) or confer tolerance would be missed in conventional drug resistance screens because they do not increase the MIC.
A potentially more informative possibility is that such mutations may have been found but not recognized as pertinent for AMR or antioxidant defense because a bacterial cell’s ability to generate less ROS or more reducing power during stress is under complex control and incompletely understood. In E. coli, 133 enzyme reactions are predicted to have the potential to generate ROS (54). In the yeast Saccharomyces cerevisiae, at least 102 genes are required to maintain redox balance, including 12 genes of unknown function (55). Genes of unknown function are prevalent in even the most well-studied bacterial pathogens. For example, 27% of genes in Mtb have unassigned functions (56), and a similar proportion bear unverified annotations. Mutations in genes of unknown function and genes whose known functions are not obviously related to oxidant stress or antioxidant defense in a drug-resistant bacterial pathogen might be dismissed from consideration as candidates for causing AMR. For example, in yeast, it was not anticipated that lysine uptake would markedly affect antioxidant defense (57).
Although ROS contribute to antibiotic actions, they can also contribute to emergence of genetic AMR. For example, Sebastian et al. (58) applied Luria-Delbrück analysis to Mtb exposed to the antibiotic rifampin. This method establishes that mutations that confer resistance to a given antibiotic existed in a bacterial population before that antibiotic was administered. They found that conditions similar to those that generate differentially detectable Mtb in vitro in response to rifampin led to elevated production of ROS in Mtb in association with de novo mutations in the rpoB and gyr genes, producing genetic AMR to rifampin and moxifloxacin, respectively. Likewise, Swaminath et al. found that M. smegmatis persisters to moxifloxacin generated ROS that led to mutations conferring resistance to moxifloxacin, ethambutol, and isoniazid (59). Similarly, a subset of fluoroquinolone-treated E. coli generated ROS that activated a process of mutagenic DNA strand break repair that led to heritable mutations imparting resistance to rifampin and ampicillin (60). Thus, the secondary ROS-mediated actions of one antibiotic can promote genetic AMR to itself as well as to other antibiotics.
The importance and difficulty of understanding phenotypic AMR
To cut off genetic AMR at its roots, we need to understand phenotypic AMR (61), which is more common than genetic AMR and predisposes to its emergence (18, 62–65). The assertion that phenotypic AMR is more common than genetic AMR is justified clinically by the prevalence of phenotypically resistant bacteria in biofilms and the prevalence of bacterial biofilms in many clinical settings (19). Further, it can be speculated that the empirically determined duration recommended for treatment of a given infection with a given antibiotic reflects the degree of phenotypic AMR displayed by members of the infecting bacterial population. From a basic science perspective, de novo mutations that confer genetic AMR arise in vitro with frequencies on the order of 10−6 to 10−9 or even lower. This range is defined in part by the fidelity of genome replication in bacterial pathogens and in part by the selection criteria for clinical progression of candidate antibiotics, because antibiotics with higher frequencies of bacterial resistance are discarded. In contrast, phenotypic AMR occurs with a frequency that can approach 100 in the following two senses. First, application of most antibiotics to a population of replicating bacteria often leads to the survival of a small proportion, typically about 10−6, that is genetically identical to the bacteria that were killed and that, after removal of the antibiotic, give rise to descendants with the same MIC as the original population (often called persisters). In that sense, a phenotypically resistant minority may be present on many or most of the occasions that an antibiotic is applied to a bacterial population. Second, application of most antibiotics to a population of nonreplicating bacteria often leads to survival of many, most, or even all of them, again without mutation and without a change in MIC when the survivors are tested under replicating conditions (7, 66–70).
Phenotypic AMR can predispose to genetic AMR by several mechanisms (62). Examples include Mtb, M. smegmatis, and E. coli persisters that survived one antibiotic that generated ROS. The ROS or the bacterial cells’ response to it was mutagenic, giving rise to genetic AMR against the same antibiotic and others (58–60). In E. coli, mutations that increased the frequency of phenotypic AMR preceded those conferring genetic AMR, likely because they increased the number of survivors of a given cycle of antibiotic exposure in which subsequent mutations could confer genetic AMR (18). In addition, some mechanisms of phenotypic AMR lead to suppression of DNA repair, allowing the emergence of mutations that confer genetic AMR (63). Moreover, the survival of phenotypically drug-resistant bacteria allows them to transform other bacteria with plasmids they may contain that confer genetic AMR to other antibiotics (71).
Research into phenotypic AMR was stymied for decades not only because the phenomenon is transient but also because when it is studied under standard laboratory conditions, which support bacterial replication, it typically involves a very small fraction of the bacterial population, the persisters. Such cells are difficult to distinguish and separate from those expressing genetic AMR. Recently, floodgates to understanding have opened with the advent of improved techniques for single-cell analysis, cell sorting, genomics, and metabolomics. The field now enjoys a healthy ferment of ideas that are described with sometimes confusing terminology. The terms were introduced earlier in this Review, but an additional challenge in the field is that some of them have been used in different ways at different times (Table 2).
In much of the literature, “phenotypic resistance” is used as a synonym for “phenotypic tolerance,” a term that predominated for many years after its introduction in 1986 (72). However, both of these terms were preceded by the term “persisters,” which Bigger introduced in 1944 (67) to refer to the small fraction of Staphylococcus pyogenes bacteria that he observed surviving exposure to penicillin and that subsequently gave rise to penicillin-sensitive populations after penicillin was removed. In Bigger’s usage, persistence is not a mechanism of phenotypic AMR but a consequence of it, whether the phenotypic AMR is of the type that is now often called persistence or the type called tolerance.
In literature that views persistence and tolerance as contrasting phenomena (6) rather than synonyms, the central focus is on the kinetics of killing of a bacterial population exposed to an antibiotic in vitro. If killing is initially fast but becomes slow when few bacteria remain, the survivors are called “persistent,” whereas if the whole population is killed at a constant rate but more slowly than usual, the bacteria are called “tolerant” (6). In this terminology, tolerance can be characterized by another metric besides MIC and MBC, namely, the minimum duration for killing (6). This usage limits persistence to the behavior of what is usually a minor subset of cells in a population and tolerance to a population whose behavior is homogeneous (6). Further, as mentioned above, stochastically arising persistence has been called type 2 persistence (10, 11), whereas stress-induced persistence has been termed type 1 persistence (10, 11). Originally, however, the terms “type 1” and “type 2” persistence had narrower definitions: The former designated individual bacteria that had ceased replicating in stationary phase and remained in that state when inoculated into fresh medium, whereas the latter designated a slow-growing subpopulation (10). Tolerance has also been categorized into “tolerance by slow growth” and “tolerance by lag” (6, 64). For the former, slower killing is attributed to a genetically determined or condition-dependent drop in growth rate, whereas for the latter, it is ascribed to a delayed onset of growth when bacteria are transferred to new culture conditions.
An emerging understanding of phenotypic AMR
Complex biology understandably and constructively leads to contending ideas. What follows is an effort to describe and reconcile some of them.
In 1942, Hobby and colleagues reported finding surviving streptococci at a frequency of 10−6 after exposure to penicillin at 37°C (a replicating condition) and at a frequency of 100 at 4°C (a nonreplicating condition) (68). They concluded from these results that “penicillin is capable of destroying bacteria only if multiplication takes place” (68). Two years later, Bigger reported similar results in staphylococci: The bacteria survived exposure to penicillin at a frequency of 10−6 at 37°C and at a frequency of 100 at 4°C, at low osmolarity without a carbon source and at low pH (each a nonreplicating condition) (67). Bigger showed that the survivors lacked heritable resistance and dubbed them persisters. He made the same inference as Hobby et al. regarding the nature of these persisters, concluding that they were “cocci which survive contact with penicillin because they are in dormant, non-dividing phase” (67). Others quickly confirmed these observations (73). Seventy-five years later, many reports on phenotypic AMR continue to embrace the assumptions of Hobby and colleagues and Bigger, who assumed that what was true for most of the bacteria in a nonreplicating population must have been true for the rare survivors in a replicating population, namely, that “dormancy”—which in Bigger’s sense meant nonreplication—was the mechanism for temporary resistance to the antibiotic. This assumption gave rise to the extension that nonreplication (or slow replication) is the mechanism for phenotypic AMR of all bacteria to any antibiotic, the underlying reasoning being that the primary targets of antibiotics are inactive (or less active) and therefore incorruptible (or less corruptible) in nonreplicating (or slowly replicating) bacteria.
Several studies lent support to this idea. A 1986 study showed that the rate of killing of E. coli by β-lactam antibiotics was directly proportional to their rate of growth (74). A landmark 2004 paper applying time-lapse microscopy to antibiotic-treated E. coli confined in microfluidic chambers demonstrated that ampicillin preferentially spared individual bacteria that were in a nonreplicating or slowly replicating state when the drug was applied (59). The linkage between slow growth and phenotypic AMR was recently extended to Salmonella enterica var. Typhimurium (75) upon reducing their growth rate through incubation in culture medium with a low concentration of magnesium ions (75). In addition, the argument that phenotypic AMR is the result of metabolic inactivity as a consequence of nonreplication is commonly defended by noting an association of AMR with a fall in ATP. ATP was reduced by 90% in Staphylococcus aureus in stationary phase, a setting that promotes phenotypic AMR, compared to the amount of ATP in replicating S. aureus; ATP-depleting metabolic poisons also increased the frequency of persisters (76). However, this is not always the case—in the Salmonella study mentioned above, no fall in ATP was observed (75).
In contrast, a rapidly growing body of research reveals a much broader view of mechanisms underlying phenotypic AMR. The concept of a single mechanism for phenotypic AMR, such as nonreplication, is belied by the observation that different individual bacteria are often phenotypically resistant to different antibiotics (77, 78). The presumption that nonreplicating bacteria are metabolically inactive has been challenged by many studies that reveal that, except for spores, nonreplicating bacteria do not stop transcribing, translating, metabolizing, or pumping. Instead, they express a different transcriptome (24, 79–81), a different proteome (82), a different metabolome (79, 82, 83), and a different lipidome (79, 84) than their replicating counterparts while maintaining membrane potential (79, 83, 85). Consistent with this view, the first compounds found to kill nonreplicating bacteria selectively, regardless of what conditions imposed nonreplication, were inhibitors of Mtb’s dihydrolipoamide acyltransferase, a component of three α-ketoacid dehydrogenase complexes in central carbon metabolism and of a peroxynitrite reductase/peroxidase complex mediating antioxidant defense (86). The same compounds spared replicating Mtb, presumably because the replicating bacteria were less reliant on the α-ketoacid dehydrogenases or had other means of coping with oxidative stress. Also supporting maintenance of metabolic activity in nonreplicating bacteria are a study that showed that E. coli maintained a constant, albeit reduced, rate of protein synthesis for at least 60 hours after entry into stationary phase (87) and another that demonstrated substantial throughput in the ATP pool in some persisters (82). In addition, inhibitors of DNA synthesis or transcription have been observed to kill almost all (90 to 99.9%) of some nonreplicating bacteria (7, 69, 88), even if this pales in logarithmic terms compared to the killing of 99.9999% of a replicating population. These results suggest that the primary targets of these antibiotics are both active and lethally corruptible in nonreplicating bacteria.
Still other studies have dissociated growth rate from phenotypic AMR. In contrast to the results in the 2004 microfluidics study (10), others found that phenotypically resistant E. coli cells were distributed in both the rapidly replicating and nonreplicating subpopulations (89). Moreover, 99% of nonreplicating cells were not phenotypically resistant, leading to the conclusion that nonreplication “is not necessary or sufficient for bacterial persistence” (89). A likely reason for this apparent discrepancy is that whereas the latter study examined wild-type populations of E. coli, in which persisters may form by diverse mechanisms, the former used E. coli hip mutants (hipA7 and hipQ) whose mutations lead to an increase in the proportion of persisters by several orders of magnitude. This ensured that almost all the persisters observed as individual cells in populations of hip mutants were generated by the same mutation-dependent mechanism. Nonreplication and slow replication happened to be features of this mechanism for the hipA7 and hipQ mutants, but this does not establish that these features are present in all persisters nor that reduction of replication rate constitutes a mechanistic explanation for persistence.
Another recent study of E. coli demonstrated that ofloxacin persisters were metabolically active cells with individually heterogeneous rates of growth and heterogeneous expression of stress responses, suggesting that different cells survive exposure to the same antibiotic via different mechanisms and that these mechanisms are not necessarily dependent on growth rate (90). Nonreplicating Mtb continued to transcribe ribosomal RNA, a marker of metabolic activity, in a subpopulation of bacteria in the lungs of chronically infected mice (91). Phenotypic AMR to the prodrug isoniazid in M. smegmatis did not correlate with single-cell growth rates; instead, it was negatively associated with single-cell expression of catalase, which is required to activate isoniazid (92). In M. smegmatis mutants with high mistranslation rates, phenotypic resistance to rifampicin was independent of replication state and attributable instead to the presence of enough mistranslated copies of RNA polymerase, rifampicin’s target, in individual bacteria to frustrate binding of the drug while preserving the enzyme’s function (93, 94). Phenotypic AMR of replicating mycobacteria in macrophages has been ascribed both to transient induction of antibiotic efflux pumps (95) and to enhanced antioxidant capacity (96). Yet another mechanism of phenotypic AMR unrelated to replication is bacterial production of nitric oxide, which inactivates some antibiotics (97). Bacterial production of nitric oxide (97) or hydrogen sulfide (96, 98) can also give rise to phenotypic AMR through induction of antioxidant defenses.
Lopatkin and colleagues recently tested the lethality of nine different bactericidal drugs for a diverse set of bacterial species (E. coli, Acinetobacter baumannii, and S. aureus) across a wide variety of coupled conditions (i.e., those in which growth and metabolism both positively correlated with nutrient availability) and uncoupled conditions (i.e., those in which growth, but not metabolism, positively correlated with nutrient availability) (99). Metabolic activity, as opposed to growth rate, was the major determinant of antibiotic lethality (99).
In sum, the picture emerging from recent studies with diverse organisms is that nonreplication (or slow replication) is not a requirement for phenotypic AMR and, when present, is not necessarily a mechanistic explanation for it. Nonreplicating or slowly replicating bacterial states are characterized by alternative metabolic landscapes, not barren ones. It has been proposed that phenotypic AMR can arise from any mechanism that imparts genetic AMR, provided that the result is achieved without dependence on a stably heritable mutation in a gene directly related to the antibiotic’s mechanism of action (9). We need more insight into mechanisms of phenotypic AMR before we can judge the validity of that hypothesis. Studies of the genetic control of phenotypic AMR offer a productive research avenue.
Genetic control of phenotypic AMR: High- and low-persistence mutants
By definition, phenotypic AMR is not stably heritable. However, the proportion of phenotypically resistant bacteria in a population is under genetic control. This is evidenced by the existence of hip (high persistence) mutations, which increase the proportion of individual bacterial cells displaying phenotypic AMR within a population of bacteria. In bacterial populations that are replicating, the proportion of phenotypically resistant bacteria is, by definition, lower than the proportional limit of sensitivity of the assay used to characterize the potency of the antibiotic. Consider an example where the MIC of a given antibiotic for a given bacterial population corresponds to the MBC99. If the fraction of the population that is phenotypically resistant to the antibiotic is more than 10−2, the MBC99 will rise. A hip mutation could increase this proportion from the typical 10−6 by up to four orders of magnitude without raising the MBC99. Such hip mutations have great potential to teach us the mechanisms of phenotypic AMR.
In 1983, Moyed and Bertrand, working with E. coli, reported the first mutations that increase the proportion of phenotypically resistant bacteria in an isogenic population (100). Study of these and many subsequent hip mutations has identified pathways and partial mechanisms involved in phenotypic AMR. The original hipA mutations led to a 1 to 4 log10 increase in survival against cell wall synthesis inhibitors (100). HipA was later found to encode a toxin, HipA, that acts as a kinase and arrests cell growth when released from its antitoxin, HipB (101). Moyed and Bertrand’s hipA7 mutant contained two mutations in hipA that ablated its ability to arrest cell growth (101), reduced its binding affinity for the antitoxin HipB (102), and altered the pool of substrates that the hipA kinase phosphorylated (103). Both HipA and HipA7 phosphorylate the glutamate–transfer RNA (tRNA) ligase GltX, resulting in a deficiency of charged glutamate-tRNAs, ribosome stalling, synthesis of the alarmones (p)ppGpp, and activation of the stringent response, which reconfigures bacterial transcription in such a way as to postpone cell division in favor of restoring amino acid synthesis. This leads to phenotypic AMR (101, 104). The higher proportion of persisters in the hipA7 mutant population can be explained by the reduced inhibition of HipA7 by HipB (102). Several other toxin-antitoxin molecular pairs have since been implicated in phenotypic AMR in diverse bacterial species (105).
In addition to toxin-antitoxin modules, hip mutations and high-tolerance mutations have also been identified in genes encoding sigma factors and other genetic regulators or genes subserving pathways of lipid metabolism, propionate metabolism, quorum sensing, or amino acid biosynthesis and catabolism (106, 107). Identification of further hip mutants and their mechanistic characterization is needed for a broader and deeper understanding of phenotypic AMR.
The converse of hip mutants are mutations that decrease the frequency of phenotypically resistant members of a bacterial population. Although not relevant from a clinical perspective, these mutations can also lend basic insights into the mechanisms of phenotypic AMR. Many of the pathways identified through the study of low-persistence mutations overlap with those affected by hip mutations, such as toxin-antitoxin modules, the stringent response, quorum sensing, sigma factors, and lipid metabolism (107).
Other low-persistence mutations affect the proteostasis pathway. In E. coli, the frequency of persisters to diverse antibiotics was drastically reduced by knocking out each of the chaperones or proteases dnaK, clpB, and lon (108). Two studies offered mechanistic insight into these effects. In one study, individually studied E. coli cells that were phenotypically resistant to antibiotics suspended replication while forming intrabacterial aggresomes of insoluble proteins. Resumption of replication was preceded by resolution of the aggresomes, which required expression of both DnaK and ClpB (109). This suggested that the chaperones were necessary for recovery of persisters after antibiotic exposure, such that knocking them out would reduce persister frequency. In the second study, treatment of mycobacteria with kanamycin or isoniazid caused aggregates of irreversibly oxidized proteins to form. As inferred from the phenotype of the knockout of ClpB, the chaperone was required for the collection, coalescence, and sequestration of these aggregates at one pole of the surviving bacterial cells. After removal of the antibiotic, the survivors resumed replication, leaving one sibling with a large burden of aggregates. The less burdened siblings were more likely to display phenotypic AMR during subsequent rounds of exposure to the antibiotic (47), supporting a role for protein chaperones in enabling some cells to display phenotypic AMR.
The propensity of ROS to cause protein misfolding and the high representation of loss of function mutations in proteostasis pathway components among low-persistence mutants highlight the importance of secondary ROS generation in the action of antibiotics and their understudied role in the biology of phenotypic AMR. Adding further to that view is the ability of the redox-active, ROS-generating agents N-acetylcysteine and ascorbic acid to kill Mtb bacteria that are phenotypically resistant to isoniazid (110). Their action can be seen as boosting the secondary action of an antibiotic.
Bacterial persistence in vivo and therapeutic implications
The term persisters is often applied to bacteria that remain viable in an experimental participant or patient who has been treated with antibiotics in a way that was expected to kill the bacteria based on in vitro studies, such as achieving blood concentrations of the antibiotic that remained above the MBC throughout the treatment interval. The usages of the term persisters for bacteria in vitro and in vivo are distinct (6) but are commonly conflated by the assumption that whatever explains persistence in vitro explains it in vivo.
In vivo persistence is a major clinical problem (61, 111), but drug-sensitive bacteria may persist in a drug-treated host for reasons other than phenotypic AMR. For example, genetic heteroresistance can give rise to in vivo persistence (14). Moreover, some bacteria may reside in a site where the antibiotic in question does not reach or sustain bactericidal concentrations (88, 112). Nonetheless, it is plausible that phenotypic AMR often accounts for persistence of bacteria in the host. Many physiological conditions found in the host can bolster phenotypic AMR. These include a shift in carbon source (8, 113); deprivation of iron, oxygen, or preferred sources of carbon (79); and actions of the immune system that stress bacteria but fall short of killing them (9, 79). Such immune system actions include residence in phagocytes (114) and exposure to ROS, reactive nitrogen species, antimicrobial peptides, or intraphagosomal or extracellular acidity. Moreover, stress imposed by exposure to sublethal concentrations of an antibiotic can increase the proportion of a bacterial population displaying phenotypic AMR to other antibiotics. This could undermine a combination therapy if one drug fails to penetrate a particular pathological niche well enough to achieve concentrations above the MBC for the bacteria in that environment and instead induces phenotypic AMR to other drugs in the combination. In short, what might be expected to be synergy between host immunity and antibiotic therapy or between the use of one antibiotic and another may be antagonism, because one can induce phenotypic AMR to the other (115).
Whereas most acute infections caused by drug-sensitive bacteria are readily treated with a single antibiotic, rapid and curative treatments of lingering, life-threating infections that are refractory to treatment with a single agent can hopefully be advanced by drug regimens designed to overcome phenotypic AMR. The diversity of routes by which bacteria can become phenotypically resistant makes it challenging to propose such regimens.
In replicating bacterial populations, phenotypic AMR is typically exhibited by a small proportion of bacteria, and different individual bacteria are often phenotypically resistant to different antibiotics. Here, a combination of antibiotics may kill the entire population. As noted earlier, in bacterial populations whose replication is impaired by the stringencies of the host environment or sublethal impact of other antibiotics, phenotypic AMR is often expressed by most or all the bacteria and extends to multiple conventional antibiotics (7, 66–70). In this case, combining the drugs is unlikely to help (9). Instead, we might do well to identify compounds selected by an unconventional criterion, for example, their ability to kill nonreplicating bacteria (7, 86). On the basis of the role of ROS in amplifying antibiotic action, one set of targets for such drugs could be enzymes that mediate antioxidant defense, as was the case for the first antibacterial compound found that killed bacteria only when they were nonreplicating under diverse conditions (86). Additional approaches could be to augment bacterial ROS production (54), to inhibit bacterial “evolvability” (116) or to block the ability of bacterial populations to generate the phenotypic heterogeneity that underlies phenotypic AMR (117). This sets up the question of how we can more efficiently identify appropriate bacterial targets for antibiotics, progress antibiotics toward clinical utility, and predict which combinations of antibiotics will act synergistically rather than antagonistically.
NEW TOOLS FOR ANTIBIOTIC DISCOVERY
Target selection
The rising incidence of genetic AMR calls for multiple societal responses (4). Among them is the urgent need to discover new antibiotics, including antibiotics of new types. How can we do so, when antibiotic discovery efforts have been yielding diminishing returns (118)?
Until recently, the preferred route for antibiotic discovery has been to make new inhibitors of old targets—that is, those targets that are inhibited by clinically approved antibiotics—and hope that the new compounds are not subject to the same causes of genetic AMR as the old ones. This strategy can lead to the discovery of antimicrobials that circumvent genetic AMR (119, 120), but the approach markedly constricts the potential anti-infective drug space (121).
The second conventional criterion for target selection has been that the bacterial target must lack a human homolog, so as to minimize mechanism-based host toxicity. However, this wastes opportunity, because tweaks as small as altering the side chain of a single active site amino acid residue (122) or those of amino acids a few residues distal from the active site (123) may suffice to confer species-specific reactivity on an inhibitor of a bacterial enzyme, sparing its human counterpart.
Last, when new targets are sought, it has been customary to choose targets that are known to be essential to the pathogen under standard laboratory conditions. The focus on these conventionally essential targets (that is, those required for the bacterium’s replication in vitro in the medium that allows its fastest replication) can lead to disappointment when a gene product that is essential under standard laboratory conditions turns out not to be essential in an infected host (124), an example of conditional essentiality. Moreover, this criterion continues to waste opportunity by ignoring targets that are essential for the bacterium to cause disease in the host but are not essential for the bacterium to replicate in vitro.
Essentiality of a candidate target is usually judged from the phenotype of its genetic knockout. However, the conventional approach to generating gene knockouts only allows the candidacy of putative targets to be tested one at a time, whereas bacterial pathogens encode thousands of enzymes, channels, and other potential targets. This approach is blind to epistasis (the impact that mutations in other genes have on the phenotype resulting from mutation in the gene under study) and so teaches nothing about possible synergies or antagonisms. Moreover, the gene knockout strategy used to define essentiality fails to inform on the degree of target vulnerability, that is, whether bacterial death would ensue from a pharmacologically attainable degree of inhibition. Last, if the target is essential in vitro, a mutant lacking it cannot be grown to infect an experimental host to determine whether the target is also essential in vivo. New techniques, however, are changing the landscape. The following discussion on new techniques is centered on Mtb, but many of the approaches should be applicable to other life-threatening bacterial infections.
Artificially regulated promoters (125), artificially regulated posttranslational degradation systems (125), and inducible CRISPR interference platforms (126) are allowing the characterization of both the conditional essentiality of candidate bacterial targets and their vulnerability (127). Moreover, mutagenesis with bar-coded transposons followed by deep sequencing to establish baseline abundances of all mutants present, incubation under conditions of interest, and further deep sequencing to determine relative fitness of each mutant under those conditions (TnSeq) allows prediction of the essentiality of all genes of a pathogen simultaneously, with the exception of genes whose functions are required for growth of the bacteria during preparation of the transposon-mutagenized library (128, 129). The inducible CRISPR platform combines these methods (126). This allows for prediction of both essentiality and vulnerability of all genes in the pathogen simultaneously under any condition chosen for testing and also allows for multiplexing to investigate genetic interactions. In theory, these methods could be applied under pathophysiologically relevant conditions, such as within host cells or in a medium whose composition resembles that of a pathological site, such as a lung cavity in TB (130). Such methods hold promise for identifying targets whose inhibition in vivo is most likely to be lethal for the pathogen.
Although antibiotic discovery has focused on inhibiting targets, forced activation of certain pathways can also be bactericidal. For example, activation of the protease ClpP by an acyldepsipeptide killed S. aureus in biofilms both in vitro and in vivo (76). Similarly, the natural product ecumicin, which activates the adenosine triphosphatase activity of ClpC1 in Mtb, killed Mtb in vitro and in mice (131). A small molecule that activated the HssRS heme sensor system killed S. aureus under anaerobic conditions (132). Systematic identification of targets for bactericidal activation might be achieved by studying gene libraries in which each gene in a bacterial pathogen is placed under control of an inducible promoter (133).
Compound profiling
Until recently, hit-to-lead progression for antibiotic development involved identifying structure-activity relationships along three axes: potency against the target; activity against the pathogen; and a constellation of pharmacological properties, such as absorption, tissue distribution, metabolism, excretion, and toxicology. To optimize these parameters, medicinal chemists typically synthesize hundreds of variants of the inhibitor. However, the effort remains largely empirical without knowing why certain changes to the compound lead to changes in activity against the pathogen. Does a compound with a given modification no longer enter the bacterium or remain there? Does the bacterium transform it? Can the compound inhibit the target in its natural milieu?
Recent advances have now added two more axes to antibiotic structure-activity relationships to answer those questions: intrabacterial pharmacokinetics, which refers to what happens to the drug inside the bacterial cell (what the bacterium does to the drug), and intrabacterial pharmacodynamics, the biochemical and physiological effects of the drug and its metabolites on the bacterium (what the drug does to the bacterium) (52, 134, 135). Liquid chromatography–mass spectrometry (LC-MS) allows quantification of the intrabacterial concentration of the antimicrobial compound and of its conversion to other products, identification of which can guide structural modifications to block the compound’s catabolism. Such information can also be used to develop predictive algorithms to guide compound selection and design. For example, LC-MS–based assessment of the degree of accumulation of diverse compounds in E. coli allowed development of an algorithm that successfully predicted how to modify an antibiotic to extend its spectrum from Gram-positive to Gram-negative bacteria (136). LC-MS can also be applied to investigate intrabacterial pharmacodynamics through delineation of a compound’s impact on the metabolome and lipidome (52, 135, 137) along with comparison of these profiles to the impact of antibiotics with known actions (134).
A frequent challenge facing target-based screens for antimicrobial compounds is that a compound that inhibits the enzyme in question and kills the pathogen encoding the enzyme may kill the pathogen for other reasons. The use of regulated hypomorphs for the enzyme in question (bacterial strains in which expression of the putative target is partially and selectively suppressed) can provide powerful evidence for on-target whole-cell activity (52). Further, hypomorph libraries can be multiplexed to allow compounds to be profiled for activity against hundreds of targets simultaneously (138). This strategy can be leveraged to identify targets of compounds with unknown mechanisms of action and additional targets for compounds with a known target. Meanwhile, methods are improving to profile hundreds of millions of compounds for selective binding to a given target through the use of DNA-encoded libraries (139) and to use target structure to guide compound design (140).
Drug regimen design
Antibiotic candidates are developed one at a time without regard to others that are already approved or in development, especially as the latter are usually industrial secrets. However, antibiotics are often used in combination for difficult-to-treat infections like TB, for polymicrobial infections, and in critically ill patients when there is not time to identify the causative agent and its antibiotic resistance profile before initiating treatment. Until recently, determining which compounds will work together effectively has resembled working on a Rubik’s cube. New approaches offer hope for greater efficiency.
Prediction of antibiotic synergy or antagonism has advanced with new genetic, metabolomic, and computational tools. Genetic approaches include systematic identification of gene-gene interactions by TnSeq (128), inference of control points in critical pathways from the conjoint analysis of multiple RNA sequencing experiments (141), and prediction of drug-drug interactions via a computational algorithm based on transcriptomic profiles of drug-treated pathogens (142). Another study combined the pairwise impacts of 14 antibiotics on the growth of E. coli with fitness scores for E. coli gene deletion strains treated with each drug individually to train a machine-learning algorithm. The algorithm predicted synergistic or antagonistic interactions of additional antibiotics against E. coli and also against S. aureus and Mtb, suggesting that genetic determinants of these interactions were conserved (143). Comparison of the metabolomic impact of antibiotics on E. coli with the metabolomic impact of 3807 gene deletions also allowed prediction of synergistic drug combinations (40). Metabolomic profiling of Mtb treated with one antibiotic identified a nontargeted enzyme whose reaction was made more vulnerable to inhibition, enabling rational selection of a second drug to inhibit the second target for a synergistic effect (144). Computational methods that enable evaluation of the complex effects of higher-order drug combinations also hold promise (145, 146).
Hollow-fiber flow systems test the impact of compound combinations in vitro while simulating each compound’s pharmacokinetic profile (147). Laser capture microdissection coupled with mass spectrometry enables intralesional pharmacokinetics and its sometimes striking distinction from blood pharmacokinetics, revealing which compounds are present in a given lesion at a given time and at what concentrations (112, 148).
Last, efforts known as quantitative systems pharmacology are under way to systematically integrate diverse sets of pharmacokinetic and pharmacodynamic information, such as can be obtained by the approaches described above, into algorithms for predicting drug interactions, safety margins, and dosing regimens (149).
Knowledge and tools still lacking
A great deal of basic and translational research remains to be done before we can discover new antibiotics efficiently. Given that TB was the primary example used here to illustrate technological advances, it is fitting that TB illustrates persistent shortcomings in our knowledge and tools.
We do not yet understand the function of nearly half of the genes in Mtb. We do not know what physiological conditions in vitro or in animal models would reveal the phenotype of knockouts or knockdowns of these genes of unknown function. Further, a few inbred mouse models presently serve as gateways through which an antibiotic must pass on its way to clinical candidacy for the treatment of TB. However, knockout of Mtb genes is now known to attenuate the pathogen in some mouse strains, but not in others constructed to mimic the genetic diversity of outbred mouse populations (150, 151). Such an outcome could invalidate the choice of a target long after extensive efforts have been invested in developing a drug that inhibits it. Perhaps a more reliable model to test TB drug candidates will be mice transplanted with the microbiota of wild mice. Responses of such mice to immune-targeting drugs better mimicked responses seen in clinical trials than did responses in mice with the less diverse microbiota associated with conventional husbandry (152).
We do not know how to prevent phenotypic AMR to TB drugs or how to eliminate Mtb persistence in vivo. We have no widely reproducible in vivo assay to identify compounds that selectively kill nonreplicating Mtb. We do not know how to target processes involved in Mtb transmission. We have no measure of the total body burden of Mtb in people. No clinically deployable test is available for enumerating differentially detectable Mtb. We lack reliable biomarkers with which to make an accurate diagnosis of TB or individualize its therapy, including determining when treatment can be stopped.
More generally, we have much to learn about the features of antibiotics that favor their accumulation in different pathogens, how they alter bacterial metabolism, and how bacteria catabolize them. Most fundamentally, we need a deeper understanding of how antibiotic-treated bacteria survive or succumb.
Conclusions
The recent explosion of knowledge about AMR reviewed here is cause for celebration. However, this new knowledge reveals that we have much farther to go than believed before we can undermine bacterial resilience as adeptly as bacteria “understand” human vulnerability, in the sense of their evolved success in exploiting humans as hosts and vectors. Our potential advantage in this battle is that bacterial pathogens must survive not only our evolved and individual immune responses but also our collective efforts to compensate for our fundamental immunodeficiency (6) by administering antibiotics. Our scientific understanding of genetic AMR is robust. In contrast, our understanding of phenotypic AMR is nascent. This is a key area for further research, but research will not be enough. Society needs to take action to use insights about AMR to combat the ever-present threat of drug-resistant bacterial pathogens.
Acknowledgments:
We thank K. Rhee, K. Saito, B. Gold, and K. Burns-Huang for insightful comments.
Funding:
The authors are supported by the NIH TB Research Unit grant number U19 AI11143 and the Milstein Program in Chemical Biology and Translational Medicine. S.M.S. is supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences under NIH award number T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program, by an F30 Predoctoral Fellowship from the National Institute of Allergy and Infectious Diseases under NIH award number F30AI140623, and by a National Defense Science and Engineering Graduate Fellowship from the U.S. Department of Defense. J.V. is supported by a grant from the Potts Memorial Foundation and by the Department of Infectious Disease, Imperial College London. The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing interests: C.N. is on the board of directors of the Tri-Institutional Therapeutics Discovery Institute and the Tres Cantos Open Lab Foundation and is on the scientific advisory boards of the Global Alliance for TB Drug Development, the Cancer Research Institute, the Rita Allen Foundation, Bridge Medicines, Leap Therapeutics, Triterpenoid Therapeutics, and the Pfizer Centers for Therapeutic Innovation. The other authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Theuretzbacher U, Gottwalt S, Beyer P, Butler M, Czaplewski L, Lienhardt C, Moja L, Paul M, Paulin S, Rex JH, Silver LL, Spigelman M, Thwaites GE, Paccaud J-P, Harbarth S, Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis 19, e40–e50 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Roope LSJ, Smith RD, Pouwels KB, Buchanan J, Abel L, Eibich P, Butler CC, Tan PS, Walker AS, Robotham JV, Wordsworth S, The challenge of antimicrobial resistance: What economics can contribute. Science 364, eaau4679 (2019). [DOI] [PubMed] [Google Scholar]
- 3.O’Neill J, Tackling drug-resistant infections globally: Final report and recommendations, in The Review on Antimicrobial Resistance (2016), pp. 1–81. [Google Scholar]
- 4.Nathan C, Cars O, Antibiotic resistance—Problems, progress, and prospects. N. Engl. J. Med 371, 1761–1763 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Nathan C, Kunkel Lecture: Fundamental immunodeficiency and its correction. J. Exp. Med 214, 2175–2191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balaban NQ, Helaine S, Lewis K, Ackermann M, Aldridge B, Andersson DI, Brynildsen MP, Bumann D, Camilli A, Collins JJ, Dehio C, Fortune S, Ghigo J-M, Hardt W-D, Harms A, Heinemann M, Hung DT, Jenal U, Levin BR, Michiels J, Storz G, Tan M-W, Tenson T, Van Melderen L, Zinkernagel A, Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol 17, 441–448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gold B, Nathan C, Targeting phenotypically tolerant Mycobacterium tuberculosis. Microbiol. Spectr 5, 10.1128/microbiolspec.TBTB2-0031-2016, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kotte O, Volkmer B, Radzikowski JL, Heinemann M, Phenotypic bistability in Escherichia coli’s central carbon metabolism. Mol. Syst. Biol 10, 736 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nathan C, Fresh approaches to anti-infective therapies. Sci. Transl. Med 4, 140sr2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S, Bacterial persistence as a phenotypic switch. Science 305, 1622–1625 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Brauner A, Fridman O, Gefen O, Balaban NQ, Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol 14, 320–330 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Bellerose MM, Baek S-H, Huang C-C, Moss CE, Koh E-I, Proulx MK, Smith CM, Baker RE, Lee JS, Eum S, Shin SJ, Cho S-N, Murray M, Sassetti CM, Common variants in the glycerol kinase gene reduce tuberculosis drug efficacy. MBio 10, e00663–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Safi H, Gopal P, Lingaraju S, Ma S, Levine C, Dartois V, Yee M, Li L, Blanc L, Ho Liang H-P, Husain S, Hoque M, Soteropoulos P, Rustad TR, Sherman DR, Dick T, Alland D, Phase variation in Mycobacterium tuberculosis glpK produces transiently heritable drug tolerance. Proc. Natl. Acad. Sci. U.S.A 116, 19665–19674 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersson DI, Nicoloff H, Hjort K, Mechanisms and clinical relevance of bacterial heteroresistance. Nat. Rev. Microbiol 17, 479–496 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Band VI, Hufnagel DA, Jaggavarapu S, Sherman EX, Wozniak JE, Satola SW, Farley MM, Jacob JT, Burd EM, Weiss DS, Antibiotic combinations that exploit heteroresistance to multiple drugs effectively control infection. Nat. Microbiol 4, 1627–1635 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dewachter L, Fauvart M, Michiels J, Bacterial heterogeneity and antibiotic survival: Understanding and combatting persistence and heteroresistance. Mol. Cell 76, 255–267 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Sakatos A, Babunovic GH, Chase MR, Dills A, Leszyk J, Rosebrock T, Bryson B, Fortune SM, Posttranslational modification of a histone-like protein regulates phenotypic resistance to isoniazid in mycobacteria. Sci. Adv 4, eaao1478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ, Antibiotic tolerance facilitates the evolution of resistance. Science 355, 826–830 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Davies D, Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov 2, 114–122 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Crabbé A, Jensen PØ, Bjarnsholt T, Coenye T, Antimicrobial tolerance and metabolic adaptations in microbial biofilms. Trends Microbiol 27, 850–863 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Amini S, Hottes AK, Smith LE, Tavazoie S, Fitness landscape of antibiotic tolerance in Pseudomonas aeruginosa biofilms. PLOS Pathog 7, e1002298 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colangeli R, Jedrey H, Kim S, Connell R, Ma S, Chippada Venkata UD, Chakravorty S, Gupta A, Sizemore EE, Diem L, Sherman DR, Okwera A, Dietze R, Boom WH, Johnson JL, Mac Kenzie WR, Alland D; DMID 01–009/Tuberculosis Trials Consortium Study 22 Teams, Bacterial factors that predict relapse after tuberculosis therapy. N. Engl. J. Med 379, 823–833 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dhar N, McKinney JD, Mycobacterium tuberculosis persistence mutants identified by screening in isoniazid-treated mice. Proc. Natl. Acad. Sci. U.S.A 107, 12275–12280 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, Britigan BE, Singh PK, Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334, 982–986 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mok WWK, Brynildsen MP, Timing of DNA damage responses impacts persistence to fluoroquinolones. Proc. Natl. Acad. Sci. U.S.A 115, E6301–E6309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saito K, Warrier T, Somersan-Karakaya S, Kaminski L, Mi J, Jiang X, Park S, Shigyo K, Gold B, Roberts J, Weber E, Jacobs WR Jr., C. F. Nathan, Rifamycin action on RNA polymerase in antibiotic-tolerant Mycobacterium tuberculosis results in differentially detectable populations. Proc. Natl. Acad. Sci. U.S.A 114, E4832–E4840 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu H-S, Roberts N, Singleton FL, Attwell RW, Grimes DJ, Colwell RR, Survival and viability of nonculturable Escherichia coli and Vibrio cholerae in the estuarine and marine environment. Microb. Ecol 8, 313–323 (1982). [DOI] [PubMed] [Google Scholar]
- 28.Li L, Mendis N, Trigui H, Oliver JD, Faucher SP, The importance of the viable but non-culturable state in human bacterial pathogens. Front. Microbiol 5, 258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chengalroyen MD, Beukes GM, Gordhan BG, Streicher EM, Churchyard G, Hafner R, Warren R, Otwombe K, Martinson N, Kana BD, Detection and quantification of differentially culturable tubercle bacteria in sputum from patients with tuberculosis. Am. J. Respir. Crit. Care Med 194, 1532–1540 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McAulay K, Saito K, Warrier T, Walsh KF, Mathurin LD, Royal-Mardi G, Lee MH, Ocheretina O, Pape JW, Fitzgerald DW, Nathan CF, Differentially detectable Mycobacterium tuberculosis cells in sputum from treatment-naive subjects in Haiti and their proportionate increase after initiation of treatment. MBio 9, e02192–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan SR, Venugopal U, Chandra G, Bharti S, Maurya RK, Krishnan MY, Effect of various drugs on differentially detectable persisters of Mycobacterium tuberculosis generated by long-term lipid diet. Tuberculosis 115, 89–95 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Hu Y, Pertinez H, Liu Y, Davies G, Coates A, Bedaquiline kills persistent Mycobacterium tuberculosis with no disease relapse: An in vivo model of a potential cure. J. Antimicrob. Chemother 74, 1627–1633 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Scanga CA, Mohan VP, Joseph H, Yu K, Chan J, Flynn JL, Reactivation of latent tuberculosis: Variations on the Cornell murine model. Infect. Immun 67, 4531–4538 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukamolova GV, Turapov O, Malkin J, Woltmann G, Barer MR, Resuscitation-promoting factors reveal an occult population of tubercle bacilli in sputum. Am. J. Respir. Crit. Care Med 181, 174–180 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosser A, Pareek M, Turapov O, Wiselka MJ, Mukamolova GV, Differentially culturable tubercule bacilli are generated during nonpulmonary tuberculosis infection. Am. J. Respir. Crit. Care Med 197, 818–821 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasouly A, Nudler E, Reactive oxygen species as the long arm of bactericidal antibiotics. Proc. Natl. Acad. Sci. U.S.A 116, 9696–9698 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stokes JM, Lopatkin AJ, Lobritz MA, Collins JJ, Bacterial metabolism and antibiotic efficacy. Cell Metab 30, 251–259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K, Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339, 1213–1216 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Imlay JA, Cell death from antibiotics without the involvement of reactive oxygen species. Science 339, 1210–1213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campos AI, Zampieri M, Metabolomics-driven exploration of the chemical drug space to predict combination antimicrobial therapies. Mol. Cell 74, 1291–1303. e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CTY, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ, Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. U.S.A 111, E2100–E2109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC, Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336, 315–319 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong Y, Zeng J, Wang X, Drlica K, Zhao X, Post-stress bacterial cell death mediated by reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A 116, 10064–10071 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ, A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Nandakumar M, Nathan C, Rhee KY, Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat. Commun 5, 4306 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Yang JH, Wright SN, Hamblin M, McCloskey D, Alcantar MA, Schrübbers L, Lopatkin AJ, Satish S, Nili A, Palsson BO, Walker GC, Collins JJ, A white-box machine learning approach for revealing antibiotic mechanisms of action. Cell 177, 1649–1661. e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaubourgeix J, Lin G, Dhar N, Chenouard N, Jiang X, Botella H, Lupoli T, Mariani O, Yang G, Ouerfelli O, Unser M, Schnappinger D, McKinney J, Nathan C, Stressed mycobacteria use the chaperone ClpB to sequester irreversibly oxidized proteins asymmetrically within and between cells. Cell Host Microbe 17, 178–190 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clatworthy AE, Pierson E, Hung DT, Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol 3, 541–548 (2007). [DOI] [PubMed] [Google Scholar]
- 49.O’Dwyer K, Spivak AT, Ingraham K, Min S, Holmes DJ, Jakielaszek C, Rittenhouse S, Kwan AL, Livi GP, Sathe G, Thomas E, Van Horn S, Miller LA, Twynholm M, Tomayko J, Dalessandro M, Caltabiano M, Scangarella-Oman NE, Brown JR, Bacterial resistance to leucyl-tRNA synthetase inhibitor GSK2251052 develops during treatment of complicated urinary tract infections. Antimicrob. Agents Chemother 59, 289–298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Boeckel TP, Brower C, Gilbert M, Grenfell BT, Levin SA, Robinson TP, Teillant A, Laxminarayan R, Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci. U.S.A 112, 5649–5654 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang H, Wang B, Zhao Q, Zhao Y, Fu C, Feng X, Wang N, Su M, Tang C, Jiang F, Zhou Y, Chen Y, Jiang Q, Antibiotic body burden of Chinese school children: A multisite biomonitoring-based study. Environ. Sci. Technol 49, 5070–5079 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Ballinger E, Mosior J, Hartman T, Burns-Huang K, Gold B, Morris R, Goullieux L, Blanc I, Vaubourgeix J, Lagrange S, Fraisse L, Sans S, Couturier C, Bacqué E, Rhee K, Scarry SM, Aubé J, Yang G, Ouerfelli O, Schnappinger D, Ioerger TR, Engelhart CA, McConnell JA, McAulay K, Fay A, Roubert C, Sacchettini J, Nathan C, Opposing reactions in coenzyme A metabolism sensitize Mycobacterium tuberculosis to enzyme inhibition. Science 363, eaau8959 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martínez JL, Rojo F, Metabolic regulation of antibiotic resistance. FEMS Microbiol. Rev 35, 768–789 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Brynildsen MP, Winkler JA, Spina CS, MacDonald IC, Collins JJ, Potentiating antibacterial activity by predictably enhancing endogenous microbial ROS production. Nat. Biotechnol 31, 160–165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ayer A, Fellermeier S, Fife C, Li SS, Smits G, Meyer AJ, Dawes IW, Perrone GG, A genome-wide screen in yeast identifies specific oxidative stress genes required for the maintenance of sub-cellular redox homeostasis. PLOS ONE 7, e44278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Z, Zeng X, Tsui SK-W, Investigating function roles of hypothetical proteins encoded by the Mycobacterium tuberculosis H37Rv genome. BMC Genomics 20, 394 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olin-Sandoval V, Yu JSL, Miller-Fleming L, Alam MT, Kamrad S, Correia-Melo C, Haas R, Segal J, Peña Navarro DA, Herrera-Dominguez L, Méndez-Lucio O, Vowinckel J, Mülleder M, Ralser M, Lysine harvesting is an antioxidant strategy and triggers underground polyamine metabolism. Nature 572, 249–253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sebastian J, Swaminath S, Nair RR, Jakkala K, Pradhan A, Ajitkumar P, De novo emergence of genetically resistant mutants of Mycobacterium tuberculosis from the persistence phase cells formed against antituberculosis drugs in vitro. Antimicrob. Agents Chemother 61, e01343–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swaminath S, Paul A, Pradhan A, Sebastian J, Nair RR, Ajitkumar P, Mycobacterium smegmatis moxifloxacin persister cells produce high levels of hydroxyl radical, generaitng genetic resisters selectable not only with moxifloxacin, but also with ethambutol and isoniazid. Microbiology 166, 180–198 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Pribis JP, García-Villada L, Zhai Y, Lewin-Epstein O, Wang AZ, Liu J, Xia J, Mei Q, Fitzgerald DM, Bos J, Austin RH, Herman C, Bates D, Hadany L, Hastings PJ, Rosenberg SM, Gamblers: An antibiotic-induced evolvable cell subpopulation differentiated by reactive-oxygen-induced general stress response. Mol. Cell 74, 785–800. e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fisher RA, Gollan B, Helaine S, Persistent bacterial infections and persister cells. Nat. Rev. Microbiol 15, 453–464 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Cohen NR, Lobritz MA, Collins JJ, Microbial persistence and the road to drug resistance. Cell Host Microbe 13, 632–642 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.El Meouche I, Dunlop MJ, Heterogeneity in efflux pump expression predisposes antibiotic-resistant cells to mutation. Science 362, 686–690 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fridman O, Goldberg A, Ronin I, Shoresh N, Balaban NQ, Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 513, 418–421 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Girgis HS, Harris K, Tavazoie S, Large mutational target size for rapid emergence of bacterial persistence. Proc. Natl. Acad. Sci. U.S.A 109, 12740–12745 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K, Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol 43, 717–731 (2002). [DOI] [PubMed] [Google Scholar]
- 67.Bigger JW, Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244, 497–500 (1944). [Google Scholar]
- 68.Hobby GL, Meyer K, Chaffee E, Observations on the mechanism of action of penicillin. Proc. Soc. Exp. Biol. Med 50, 281–285 (1942). [Google Scholar]
- 69.Sala C, Dhar N, Hartkoorn RC, Zhang M, Ha YH, Schneider P, Cole ST, Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother 54, 4150–4158 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie Z, Siddiqi N, Rubin EJ, Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother 49, 4778–4780 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bakkeren E, Huisman JS, Fattinger SA, Hausmann A, Furter M, Egli A, Slack E, Sellin ME, Bonhoeffer S, Regoes RR, Diard M, Hardt W-D, Salmonella persisters promote the spread of antibiotic resistance plasmids in the gut. Nature 573, 276–280 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tuomanen E, Phenotypic tolerance: The search for beta-lactam antibiotics that kill nongrowing bacteria. Rev. Infect. Dis 8 (suppl. 3), S279–S291 (1986). [DOI] [PubMed] [Google Scholar]
- 73.Chain E, Duthie ES, Bactericidal and bacteriolytic action of penicillin on the staphylococcus. Lancet 245, 652–657 (1945). [Google Scholar]
- 74.Tuomanen E, Cozens R, Tosch W, Zak O, Tomasz A, The rate of killing of Escherichia coli by beta-lactam antibiotics is strictly proportional to the rate of bacterial growth. J. Gen. Microbiol 132, 1297–1304 (1986). [DOI] [PubMed] [Google Scholar]
- 75.Pontes MH, Groisman EA, Slow growth determines nonheritable antibiotic resistance in Salmonella enterica. Sci. Signal 12, eaax3938 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conlon BP, Rowe SE, Gandt AB, Nuxoll AS, Donegan NP, Zalis EA, Clair G, Adkins JN, Cheung AL, Lewis K, Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol 1, 16051 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Stewart B, Rozen DE, Genetic variation for antibiotic persistence in Escherichia coli. Evolution 66, 933–939 (2012). [DOI] [PubMed] [Google Scholar]
- 78.Harms A, Maisonneuve E, Gerdes K, Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 354, aaf4268 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Rittershaus ESC, Baek S-H, Sassetti CM, The normalcy of dormancy: Common themes in microbial quiescence. Cell Host Microbe 13, 643–651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pu Y, Zhao Z, Li Y, Zou J, Ma Q, Zhao Y, Ke Y, Zhu Y, Chen H, Baker MAB, Ge H, Sun Y, Xie XS, Bai F, Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol. Cell 62, 284–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Keren I, Minami S, Rubin E, Lewis K, Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. MBio 2, e00100–11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Radzikowski JL, Vedelaar S, Siegel D, Ortega ÁD, Schmidt A, Heinemann M, Bacterial persistence is an active σS stress response to metabolic flux limitation. Mol. Syst. Biol 12, 882 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eoh H, Rhee KY, Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A 110, 6554–6559 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eoh H, Wang Z, Layre E, Rath P, Morris R, Moody DB, Rhee KY, Metabolic anticipation in Mycobacterium tuberculosis. Nat. Microbiol 2, 17084 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.de Carvalho LPS, Darby CM, Rhee KY, Nathan C, Nitazoxanide disrupts membrane potential and intrabacterial pH homeostasis of Mycobacterium tuberculosis. ACS Med. Chem. Lett 2, 849–854 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bryk R, Gold B, Venugopal A, Singh J, Samy R, Pupek K, Cao H, Popescu C, Gurney M, Hotha S, Cherian J, Rhee K, Ly L, Converse PJ, Ehrt S, Vandal O, Jiang X, Schneider J, Lin G, Nathan C, Selective killing of nonreplicating mycobacteria. Cell Host Microbe 3, 137–145 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gefen O, Fridman O, Ronin I, Balaban NQ, Direct observation of single stationary-phase bacteria reveals a surprisingly long period of constant protein production activity. Proc. Natl. Acad. Sci. U.S.A 111, 556–561 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sarathy JP, Via LE, Weiner D, Blanc L, Boshoff H, Eugenin EA, Barry CE III, Dartois VA, Extreme drug tolerance of Mycobacterium tuberculosis in caseum. Antimicrob. Agents Chemother 62, e02266–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Orman MA, Brynildsen MP, Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob. Agents Chemother 57, 3230–3239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goormaghtigh F, Fraikin N, Putrinš M, Hallaert T, Hauryliuk V, Garcia-Pino A, Sjödin A, Kasvandik S, Udekwu K, Tenson T, Kaldalu N, Van Melderen L, Reassessing the role of Type II toxin-antitoxin systems in formation of Escherichia coli Type II persister cells. MBio 9, e00640–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Manina G, Dhar N, McKinney JD, Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17, 32–46 (2015). [DOI] [PubMed] [Google Scholar]
- 92.Wakamoto Y, Dhar N, Chait R, Schneider K, Signorino-Gelo F, Leibler S, McKinney JD, Dynamic persistence of antibiotic-stressed mycobacteria. Science 339, 91–95 (2013). [DOI] [PubMed] [Google Scholar]
- 93.Javid B, Sorrentino F, Toosky M, Zheng W, Pinkham JT, Jain N, Pan M, Deighan P, Rubin EJ, Mycobacterial mistranslation is necessary and sufficient for rifampicin phenotypic resistance. Proc. Natl. Acad. Sci. U.S.A 111, 1132–1137 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Su H-W, Zhu J-H, Li H, Cai R-J, Ealand C, Wang X, Chen Y-X, Kayani MUR, Zhu TF, Moradigaravand D, Huang H, Kana BD, Javid B, The essential mycobacterial amidotransferase GatCAB is a modulator of specific translational fidelity. Nat. Microbiol 1, 16147 (2016). [DOI] [PubMed] [Google Scholar]
- 95.Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L, Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145, 39–53 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mishra R, Kohli S, Malhotra N, Bandyopadhyay P, Mehta M, Munshi M, Adiga V, Ahuja VK, Shandil RK, Rajmani RS, Seshasayee ASN, Singh A, Targeting redox heterogeneity to counteract drug tolerance in replicating Mycobacterium tuberculosis. Sci. Transl. Med 11, eaaw6635 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gusarov I, Shatalin K, Starodubtseva M, Nudler E, Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 325, 1380–1384 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shatalin K, Shatalina E, Mironov A, Nudler E, H2S: A universal defense against antibiotics in bacteria. Science 334, 986–990 (2011). [DOI] [PubMed] [Google Scholar]
- 99.Lopatkin AJ, Stokes JM, Zheng EJ, Yang JH, Takahashi MK, You L, Collins JJ, Bacterial metabolic state more accurately predicts antibiotic lethality than growth rate. Nat. Microbiol 4, 2109–2117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moyed HS, Bertrand KP, hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol 155, 768–775 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Korch SB, Henderson TA, Hill TM, Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol. Microbiol 50, 1199–1213 (2003). [DOI] [PubMed] [Google Scholar]
- 102.Schumacher MA, Piro KM, Xu W, Hansen S, Lewis K, Brennan RG, Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323, 396–401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Semanjski M, Germain E, Bratl K, Kiessling A, Gerdes K, Macek B, The kinases HipA and HipA7 phosphorylate different substrate pools in Escherichia coli to promote multidrug tolerance. Sci. Signal 11, eaat5750 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Kaspy I, Rotem E, Weiss N, Ronin I, Balaban NQ, Glaser G, HipA-mediated antibiotic persistence via phosphorylation of the glutamyl-tRNA-synthetase. Nat. Commun 4, 3001 (2013). [DOI] [PubMed] [Google Scholar]
- 105.Ronneau S, Helaine S, Clarifying the link between toxin–antitoxin modules and bacterial persistence. J. Mol. Biol 431, 3462–3471 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Hicks ND, Yang J, Zhang X, Zhao B, Grad YH, Liu L, Ou X, Chang Z, Xia H, Zhou Y, Wang S, Dong J, Sun L, Zhu Y, Zhao Y, Jin Q, Fortune SM, Clinically prevalent mutations in Mycobacterium tuberculosis alter propionate metabolism and mediate multidrug tolerance. Nat. Microbiol 3, 1032–1042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fauvart M, De Groote VN, Michiels J, Role of persister cells in chronic infections: Clinical relevance and perspectives on anti-persister therapies. J. Med. Microbiol 60, 699–709 (2011). [DOI] [PubMed] [Google Scholar]
- 108.Wu N, He L, Cui P, Wang W, Yuan Y, Liu S, Xu T, Zhang S, Wu J, Zhang W, Zhang Y, Ranking of persister genes in the same Escherichia coli genetic background demonstrates varying importance of individual persister genes in tolerance to different antibiotics. Front. Microbiol 6, 1003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pu Y, Li Y, Jin X, Tian T, Ma Q, Zhao Z, Lin S.-y., Chen Z, Li B, Yao G, Leake MC, Lo C-J, Bai F, ATP-dependent dynamic protein aggregation regulates bacterial dormancy depth critical for antibiotic tolerance. Mol. Cell 73, 143–156. e4 (2019). [DOI] [PubMed] [Google Scholar]
- 110.Vilchèze C, Jacobs WR Jr., The isoniazid paradigm of killing, resistance, and persistence in Mycobacterium tuberculosis. J. Mol. Biol 431, 3450–3461 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Van den Bergh B, Fauvart M, Michiels J, Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol. Rev 41, 219–251 (2017). [DOI] [PubMed] [Google Scholar]
- 112.Strydom N, Gupta SV, Fox WS, Via LE, Bang H, Lee M, Eum S, Shim T, Barry CE III, Zimmerman M, Dartois V, Savic RM, Tuberculosis drugs’ distribution and emergence of resistance in patient’s lung lesions: A mechanistic model and tool for regimen and dose optimization. PLOS Med 16, e1002773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Amato SM, Brynildsen MP, Nutrient transitions are a source of persisters in Escherichia coli biofilms. PLOS ONE 9, e93110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stapels DAC, Hill PWS, Westermann AJ, Fisher RA, Thurston TL, Saliba A-E, Blommestein I, Vogel J, Helaine S, Salmonella persisters undermine host immune defenses during antibiotic treatment. Science 362, 1156–1160 (2018). [DOI] [PubMed] [Google Scholar]
- 115.Liu Y, Tan S, Huang L, Abramovitch RB, Rohde KH, Zimmerman MD, Chen C, Dartois V, VanderVen BC, Russell DG, Immune activation of the host cell induces drug tolerance in Mycobacterium tuberculosis both in vitro and in vivo. J. Exp. Med 213, 809–825 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ragheb MN, Thomason MK, Hsu C, Nugent P, Gage J, Samadpour AN, Kariisa A, Merrikh CN, Miller SI, Sherman DR, Merrikh H, Inhibiting the evolution of antibiotic resistance. Mol. Cell 73, 157–165. e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rego EH, Audette RE, Rubin EJ, Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546, 153–157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL, Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov 6, 29–40 (2007). [DOI] [PubMed] [Google Scholar]
- 119.McNeil MB, Dennison D, Shelton C, Flint L, Korkegian A, Parish T, Mechanisms of resistance against NITD-916, a direct inhibitor of Mycobacterium tuberculosis InhA. Tuberculosis 107, 133–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maffioli SI, Zhang Y, Degen D, Carzaniga T, Del Gatto G, Serina S, Monciardini P, Mazzetti C, Guglierame P, Candiani G, Chiriac AI, Facchetti G, Kaltofen P, Sahl HG, Dehò G, Donadio S, Ebright RH, Antibacterial nucleoside-analog inhibitor of bacterial RNA polymerase. Cell 169, 1240–1248. e23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nathan C, Making space for anti-infective drug discovery. Cell Host Microbe 9, 343–348 (2011). [DOI] [PubMed] [Google Scholar]
- 122.Bryk R, Arango N, Maksymiuk C, Balakrishnan A, Wu Y-T, Wong C-H, Masquelin T, Hipskind P, Lima CD, Nathan C, Lipoamide channel-binding sulfonamides selectively inhibit mycobacterial lipoamide dehydrogenase. Biochemistry 52, 9375–9384 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lin G, Li D, de Carvalho LPS, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C, Inhibitors selective for mycobacterial versus human proteasomes. Nature 461, 621–626 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pethe K, Sequeira PC, Agarwalla S, Rhee K, Kuhen K, Phong WY, Patel V, Beer D, Walker JR, Duraiswamy J, Jiricek J, Keller TH, Chatterjee A, Tan MP, Ujjini M, Rao SP, Camacho L, Bifani P, Mak PA, Ma I, Barnes SW, Chen Z, Plouffe D, Thayalan P, Ng SH, Au M, Lee BH, Tan BH, Ravindran S, Nanjundappa M, Lin X, Goh A, Lakshminarayana SB, Shoen C, Cynamon M, Kreiswirth B, Dartois V, Peters EC, Glynne R, Brenner S, Dick T, A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat. Commun 1, 57 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schnappinger D, Ehrt S, Regulated expression systems for mycobacteria and their applications. Microbiol. Spectr 2, MGM2-0018-2013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rock JM, Hopkins FF, Chavez A, Diallo M, Chase MR, Gerrick ER, Pritchard JR, Church GM, Rubin EJ, Sassetti CM, Schnappinger D, Fortune SM, Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat. Microbiol 2, 16274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kim J-H, O’Brien KM, Sharma R, Boshoff HIM, Rehren G, Chakraborty S, Wallach JB, Monteleone M, Wilson DJ, Aldrich CC, Barry CE III, Rhee KY, Ehrt S, Schnappinger D, A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc. Natl. Acad. Sci. U.S.A 110, 19095–19100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, Rubin EJ, Schnappinger D, Ehrt S, Fortune SM, Sassetti CM, Ioerger TR, Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. MBio 8, e02133–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xu W, DeJesus MA, Rücker N, Engelhart CA, Wright MG, Healy C, Lin K, Wang R, Park SW, Ioerger TR, Schnappinger D, Ehrt S, Chemical genetic interaction profiling reveals determinants of intrinsic antibiotic resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother 61, e01334–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sarathy JP, Liang H-PH, Weiner D, Gonzales J, Via LE, Dartois V, An in vitro caseum binding assay that predicts drug penetration in tuberculosis lesions. J. Vis. Exp, 55559 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gao W, Kim J-Y, Anderson JR, Akopian T, Hong S, Jin Y-Y, Kandror O, Kim J-W, Lee I-A, Lee S-Y, McAlpine JB, Mulugeta S, Sunoqrot S, Wang Y, Yang S-H, Yoon T-M, Goldberg AL, Pauli GF, Suh J-W, Franzblau SG, Cho S, The cyclic peptide ecumicin targeting ClpC1 is active against Mycobacterium tuberculosis in vivo. Antimicrob. Agents Chemother 59, 880–889 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mike LA, Dutter BF, Stauff DL, Moore JL, Vitko NP, Aranmolate O, Kehl-Fie TE, Sullivan S, Reid PR, DuBois JL, Richardson AR, Caprioli RM, Sulikowski GA, Skaar EP, Activation of heme biosynthesis by a small molecule that is toxic to fermenting Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A 110, 8206–8211 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Melief E, Kokoczka R, Files M, Bailey MA, Alling T, Li H, Ahn J, Misquith A, Korkegian A, Roberts D, Sacchettini J, Parish T, Construction of an overexpression library for Mycobacterium tuberculosis. Biol. Methods Protoc 3, bpy009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jansen RS, Rhee KY, Emerging approaches to tuberculosis drug development: At home in the metabolome. Trends Pharmacol. Sci 38, 393–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chakraborty S, Gruber T, Barry III CE, Boshoff HI, Rhee KY, Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339, 88–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Parker EN, Drown BS, Geddes EJ, Lee HY, Ismail N, Lau GW, Hergenrother PJ, Implementation of permeation rules leads to a FabI inhibitor with activity against Gram-negative pathogens. Nat. Microbiol 5, 65–75 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Venugopal A, Bryk R, Shi S, Rhee K, Rath P, Schnappinger D, Ehrt S, Nathan C, Virulence of Mycobacterium tuberculosis depends on lipoamide dehydrogenase, a member of three multienzyme complexes. Cell Host Microbe 9, 21–31 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Johnson EO, LaVerriere E, Office E, Stanley M, Meyer E, Kawate T, Gomez JE, Audette RE, Bandyopadhyay N, Betancourt N, Delano K, Da Silva I, Davis J, Gallo C, Gardner M, Golas AJ, Guinn KM, Kennedy S, Korn R, McConnell JA, Moss CE, Murphy KC, Nietupski RM, Papavinasasundaram KG, Pinkham JT, Pino PA, Proulx MK, Ruecker N, Song N, Thompson M, Trujillo C, Wakabayashi S, Wallach JB, Watson C, Ioerger TR, Lander ES, Hubbard BK, Serrano-Wu MH, Ehrt S, Fitzgerald M, Rubin EJ, Sassetti CM, Schnappinger D, Hung DT, Large-scale chemical-genetics yields new M. tuberculosis inhibitor classes. Nature 571, 72–78 (2019). [DOI] [PubMed] [Google Scholar]
- 139.Kunig V, Potowski M, Gohla A, Brunschweiger A, DNA-encoded libraries - an efficient small molecule discovery technology for the biomedical sciences. Biol. Chem 399, 691–710 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Waman VP, Vedithi SC, Thomas SE, Bannerman BP, Munir A, Skwark MJ, Malhotra S, Blundell TL, Mycobacterial genomics and structural bioinformatics: Opportunities and challenges in drug discovery. Emerg. Microbes Infect 8, 109–118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Peterson EJR, Ma S, Sherman DR, Baliga NS, Network analysis identifies Rv0324 and Rv0880 as regulators of bedaquiline tolerance in Mycobacterium tuberculosis. Nat. Microbiol 1, 16078 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ma S, Jaipalli S, Larkins-Ford J, Lohmiller J, Aldridge BB, Sherman DR, Chandrasekaran S, Transcriptomic signatures predict regulators of drug synergy and clinical regimen efficacy against tuberculosis. MBio 10, e02627–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chandrasekaran S, Cokol-Cakmak M, Sahin N, Yilancioglu K, Kazan H, Collins JJ, Cokol M, Chemogenomics and orthology-based design of antibiotic combination therapies. Mol. Syst. Biol 12, 872 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang Z, Soni V, Marriner G, Kaneko T, Boshoff HIM, Barry III CE, Rhee KY, Mode-of-action profiling reveals glutamine synthetase as a collateral metabolic vulnerability of M. tuberculosis to bedaquiline. Proc. Natl. Acad. Sci. U.S.A 116, 19646–19651 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cokol M, Kuru N, Bicak E, Larkins-Ford J, Aldridge BB, Efficient measurement and factorization of high-order drug interactions in Mycobacterium tuberculosis. Sci. Adv 3, e1701881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Clemens DL, Lee B-Y, Silva A, Dillon BJ, Masleša-Galić S, Nava S, Ding X, Ho C-M, Horwitz MA, Artificial intelligence enabled parabolic response surface platform identifies ultra-rapid near-universal TB drug treatment regimens comprising approved drugs. PLOS ONE 14, e0215607 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gumbo T, Pasipanodya JG, Romero K, Hanna D, Nuermberger E, Forecasting accuracy of the hollow fiber model of tuberculosis for clinical therapeutic outcomes. Clin. Infect. Dis 61 (suppl. 1), S25–S31 (2015). [DOI] [PubMed] [Google Scholar]
- 148.Greenwood DJ, Dos Santos MS, Huang S, Russell MRG, Collinson LM, MacRae JI, West A, Jiang H, Gutierrez MG, Subcellular antibiotic visualization reveals a dynamic drug reservoir in infected macrophages. Science 364, 1279–1282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nijsen MJMA, Wu F, Bansal L, Bradshaw-Pierce E, Chan JR, Liederer BM, Mettetal JT, Schroeder P, Schuck E, Tsai A, Xu C, Chimalakonda A, Le K, Penney M, Topp B, Yamada A, Spilker ME, Preclinical QSP modeling in the pharmaceutical industry: An IQ Consortium survey examining the current landscape. CPT Pharmacometrics Syst. Pharmacol 7, 135–146 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Smith CM, Proulx MK, Olive AJ, Laddy D, Mishra BB, Moss C, Gutierrez NM, Bellerose MM, Barreira-Silva P, Phuah JY, Baker RE, Behar SM, Kornfeld H, Evans TG, Beamer G, Sassetti CM, Tuberculosis susceptibility and vaccine protection are independently controlled by host genotype. MBio 7, e01516–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Smith CM, Sassetti CM, Modeling diversity: Do homogeneous laboratory strains limit discovery? Trends Microbiol 26, 892–895 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, McCulloch JA, Anastasakis DG, Sarshad AA, Leonardi I, Collins N, Blatter JA, Han S-J, Tamoutounour S, Potapova S, Foster St. Claire MB, Yuan W, Sen SK, Dreier MS, Hild B, Hafner M, Wang D, Iliev ID, Belkaid Y, Trinchieri G, Rehermann B, Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, eaaw4361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]