

SUMMARY
Autophagy dysregulation is implicated in metabolic diseases, including type 2 diabetes. However, the mechanism by which the autophagy machinery regulates metabolism is largely unknown. Autophagy is generally considered a degradation process via lysosomes. Here, we unveil a metabolically important non-cell-autonomous, non-degradative mechanism regulated by the essential autophagy protein Becn1 in adipose tissue. Upon high-fat diet challenge, autophagy-hyperactive Becn1F121A mice show systemically improved insulin sensitivity and enhanced activation of AMP-activated protein kinase (AMPK), a central regulator of energy homeostasis, via a non-cell-autonomous mechanism mediated by adiponectin, an adipose-derived metabolic hormone. Adipose-specific Becn1F121A expression is sufficient to activate AMPK in non-adipose tissues and improve systemic insulin sensitivity by increasing adiponectin secretion. Further, Becn1 enhances adiponectin secretion by interacting with components of the exocyst complex via the coiled-coil domain. Together, our study demonstrates that Becn1 improves insulin sensitivity by facilitating adiponectin secretion through binding the exocyst in adipose tissue.
In brief
Kuramoto et al. demonstrate a non-degradative and non-cell-autonomous mechanism by which the autophagy protein Becn1 regulates systemic energy metabolism. Becn1 interacts with exocyst proteins in white adipose tissue to facilitate adiponectin secretion, resulting in systemic insulin sensitization via the adiponectin receptor-AMPK signaling pathway in non-adipose tissues.
Graphical Abstract
INTRODUCTION
Autophagy is a key lysosomal degradation pathway induced by stress, such as fasting and exercise, which breaks down damaged or unnecessary structures by fusion of autophagosome vesicles with lysosomes (Kaur and Debnath, 2015; Rocchi and He, 2017b). Deletion of autophagy genes in mice, either globally or specifically in metabolic tissues, including pancreatic β cells and liver, has been linked with metabolic dysfunctions, such as hyperglycemia and insulin resistance, the hallmarks of type 2 diabetes (T2D) (Ebato et al., 2008; He et al., 2013; Jung et al., 2008; Rocchi and He, 2015; Yang et al., 2010). In contrast, recent data indicate that both physiological and genetic activation of autophagy increases insulin sensitivity (He et al., 2012; Yamamoto et al., 2018). However, the mechanism by which the autophagy machinery regulates insulin sensitivity and T2D pathogenesis remains obscure. Autophagy is generally considered a destruction and degradation process. Although emerging evidence suggests that autophagy may have an unconventional secretory function in immunity and inflammation (Bel et al., 2017; Zhang et al., 2015), whether autophagy-related proteins play non-degradative roles in the regulation of metabolism, as well as the underlying mechanisms, is unknown.
Besides serving as lipid storages, adipose tissue is an important endocrine organ that regulates energy metabolism in various metabolic tissues via secreting adipokines (adipose-derived hormones), including adiponectin. Adiponectin regulates a spectrum of metabolic parameters in the muscle, liver, and vascular system, including maintaining insulin sensitivity, promoting free fatty acid oxidation, reducing hepatic glucose and triglyceride (TG) production, and inhibiting vascular inflammation (Kadowaki et al., 2006; Ouchi et al., 2011; Stern et al., 2016; Yamauchi et al., 2001). The expression and plasma concentration of adiponectin positively correlates with insulin sensitivity and is reduced in obesity and T2D in both animal models and humans (Arita et al., 1999; Hotta et al., 2001; Li et al., 2009). Low blood adiponectin caused by SNPs (single-nucleotide polymorphisms) or mutations in the Adiponectin gene in humans, also known as hypoadiponectinemia, has been associated with insulin resistance and T2D (Hara et al., 2002; Kondo et al., 2002; Menzaghi et al., 2002; Stumvoll et al., 2002). Adiponectin knockout (KO) mice phenocopy the human phenotypes and develop insulin resistance (Kubota et al., 2002; Nawrocki et al., 2006), demonstrating the key role of adiponectin in the prevention of T2D. Thus, understanding factors that regulate the endocrine pathway of adiponectin secretion in adipose tissue will help elucidate the pathogenesis of insulin resistance and metabolic disorders. However, despite the importance of adiponectin in metabolic homeostasis, the molecular mechanism of adiponectin secretion from adipose tissue is unclear. Although extracellular signals, such as insulin, adrenergic stimuli, and fibroblast growth factor 21 (FGF21) (Blümer et al., 2008; Cong et al., 2007; Geng et al., 2019; Hajri et al., 2011; Holland et al., 2013; Komai et al., 2016; Lim et al., 2015; Lin et al., 2013), have been reported to increase adiponectin release in adipocytes via corresponding receptors, the intracellular mechanism that controls the secretion of adiponectin after it is folded and packaged in the endoplasmic reticulum (ER) is poorly defined.
Beclin 1/Becn1 is an essential autophagy protein that binds the class III Vps34 phosphatidylinositol 3-kinase (PtdIns3K) complex and regulates its stability and kinase activity during autophagy initiation (He and Levine, 2010). Becn1 is reported to be required for exercise-induced activation of AMPK (AMP-activated protein kinase), a central positive regulator of insulin sensitivity and glucose uptake, in muscle via binding with the Toll-like receptor 9 (TLR9) (He et al., 2012; Liu et al., 2020). We recently established a Becn1 mutant mouse model of hyperactive autophagy, Becn1F121A knockin (KI) mice, to study the role and mechanism of autophagy in metabolic regulation (Rocchi et al., 2017; Yamamoto et al., 2018). The Becn1F121A point mutant cannot be bound and inhibited by the anti-apoptotic and anti-autophagy protein Bcl-2 and leads to high autophagy. Becn1F121A-mediated genetic upregulation of autophagy improves systemic insulin sensitivity, but the mechanism by which autophagy regulates insulin sensitivity is unclear.
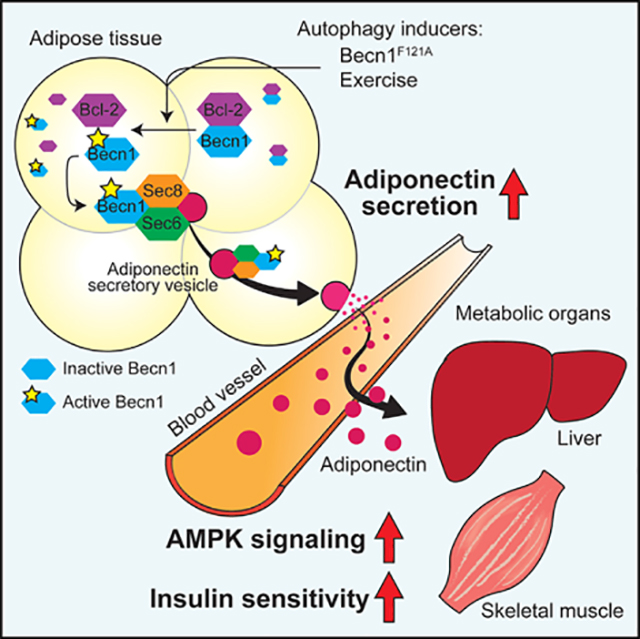
In this study, we found that Becn1 regulates metabolism through a previously uncharacterized non-degradative, non-cell-autonomous mechanism by regulating adiponectin secretion from adipocytes. The autophagy-hyperactive Becn1F121A mutation mimics the effect of exercise on AMPK activation and leads to systemic AMPK activation at resting conditions. This effect is exerted by a non-cell-autonomous mechanism mediated by autophagy-induced circulating adiponectin from adipocytes, because adipose tissue-specific expression of Becn1F121A is sufficient to increase liver and muscle AMPK activation, circulating adiponectin levels, and systemic insulin sensitivity, which is abolished by reduced adiponectin expression. Mechanistically, we discovered that an interaction between Becn1 and the exocyst components in adipocytes is required for Becn1F121A-induced secretion of adiponectin. Altogether, we establish a model of Becn1-facilitated adiponectin secretion in adipose tissue: when the essential autophagy protein Becn1 is released from the inhibitory binding of Bcl-2, it promotes adiponectin secretion via binding to the exocyst complex in adipose tissue, which elevates circulating adiponectin, activates AMPK in target metabolic tissues, and increases insulin sensitivity systemically (Figure 7G).
Figure 7. Adipose-specific Becn1F121A non-cell-autonomously improves systemic insulin sensitivity via promoting adiponectin secretion.

(A) WB analysis of serum adiponectin levels in Adipoq-Cre or control (WT) mice injected with AAV2/8-FLEX-Becn1F121A and then fed with HFD for 12 weeks. n = 4. t test.
(B) ELISA of serum levels of adiponectin, resistin, and leptin in Adipoq-Cre or WT mice injected with AAV2/8-FLEX-Becn1F121A and then fed with HFD for 12 weeks. WT+AAV-Becn1F121A, n = 11; Adipoq-Cre+AAV-Becn1F121A, n = 9. t test.
(C and D) Reducing adiponectin in adipose-specific Becn1F121A mice abolishes the insulin-sensitizing effects of adipose Becn1F121A. GTT and ITT (C) and ELISA of serum adiponectin (D) of WT and adipose-specific Becn1F121A mice expressing both or one copy of Adiponectin after 8 weeks of HFD feeding. For GTT and ITT: WT+AAV-Becn1F121A, n = 9; Adipoq-Cre+AAV-Becn1F121A, n = 9; Adipoq+/− KO+AAV-Becn1F121A, n = 6; Adipoq+/− KO Adipoq-Cre+AAV-Becn1F121A, n = 5. For adiponectin ELISA: n = 5–6. One-way ANOVA with Dunnett’s test.
(E) Inhibiting early (upper), but not late (lower), stages of autophagy reduces adiponectin release to circulation in mice. WB analysis of adiponectin in the serum of HFD-fed Becn1F121A mice treated with SBI-0206965 (SBI) or Spautin-1 (early-stage inhibitors, inhibiting autophagosome formation) or chloroquine (CQ; late-stage inhibitor, inhibiting lysosomal degradation). n = 4–6. t test.
(F) ELISA of serum adiponectin in HFD-fed Becn1F121A mice treated with SBI-0206965, Spautin-1, or CQ. n = 5–8. One-way ANOVA with Dunnett’s test. Data represent mean ± SEM. *,#,¶p < 0.05; **,##,¶¶p < 0.01; ***,###p < 0.001.
(G) Model of Becn1-regulated adiponectin secretion. When the essential autophagy protein Becn1 is released from its inhibitor Bcl-2 in adipose tissue, triggered by exercise or the active Becn1F121A mutation, it interacts with Sec6 and other exocyst components to increase adiponectin secretion, systemic AMPK activation, and insulin sensitivity.
RESULTS
Autophagy-hyperactive Becn1F121A mice show systemic AMPK activation in metabolic tissues caused by circulator factors
To study how autophagy regulates metabolism, we previously generated an autophagy-hyperactive KI mouse model carrying a single amino acid substitution (F121A) in the Becn1 protein (Becn1F121A), which disrupts inhibitory binding between Becn1 and Bcl-2 and leads to constitutive activation of autophagy (Rocchi et al., 2017). We found that in response to 8- or 12-week high-fat diet (HFD) feeding, while Becn1F121A mice showed comparable body weight to wild-type (WT) mice, they are less glucose tolerant yet more insulin sensitive than WT mice (Yamamoto et al., 2018) (Figure S1A). Our previous findings suggest that after HFD challenge, the Becn1F121A mice showed excessive autophagic degradation of insulin granule vesicles (vesicophagy) in β cells, leading to impaired insulin secretion and glucose intolerance. However, it does not explain why Becn1F121A mice are paradoxically more insulin sensitive. To solve this puzzle, we analyzed various metabolic pathways in their metabolic tissues and found that compared with WT mice, metabolic tissues (liver and skeletal muscle) of Becn1F121A mice showed enhanced phosphorylation/activation of AMPK, which plays a central beneficial role in energy homeostasis and insulin sensitization (Garcia and Shaw, 2017), after feeding of either regular diet (RD) (Figure 1A) or HFD (Figure 1B). Further, in support of AMPK activation, Becn1F121A mouse tissues showed a spectrum of changes downstream of AMPK, including increased phosphorylation of Raptor (Regulatory-associated protein of mTOR [mechanistic target of rapamycin]), decreased phosphorylation of p70S6K and IRS-1 (Insulin receptor substrate 1), and stabilization of IRS-1 (Figure 1B). AMPK phosphorylates Raptor at S792, which leads to inhibition of mTOR and its substrate p70S6K (Gwinn et al., 2008). The latter can phosphorylate IRS-1 at several serine residues, including S302 and S632/635 (Haruta et al., 2000; Ozes et al., 2001; Ueno et al., 2005; Werner et al., 2004), which promotes IRS-1 degradation via proteasomes (Pederson et al., 2001; Sun et al., 1999; Takano et al., 2001). Because IRS-1 is a key docking protein downstream of the activated insulin receptor (IR) and insulin-like growth factor-1 receptor (IGF-1R) to form scaffolding complexes and initiate insulin signaling cascades, stabilization of IRS-1 by AMPK activation in part enhances insulin sensitivity. Thus, more Raptor S792 phosphorylation, less p70S6K T389 phosphorylation, less IRS1 S302 and S632/635 phosphorylation, and higher levels of IRS-1 in the metabolic tissues of Becn1F121A mice suggest an activated AMPK signaling pathway in these mice (Figure 1B).
Figure 1. Autophagy-hyperactive Becn1F121A mice show enhanced AMPK activation in tissues, caused by factors in circulation.

(A) Metabolic tissues of Becn1F121A KI mice show increased AMPK activation. Western blot (WB) analysis and quantification of AMPK phosphorylation in liver and muscle of Becn1F121A mice. n = 7.
(B) WB analysis and quantification of AMPK activation and signaling pathways downstream of AMPK in the liver of HFD-fed WT and Becn1F121A mice. n = 7.
(C) Circulating factors in Becn1F121A mice play a role in activating AMPK. WB analysis of AMPK phosphorylation in cell lines cultured for 1 h in medium containing 10% mouse serum from WT or Becn1F121A mice. n = 4.
Data represent mean ± SEM, t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Notably, WT Becn1 is reported to be required for exercise-induced activation of AMPK in muscle via binding with TLR9 (He et al., 2012; Liu et al., 2020). Thus, the hyperactive Becn1F121A mutation mimics the effect of exercise on AMPK activation at resting conditions. Intriguingly, this effect is not due to differences in the level of TLR9 (Figure S1B) and is not limited to muscle (Figures 1A and 1B). To reveal the mechanism underlying AMPK hyperactivation in Becn1F121A mice, we asked whether AMPK activation is caused by a cell-autonomous or non-cell-autonomous mechanism. We added 10% serum collected from either WT or Becn1F121A mice to the medium of WT cells of metabolic tissue origins. We found that when added to cell culture media, serum from Becn1F121A mice also leads to enhanced AMPK activation in various cell lines (including NIH 3T3 fibroblasts, HepG2 and Huh7 hepatocytes, C2C12 myotubes, and 3T3-L1 preadipocyte-differentiated adipocytes), compared with serum from WT mice or the PBS control (Figures 1C and S1C). In contrast, primary cells isolated from WT and Becn1F121A mice, including primary MEFs (mouse embryonic fibroblasts) and primary hepatocytes, did not show differences in AMPK activation in the absence or presence of the AMPK activator AICAR (Figure S1D). These data suggest that Becn1F121A-induced AMPK-activating, as well as insulin-sensitizing, effects are likely mediated by a non-cell-autonomous mechanism through circulating factors.
Becn1F121A mice have increased circulating levels of adiponectin
To identify the Becn1-controlled circulating factor that has an AMPK-activating and potentially insulin-sensitizing effect, we did a Luminex array (Millipore) on metabolic hormones in the serum of WT and Becn1F121A mice (Figure S2A, showing partial data). We detected a higher circulating level of adiponectin, but not other metabolic hormones tested, in Becn1F121A mice than in WT littermates in response to either RD or HFD feeding (Figure 2A). We confirmed that Becn1F121A mice have elevated circulating adiponectin by western blot (WB) analysis (Figure 2B), despite comparable body weight, white adipose tissue (WAT) mass (Figure S2B), intra-tissue adiponectin levels (Figure S2C), and circulating levels of the adiponectin secretagogue FGF21 (Figure S2D) with those of WT mice. In addition, we found that Becn1 heterozygous KO mice have reduced serum adiponectin compared with WT littermates (Figure 2C), further supporting the role of Becn1 in the regulation of circulating adiponectin levels.
Figure 2. Becn1 positively regulates adiponectin secretion in vivo and in vitro.

(A) ELISA of total adiponectin and HMW adiponectin in the serum of WT (+/+) mice and heterozygous (FA/+) or homozygous (FA/FA) Becn1F121A KI mice fed with RD (regular diet) or HFD for 8 weeks. Total adiponectin, n = 8–17; HMW adiponectin, n = 7–11. One-way ANOVA with Dunnett’s test.
(B) WB analysis of adiponectin in the serum of WT and Becn1F121A mice fed with HFD for 8 weeks. n = 6.
(C) ELISA of serum adiponectin in WT littermates (+/+) and Becn1+/− KO (+/−) mice fed with HFD for 8 weeks. +/+, n = 12; +/−, n = 10.
(D) Total and HMW adiponectin are increased in Becn1F121A mice. WB analysis of HMW and total adiponectin in the serum of WT and Becn1F121A mice using a mouse anti-adiponectin antibody (MA1–054; Thermo Fisher Scientific). Total protein loading is shown by Ponceau S staining. n = 7.
(E) Becn1F121A-induced adiponectin secretion is cell autonomous. Primary adipocytes (adipose-derived stromal vascular fraction [SVF] cells) isolated from Becn1F121A mice secrete more adiponectin into the conditioned medium than those from WT mice, quantified by WB analysis after 24-h culture. Total protein loading is shown by Ponceau S staining. n = 3 mice.
Data represent mean ± SEM, t test. *p < 0.05; **p < 0.01; ***p < 0.001.
Adiponectin exists in the circulation as different multimer complexes, categorized as high-molecular-weight (HMW) multimers, medium-molecular-weight (MMW) hexamers, and low-molecular-weight (LMW) trimers, with the HMW adiponectin being the most biologically active form (Basu et al., 2007; Lara-Castro et al., 2006; Murdolo et al., 2009; Pajvani et al., 2004). We found that the HMW and MMW forms of adiponectin show a significant increase in the serum of Becn1F121A mice, using ELISA (Figure 2A) and an antibody that detects the total and HMW adiponectin (Figure 2D) or another antibody that detects the MMW, but not the HMW, form of adiponectin (Figure S2E). Further, we found that increased adiponectin secretion from Becn1F121A adipocytes is cell autonomous, because there is more adiponectin secreted into the medium from in vitro-cultured primary adipocytes isolated from Becn1F121A mice (Figure 2E) or from Becn1F121A-expressing 3T3-L1-differentiated adipocytes (Figure S2F). Together, these data suggest that Becn1F121A induces adiponectin secretion, mediated by a cell-autonomous mechanism.
Becn1F121A-induced adiponectin signaling is essential for non-cell-autonomous AMPK activation
Adiponectin activates several intracellular targets, including AMPK signaling, via binding and signaling through the membrane receptors adiponectin receptor 1 (AdipoR1) and AdipoR2 in metabolic tissues, such as liver and muscle (Wu et al., 2003; Yamauchi et al., 2002; Yoon et al., 2006). To determine whether AMPK activation in Becn1F121A mice is dependent on increased secretion of adiponectin, we asked whether depleting adiponectin receptors in non-adipose cells abolishes AMPK activation induced by Becn1F121A mouse serum. We found that compared with WT mouse serum, serum from Becn1F121A mice led to higher AMPK activation in WT C2C12 myotubes but failed to do so in C2C12 myotubes deleted for the adiponectin receptor AdipoR1 (the major adiponectin receptor expressed in myotubes) by short hairpin RNAs (shRNAs) (Figure 3A). Similarly, we also found that the adiponectin receptors are required for Becn1F121A mouse serum-induced AMPK activation in NIH 3T3 fibroblasts (primarily expressing AdipoR1) (Figure 3B) and 3T3L1-differentiated adipocytes (expressing both AdipoR1 and AdipoR2) (Figure S3). These data suggest that adiponectin signaling is required for AMPK activation in target metabolic cells induced by Becn1F121A mouse serum.
Figure 3. Non-cell-autonomous activation of AMPK by Becn1F121A mouse serum is mediated by adiponectin signaling.

(A and B) C2C12 myotubes (A) or NIH 3T3 fibroblasts (B) stably expressing scrambled (Scr) or shRNAs against adiponectin receptor 1 (AdipoR1) were cultured for 1 h in medium containing 10% mouse serum from WT or Becn1F121A mice, and AMPK phosphorylation was analyzed and quantified by WB studies. AdipoR1 KD efficiency is shown on the right. n = 3 mice.
Data represent mean ± SEM, t test. *p < 0.05; **p < 0.01. NS, not significant.
The exocyst components Sec5, Sec6, and Sec8 are binding partners of Becn1 and preferentially interact with the hyperactive Becn1F121A mutant compared with WT Becn1
We next aimed to understand how the autophagy machinery cell autonomously regulates adiponectin secretion in adipocytes. Indeed, we found that the WAT of the Becn1F121A mice shows disrupted Becn1-Bcl-2 interaction, assayed by co-immunoprecipitation (coIP) (Figure 4A), as well as increased autophagy activity, evidenced by elevated LC3-II and p62 flux using a lysosomal inhibitor chloroquine (Figure S4A). Nonetheless, we did not detect elevated or accelerated adipogenesis in the WAT of Becn1F121A mice (Figure 4B), in primary adipocytes from Becn1F121A mice (Figure S4B), or in Becn1F121A-expressing 3T3-L1-differentiated adipocytes (Figures 4C and S4C), by analyzing the expression of a variety of adipogenesis markers. The data suggest that upregulating autophagy by Becn1F121A does not affect adipose development and differentiation. We then suspected that autophagosomes may directly sequestrate adiponectin and promote its secretion, because we previously found that under certain conditions hormones can become an autophagy cargo (Kuramoto and He, 2018; Yamamoto et al., 2018). However, our imaging and biochemical data ruled out this possibility as well. We did not observe significant colocalization between adiponectin and the autophagosome marker LC3 in primary adipocytes of either WT GFP-LC3 or Becn1F121A GFP-LC3 reporter mice by confocal microscopy (Figure S4D). Moreover, using a biochemical method we developed to immunoisolate intact autophagosomes from cells and tissues (Yamamoto et al., 2018), we did not detect adiponectin inside autophagosomes purified from 3T3-L1-differentiated adipocytes (Figure S4E). Thus, these data suggest that adiponectin elevation in Becn1F121A KI mice is not due to altered adipogenesis or enhanced sequestration and secretion of adiponectin directly by autophagosome vesicles.
Figure 4. Increasing autophagy by Becn1F121A does not promote adipogenesis in vivo or in vitro.

(A) CoIP of endogenous Bcl-2 and Becn1 in the iWAT of WT and Becn1F121A mice. n = 3 mice.
(B) Adipogenesis markers in gWAT and iWAT of RD-fed WT and Becn1F121A mice were analyzed by WB. n = 6 mice. t test. Data represent mean ± SEM.
(C) 3T3-L1 preadipocytes stably expressing hemagglutinin (HA)-tagged WT Becn1 or Becn1F121A were cultured in the adipocyte-differentiation medium for a 10-day time course, and adipogenesis markers were analyzed every 2 days by WB.
Nonetheless, we detected colocalization between adiponectin and Becn1 in primary adipocytes and a higher level of colocalization of adiponectin with Becn1F121A than with WT Becn1 (Figure 5A), supporting that Becn1 may play a direct role in regulating adiponectin secretion. To investigate the potential mechanism of Becn1-regulated adiponectin release, we generated a HEK293 cell line stably expressing a tetracycline-inducible cleavable zz tag-Becn1–3XFlag fusion protein (Sun et al., 2008). Using this cell line, we pulled down several subunits of the exocyst as Becn1 binding partners, including Sec5/Exoc2, Sec6/Exoc3, and Sec8/Exoc4 (Figure S5A). In the meantime, when we interrogated published databases and reports of the Becn1 interactome, we found that another exocyst component Exo84/Exoc8 has been reported as a Becn1-binding protein (Bodemann et al., 2011). The exocyst is a multiprotein complex involved in plasma-membrane docking of secretory vesicles, containing eight components: Sec3/Exoc1, Sec5/Exoc2, Sec6/Exoc3, Sec8/Exoc4, Sec10/Exoc5, Sec15/Exoc6, Exo70/Exoc7, and Exo84/Exoc8 (Heider and Munson, 2012; Wu and Guo, 2015). By coIP, we confirmed the interaction of the key exocyst component Sec6 with both WT Becn1 and Becn1F121A in HEK293 cells (Figure 5B) and found that the hyperactive mutant form, Becn1F121A, shows a higher affinity with Sec6 than WT Becn1 in HEK293 cells stably expressing WT Becn1 or Becn1F121A (Figures 5B and 5C), in primary adipocytes isolated from WT or Becn1F121A mice (Figure 5D), and in 3T3-L1-differentiated adipocytes (Figure S5B). Different from the previous report (Bodemann et al., 2011), we were not able to detect an interaction between Becn1 and the exocyst components Exo84 or Exo70 in HEK293 cells (Figure 5B), but we found that in the presence of Becn1F121A, there is a stronger binding between Sec6 and several other exocyst components, such as Sec5 and Sec8 (Figure 5C). Further, in addition to Sec6, the exocyst components Sec5 and Sec8, as well as PtdIns3K complex subunits ATG14, VPS34, and UVRAG, also show stronger binding with Becn1F121A than with WT Becn1 analyzed by coIP in primary adipocytes (Figure 5D). Together, these data suggest that Becn1 binds the exocyst complex and facilitates exocyst assembly, and the exocyst machinery preferentially interacts with the hyperactive Becn1F121A mutant compared with WT Becn1.
Figure 5. Becn1 forms a complex with the exocyst, and the Becn1-exocyst interaction is essential to increase secretion of adiponectin, but not leptin, in Becn1F121A adipocytes.

(A) Immunofluorescence staining of adiponectin (green) and Becn1 (red) in SVF primary adipocytes isolated from WT or Becn1F121A mice. The intensity line profile measurements show intensity distribution on dashed arrows in the images above. Pearson correlation coefficient is used to quantify colocalization between adiponectin and Becn1. Low magnification, scale bars: 10 μm; high magnification, scale bars: 5 μm. n = 7 cells. t test.
(B) The exocyst component Sec6, but not Exo84 or Exo70, is pulled down by Becn1F121A more strongly than WT Becn1. CoIP of FLAG-Sec6 by HA-Becn1 in HEK293 cells transfected with Sec6 and WT Becn1 or Becn1F121A.
(C) Becn1F121A promotes exocyst assembly. CoIP of HA-Becn1, Sec8, and Sec5 by FLAG-Sec6 in HEK293 cells transfected with Sec6 and WT Becn1 or Becn1F121A.
(D) CoIP of endogenous exocyst components (Sec6, Sec5, and Sec8) and autophagy-related PtdIns3K proteins (Atg14, Vps34, and UVRAG) with the endogenous Becn1 protein in primary adipocytes isolated from WT or Becn1F121A mice. One-way ANOVA with Tukey-Kramer test.
(E) CoIP of HA-hSec6 (human Sec6) and indicated full-length (FL) and deletion mutants of FLAG-hBecn1F123A (human Becn1F123A, mouse Becn1F121A equivalent) in HEK293 cells.
(F) WB analysis of adiponectin levels in the conditioned medium secreted from WT 3T3-L1-differentiated adipocytes stably expressing HA-tagged FL Becn1F121A or the Becn1F121A ΔCCD deletion mutant after 24-h culture. Intracellular adiponectin and total protein loading (Ponceau S staining) are also shown. n = 3.
(G) ELISA of adiponectin and leptin in the conditioned medium of WT 3T3-L1-differentiated adipocytes stably expressing HA-tagged FL Becn1F121A or the Becn1F121A ΔCCD deletion mutant after 24-h culture. n = 6.
(H) Leptin and adiponectin vesicle pools have limited colocalization in adipocytes. Immunofluorescence staining of adiponectin and leptin in primary adipocytes of WT mice. Low magnification, scale bars: 10 μm; high magnification, scale bars: 5 μm. n = 12 regions.
Data represent mean ± SEM. One-way ANOVA with Tukey-Kramer test. *p < 0.05; **p < 0.01.
The Becn1-exocyst interaction is essential for promoting adiponectin secretion
To further determine whether the Becn1-Sec6 interaction is important for adiponectin secretion, we aimed to generate a loss-of-Sec6 interaction mutant of Becn1. Becn1 contains three major domains: the BH3 (Bcl-2 homology 3) domain that binds Bcl-2, the coiled-coil domain (CCD), and the evolutionally conserved domain (ECD) (He and Levine, 2010; Levine et al., 2015). We generated deletion mutants of Becn1F121A lacking the N-terminal intrinsically disordered region, the BH3 domain, the CCD, the ECD, or the C-terminal regions, respectively (Figure 5E). We found that the Becn1ΔCCD mutant (lacking the CCD) disrupts the Becn1-Sec6 binding by coIP (Figure 5E), suggesting that the CCD of Becn1 is important for exocyst binding. We subsequently stably expressed full-length BecnF121A and the loss-of-Sec6 interaction Becn1F121AΔCCD mutant in WT 3T3-L1 preadipocytes, induced adipocyte differentiation of the 3T3-L1 cells, and analyzed adiponectin levels in the conditioned medium. We found that Becn1F121AΔCCD abolishes the increase in the medium adiponectin level induced by Becn1F121A and by WB (Figure 5F) and ELISA (Figure 5G) analyses, suggesting that disrupting Becn1-Sec6 binding interferes with Becn1-regulated adiponectin secretion in adipocytes. Thus, we conclude that Becn1 regulates adiponectin secretion by interacting with the exocyst.
We also demonstrated that the exocyst machinery functions in adiponectin release from adipocytes, which has not been characterized before. We found that shRNA knockdown (KD) of Sec6 significantly decreases the adiponectin level in the conditioned medium of 3T3-L1 preadipocyte-differentiated adipocytes (Figure S5C), suggesting that Sec6 is required for adiponectin secretion in adipocytes. Yet KD of another exocyst component Sec5 does not affect adiponectin secretion (Figure S5D). This is in line with reported phenomena in Drosophila, where mutating Sec5 does not impair the secretion of neurotransmitters (Murthy et al., 2003). Based on the cryoelectron microscopy (cryo-EM) structure of the exocyst (Mei et al., 2018), we speculate that it is possible that Becn1 and Sec5 may be partially functionally redundant in adiponectin secretion, and binding of Becn1 to Sec6 or the exocyst may compensate for the loss of Sec5 in the secretion. We found that KD of Sec8 reduces adiponectin secretion (Figure S5D); yet, depleting Sec8 also reduces the stability of Sec6, consistent with the structural data suggesting that Sec8 forms a paired subcomplex with Sec6 in the exocyst (Mei et al., 2018). These data demonstrate that Sec6 needs to cooperate with Sec8, but not Sec5, to exert its effects on adiponectin secretion.
We also found that Sec6 is partially required for the secretion of another adipokine leptin (Figure S5C). However, intriguingly, we found that different from adiponectin, expressing Becn1F121A or blocking the Becn1-Sec6 binding does not affect the secretion of leptin from 3T3-L1-differentiated adipocytes (Figure 5G). Our microscopy data suggest that this specificity is potentially caused by subcellular compartmentation and distinct trafficking pathways of different types of secretory vesicles. We found that as previously reported (Xie et al., 2008), although adiponectin-containing vesicles are enriched in the internal subcellular pool (center), leptin vesicles mostly localize in proximity to the cell surface (periphery) and show limited colocalization with adiponectin (Figure 5H). In addition, compared with adiponectin vesicles, leptin vesicles have reduced colocalization with Becn1, and such colocalization is not increased by hyperactive Becn1F121A (Figure S5E). Thus, it is likely that various types of vesicles are originated from distinct compartments of the Golgi and trafficked to different subcellular locations that are Becn1 positive or negative. We propose that the Becn1-Sec6 complex forms approximate to adiponectin vesicles at the center of the adipocyte to promote their transport toward the plasma membrane.
Adipose tissue-specific expression of Becn1F121A is sufficient to improve systemic insulin sensitivity, glucose tolerance, and insulin signaling in liver and muscle
To further demonstrate that Becn1 in the WAT is key for adiponectin secretion and metabolic improvement, we compared the metabolic phenotypes of mice expressing Becn1F121A specifically in the WAT or globally in the whole body, to determine whether adipose tissue-specific Becn1F121A expression is sufficient to activate AMPK in non-adipose tissues and improve insulin sensitivity systemically. We utilized the FLEX switch system to deliver Becn1F121A to adipose tissue via adeno-associated virus 2/8 (AAV2/8) (Jimenez et al., 2013) that can efficiently target adipose tissues. In the FLEX system, the Becn1F121A reading frame is reversed and flanked by loxP and lox2272 sites (Figure 6A). When AAV2/8-FLEX-Becn1F121A is injected into WT mice (without Cre expression), Becn1F121A is not expressed, while when AAV2/8-FLEX-Becn1F121A is injected into an adipose-specific Adiponectin-Cre (Adipoq-Cre) mouse line (Cre driven by the adiponectin promoter), the Becn1F121A reading frame is flipped following Cre-Lox recombination and expresses specifically in adipose tissues. Using this system, we confirmed the expression specificity of Becn1F121A after AAV delivery in both inguinal WAT (iWAT) and gonadal WAT (gWAT), but not liver, of Adipoq-Cre mice (Figures 6A and S6A).
Figure 6. Adipose-specific Becn1F121Aimproves both insulin sensitivity and glucose tolerance and regulates insulin signaling, AMPK activity, and lipid metabolism in non-adipose tissues.

(A) Adipose-specific expression of Becn1F121A via injection of AAV2/8-FLEX-3XFlag-Becn1F121A to Adipoq-Cre mice.
(B) GTT and ITT in Adipoq-Cre or control (WT) mice injected with AAV2/8-FLEX-3XFlag-Becn1F121A and then fed with HFD for 12 weeks. WT+AAV-Becn1F121A, n = 11; Adipoq-Cre+AAV-Becn1F121A, n = 9.
(C) ELISA of serum insulin levels before and after intraperitoneal (i.p.) glucose injection in Adipoq-Cre or control (WT) mice injected with AAV2/8-FLEX-3XFlagBecn1F121A and fed with HFD for 12 weeks. WT+AAV-Becn1F121A, n = 11; Adipoq-Cre+AAV-Becn1F121A, n = 9. One-way ANOVA with Tukey-Kramer test.
(D) WB analysis of insulin-induced Akt phosphorylation in the liver and muscle of Adipoq-Cre or control (WT) mice injected with AAV2/8-FLEX-3XFlag-Becn1F121A and fed with HFD for 12 weeks. n = 3. t test.
(E) WB analysis of AMPK activation and signaling pathways downstream of AMPK in the liver of Adipoq-Cre or WT mice injected with AAV2/8-FLEX-Becn1F121A and then fed with HFD for 12 weeks. n = 4. t test.
(F) TG levels in the liver of Adipoq-Cre or WT mice injected with AAV2/8-FLEX-Becn1F121A and then fed with HFD for 12 weeks. WT+AAV-Becn1F121A, n = 12; Adipoq-Cre+AAV-Becn1F121A, n = 9. t test.
(G) WB analysis of PLIN2 and ACC in the liver of Adipoq-Cre or WT mice injected with AAV2/8-FLEX-Becn1F121A and then fed with HFD for 12 weeks. n = 4.
Data represent mean ± SEM, t test. *p < 0.05; **p < 0.01; ***p < 0.001. AUC, area under the curve.
With RD feeding, we found that Adipoq-Cre mice injected with AAV-FLEX-Becn1F121A (Adipoq-Cre AAV-Becn1F121A mice) have similar body weight (Figure S6B), insulin sensitivity (by insulin tolerance test [ITT]), and glucose tolerance (by glucose tolerance test [GTT]) (Figure S6C) as the control (WT AAV-Becn1F121A) mice. Yet, in response to HFD feeding, although Adipoq-Cre AAV-Becn1F121A mice have comparable body weight (Figure S6B) and WAT mass (Figure S6D) with the control mice, they show significantly improved insulin sensitivity similar to the global Becn1F121A KI mice (Figure 6B), suggesting that adipose-specific expression of Becn1F121A is sufficient to enhance systemic insulin sensitivity. Importantly, different from global expression of Becn1F121A, which improves insulin sensitivity but impairs glucose tolerance as a result of reducing insulin storage and secretion in islets (Figure S1A) (Yamamoto et al., 2018), adipose tissue-specific Becn1F121A expression improves both insulin sensitivity (ITT) and glucose tolerance (GTT) (Figure 6B, compared with Figure S1A), suggesting an anti-diabetic function of Becn1F121A in adipose tissue.
Along this line, we found that after HFD challenge, control mice showed an abnormally high level of insulin in the serum (hyperinsulinemia) under basal conditions (time 0 min), whereas Adipoq-Cre AAV-Becn1F121A mice showed significantly reduced HFD-induced hyperinsulinemia (Figure 6C). Further, different from whole-body Becn1F121A KI (Yamamoto et al., 2018), adipose tissue-specific expression of Becn1F121A does not cause a reduction in the insulinogenic index (a measure of glucose-stimulated insulin secretion) (Figure S6E), further supporting that previously observed Becn1F121A-induced reduction in insulin secretion is due to its role specifically in β cells. In addition, in support of systemic insulin sensitization, we detected higher phosphorylation of Akt at S473, a phosphorylation target downstream of the activated IR, in both liver and muscle of Adipoq-Cre AAV-Becn1F121A mice than in the control mice, after exogenous insulin stimulation after HFD feeding (Figure 6D), suggesting that Becn1F121A in adipose tissue improves insulin signaling in non-adipose tissues. Thus, together, these findings demonstrate that Becn1F121A is metabolically beneficial and improves systemic insulin sensitivity when specifically expressed in the WAT, and highlight that autophagy proteins may play different roles in different metabolic tissues.
Adipose-specific Becn1F121A regulates AMPK activity and lipid metabolism in non-adipose tissues
We further found that similar to the effect of global Becn1F121A expression (Figure 1B), adipose tissue-specific expression of Becn1F121A is sufficient to activate AMPK in the liver (Figure 6E) and muscle (Figure S6F), evidenced by activated AMPK phosphorylation and downstream signaling of AMPK, including increased phosphorylation of Raptor, decreased phosphorylation of p70S6K and IRS-1, and stabilization of IRS-1. Notably, upon IR activation, IRS-1 is phosphorylated at Y612, which is essential for downstream PtdIns3K activation and GLUT4 translocation. We detected a higher cellular level of Y612 phosphorylation in IRS-1 in Adipoq-Cre AAV-Becn1F121A mice than in control mice (Figure S6G), further supporting liver insulin sensitization mediated non-cell autonomously by adipose Becn1F121A.
Further, adiponectin systemically regulates lipid metabolism by activating AMPK and peroxisome proliferator-activated receptor (PPAR) a signaling pathways via AdipoR2 in the liver (Kadowaki et al., 2006; Yamauchi et al., 2002; Yoon et al., 2006). Activated AMPK inhibits SREBP1 and SREBP2 (sterol regulatory element binding proteins), the master transcription factors of fatty acid biosynthesis (Li et al., 2011). Activation of PPARα induces gene expression involved in fatty acid oxidation (Haemmerle et al., 2011; Rakhshandehroo et al., 2010). Thus, as the outcome of elevated circulating adiponectin, decreased fatty acid biosynthesis and increased fatty acid oxidation together will lead to reduced liver TG levels. Indeed, compared with control mice, we found that Adipoq-Cre AAV-Becn1F121A mice have significantly decreased liver TG contents (Figure 6F) and, as a result, a reduced level of the lipid droplet coating protein PLIN2 (Perilipin-2) in the liver (Figure 6G), because PLIN2 is stable only on lipid droplets, and cytosolic PLIN2 is degraded by the proteasome pathway (Masuda et al., 2006; Xu et al., 2005). Such reduction in TG and PLIN2 is not due to higher lipophagy in the liver of these mice because the Becn1F121A expression is adipose specific. Rather, in line with increased circulating adiponectin, in response to HFD feeding, we detected a significantly reduced level of the SREBP-target key lipogenesis enzyme ACC (acetyl coenzyme A carboxylase) by WB analysis (Figure 6G) and a trend of increased expression of PPARα-target fatty acid oxidation genes by qPCR (including peroxisomal acyl-coenzyme A oxidase 1 [Acox1], carnitine palmitoyltransferase 1A [Cpt1a], uncoupling protein 2 [Ucp2], and fatty acid binding protein 1 [Fabp1]) (Figure S6H) in the liver of Adipoq-Cre AAV-Becn1F121A mice compared with control mice. These data support that adipose Becn1 non-cell-autonomously regulates AMPK and PPARα signaling pathways and lipid metabolism in the liver. Together, the findings demonstrate that the activity of Becn1 in the WAT plays an important role in the regulation of systemic glucose and lipid metabolism.
Adipose-specific Becn1F121A increases adiponectin secretion and non-cell-autonomously regulates systemic insulin sensitivity via enhancing adiponectin secretion
We further detected a significantly higher level of circulating adiponectin in Adipoq-Cre AAV-Becn1F121A mice than in control mice in response to HFD treatment, measured by WB analysis (Figure 7A) and ELISA (Figure 7B). The circulating adiponectin level is also increased in adipose-specific Becn1F121A mice fed with RD (Figure S7A), which is similar to global Becn1F121A mice (Figure 2A). Consistent with cell culture studies (Figure 5G), we found that except adiponectin, circulating levels of two other adipokines, leptin and resistin, are comparable in control and adipose-specific Becn1F121A mice after HFD treatment (Figure 7B), further supporting a level of specificity of Becn1 in the regulation of adipokine secretion. These data suggest that adipose tissue-specific Becn1F121A expression is sufficient to increase adiponectin secretion, in support of the cell-autonomous effect of Becn1F121A on adiponectin secretion in primary adipocytes (Figure 2E).
To directly illustrate whether the metabolic effects of adipose-specific Becn1F121A are dependent on elevated adiponectin secretion, we asked whether reduced adiponectin expression abolishes Becn1F121A-induced insulin sensitization in response to HFD feeding, using the Adiponectin (Adipoq) KO mice. Consistent with previous literature (Nawrocki et al., 2006), we confirmed that the serum adiponectin level in Adipoq+/− heterozygous KO mice is approximately half of the WT level after either RD (Figure S7B) or HFD (Figure S7C) feeding. Thus, we generated heterozygous Adipoq+/−; Adipoq-Cre/+ mice and then injected them with AAV-Becn1F121A to obtain adipose-specific Becn1F121A mice expressing only one copy of the Adiponectin gene. We used AAV-Becn1F121A-injected Adipoq+/+; Adipoq-Cre/+ mice as the positive control, and AAV-Becn1F121A-injected Adipoq+/− KO and WT mice without Cre expression as the negative control. We found that under RD feeding conditions, WT and adipose-Becn1F121A mice expressing both or one copy of Adiponectin showed similar body weight, glucose tolerance, and insulin tolerance (Figures S7D and S7E). After HFD feeding, although all four mouse strains had comparable body weight (Figure S7F), only adipose-Becn1F121A mice expressing both copies of Adiponectin, but not those expressing just one copy of Adiponectin, showed improved glucose tolerance (GTT) and insulin tolerance (ITT) compared with non-Becn1F121A-expressing WT mice (Figure 7C), suggesting that reducing adiponectin in adipose-specific Becn1F121A mice abolishes the systemic insulin-sensitizing effects of adipose Becn1F121A in response to HFD feeding. Indeed, we found that adipose Becn1F121A-induced adiponectin secretion to the circulation is abolished when there is a 50% reduction in adiponectin expression in Adipoq+/− mice (Figure 7D). These data further support that the metabolic-improving effects of Becn1F121A are mediated through the adiponectin pathway. Altogether, these findings demonstrate that adipose Becn1 improves systemic glucose tolerance and insulin sensitivity by promoting adiponectin secretion into the circulation.
Model of Becn1-facilitated adiponectin secretion in adipocytes
In addition to the hyperactive genetic mutant Becn1F121A, we found additional upstream physiological and pharmacological modulators of autophagy that also regulate adiponectin secretion. Exercise is a potent physiological inducer of autophagy that disrupts the Becn1-Bcl-2 inhibitory binding (He et al., 2012; Rocchi and He, 2017a). We found that 90-min exercise increases the level of adiponectin in the serum of both male and female WT mice (Figure S7G), suggesting that adiponectin secretion is enhanced by a single bout of exercise. This result is in line with our earlier finding that chronic exercise also increases circulating adiponectin in WT mice, but not in Becn1-inhibited Bcl-2AAA mutant mice (He et al., 2012). This is likely mediated by exercise-induced release of Becn1 from the inhibitory binding of Bcl-2. Via coIP, we found that exercise disrupts the binding of Becn1 with its inhibitor Bcl-2 in the WAT of WT mice (Figure S7H). Although we also detected weak interactions between Becn1 and TLR9 in both WAT and muscle (Figure S7I), different from Becn1-TLR9 binding in muscle, which is enhanced by exercise (Liu et al., 2020), Becn1-TLR9 interaction in the WAT is constitutive and not regulated by exercise (Figure S7I). Thus, we propose that exercise promotes adiponectin secretion by releasing Becn1 from the inhibitory Bcl-2 binding in the WAT.
Conversely, we found that inhibiting the early phase of autophagy imposes an opposite effect. SBI-0206965 and Spautin-1, two inhibitors that block the initiating step of autophagy via inactivating the ULK1 kinase (Egan et al., 2015) or the Vps34 kinase complex (Liu et al., 2011), respectively, reduce the adiponectin level in the serum of WT mice; yet, inhibiting the late stages of autophagy, such as autophagosome-lysosome fusion or autolysosome degradation by chloroquine, does not significantly alter circulating adiponectin levels, measured by WB (Figure 7E) and ELISA analyses (Figure 7F). Given the lack of detection of adiponectin inside autophagosomes (Figures S4D and S4E), these data support that the upstream PtdIns3K-Becn1 complex, rather than the sequestrating and degradative steps of autophagy, is important for adiponectin regulation. Because compared with WT Becn1, Becn1F121A shows reduced binding to its inhibitor Bcl-2 in the WAT (Figure 4A), altogether, we propose the model of Becn1-facilitated adiponectin secretion: under autophagy-inducing conditions that release Becn1 from its inhibitor Bcl-2 (by exercise or the active Becn1F121A mutant), Becn1 interacts with Sec6 and other exocyst components to enhance adiponectin secretion from adipose tissue, which activates AMPK in non-adipose metabolic tissues and systemically increases insulin sensitivity via circulation (Figure 7G).
DISCUSSION
Although emerging evidence suggests that autophagy abnormality is associated with T2D, the mechanism by which autophagy regulates insulin sensitivity is largely unknown. Autophagy is generally thought to be a cellular self-degradation process via lysosomes. In this study, we discovered a previously unknown secretory function regulated by the essential autophagy protein Becn1 in the WAT, which plays a pivotal role in metabolic regulation. We previously found that autophagy-hyperactive Becn1F121A KI mice are protected from HFD-induced insulin resistance (Figure S1A) (Yamamoto et al., 2018). Through analyzing the metabolic signaling pathways in their metabolic tissues, we found that the global Becn1F121A KI mice have higher activation of AMPK, a central player beneficial for energy homeostasis (Figure 1). It is normally believed that autophagy proteins regulate metabolism through cell-autonomous mechanisms, for example, by alleviation of ER stress or improvement of mitochondrial function, because both have been previously linked with insulin sensitization (Patti and Corvera, 2010; Salvadó et al., 2015; Zhang et al., 2013). However, this seems not to be the case in Becn1F121A mice. We found that when added to cell culture media, serum from Becn1F121A mice also enhances AMPK activation in the cells, suggesting that circulating factors are key for Becn1F121A-induced metabolic benefits. To identify the Becn1-regulated, AMPK-activating, circulating factor, we performed a Luminex metabolic hormonal screen in the serum of WT and Becn1F121A mice and detected a higher circulating level of adiponectin in Becn1F121A mice (Figure 2). Adiponectin is a key insulin-sensitizing adipokine that targets multiple metabolic tissues, and its reduction in circulation is strongly associated with T2D and metabolic syndrome. We found that AMPK activation in Becn1F121A mice is at least partially mediated by increased secretion of adiponectin, because the adiponectin receptor of target cells is essential for the AMPK-activating effects of Becn1F121A mouse serum (Figure 3). Based on the findings, we propose that improved insulin sensitivity in Becn1F121A mice is mediated through a non-cell-autonomous mechanism (through the circulating factor adiponectin).
Primary adipocytes from Becn1F121A mice also secrete more adiponectin into medium, suggesting that Becn1F121A-induced adiponectin secretion is cell autonomous. To reveal the underlying mechanism, we carried out a number of different analyses using Becn1F121A-expressing adipocytes. Inhibition of autophagy by disrupting autophagy genes Atg5 or Atg7 in pre-adipocytes impairs normal white adipocyte development and differentiation (Baerga et al., 2009; Singh et al., 2009; Zhang et al., 2009), likely caused by a defect in the removal of organelles (such as mitochondria) during adipogenesis. However, we found that elevating autophagy by Becn1F121A, in contrast, does not increase WAT mass (Figure S2) or accelerate adipogenesis (Figures 4, S4B, and S4C). Instead, we found that Becn1 interacts with the exocyst components, including Sec6, and compared with WT Becn1, the Becn1F121A mutant interacts more strongly with the exocyst (Figures 5B and 5D). Notably, a previous study performed in HEK293 cells reported that an interaction between Becn1 and another exocyst component Exo84 is important for autophagosome formation during starvation (Bodemann et al., 2011). However, we were not able to detect such interaction in HEK293 cells or adipocytes. In addition, whether and how the Becn1-exocyst interaction plays a role in the regulation of the exocyst pathway and function, especially in organs with endocrine functions (such as WAT), was still unstudied. Using a Becn1 deletion mutant that loses Sec6 binding we identified by mutational studies, we found that the Becn1-Sec6/exocyst binding is essential for Becn1F121A to promote adiponectin secretion (Figures 5E–5G). Given the exocyst complex facilitates the docking of secretory vesicles at the plasma membrane, we proposed a previously unknown role of Becn1 in the regulation of adiponectin secretion via the exocyst. This is a different mechanism from reported unconventional autophagic secretion of the leaderless cargo interleukin-1β (IL-1β) via autophagosome-like vesicles (Zhang et al., 2015) or LC3-dependent secretion of exosomes/extracellular vesicles that contain RNA-binding proteins (Leidal et al., 2020). It is possible that in addition to Becn1, additional autophagy proteins, especially other PtdIns3K complex subunits, may participate in the regulation of the exocyst assembly and adiponectin secretion in adipocytes. Future loss-of-function studies in both WT and Becn1F121A-expressing cells are needed to test these ideas.
After HFD challenge, global Becn1F121A KI mice show improved insulin sensitivity but impaired glucose tolerance (GTT) (Figure S1A) as a result of lower insulin storage in islet β cells caused by chronic autophagic degradation of insulin granules (Yamamoto et al., 2018). It should be noted that although these mice are glucose intolerant with reduced insulin secretion, their islets are not degenerated or reduced in size; instead, their β cell mass is expanded. We reasoned that this compensatory increase in β cell mass can be partly explained by the elevated level of circulating adiponectin in the Becn1F121A mice, because adiponectin has been reported to play a role in maintaining β cell mass (Holland et al., 2011). Using the AAV system, we were able to dissect and differentiate the diverse functions of Becn1F121A in different metabolic cells, for example, adipocytes versus β cells. Different from global Becn1F121A KI mice, adipose tissue-specific Becn1F121A mice show improved glucose tolerance, as well as improved insulin sensitivity (Figure 6), highlighting different physiological outcomes of autophagy manipulation in different metabolic tissues.
Intriguingly, we found that adipose-specific Becn1F121A expression is sufficient to increase circulating levels of adiponectin, but not two other adipokines, leptin and resistin (Figures 7A and 7B), suggesting that Becn1 has specificity in the regulation of adipokine secretion. This is supported by our finding demonstrating that the Becn1-Sec6 interaction is important for the secretion of adiponectin, but not leptin (Figure 5G). We reason that the specificity of the exocyst-autophagy network in the regulation of adipokine secretion is determined by subcellular compartmentation and distinct trafficking pathways of different types of secretory vesicles. Although it is possible that the autophagy machinery and upstream autophagy modulators may regulate a broader endocrine function of WAT and release of additional adipokines, how specificity is achieved at the cellular level is worthy of further investigation. The use of different autophagy mutant adipocytes and mouse lines will be useful to understand the role of the autophagy machinery in the regulation of the adipocyte secretome.
Besides the hyperactive Becn1F121A genetic mutant, we found that upstream physiological and pharmacological autophagy modulators that alter Becn1-Bcl-2 binding also regulate circulating adiponectin levels. The physiological autophagy inducer exercise and the early-stage autophagy inhibitors have opposite effects on circulating adiponectin. Exercise increases the circulating level of adiponectin (Figure S7G), whereas inhibiting the ULK1 or the Becn1-PtdIns3K complex (notably not the late-stage autolysosomal degradation) decreases serum adiponectin (Figures 7E and 7F). We propose that autophagy inducers, such as acute and chronic exercise, promotes adiponectin secretion by releasing Becn1 from inhibitory Bcl-2 binding in adipose tissue (Figure 7G), rather than through enhancing Becn1-TLR9 binding as previously reported in muscle (Liu et al., 2020), for three reasons: (1) via coIP, we found that exercise disrupts the binding of Becn1 with its inhibitor Bcl-2 in the WAT of WT mice (Figure S7H); (2) Becn1F121A does not change TLR9 levels under either RD or HFD treatment (Figure S1B); and (3) although we detected weak interactions between Becn1 and TLR9 in both WAT and muscle, different from Becn1-TLR9 binding in muscle, which is enhanced by exercise (Liu et al., 2020), Becn1-TLR9 interaction in the WAT seems constitutive and not regulated by exercise (Figure S7I). Altogether, our results unveiled a non-degradative, non-cell-autonomous mechanism of Becn1 in metabolic regulation and demonstrated that Becn1-facilitated adiponectin secretion is an important mechanism by which Becn1 regulates systemic AMPK activity and insulin sensitivity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the lead contact, Congcong He (congcong.he@northwestern.edu).
Materials availability
The plasmids and mice used in this study are available from the lead contact.
Data and code availability
This study did not generate/analyze datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal
All mouse care and procedures were performed in accordance with animal experimental protocols approved by the Northwestern University Institutional Animal Care and Use Committee (IACUC). All mice were housed on a 14-hr/10-hr light/dark cycle with ad libitum access to chow diet and water. GFP-LC3 mice, Becn1F121A KI mice, and Becn1+/− KO mice were described previously (Mizushima et al., 2004; Qu et al., 2003; Rocchi et al., 2017). C57BL/6J mice (JAX#000664), Adiponectin-Cre (Adipoq-Cre) mice (JAX#028020), and Adiponectin KO (Adipoq KO) mice (JAX#008195) were acquired from the Jackson Laboratory. All strains have been backcrossed for more than 12 generations to C57BL/6J mice. All experiments were performed with sex- and age-matched mice. Diet-induced diabetic mice were generated by HFD (D12492; Research Diets Inc) feeding for 12 weeks. SBI-0206965 (2 mg/kg body weight), Spautin-1 (2 mg/kg body weight) and chloroquine (50 mg/kg body weight) were injected intraperitoneally (i.p.) into mice for 10 consecutive days.
Cell culture
3T3-L1 preadipocytes, HEK293 cells, HEK293FT cells, Flp-In T-REx 293 cells, NIH 3T3 cells, C2C12 myoblasts, HepG2 cells, Huh7 cells, WI-38 cells, GP2–293 cells, and stromal vascular fraction (SVF) cells were cultured in culture medium: Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin. For 3T3-L1 preadipocytes differentiation to mature adipocytes, two days after full confluency, 3T3-L1 cells (day 0) were cultured in culture medium supplemented with 0.5 mM IBMX, 1 μM dexamethasone, 1 μg/ml insulin for 2 days. Then cells were cultured in culture medium containing 1 μg/ml insulin and refed every two days. At day 10 of culture, 3T3-L1 adipocytes were utilized for experiments. For C2C12 myoblast differentiation to myotubes, fully confluent C2C12 myoblasts (day 0) were cultured in DMEM containing 2% horse serum, 100 units/ml penicillin and 100 μg/ml streptomycin, and refed with fresh medium every day. At day 6 after differentiation, cells were utilized for experiments.
SVF cells containing fibroblastic preadipocytes were isolated from iWAT of 8 week-old mice by collagenase digestion. Briefly, the iWAT was harvested and washed with ice-cold Hank’s balanced salt solution (HBSS). Then iWAT was minced in HBSS containing 300 U/ml type I collagenase and incubated at 37°C for 1 hour with shaking (150 rpm). The digestion was stopped by adding culture medium. The digest was centrifuged at 500 × g for 5 min, and resuspended in the culture medium. After repeating this wash step once again, the cell suspension was filtered through a 70-μm cell strainer and plated in a gelatin-coated dish. To remove non-preadipocytes including red blood cells and immune cells, cells were washed with PBS after 2 hours incubation and then cultured in fresh culture medium. Two days after full confluency, the SVF-derived fibroblastic preadipocytes were cultured in culture medium supplemented with 0.5 mM IBMX, 1 μM dexamethasone, 2 μM rosiglitazone, 1 μg/ml insulin for 2 days. Cells were then cultured in the same way as 3T3-L1 preadipocyte differentiation.
Primary MEFs were isolated from E13.5 embryos by mechanical and enzymatic dissociation method and cultured in MEF culture medium (DMEM with 15% FBS, 2 mM glutamine, 1 x MEM nonessential amino acid, 0.1 mM 2-mercaptoethanol, 100 unit/ml penicillin, and 100 ug/ml streptomycin). Briefly, after the head and red-colored organs were removed from the embryo, tissues were dissociated by pipetting and incubating with 1 mL of 0.05% trypsin-EDTA including 100 kU DNase I (D5025, Millipore Sigma) for 5 min at room temperature. Cell suspension (supernatant) was collected and the digestion was stopped by addion of the MEF culture medium. This digestion step was repeated with the remaining tissues (precipitate). The digest was centrifuged at 300 x g for 5 min and resuspend in the MEF culture medium. The cell suspension was filtered through the 70 μm cell strainer and plated in the culture dish.
Primary hepatocytes were isolated from the adult male mice by perfusing the collagenase solution from the portal vein. Briefly, the mouse was euthanized and perfused with 50 mL of calcium and magnesium ions-free HBSS including 0.5 mM EDTA, followed by 40 mL collagenase solution (HBSS including 200 U/ml type II collagenase [LS004176, Worthington], and 2% BSA) at 5 ml/min using peristaltic pump. After perfusion, the liver was cut into small pieces in the culture medium and passed through the 70 μm cell strainer. The digest was centrifuged at 40 x g for 2 min and resuspend in the culture medium. To eliminate non-parenchymal cells, dead cells and cell debris, the cell suspension was layered over the 1.077 g/ml Histopaque (10771, Millipore Sigma) and centrifuged at 200 x g for 5 min. Then primary hepatocytes were resuspended in the culture medium and plated in the gelatin-coated dish.
Plasmid
Human Sec6 was cloned from cDNA generated by reverse transcription of the total RNA of WI-38 cells. HA-tagged human SEC6 (HA-hSEC6) and Flag-tagged human BECN1 (Flag-hBECN1) were subcloned into the EcoRI restriction enzyme site of the pcDNA 3.1 (−) vector using In-Fusion cloning kit. Flag-tagged human SEC6 (Flag-hSEC6) and HA-tagged human BECN1 (HA-hBECN1) were subcloned into the EcoRI restriction enzyme site of the pCDH-CMV-MCS-EF1-Puro lentivector using In-Fusion cloning kit. HA-tagged mouse BECN1 (HA-mBECN1) was amplified by PCR from BECLIN-HA WT (Addgene plasmid #46993) (Russell et al., 2013) and ligated into the XbaI and EcoRI restriction enzyme sites of pCDH-CMV-MCS-EF1-Puro lentivector. Flag-hBECN1 mutants and HA-mBECN1 mutants were generated by site-direct mutagenesis. Double-strand oligos encoding shRNAs against the target genes (Table S1) were cloned into pLKO.1-TRC cloning vector lentivector (Addgene plasmid #10878) (Moffat et al., 2006). A scramble shRNA lentivector (Addgene plasmid #1864) (Sarbassov et al., 2005) was used as a negative control.
METHOD DETAILS
AICAR treatment
Prior to the AICAR treatment, cells were incubated in DMEM containing 0.5% FBS for 6 hours. Cells were treated with 1 mM AICAR (10010241, Cayman Chemical) for 2 hours. Then cells were washed with PBS and lysed with hot-Laemmli buffer. The cell lysate was heated at 95°C for 5 min and used for immunoblotting.
GTT and ITT
GTT and ITT were performed as previously described (Yamamoto et al., 2018). Briefly, mice were fasted for 6 hours and 4 hours prior to GTT and ITT, respectively. Glucose was i.p. injected at 1.5 g/kg body weight. Insulin was i.p. injected at 0.75 U/kg in RD-fed mice and 0.5 U/kg in HFD-fed mice. Blood was collected from the tail vein, and the blood glucose levels were measured using glucose meters (Counter Next EZ; Ascensia Diabetes Care). The insulinogenic index was calculated as follows: insulinogenic index = (blood insulin at 15 min - blood insulin at 0 min [picomoles per liter])/(blood glucose at 15 min - blood glucose at 0 min [milligrams per deciliter]).
Hormone ELISA
High molecular weight (HMW) adiponectin levels and total adiponectin levels in mouse serum were determined using ELISA (47-ADPMS-E01, ALPCO; and DY1119, R&D Systems), according to the manufacture’s protocol. For serum insulin measurement, mice were i.p. injected with glucose (1.5 g/kg) after 6 hours of fasting, sera were collected at 0, 15, and 30 min after glucose injection, and serum insulin levels were measured by ELISA (90080; Crystal Chem) according to the manufacture’s protocol. Leptin levels in the mouse serum and 3T3-L1 medium were measured by the mouse leptin ELISA kit (DY498–05, R&D Systems). Resistin levels were measured using the resistin ELISA kit (ELM-Resistin, RayBiotech, Inc.). Serum FGF-21 levels were determined using the FGF-21 ELISA kit (KE10042, Proteintech).
Lentivirus production and infection
HEK293FT cells were transfected with a lentiviral expression vector together with a pCMV-VSV-G (Addgene plasmid #8454) (Stewart et al., 2003) and a psPAX2 (Addgene plasmid #12260, a gift from Didier Trono) using the lipofectamine 3000 transfection reagent. Thirty-six hours after transfection, the medium containing the lentivirus was collected and filtered. 3T3-L1 cells, C2C12 cells and NIH 3T3 cells were infected with lentivirus by cell culture in the lentivirus-containing medium with 8 μg/ml polybrene.
Retrovirus production and infection
GP2–293 cells were transfected with pCMV-VSV-G and pBABEpuro GFP-LC3 (Addgene plasmid #22405) (Fung et al., 2008) plasmids using lipofectamine 3000 transfection reagents. Forty-eight hours after transfection, the medium containing the retrovirus was collected and filtered. 3T3-L1 cells were infected with retrovirus by cell culturing in the retrovirus-containing medium with 8 μg/ml polybrene.
Immunopurification of autophagosomes
Autophagosomes were immunopurified from GFP-LC3-expressing 3T3-L1 adipocytes as described previously (Yamamoto et al., 2018). Briefly, cells were homogenized in lysis buffer (0.25 M sucrose, 1 mM EDTA, 20 mM HEPES [pH7.4], 1 x Halt protease and phosphatase inhibitor cocktail) by passing through 27G (5 times) and 30G (10 times) needles. Lysates were centrifuged at 1, 000 × g for 10 min at 4°C. The supernatants were recovered and centrifuged at 20, 000 × g for 20 min at 4°C. Pellets were resuspended in lysis buffer and incubated with anti-GFP antibody for 2 hours at 4°C. The beads were washed for 4 times with lysis buffer, and the immunoprecipitated proteins were eluted in Laemmli buffer and detected by immunoblotting.
Immunofluorescence staining
Cells were plated on a gelatin-coated cover glass, and were fixed with 4% paraformaldehyde in PBS for 30 min and washed with PBS. The cells were then permeabilized with 0.01% digitonin in PBS for 10 min and incubated with primary antibody in PBS containing 5% normal goat serum. After overnight incubation at 4°C, the cells were washed with PBS and incubated with Alexa Fluor-conjugated secondary antibodies in PBS for 1 hour. After washing with PBS, the cells were mounted in a ProLong Diamond Antifade mountant. Fluorescence images were acquired using a spinning-disk confocal microscope [CSU-W1 spinning disk field scanning confocal system (Yokogawa Electric Corp.) with Hamamatsu Flash 4 camera (Hamamatsu Photonics) mounted to a Nikon Ti2 microscope (Nikon)].
Tandem affinity purification of the Becn1 complex
HEK293 cells stably expressing tetracycline-inducible ZZ tag-hBECN1–3 × Flag were generated using the pZZ-3XFlag construct (Sun et al., 2008) and the Flp-In T-REx 293 cell system according to the manufacture’s protocol. Cells were incubated in culture medium containing 1 μg/ml tetracycline for 24 hours. After washing with PBS, the cells were lysed in Triton X-100 cell lysis buffer. The lysate was centrifuged at 15,000 × g for 10 min at 4°C, and the supernatant was incubated with IgG Sepharose for 3 hours at 4°C. The bound proteins were then eluted by TEV protease cleavage. The eluate was incubated with anti-Flag antibody-conjugated beads for 14–16 hours at 4°C, and the bound proteins were eluted in Laemmli buffer and detected by immunoblotting.
Co-immunoprecipitation (co-IP)
HEK293 cells transfected with plasmids by the lipofectamine 3000 transfection reagent, SVF-derived adipocytes, or mouse WAT and muscle were lysed in Triton X-100 cell lysis buffer (1% Triton X-100, 20 mM HEPES [pH7.4], 150 mM NaCl, 1 mM EDTA). The lysate was centrifuged at 15,000 × g for 10 min at 4°C, the supernatant was incubated with the target protein-specific antibody and Protein A/G beads for 14–16 hours at 4°C. After bead washing for 4 times with Triton X-100 cell lysis buffer, immunoprecipitated proteins were eluted in Laemmli buffer and detected by immunoblotting.
AAV delivery of pAAV-FLEX-3xFlag-mBecn1F121A
The EGFP ORF region of the pAAV-FLEX-GFP plasmid (Addgene plasmid #28304, a gift from Edward Boyden) was replaced by 3xFlag-mBecn1F121A ORF using KpnI and XhoI restriction enzymes (NEB) and the In-Fusion HD Plus cloning kit. The AAV2/8 virus was produced by the Sanford Burnham Prebys Medical Discovery Institute Viral Vector Core Facility. The virus titer was 9 × 10e12 (virus genome (VG)/ml). Viral aliquots were stored at −80°C until injection.
6-week old mice were anesthetized with ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight). Hairs were removed from the iWAT area using clippers and hair depilatory cream. The surgical area was disinfected with 5% povidone-iodine solution followed by 70% ethanol. A small incision was made on the skin using surgical scissors, and exposed the iWAT. 5 × 10e10 VG per 30 μl phosphate-buffered saline (PBS) virus solution was injected into each iWAT lobe using a 31 G insulin syringe (BD). The skin was closed with an absorbable suture. For gWAT delivery, 5 × 10e10 VG per 100 μl PBS virus solution was i.p. injected into the gWAT of the same mouse. For postoperative care, mice were injected subcutaneously with meloxicam (0.5 mg/kg body weight) for two consecutive days.
Total lipid extraction and TG measurement
Total lipids were extracted from the mouse liver using the Bligh and Dyer method (Bligh and Dyer, 1959). Extracted total lipids were resuspended in isopropanol. TG levels were measured using Infinity Triglycerides Liquid Stable Reagent (TR22421, Thermo Fisher Scientific) according to the manufacturer’s protocol.
Treadmill exercise
Exercise was performed as described previously (He et al., 2012) using a 10° uphill Exer 3/6 open treadmill (Columbus Instruments), with some modifications. Briefly, for acclimation, on day 1, mice ran for 5 min at 8 m/min, and on day 2, mice ran for 5 min at 8 m/min followed by another 5 min at 10 m/min. On day 3, mice were subject to running exercise starting at 12 m/min for 40 min. After 40 min, the treadmill speed was increased at the rate of 1 m/min every 10 min for 30 min and then every 5 min for 20 min.
RT-PCR and qPCR
Total RNA was isolated from the 3T3-L1 adipocytes and the mouse liver using the Trizol Reagent. Two micrograms of total RNA was reverse-transcribed using High-Capacity cDNA Reverse Transcription kit (4368814, Thermo Fisher Scientific) according to the manufacturer’s protocol. RT-PCR (reverse transcription-PCR) was performed using SapphireAmp Fast PCR Master Mix (RR350, Takara Bio Inc.). The amplicons were separated and detected by agarose gel electrophoresis and ethidium bromide staining. The band intensity was measured using ImageJ software. qPCR (quantitative PCR) was performed using the PowerUP SYBR Green Master Mix and QuantStudio 7 Flex Real-Time PCR system (Thermo Fisher Scientific). The relative gene expressions were quantified using the −2^(ΔΔCt) method and normalized to Actb. The sequences of the primers used for PCR are listed in Table S2.
Immunoblotting
For Akt phosphorylation in the liver, mice were i.p. injected with insulin (1.5 U/kg) after 4 hours fasting, and tissues were collected 15 minutes after insulin injection. For all immunoblotting analyses, tissues and cells were lysed using RIPA lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.1% sodium lauryl sulfate [SDS], 0.5% sodium deoxycholate, 1 mM EDTA, 1 x Halt protease and phosphatase inhibitor cocktail). After centrifugation at 15, 000 × g for 10 min at 4°C, the supernatant was collected and protein concentration was determined using a BCA protein assay kit. Proteins were denatured in Laemmli buffer by heating at 95°C for 5 min. Samples containing the equivalent amount of protein were resolved by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane or nitrocellulose membrane. After membrane blocking by 0.5% skim milk in Tris-buffered saline containing 0.05% Tween-20 (TBS-T), the proteins were probed with primary antibody overnight at 4°C. The membrane was then washed with TBS-T for 10 min 3 times, and incubated with HRP-conjugated secondary antibody for 1 hour. After membrane washing with TBS-T, specific bands were visualized by chemiluminescence and myECL imager (Thermo Fisher Scientific). The band intensities were quantified using ImageJ software.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are shown as mean ± standard errors of the mean (SEM). Data were analyzed by Student’s t test for two groups, and by one-way analysis of variance (ANOVA) followed by Tukey-Kramer test or Dunnett’s test for more than two groups. The difference with a P value of < 0.05 was considered statistically significant. Please note that statistical details are found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-LC3B | Novus Biologicals | Cat#NB100-2220; RRID:AB_10003146 |
| anti-Leptin | Novus Biologicals | Cat#NB300-611; RRID:AB_2136214 |
| anti-TLR9 | Novus Biologicals | Cat#NBP2-24729 |
| anti-p62 | BD Biosciences | Cat#610833; RRID:AB_398152 |
| anti-PLIN2 | Progen | Cat#GP40 |
| anti-acetyl-CoA carboxylase | Cell Signaling Technology | Cat#3676; RRID:AB_2219397 |
| anti-fatty acid synthase | Cell Signaling Technology | Cat#3180, RRID:AB_2100796 |
| anti-PPARγ | Cell Signaling Technology | Cat#2435; RRID:AB_2166051 |
| anti-C/EBPα | Cell Signaling Technology | Cat#8178; RRID:AB_11178517 |
| anti-PLIN1 | Cell Signaling Technology | Cat#9349; RRID:AB_10829911 |
| anti-AMPK | Cell Signaling Technology | Cat#2532; RRID:AB_330331 |
| anti-phospho-AMPK (Thr172) | Cell Signaling Technology | Cat#2535; RRID:AB_331250 |
| anti-phospho-AMPK (Thr172) | Cell Signaling Technology | Cat#2531; RRID:AB_330330 |
| anti-Raptor | Cell Signaling Technology | Cat#2280; RRID:AB_561245 |
| anti-phospho-Raptor (Ser792) | Cell Signaling Technology | Cat#2083; RRID:AB_2249475 |
| anti-p70S6K | Cell Signaling Technology | Cat#2708; RRID:AB_390722 |
| anti-phospho-p70S6K (Thr389) | Cell Signaling Technology | Cat#9234; RRID:AB_2269803 |
| anti-IRS-1 | Cell Signaling Technology | Cat#3407; RRID:AB_2127860 |
| anti-phospho-IRS-1 (Ser302) | Cell Signaling Technology | Cat#2491; RRID:AB_823551 |
| anti-phospho-IRS-1 (Ser636/639) | Cell Signaling Technology | Cat#2388; RRID:AB_330339 |
| anti-Akt | Cell Signaling Technology | Cat#4691; RRID:AB_915783 |
| anti-phospho-Akt (Ser473) | Cell Signaling Technology | Cat#4060; RRID:AB_2315049 |
| anti-ATG14 | MBL International | Cat#PD026; RRID:AB_1953054 |
| anti-UVRAG | Millipore Sigma | Cat#SAB4200005; RRID:AB_10603351 |
| anti-GFP | Millipore Sigma | Cat#G1544; RRID:AB_439690 |
| horseradish peroxidase (HRP)-conjugated anti-Flag M2 | Millipore Sigma | Cat#A8592; RRID:AB_439702 |
| anti-VPS34 antibody | Echelon Biosciences | Cat#Z-R016; RRID:AB_11128208 |
| anti-GAPDH | Thermo Fisher Scientific | Cat#MA5-15738; RRID:AB_10977387 |
| anti-Phospho-IRS1 (Tyr612) | Thermo Fisher Scientific | Cat#44-816G; RRID:AB_2533768 |
| anti-BECN1 | Santa Cruz Biotechnology | Cat#sc-11427; RRID:AB_2064465 |
| anti-BECN1 | Santa Cruz Biotechnology | Cat#sc-48341; RRID:AB_626745 |
| anti-α-Tubulin | Santa Cruz Biotechnology | Cat#sc-53029; RRID:AB_793541 |
| anti-Bcl-2 | Santa Cruz Biotechnology | Cat#sc-7382; RRID:AB_626736 |
| HRP-conjugated anti-β-Actin | Santa Cruz Biotechnology | Cat#sc-47778 HRP; RRID:AB_2714189 |
| anti-SEC6 | Santa Cruz Biotechnology | Cat#sc-374054; RRID:AB_10916711 |
| anti-SEC5 | Santa Cruz Biotechnology | Cat#sc-393230 |
| anti-SEC8 | Santa Cruz Biotechnology | Cat#sc-514215 |
| anti-EXO84 | Santa Cruz Biotechnology | Cat#sc-515532 |
| anti-EXO70 | Santa Cruz Biotechnology | Cat#sc-365825; RRID:AB_10843358 |
| anti-AdipoR1 | Santa Cruz Biotechnology | Cat#sc-518030 |
| anti-HA | Cell Signaling Technology | Cat#3724; RRID:AB_1549585 |
| anti-HA | Santa Cruz Biotechnology | Cat#sc-7392; RRID:AB_627809 |
| anti-adiponectin | Cell Signaling Technology | Cat#2789; RRID:AB_2221630 |
| anti-adiponectin | Thermo Fisher Scientific | Cat#MA1-054; RRID:AB_557516 |
| HRP-conjugated anti-rabbit IgG | Jackson ImmunoResearch Labs | Cat#111-035-003; RRID:AB_2313567 |
| HRP-conjugated anti-mouse IgG | Jackson ImmunoResearch Labs | Cat#115-035-003; RRID:AB_10015289 |
| HRP-conjugated anti-guinea pig IgG | Thermo Fisher Scientific | Cat#A18769; RRID:AB_2535546 |
| Alexa Fluor 594-conjugated anti-mouse IgG | Thermo Fisher Scientific | Cat#A-11005 RRID:AB_2534073 |
| Alexa Fluor Plus 488-conjugated anti-mouse IgG | Thermo Fisher Scientific | Cat#A32723; RRID:AB_2633275 |
| Alexa Fluor Plus 594-conjugated anti-rabbit IgG | Thermo Fisher Scientific | Cat#A32740; RRID:AB_2762824 |
| Mouse IgG Isotype Control | Thermo Fisher Scientific | Cat#02-6502; RRID:AB_2532951 |
| Bacterial and virus strains | ||
| Lentivirus: shRNA (pLKO.1) | This paper | N/A |
| Retrovirus: GFP-LC3 | This paper | N/A |
| Adeno-associated virus (AAV): CAG-FLEX-3xFlag-mBECN1F121A | This paper | N/A |
| Lentivirus: HA-tagged mouse BECN1 (HA-mBECN1) | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Dexamethasone | Millipore Sigma | Cat#D4902; CAS: 50-02-2 |
| Polybrene | Millipore Sigma | Cat#TR-1003-G |
| anti-Flag M2 affinity gel | Millipore Sigma | Cat#A2220 |
| Type I collagenase | Worthington Biomedical Corp. | Cat#LS004196 |
| Type II collagenase | Worthington Biomedical Corp. | Cat#LS004176 |
| 3-isobutyl-1-methylxanthine (IBMX) | MP biomedicals | Cat#195262; CAS: 28822-58-4 |
| Rosiglitazone | Tokyo Chemical Industry Co. | Cat#R0106; CAS: 122320-73-4 |
| Halt Protease and Phosphatase inhibitor cocktail | Thermo Fisher Scientific | Cat#78446 |
| ProLong Diamond Antifade Mountant | Thermo Fisher Scientific | Cat#P36961 |
| Lipofectamine 3000 transfection reagent | Thermo Fisher Scientific | Cat#L3000150 |
| PowerUP SYBR Green Master Mix | Thermo Fisher Scientific | Cat#A25742 |
| Protein A/G Plus-Agarose | Santa Cruz Biotechnology | Cat#sc-2003 |
| IgG Sepharose 6 Fast Flow | Cytiva | Cat#17-0969-01 |
| ProTEV Plus | Promega | Cat#V6101 |
| SBI-0206965 | ApexBio | Cat#A8715; CAS: 1884220-36-3 |
| DNase I | Millipore Sigma | Cat#D5025; CAS: 9003-98-9 |
| AICAR | Cayman Chemical | Cat#10010241; CAS: 2627-69-2 |
| Infinity Triglycerides Liquid Stable Reagent | Thermo Fisher Scientific | Cat#TR22421 |
| SapphireAmp Fast PCR Master Mix | Takara Bio Inc. | Cat#RR350B |
| Spautin-1 | AdooQ Bioscience | Cat#A12942; CAS: 1262888-28-7 |
| Chloroquine (phosphate) | Cayman Chemical | Cat#14194; CAS: 50-63-5 |
| Insulin | Millipore Sigma | Cat#I0516-5ML; CAS: 11070-73-8 |
| Histopaque-1077 | Millipore Sigma | Cat#10771 |
| Critical commercial assays | ||
| Mouse HMW & Total Adiponectin ELISA | ALPCO | Cat#47-ADPMS-E01 |
| Mouse Adiponectin/Acrp30 DuoSet ELISA | R&D Systems | Cat#DY1119 |
| Ultra Sensitive Mouse Insulin ELISA Kit | Crystal Chem | Cat#90080 |
| Mouse Leptin DuoSet ELISA | R&D Systems | Cat#DY498-05 |
| Mouse Resistin ELISA Kit | RayBiotech, Inc. | Cat#ELM-Resistin-1 |
| Mouse/Rat FGF-21 ELISA Kit | Proteintech | Cat#KE10042 |
| In-Fusion HD cloning Plus kit | Takara Bio Inc. | Cat#638910 |
| BCA protein assay kit | Thermo Fisher Scientific | Cat#23225 |
| High-Capacity cDNA Reverse Transcription kit | Thermo Fisher Scientific | Cat#4368814 |
| Experimental models: Cell lines | ||
| Human: HEK293 cells | ATCC | Cat#CRL-1573 |
| Human: HEK293FT cells | Thermo Fisher Scientific | Cat#R70007 |
| Human: T-REx 293 cells | Thermo Fisher Scientific | Cat#R71007 |
| Human: GP2-293 cells | Takara Bio Inc. | Cat#631458 |
| Human: WI-38 cells | ATCC | Cat#CCL-75 |
| Human: Huh7 cells | JCRB Cell Bank | Cat#JCRB0403 |
| Mouse: 3T3-L1 cells | ATCC | Cat#CL-173 |
| Human: HepG2 cells | ATCC | Cat#HB-8065 |
| Mouse: NIH 3T3 cells | ATCC | Cat#CRL-1658 |
| Mouse: C2C12 cells | ATCC | Cat#CRL-1772 |
| Experimental models: Organisms/Strains | ||
| Mouse: Becn1F121A knockin mice: B6.129(Cg)-Becn1tm1.1Hec/J | The Jackson Laboratory | Cat#033360 |
| Mouse: Adipoq-Cre: B6.FVB-Tg(Adipoq-cre)1Evdr/J | The Jackson Laboratory | Cat#028020 |
| Mouse: Adipoq KO: B6;129-Adipoqtm1Chan/J | The Jackson Laboratory | Cat#008195 |
| Mouse: Becn1 heterozygous KO mice: B6.129X1-Becn1tm1Blev/J | The Jackson Laboratory | Cat#018429 |
| Mouse: GFP-LC3 mice: Tg(CAG-EGFP/Map1lc3b)53Nmz | RIKEN BioResource Research Center | Cat#RBRC00806 |
| Mouse: C57BL/6J mice | The Jackson Laboratory | Cat#000664 |
| Oligonucleotides | ||
| Primers for shRNA, see Table S1 | This paper | N/A |
| Primers for PCR, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| pcDNA3.1(-) | Thermo Fisher Scientific | Cat#V79520 |
| pCDH-CMV-MCS-EF1-Puro | System Biosciences | Cat#CD510B-1 |
| BECLIN-HA WT | Russell et al., 2013 | Addgene plasmid Cat#46993; RRID:Addgene_46993 |
| pLKO.1 - TRC cloning vector | Moffat et al., 2006 | Addgene plasmid Cat #10878; RRID:Addgene_10878 |
| pLKO.1 scramble shRNA | Sarbassov et al., 2005 | Addgene plasmid Cat #1864; RRID:Addgene_1864 |
| pCMV-VSV-G | Stewart et al., 2003 | Addgene plasmid Cat #8454; RRID:Addgene_8454 |
| psPAX2 | Gift of Didier Trono lab | Addgene plasmid Cat #12260; RRID:Addgene_12260 |
| pBABEpuro GFP-LC3 | Fung et al., 2008 | Addgene plasmid Cat #22405; RRID:Addgene_22405 |
| pAAV-FLEX-GFP | Gift of Edward Boyden lab | Addgene plasmid Cat #28304; RRID:Addgene_28304 |
| pZZ-Flag | Sun et al., 2008 | N/A |
| pAAV-FLEX-3xFlag-mBecn1F121A | This Paper | N/A |
| pcDNA3.1 (-) HA-hSec6 | This Paper | N/A |
| pcDNA3.1 (-) Flag-hBECN1 | This Paper | N/A |
| pCDH-Flag-hSec6 | This Paper | N/A |
| pCDH-HA-hBECN1 | This Paper | N/A |
| pCDH-HA-mBECN1 | This Paper | N/A |
| Software and algorithms | ||
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| NIS-Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
Highlights.
Autophagy-hyperactive Becn1F121A mice show systemic activation of AMPK
Becn1 positively regulates the secretion of an adipokine, adiponectin
Becn1-exocyst interaction is essential for facilitating adiponectin secretion
Adipose Becn1F121A is sufficient to improve metabolism by elevating serum adiponectin
ACKNOWLEDGMENTS
We thank Dr. Qing Zhong for providing the pZZ-FLAG construct; Dr. Noboru Mizushima for providing the GFP-LC3 mice; Sanford Burnham Prebys Medical Discovery Institute Viral Vector Core Facility for AAV production; and the following facilities at Northwestern University Feinberg School of Medicine for technical support: the Comprehensive Metabolic Core for metabolic hormone multiplex analyses, the Center for Advanced Microscopy for confocal microscopy studies, the NUSeq Core of Center for Genetic Medicine for qPCR analyses, and the Sanger Sequencing Core for sequencing. The study is supported by NIH R01-DK113170 and Northwestern University Lurie Cancer Center Translational Bridge Award (to C.H.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109184.
REFERENCES
- Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, et al. (1999). Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun 257, 79–83. [DOI] [PubMed] [Google Scholar]
- Baerga R, Zhang Y, Chen PH, Goldman S, and Jin S (2009). Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy 5, 1118–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, Pajvani UB, Rizza RA, and Scherer PE (2007). Selective downregulation of the high molecular weight form of adiponectin in hyperinsulinemia and in type 2 diabetes: differential regulation from nondiabetic subjects. Diabetes 56, 2174–2177. [DOI] [PubMed] [Google Scholar]
- Bel S, Pendse M, Wang Y, Li Y, Ruhn KA, Hassell B, Leal T, Winter SE, Xavier RJ, and Hooper LV (2017). Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 357, 1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Blu€mer RM, van Roomen CP, Meijer AJ, Houben-Weerts JH, Sauerwein HP., and Dubbelhuis PF (2008). Regulation of adiponectin secretion by insulin and amino acids in 3T3-L1 adipocytes. Metabolism 57, 1655–1662. [DOI] [PubMed] [Google Scholar]
- Bodemann BO, Orvedahl A, Cheng T, Ram RR, Ou YH, Formstecher E, Maiti M, Hazelett CC, Wauson EM, Balakireva M, et al. (2011). RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 144, 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Chen K, Li J, Gao P, Li Q, Mi S, Wu X, and Zhao AZ (2007). Regulation of adiponectin and leptin secretion and expression by insulin through a PI3K-PDE3B dependent mechanism in rat primary adipocytes. Biochem. J 403, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, et al. (2008). Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 8, 325–332. [DOI] [PubMed] [Google Scholar]
- Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, et al. (2015). Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 59, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung C, Lock R, Gao S, Salas E, and Debnath J (2008). Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 19, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia D, and Shaw RJ (2017). AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 66, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L, Liao B, Jin L, Huang Z, Triggle CR, Ding H, Zhang J, Huang Y, Lin Z, and Xu A (2019). Exercise Alleviates Obesity-Induced Metabolic Dysfunction via Enhancing FGF21 Sensitivity in Adipose Tissues. Cell Rep. 26, 2738–2752.e4. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, and Shaw RJ (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemmerle G, Moustafa T, Woelkart G, Büttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, et al. (2011). ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med 17, 1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajri T, Tao H, Wattacheril J, Marks-Shulman P, and Abumrad NN (2011). Regulation of adiponectin production by insulin: interactions with tumor necrosis factor-α and interleukin-6. Am. J. Physiol. Endocrinol. Metab. 300, E350–E360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Boutin P, Mori Y, Tobe K, Dina C, Yasuda K, Yamauchi T, Otabe S, Okada T, Eto K, et al. (2002). Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes 51, 536–540. [DOI] [PubMed] [Google Scholar]
- Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, and Kobayashi M (2000). A rapamycin-sensitive pathway downregulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol. Endocrinol 14, 783–794. [DOI] [PubMed] [Google Scholar]
- He C, and Levine B (2010). The Beclin 1 interactome. Curr. Opin. Cell Biol 22, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al. (2012). Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Wei Y, Sun K, Li B, Dong X, Zou Z, Liu Y, Kinch LN, Khan S, Sinha S, et al. (2013). Beclin 2 functions in autophagy, degradation of G protein-coupled receptors, and metabolism. Cell 154, 1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heider MR, and Munson M (2012). Exorcising the exocyst complex. Traffic 13, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, et al. (2011). Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med 17, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, Bauer SM, Wade M, Singhal E, Cheng CC, et al. (2013). An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 17, 790–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta K, Funahashi T, Bodkin NL, Ortmeyer HK, Arita Y, Hansen BC, and Matsuzawa Y (2001). Circulating concentrations of the adipocyte protein adiponectin are decreased in parallel with reduced insulin sensitivity during the progression to type 2 diabetes in rhesus monkeys. Diabetes 50, 1126–1133. [DOI] [PubMed] [Google Scholar]
- Jimenez V, Muñoz S, Casana E, Mallol C, Elias I, Jambrina C, Ribera A, Ferre T, Franckhauser S, and Bosch F (2013). In vivo adeno-associated viral vector-mediated genetic engineering of white and brown adipose tissue in adult mice. Diabetes 62, 4012–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, et al. (2008). Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 8, 318–324. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, and Tobe K (2006). Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest 116, 1784–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur J, and Debnath J (2015). Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 16, 461–472. [DOI] [PubMed] [Google Scholar]
- Komai AM, Musovic S, Peris E, Alrifaiy A, El Hachmane MF, Johansson M, Wernstedt Asterholm I, and Olofsson CS (2016). White Adipocyte Adiponectin Exocytosis Is Stimulated via b3-Adrenergic Signaling and Activation of Epac1: Catecholamine Resistance in Obesity and Type 2 Diabetes. Diabetes 65, 3301–3313. [DOI] [PubMed] [Google Scholar]
- Kondo H, Shimomura I, Matsukawa Y, Kumada M, Takahashi M, Matsuda M, Ouchi N, Kihara S, Kawamoto T, Sumitsuji S, et al. (2002). Association of adiponectin mutation with type 2 diabetes: a candidate gene for the insulin resistance syndrome. Diabetes 51, 2325–2328. [DOI] [PubMed] [Google Scholar]
- Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, et al. (2002). Disruption of adiponectin causes insulin resistance and neointimal formation. J. Biol. Chem 277, 25863–25866. [DOI] [PubMed] [Google Scholar]
- Kuramoto K, and He C (2018). The BECN1-BCL2 complex regulates insulin secretion and storage in mice. Autophagy 14, 2026–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Castro C, Luo N, Wallace P, Klein RL, and Garvey WT (2006). Adiponectin multimeric complexes and the metabolic syndrome trait cluster. Diabetes 55, 249–259. [PubMed] [Google Scholar]
- Leidal AM, Huang HH, Marsh T, Solvik T, Zhang D, Ye J, Kai F, Goldsmith J, Liu JY, Huang YH, et al. (2020). The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat. Cell Biol. 22, 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Liu R, Dong X, and Zhong Q (2015). Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 25, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Shin HJ, Ding EL, and van Dam RM (2009). Adiponectin levels and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA 302, 179–188. [DOI] [PubMed] [Google Scholar]
- Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al. (2011). AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CY, Hong W, and Han W (2015). Adiponectin is released via a unique regulated exocytosis pathway from a pre-formed vesicle pool on insulin stimulation. Biochem. J 471, 381–389. [DOI] [PubMed] [Google Scholar]
- Lin Z, Tian H, Lam KS, Lin S, Hoo RC, Konishi M, Itoh N, Wang Y, Bornstein SR, Xu A, and Li X (2013). Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 17, 779–789. [DOI] [PubMed] [Google Scholar]
- Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, et al. (2011). Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Nguyen PT, Wang X, Zhao Y, Meacham CE, Zou Z, Bordieanu B, Johanns M, Vertommen D, Wijshake T, et al. (2020). TLR9 and beclin 1 crosstalk regulates muscle AMPK activation in exercise. Nature 578, 605–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda Y, Itabe H, Odaki M, Hama K, Fujimoto Y, Mori M, Sasabe N, Aoki J, Arai H, and Takano T (2006). ADRP/adipophilin is degraded through the proteasome-dependent pathway during regression of lipid-storing cells. J. Lipid Res. 47, 87–98. [DOI] [PubMed] [Google Scholar]
- Mei K, Li Y, Wang S, Shao G, Wang J, Ding Y, Luo G, Yue P, Liu JJ, Wang X, et al. (2018). Cryo-EM structure of the exocyst complex. Nat. Struct. Mol. Biol 25, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzaghi C, Ercolino T, Di Paola R, Berg AH, Warram JH, Scherer PE, Trischitta V, and Doria A (2002). A haplotype at the adiponectin locus is associated with obesity and other features of the insulin resistance syndrome. Diabetes 51, 2306–2312. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, and Ohsumi Y (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Murdolo G, Hammarstedt A, Schmelz M, Jansson PA, and Smith U (2009). Acute hyperinsulinemia differentially regulates interstitial and circulating adiponectin oligomeric pattern in lean and insulin-resistant, obese individuals. J. Clin. Endocrinol. Metab 94, 4508–4516. [DOI] [PubMed] [Google Scholar]
- Murthy M, Garza D, Scheller RH, and Schwarz TL (2003). Mutations in the exocyst component Sec5 disrupt neuronal membrane traffic, but neurotransmitter release persists. Neuron 37, 433–447. [DOI] [PubMed] [Google Scholar]
- Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, et al. (2006). Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J. Biol. Chem 281, 2654–2660. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Parker JL, Lugus JJ, and Walsh K (2011). Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol 11, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE, and Donner DB (2001). A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc. Natl. Acad. Sci. USA 98, 4640–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajvani UB, Hawkins M, Combs TP, Rajala MW, Doebber T, Berger JP, Wagner JA, Wu M, Knopps A, Xiang AH, et al. (2004). Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J. Biol. Chem 279, 12152–12162. [DOI] [PubMed] [Google Scholar]
- Patti ME, and Corvera S (2010). The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev 31, 364–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson TM, Kramer DL, and Rondinone CM (2001). Serine/threonine phosphorylation of IRS-1 triggers its degradation: possible regulation by tyrosine phosphorylation. Diabetes 50, 24–31. [DOI] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. (2003). Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest 112, 1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhshandehroo M, Knoch B, Mu€ller M, and Kersten S (2010). Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocchi A, and He C (2015). Emerging roles of autophagy in metabolism and metabolic disorders. Front. Biol. (Beijing) 10, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocchi A, and He C (2017a). Activating Autophagy by Aerobic Exercise in Mice. J. Vis. Exp. 2017, 55099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocchi A, and He C (2017b). Regulation of Exercise-Induced Autophagy in Skeletal Muscle. Curr. Pathobiol. Rep 5, 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocchi A, Yamamoto S, Ting T, Fan Y, Sadleir K, Wang Y, Zhang W, Huang S, Levine B, Vassar R, and He C (2017). A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLoS Genet. 13, e1006962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, and Guan KL (2013). ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 15, 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadó L, Palomer X, Barroso E, and Vázquez-Carrera M (2015). Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. Metab. 26, 438–448. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, and Czaja MJ (2009). Autophagy regulates adipose mass and differentiation in mice. J. Clin. Invest 119, 3329–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern JH, Rutkowski JM, and Scherer PE (2016). Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 23, 770–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumvoll M, Tschritter O, Fritsche A, Staiger H, Renn W, Weisser M, Machicao F, and Häring H (2002). Association of the T-G polymorphism in adiponectin (exon 2) with obesity and insulin sensitivity: interaction with family history of type 2 diabetes. Diabetes 51, 37–41. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Goldberg JL, Qiao LY, and Mitchell JJ (1999). Insulin-induced insulin receptor substrate-1 degradation is mediated by the proteasome degradation pathway. Diabetes 48, 1359–1364. [DOI] [PubMed] [Google Scholar]
- Sun Q, Fan W, Chen K, Ding X, Chen S, and Zhong Q (2008). Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 105, 19211–19216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano A, Usui I, Haruta T, Kawahara J, Uno T, Iwata M, and Kobayashi M (2001). Mammalian target of rapamycin pathway regulates insulin signaling via subcellular redistribution of insulin receptor substrate 1 and integrates nutritional signals and metabolic signals of insulin. Mol. Cell. Biol 21, 5050–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Carvalheira JB, Tambascia RC, Bezerra RM, Amaral ME, Carneiro EM, Folli F, Franchini KG, and Saad MJ (2005). Regulation of insulin signalling by hyperinsulinaemia: role of IRS-1/2 serine phosphorylation and the mTOR/p70 S6K pathway. Diabetologia 48, 506–518. [DOI] [PubMed] [Google Scholar]
- Werner ED, Lee J, Hansen L, Yuan M, and Shoelson SE (2004). Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. J. Biol. Chem 279, 35298–35305. [DOI] [PubMed] [Google Scholar]
- Wu B, and Guo W (2015). The Exocyst at a Glance. J. Cell Sci 128, 2957–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Motoshima H, Mahadev K, Stalker TJ, Scalia R, and Goldstein BJ (2003). Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes 52, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Xie L, O’Reilly CP, Chapes SK, and Mora S (2008). Adiponectin and leptin are secreted through distinct trafficking pathways in adipocytes. Biochim. Biophys. Acta 1782, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Sztalryd C, Lu X, Tansey JT, Gan J, Dorward H, Kimmel AR, and Londos C (2005). Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J. Biol. Chem 280, 42841–42847. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Kuramoto K, Wang N, Situ X, Priyadarshini M, Zhang W, Cordoba-Chacon J, Layden BT, and He C (2018). Autophagy Differentially Regulates Insulin Production and Insulin Sensitivity. Cell Rep. 23, 3286–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, et al. (2001). The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med 7, 941–946. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, et al. (2002). Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med 8, 1288–1295. [DOI] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, and Hotamisligil GS (2010). Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH, and Kim JB (2006). Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes 55, 2562–2570. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, and Jin S (2009). Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 106, 19860–19865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Hietakangas V, Wee S, Lim SC, Gunaratne J, and Cohen SM (2013). ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 27, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Kenny SJ, Ge L, Xu K, and Schekman R (2015). Translocation of interleukin-1b into a vesicle intermediate in autophagy-mediated secretion. eLife 4, e11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.