

This study highlights the molecular mechanisms of four assembly factors in folding the ribosomal central decoding region.
Abstract
While a structural description of the molecular mechanisms guiding ribosome assembly in eukaryotic systems is emerging, bacteria use an unrelated core set of assembly factors for which high-resolution structural information is still missing. To address this, we used single-particle cryo–electron microscopy to visualize the effects of bacterial ribosome assembly factors RimP, RbfA, RsmA, and RsgA on the conformational landscape of the 30S ribosomal subunit and obtained eight snapshots representing late steps in the folding of the decoding center. Analysis of these structures identifies a conserved secondary structure switch in the 16S ribosomal RNA central to decoding site maturation and suggests both a sequential order of action and molecular mechanisms for the assembly factors in coordinating and controlling this switch. Structural and mechanistic parallels between bacterial and eukaryotic systems indicate common folding features inherent to all ribosomes.
INTRODUCTION
Ribosome biogenesis is an essential, energy-demanding process in both prokaryotic and eukaryotic cells (1). In humans, defects in ribosome biogenesis are linked to diverse pathologies, called ribosomopathies, that manifest as a broad range of developmental disorders (2, 3). In bacteria, protein factors involved in ribosome biogenesis are critical for cell growth and pathogenesis (4, 5), making them potential antimicrobial targets (6, 7). Detailed knowledge of the molecular mechanisms driving ribosome assembly, as further developed in this work, is essential for understanding the mechanistic basis of ribosomopathies and opening new pharmaceutical approaches to combat multidrug resistance in bacteria.
In bacterial systems, similar to Escherichia coli, ribosome biogenesis involves the assembly of two ribosomal subunits, where the smaller 30S subunit is formed by the 16S ribosomal RNA (rRNA) and 21 ribosomal proteins (r-proteins), while the larger 50S subunit comprises the 5S rRNA, the 23S rRNA, and 33 r-proteins. Ribosome assembly occurs cotranscriptionally and RNA folding starts immediately upon synthesis of the 5′-end of the transcript. Moreover, r-proteins associate with the folding rRNA transcript even as it is being processed by ribonucleases (i.e., excised and trimmed from a longer transcript) and chemically modified by transacting factors, such as the methyl transferase RsmA (also known as KsgA). Despite this complexity, prior biochemical and structural work has shown that assembly of the 30S subunit is a robust process proceeding via multiple redundant parallel pathways, where the 5′-body domain forms first, followed by the central platform and head domains, and lastly the 3′ minor domain with the functionally important central decoding region (CDR) (8–11). During the later assembly phases including CDR folding, kinetic traps are prevalent. These are local minima in the ribosome’s folding landscape and correspond to alternative RNA conformations that result from degenerate local interactions and rearrange slowly into the native structure at physiological conditions (12). Ribosomal assembly factors are presumed to intervene and promote 30S biogenesis by avoiding such kinetic traps, thus allowing the CDR to adopt a functional fold. These factors include RimP, RsmA, RsgA, and RbfA, which play intertwined roles and assist in CDR folding (13, 14) by ensuring the correct placement of the 3′ minor domain [i.e., helices 44 (h44) and h45] between the rRNA domains forming the 30S head, body, and platform (see Fig. 1, state M). The dedication of several assembly factors to CDR folding underscores its central function as an integral part of the 30S A and P sites where transfer RNAs (tRNAs) recognize and translate the mRNA sequence (15). In the CDR, h44 along with its connecting linker regions participate in mRNA-tRNA binding while the 16S 3′-end harbors the anti–Shine-Dalgarno sequence to recruit mRNA to the 30S subunit (15, 16). While cryo–electron microscopy (cryo-EM) has described the binding positions of the 30S assembly factors RsgA, RbfA, and RsmA at intermediate resolution (13, 17–19), the molecular mechanisms by which they avoid kinetic traps and promote CDR biogenesis remain unclear. Here, we present eight cryo-EM structures of 30S assembly factor complexes that address fundamental molecular mechanisms governing assembly factor–mediated folding of the CDR.
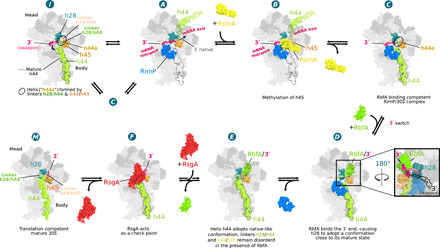
Fig. 1. Cryo-EM structures of 30S subunits in complex with different assembly factors.
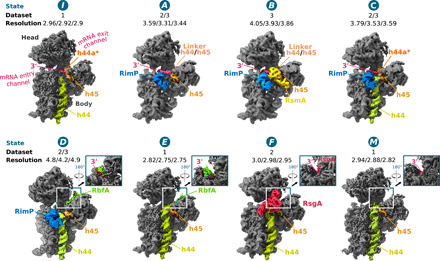
Cryo-EM reconstructions (states I, A to F, and M) of the 30S assembly complexes are shown from the front (intersubunit side), with insets showing the backside (solvent side) to highlight the position of RbfA and/or the 16S 3′-end in the mRNA exit channel in states D to M. The assigned 30S state is shown above each reconstruction along with the originating dataset and resolution (overall/body/head). Dataset 1: 30S+RbfA; dataset 2: 30S+RbfA+RsgA+RimP+RimM; dataset 3: 30S+RbfA+RimP+RsmA. Cryo-EM maps are derived from the multibody refinement and shown as composite maps [phenix.combine_focused_maps (43)] and are unsharpened and filtered to their estimated global resolution.
RESULTS
Overall cryo-EM characterization of 30S complexes with late-stage ribosomal assembly factors
Conformational heterogeneity in isolated ribosomal subunits was first described over 50 years ago by Zamir et al. and Moazed et al. (20, 21) when they demonstrated that isolated 30S exist in an “active” and “inactive” conformation and more recently by McGinnis et al. (22), who showed that an inactive 30S conformation is a bona fide in vivo state. Both in vitro and in vivo work agree that a conformational change in the CDR differentiates the two 30S conformations. We surmise that the inactive population in the isolated 30S subunits mimics a trapped immature-like state allowing, as previously demonstrated (13, 17, 19), late-stage ribosome assembly factors to interact with purified subunits. Therefore, using natively purified mature and inactive 30S subunits, we first investigated the 30S-RbfA complex (dataset 1) and observed 30S subunits with weakened density for 16S rRNA helix 44 (h44), resembling those that accumulate in strains where assembly factors are deleted (23, 24). On the basis of this observation, we surmised that the binding sites of other late-stage ribosomal assembly factors influencing h44 placement might also be exposed. Accordingly, we prepared two other complexes (datasets 2 and 3) using combinations of RbfA, RsgA, RimP, RimM, RsmA, and 30S subunits as described in the Materials and Methods (Fig. 1 and fig. S1). From these three datasets, eight high-resolution structures were determined using single-particle cryo-EM (table S4 and figs. S2 to S4), including two apo 30S structures with no assembly factors bound that show an inactive (state I) and mature state (state M), consistent with those proposed by the biochemical work half a century ago (Fig. 1). The remaining six structures, with up to two different assembly factors bound simultaneously (Fig. 1), can be ordered by the increasing maturation, i.e., native structure content, in the CDR (states A to F; Fig. 1 and Table 1), suggesting a sequence of action for the tested assembly factors and disclosing intermediate conformations in the late stages of CDR folding.
Table 1. Overview of structural features observed in various states during late-stage ribosome assembly.
| 30S state | 16S 3′-end position | H28 conformation | h44 position* |
h44 linker conformation† |
Assembly factor bound |
| I | Entry channel | Immature | Front | h44a | – |
| A | Entry channel | Immature | Exit | Disordered | RimP |
| B | Entry channel | Immature | Exit | Disordered | RimP + RsmA |
| C | Entry channel | Immature | Front | h44a | RimP |
| D | Exit channel | Mature | Front | Disordered | RimP + RbfA |
| E | Exit channel | Mature | Front | Disordered | RbfA |
| F | Exit channel | Mature | Front | Mature | RsgA |
| M | Exit channel | Mature | Front | Mature | – |
*In the front position, h44 localizes on the intersubunit surface of the 30S subunit. In the exit position, partial cryo-EM density attributed to h44 is observed to project out of the exit channel (see discussion in fig. S8).
†Proposed secondary structure and description of disordered/unmodeled 16S rRNA residues in the h44 linkers are shown in fig. S7.
When determining the structures, refining the entire 30S subunit as a single body [by three-dimensional (3D) classification and consensus refinement] led to low local resolution in the head region, indicating its variable orientation relative to the body region (figs. S5 and S6). Therefore, we refined the 30S head and body regions independently using a multibody approach (25), which yielded cryo-EM maps with overall resolutions between 2.75 and 4.9 Å. Local resolution in the assembly factor regions ranged between 2.6 and >5 Å (fig. S5) enabling their modeling in the 30S-bound state (tables S4 to S11 and Fig. 1). In the presence of RbfA (states D and E), the cryo-EM map indicates that r-protein S21 is missing, while in the other states, the map around S2 and the C terminus of S7 is weaker, reflecting their increased conformational flexibility or substoichiometric binding (13, 19). In all structures, except for state M, the CDR is immature in terms of its 16S rRNA fold (Table 1 and fig. S7), while the cryo-EM maps indicate that its 5′ and 3′ 16S termini are processed. Accordingly, the structures are interpreted here in terms of CDR folding, a final step in 30S maturation, where the assembly factors play intertwined roles to avoid kinetic traps (12–14).
An rRNA switch in the 16S rRNA delineates the CDR transition into a mature-like state
Comparing the structures of the inactive (state I; Fig. 2A) and mature 30S subunit (state M; Fig. 2C) maps the most pronounced differences primarily to the region around rRNA helix h28 that forms the so-called neck connecting the head and body regions. Here, the most substantial change involves the 16S 3′-end (pink) swapping from the mRNA entry channel in state I to its canonical position within the mRNA exit channel in state M (arrow 1 in Fig. 2, D to F), by hinging around G1530 that is fixed by stacking onto A1507 in h45.
Fig. 2. An rRNA switch is central to CDR folding.
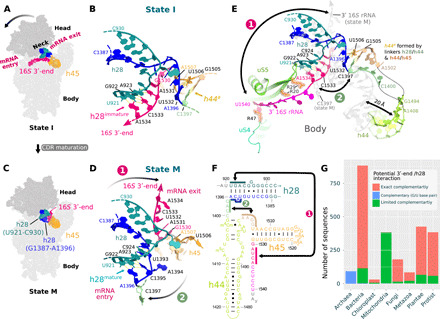
Two conformations of h28 are observed in states I (inactive 30S) (A and B) and M (mature 30S) (C and D) and define an rRNA secondary structure in the CDR. The conformational change is shown schematically (A and C) and in models of the neck region (h28; B and D). The 16S 3′-end swaps between the mRNA entry (B) and exit channels (D; arrow 1) and h28 switches from h28immature (B; U921-A923:U1532-A1534) to h28mature (D; U921-A923:U1393-A1396). In state I (B), U1393-A1396 and subsequent residues in the h28/44 linker (green) can interact with the h44/45 linker (brown) to form a labile helix-like h44a (poorly defined in the cryo-EM map; see fig. S8C). (E) Superimposed CDR structures in states I (colored) and M (gray) highlight changes outside the h28 region, for example, a displacement of h44. The CDR in all states is shown in fig. S7, with supporting cryo-EM maps in fig. S8. (F) Residues involved in the rRNA secondary structure switch are indicated by solid-colored bars where arrow 1 indicates base pairing in h28immature and arrow 2 the formation of h44a. (G) Bar graph comparing the number of 16S rRNA sequences, in various phylogenetic groups, with exact complementarity between the 16S 3′-end (U1532 to A1534) and h28 (residues U921 to A923; pink barks) and those with limited complementarity (≥1 mismatches; green bars). Archaea sequences are complementary in these regions but are colored distinctly to reflect the fact that the complementarity is formed by a non–Watson-Crick, G923:U1532, base pair (41).
This 3′-end swap is accompanied by a reorganization of helix h28: In state I, h28 shows a nonnative secondary structure, termed h28immature, where residues U921 to C924 base pair with the 16S 3′-end residues U1531 to A1534 (Fig. 2B), thus stabilizing the latter in the mRNA entry channel. In the mature state M, with the 3′-end swapped to the mRNA exit channel, h28 adopts its native secondary structure, termed h28mature, where residues U921 to C924 instead base pair with residues U1393 to A1396 (Fig. 2D) that precede the h28/h44 linker (Fig. 2F). This rRNA switch is largely consistent with the aforementioned chemical probing studies by the Noller and Weeks groups that showed “a reciprocal interconversion between two differently structured states” with chemical reactivity changes “almost exclusively confined to the decoding site” (20, 21). We, therefore, surmise that the alternative inactive CDR conformation in state I is characteristic of the inactive 30S conformation in vivo and represents a stable kinetic trap, accessed by interconverting with the mature 30S subunit or during folding of the CDR where the assembly factors are observed to play a role in stabilizing specific switch conformations (see below). This RNA switch could widely occur throughout all kingdoms of life, as our analysis of small subunit rRNA sequences indicates that the potential for the 3′-end to base pair and form helix h28immature is largely preserved across all phylogenetic groups except in mitochondria (Fig. 2G).
Our cryo-EM structures indicate that this rRNA switch has far ranging effects on the 30S subunit. For example, after multibody refinement, a principal components analysis (PCA) of the 30S head position relative to the body shows that the first principal component separates the dataset in a manner that reflects the h28 conformation (fig. S6). This corroborates the role of h28 as the main connection, or neck, between the 30S head and body regions (Fig. 2A) such that the rRNA switch in h28 also influences the head position. Moreover, base pairing in the h28mature conformation restrains residues U1393-A1396 that connect h28 to the long decoding helix h44 and can, thus, influence h44 conformation and dynamics. In the most immature states A and B, for example, residues U1393 to A1396 (blue) show no native base pairing interactions in h28immature and effectively elongate the h28/h44 linker (residues C1397 to C1399; green), thus conveying more flexibility to h44. Accordingly, in these states, h44 is not observed in its native position on the front of the 30S (Fig. 1 and Table 1), and additional density in the cryo-EM map instead suggests that it is repositioned into the mRNA exit channel (fig. S8). In states I and C, the elongated h28/h44 linker alternatively forms a labile helix-like structure, termed h44a, with residues in the h44/h45 linker (residues A1502 to G1505, brown; Fig. 2, B and E). Formation of h44a by both h44 linker regions allows the decoding helix to access a native-like position on the front side of the 30S subunit, but with its upper region still displaced by some 20 Å in state I relative to its position in the mature state M (Fig. 2E).
Together, this suggests that, during CDR maturation, the secondary structure switch in h28 reins in residues U1393 to A1396, thus shortening the h28/h44 linker and confining h44 to a more canonical position on the front of the 30S subunit (Fig. 1). Such folding of both h44 linker regions into a helix-like h44a and displacement of the h44 top end is also observed in eukaryotic 40S assembly complexes (27), further indicating mechanistic parallels between eukaryotic and prokaryotic ribosome assembly and supporting the idea that the natively isolated 30S inactive state mimics immature assembly intermediates as well.
Roles for late-stage assembly factors in maturation of the CDR
In the cryo-EM reconstructions (Fig. 1), we were able to visualize four of the five ribosome assembly factors added to the in vitro 30S complex, except for RimM that binds outside the CDR near uS19 and uS13 in the 30S head (23). Structural models for the 30S-bound assembly factors RsmA, RimP, RbfA, and RsgA were generated by refining template coordinates, taken from the Protein Data Bank (PDB) (RsmA PDB ID: 1QYR; RsgA PDB ID: 5NO3) or generated de novo by solution-state nuclear magnetic resonance (NMR) (RimP and RbfA; fig. S9 and table S13), into the cryo-EM map.
RimP and RsmA delay h44 positioning on the front of the 30S subunit
In the presented series of ribosomal complexes (Fig. 1), RimP and RsmA are observed bound to the 30S subunits showing the most nonnative structure in the CDR. RsmA (KsgA/Dim1 protein family) is a universally conserved rRNA methylase that modifies residues A1518 and A1519 in h45 (28), and RimP is essential for survival under stress conditions in Mycobacterium (5, 29). Among all factors studied here, RimP is the most versatile and is seen in four assembly states (states A to D) bound on the front of the 30S subunit (blue in Fig. 1). During ribosome biogenesis, RimP is known to promote binding of the r-protein uS12 (29–31). Accordingly, our cryo-EM data show it to interact directly with uS12 by forming an intermolecular β sheet via antiparallel pairing between uS12 strand β1 and RimP strand β3 (Fig. 3A). This results in a conformational change in loop β1/β2 of uS12, which contains the universally conserved PNSA motif interacting with the top of h44 during decoding (32). Reorientation of this loop may therefore contribute to the disorder in the top of h44 when RimP is bound (states A to D). In state B (Fig. 3B), RimP binds adjacent to RsmA that interacts with h24, h27, and h45, as reported previously (18). Both RimP and RsmA bind such that h44 cannot access its native position (indicated in gray) on the front of the 30S subunit. Specifically, the N-terminal domain of RimP (loop β1/β2) obstructs h44 from approaching uS12 (purple), while loop β6/β7 of RsmA keeps h44 from contacting the h45 loop (orange). In state C (Fig. 3C), after dissociation of RsmA, the C-terminal RimP domain retracts from the native h44 binding site by ca. 4 Å (arrow) and the lower part of h44 (green) docks onto the front of the 30S subunit. Yet, the N-terminal RimP domain (loop β1/β2) still prevents the tip of h44 (residues A1492 and A1493) from approaching uS12 and assuming its mature fold, thus keeping the CDR in a nonnative conformation. This remaining disorder and flexibility in both h44 linker regions may facilitate the swap of the 16S 3′-end, by keeping an open path for it to leave the mRNA entry channel.
Fig. 3. Roles for the assembly factors RsmA and RimP in CDR maturation.
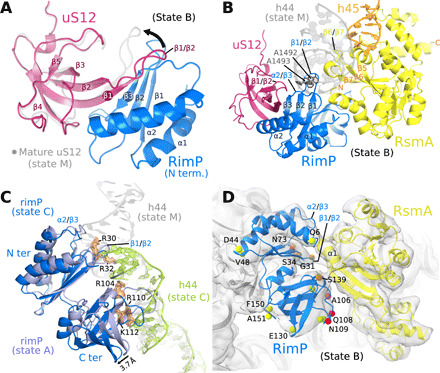
(A) The intermolecular β sheet formed by RimP and uS12, as observed in states A to D (state C shown), superimposed on uS12 of the mature 30S subunit (state M; gray), to highlight the conformational change in the β1/β2 loop of uS12 (arrow). (B) In state B, RimP (blue) and RsmA (yellow) bind adjacent on the 30S subunit and occupy the position of h44 in the mature 30S subunit (gray). (C) In state C, the C-terminal domain of RimP (blue; superposed on state A in violet) withdraws from the h44 binding site, such that the lower part of h44 (green; residues C1409 to G1491) repositions as seen in the mature 30S subunit (gray ribbon), while the upper part and linker region are disordered (not visible in the cryo-EM map). (D) Zoom on the RimP/RsmA interface seen by cryo-EM (state B). Indicated residues (spheres) were observable in the CLEANEX NMR spectrum of free RimP and are colored by the ratio κ of their HN/H2O exchange rates in the absence (kex,free) and presence (kex,+RsmA) of RsmA, κ = kex,free /kex,+RsmA: green (κ = 0.8 to 1.2), orange (κ = 1.6 to 2.0), and red (κ > 2). All RimP residues with significant solvent protection after RsmA addition (κ ≥ 1.6) localize to the RimP/RsmA interface seen in the cryo-EM structure of their 30S complex. See fig. S12 for RimP CLEANEX spectra and derived HN/H2O exchange rates.
RimP and RsmA were not previously known to directly interact, as suggested by our cryo-EM structures that show them binding adjacently on the ribosome. However, both proteins appear dynamic, showing reduced local resolution in the cryo-EM map (fig. S5) that limits the characterization of their shared interface. We, therefore, used solution-state NMR to probe a direct RimP/RsmA interaction, which was indeed corroborated by several observables. First, the 2D 1H, 15N BEST-TROSY (band-selective excitation short transients - transverse relaxation optimized spectroscopy) spectrum of [U15N]-labeled RimP showed significant global signal broadening and attenuation after adding only 0.25 mol equivalent of RsmA (fig. S10), indicating slowed molecular tumbling of RimP (16.7 kDa) from association with the considerably larger RsmA (30.4 kDa). Yet, the absence of discernible chemical shift changes point to only weak association (KA < 104 M−1) and precluded a straightforward elucidation of the binding interface. Second, the NMR-derived apparent translational diffusion coefficient of RimP decreased from 1.09 ± 0.01·10−10 m2/s to 0.98 ± 0.03·10−10 m2/s (in aqueous buffer at room temperature) in the presence of 0.25 mol equivalents of RsmA, implying a ca. 11% increase in the averaged hydrodynamic radius of RimP from association with RsmA (fig. S11). Third, the addition of RsmA also caused a clear reduction of fast HN/H2O exchange rates (by >30%, i.e., kex,bound /kex,free ≤ 0.625) for 30% (8 of 27) of the solvent-exposed amide protons observed in the CLEANEX NMR spectrum (33) of RimP (fig. S11 and table S1). These significantly solvent shielded amide groups cluster conspicuously near the RimP/RsmA interface seen in the cryo-EM model of the ternary RimP/RsmA/30S complex (Fig. 3D), suggesting a similar arrangement of RimP/RsmA in solution. This was further tested by comparing the impact of RsmA versus RbfA addition on RimP NMR observables. To accentuate any unspecific effects, an excess of the nonbinding RbfA (2 mol equivalent) was added to RimP first, which caused uniform decay in RimP signal (−20%) except in highly mobile loop regions and some side chains (fig. S12A) presumably due to a general restriction of molecular motion from the engendered crowding effect (34). Subsequent addition of eight times less RsmA (0.25 mol equivalent) induced, on average, another 20% RimP amide signal attenuation, with disproportionally stronger signal decay clustering in specific inherently flexible loop regions (fig. S12B). Thus, several signals stand out from residues in loops β4-β5, β5-β6, and in the C-terminal part of strand β7, indicating a strong dampening of their local motions in agreement with a RsmA contact. Moreover, residues in the extended β4-β5 and nearby loop β7-β8 show significant protection from fast HN/H2O exchange in the RimP CLEANEX spectrum in the presence of RsmA (fig. S12B), as opposed to RbfA (fig. S12A). Together, these residues delineate a contiguous surface on the C-terminal RimP domain (fig. S12, C and D) that mediates a weak but site-selective RimP/RsmA association similar to their arrangement on the 30S subunit. The RsmA side of the interface seen in the cryo-EM map includes its N-terminal helix 1 and the β6-β7 loop, which may have a functional significance as this loop contains a motif (FXPXPXVXS) common to Erm and KsgA methyltransferases where the conserved phenylalanine stabilizes the substrate base through stacking interactions (28).
Extending the previously described role of RimP in promoting S12 binding (29–31), our results also indicate a broader role in 30S subunit assembly where RimP delays h44 positioning on the front of the 30S subunit, thus exposing the RsmA binding site, and may even preassociate with RsmA to facilitate its recruitment to the subunit. Moreover, by keeping the upper part of h44 and its linker regions disordered, RimP maintains an open unstructured channel in the 30S neck region for the 16S 3′-end to swap.
RbfA promotes the h28immature to h28mature rRNA switch
While RimP binding is compatible with either position of the 16S 3′-end and binds similarly in states C and D, RbfA is visualized on the 30S subunit (states D and E) only after the 3′-end has swapped from the entry to the exit channel. This swapped 16S 3′-end is an integral part of the RbfA binding site in the mRNA exit channel and specifically interacts with the (type II) KH domain of RbfA (Fig. 4A): The 16S rRNA residues G1530 to A1536 run along the RNA binding surface of RbfA, where residues A1531 to C1533 interact near the GXXG sequence motif [AXG in RbfA (35)] characteristic of KH domain proteins while A1534-C1535 extend into a pocket on RbfA and π stack between the highly conserved Phe78 of RbfA and Arg43 of r-protein bS18 (Fig. 4A). Comparing the ribosome-bound (cryo-EM) and free (NMR) solution structures reveals some prominent structural adaptions in RbfA that promote this interaction with the 16S 3′-end. Namely, helix α1 rotates by about 20° and the subsequent loop α1/β1 rearranges such that the 310 helix present in the apo RbfA structure unfolds to reposition near nucleotides A1534 to C1536 (Fig. 4B).
Fig. 4. Effects of RbfA on the conformation of the CDR.
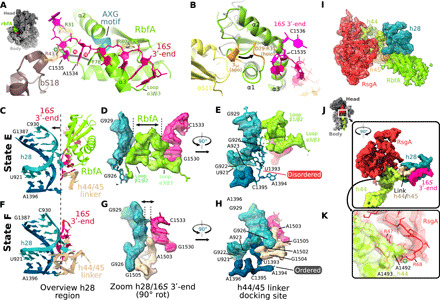
(A) RbfA is bound within the mRNA exit channel where it captures the 16S 3′-end, e.g., via a Phe78 (RbfA)-A1534-C1535-R43(bS18) π-stacking interaction (dotted lines). (B) Structural differences between the NMR solution structure of apo RbfA (gray) and 30S-bound holo RbfA (state E; green) include the unfolding of a 310 helix (orange) in the α1/β1 loop such that the loop closes around the 16S 3′-end (near A1534-C1535). (C to H) The h28 region in the RbfA (state E; C to E) and RsgA (state F; F to H) bound 30S structures. RbfA inserts between 16S 3′-end and h28, displacing the upper part of h28 [arrow in (C), compare with (F)] while destabilizing its lower end according to disorder in the cryo-EM map around residues U1393 to A1394 [compare map around residues U1393 to A1394 in (E) and (H)], thus keeping h28 from adopting a fully matured conformation. (I) The superimposed RbfA (green; state D) and RsgA (red; state F) cryo-EM maps (segmented density for RsgA, h44, and h28) show no overlap. (J) Close-up view of RsgA (red) clamping around the top of h44 in the CDR. (K) Corresponding detailed view showing a cation-π interaction that sandwiches A1492 (in h44; green) between Arg47 and Arg68 (in RsgA; red). The cryo-EM density shown in all panels corresponds to the consensus or multibody refinement maps for the body region and was segmented using the underlying model (radius, ∼2.5 Å).
The interaction of RbfA with the 16S 3′-end in the exit channel is inconsistent with previous cryo-EM reconstructions that localized RbfA on the front of the 30S subunit (17), where it cannot interact with the 16S 3′-end. Therefore, we again used NMR to confirm this interaction using mimics of the 16S 3′-end to define its binding site on RbfA from induced chemical shift perturbations (CSPs) (36) in the well-dispersed 2D 1H, 15N heteronuclear single-quantum coherence (HSQC) fingerprint spectrum (fig. S13A). Mapping the amide groups with significant CSP on the cryo-EM structure of RbfA in the 30S complex indeed corroborates the rRNA binding surface and structural rearrangement of loop α1/β1 observed in states D and E (fig. S13C). These NMR titration experiments also show that interactions near the consensus AXG motif in RbfA are unspecific since local CSPs are induced by adding either 3′-end mimics or poly(U) control; in contrast, only 16S 3′-end mimics induce CSPs in loop α1/β1, revealing its interaction with rRNA to be specific (fig. S13B). Our NMR and cryo-EM results, therefore, indicate that RbfA facilitates the RNA secondary structure switch by sequestering residues U1531 to C1535, preventing their base pairing in h28immature, and holding the 16S 3′-end in the mRNA exit channel. This function might explain the importance of RbfA for cell growth at low temperatures (37, 38), where an h28immature to h28mature conversion may otherwise be kinetically unfavored.
The RbfA-promoted conversion from h28immature to h28mature is not sufficient, however, to complete CDR folding, and RbfA appears to even delay folding of the h44/45 linker. Besides interacting with the 16S 3′-end, RbfA packs against and distorts the upper part of h28 (arrow in Fig. 4C), with its loops β1/β2 and α3/β3 acting as a wedge to prevent the tertiary interaction seen in the mature CDR that sandwiches A1503 of the h44/h45 linker between h28 and the 16S 3′-end (compare Fig. 4, D and G). Moreover, in the presence of RbfA, the lower part of h28 around the A923:U1393 base pair is destabilized since the map around residues U1393 to A1394 is weak and fragmented as compared to the same region in the RsgA/30S complex (compare Fig. 4, E and H). These disordered residues play an important role in stabilizing the h44/45 linker in the mature 30S where A1502-G1505 in the linker form a compact turn that packs into the minor groove of h28 around residues U1393 to A1394 (Fig. 4H and fig. S14).
RbfA was originally isolated as a suppressor of a cold-sensitive mutation in the 16S rRNA [C23U (38)]. While not proximal to RbfA, this residue is a component of h1 that is adjacent to h28; thus, RbfA and h1 could influence each other allosterically via h28, or RbfA could have alternative modes of interaction (17). The interaction between RbfA and the 16S 3′-end is consistent with the finding that mammalian (mitochondrial) RbfA homologs interact with the 3′-minor domain of the small ribosomal subunit rRNA (39). Moreover, this interaction is similar to that observed between Pno1 and the 3′-end of the 18S rRNA during the late stages of eukaryotic ribosome biogenesis (27, 40). For example, both proteins bind within the exit channel and use a KH domain to fix the position of the 3′-end of the rRNA within the exit channel. Pno1, however, contains two KH domains to protect and position the entire 3′-end during rRNA trimming. In contrast, RbfA with its single KH domain fixes only residues A1531 to C1536 (of 1540 residues), and we therefore speculate that RbfA may cooperate with another still unknown protein to play a role similar to Pno1 in protecting and positioning the 3′-end during rRNA trimming. This would agree with our hypothesis that a conserved CDR core folding pathway exists across phylogenetic groups, although the protein factors involved in subunit biogenesis maybe be unrelated by sequence.
RsgA checks CDR maturation
RsgA is one of the last assembly factors to interact with the 30S subunit, removing RbfA and acting as a checkpoint for the entry of the assembling subunit into the pool of translating ribosomes (13, 19, 41). In the RsgA-bound state F (Fig. 1), the guanosine triphosphatase (GTPase) domain of RsgA clamps around the CDR, while its zinc-binding domain forms a bridge with the 30S head near h29, as seen in previous cryo-EM reconstructions (13, 19). The RsgA and RbfA binding sites do not overlap (Fig. 4I), indicating that an allosteric effect, rather than steric hindrance, is responsible for the RsgA-induced release of RbfA (41). Regarding the nature of this allosteric change, we only observe one RsgA-bound state, while PCA reveals that RsgA induces a 30S head conformation distinct from all other states (fig. S6, E to H). Noller and colleagues showed that 30S head movement is related, in part, to a hinge that lies at a weak point in h28 near the bulged G926 (26). This is the same region pictured in Fig. 4 (C and D), where RbfA loops β1/β2 and α3/β3 wedge apart h28 and the 16S 3′-end. Accordingly, RsgA-induced allosteric changes in the 30S head could promote an h28 conformation incompatible with RbfA binding, thus leading to its release. The 16S 3′-end would then be free to reposition near h28 and interact with A1503 (Fig. 4G) to stabilize the h44/45 linker. Moreover, by clamping around the CDR (Fig. 4J), RsgA constrains its folding landscape and further promotes h44/45 linker maturation via extensive interactions along the minor groove of h44. For instance, Arg47 and Arg68 in the OB domain of RsgA form a pocket to fix the position of A1492 at the top of h44 presumably via cation-π stacking (Fig. 4K). Such direct interactions with the CDR substantiate the previous hypothesis that RsgA acts as a checkpoint and probes the maturation state of the 30S subunit, making its GTPase activity dependent on CDR conformation (13, 19).
DISCUSSION
The high-resolution cryo-EM structures presented here can be arranged by increasing native structure content in the CDR to delineate a putative sequence of states (states A to F; Fig. 5) through which the four analyzed late-stage assembly factors RimP, RsmA, RbfA, and RsgA guide rRNA folding during CDR maturation. Variations in this pathway are possible given that 30S assembly has been predicted to follow multiple redundant parallel pathways (8–11). Moreover, individual steps may also occur in the absence of their associated factors, considering that the latter are not individually essential, and play an indispensable role only under certain growth conditions where specific conformational rearrangement might be unfavorable (e.g., during cold shock) (5, 29, 37). A central feature of our model is a conserved rRNA secondary structure switch (h28immature > h28mature conversion) with an accompanying swap of the 16S 3′-end from the mRNA entry to the exit channel. This occurs between states C and D and is assisted by RbfA that stabilizes the 16S 3′-end in its canonical position inside the mRNA exit channel, thus preventing a reversion to the alternative h28immature arrangement. RimP and RsmA, in turn, likely facilitate the rRNA switch during the preceding states (states A to C) by delaying the top of h44 and its linker regions from accessing their canonical position on the front of the 30S subunit. In this regard, we suggest that state I represents an inactive, kinetically trapped state where h44 prematurely assumes a noncanonical conformation on the front of the 30S subunit, with its linkers extended or forming a labile, nonnative helix-like h44a. The structure of this inactive state is consistent with that inferred from biochemical analyses half a century ago and, more recently, from in vivo studies and studies on purified subunits (20, 21, 42). This opens the possibility for assembly factors to have parallel roles in canonical ribosome assembly and in resolving trapped inactive states, allowing them to acquire a mature fold. Last, there are several apparent parallels between the prokaryotic system studied here and eukaryotic systems. For instance, RbfA may have a role analogous to the eukaryotic Pno1 in binding and protecting the 3′ rRNA end (27), while common rRNA sequence features indicate that the observed reorganization of h28 can occur in all phylogenetic kingdoms except in the more distinct mitochondria. This suggests that the rRNA switch mechanism observed in our 30S complex structures is inherent to small subunit assembly throughout life.
Fig. 5. Schematic model for the stepwise folding of the CDR.
The well-defined cryo-EM structures ordered according to increasing 16S rRNA native structure content illustrates a potential folding pathway for the CDR. State A has the most nonnative CDR fold and shows RimP bound, the 16S 3′-end inside the mRNA entry channel (h28immature), and the decoding h44 positioned in the mRNA exit channel (see fig. S8D discussion). This 30S configuration enables RsmA binding in state B and exposes residues A1518 to A1519 in the h45 loop for methylation by RsmA (28). RsmA release (state C) partially opens the canonical binding site of h44, allowing it to drop into place while its two linkers form a poorly defined helix similar to h44a in state I. Upon RbfA binding (state D) the 3′-end swaps into the exit channel (see inset), stabilized by interactions with the KH domain of RbfA, allowing h28mature to form. After RimP dissociation (state E), h44 adopts a more native-like conformation although both linker regions remain largely disordered. After RsgA binding and release of RbfA (state F), the h44 linkers fold into a nearly native conformation. Last, after guanosine triphosphate hydrolysis, RsgA leaves the ribosome in a mature conformation (state M). In this model, state I represents an inactive state (or kinetic trap) accessed, for instance, when RimP prematurely dissociates from states A or C and h44 drops into its mature position before the 3′-end can swap into the exit channel.
MATERIALS AND METHODS
Ribosome preparation
E. coli CAN/20-E12 cells were grown in a 150-liter fermenter (Bioprocess Technology) in LB medium at 37°C, 300 rpm, and a constant sterile air flux of 85 liter min−1. Growth was monitored until the exponential phase was reached [optical density at 600 nm (OD600) = 0.6], when the temperature was lowered to 20°C, and cells were harvested using a high-speed tubular centrifuge (CEPA Z-41) to yield 89 g of dried pellet. Cells were washed two times with TICO buffer [10 mM Hepes-KOH (pH 7.6), 6 mM MgCl2, 30 mM NH4CI, and 6 mM β-mercaptoethanol] to eliminate traces of the medium and then resuspended in 2 ml of TICO buffer supplemented with 0.25 mM phenylmethylsulfonyl fluoride per 1 g of cells, at 4°C, for further disruption using 600 bar in an APV Gaulin homogenizator. Lysate was clarified by ultracentrifugation (Optima L-90, Beckman Coulter) in two steps, first for 45 min at 42,000g, then for 20 hours at 72,600g. The pellet obtained in the second centrifugation step, considered crude ribosomes, was resuspended in TICO buffer, its concentration verified by measuring absorbance at 260 nm (Ultraspec 3100 Pro spectrophotometer, Amersham Biosciences), and stored in 4000 A260 (absorbance at 260 nm) aliquots. A fraction containing 4000 A260 units of these crude ribosomes was loaded into a 5.7 to 40% sucrose gradient [in 10 mM Hepes-KOH (pH 7.6), 1 mM MgCl2, 100 mM NH4Cl, and 6 mM β-mercaptoethanol buffer], prepared in a 15 Ti Zonal rotor, and centrifuged 17 hours at 23,000 rpm and 4°C. The gradient was fractioned by pumping in a 50% sugar solution, at 2000 rpm (Ultrarac 7000 fraction collector, LKB Bromma); fractions containing 30S and 50S particles were pooled separately and centrifuged at 118,000g for 22 hours. Pellets were washed to remove sucrose and then resuspended in TICO buffer to get a final concentration of 600 A260/ml.
Cloning and purification of assembly factors for cryo-EM analysis
RbfA
The coding sequence for E. coli RbfA was amplified from genomic DNA using the primers fxf-RbfA-0326 and fxr-RbfA-0327 (table S2) and the resulting fragment cloned using the Fx-cloning methodology (44) into pINIT-cat (pINIT_cat-Rbfa; table S3) for sequence validation and subsequently into p7XC3GH plasmid (table S3) to yield the expression plasmid p7XC3GH-RbfA (table S3). For overexpression, E. coli BL21 (DE3) cells were transformed with the plasmid p7XC3GH-RbfA, used to directly inoculate liquid LB medium supplemented with kanamycin (50 μg/ml), and grown at 37°C (200 rpm) overnight, in an orbital shaker (New Brunswick, Eppendorf), up to an OD600 of about 3.0. This culture was used to inoculate a large-scale culture at an initial OD600 = 0.05. Cells were then grown under same conditions described above until reaching exponential growth phase (OD600 = 0.5 to 0.6). At this point, the temperature was lowered to 20°C, and isopropyl-β-d-thiogalactopyranoside was added to a final concentration of 0.5 mM for induction. After 16 hours at 20°C and 200 rpm, the cells were harvested by centrifugation at 5000g and 4°C for 30 min in an Avanti J20-XP centrifuge (Beckman Coulter), flash frozen in liquid nitrogen, and stored at −80°C until further use. For protein purification, the cell pellet was thawed and resuspended in 50 mM Hepes (pH 7.8), 300 mM NaCl, 5 mM β-mercaptoethanol, 1% Triton X-100, lysozyme (0.3 mg/ml), and 1× EDTA free protease inhibitor cocktail and lysed by sonication on ice for a total time of 10 min (Vibra-Cell VC 505 sonicator, 14-mm-diameter probe). The lysate was clarified by ultracentrifugation at 186,000g for 60 min at 4°C in an Optima L-90 ultracentrifuge (Beckman Coulter). The soluble protein fraction was applied to a Ni2+ NTA HP column (GE Healthcare) equilibrated in 50 mM Hepes (pH 7.8), 300 mM NaCl, and 5 mM β-mercaptoethanol and released from the column by stepwise elution, such that the protein was recovered at an imidazole concentration of 225 mM. The sample was then concentrated by centrifugation using an AMICON concentrator [5000 molecular weight cutoff (MWCO)] until a final volume of 1 ml was obtained. To reduce the imidazole concentration, this sample was diluted with 4 ml of 50 mM tris-HCl (pH 7.8), 150 mM NaCl, and 2 mM β-mercaptoethanol buffer and concentrated twice. The His-GFP tag was then cleaved using 3C protease at a final concentration of 0.020 mg of C3 protease/mg of fusion protein and incubated at 4°C for 2 hours with mild agitation. Cleavage was confirmed by SDS–polyacrylamide gel electrophoresis (PAGE), and the sample was then applied to Ni2+ NTA HP and HiLoad 16/600 Superdex 75 size exclusion columns (GE Healthcare) connected in series and equilibrated with 50 mM tris-HCl (pH 7.8), 150 mM NaCl, and 2 mM β-mercaptoethanol buffer. The Ni2+-NTA HP column was used as a reverse HisTrap column to remove the C3 protease and cleaved tag. The eluted protein was collected and stored at −80°C. The purity, integrity, and identity of the protein were analyzed using SDS-PAGE, matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF), and liquid chromatography–tandem mass spectrometry (LC-MS/MS) (CIC bioGUNE, proteomic platform). Protein concentration was determined spectrophotometrically at 280 nm using an extinction coefficient of 4470 M−1 cm−1. Before complex preparation, the protein’s aggregation was checked, and the buffer was exchanged to 20 mM Hepes (pH 7.8), 10 mM MgCl2, 60 mM NH4Cl, and 6 mM β-mercaptoethanol buffer using a Superdex 75 10/300 GL column.
RsgA
The coding sequence for E. coli RsgA was amplified from genomic DNA using the primers fxf-RsgA-0328 and fxr-RsgA-0329 (table S2). The resulting fragment was cloned by Fx-cloning methodology (44) into pINIT-cat (pINIT_cat-RsgA; table S3) for sequence validation and subsequently into p7XNH3 plasmid (table S3) to yield the expression plasmid p7XNH3-RsgA (table S3). For overexpression, E. coli BL21 (DE3) cells were transformed freshly with p7XNH3-RsgA and grown as described for RbfA. For protein purification, the cell pellet was thawed and resuspended in lysis buffer [50 mM tris-HCl (pH 7.8), 300 mM NaCl, and 5 mM β-mercaptoethanol], supplemented with 1% Triton X-100, lysozyme (0.3 mg/ml), and 1× EDTA free protease inhibitor cocktail and benzonase. Lysis was performed by sonication on ice for a total time of 2.5 min (Vibra-Cell VC 505 sonicator, 14-mm-diameter probe). The lysate was clarified by ultracentrifugation at 186,000g for 40 min at 4°C in an Optima L-90 ultracentrifuge (Beckman Coulter). The soluble protein fraction was applied to a Ni2+ NTA HP column (GE Healthcare) equilibrated in 50 mM Hepes (pH 7.8), 300 mM NaCl, and 5 mM β-mercaptoethanol and released from the column by linearly increasing imidazole concentration up to 500 mM. Fractions containing RsgA were pooled together and desalted using an Econo-Pac desalting column (Bio-Rad) equilibrated with 50 mM tris-HCl (pH 7.8), 300 mM NaCl, and 5 mM β-mercaptoethanol. Subsequently, the protein was incubated with 3C protease at a ratio of 0.02 mg of 3C/mg of fusion protein and cleaved at 4°C overnight under mild agitation. During this incubation, some precipitation appeared and was eliminated by centrifugation at 10,000 rpm and 4°C for 10 min. The cleaved sample was then applied to a Ni2+ NTA HP column and HiLoad 16/600 Superdex 75 size exclusion column (GE Healthcare), connected in series and equilibrated with 50 mM tris-HCl (pH 7.8), 300 mM NaCl, and 2 mM β-mercaptoethanol. The Ni2+ NTA HP column was used as a reverse HisTrap column to remove the C3 protease and cleaved tag. The eluted protein was collected and stored at −80°C. Protein purity, integrity, and identity were analyzed by SDS-PAGE, MALDI-TOF, and LC-MS/MS (CIC bioGUNE, proteomic platform). Protein concentration was determined spectrophotometrically at 280 nm using an extinction coefficient of 24,660 M−1 cm−1. Before complex preparation, the protein’s aggregation state was checked, and the buffer was exchanged to 20 mM Hepes (pH 7.8), 10 mM MgCl2, 60 mM NH4Cl, and 6 mM β-mercaptoethanol using a Superdex 75 10/300 GL column.
RimM
The coding sequence for E. coli RimM was amplified from genomic DNA using the primers fxf-RimM-0324 and fxr-RimM-0325 (table S2). The resulting fragment was cloned by Fx-cloning methodology (44) into pINIT-cat (pINIT_cat-RimM; table S3) for sequence validation and subsequently into p7XC3GH plasmid (table S3) to yield the expression plasmid p7XC3GH-RimM (table S3). For overexpression, E. coli BL21 (DE3) cells were transformed freshly with p7XC3GH-RimM and grown as described for RbfA. For protein purification, the cell pellet was thawed and resuspended in lysis buffer, supplemented with lysozyme (0.3 mg/ml), 1× EDTA-free protease inhibitor cocktail, and deoxyribonuclease I (DNase I) (0.1 mg/ml). Cells were lysed using an APV Gaulin homogenizer at 850 bar (two times). The lysate was clarified by ultracentrifugation at 186,000g for 30 min at 4°C in an Optima L-90 centrifuge (Beckman Coulter). The soluble protein fraction was applied to a Ni2+ HisTrap FF crude column (GE Healthcare) equilibrated with 50 mM tris-HCl (pH 7.8), 300 mM NaCl, 2 mM β-mercaptoethanol buffer and released from the column by stepwise elution, with the protein completely eluting at 250 mM imidazole. The protein was concentrated using an AMICON concentrator (5,000 MWCO) and applied to a HiLoad 26/10 desalting column (GE Healthcare) equilibrated with 50 mM tris-HCl (pH 7.8), 300 mM NaCl, and 2 mM β-mercaptoethanol. Fractions containing protein were pooled and His-GFP tag was cleaved with 3C protease (0.02 mg/mg of fusion protein) at 4°C with mild agitation. Cleavage was confirmed by SDS-PAGE, and the cleaved protein was concentrated using an AMICON concentrator (5,000 MWCO) and then applied to a Ni2+ NTA HP and a HiLoad 16/600 Superdex 75 size exclusion column (GE Healthcare) connected in series and equilibrated with 20 mM Hepes (pH 7.8), 10 mM MgCl2, 60 mM NH4Cl, and 6 mM β-mercaptoethanol. Eluted sample was concentrated using an AMICON concentrator (10,000 MWCO), and the concentration was estimated using 40,575 M−1 cm−1 as a molar extinction coefficient. Protein purity, integrity, and identity were analyzed by SDS-PAGE, MALDI-TOF, and LC-MS/MS (CIC bioGUNE, proteomic platform). Before complex preparation, the protein’s aggregation state was checked, and the buffer was exchanged to 20 mM Hepes (pH 7.8), 10 mM MgCl2, 60 mM NH4Cl, and 6 mM β-mercaptoethanol using a Superdex 75 10/300 GL column.
RimP
The coding sequence for E. coli RimP was amplified from genomic DNA using the primers Fxf-RimP-0427 and Fxr-RimP-0428 (table S2); the resulting fragment was cloned by Fx-cloning methodology (44) into pINIT-cat (pINIT_cat-RimP; table S3) for sequence validation and subsequently into p7XNH3 plasmid to yield the expression plasmid p7XNH3-RimP (table S3). For overexpression, E. coli BL21 (DE3) cells were transformed freshly with p7XNH3-RimP and grown as described for RbfA. The cell pellet was thawed and resuspended in lysis buffer, supplemented with lysozyme (0.3 mg/ml), 1× EDTA free protease inhibitor cocktail, and DNase I (0.1 mg/ml). Cells were lysed using an APV Gaulin homogenizator at 850 bar (two times). The lysate was clarified by ultracentrifugation at 186,000g for 30 min at 4°C in an Optima L-90 centrifuge (Beckman Coulter). The soluble protein fraction was applied to a Ni2+ HisTrap FF crude column (GE Healthcare) equilibrated with 50 mM tris-HCl (pH 7.8), 300 mM NaCl, and 2 mM β-mercaptoethanol buffer and eluted by increasing imidazole concentration stepwise to 250 mM. The eluted protein was concentrated and equilibrated with 50 mM tris-HCl (pH 7.8), 300 mM NaCl, and 2 mM β-mercaptoethanol. Fractions containing protein were pooled and cleaved overnight with 3C protease, at 0.020 mg/mg of fusion protein, at 4°C with mild shaking. The cleaved protein was concentrated using an AMICON concentrator (5,000 MWCO) before being applied to a Ni2+ NTA HP and a HiLoad 16/600 Superdex 75 size exclusion column (GE Healthcare) connected in series and equilibrated with 20 mM Hepes (pH 7.8), 10 mM MgCl2, 60 mM NH4Cl, and 6 mM β-mercaptoethanol. Eluted sample was pooled, and the concentration was estimated using 9970 M−1 cm−1 as molar extinction coefficient. Protein purity, integrity, and identity were analyzed by SDS-PAGE, MALDI-TOF, and LC-MS/MS (CIC bioGUNE, proteomic platform). Before complex preparation, the protein’s aggregation state was checked, and the buffer was exchanged to 20 mM Hepes (pH 7.8), 10 mM MgCl2, 60 mM NH4Cl, and 6 mM β-mercaptoethanol using a Superdex 75 10/300 GL column.
RsmA
The coding sequence for E. coli RsmA was amplified from genomic DNA using the primers fxf-RsmA-0431 and Fxr-RsmA-0432 (table S2); the resulting fragment was cloned by Fx-cloning methodology (44) into pINIT-cat (pINIT_cat-RsmA; table S3) for sequence validation and subsequently into p7XC3GH plasmid (table S3) to yield the expression plasmid p7XC3GH-RsmA (table S3). For overexpression, E. coli BL21 (DE3) cells were transformed freshly with p7XC3GH-RsmA and grown as described for RbfA. For protein purification, the cell pellet was thawed and resuspended in lysis buffer supplemented with lysozyme (0.3 mg/ml), 1% Triton X-100, and DNase I (0.1 mg/ml) and lysed by sonication on ice for 2.5 min (Vibra-Cell VC 505 sonicator, 14-mm-diameter probe). The lysate was clarified by ultracentrifugation at 186,000g for 40 min at 4°C in an Optima L-90 centrifuge (Beckman Coulter). The soluble protein fraction was applied to a Ni2+ HisTrap FF crude column (GE Healthcare) equilibrated with 50 mM tris-HCl (pH 8.0), 300 mM NaCl, and 2 mM β-mercaptoethanol buffer and eluted by a linearly increasing imidazole concentration. Fractions containing protein were pooled and cleaved overnight with 3C protease, at 0.0066 mg/mg of fusion protein, at 4°C with mild shaking. As this protein showed tendency to precipitate when concentrated, the protein was dialyzed overnight at 4°C against 50 mM tris-HCl (pH 7.8), 150 mM NaCl, and 5 mM β-mercaptoethanol. During this dialysis, the protein was cleaved by including 3C protease, at 0.0066 mg/mg of fusion protein. The buffer-exchanged protein was then collected, concentrated using an AMICON filter with 10,000 MWCO, and applied to a Ni2+ NTA HP and a HiLoad 16/60 Superdex 75 size exclusion column (GE Healthcare) connected in series and equilibrated in 50 mM tris-HCl (pH 7.8), 150 mM NaCl, and 5 mM β-mercaptotheanol. Eluted protein was pooled and concentrated using an AMICON concentrator (10,000 MWCO). The concentration was calculated using 12,045 M−1 cm−1 as molar extinction coefficient. Protein purity, integrity, and identity were analyzed by SDS-PAGE, MALDI-TOF, and LC-MS/MS (CIC bioGUNE, proteomic platform). Before complex preparation, the protein’s aggregation state was checked, and the buffer was exchanged to 20 mM Hepes (pH 7.8), 10 mM MgCl2, 60 mM NH4Cl, and 6 mM β-mercaptoethanol using a Superdex 75 10/300 GL column.
Sample preparation for solution-state NMR
Preparation of 15N/13C-labeled RimP and RbfA
Labeled proteins were prepared as described previously (45). Briefly, transformed cells were grown in [U-13C,15N]-enriched M9 media containing 13C6 d-glucose (2 g/liter) (98%) and 15NH4Cl (1 g/liter) (99%) as the sole carbon and nitrogen source, respectively. Cells were grown to an OD600 of 0.6 before induction with Isopropyl b- d-1-thiogalactopyranoside (ITPG) (final concentration, 0.5 mM) for 36 to 40 hours at 18°C and harvested by centrifugation for 30 min at 5000 rpm. The resulting pellet was resuspended in lysis buffer [100 mM tris, 1 M NaCl, 10% glycerol, 100 μM tris(2-carboxyethyl)phosphine (TCEP), 0.5% Triton X-100, and one tablet of cOmplete EDTA-free PIC (Roche), at pH 8] and lysed by sonication. Subsequently, the proteins were purified as described in the previous section. Last, the proteins were transferred into a buffer [10 mM Hepes (pH 7.6), 6 mM MgCl2, 150 mM NH4Cl, 75 μM TCEP, and 7% D2O or 100% D2O] suitable for subsequent NMR experiments in presence of 30S ribosomes.
NMR spectroscopy
A set of complementary 3D HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, HN(CO)CACB, HN(CA)HA, and HN(COCA)HA BEST-TROSY experiments (11) for sequential backbone assignment, supplemented by a complete set of 3D (H)C,CH, H,CH, H,NH, and (H)C,NH edited nuclear Overhauser effect spectroscopy (NOESY) experiments (all with 150 ms of mixing time) for structure analysis, recorded on a 800-MHz BRUKER AVANCE III spectrometer equipped with 5 mm of TCI CryoProbe or a 600-MHz BRUKER AVANCE III spectrometer equipped with 5 mm of TXI probe. Proton 1H chemical shifts were directly referenced to added DSS (2,2-dimethyl-2-silapentane-5-sulphonic acid). The 13C and 15N chemical shifts were indirectly referenced relative to 1 H according to International Union of Pure and Applied Chemistry ratios. Acquisition temperatures were 293 K for RbfA and 298 K for RimP. The complete set of assignments was deposited in the Biological Magnetic Resonance Data Bank (BMRB).
NMR data processing and analysis
All NMR data were processed with NMRPipe (46) and analyzed using National Magnetic Resonance Facility at Madison - sparky (47) or collaborative computing project for NMR (CCPNMR) (48). The propensities for the formation of regular secondary structure in both proteins were evaluated using TALOS+ (49).
Structure determination by NMR
3D structure models were generated by distance geometry calculations with simulated annealing in vacuo using the XPLOR-NIH Package (50). The nuclear Overhauser effect (NOE)–based distance restraints were extracted from the described full set of 3D edited NOESY spectra. Backbone torsion angle restraints were estimated from backbone secondary chemical shifts using the TALOS+ software package. Hydrogen bonds within α helices and between adjacent β strands were inferred from NOE pattern analysis and implemented in the refinement protocol as distance restraints between backbone amide protons HiNi and carbonyl oxygen OjCj atoms or carbonyl carbon CjOj atoms, respectively. The 10 best-fit models without NOE violations >0.5 Å and dihedral angle violations >10° were selected on the basis of XPLOR target function values. For subsequent refinement in explicit solvent using the Amber force field 99SB in GROMACS (51), each protein was embedded in a cubic box filled by a static TIP3P water model. The system was neutralized and adjusted to a salt concentration of approximately 20 mM (to approximate the experimental sample conditions) by adding an appropriate number of sodium and chloride ions at least 8 Å apart from the solute. A leap-frog algorithm was used to integrate the equations of motion, using a time step of 2 fs. Position restraints with a force constant of 1000 kcal/mol for all protein atoms were applied during all subsequent equilibration stages. Bond lengths were constrained with the linear constraints solver (52), using a normal order of 4 in the expansion of the constraint coupling matrix. The particle mesh Ewald method was used for the treatment of long-range electrostatic effects (applying fourth order for spline interpolation and a grid spacing of 1.6 Å along each axis), whereas a 9-Å cutoff was chosen for short-range van der Waals and Coulomb interactions. After initial steepest descent minimization, the system was equilibrated for 100 ps to a temperature of ca. 298 K under a canonical ensemble using Bussi thermostat with 0.1 ps of coupling time and separate temperature baths for the protein and the solvent. Subsequently, the system was relaxed to an isothermal-isobaric (NPT) ensemble until density stabilization using the Berendsen pressure coupling method prior switching to an extended ensemble pressure coupling scheme according to Parrinello-Rahman for final structure refinement. For this, 500 ps of MD (Molecular Dynamics) runs were performed under NPT ensemble at 298 K of target temperature, defining the NOE-based distance restraints as time averaged (more than 20 ps) to allow a larger conformational space to be sampled. During further 100 ps of MD, all distance restraints (NOE contacts and hydrogen bonds) were incorporated as simple harmonic potentials. The extent of restraints used for refinement is listed in table S13. Force constants for all distance and torsion angle constraints were set to 1000 kJ/mol·nm−2 and 200 kJ/mol·rad−2, respectively. A flexible SPC water model was used during final minimization by the conjugate gradient (CG) method, with a steepest descent minimization after every 500 CG steps. The resulting models were sorted by overall potential energy and validated using PDB software (http://deposit.rcsb.org/validate/), PROSA (https://prosa.services.came.sbg.ac.at/prosa.php) (53), and MolProbity (http://molprobity.biochem.duke.edu). The structural models were visualized by PyMOL (The PyMOL Molecular Graphics System, Version 2.3.0, Schrödinger LLC).
Diffusion experiments
Translational diffusion was measured using the stimulated echo NMR method with bipolar pulses, variable gradients, and selective water presaturation (modified BRUKER pulse program stebpgp1d) (54) on an 800-MHz spectrometer (see above). The total diffusion time Δ and encoding gradient duration δ were set to 220 and 4 ms, respectively; the calibrated z gradient strengths increased from 1.45 to 27.58 G/cm. Translational diffusion coefficients D were obtained by fitting selected 1H signals to a monoexponential decay function (Stejskal-Tanner)
| (1) |
where G is the applied field strength of the encoding/decoding gradients, I is the peak intensity measured at field strength G, I0 is the peak intensity at G = 0, γ is the gyromagnetic ratio of the protons (2.675·104 rad G−1 s−1), and the delays δ and Δ during which diffusion is monitored as defined by the pulse sequence were set to 4 and 220 ms, respectively.
Hydrogen exchange experiments
Proton/deuteron (H/D) exchange
For the measurement of slow proton/deuteron (H/D) exchange, a 960 μM sample of 15N-labeled RimP was diluted 1:10 in buffer solution containing 10 mM Hepes-d18, 6 mM MgCl2, and 150 mM NH4Cl, in 99% D2O (pH 7.6), and a series of eight consecutive 15N TROSY experiments were acquired 0, 32, 64, 96, 128, 213, 245, and 277 min after sample preparation. The signal intensities were fitted by monoexponential decay function using NLS (‘Non Linear equations Solver) algorithm in the R software package to derive amide H/D exchange rates for semiquantitative analysis.
CLEANEx experiment
Fast HN/H2O exchange rates (kex ∼ 0.5 to 50 s−1) were sampled using the CLEANEX (Phase Modulated CLEAN chemical EXchange Spectroscopy)–PM experiment with fast 15N-HSQC implementation (33) and semi-interleaved acquisition of three mixing times (τmix = 25, 50, 75 ms) with the reference HSQC. To derive semiquantitative kex rates, the recovery of each observed signal was fitted to
| (2) |
where Iτm is the peak intensity at mixing time τm, and I0 is the pertaining intensity in the reference HSQC; R1,app (the effective longitudinal proton relaxation rate) and kex (HN/H2O exchange rate) are derived by nonlinear optimization using R software for statistical computing. Efficient suppression of radiation damping during τm (using a weak continuous gradient) allowed to neglect the R1,app of H2O also used in the original equation (33).
Assembly factor complex preparation and vitrification
Our initial low-resolution characterization of isolated 30S subunits indicated that the region around h44 was variable and reasoned that the RbfA binding site proposed by Datta et al. (17) might be exposed. Therefore, the first sample we prepared was 30S-RbfA, as described below. Characterization of this sample indicated the presence of 30S subunits resembling the 30S assembly states that accumulate in various assembly factor deletion strains (23, 24). This suggested that isolated 30S contained conformations that might also be substrates for these factors. Accordingly, we assayed the ability of assembly factors implicated in the placement of h44 to bind the natively isolated 30S states (RbfA, RsgA, RimM, and RimP; dataset 2). In this dataset, we did not see RimM but realized that RimP was positioned adjacent to the binding site expected for RsmA, so in the third dataset, RsmA was added.
Dataset 1: 30S-RbfA
To isolate the 30S-RbfA complex, we coincubated 1 μM 30S subunits (E. coli) with 25 μM RbfA in a buffer containing 20 mM Hepes-KOH (pH 7.8), 10 mM MgCl2, 60 mM NH4CH3CO2, and 6 mM β-mercaptoethanol at 37°C for 30 min. The resulting complex was diluted 1:3 in the same buffer and subsequently plunge-frozen in liquid ethane on glow discharged Quantifoil R2/2 grids using a Vitrobot (FEI) set to 4°C and 100% humidity with a 30-s incubation and 3- to 3.5-s blot time. Grid quality and complex integrity were assayed before high-resolution data collection by screening and single-particle analysis of data collected at the CIC bioGUNE electron microscopy platform (JEOL 2200FS and UltraScan 4000 SP).
Dataset 2: 30S-RbfA-RimM-RimP-RsgA
One micromolar of 30S subunits (E. coli) were coincubated with 6 μM RbfA, 7 μM RimM, 12 μM RimP, 3 μM RsgA, and 250 μM GMPPNP in a buffer containing 20 mM Hepes-KOH (pH 7.8), 10 mM MgCl2, 60 mM NH4CH3CO2, and 6 mM β-mercaptoethanol at 37°C for 10 min. The resulting complex was diluted 3:1 (complex:buffer) in the same buffer and subsequently plunge-frozen in liquid ethane on glow discharged Quantifoil R2/2 grids using a Vitrobot (FEI) set to 4°C and 100% humidity with a 30-s incubation and 3- to 3.5-s blot time. Grid quality and complex integrity were assayed before high-resolution data collection by screening and single-particle analysis of data collected at the CIC bioGUNE electron microscopy platform (JEOL 2200FS and UltraScan 4000 SP).
Dataset 3: 30S-RbfA-RimP-RsmA
One micromolar of 30S subunits (E. coli) were coincubated with 4 μM RbfA, 4 μM RimP, and 4 μM RsmA, in a buffer containing 20 mM Hepes-KOH (pH 7.8), 10 mM MgCl2, 60 mM NH4CH3CO2, and 6 mM β-mercaptoethanol at 37°C for 10 min. The resulting complex was diluted 3:1 (complex:buffer) in the same buffer and subsequently plunge-frozen in liquid ethane on glow discharged Quantifoil R2/2 grids using a Vitrobot (FEI) set to 4°C and 100% humidity with a 30-s incubation and 3-s blot time. Grid quality and complex integrity were assayed before high-resolution data collection by screening and single-particle analysis of data collected at the CIC bioGUNE electron microscopy platform (JEOL 2200FS + UltraScan 4000 SP). In this complex, the ribosomes were isolated from a wild-type strain, and no SAM-e (S-Adenosyl methionine) substrate was present for RsmA and, therefore, represents a postmethylation complex.
Electron microscopy
Dataset 1: 30S-RbfA
Automated data acquisition (EPU software, Thermo Fisher Scientific) was performed at eBIC (Diamond Light Source, UK; EM15422; M02) with a Titan Krios microscope (FEI) at 300 kV equipped with an energy filter (zero loss) and K2 direct detector (FEI; table S4). In total, 3415 movies were collected with each movie containing 20 frames over an 8-s exposure at a magnification of ×133,333 (yielding a pixel size of 1.05 Å). The total exposure was 38.8 electrons/Å (1.94 electrons/Å per fraction), and defocus values from −1.2 to −3.0 μm were used. Dataset 1 showed good particle density.
Dataset 2: 30S-RbfA-RimM-RimP-RsgA
Automated data acquisition (EPU software, FEI) was performed at eBIC (Diamond Light Source, UK; EM-17171-3; M03) with a Titan Krios microscope (FEI) at 300 kV equipped with an energy filter (zero loss) and Falcon III direct detector (FEI; linear mode; table S4). In total, 6736 movies were collected with each movie containing 19 frames over a 0.5-s exposure at a magnification of ×129,032 (yielding a pixel size of 1.085 Å). The total exposure was 46.1 electrons/Å (2.43 electrons/Å per fraction), and defocus values from −1.0 to −2.5 μm were used. Dataset 2 showed lower particle density and general contamination throughout.
Dataset 3: 30S-RbfA-RimP-RsmA
Automated data acquisition (EPU software, FEI) was performed at eBIC (Diamond Light Source, UK; EM-17171-12; M03) with a Titan Krios microscope (FEI) at 300 kV equipped with an energy filter (zero loss) and Falcon III direct detector (FEI; linear mode; table S4). In total, 4395 movies were collected with each movie containing 27 frames over a 0.74-s exposure at a magnification of ×129,032 (yielding a pixel size of 1.085 Å). The total exposure was 42 electrons/Å (1.556 electrons/Å per fraction), and defocus values from −1.0 to −2.25 μm were used.
Image processing and structure determination
Unless otherwise stated, all image processing steps were performed within the RELION 3.0 GUI (28, 29).
Dataset 1: 30S-RbfA
Motion correction was performed with the MotionCor2-like algorithm in RELION 3.0 (55, 56) using the dose weighting and patch (5 × 5) options. Contrast transfer function (CTF) estimation for each aligned micrograph was performed using Gctf and the equiphase averaging option (57). A total of 231,521 projection images of 30S particles were picked using SPHIRE-crYOLO (SPHIRE, Sparx for High Resolution Electron Microscopy; cr, cryo; YOLO, You Only Look Once) (58). After these steps, micrographs with outlying values for total motion, defocus, or astigmatism were removed from the dataset (leaving 3369 micrographs). Initially, particles were rescaled and extracted with a pixel size of 2.19 Å (box size of 192 pixels by 192 pixels) and the dataset cleaned by using a combination of RELION Initial Model (1 class) (55), RELION 3D Classification (3 classes), and RELION 2D Classification (100 classes). Subsequently, the well-aligning particle projections (total of 141,113) were recentered and reextracted with a pixel size of 1.05 Å and refined starting from the de novo initial model using RELION 3D autorefine first without and after with a mask to generate a reconstruction at 3.02 Å (B-factor, −84) as determined by RELION postprocessing. This reconstruction was used to initiate CTF refinement (per particle defocus fitting) and Bayesian polishing (55). The polished particle projections where again subjected to 3D autorefinement (first without and then with a mask) to generate a reconstruction at 2.68 Å (B-factor, −52), as determined by RELION postprocessing. Again, the reconstruction was used to initiate CTF refinement with both per particle defocus fitting and beam tilt estimation (five beam tilt classes assigned with EPU_beamtiltclasses.py; https://github.com/dzyla/EPU_beamtiltclasses) resulting in a reconstruction at 2.61 Å (B-factor, −46; Vol-1; fig. S2) after 3D autorefinement with a mask. The data were then refined using RELION multibody refinement (body 1 = 30S body/platform and body 2 = 30S head) (25), and, subsequently, the subtracted projections (relion_flex_analyse) containing signal for 30S body were subjected to a 3D classification (no image alignment, four classes) under a mask corresponding to RbfA and h44, which showed high local resolution. Two of the four classes resulted in interpretable density (with and without RbfA) that after reverting back to the unsubtracted projections were refined to 2.69 Å (B-factor, −46; Vol-1A) and 2.96 Å (B-factor, −45; Vol-1B, state I; fig. S2) resolution, respectively. Vol-1B was multibody refined again and using the subtracted projections was subjected to a 3D classification (no image alignment, three classes) under a mask corresponding to the CDR. The single well-defined class was then finally subjected to a consensus and multibody refinement yielding state I (consensus 2.96 Å B-factor, −54). In Vol-1A, the density corresponding to RbfA was weak with respect to the surrounding regions, and therefore, the dataset was multibody refined again and using the subtracted projections was subjected to a 3D classification (no image alignment, three classes) under a mask corresponding to the RbfA region. This yielded two well-defined classes that were finally subjected to a consensus and multibody refinement yielding state E (consensus 2.82 Å B-factor, −44) and state M (consensus 2.94 Å B-factor, −42). When RELION multibody refinement was used to yield separate maps for each region, the maps were merged using phenix.combine_focused_maps and aligned to the consensus refinement for illustration purposes only. The FSC (fourier shell correlation) plots for the consensus refinement and the multibody refinements corresponding to states I, E, and M, as well as the local resolution maps for the multibody refinements, are shown in fig. S5.
Dataset 2: 30S-RbfA-RimM-RimP-RsgA
Motion correction was performed on the 6736 collected movies with the MotionCor2-like algorithm in RELION 3.0 (55, 56) using the dose weighting and patch (5 × 5) options. CTF estimation for each aligned micrograph was performed using Gctf and the equiphase averaging option (57). Micrographs with outlying values for total motion, defocus, or CTF figure of merit were removed from the dataset. A total of 200,953 projection images of 30S particles were picked using SPHIRE-crYOLO (58) and extracted. This initial dataset was cleaned using the RELION 2D Classification (100 classes), RELION Initial Model (3 classes) (55), and RELION 3D Classification (3 classes) to select particles that yielded well-defined volumes, such that 92,491 particles were retained and pooled together. These well-aligning particle projections were reextracted and recentered with a pixel size of 1.085 Å and refined starting from the de novo initial model to generate a reconstruction at 3.42 Å (B-factor, −93), as determined by RELION postprocessing. This reconstruction was used to initiate a CTF refinement (per particle defocus fitting and beam tilt estimation) and Bayesian polishing (55). The polished particle projections where again subjected to 3D autorefinement (first without and then with a mask) to generate a reconstruction at 3.06 Å (B-factor, −104), as determined by RELION postprocessing (Vol-2; fig. S3). This map showed high local resolution in the density corresponding to the regions around RsgA, uS12, and h44, and accordingly, we used 3D classification (four classes; mask covering the entire subunit) to separate the dataset into subsets, three of which yielded well-defined volumes and one which represented poorly aligning projections. The first subset showed strong well-defined density for RsgA and after a second CTF refinement (with five beam tilt classes assigned with EPU_beamtiltclasses.py; https://github.com/dzyla/EPU_beamtiltclasses) refined to a resolution of 3.00 Å (B-factor, −85; 21,573 projections), yielding state F. This final consensus refinement showed high local resolution in the head region relative to the body/platform (see fig. S3), which could not be accounted for by a global 3D classification, suggesting that the head moves independently of the body. Accordingly, data corresponding to state F were refined using RELION multibody refinement (body 1 = 30S body/platform and body 2 = 30S head) (25). The second and third subsets both showed additional density on uS12 and weak fragmented density for h44, and therefore, these two subsets were grouped and re-refined together to yield Vol-2B (3.09 Å; fig. S3). This volume was subjected to a 3D classification using a mask to focus on the 30S body/plat regions, yielding two well-defined volumes and one poorly resolved volume. Projections corresponding to the well-resolved volumes were selected and refined separately, including an additional CTF refinement and a 3D classification focused on the RimP density (three classes, no image alignment) to select a subset with strong well-defined density for RimP. These volumes were refined to 3.15 Å (Vol-2B-1; 22,735 projections) and 3.9 Å (Vol-2B-2; 9900 projections) and differed primarily with respect to the presence or absence of h44. As these two volumes had counterparts in dataset 3 in terms of their composition (Vol-2B-1 corresponds to Vol-3B-1 and Vol-2B-2 to Vol-3D-1) and because the two datasets were collected on the same microscope/detector at the same magnification (table S4), the identical subsets were merged. In the case Vol-2B-2 and Vol-3D-1, the datasets were refined together, multibody refined, and subjected to 3D classification using subtracted projections and a mask corresponding to RbfA (three classes, no alignment). This yielded two well-defined classes that were finally subjected separately to a consensus and multibody refinement yielding state C (consensus 3.78 Å B-factor, −108) and state D (consensus 4.8 Å B-factor, −130). In the case Vol-2B-1 and Vol-3B-1, the datasets were refined together, multibody refined, and subjected to 3D classification using subtracted projections and a mask corresponding to the CDR (three classes, no alignment). This yielded a well-defined class that was finally subjected to a consensus and multibody refinement yielding state A (consensus 3.59 Å B-factor, −103). When RELION multibody refinement was used to yield separate maps for each region, the maps were merged using phenix.combine_focused_maps and aligned to the consensus refinement for illustration purposes only. Although RimM was present in the sample, it was not observed bound to the 30S in any of the resulting cryo-EM maps. The FSC plots for the consensus refinement and the multibody refinements corresponding to states A, C, D, and F as well as the local resolution maps for the multibody refinements are shown in fig. S5.
Dataset 3: 30S-RbfA-RimP-RsmA
Motion correction was performed on the 4395 collected movies with the MotionCor2-like algorithm in RELION 3.0 (55, 56) using the dose weighting and patch (5 × 5) options. CTF estimation for each aligned micrograph was performed using Gctf and the equiphase averaging option (57). Micrographs with outlying values for total motion, defocus, or CTF figure of merit were removed from the dataset. A total of 406,522 projection images of 30S particles were picked using SPHIRE-crYOLO (58) and extracted. This initial dataset was cleaned using the RELION 2D Classification (100 classes) and RELION 3D Classification (4 classes) to select particles that yielded well-defined volumes, such that 156,287 particles were retained and pooled together. These well-aligning particle projections were reextracted and recentered with a pixel size of 1.085 Å and refined starting from the de novo initial model to generate a reconstruction at 3.89 Å (B-factor, −193), as determined by RELION postprocessing. This reconstruction was used to initiate a CTF refinement (per particle defocus fitting + beam tilt estimation) and Bayesian polishing (55). The polished particle projections were again subjected to 3D autorefinement (first without and then with a mask) to generate a reconstruction, Vol-3 at 3.41 Å (B-factor, −133; fig. S4). This map showed high local resolution and accordingly was subjected to two rounds of 3D classification, where the first used a mask corresponding to the entire subunit (with image alignment) and the second used a mask corresponding to CDR (no image alignment, four classes). The four classes were then refined, yielding four well-defined volumes, Vol-3A to Vol-3D (fig. S4). Volume Vol-3A showed density for RimP and RsmA and was further classified under a mask first for the CDR and then for RimP/RsmA and lastly refined to yield state B (consensus 4.05 Å B-factor, −138; fig. S4). Although the resolution decreased through these last steps, density for the CDR improved in interpretability. Vol-3B showed density for RimP (no h44) similar to volume Vol-2B-1 (fig. S3) and, therefore, after a single 3D classification using a mask for the entire subunit, was joined with projections corresponding to Vol-2B-1. The merged data were refined as described above, yielding state A (consensus 3.59 Å B-factor, −103; fig. S3). Vol-3C showed density for RbfA but was not followed further, as state E in dataset 1 was similar and of higher quality. Vol-3D showed density for RimP (and h44) similar to volume Vol-2B-2 (fig. S3) and, therefore, after a single 3D classification under a mask for RimP, was joined with projections corresponding to Vol-2B-2. The merged data were refined as described above, yielding states C and D (fig. S3). When RELION multibody refinement was used to yield separate maps for each region, the maps were merged using phenix.combine_focused_maps and aligned to the consensus refinement for illustration purposes only. The FSC plots for the consensus refinement and the multibody refinements corresponding to state B as well as the local resolution maps for the multibody refinement are shown in fig. S5.
Cryo-EM model building
As starting models, the PDB structures 4YBB (crystal structure of E. coli 30S subunit), 1QYR (crystal structure of RNA adenine dimethyltransferase), and 5NO3 (cryo-EM structure of RsgA-GDPNP) were used, while the models solved by NMR (described above) served as templates for RbfA and RimP. For refinement and model building, the cryo-EM maps originating from the RELION multibody refinement (low-pass filtered to the global resolution) were used, such that separate models for the head and body were built (containing rRNA residues C931 to G1386 and the r-proteins S3, S7, S9, S10, S13, S14, and S19 for the head domain and rRNA residues A1 to C930 and G1387 to A1542 and the r-proteins S3, S7, S9, S10, S13, S14, and S19 for the body domain of the 30S subunit, respectively). Parts of the ribosomal structure not accounted by the cryo-EM density due to their absence or local disorder were omitted from the final models. For examples, see secondary structure plots in fig. S7. Because of the quality of the cryo-EM maps obtained, some differences in the 30S subunit relative to the starting model 4YBB were observed. First, in chain S3, the C-terminal residues 207 to 212 could be added to the 30S model. In the C-terminal segment of S5, both the backbone and side chain conformation of residues G158 to L165 were modeled differently. Furthermore, a difference in the sequence register was observed for residues Tyr20-Asn43 of chain S14. In addition, for chain S19, additional density accounting for residue Gly82-Ala84 was observed. Model building was started with a preliminary rigid body refinement, followed by several cycles of manual model building using Coot (59) and real-space refinement in Phenix (43) (with secondary structure restraints and Ramachandran restraints). To combine the individual 30S head and body models into a single model, corresponding to the consensus cryo-EM map derived from the RELION 3D autorefinement, the separate models for the head and body region where rigid body fitted and few side chains of residue type Arg and Lys were at the interface were manually remodeled to avoid clashes. Note that as the 30S head in the consensus cryo-EM map shows much reduced local resolution (fig. S5), these consensus structures should be considered as a model owing to the fact that the interface does not account for the structural changes that allow the flexibility in the head. Validation statistics were derived using MolProbity as a part of the Phenix validation tools (1), and the guanidino carboxy denotation issues were resolved by in-house script (tables S4 to S11). For figure preparation, University of California San Francisco Chimera (60), ChimeraX (61), or PyMOL 2.3 (The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger LLC) were used.
Analysis of 16S rRNA sequences
Aligned rRNA sequences representing the three phylogenetic domains and two organelles (16S.T.alnfasta) were downloaded from “The Comparative RNA Web (CRW) Site.” Before using Biopython to categorize sequences by the complementarity between h28 and the 16S 3′-end (Fig. 2G), ambiguous or incomplete sequences were removed from the alignment by omitting any sequences (i) that lacked residues corresponding to the highly conserved KsgA/Dim1 methylation site in the loop of h45 (GAA 1517-1519 in E. coli or positions 8864, 8868, and 8870 in the 16S.T.alnfasta), (ii) where the two strands of h28 were not complementary (i.e., mismatch between UGA 921-923 and UCA 1393,1395-1396), (iii) where the 16S 3′-end (UCA 1532-1534) sequence include ambiguous sequence (Ns) or was completely missing (−), and (iv) that lacked sufficient information to retrieve taxonomy data.
Acknowledgments
Funding: This work was supported by grants from the European Union’s Seventh Framework Program (Marie Curie Actions; COFUND; to S.R.C. and A.S.), Marie Curie Action Career Integration Grant (PCIG14-GA-2013-632072 to P.F.), and Ministerio de Economía Y Competitividad Grant (MINECO; CTQ201782222-R to P.F. and S.R.C.). We thank MINECO for the Severo Ochoa Excellence Accreditation (SEV-2016-0644) and the proteomics platform of CIC bioGUNE for sample analysis by MALDI-TOF mass spectroscopy. We acknowledge Diamond for access and support of the cryo-EM facilities at the U.K. National Electron Bio-imaging Centre (eBIC), proposals EM171713, EM17171-12, and EM15422, funded by the Wellcome Trust, MRC, and BBSRC. Author contributions: A.S., I.I., B.O.-L., R.C., N.D., and E.A. were responsible for cloning/purification of ribosomes and proteins. A.S., T.D., J.P.L.-A., J.L.L.., T.K., P.F., and S.R.C. were involved in the structural analysis and sequence analysis. D.G.-C. was responsible for cryo-EM data collection at the CIC bioGUNE. A.S. and T.D. were responsible for NMR data collection. All authors were involved in the writing/review of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Coordinates for the 30S head, body, and consensus structures for states I, A to F, and M have been deposited in the PDB with the following accession numbers: 7AF5,7BOF, 7NAX, 7AFD, 7NAS, 7NAT, 7AFN, 7AFO, 7NAW, 7AFH, 7AFI, 7NAU, 7AFK, 7AFL, 7NAV, 7AF8, 7BOG, 7BOH, 7AFA, 7BOI, 7NAR, 7AF3, 7BOD, and 7BOE. The corresponding cryo-EM maps have been deposited in the EMDB with the following accession numbers: 11753, 12241, 12251, 11761, 12246, 12247, 11771, 11772, 12250, 11765, 11766, 12248, 11768, 11769, 12249, 11756, 12242, 12243, 11758, 12244, 12245, 11751, 12239, and 12240. The coordinates of the isolated assembly factors studied by NMR have been deposited in the PDB with the accession codes 7AFQ and 7AFR and their full NMR signal assignments in the BMRB with the accession codes 34385 and 28014. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/23/eabf7547/DC1
REFERENCES AND NOTES
- 1.Warner J. R., The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24, 437–440 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Hetman M., Slomnicki L. P., Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J. Neurochem. 148, 325–347 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farley K. I., Baserga S. J., Probing the mechanisms underlying human diseases in making ribosomes. Biochem. Soc. Trans. 44, 1035–1044 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu L., Gunasekera A. H., Mack J., Olejniczak E. T., Chovan L. E., Ruan X., Towne D. L., Lerner C. G., Fesik S. W., Solution structure and function of a conserved protein SP14. 3 encoded by an essential Streptococcus pneumoniae gene. J. Mol. Biol. 311, 593–604 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Poonam, Yennamalli R. M., Bisht G. S., Shrivastava R., Ribosomal maturation factor (RimP) is essential for survival of nontuberculous mycobacteria, Mycobacterium fortuitum under in vitro acidic stress conditions. 3 Biotech 9, 127 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maguire B. A., Inhibition of bacterial ribosome assembly: A suitable drug target? Microbiol. Mol. Biol. Rev. 73, 22–35 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stokes J. M., Brown E. D., Chemical modulators of ribosome biogenesis as biological probes. Nat. Chem. Biol. 11, 924–932 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Talkington M. W. T., Siuzdak G., Williamson J. R., An assembly landscape for the 30S ribosomal subunit. Nature 438, 628–632 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adilakshmi T., Bellur D. L., Woodson S. A., Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature 455, 1268–1272 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dutca L. M., Culver G. M., Assembly of the 5′ and 3′ minor domains of 16S ribosomal RNA as monitored by tethered probing from ribosomal protein S20. J. Mol. Biol. 376, 92–108 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulder A. M., Yoshioka C., Beck A. H., Bunner A. E., Milligan R. A., Potter C. S., Carragher B., Williamson J. R., Visualizing ribosome biogenesis: Parallel assembly pathways for the 30S subunit. Science 330, 673–677 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodson S. A., Recent insights on RNA folding mechanisms from catalytic RNA. Cell. Mol. Life Sci. 57, 796–808 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Razi A., Guarné A., Ortega J., The cryo-EM structure of YjeQ bound to the 30S subunit suggests a fidelity checkpoint function for this protein in ribosome assembly. Proc. Natl. Acad. Sci. U.S.A. 114, E3396–E3403 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Razi A., Britton R. A., Ortega J., The impact of recent improvements in cryo-electron microscopy technology on the understanding of bacterial ribosome assembly. Nucleic Acids Res. 45, 1027–1040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter A. P., Clemons W. M., Brodersen D. E., Morgan-Warren R. J., Wimberly B. T., Ramakrishnan V., Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407, 340–348 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Kaminishi T., Wilson D. N., Takemoto C., Harms J. M., Kawazoe M., Schluenzen F., Hanawa-Suetsugu K., Shirouzu M., Fucini P., Yokoyama S., A snapshot of the 30S ribosomal subunit capturing mRNA via the Shine-Dalgarno interaction. Structure 15, 289–297 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Datta P. P., Wilson D. N., Kawazoe M., Swami N. K., Kaminishi T., Sharma M. R., Booth T. M., Takemoto C., Fucini P., Yokoyama S., Agrawal R. K., Structural aspects of RbfA action during small ribosomal subunit assembly. Mol. Cell 28, 434–445 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boehringer D., O’Farrell H. C., Rife J. P., Ban N., Structural insights into methyltransferase KsgA function in 30S ribosomal subunit biogenesis. J. Biol. Chem. 287, 10453–10459 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.López-Alonso J. P., Kaminishi T., Kikuchi T., Hirata Y., Iturrioz I., Dhimole N., Schedlbauer A., Hase Y., Goto S., Kurita D., Muto A., Zhou S., Naoe C., Mills D. J., Gil-Carton D., Takemoto C., Himeno H., Fucini P., Connell S. R., RsgA couples the maturation state of the 30S ribosomal decoding center to activation of its GTPase pocket. Nucleic Acids Res. 45, 6945–6959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moazed D., Van Stolk B. J., Douthwaite S., Noller H. F., Interconversion of active and inactive 30 S ribosomal subunits is accompanied by a conformational change in the decoding region of 16 S rRNA. J. Mol. Biol. 191, 483–493 (1986). [DOI] [PubMed] [Google Scholar]
- 21.Zamir A., Miskin R., Elson D., Interconversions between inactive and active forms of ribosomal subunits. FEBS Lett. 3, 85–88 (1969). [DOI] [PubMed] [Google Scholar]
- 22.McGinnis J. L., Liu Q., Lavender C. A., Devaraj A., McClory S. P., Fredrick K., Weeks K. M., In-cell SHAPE reveals that free 30S ribosome subunits are in the inactive state. Proc. Natl. Acad. Sci. U.S.A. 112, 2425–2430 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo Q., Goto S., Chen Y., Feng B., Xu Y., Muto A., Himeno H., Deng H., Lei J., Gao N., Dissecting the in vivo assembly of the 30S ribosomal subunit reveals the role of RimM and general features of the assembly process. Nucleic Acids Res. 41, 2609–2620 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leong V., Kent M., Jomaa A., Ortega J., Escherichia coli rimM and yjeQ null strains accumulate immature 30S subunits of similar structure and protein complement. RNA 19, 789–802 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakane T., Kimanius D., Lindahl E., Scheres S. H., Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. eLife 7, e36861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohan S., Donohue J. P., Noller H. F., Molecular mechanics of 30S subunit head rotation. Proc. Natl. Acad. Sci. U.S.A. 111, 13325–13330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ameismeier M., Cheng J., Berninghausen O., Beckmann R., Visualizing late states of human 40S ribosomal subunit maturation. Nature 558, 249–253 (2018). [DOI] [PubMed] [Google Scholar]
- 28.O’Farrell H. C., Scarsdale J. N., Rife J. P., Crystal structure of KsgA, a universally conserved rRNA adenine dimethyltransferase in Escherichia coli. J. Mol. Biol. 339, 337–353 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Chu T., Weng X., Law C. O. K., Kong H.-K., Lau J., Li S., Pham H. Q., Wang R., Zhang L., Kao R. Y. T., Lau K.-F., Ngo J. C. K., Lau T. C. K., The ribosomal maturation factor P from Mycobacterium smegmatis facilitates the ribosomal biogenesis by binding to the small ribosomal protein S12. J. Biol. Chem. 294, 372–378 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sashital D. G., Greeman C. A., Lyumkis D., Potter C. S., Carragher B., Williamson J. R., A combined quantitative mass spectrometry and electron microscopy analysis of ribosomal 30S subunit assembly in E. coli. eLife 3, e04491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bunner A. E., Nord S., Wikström P. M., Williamson J. R., The effect of ribosome assembly cofactors on in vitro 30S subunit reconstitution. J. Mol. Biol. 398, 1–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma I. M., Woodson S. A., RbfA and IF3 couple ribosome biogenesis and translation initiation to increase stress tolerance. Nucleic Acids Res. 48, 359–372 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hwang T.-L., van Zijl P. C. M., Mori S., Accurate quantitation of water-amide proton exchange rates using the phase-modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme. J. Biomol. NMR 11, 221–226 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Schreiber G., Haran G., Zhou H.-X., Fundamental aspects of protein-protein association kinetics. Chem. Rev. 109, 839–860 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y. J., Swapna G. V. T., Rajan P. K., Ke H., Xia B., Shukla K., Inouye M., Montelione G. T., Solution NMR structure of ribosome-binding factor A (RbfA), a cold-shock adaptation protein from Escherichia coli. J. Mol. Biol. 327, 521–536 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Williamson M. P., Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Jones P. G., Inouye M., RbfA, a 30S ribosomal binding factor, is a cold-shock protein whose absence triggers the cold-shock response. Mol. Microbiol. 21, 1207–1218 (1996). [DOI] [PubMed] [Google Scholar]
- 38.Dammel C. S., Noller H. F., Suppression of a cold-sensitive mutation in 16S rRNA by overexpression of a novel ribosome-binding factor, RbfA. Genes Dev. 9, 626–637 (1995). [DOI] [PubMed] [Google Scholar]
- 39.Rozanska A., Richter-Dennerlein R., Rorbach J., Gao F., Lewis R. J., Chrzanowska-Lightowlers Z. M., Lightowlers R. N., The human RNA-binding protein RBFA promotes the maturation of the mitochondrial ribosome. Biochem. J. 474, 2145–2158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scaiola A., Peña C., Weisser M., Böhringer D., Leibundgut M., Klingauf-Nerurkar P., Gerhardy S., Panse V. G., Ban N., Structure of a eukaryotic cytoplasmic pre-40S ribosomal subunit. EMBO J. 37, e98499 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goto S., Kato S., Kimura T., Muto A., Himeno H., RsgA releases RbfA from 30S ribosome during a late stage of ribosome biosynthesis. EMBO J. 30, 104–114 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jahagirdar D., Jha V., Basu K., Gomez-Blanco J., Vargas J., Ortega J., Alternative conformations and motions adopted by 30s ribosomal subunits visualized by cryo-electron microscopy. bioRxiv 2020.03.21.001677 , (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liebschner D., Afonine P. V., Baker M. L., Bunkóczi G., Chen V. B., Croll T. I., Hintze B., Hung L. W., Jain S., McCoy A. J., Moriarty N. W., Oeffner R. D., Poon B. K., Prisant M. G., Read R. J., Richardson J. S., Richardson D. C., Sammito M. D., Sobolev O. V., Stockwell D. H., Terwilliger T. C., Urzhumtsev A. G., Videau L. L., Williams C. J., Adams P. D., Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Struct. Biol. 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geertsma E. R., Dutzler R., A versatile and efficient high-throughput cloning tool for structural biology. Biochemistry 50, 3272–3278 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Schedlbauer A., Ochoa-Lizarralde B., Iturrioz I., Çapuni R., Diercks T., de Astigarraga E., Fucini P., Connell S. R., Backbone and sidechain NMR assignments for the ribosome maturation factor RimP from Escherichia coli. Biomol. NMR Assign. 14, 189–193 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 47.Lee W., Tonelli M., Markley J. L., NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skinner S. P., Fogh R. H., Boucher W., Ragan T. J., Mureddu L. G., Vuister G. W., CcpNmr AnalysisAssign: A flexible platform for integrated NMR analysis. J. Biomol. NMR 66, 111–124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen Y., Delaglio F., Cornilescu G., Bax A., TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwieters C. D., Bermejo G. A., Clore G. M., Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Sci. 27, 26–40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015). [Google Scholar]
- 52.Hess B., P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Wiederstein M., Sippl M. J., ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang X., Li C. G., Ye C.-H., Liu M.-L., Determination of molecular self-diffusion coefficient using multiple spin-echo NMR spectroscopy with removal of convection and background gradient artifacts. Anal. Chem. 73, 3528–3534 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Zivanov J., Nakane T., Forsberg B. O., Kimanius D., Hagen W. J. H., Lindahl E., Scheres S. H. W., New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng S. Q., Palovcak E., Armache J. P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang K., Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wagner T., Merino F., Stabrin M., Moriya T., Antoni C., Apelbaum A., Hagel P., Sitsel O., Raisch T., Prumbaum D., Quentin D., Roderer D., Tacke S., Siebolds B., Schubert E., Shaikh T. R., Lill P., Gatsogiannis C., Raunser S., SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2, 218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. Sec. D Struct. Biol. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera––A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., Ferrin T. E., UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan Y. Z., Baldwin P. R., Davis J. H., Williamson J. R., Potter C. S., Carragher B., Lyumkis D., Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A., Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grishin N. V., KH domain: One motif, two folds. Nucleic Acids Res. 29, 638–643 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/23/eabf7547/DC1