

Abstract
Mantle cell lymphoma (MCL) is a rare subtype of non‐Hodgkin's lymphoma, which is characterized by overexpression of cyclin D1. Although novel drugs, such as ibrutinib, show promising clinical outcomes, relapsed MCL often acquires drug resistance. Therefore, alternative approaches for refractory and relapsed MCL are needed. Here, we examined whether a novel inhibitor of enhancer of zeste homologs 1 and 2 (EZH1/2), OR‐S1 (a close analog of the clinical‐stage compound valemetostat), had an antitumor effect on MCL cells. In an ibrutinib‐resistant MCL patient–derived xenograft (PDX) mouse model, OR‐S1 treatment by oral administration significantly inhibited MCL tumor growth, whereas ibrutinib did not. In vitro growth assays showed that compared with an established EZH2‐specific inhibitor GSK126, OR‐S1 had a marked antitumor effect on MCL cell lines. Furthermore, comprehensive gene expression analysis was performed using OR‐S1–sensitive or insensitive MCL cell lines and showed that OR‐S1 treatment modulated B‐cell activation, differentiation, and cell cycle. In addition, we identified Cyclin Dependent Kinase Inhibitor 1C (CDKN1C, also known as p57, KIP2), which contributes to cell cycle arrest, as a direct target of EZH1/2 and showed that its expression influenced MCL cell proliferation. These results suggest that EZH1/2 may be a potential novel target for the treatment of aggressive ibrutinib‐resistant MCL via CDKN1C‐mediated cell cycle arrest.
Keywords: CDKN1C, EZH1/2, ibrutinib, mantle cell lymphoma
This study reveals that oral administration of OR‐S1 significantly suppressed tumor growth in an ibrutinib‐resistant mantle cell lymphoma (MCL) patient–derived xenograft mouse model. OR‐S1 induces cell cycle arrest via the upregulation of CDKN1C. CDKN1C expression was directly regulated by enhancer of zeste homologs 1 and 2 (EZH1/2).
1. INTRODUCTION
Mantle cell lymphoma (MCL) is a malignancy with poor prognosis and comprises about 3% of non‐Hodgkin's lymphoma (NHL). 1 In the majority of cases, the molecular mechanism initiating MCL is the t(11:14)(q13;q32) translocation resulting in overexpression of cyclin D1 driven by the immunoglobulin heavy chain locus. 2 , 3 Subsequently, secondary genetic alterations in pathways, such as CDKN2A/CDK4/RB1 and ARF/MDM2/p53, occur in MCL leading to tumor growth, as in other cancers. 2 , 3
R‐CHOP, a standard chemotherapy for B‐cell lymphoma, is commonly selected as first‐line MCL treatment; however, many MCL patients eventually relapse. Several new drugs such as ibrutinib have been approved for MCL treatment. In particular, ibrutinib, an inhibitor of Bruton's tyrosine kinase (BTK), is a drug that is effective in inducing antitumor effects on chronic lymphocytic leukemia and MCL cells, 4 , 5 , 6 and the combination of ibrutinib with R‐CHOP has shown a synergistic effect in NHL patients. 7 However, almost 30% of MCL patients are resistant to ibrutinib, and postibrutinib outcomes in patients with MCL are poor. 8 , 9 The C481S mutation at the ibrutinib‐binding site of BTK enhances BTK and protein kinase B activation and tissue‐specific proliferation of ibrutinib‐resistant MCL cells. 10 These findings accelerated the search for drugs effective on ibrutinib‐resistant MCL; nevertheless, of those with potential, none that were uniquely successful in the postibrutinib setting were identified. 9
As it is becoming clear that dysregulation of a variety of epigenetic mechanisms is associated with hematological malignancy including lymphoma, drugs targeting epigenetic factors have been developed extensively. 11 Enhancer of zeste homologs 1 and 2 (EZH1/2), catalytic components of polycomb repressive complex 2 (PRC2), catalyze histone H3 lysine 27 trimethylation (H3K27me3). Ezh1/2 is reported to play important roles in hematopoietic stem cell (HSC) function. Previous analysis of Ezh1‐KO mice suggests that deletion of Ezh1 increases cellular senescence in HSC and results in bome marrow (BM) failure. 12 Ezh2 overexpression in HSCs prevents exhaustion of the long‐term repopulating potential of HSCs during serial transplantation. 13 In the context of B cells, Ezh2 is essential for B‐cell development and required for germinal center formation, 14 , 15 , 16 and dysregulation of Ezh2 is associated with B‐cell tumorigenesis. The gain‐of‐function mutation EZH2Y641, which occurs most frequently in follicular lymphoma and diffuse large B‐cell lymphoma (DLBCL), has been reported to induce DLBCL‐like disease in cooperation with BCL2 in mice 15 , 17 and, to target this mutation, a number of EZH2‐specific inhibitors, including GSK126, have been developed. 18 , 19 , 20 , 21 , 22 EZH2 is overexpressed in proliferating MCL cells. 23 , 24 , 25 Some reports have shown that the microRNA miR26A1 negatively regulates EZH2 expression and that miR26A1 expression is suppressed by DNA hypermethylation in MCL. 26 , 27 , 28 In addition, the complementary role of EZH1 in the epigenetic activity of EZH2 has been demonstrated in hematological malignancies. 29 , 30 , 31 Taken together, these findings indicate that inhibition of both EZH1 and EZH2 enzymatic activity may be effective in treating aggressive MCL.
In this study, we assessed the effect of a novel EZH1/2 dual inhibitor OR‐S1, a close analog of valemetostat, also known as DS‐3201 or (R)‐OR‐S2, 32 on MCL tumor growth. Oral treatment with OR‐S1 suppressed ibrutinib‐resistant MCL tumor growth in a patient‐derived xenograft (PDX) mouse model. Compared with an established EZH2‐specific inhibitor GSK126, an over 10‐fold lower dose of OR‐S1 was required for a 50% reduction in proliferation (half maximal growth inhibition; GI50) of MCL cell lines. Furthermore, our findings indicated that direct upregulation of Cyclin Dependent Kinase Inhibitor 1C (CDKN1C) expression by OR‐S1 negatively affects MCL cell proliferation. Thus, dual inhibition of EZH1 and EZH2 may provide a new therapeutic strategy for MCL.
2. MATERIALS AND METHODS
2.1. Compounds
The synthesis and characterization of OR‐S1 (Daiichi Sankyo) are described in a Patent Cooperation Treaty application (publication number: WO/2015/141616) on the World Intellectual Property Organization website (https://patentscope.wipo.int/).
2.2. Human MCL samples
Fresh heparinized blood was obtained from patients at the National Cancer Center Hospital (Tokyo, Japan) with informed consent and institutional review board approval. Mononuclear cells from blood were purified in accordance with the Ficoll manufacturer's instructions.
2.3. Cell lines
Human MCL cell lines Mino, JeKo‐1, and REC‐1 were purchased from the American Type Culture Collection (ATCC) and cultured according to the instructions provided. For growth assays, 1 × 105 cells of each cell line were initially cultured in 24‐well plates and then treated with various concentrations of OR‐S1 or GSK126, ranging in concentration from 3000 to 0.3 nmol/L or 3000 to 100 nmol/L, respectively. Absolute cell number was evaluated for up to 15 days with two or three cell passages.
2.4. Patient and mouse xenograft experiments
A patient with MCL who achieved partial response (PR) after eight cycles of R‐CHOP experienced progressive disease (PD) after 1 year and was treated with four courses of bendamustine. The patient experienced PD again and was treated with ibrutinib for 5 months, but again experienced PD. The BTK C481S mutation was not detected in a peripheral blood sample from the patient after ibrutinib failure. The primary MCL cells were transplanted via subcutaneous injection into 6‐week‐old SCID‐Beige (SCIDbg) mice (CB17.Cg‐Prkdc scid Lyst bg‐J/CrlCrlj; Charles River). Studies were approved by the Institutional Review Boards of the National Cancer Center and conducted in accordance with the Declaration of Helsinki.
2.5. Statistical analysis
Unpaired two‐tailed Student's t‐tests were used for comparisons.
Details of the materials and methods can be found in the Supplementary Files.
3. RESULTS
3.1. OR‐S1 has an antitumor effect on MCL cells
OR‐S1 (Figure 1A) is known to strongly and selectively inhibit the methyltransferase activity of both EZH1 and EZH2. 32 To examine the effect of OR‐S1 on MCL, we transplanted patient‐derived MCL tumor into immunodeficient mice to create a PDX model. The donated sample came from a patient refractory to ibrutinib. One week after transplantation, ibrutinib or OR‐S1 were orally administered twice a day for 3 weeks (Figure 1B). In accordance with the clinical findings, treatment with ibrutinib did not inhibit tumor in mice (Figure 1C, left). However, treatment with OR‐S1 significantly suppressed the tumor growth (Figure 1C, right), and all OR‐S1–treated mice survived to the end of the study period. Additionally, an in vitro growth inhibition assay suggested that the EZH2 inhibitor GSK126 was less effective than OR‐S1 (Figure S1A). Thus, dual inhibition of EZH1 and EZH2 was an effective treatment in vivo for an MCL tumor resistant to ibrutinib.
FIGURE 1.
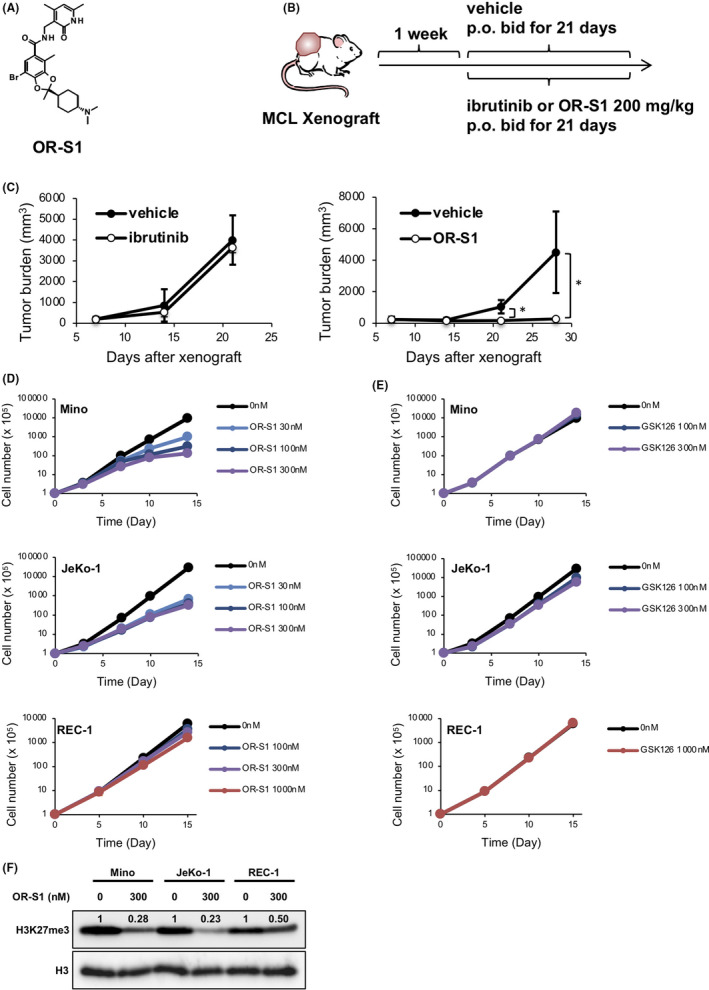
OR‐S1 is an effective suppressor of mantle cell lymphoma (MCL) tumor growth in vitro and in vivo. A, The chemical structure of OR‐S1. B, Protocol for the in vivo study of OR‐S1 efficacy of using an MCL patient–derived xenograft (PDX) mouse model. A patient‐derived tumor was xenografted into SCIDbg mice, and after 1 wk vehicle control or 200 mg/kg of ibrutinib or OR‐S1 was orally administered twice a day for 3 wk. C, Tumor burden of mice treated with ibrutinib (left) or OR‐S1 (right) compared with vehicle control. When tumor volumes exceeded 2000 mm3, the mice were euthanized. Data represent the mean of triplicates ± SD. *P <.05. D, E, Growth curves of three MCL cell lines (Mino, JeKo‐1, and REC‐1) treated with OR‐S1 (D) or GSK126 (E). Cell number was calculated on days 3, 7, 10, and 14 (Mino and JeKo‐1), or days 5, 10, and 15 (REC‐1). F, Western blotting for H3 lysine 27 trimethylation (H3K27me3). Cells were treated with vehicle alone or 300 nmol/L OR‐S1 for 5 d, and total histones were extracted in 0.4 N HCl. Band intensity was measured by ImageJ
Because of the direct effect of ibrutinib on the survival of MCL cells, ibrutinib sharply suppressed the cell growth of the three MCL cell lines (Figure S1B). EZH2 represents a promising therapeutic target in multiple cancers, and its inhibitors have already been established. 33 To compare the efficacy of OR‐S1 with that of an EZH2‐specific inhibitor, we treated three MCL cell lines (Mino, JeKo‐1, and REC‐1) with OR‐S1 or GSK126. OR‐S1 at concentrations below 100 nmol/L sharply suppressed the growth of Mino and JeKo‐1, whereas OR‐S1 concentrations above 100 nmol/L were required to inhibit REC‐1 growth (Figures 1D and S1C). On the other hand, more than 1000 nmol/L GSK126 was required to achieve GI50 of Mino and REC‐1 cells, which was not reached until day 7 after the beginning of treatment, whereas approximately 300 nmol/L GSK126 was required to achieve GI50 of Jeko‐1 cells (Figures 1E and S1D). To analyze the effect of OR‐S1 on the level of H3K27me3, we performed Western blotting and showed that prior to treatment with OR‐S1, H3K27me3 was abundant in all three cell lines, and that the level of H3K27me3 in Mino and JeKo‐1 cells decreased to 28 and 23% of those of their respective untreated controls after OR‐S1 exposure; however, the level of H3K27me3 in REC‐1 cells decreased to only approximately 50% after exposure to OR‐S1 (Figure 1E). The inhibition of H3K27me3 by OR‐S1 was stronger than that by GSK‐126 (Figure S1E). These results suggested that EZH1/2 dual inhibition was effective in inhibiting MCL cell proliferation, and that Mino and JeKo‐1 were OR‐S1–sensitive cell lines, while REC‐1 was an OR‐S1–insensitive cell line.
3.2. Altering gene expression in MCL cells treated with OR‐S1
To investigate molecular mechanisms underlying the effect of OR‐S1 on MCL cell viability, we isolated mRNA from the three MCL cell lines treated with vehicle control, 300 nmol/L, or 1000 nmol/L OR‐S1 for 5 days and 7 days, and performed RNA‐seq (Gene Expression Omnibus [GEO] accession: GSE123518). Gene Set Enrichment Analysis (GSEA) analysis showed that expression of PRC2 target genes or H3K27me3‐related genes in all three cell lines significantly changed after exposure to OR‐S1 (Figure S2A, Table S1). Gene expression profiling showed that on day 5 and day 7 after treatment with 300 nmol/L or 1000 nmol/L OR‐S1, the number of over‐2‐fold differentially expressed genes (DEGs) between vehicle‐treated and each concentration of OR‐S1–treated cells was much higher for the two OR‐S1–sensitive MCL cell lines than for the REC‐1 line (Figure 2A). Notably, upregulated genes accounted for over three quarters of the DEGs in both Mino and JeKo‐1 (Figure 2A). We extracted common DEGs in both Mino and JeKo‐1 treated with each concentration of OR‐S1, and 130 genes were identified (Figure 2B and Table S1). Of the 130 genes, we confirmed that 41 genes were known targets of EZH2, which were identified from ENCODE ChIP‐Seq data (Figure 2B table and Table S2). These results indicated that the transcriptional repressive activity of EZH was inhibited by OR‐S1 treatment.
FIGURE 2.
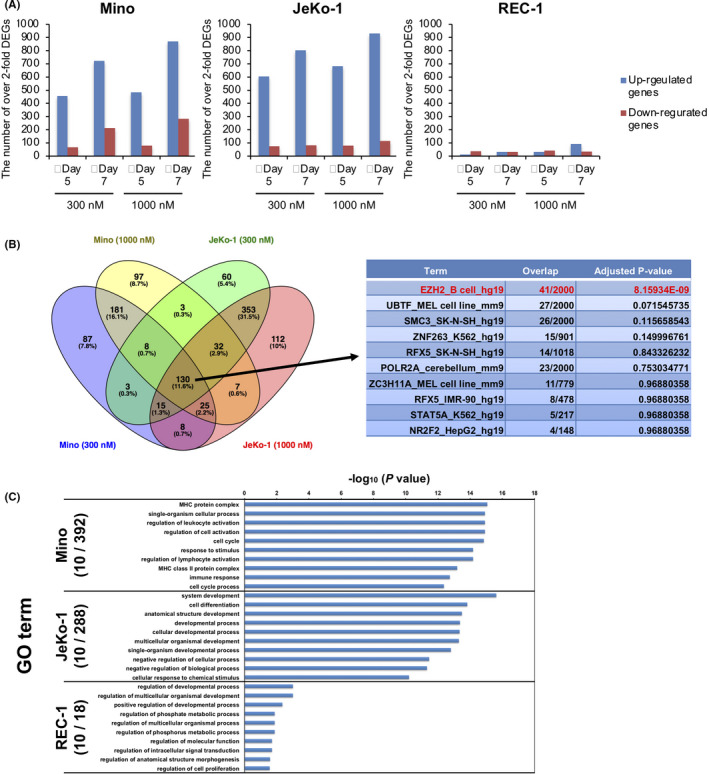
A, The number of over‐2‐fold differentially expressed genes (DEGs) in each of the mantle cell lymphoma (MCL) cell lines on day 5 and day 7 after treatment with 300 nmol/L or 1000 nmol/L OR‐S1. RNA‐seq data were deposited in GEO (GEO accession: GSE123518). B, Candidate genes directly related to sensitivity to OR‐S1. The left Venn diagram represents the number of overlapping DEGs in 300 nmol/L and 1000 nmol/L OR‐S1–treated Mino and JeKo‐1 on day 5. The right table shows Enrichr analysis of the ENCODE ChIP‐seq database using 130 common DEGs in all samples. C, The top 10 gene ontology (GO) analysis terms in which over‐2‐fold DEGs are enriched in each of the MCL cell lines on day 5 of treatment with 300 nmol/L OR‐S1. The enriched GO terms obtained from GO analysis and corresponding P‐values are visualized as a bar graph
Next, to identify significant gene sets modulated by EZH1/2 dual inhibition in each of the MCL cell lines, we investigated gene ontology (GO) terms associated with the over‐2‐fold DEGs. On day 5 after treatment initiation, 392 and 288 GO terms were correlated with the DEGs in Mino and JeKo‐1, respectively, although only 18 GO terms were correlated with the DEGs in REC‐1. When focusing on the top ten in the lists of significant GO terms obtained from the DEGs in the OR‐S1–sensitive MCL cell lines, we found that those from Mino were mainly composed of GO terms associated with immune response, cell activation, and cell cycle, while those from JeKo‐1 were mainly composed of GO terms associated with cellular development and differentiation (Figure 2C). The list from REC‐1 was composed of multiple pathways and had lower correlation values than those of Mino and JeKo‐1. These results indicated that OR‐S1 induced B‐cell activation, differentiation, and cell cycle.
3.3. OR‐S1 induces differentiation marked by CD138 expression on MCL cells
EZH2 regulates B‐cell development and is required for germinal center formation. 14 , 15 , 16 To investigate the effect of OR‐S1 on MCL cell differentiation, CD138 mRNA expression, which is a hallmark of plasma cells, was analyzed by quantitative PCR. Exposure to OR‐S1 suppressed increases in the level of CD138 mRNA in REC‐1, but induced over‐2‐ and 3‐fold increases in its level in Mino and JeKo‐1, respectively (Figure S2B). There were obvious differences in expression of B‐cell– and plasma cell–associated genes (CD20, CD79A, PAX5, OCT2, BOB1, CD38, PRDM1, and IRF4) in Mino cells, but not in REC‐1 cells (Figure S2C). Furthermore, flow cytometric analysis showed that the CD138 protein expression level was also elevated in Mino and JeKo‐1, but not in REC‐1 (Figure S2B). To validate the effect on B‐cell differentiation in vivo, we administered OR‐S1 to ibrutinib‐resistant MCL‐PDX model mice for 7 days and then, using quantitative PCR, analyzed CD138 expression in the harvested tumors (Figure S2D). As expected, the CD138 expression level in the tumors was significantly upregulated in mice treated with OR‐S1, compared with those treated with vehicle control (Figure S2D). These results showed that OR‐S1 activated the terminal B‐cell differentiation program.
3.4. OR‐S1 induces cell cycle arrest via the upregulation of CDKN1C expression
EZH2 inhibition is known to promote cell cycle arrest in lymphoma cell lines. 18 , 20 To examine whether OR‐S1 induces cell cycle arrest in MCL cell lines, we performed cell cycle analysis by propidium iodide (PI) staining. Cell cycle progression of Mino and JeKo‐1 was arrested by OR‐S1 treatment, with an increase in the number of cells in the G1/G0 phase, whereas REC‐1 was unaffected (Figure 3A). We next focused on the GO gene set related to cell cycle arrest. We identified CDKN1C as especially upregulated in the OR‐S1–treated Mino and JeKo‐1 MCL cell lines (Figure 3B). CDKN1C is known to be a direct target of EZH2 and is downregulated in breast cancer cells. 34 , 35 , 36 The time‐dependent induction of CDKN1C expression was confirmed in Mino and JeKo‐1 by quantitative PCR, and protein expression was confirmed by Western blotting of a total cell lysate from JeKo‐1 treated with OR‐S1 (Figure 3C‐D). We also assessed the expression of CDKN2A, a well‐known target of EZH2 in B cells, 20 , 21 , 37 , 38 but the level of the CDKN2A expression in OR‐S1–treated MCL cell lines versus controls was either unchanged or undetected (Figure S3). Next, we administered OR‐S1 or vehicle to ibrutinib‐resistant MCL‐PDX mice and isolated total RNA from the MCL tumors (Figure S2C) for CDKN1C expression analysis. Compared with the tumor cells exposed to vehicle alone, OR‐S1 significantly induced CDKN1C expression (Figure 3E).
FIGURE 3.
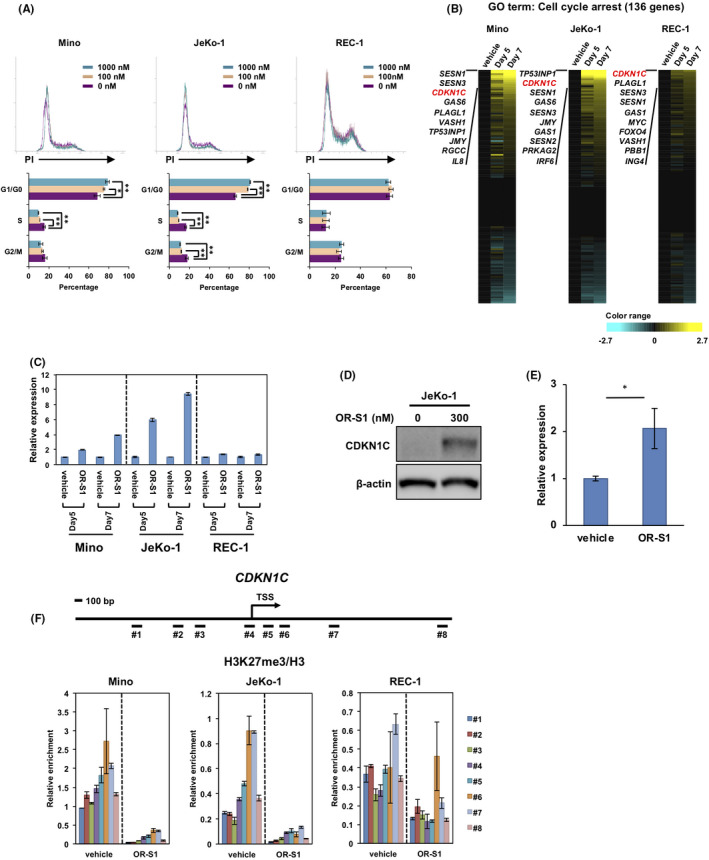
Treatment of OR‐S1 induces cell cycle arrest in mantle cell lymphoma (MCL) cells. A, Propidium iodide (PI) staining for cell cycle analysis. Cells were treated with vehicle alone or OR‐S1 (100 nmol/L or 1000 nmol/L) for 4 d. Representative flow cytometric analyses (upper panel) are shown of three independent experiments, and histogram data (lower panel) represent the mean percentage of cells ± SD in each cell cycle phase. *P < .05, **P < .01. B, Heat map represents log‐transformed relative expression values of 136 genes associated with the gene ontology (GO) term “cell cycle arrest” in three MCL cell lines treated with vehicle alone or 300 nmol/L OR‐S1 for 5 and 7 d. Yellow indicates upregulation and blue indicates downregulation of gene expression. C, Quantitative PCR validation of the CDKN1C expression found by RNA‐seq. Cells were treated with vehicle alone or 300 nmol/L OR‐S1. CDKN1C expression levels were normalized to GAPDH expression, and the relative expression level of each vehicle control–treated MCL cell line was defined as 1. Data represent the mean of triplicates ± SD. D, Protein expression of CDKN1C evaluated by Western blotting. Lysates were obtained from vehicle control– or 300 nmol/L OR‐S1–treated JeKo‐1. β‐actin was used as a loading control. E, Quantitative PCR for the expression of CDKN1C in MCL patient–derived xenograft (PDX) tumors. Tumors were obtained from mice treated with vehicle control or OR‐S1 (n = 3), and total RNA was purified. Relative CDKN1C expression was calculated as the ratio of CDKN1C to GAPDH expression. Data represent the mean of triplicates ± SD. F, ChIP assay at the CDKN1C locus. Fragmented DNAs (#1‐#8) were obtained from vehicle control– or 300 nmol/L OR‐S1–treated MCL cell lines. The levels of H3 lysine 27 trimethylation (H3K27me3) were normalized to total H3. Data represent the mean of triplicates ± SD
To confirm that the CDKN1C expression was directly regulated by EZH1/2 in MCL cells, we performed a ChIP assay for H3K27me3 at the CDKN1C locus. Around the transcription start site (TSS) of CDKN1C, peaks of H3K27me3 were dramatically decreased by OR‐S1 treatment in the two OR‐S1–sensitive MCL cell lines but less affected in REC‐1 (Figure 3F). These results indicated that EZH1/2 dual inhibition induces cell cycle arrest with direct upregulation of CDKN1C expression.
3.5. CDKN1C expression is responsible for MCL tumor growth
To investigate whether CDKN1C is responsible for growth inhibition of MCL cells, we generated a CDKN1C knockout version of JeKo‐1 by CRISPR/CAS9 editing and treated it with OR‐S1 (Figure 4A). CDKN1C‐knockout JeKo‐1 showed a slight but significant OR‐S1 resistance (Figure 4B). Then, to elucidate whether upregulation of CDKN1C inhibits MCL cell proliferation, we transduced CDKN1C into REC‐1 using a retroviral vector. Enforced CDKN1C expression in REC‐1 caused significant growth inhibition (Figure 4C).
FIGURE 4.
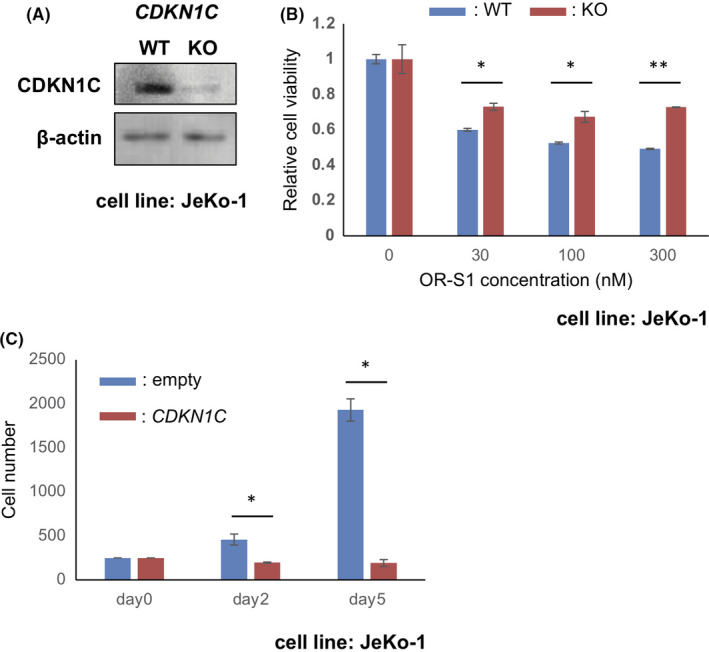
The influence of CDKN1C expression on mantle cell lymphoma (MCL) cell proliferation. A, Western blotting confirmation of loss of CDKN1C in the CRISPR/Cas9 CDKN1C knockout of JeKo‐1. Lysates were obtained from control (WT) or CDKN1C‐knockout (KO) JeKo‐1 that had been exposed to 300 nmol/L OR‐S1 for 5 d. β‐actin was used as a loading control. B, Partial acquisition of resistance to OR‐S1 in CDKN1C‐knockout JeKo‐1. Control (WT) and CDKN1C‐knockout (KO) JeKo‐1 were treated with vehicle or several concentrations of OR‐S1 (30, 100, and 300 nmol/L) for 4 d. Data are shown as relative cell viability (the mean of triplicates ± SD) compared with each vehicle control defined as 1. *P < .05, **P < .01. C, Inhibitory effect of enforced CDKN1C expression on REC‐1 cell proliferation. CDKN1C was overexpressed in REC‐1 using retroviral transduction. The stable transfectant was cultured until day 5 (counted on day 2 and day 5). Data represent the mean of triplicates ± SD. *P < .05
Finally, a previous report has shown that CDKN1C inhibits cyclin‐CDK complex activity, which in turn negatively affects cell cycle progression via the Retinoblastoma (RB)‐E2F pathway. 39 To investigate whether EZH dual inhibition modulates cell cycle machinery, we analyzed phosphorylation of RB as a measure of its activation and expression of cyclin D1 in OR‐S1–treated cells. In Mino cells, 300 nmol/L OR‐S1 significantly suppressed phosphorylation of RB at S780 but not at S807/811 (Figure 5A). In JaKo‐1 cells, OR‐S1 suppressed phosphorylation of RB at both S780 and S807/811. On the other hand, cyclin D1 expression was not associated with the MCL growth inhibition caused by exposure to OR‐S1 (Figure 5A). REC‐1 required a much higher dose of OR‐S1 for suppression of RB phosphorylation. GSK126 also induced suppression of RB phosphorylation in Mino and JeKo‐1, but a higher dose was required. Collectively, these results indicated that EZH1/2 dual inhibition induced cell cycle arrest via direct upregulation of CDKN1C expression to inhibit tumor cell proliferation (Figure 5B).
FIGURE 5.
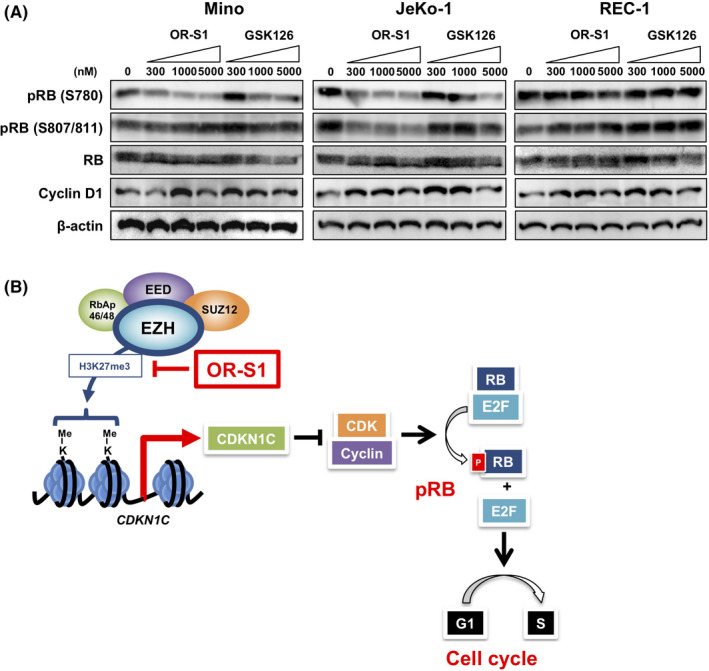
Treatment with OR‐S1 leads to inhibition of Retinoblastoma (RB) phosphorylation in patient‐derived xenograft (MCL) cells. A, Western blotting for levels of RB phosphorylation (at S780 and S807/811) and cyclin D1 expression. Cells were treated with vehicle control, GSK126, or OR‐S1 at three different concentrations (300, 1000, and 5000 nmol/L) for 5 d and then lysed for analysis. β‐actin was used as a loading control. pRB, phosphorylated RB. B, Model showing the molecular mechanisms underlying the efficacy of OR‐S1 as a growth inhibitor of MCL cells. Findings in this study are highlighted in red font, a bar, and an arrow
4. DISCUSSION
In this study, we examined whether EZH1/2 was a suitable target for MCL therapy. We showed the safety of continuous oral administration of OR‐S1 using an MCL‐PDX model, and significant inhibition of tumor growth in mice, with no lethal side effects during the study period. We tried developing MCL‐PDX models derived from several MCL tumor samples. Only one tumor sample, derived from an MCL patient who acquired ibrutinib resistance, was transplantable. We administered ibrutinib to the MCL‐PDX mice and confirmed that tumor growth was not suppressed. Our in vivo study suggests that OR‐S1 overcomes the resistance of MCL cells to ibrutinib in a BTK‐independent manner.
OR‐S1 strongly decreases the level of H3K27me3 via inhibition of both EZH1 and EZH2 activity. 32 Our in vitro assays showed that OR‐S1 sharply inhibited cell proliferation, which was accompanied by cell cycle arrest and B‐cell differentiation. Comparison of growth inhibition caused by OR‐S1 and GSK126 showed that inhibition of both EZH1 and EZH2 by OR‐S1 was over 10‐fold more effective than inhibition of EZH2 alone with GSK126 in Mino and JeKo‐1 (although each had GI50 values of <100 nmol/L). These results indicate that cooperative activity of EZH1 and EZH2 is necessary for the viability of MCL cells. Although the therapeutic potential of EZH2 as a molecular target is well known, that of EZH1 has not been demonstrated in detail. In contrast to EZH2, expression of EZH1 is low in pro‐B and pre‐B cells but increased in mature and recirculating B cells including naïve B cells (the origin of MCL). 14 The present study provides evidence of the importance not only of EZH2 but also of EZH1 as epigenetic regulators in MCL tumor growth.
OR‐S1 was less effective in inhibiting the growth of REC‐1 and in lowering the level of H3K27me3 in REC‐1 than in Mino and JeKo‐1. This result indicates that REC‐1 had acquired a mechanism that made it insensitive to OR‐S1. We suggest three possible explanations for the relative insensitivity of REC‐1 to OR‐S1.
First, REC‐1 might have acquired a drug efflux mechanism that reduces the nuclear concentration of OR‐S1 or mutations in EZH1/2 that results in resistance to OR‐S1. A recent report has shown differences in the resistance mechanisms of EZH2 inhibitors. 40 Thus, OR‐S1–resistant REC‐1 cells might be sensitive to other EZH1/2 inhibitors.
Second, levels of H3K27me3 at the CDKN1C locus in REC‐1 were less affected by treatment with OR‐S1 than those in Mino and JeKo‐1. As we have shown in the present study, expression of CDKN1C, but not of CDKN2A, was associated with OR‐S1 sensitivity, and it has been previously shown that the level of CDKN1C expression correlates with the level of H3K27me3 around the TSS. 34 As levels of H3K27me3 around the TSS in REC‐1 remained relatively stable in the presence of 300 nmol/L OR‐S1 for 5 days, there was no strong induction of CDKN1C expression, and, therefore, growth inhibition could not occur. On the other hand, when REC‐1 was exposed to 1000 nmol/L OR‐S1 for 7 days, the RNA‐seq data showed that when growth was inhibited by 23%, CDKN1C expression was over 2‐fold upregulated (Figure S1A), indicating that upregulation of CDKN1C may be associated with the inhibition of cell growth. Recently, downregulation of CDKN1C was reported to drive tumorigenesis and to be associated with poor overall survival of breast cancer patients. 36 In the present study, experiments on CDKN1C‐overexpressing cells showed that downregulated CDKN1C expression is crucial for MCL cell proliferation. CDKN2A expression is well recognized as a parameter of tumor suppression by EZH2‐specific inhibitors. 20 , 21 , 37 , 38 However, we did not observe the induction of CDKN2A expression in MCL cell lines by EZH1/2 dual inhibition, indicating that PRC2 target genes in MCL may differ from those in other B‐cell malignancies.
Third, OR‐S1 did not induce the surface expression of CD138 on REC‐1 cells, suggesting that OR‐S1 did not trigger differentiation. DEGs in REC‐1 treated with OR‐S1 were less associated with cell activation and differentiation than those in Mino and JeKo‐1. A recent report has shown that SOX11 promotes tumor growth of MCL cells and represses the plasma cell gene program, 41 indicating the correlation between the terminal B‐cell differentiation program and growth inhibition in MCL. Furthermore, dedifferentiated transformation in MCL cells is correlated with drug resistance. 42 Our data suggest that the block in the B‐cell terminal differentiation program in REC‐1 may lead to the maintenance of cell proliferation regardless of OR‐S1 exposure and link to OR‐S1 insensitivity.
Our findings, along with those of others showing that EZH2 expression is associated with MCL proliferation, 23 , 24 , 25 provide evidence that EZH1/2 could have potential as novel therapeutic targets for the treatment of aggressive MCL. Furthermore, our study demonstrated the potential efficacy of OR‐S1, or its close analog valemetostat, currently under study in a variety of clinical settings for the treatment of patients with ibrutinib‐resistant MCL. Our study should advance the prospect of treating MCL patients with dual EZH1/2‐targeted therapies, which currently remain unapproved for use in the clinic.
DISCLOSURE
DH, NA, and KA are employees of Daiichi Sankyo, Co., Ltd. AK, KI, and YO are employees of Daiichi Sankyo RD Novare Co., Ltd. The other authors have no conflicts of interest to declare.
Supporting information
Figure S1‐S3
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGMENTS
The authors thank Yutaka Shima, Kazutsune Yamagata, and Yukiko Aikawa for technical advice. This work was supported by an Acceleration Transformative Research for Medical Innovation grant from the Japan Agency for Medical Research and Development and by the National Cancer Center Research and Development Fund.
Kagiyama Y, Fujita S, Shima Y, et al. CDKN1C‐mediated growth inhibition by an EZH1/2 dual inhibitor overcomes resistance of mantle cell lymphoma to ibrutinib. Cancer Sci. 2021;112:2314–2324. 10.1111/cas.14905
REFERENCES
- 1. Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer. 2008;113:791‐798. [DOI] [PubMed] [Google Scholar]
- 2. Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7:750‐762. [DOI] [PubMed] [Google Scholar]
- 3. Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest. 2012;122:3416‐3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI‐32765 blocks B‐cell activation and is efficacious in models of autoimmune disease and B‐cell malignancy. Proc Natl Acad Sci USA. 2010;107:13075‐13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI‐32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma J, Lu P, Guo A, et al. Characterization of ibrutinib‐sensitive and ‐resistant mantle lymphoma cells. Br J Haematol. 2014;166:849‐861. [DOI] [PubMed] [Google Scholar]
- 7. Younes A, Thieblemont C, Morschhauser F, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R‐CHOP) for treatment‐naive patients with CD20‐positive B‐cell non‐Hodgkin lymphoma: a non‐randomised, phase 1b study. Lancet Oncol. 2014;15:1019‐1026. [DOI] [PubMed] [Google Scholar]
- 8. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle‐cell lymphoma. N Engl J Med. 2013;369:507‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martin P, Maddocks K, Leonard JP, et al. Postibrutinib outcomes in patients with mantle cell lymphoma. Blood. 2016;127:1559‐1563. [DOI] [PubMed] [Google Scholar]
- 10. Chiron D, Di Liberto M, Martin P, et al. Cell‐cycle reprogramming for PI3K inhibition overrides a relapse‐specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4:1022‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ntziachristos P, Abdel‐Wahab O, Aifantis I. Emerging concepts of epigenetic dysregulation in hematological malignancies. Nat Immunol. 2016;17:1016‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hidalgo I, Herrera‐Merchan A, Ligos JM, et al. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence‐like cell cycle arrest. Cell Stem Cell. 2012;11:649‐662. [DOI] [PubMed] [Google Scholar]
- 13. Kamminga LM, Bystrykh LV, de Boer A, et al. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood. 2006;107:2170‐2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Su IH, Basavaraj A, Krutchinsky AN, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003;4:124‐131. [DOI] [PubMed] [Google Scholar]
- 15. Beguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Velichutina I, Shaknovich R, Geng H, et al. EZH2‐mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010;116:5247‐5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B‐cell lymphomas of germinal‐center origin. Nat Genet. 2010;42:181‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2‐activating mutations. Nature. 2012;492:108‐112. [DOI] [PubMed] [Google Scholar]
- 19. Knutson SK, Wigle TJ, Warholic NM, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890‐896. [DOI] [PubMed] [Google Scholar]
- 20. Qi W, Chan H, Teng L, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA. 2012;109:21360‐21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110(19):7922‐7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bradley WD, Arora S, Busby J, et al. EZH2 inhibitor efficacy in non‐Hodgkin's lymphoma does not require suppression of H3K27 monomethylation. Chem Biol. 2014;21:1463‐1475. [DOI] [PubMed] [Google Scholar]
- 23. Visser HP, Gunster MJ, Kluin‐Nelemans HC, et al. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br J Haematol. 2001;112:950‐958. [DOI] [PubMed] [Google Scholar]
- 24. van Kemenade FJ, Raaphorst FM, Blokzijl T, et al. Coexpression of BMI‐1 and EZH2 polycomb‐group proteins is associated with cycling cells and degree of malignancy in B‐cell non‐Hodgkin lymphoma. Blood. 2001;97:3896‐3901. [DOI] [PubMed] [Google Scholar]
- 25. Kanduri M, Sander B, Ntoufa S, et al. A key role for EZH2 in epigenetic silencing of HOX genes in mantle cell lymphoma. Epigenetics. 2013;8:1280‐1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kopparapu PK, Bhoi S, Mansouri L, et al. Epigenetic silencing of miR‐26A1 in chronic lymphocytic leukemia and mantle cell lymphoma: Impact on EZH2 expression. Epigenetics. 2016;11:335‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sander S, Bullinger L, Klapproth K, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR‐26a. Blood. 2008;112:4202‐4212. [DOI] [PubMed] [Google Scholar]
- 28. Lu J, He ML, Wang L, et al. MiR‐26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res. 2011;71:225‐233. [DOI] [PubMed] [Google Scholar]
- 29. Mochizuki‐Kashio M, Aoyama K, Sashida G, et al. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1‐dependent manner. Blood. 2015;126:1172‐1183. [DOI] [PubMed] [Google Scholar]
- 30. Garapaty‐Rao S, Nasveschuk C, Gagnon A, et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem Biol. 2013;20:1329‐1339. [DOI] [PubMed] [Google Scholar]
- 31. Fujita S, Honma D, Adachi N, et al. Dual inhibition of EZH1/2 breaks the quiescence of leukemia stem cells in acute myeloid leukemia. Leukemia. 2018;32:855‐864. [DOI] [PubMed] [Google Scholar]
- 32. Honma D, Kanno O, Watanabe J, et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017;108(10):2069‐2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang X, Karuturi RK, Sun F, et al. CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS One. 2009;4:e5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mahara S, Lee PL, Feng M, Tergaonkar V, Chng WJ, Yu Q. HIFI‐alpha activation underlies a functional switch in the paradoxical role of Ezh2/PRC2 in breast cancer. Proc Natl Acad Sci USA. 2016;113:E3735‐E3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qiu Z, Li Y, Zeng B, Guan X, Li H. Downregulated CDKN1C/p57(kip2) drives tumorigenesis and associates with poor overall survival in breast cancer. Biochem Biophys Res Comm. 2018;497:187‐193. [DOI] [PubMed] [Google Scholar]
- 37. Caganova M, Carrisi C, Varano G, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest. 2013;123:5009‐5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mohammad F, Weissmann S, Leblanc B, et al. EZH2 is a potential therapeutic target for H3K27M‐mutant pediatric gliomas. Nat Med. 2017;23:483‐492. [DOI] [PubMed] [Google Scholar]
- 39. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev. 1999;13:1501‐1512. [DOI] [PubMed] [Google Scholar]
- 40. Bisserier M, Wajapeyee N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B‐cell lymphomas. Blood. 2018;131:2125‐2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vegliante MC, Palomero J, Perez‐Galan P, et al. SOX11 regulates PAX5 expression and blocks terminal B‐cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121:2175‐2185. [DOI] [PubMed] [Google Scholar]
- 42. Sun B, Shah B, Fiskus W, et al. Synergistic activity of BET protein antagonist‐based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood. 2015;126:1565‐1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1‐S3
Table S1
Table S2
Table S3
Table S4