

Abstract
Despite a remarkable increase in the genomic profiling of cancer, integration of genomic discoveries into clinical care has lagged behind. We report the feasibility of rapid identification of targetable mutations in 153 pediatric patients with relapsed/refractory or high-risk leukemias enrolled on a prospective clinical trial conducted by the LEAP Consortium. Eighteen percent of patients had a high confidence, Tier 1 or 2, recommendation. We describe clinical responses in the 14% of patients with relapsed/refractory leukemia who received the matched targeted therapy. Further, in order to inform future targeted therapy for patients, we validated variants of uncertain significance (VUS), performed ex vivo drug sensitivity testing in patient leukemia samples, and identified new combinations of targeted therapies in cell lines and patient-derived xenograft models. These data and our collaborative approach should inform the design of future precision medicine trials.
Introduction:
Since primary patient samples are easy to obtain in patients with leukemia, the study of leukemia has often been at the leading edge of technology advances. For example, the first characterization of gene expression programs in cancer and the first sequencing of a human cancer genome were performed in the context of leukemia(1–3). Subsequently, the last decade has seen the deep genomic characterization of childhood leukemias, including acute myeloid leukemia (AML)(4), B-cell precursor lymphoblastic leukemia (B-ALL)(5), T-cell precursor lymphoblastic leukemia (T-ALL)(6) and myelodysplastic syndrome (MDS)(7). However, the feasibility of applying these discoveries to clinical care has not been tested prospectively. Indeed, most early precision medicine studies in pediatrics in the United States have focused on solid tumors. Rationale for this solid tumor centric approach have included arguments that pediatric leukemia is too aggressive for therapeutic decisions to be delayed by genomic data wait times and that single agent therapies are unlikely to be effective for leukemia. Pediatric leukemia, however, remains the second leading cause of cancer-related death in children, and while exciting advances have been made in the recent application of chimeric antigen receptor T-cell (CAR T) therapy for the treatment of children with relapsed/refractory B-ALL, 50% of patients treated with this modality will also relapse(8–10).
A number of studies have evaluated the feasibility of identifying targetable mutations in pediatric patients with relapsed or refractory cancers through single institution studies or multi-institutional consortiums. These studies have largely evaluated patients with solid tumors and brain tumors and have not comprehensively addressed the feasibility of identifying targetable mutations, in real time, in patients with acute leukemia(11). Two multi-institutional studies focused exclusively on children with solid tumors. For example, the Baylor College of Medicine Advancing Sequencing into Childhood Cancer Care (BASIC3) study examined tumor/germline Whole Exome Sequencing (WES) in 150 children with solid tumors, with potentially actionable findings in 40% of newly diagnosed patients(12). The multi-institutional Individualized Cancer Therapy (iCAT) study, enrolling pediatric/young adult patients with solid tumors from four institutions, reported that among 100 patients, 31% received a targeted therapy recommendation, but only three of these patients actually received the therapy(13). For two of the larger single institution studies, while children with leukemia were enrolled, they represented a minority of the participants. The University of Michigan reported a single site study of 102 pediatric patients with relapsed, refractory or rare cancers. Of these, 46% had actionable findings that changed their management, ranging from change in therapy to genetic counseling for a cancer predisposition(14). Only 28 of the children enrolled in this study had a hematologic malignancy. In Columbia University’s Precision in Pediatric Sequencing (PIPSeq) program, 101 high-risk patients, including 38 with hematologic diseases, were evaluated with a combination of WES, RNA sequencing and targeted panels. Thirty-eight percent of patients in the PIPSeq program were reported to have at least one potentially actionable finding, including 17 patients with hematologic conditions(15).
In addition to the lack of pediatric leukemia data generated in prospective cancer precision medicine studies, other gaps have been noted in these initial clinical trials. First, many variants of uncertain significance (VUS) have been identified, with limited efforts to better characterize these variants. Second, there remains a paucity of preclinical response data in the specific pediatric disease context of interest for some of the relevant mutational events. For example, preclinical response to PI3K inhibitors in adult PIK3CA p.E545K mutant breast cancer has been reported(16), but data on preclinical response in PIK3CA p.E545K mutant embryonic rhabdomyosarcoma are lacking(13). Third, drug access remains a major problem for these children with the median lag time between first in human and first in child clinical trials of 6.5 years(17).
To address these gaps, we established a pediatric leukemia clinical genomics consortium in the United States, known as the Leukemia Precision-based Therapy (LEAP) Consortium, which includes 15 major pediatric cancer institutions. We hypothesized that it is feasible to identify and match, in real-time, actionable alterations with a targeted therapy for pediatric patients with relapsed, refractory or high-risk leukemias or MDS. Using the combination of a DNA-based next-generation sequencing (NGS) panel and RNA-based gene fusion testing, followed by data review by our multidisciplinary molecular tumor board, we conducted a clinical trial to test this hypothesis. Given the limited availability of targeted therapies for pediatric patients and the delays in testing novel drugs in this patient population in clinical trials(17), we expected that few patients would be able to access the targeted therapy. Using the power of a collaborative effort and the relative ease of access to leukemia samples, we tested recommended therapies ex vivo in primary patient samples, explored possibilities for synergistic drug combinations, and utilized an approach for novel variant validation with the goal of increasing the potential for high confidence matched therapies. This multi-faceted approach may inform the future practice of precision medicine for patients with leukemia.
Results:
LEAP Consortium Clinical Trial
Between July 2016 and September 2018, we enrolled 153 patients across 13 pediatric institutions. All subjects had relapsed/refractory or high-risk leukemia or MDS. Patients were enrolled in two cohorts; the distribution of diagnoses is shown in Table 1. These patients represented 41% of eligible patients for 11 of the 13 institutions for which baseline clinical data was available and were enriched for patients with relapsed/refractory leukemias. Patients had a diagnostic bone marrow sample submitted for sequencing at the time of enrollment or a peripheral blood sample with at least 20% leukemia involvement. There was no blast percentage requirement for patients with MDS or juvenile myelomonocytic leukemia (JMML). The samples were profiled using a CLIA-certified DNA-based NGS panel targeting selected exons across 95 genes(18). The panel was not designed to target splice variants or alterations in regulatory regions. Samples from patients with B-ALL were also profiled for the presence of Philadelphia chromosome-like (Ph-like) fusions initially using multiplex RT-PCR, but we subsequently transitioned to a targeted RNA-based fusion detection panel (AMPSeq) when this test became available for clinical application(19). Five patients enrolled at Columbia University Medical Center also had clinical RNASeq data that were submitted and analyzed as part of this trial(20). All data used for the matched targeted therapy (MTT) recommendations and returned to the treating oncologist were obtained in CAP-CLIA approved labs.
Table 1.
Patient Characteristics
| Total N (%) | Cohort 1: Relapsed/Refractory N (%) | Cohort 2: Newly Diagnosed N (%) | |
|---|---|---|---|
| N | 153 | 101 | 52 |
| Sex | |||
| Female | 73 (48) | 46 (46) | 27 (51) |
| Male | 80 (52) | 55 (54) | 25 (48) |
| Age (yrs.) at sample, median (range) | 11 (<1, 26) | 11 (1, 25) | 7 (<1, 26) |
| < 1 yrs. | 6 (11) | 0 (0) | 6 (11) |
| 1 – 9 yrs. | 64 (42) | 41 (41) | 23 (44) |
| 10 – 17 yrs. | 54 (35) | 38 (38) | 16 (31) |
| ≥ 18 yrs. | 29 (19) | 22 (22) | 7 (13) |
| Disease | |||
| AML | 67 (44) | 34 (34) | 33 (63) |
| MDS | 1 (<1) | 0 (0) | 1 (2) |
| JMML | 3 (2) | 1 (1) | 2 (4) |
| ALL | 59 (39) | 56 (56) | 3 (6) |
| B-cell | 49 | 48 | 1 |
| T-cell | 10 | 8 | 2 |
| Infant KMT2A-rearranged ALL | 3 (2) | 1 (1) | 2 (4) |
| Leukemia of ambiguous lineage | 9 (6) | 4 (4) | 5 (9) |
| Other rare leukemia | 4 (3) | 2 (2) | 2 (4) |
| Therapy-related leukemia | 7 (5) | 3 (3) | 4 (8) |
The average turnaround time to results for the DNA sequencing panel was 5.25 (range 2-15) days and 16.6 (range 4-35) days for the RNA-based fusion panel; these data were immediately communicated to the enrolling site. The sequencing data, in combination with clinical data and any available local sequencing data, were then reviewed as part of an expert virtual biweekly-weekly panel meeting. The average time to expert panel review was 15 days from the time of return of DNA sequencing data, and a summary of targetable lesions was issued to the site principal investigator (PI) and treating oncologist. Panel reviews were prioritized for patients with relapsed/refractory disease in immediate need of a next therapy and were also scheduled to accommodate the participation of the treating oncologist or site PI. The treatment recommendations were tiered based on the level of evidence for the genomic lesion and the drug target. Recommendations for targeted therapy were for a drug target/pathway or for a specific drug, depending on the type of genomic alteration that was present. The tiering system is listed in Supplementary Table 1 and was based on the iCat study(13). For patients with multiple targetable alterations, each was tiered according to the tiering scale provided in Supplementary Table 1 but was not further prioritized. Final treatment decisions were at the discretion of the treating oncologist. The trial schema is summarized in Figure 1.
Figure 1:
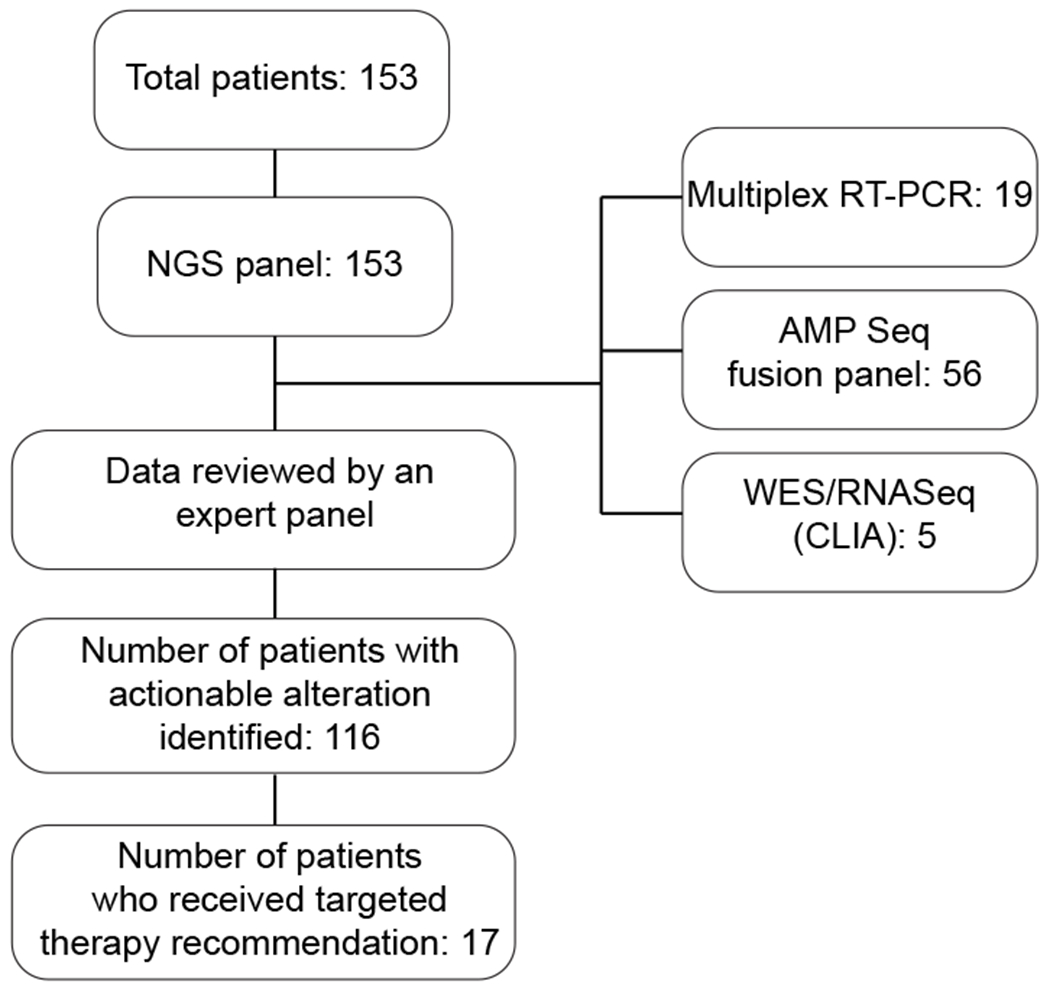
LEAP Consortium clinical trial schema. The diagram outlines the clinical genomics data that was obtained during this trial and informed matched targeted therapy recommendations.
Use of additional NGS data and correlation among panels
There has been an increased availability of NGS panel sequencing; however, the correlation of findings among panels is not known. Currently, the NCI-COG MATCH trial requires tissue for central laboratory sequencing for trial enrollment and matching of the genomic alteration to targeted therapy. With increased availability of local sequencing, we reasoned it would be prudent to know if there is significant correlation among different laboratory tests, as availability of local sequencing may shorten time to results and thus increase clinical trial eligibility and accrual. For the LEAP trial, sequencing using the central DNA panel was required, but 32 of 153 patients (21%) enrolled on this trial had additional panel sequencing performed at their referring institution. For genes that were covered by both panels, we asked if there were correlations in 1) mutations discovered and 2) variant allele frequencies for these mutations. This comparison included 20 genes, encompassing 59 mutations (37 missense, 12 frameshift and 10 nonsense). There was significant correlation between variant allele frequency (VAF) percentages detected by the assays, with r=0.92 (Supplementary Figure 1). There were three missense mutations that were discordant between panels, all with low VAFs of 3-6%, at or near the limit of detection for both assays where small stochastic differences may result in discordant calls. These discrepancies may have also been influenced by differences in bone marrow sampling. The pathologists’ interpretations of the mutations as pathogenic/likely pathogenic versus variants of unknown significance were concordant for 86% of the mutations.
Targeted sequencing identifies therapeutic targets, alters diagnosis, and informs germline evaluation
We broadly defined actionable alterations as a cancer-associated genomic event for which there was a targeted drug available. The actionability of the alteration was tiered based on the level of evidence for the targeted drug and the specific alteration in the disease context (Table 2 and Supplementary Table 1). The actionable genetic alterations were identified by a combination of standard-of-care cytogenetics/FISH and sequencing performed as part of the study. The co-mutation plot in Figure 2 summarizes the genomic findings that led to the recommendation, and the full mutation data are included in the Supplementary Data. For 116 (76%) of patients enrolled on this trial, an actionable genetic alteration was identified (Table 2), although the majority of these were not high confidence (Tier 1 or 2) matches. Indeed, the majority of recommendations were in Tier 3, where only pre-clinical evidence was present for inhibition of the specific target in the specific tumor type. Twenty-one (18%) of recommendations were categorized as Tier 1 or 2.
Table 2.
Actionable Alteration and Inhibitor Recommendations
| Total N (%) | Cohort 1: Relapsed/Refractory N (%) | Cohort 2: Newly Diagnosed N (%) | |
|---|---|---|---|
| N | 153 | 101 | 52 |
| Actionable Alteration Identified [90% CI]* | 116 (76) [69-81] | 77 (77) [69-84] | 39 (74) [62-83] |
| Alteration Tier | |||
| 1 | 16 (14) | 9 (11) | 7 (18) |
| 2 | 6 (4) | 6 (8) | 0 (0) |
| 3 | 72 (62) | 46 (56) | 26 (68) |
| 4 | 7 (6) | 5 (6) | 2 (5) |
| 5 | 16 (14) | 13 (16) | 3 (8) |
| Additional Recommendation† | 3 (2) | 3 (3) | 0 (0) |
| Recommended Inhibitor Target [90% CI]** | 114 (75) [68-80] | 77 (76) [68-83] | 37 (71) [59-81] |
| MEK | 62 (41) | 39 (39) | 23 (44) |
| DOT1L/Menin | 23 (15) | 11 (11) | 12 (23) |
| FLT3 | 14 (9) | 9 (12) | 5 (9) |
| MTOR | 13 (8) | 9 (9) | 4 (8) |
| JAK1/2 | 14 (9) | 10 (10) | 4 (8) |
| JAK3 | 1 (1) | 1 (1) | 0 (0) |
| WEE1 | 6 (4) | 5 (5) | 1 (2) |
| HDAC | 5 (3) | 4 (4) | 1 (2) |
| ABL | 5 (3) | 5 (5) | 0 (0) |
| Gamma Secretase | 4 (3) | 2 (2) | 2 (4) |
| IDH1 | 4 (3) | 2 (2) | 2 (4) |
| CDK4/6 | 3 (2) | 2 (2) | 1 (2) |
| KIT | 3 (2) | 0 (0) | 3 (6) |
| PI3K | 3 (2) | 3 (3) | 0 (0) |
| TP53 | 2 (1) | 1 (1) | 1 (2) |
| AKT | 2 (1) | 2 (2) | 0 (0) |
| BCL2 | 2 (1) | 2 (2) | 0 (0) |
| BRAF | 2 (1) | 0 (0) | 2 (4) |
| CRM1 | 2 (1) | 1 (1) | 1 (2) |
| SRC | 2 (1) | 2 (2) | 0 (0) |
| PDGFRB | 1 (1) | 1 (1) | 0 (0) |
| CXCR4 | 1 (1) | 1 (1) | 0 (0) |
One patient had an actionable mutation found, but the patient died prior to having a treatment recommendation sent (case 100) and one patient had a BCR-ABL1 fusion identified but was already being treated with dasatinib so no additional recommendation was provided (case 90).
Patients may have had multiple recommended inhibitors.
Additional features of interest added retrospectively based on targetability during the course of the clinical trial (ex. TP53, CDKN2A)
Figure 2:
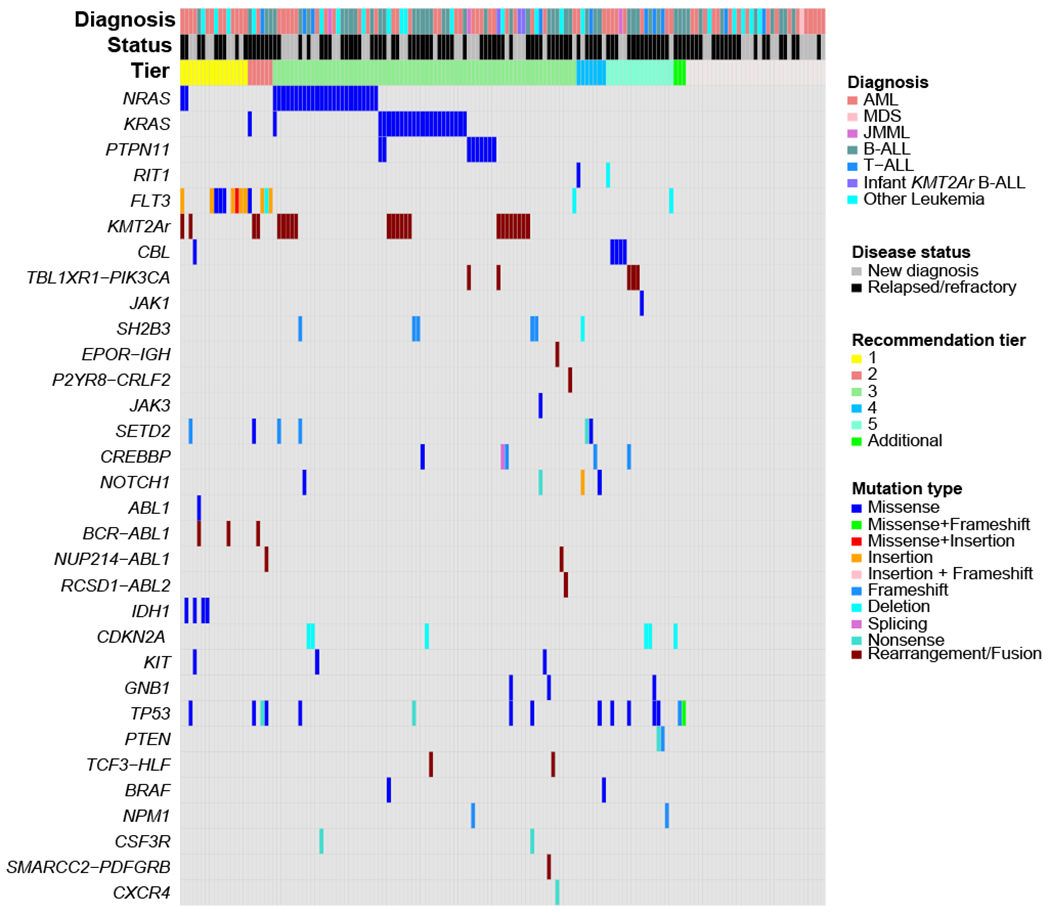
Landscape of targetable genomic alterations for the LEAP Consortium Clinical Trial. Co-mutation plot showing targetable genomic alterations for the 153 patients enrolled on the LEAP trial. Samples are sorted according to the recommendation tier (across top) and grouped by drug target along left axis. For patients with more than one targetable alteration, the highest tier recommendation is used for diagram order.
The exact mutation and leukemia disease subtype informed the tiering classification as described in Supplementary Table 1. As an example, identified mutations in FLT3 were classified in Tier 1, 2, and 5. Midostaurin, sorafenib, and gilteritinib have all been demonstrated to have clinical efficacy in pediatric and/or adult patients with FLT3-mutant AML(21–24); thus a recommendation for using a FLT3 inhibitor in a patient with FLT3-mutant AML was classified as Tier 1. Conversely, while FLT3 mutations have been described in B-ALL(25,26) and T-ALL(6,27), it is not known whether FLT3 inhibitors have efficacy in patients with these leukemias. Based on the evidence in AML, we considered a recommendation for a FLT3 inhibitor in patients with ALL as Tier 2. Finally, we identified a rare FLT3 p.D600del juxtamembrane domain mutation in patient #61 with relapsed AML. This mutation is predicted to interrupt the inhibitory conformation of the FLT3 protein, thereby potentially leading to increased FLT3 signaling, although it remains unclear whether this mutation is activating(4). Given the importance of FLT3 in AML and the limited available treatment options, the expert panel tiered this mutation as Tier 5.
Seventeen patients received the recommended targeted therapy, 14 of whom had relapsed/refractory disease (Table 3 and Supplementary Table 2). Of the 17 patients who received targeted therapy, 11 of the targetable genomic changes would not have been identified with standard-of-care clinical evaluation performed by most institutions (Table 3, “Detected with NGS only” column). The top reasons for not using the targeted therapy recommendation included: 1) having a standard therapy option (59%), 2) having another clinical trial available (19%), and 3) patient too ill to receive the targeted therapy (11%) and are all categorized in Supplementary Table 3.
Table 3:
Targeted therapy used
| Patient number | Disease | Target | Proposed drug or target/pathway inhibitor | Drug used | Tier | Toxicity attributed to targeted therapy | How targeted therapy was accessed? | Detected with NGS only |
|---|---|---|---|---|---|---|---|---|
| 1 | AML, new diagnosis | FLT3 ITD | FLT3 inhibitor | sorafenib | 1 | * | off label | |
| 121 | AML, new diagnosis | FLT3 p.I836del | FLT3 inhibitor | sorafenib | 3 | none | off label | X |
| 140 | AML, new diagnosis | FLT3 ITD | FLT3 inhibitor | sorafenib | 1 | * | off label | |
| 15 | AML, relapsed | FLT3 ITD | FLT3 inhibitor | gilteritinib | 1 | * | single patient IND (compassionate use) | |
| 31 | AML, relapsed | NRAS | MEK inhibitor | trametinib | 3 | * | off label | X |
| 66 | AML, relapsed | BCR-ABL1 | imatinib/dasatinib | dasatinib | 2 | * | off label | |
| 72 | AML, relapsed | NRAS, PTPN11 | MEK inhibitor | trametinib | 3 | none | off label | X |
| 144 | AML, relapsed | FLT3 ITD | FLT3 inhibitor | midostaurin, gilteritinib | 1 | * | off label (midostaurin); expanded access program (gilteritinib) | |
| 152 | AML, relapsed | CSF3R | dasatinib | dasatinib | 3 | * | off label | X |
| 18 | B-ALL, refractory | EPOR-IGH | ruxolitinib | ruxolitinib | 3 | none | clinical trial (NCT02723994) | X |
| 16 | B-ALL, relapsed | NRAS | MEK inhibitor | everolimus | 3 | X | ||
| mTOR inhibitor | 5 | none | off label | |||||
| 37 | B-ALL, relapsed | NUP214-ABL1 | imatinib/dasatinib | dasatinib | 3 | none | off label | X |
| 38 | B-ALL, relapsed | RCSD1-ABL2 | imatinib/dasatinib | imatinib | 3 | * | off label | X |
| 43 | B-ALL, relapsed | NRAS, KRAS | MEK inhibitor | trametinib | 3 | none | off label | X |
| 55 | B-ALL, relapsed | ABL1 p.T315I | ponatinib | ponatinib | 1 | none | FDA authorized indication | X |
| 153 | B-ALL, relapsed | TCF3-HLF | venetoclax | venetoclax | 3 | * | off label | |
| 23 | MPAL, refractory | NUP214-ABL1 | imatinib/dasatinib | imatinib, dasatinib | 3 | * | off label | X |
Please see full description of toxicity within the case vignettes in the supplementary data
Clinical cases for all patients who received targeted therapy are summarized in the Supplementary Data. For patients who received targeted therapy recommendations, clinical responses were variable. For example, patient #152 was a 14-year-old boy who presented with therapy-related MDS approximately 6 months after completing therapy for Ph+ ALL. Cytogenetic analysis showed monosomy 7, and the patient underwent a matched unrelated donor stem cell transplant 5 months following diagnosis. He relapsed approximately 90 days after transplant with AML that responded poorly to treatment with azacitidine. He was experiencing rapid progression of his AML when leukemia profiling with the NGS panel demonstrated a CSF3R p.Q776* in 46% of reads, predicted to result in increased signaling of the SRC family kinases(28). Dasatinib was started and his platelet count normalized. After 3 weeks of dasatinib, 6-mercaptopurine was added to the treatment regimen due to rising peripheral blasts, and blood counts stabilized again. Dasatinib was discontinued after a total of 9 weeks of treatment due to development of bloody diarrhea.
The development of more sensitive clinical tests for fusion detection over the course of this trial also enhanced targeted therapy recommendations. This was especially evident in the treatment of patient # 38, who did not have a fusion detected using multiplex RT-PCR at the time of initial study enrollment, but was discovered to have a targetable RCSD1-ABL2 fusion detected at subsequent relapse using the then-available AMPSeq panel. At the time of fusion detection, the patient had chemotherapy refractory leukemia and sepsis. The patient received chemotherapy in combination with imatinib, which led to a durable complete remission of previously refractory disease and enabled a second stem cell transplant after 10 months of combination therapy. Thus, this fusion testing and the ability to incorporate imatinib into her treatment was life-saving for this patient, and she remains in remission now for 12 months post-transplant and 23 months from the identification of the fusion.
While the primary purpose of the study was to match somatic genomic findings to targeted therapies, the impact of clinical genomic sequencing certainly expands beyond this goal. For example, while the study utilized tumor-only sequencing that cannot differentiate germline from somatic variants, mutations in TP53, RUNX1, ETV6, CEBPA and GATA2 prompted consideration of a germline cancer predisposition and consideration for germline evaluation was included in the recommendation. Patient #65 with a PTPN11 p.T411M mutation also had a recommendation for possible germline evaluation.
This study also informed a change in diagnosis for some patients. For example, patient #53 was initially diagnosed with AML and was started on standard-of-care AML therapy. The NGS panel sequencing returned showing mutations in NOTCH1, ETV6, EZH2, and NRAS prompting re-evaluation of the leukemia histopathology and a change in diagnosis to early T-precursor ALL. The patient completed the AML induction course and was in remission after the first month. He subsequently received treatment with an ALL-directed chemotherapy regimen and remains in remission now for 34 months.
Ras pathway mutations are frequent in pediatric leukemia and show sensitivity to MEK inhibition
Ras/MAPK pathway mutations (NRAS, KRAS, PTPN11, and CBL) comprised the most frequent “targetable” pathway in our patient cohort. For patients with B-ALL, we compared the VAF for patients with relapsed/refractory disease enrolled on this LEAP consortium trial to that of patients with newly-diagnosed B-ALL contemporaneously treated at the Dana-Farber Cancer Institute and profiled with the same panel assay. A subset of patients had more than one Ras pathway mutation, and we used the mutation with the highest VAF for this analysis. Patients with relapsed ALL had a significantly higher VAF for Ras pathway mutations (Figure 3A) compared to the newly diagnosed cohort. There was a similar trend for patients with AML, all enrolled on the LEAP trial, although it did not reach statistical significance (Figure 3B). Ras pathway mutations increase signaling via the MEK/ERK signaling pathways, leading to increased cell proliferation(29). MEK inhibitors, such as selumetinib, trametinib, and PD0325901, have been shown to inhibit this signaling and are under evaluation in clinical trials(29). We tested the sensitivity of primary patient samples from our study to trametinib and selumetinib. Samples that had detectable NRAS, KRAS or PTPN11 mutations were more sensitive to selumetinib and trametinib ex vivo compared to Ras pathway wild-type controls (Figure 3C and 3D, Supplementary Table 4), although response did not correlate with VAF (Supplementary Figures 2A and 2B).
Figure 3: RAS pathway mutations are a therapeutic target in pediatric acute leukemia.
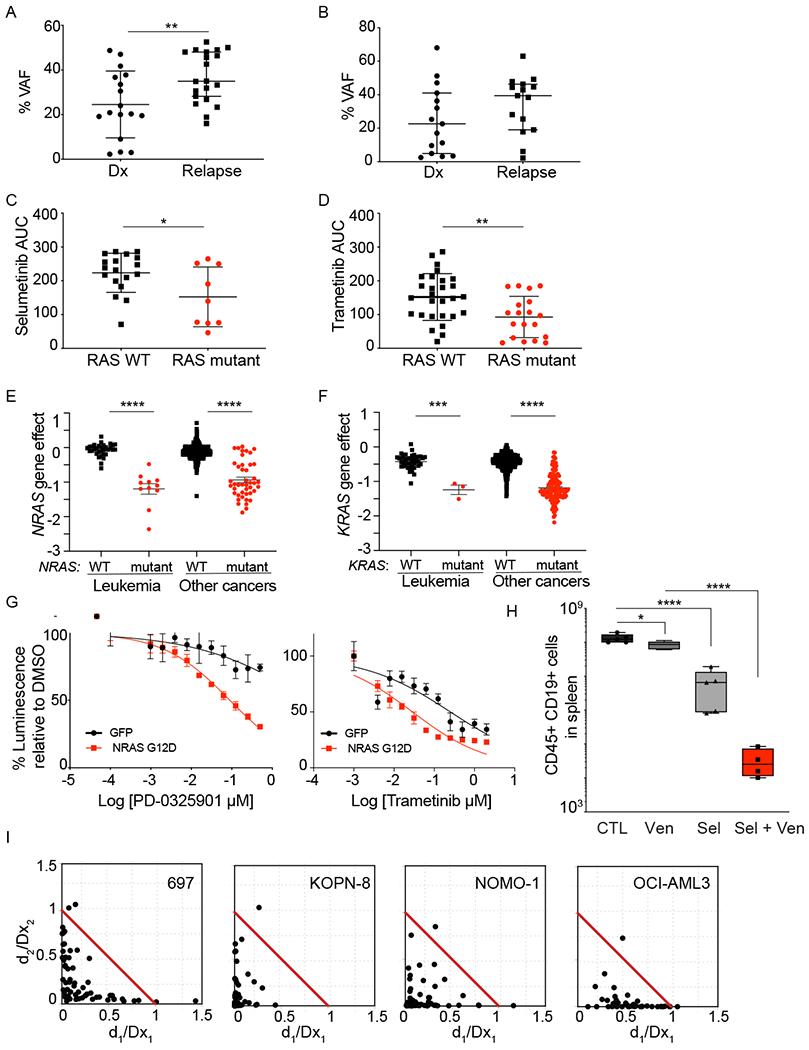
A) VAF for NRAS, KRAS, and PTPN11 mutations in patients with B-ALL on the LEAP trial compared to patients with newly-diagnosed B-ALL treated at Dana-Farber Cancer Institute. Leukemia samples were profiled using the same assay. **P<0.005 using Mann-Whitney test. B) VAF for NRAS, KRAS, and PTPN11 mutations in patients with AML on the LEAP trial. Primary leukemia samples from patients enrolled on the LEAP Consortium trial were tested ex vivo in response to MEK inhibitors. Samples with Ras pathway mutations (NRAS, KRAS, and PTPN11) were more sensitive to selumetinib (C) and trametinib (D), compared to WT samples. Shown is average area under the curve (AUC) for individual samples, with average +/− SD. *P<0.05 and **P<0.005 using unpaired t-test. Genome-scale CRISPR-Cas9 screen of 739 cell lines showed RAS mutant leukemia and other cell lines to be differentially dependent on NRAS (E) or KRAS (F) compared to RAS WT leukemia cell lines. Shown is the average gene effect +/− SEM. ****P<0.0001 using Mann-Whitney test. G) Expression of NRAS p.G12D in murine MLL-AF9 cells increased their sensitivity to MEK inhibitors, PD-0325901 and trametinib. Shown is average dose response +/− SEM, with 4 replicates. H) Patient-derived xenograft (PDX) model characterized by TCF3-HLF NRAS p.G12D was treated with venetoclax, selumetinib or venetoclax plus selumetinib. The combination of venetoclax with selumetinib decreased leukemia burden compared to mice treated with each drug individually. Data are flow cytometric quantification of total human CD45+ CD19+ ALL cells in harvested murine spleens (N=5). * P<0.01 and ****P<0.0001 using one-way ANOVA with Tukey’s post-test for multiple comparisons. I) Combination of selumetinib with venetoclax is synergistic across Ras pathway mutant acute leukemia cell lines. Synergy was assessed by Chou-Talalay combination index (CI) across the indicated cell lines. Normalized isobolograms depict CI scores over a range of concentrations. The coordinates of the CI scores are d1/Dx1 and d2/Dx2, where Dx1 is the concentration of drug 1 (selumetinib) that alone produces the fractional inhibition effect x, and Dx2 is the concentration of drug 2 (venetoclax) that alone produces the fractional inhibition effect x. The red line displayed is the line of additivity. Points below the line are synergistic and above the line are antagonistic. Cell viability was measured at 3 days of combination treatment using an ATP-based assay.
We expanded the analysis of sensitivity to Ras pathway inhibition to cancer cell lines using the Broad Institute’s Cancer Dependency Map(30). Seven hundred thirty-nine cancer cell lines were screened with a genome-scale CRISPR library(31). Focusing on the NRAS and KRAS mutant leukemia cell lines, we asked whether they are more sensitive to deletion of NRAS and KRAS, respectively. As anticipated, the presence of NRAS or KRAS activating mutations led to increased sensitivity to the genomic loss of NRAS or KRAS in the respective cell lines (Figure 3E and 3F). This pattern of sensitivity to NRAS or KRAS loss was also seen in solid tumor cell lines where inhibitors of the Ras signaling pathway are routinely used clinically(29). Analysis of the BEAT AML drug sensitivity data(32) showed the KRAS/NRAS mutant AML samples to be differentially sensitive to the MEK inhibitors selumetinib, trametinib and CI-1040 (Supplementary Figures 3A and 3B). In accordance with these results, expression of mutant NRAS sensitized MLL-AF9 murine cells to the MEK inhibitors trametinib and PD0325901 (Figure 3G).
The LEAP clinical trial enrolled two patients with B-ALL with TCF3-HLF translocations. Patients with B-ALL with TCF3-HLF account for less than 1% of pediatric B-ALL, but are considered incurable(33). Pre-clinical data suggests that ALL characterized by TCF3-HLF fusions may be particularly sensitive to BCL2 inhibition with venetoclax(33,34). Accordingly, the pediatric clinical trial of venetoclax for patients with relapsed/refractory leukemia or neuroblastoma (NCT03236857) allows patients with new diagnosis of TCF3-HLF B-ALL to be enrolled and receive venetoclax in combination with chemotherapy. The response of patient-derived xenograft (PDX) models of TCF3-HLF B-ALL to venetoclax is variable(33,35), and a large percentage of patients with TCF3-HLF B-ALL have Ras pathway mutations(33). Using a PDX model derived from a patient’s leukemia with TCF3-HLF and a KRAS p.G12D mutation, we tested the combination of the MEK inhibitor, selumetinib, with venetoclax. NSG mice were injected with cells from this PDX, and after disease was established, mice were randomized to receive vehicle, venetoclax, selumetinib or both inhibitors. Single-agent venetoclax had minimal effect in this model, but the combination of venetoclax with selumetinib led to a nearly 3 log-fold reduction in leukemia burden compared to venetoclax alone (Figure 3H). We then expanded testing of the combination of venetoclax with a MEK inhibitor to Ras pathway mutant B-ALL and AML cell lines. The combination of selumetinib or trametinib with venetoclax was synergistic in multiple cell line models (Figure 3I and Supplementary Figure 4). We further tested the combination of trametinib and venetoclax in 5 primary patient samples from the LEAP study in vitro. There was a decrease in the average area under the curve (AUC) for the combination of trametinib with venetoclax compared to each inhibitor alone (Supplementary Figure 5A and Supplementary Table 4).
Activation of ERK1/2 signaling promotes degradation of pro-apoptotic protein, BIM(36). MEK inhibition increases BIM levels, priming cells for apoptosis when combined with Bcl-2 or Bcl-XL inhibitors(37,38). This synergistic combination has been demonstrated in models of melanoma(39), colon(37,39), lung(40,41) and ovarian(38) cancer. We treated KOPN8 cells with increasing concentrations of trametinib or selumetinib. Indeed, both led to a concentration dependent increase in BIM (Supplementary Figures 5B and 5C). The combination of trametinib with venetoclax further increased levels of NOXA and cleaved BAX proteins, consistent with a loss of viability (Supplementary Figure 5D).
Patient #153 with B-ALL harboring a TCF3-HLF translocation and a KRAS mutation was enrolled on the LEAP study at time of relapse after a matched unrelated donor stem cell transplant and presented with characteristic coagulopathy and hypercalcemia(42). She was started on induction chemotherapy but had rapid progression of disease as well as pulmonary hemorrhage requiring intubation. Venetoclax was added to her chemotherapy regimen with clinical improvement in the following week. After a cycle of a combination of daunorubicin, vincristine, asparaginase, dexamethasone and venetoclax, she was in remission with low MRD. She continued to receive chemotherapy with venetoclax, followed by CD19-CAR T cell therapy, but unfortunately relapsed five months after receiving CAR T. She again received chemotherapy in combination with venetoclax after the second relapse and was able to achieve a clinical remission with low MRD. Although combining a MEK inhibitor with venetoclax was an intriguing consideration based upon preclinical data, this regimen was not given to the patient due to the lack of safety data for this combination in humans.
Validation of variants of unknown significance
A common Tier 1 genetic alteration was the presence of FLT3 mutations in patients with AML. We performed functional testing for drug sensitivity to FLT3 inhibitors across primary samples using an ex vivo drug sensitivity platform(32). As expected, FLT3 mutant samples were differentially sensitive to crenolanib, gilteritinib, midostaurin or quizartinib when compared to FLT3 wild-type samples (Figures 4A, 4B, 4C and 4D, respectively, and Supplementary Table 4). Interpretation of clinical sequencing, however, can be complicated by variants of unknown significance. For mutations in genes encoding proteins that are known drug targets, validation of this significance is particularly relevant as this may influence enrollment in targeted therapy trials. We thus used a multi-faceted approach to study the FLT3 p.A680V variant, which was identified in patient #1 and has been previously described in pediatric patients with ALL and AML(4,43,44). To investigate possible mechanisms by which the p.A680V mutation could be activating the FLT3 kinase, we performed molecular dynamics (MD) simulations. MD predicts positions of atoms in a protein subjected to thermal fluctuations and can inform hypotheses regarding structural differences between wild-type and mutant forms. Based on the crystal structure of FLT3 (Figure 4E), we built models of wild-type FLT3 and FLT3 p.A680V that included the kinase and juxta-membrane (JM) domains. Residue 680 is located in the N-terminal lobe of the kinase domain in close proximity to a hydrogen bond (H-bond) between Y599 (JM domain) and E604 (kinase domain) (Figure 4E and 4F). We hypothesized that this H-bond is important for stabilization of the JM domain in a conformation that blocks the kinase active site, thereby contributing to auto-inhibition of the kinase (Figure 4E). Short time-scale MD simulations showed that extra steric bulk from the methyl groups of A680V compared to A680 disrupts the Y599/E604 H-bond (Figure 4G and 4H). We predicted that this could destabilize the JM domain from its auto-inhibitory conformation (Figure 4I). Throughout MD simulations we observed that the prevalence of Y599/E604 H-bonds are decreased for the mutant over the wild-type, whereas the prevalence of Y599/water molecule H-bonds are increased in the mutant over the wild-type (Figure 4H). The Y599/water molecule H-bonds are between the JM domain of the protein and the solvent front and an increase in the number of these bonds in the mutant structure suggests that the JM domain in the mutant is no longer in its auto-inhibitory position close to the kinase domain (Figure 4I). These data support our structural prediction that formation of the Y599/E604 H-bond is impaired by a mutation of residue 680 from alanine to valine. We propose that destabilization of the JM domain releases auto-inhibition and shifts the conformational equilibrium toward an active-like structure (Figure 4I). Concordant with the MD simulations, expression of FLT3 p.A680V in Ba/F3 cells led to IL-3 independent growth (Figure 4J). Furthermore, cells expressing FLT3 p.A680V were sensitive to treatment with gilteritinib (Figure 4K) with a concentration-dependent reduction in FLT3 phosphorylation (Figure 4L).
Figure 4: FLT3 activating mutations increased ex vivo sensitivity to FLT3 inhibitors.
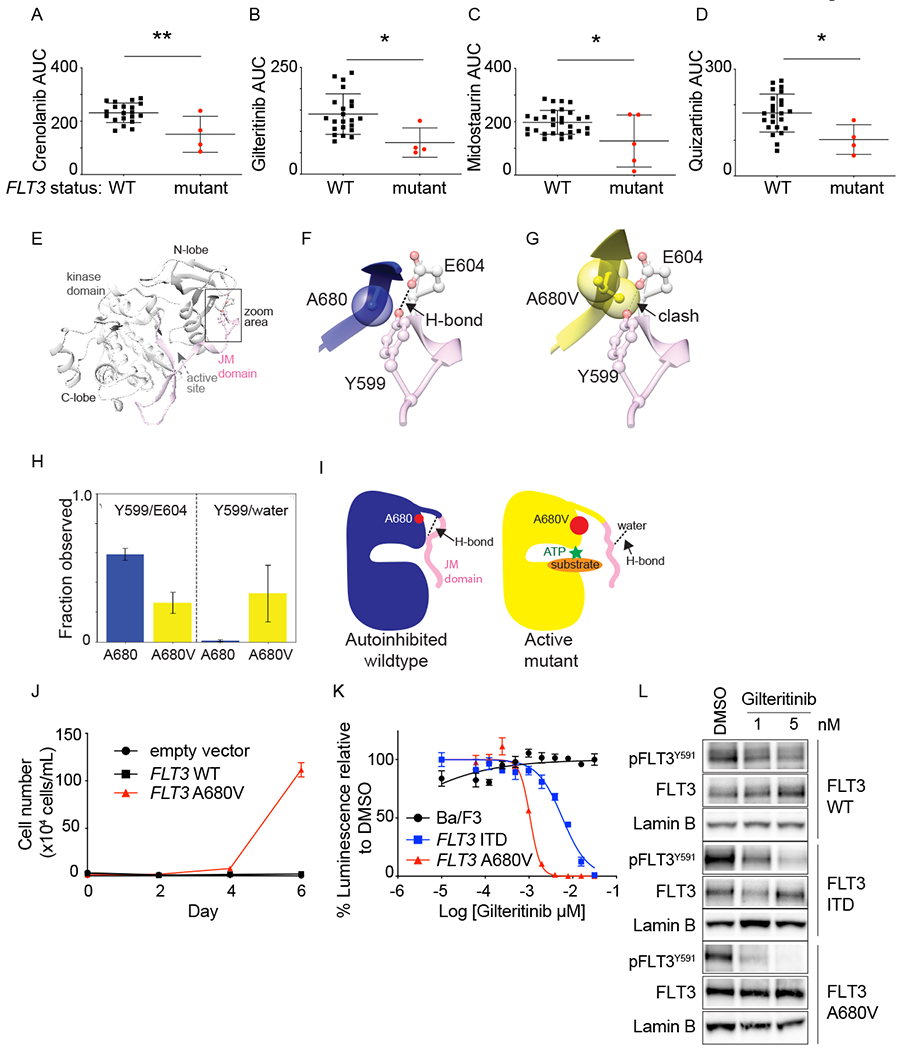
Primary leukemia samples from patients enrolled on the LEAP Consortium trial were tested ex vivo with FLT3 inhibitors. Samples with FLT3 mutations were more sensitive to crenolanib (A), gilteritinib (B), midostaurin (C), and quizartinib (D), compared to FLT3 wildtype samples. Shown is area under the curve (AUC) for individual samples, mean +/− SD. *P<0.05 and **P<0.005, using unpaired t test. The first trial patient had AML with a FLT3 p.A680V mutation, which has been described but had unknown functional significance. (E) Crystal structure of auto-inhibited FLT3 kinase domain (grey) and JM domain (pink). Area surrounding residue 680 is highlighted. (F) Zoom in on residues A680 (blue) and (G) A680V (yellow) from active-like models of FLT3 shown relative to the Y599/E604 H-bond observed in the auto-inhibited crystal structure. (H) Prevalence of H-bonds between: Y599/E604 (left) and Y599/water molecules (right) in molecular dynamics simulations of inactive-like models. (I) Proposed mechanism of activation of the A680V mutation. J) Ba/F3 cells expressing empty vector, FLT3 WT or FLT3 p.A680V were grown without IL-3, and cells counted using trypan blue exclusion. Shown is the average number of cells ± SD of 3 replicates. K) Ba/F3 cells expressing FLT3-ITD or FLT3 p.A680V were tested with a range of gilteritinib concentrations and viability evaluated at day 2 by an ATP-based assay as a percentage of cells relative to DMSO control. Shown are the mean ± SD of 4 replicates. L) Western immunoblotting showing inhibition of FLT3 phosphorylation in response to gilteritinib treatment.
Discussion
The sequencing of the first human leukemia genome was reported in 2009(1). Since that time, there have been extensive efforts to genomically characterize cancer, including leukemia, with the goal of improving diagnosis, prognostic markers and treatment. Real time prospective clinical genomic evaluation of pediatric patients with leukemia remains in the early stages, and the feasibility of applying genomic characterization to inform targeted therapy in pediatric patients with leukemia has been explored only minimally.
The targeted therapy recommendations that were part of this trial were made based on the combination of standard-of-care evaluations (cytogenetics, FISH, FLT3 testing), as well as NGS panels. The majority of targetable alterations for which the patients received therapy were detected using DNA-based NGS panels and RNA-based AMPSeq fusion analysis, rather than current assays routinely used to characterize leukemia biology clinically. For the patients who actually received the targeted therapy recommendation, all alterations leading to the therapy were detected using the NGS panels. While NGS panels are entering clinical practice, they are not universally considered to be standard-of-care for patients with leukemia. Given the results of this study, we believe that the use of NGS panels should be part of the standard leukemia diagnostic evaluation for all pediatric patients, and especially so for all patients with high-risk, rare or relapsed/refractory disease. Germline sequencing may also contribute to the value of targeted panel evaluation and inform interpretation of identified variants. Economic and health policy cost-benefit analyses will be needed in future genomically-informed targeted therapy studies that are powered to assess their efficacy.
The clinical availability of assays for genomic assessment of leukemia, as well as the landscape of available targeted therapies, are rapidly evolving and influenced therapy recommendations for subjects enrolled on the LEAP Consortium trial. During the course of this study, for example, fusion analysis was first done using multiplex RT-PCR and then changed to AMPSeq(45) with an improved ability to detect targetable fusions. With increased availability of clinical grade RNA sequencing and the expanded use of other NGS assays more broadly, the ability to identify targetable alterations in real time will improve, and systems for rapid interpretation and guidance for clinicians will be needed. The dynamic landscape of drug availability also led to differences in therapy recommendations over the course of this study. For example, at the time of initiation of the LEAP study, gamma secretase inhibitors were under early-phase clinical trial investigation for patients with NOTCH1-mutated T-ALL and were thus part of the therapy recommendation. During the course of the LEAP trial, further clinical development of these inhibitors was put on hold due to toxicity, and inclusion of GSIs in our MTT recommendations was no longer relevant. Potential targeting of TP53 mutations also evolved over the course of our trial with the development of APR-246(46) and phase 1/2 clincial trial testing of this compound in combination with azacitidine in adult patients with AML. Indeed, preclinical data have recently been reported suggesting the consideration of this drug for children with TP53-mutant ALL(47). The targeted therapy recommendations in our study were made for single agents, but a subset of patients received the targeted therapy in combination with chemotherapy. Genomically informed therapeutic trials using combinations of drugs will be needed to inform efficacy and toxicity.
An evolving understanding of driver versus passenger events and the relevance of VAFs will also influence future therapy recommendations. For example, we validated the FLT3 p.A680V mutation in AML as activating and rendering sensitivity to FLT3 inhibitors, suggesting that a FLT3 inhibitor could be recommended for future patients with FLT3 p.A680V mutant leukemia(4,43). Systematic efforts to characterize VUS as activating, such as MITE-Seq (Mutagenesis by Integrated TilEs)(48) are needed, particularly for targets with readily available inhibitors. These efforts may add pre-clinical evidence to a previously uncharacterized variant. Likewise, the targeting of a sub-clonal mutation, and its effect on the bulk leukemia population, have not been well studied. Our study was not powered to assess efficacy of specific targeted therapies in the context of variable VAFs. There was no correlation between VAF and sensitivity to MEK inhibitors in vitro, which may be due to a number of factors: differences in VAF between the patient sample that was sequenced and the viable cells that were screened, potential paracrine effects from Ras pathway mutant cells or alternative mechanisms of activating the Ras pathway. It is also possible that the relationship between VAF and sensitivity to MEK inhibitors is not linear. Notably, the MEK inhibitor trametinib had clinical efficacy only in RAS-mutant myeloid malignancies(49). Future genomically-informed therapeutic trials will need to be conducted to address the question of the level of clone that is needed for a therapeutic response. We did, however, correlate the VAF findings between clinical laboratories that performed NGS panel sequencing. This analysis showed a significant correlation between the results and supports the possibility of using institutional assays for specific genes rather than requiring a centralized test.
Ex vivo drug sensitivity testing is another potential approach to guide targeted therapy recommendations. In our study, leukemia samples with molecular alterations predicted to sensitize to a particular drug were in fact more sensitive to the inhibitor than those samples without the mutation. Unfortunately, the study was not powered to assess whether the ex vivo drug sensitivity correlated with a patient response to the targeted inhibitor. Future clinical trials using ex vivo drug sensitivity testing would need to be performed to address this question. Beyond ex vivo testing of single agents, robust preclinical testing to study drug combinations is needed as patients with acute leukemia are generally not curable with single-drug therapies. One goal of the LEAP consortium was to conduct pre-clinical studies to inform future clinical trials. The combination of a MEK inhibitor with venetoclax was synergistic in both B-ALL and AML cell lines, as well as in a highly aggressive PDX model for TCF3-HLF and KRAS-mutant B-ALL. The combination of the MEK inhibitor cobimetinib with venetoclax was recently found to be synergistic in a subset of AML cell lines and primary patient samples(50). Testing this combination in additional preclinical in vivo models would be a valuable next step toward informing biomarkers of response and testing in clinical trials.
This first-in-the-US multi-institutional prospective pediatric leukemia genomics trial brought state-of-the-art clinical genetic testing, assessment of prognostic biomarkers and targeted therapy treatment recommendations to children and young adults with high-risk, relapsed or refractory leukemias or MDS. Future studies will need to comprehensively assess the impact of genomic testing and matched targeted therapy on patient outcome, as well as the financial implications of integrating genomic testing into the clinical care for all patients. Our collaboration provided a unique opportunity to perform sophisticated patient-specific in vitro drug testing assays to impact matched targeted therapy discovery efforts, which will be further validated in patient-derived xenograft models in future studies. We believe that a model like the LEAP Consortium has the potential to transform precision medicine approaches for children with high-risk leukemias and to inform future genomics-guided therapeutic trials and drug discovery efforts.
Materials and Methods:
LEAP Consortium Clinical Trial
The patients all had a diagnosis of relapsed/refractory or high-risk leukemia or myelodysplastic syndrome. The patients were enrolled in two cohorts. Cohort 1 included patients with relapsed or refractory leukemia. Cohort 2 included pediatric patients with high-risk or rare leukemias. These were defined as AML, KMT2A-rearranged infant ALL or hypodiploid ALL, rare leukemia (e.g., JMML, MPAL), or leukemias secondary to therapy. Patient characteristics are listed in Table 1. At the time of enrollment, patients had a leukemia sample (or DNA derived from a leukemia sample) sent for sequencing using an NGS panel profiling 95 genes commonly mutated in leukemia (Rapid Heme Panel). A subset of patients also had fusion analysis using multiplex RT-PCR (19 patients), AMPSeq fusion panel (56 patients) or RNASeq (5 patients) (Figure 1). Clinical data, including cytogenetics and FISH, in combination with sequencing results, were then reviewed by an expert panel of physicians. For patients who had leukemia profiling performed at their home institution, data were also reviewed by the expert panel.
The institutional review board of each participating institution approved the protocol before enrolling patients. Written informed consent was obtained from patients and/or parents/guardians before study enrollment and initiation of therapy. The study was registered on Clinicaltrials.gov (NCT02670525).
Cell Culture and Viability Assays
The 697 cell line was obtained from Dr. Scott Armstrong. KOPN-8 cells were obtained from Dr. Sarah Tasian. NOMO-1 and OCI-AML3 were provided by Dr. Ross Levine. Cell line identity was verified using STR profiling. All cell lines were maintained in RPMI 1640 (Cellgro) supplemented with 1% penicillin/streptomycin (PS)(Cellgro) and 10% FBS (Sigma-Aldrich) at 37°C with 5% CO2. Ba/F3 cells were maintained in RPMI 1640 (Cellgro) supplemented with 1% PS, murine IL-3 and 10% FBS. After transduction with either FLT3, FLT3-ITD, or FLT3-A680V vector, selection was made by removing IL-3 from the culturing medium.
For drug sensitivity assays, viability was evaluated using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) after the indicated days of exposure to the specific drug or combination of drugs. Luminescence was measured using FLUOstar Omega from BMG Labtech. The IC50 values were determined using Prism GraphPad version 8 software. Murine KMT2A-AF9 leukemia cells were generated as previously described(51).
Drug sensitivity testing
Trametinib (#S2673), selumetinib (#S1008), venetoclax (#S8048), PD0325901 (#S1036) and gilteritinib (#S7754) used for cell line testing were purchased from Selleckchem. Selumetinib (#S-4490) and venetoclax (#V-3579) used for the mouse study were purchased from LC laboratories.
Dose response
Synergy drug testing was performed with the combination of selumetinib or trametinib and venetoclax as previously described(52).
Ex vivo drug sensitivity testing for primary patient leukemia samples was performed as previously described(32). Briefly, drug sensitivity was analyzed by incubating freshly isolated mononuclear cells from patient blood or bone marrow specimens in RPMI-1640 medium supplemented with fetal bovine serum (10% for myeloid specimens; 20% for lymphoid specimens), L-glutamine, penicillin/streptomycin, fungizone, 10−4 M beta-mercaptoethanol, and graded concentrations of respective small-molecules for 72 hours. Relative numbers of viable cells were quantified using a tetrazolium-based MTS assay (CellTiter 96 Aqueous), and the resulting seven point dose response curve data were fit using Probit to calculate the response metric, area-under the dose response curve (AUC). Data for response to gilteritinib, crenolanib, midostaurin, quizartinib, selumetinib, trametinib and venetoclax were included in the analyses for this manuscript. Combination testing for this assay was performed as previously described(53).
Immunoblotting
Cells were lysed in Cell Signaling Lysis Buffer (Cell Signaling Technology) as previously reported(54) and resolved by gel electrophoresis using Novex 4-12% Bis-Tris Gels (Invitrogen), transferred to a PVDF membrane (Bio-Rad) and blocked for one hour in 5% BSA (Sigma). Blots were incubated in primary antibody to pFLT3-Y591 (Cell Signaling #3461S), total FLT3 (Cell Signaling #3462S), pErk1/2-Thr202/Tyr204 (Cell Signaling #4370S), total ERK (Cell Signaling #4695S), BIM (Cell Signaling #2933S), Bax (Cell Signaling #5023S), NOXA (Cell Signaling #14766S) or Lamin B (Cell Signaling #12586S), followed by the secondary antibodies anti-rabbit HRP (GE Healthcare Life Sciences #NA9341ML) or anti-mouse HRP (GE Healthcare Life Sciences #NA93101ML). Bound antibody was detected using the Western Lightning Chemiluminescence Reagent (Perkin Elmer).
In silico modeling
Models of inactive FLT3 were built using the auto-inhibited crystal structure of FLT3 (1RJB) as a reference(55). Proteins were prepared using the PrepWizard tool in Maestro®. For the active-like FLT3 models, N-terminal JM domain residues were removed. A680V mutation was introduced into these models with the rotamer search in Chimera. A total of 4×40ns explicit solvent Molecular Dynamics (MD) simulations were performed with the inactive-like FLT3 model, while 2×120ns MD simulations were performed for the active-like FLT3 model. Amber14 suite was used for all MD simulations(56). ChimeraX, a virtual reality visualization software, was used for investigation of conformational changes in the models, and figures were generated with the Chimera Software(57). For more details, please see Supplemental Methods.
In vivo drug testing in a TCF3-HLF ALL PDX model
Primary ALL cells were obtained from a pediatric patient with relapsed ALL at the Children’s Hospital of Philadelphia (CHOP) Center for Childhood Cancer Research on an approved institutional research biobanking protocol. Written informed consent was obtained prior to sample acquisition in accordance with the Declaration of Helsinki. Animal studies were conducted under a CHOP Institutional Animal Use and Care Committee (IACUC)-approved protocol in accordance with the Panel on Euthanasia of the American Veterinary Medical Association’s guidelines. Patient leukemia cells were injected into NSG mice to establish the primary PDX model. Subsequently, 100,000 cells from primary PDX spleens were injected into secondary 8 week-old male NSG mice for experimental studies. After flow cytometric confirmation of ≥1% CD45+ CD19+ human ALL (fluorochrome-conjugated antibodies #17-9459-42 and #47-0199-42 from EBioscience) in murine peripheral blood, mice were randomized to treatment with vehicle, venetoclax 100 mg/kg per os (PO) once daily x 5 days/week, selumetinib 100 mg/kg PO once daily x 5 days/week, or both venetoclax and selumetinib x 5 days/week, for a total of 21-day course. Human ALL cells were quantified weekly in murine peripheral blood and in harvested spleens after 21 days of treatment by flow cytometry using a FACSVerse flow cytometer (BD Biosciences) and with data analysis in Cytobank as described(58–60).
Statistical analysis
Statistical significance was determined by two-tailed t-test or Mann-Whitney test for pair-wise comparison of groups, as indicated and considered significant if < 0.05. Pearson correlation was used to measure the strength of linear association between continuous measures. For PDX studies, human ALL cell numbers in murine spleens of vehicle- and drug-treated animals were evaluated for statistical significance using one-way ANOVA with Tukey’s post-test for multiple comparisons. Statistical calculations were performed using Prism GraphPad version 8 software.
Supplementary Material
Statement of Significance:
Patients with relapsed/refractory leukemias face limited treatment options. Systematic integration of precision medicine efforts can inform therapy. We report the feasibility of identifying targetable mutations in children with leukemia and describe correlative biology studies validating therapeutic hypotheses and novel mutations.
Acknowledgements
We are grateful to the patients and families who participated in this clinical trial. We are grateful to the late Dr. Frank Kuo for his hard work in establishing and validating the targeted sequencing panel, Rapid Heme Panel, which was used in this study. We are grateful to Dr. Marilyn Li and the Children’s Hospital of Philadelphia Division of Genomic Diagnostics for assistance with clinical genetic testing. This work is supported by a St. Baldrick’s Foundation Consortium grant and Hannah’s Heroes. NVD is a Julia’s Legacy of Hope St. Baldrick’s Foundation Fellow. This work was additionally supported by the following grants: Wong Family Fund for Translational Research (YP), R35 CA210030 (KS), K08 CA222684 (YP), 1K08CA184418 (SKT), 1U01CA232486 (SKT), Alex’s Lemonade Stand Foundation (LG), P30 CA046934 (LG), 1R01CA214428-01A1 (CET), the Children’s Leukemia Research Foundation (KS), Pan-Mass Challenge Team Crank (KS), and the 4C’s Fund (KS). MLL is the Benioff Chair of Children’s Health and the Deborah and Arthur Ablin Endowed Chair for Pediatric Molecular Oncology at Benioff Children’s Hospital. Biobanking and patient-derived xenograft modeling studies at the Children’s Hospital of Philadelphia were also supported by the SchylerStrong Foundation in memory of Schyler Anna Herman, the Simutis family childhood leukemia research fund in memory of Andrew David Simutis, and the Viands family childhood leukemia research fund in honor of Nathaniel J Viands (SKT). S.L. is a Fellow of the Leukemia and Lymphoma Society. We are grateful to the Seattle Children’s Tumor Bank for supporting this study. AP is a recipient of support from the ATIP/AVENIR French research and the ERC Starting program (758848), and is supported by the St Louis Association for leukemia research, the LNCC and the Emergence 2017 Program from the city of Paris.
Conflicts of interest:
SKT receives research funding from Incyte Corporation and Gilead Sciences and is a member of the scientific advisory board for Aleta Biotherapeutics. MPJ is a consultant to and shareholder of Schrodinger LLC, which licenses software used in this work. NVD is a current employee of Genentech, Inc., a member of the Roche Group. KAJ receives research funding from Roche and is a consultant for Bayer Pharmaceuticals. KS has consulted for Rigel Pharmaceuticals, Kronos Bio, and Auron Therapeutics, receives grant funding from Novartis for an unrelated project, and holds stock options with Auron Therapeutics. JWT has received research support from Agios, Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Petra, Seattle Genetics, Syros, and Takeda. MLL is on the advisory board of Medisix Therapeutics, Inc.
References:
- 1.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. The New England journal of medicine 2009;361:1058–66 doi 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999;286(5439):531–7 doi 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 3.Ebert BL, Golub TR. Genomic approaches to hematologic malignancies. Blood 2004;104(4):923–32 doi 10.1182/blood-2004-01-0274. [DOI] [PubMed] [Google Scholar]
- 4.Bolouri H, Farrar JE, Triche T Jr., Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 2018;24(1):103–12 doi 10.1038/nm.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mullighan CG. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program 2014;2014:174–80 doi 10.1182/asheducation-2014.1.174. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nature Genetics 2017;49:1211–8 doi 10.1038/ng.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz JR, Ma J, Lamprecht T, Walsh M, Wang S, Bryant V, et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 2017;8(1):1557 doi 10.1038/s41467-017-01590-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018;378(5):439–48 doi 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017;129(25):3322–31 doi 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015;385(9967):517–28 doi 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forrest SJ, Geoerger B, Janeway KA. Precision medicine in pediatric oncology. Curr Opin Pediatr 2018;30(1):17–24 doi 10.1097/mop.0000000000000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol 2016;2(5):616–24 doi 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris MH, DuBois SG, Glade Bender JL, Kim A, Crompton BD, Parker E, et al. Multicenter Feasibility Study of Tumor Molecular Profiling to Inform Therapeutic Decisions in Advanced Pediatric Solid Tumors: The Individualized Cancer Therapy (iCat) Study. JAMA oncology 2016. doi 10.1001/jamaoncol.2015.5689. [DOI] [PubMed] [Google Scholar]
- 14.Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, et al. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. Jama 2015;314(9):913–25 doi 10.1001/jama.2015.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oberg JA, Glade Bender JL, Sulis ML, Pendrick D, Sireci AN, Hsiao SJ, et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Med 2016;8(1):133 doi 10.1186/s13073-016-0389-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med 2019;380(20):1929–40 doi 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- 17.Neel DV, Shulman DS, DuBois SG. Timing of first-in-child trials of FDA-approved oncology drugs. Eur J Cancer 2019;112:49–56 doi 10.1016/j.ejca.2019.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kluk MJ, Lindsley RC, Aster JC, Lindeman NI, Szeto D, Hall D, et al. Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies. J Mol Diagn 2016;18(4):507–15 doi 10.1016/j.jmoldx.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang F, Lin F, Cao K, Surrey LF, Aplenc R, Bagatell R, et al. Development and Clinical Validation of a Large Fusion Gene Panel for Pediatric Cancers. J Mol Diagn 2019;21(5):873–83 doi 10.1016/j.jmoldx.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oberg JA, Glade Bender JL, Sulis ML, Pendrick D, Sireci AN, Hsiao SJ, et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Medicine 2016;8:133 doi 10.1186/s13073-016-0389-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017;377(5):454–64 doi 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. The Lancet Oncology 2017;18:1061–75 doi 10.1016/S1470-2045(17)30416-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med 2019;381(18):1728–40 doi 10.1056/NEJMoa1902688. [DOI] [PubMed] [Google Scholar]
- 24.Pollard JA, Alonzo TA, Brown PA, Gerbing RB, Fox E, Choi JK, et al. Sorafenib in Combination with Standard Chemotherapy for Children with High Allelic Ratio FLT3/ITD+ AML Improves Event-Free Survival and Reduces Relapse Risk: A Report from the Children’s Oncology Group Protocol AAML1031. Blood 2019;134:292 doi 10.1182/blood-2019-129557. [DOI] [Google Scholar]
- 25.Armstrong SA, Kung AL, Mabon ME, Silverman LB, Stam RW, Den Boer ML, et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell 2003;3:173–83. [DOI] [PubMed] [Google Scholar]
- 26.Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, et al. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood 2004;103:3544–6 doi 10.1182/blood-2003-07-2441. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012;481:157–63 doi 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. The New England Journal of Medicine 2013;368:1781–90 doi 10.1056/NEJMoa1214514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol 2018;15(11):709–20 doi 10.1038/s41571-018-0105-0. [DOI] [PubMed] [Google Scholar]
- 30.Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov 2016;6(8):914–29 doi 10.1158/2159-8290.cd-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broad D DepMap 20Q1 Public. 2020. [Google Scholar]
- 32.Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018;562(7728):526–31 doi 10.1038/s41586-018-0623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer U, Forster M, Rinaldi A, Risch T, Sungalee S, Warnatz HJ, et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 2015;47(9):1020–9 doi 10.1038/ng.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frismantas V, Dobay MP, Rinaldi A, Tchinda J, Dunn SH, Kunz J, et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood 2017;129(11):e26–e37 doi 10.1182/blood-2016-09-738070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang BH, Leonard J, Wolf J, Degnin M, Lenz K, Wilmot B, et al. Significant In Vivo Sensitivity to Aurora Kinase Inhibition in TCF3-Hlf rearranged Acute Lymphoblastic Leukemia. Blood 2018;132(Supplement 1):4026 doi 10.1182/blood-2018-99-115706. [DOI] [Google Scholar]
- 36.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem 2003;278(21):18811–6 doi 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 37.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell 2013;23(1):121–8 doi 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iavarone C, Zervantonakis IK, Selfors LM, Palakurthi S, Liu JF, Drapkin R, et al. Combined MEK and BCL-2/XL Inhibition Is Effective in High-Grade Serous Ovarian Cancer Patient-Derived Xenograft Models and BIM Levels Are Predictive of Responsiveness. Mol Cancer Ther 2019;18(3):642–55 doi 10.1158/1535-7163.MCT-18-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, Scott CL. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest 2008;118(11):3651–9 doi 10.1172/JCI35437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A 2009;106(46):19503–8 doi 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan N, Wong M, Nannini MA, Hong R, Lee LB, Price S, et al. Bcl-2/Bcl-xL inhibition increases the efficacy of MEK inhibition alone and in combination with PI3 kinase inhibition in lung and pancreatic tumor models. Mol Cancer Ther 2013;12(6):853–64 doi 10.1158/1535-7163.MCT-12-0949. [DOI] [PubMed] [Google Scholar]
- 42.Inukai T, Hirose K, Inaba T, Kurosawa H, Hama A, Inada H, et al. Hypercalcemia in childhood acute lymphoblastic leukemia: frequent implication of parathyroid hormone-related peptide and E2A-HLF from translocation 17;19. Leukemia 2007;21(2):288–96 doi 10.1038/sj.leu.2404496. [DOI] [PubMed] [Google Scholar]
- 43.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang Y- L, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. The New England Journal of Medicine 2014;371:1005–15 doi 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tarlock K, Hansen ME, Hylkema T, Ries R, Farrar JE, Auvil JG, et al. Discovery and Functional Validation of Novel Pediatric Specific FLT3 Activating Mutations in Acute Myeloid Leukemia: Results from the COG/NCI Target Initiative. Blood 2015;126:87. [Google Scholar]
- 45.Surrey LF, MacFarland SP, Chang F, Cao K, Rathi KS, Akgumus GT, et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med 2019;11(1):32 doi 10.1186/s13073-019-0644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sallman DA, DeZern AE, Steensma DP, Sweet KL, Cluzeau T, Sekeres MA, et al. Phase 1b/2 Combination Study of APR-246 and Azacitidine (AZA) in Patients with <em>TP53</em> mutant Myelodysplastic Syndromes (MDS) and Acute Myeloid Leukemia (AML). Blood 2018;132:3091. [Google Scholar]
- 47.Demir S, Boldrin E, Sun Q, Hampp S, Tausch E, Eckert C, et al. Therapeutic targeting of mutant p53 in pediatric acute lymphoblastic leukemia. Haematologica 2020;105(1):170–81 doi 10.3324/haematol.2018.199364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melnikov A, Rogov P, Wang L, Gnirke A, Mikkelsen TS. Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res 2014;42(14):e112 doi 10.1093/nar/gku511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borthakur G, Popplewell L, Boyiadzis M, Foran J, Platzbecker U, Vey N, et al. Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in RAS-mutant relapsed or refractory myeloid malignancies. Cancer 2016;122:1871–9 doi 10.1002/cncr.29986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han L, Zhang Q, Dail M, Shi C, Cavazos A, Ruvolo VR, et al. Concomitant targeting of BCL2 with venetoclax and MAPK signaling with cobimetinib in acute myeloid leukemia models. Haematologica 2020;105(3):697–707 doi 10.3324/haematol.2018.205534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cremer A, Ellegast JM, Alexe G, Frank ES, Ross L, Chu SH, et al. Resistance Mechanisms to SYK Inhibition in Acute Myeloid Leukemia. Cancer Discov 2020;10(2):214–31 doi 10.1158/2159-8290.CD-19-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pikman Y, Alexe G, Roti G, Conway AS, Furman A, Lee ES, et al. Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-cell Acute Lymphoblastic Leukemia. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 2017;23:1012–24 doi 10.1158/1078-0432.CCR-15-2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kurtz SE, Eide CA, Kaempf A, Khanna V, Savage SL, Rofelty A, et al. Molecularly targeted drug combinations demonstrate selective effectiveness for myeloid- and lymphoid-derived hematologic malignancies. Proc Natl Acad Sci U S A 2017;114(36):E7554–E63 doi 10.1073/pnas.1703094114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pikman Y, Puissant A, Alexe G, Furman A, Chen LM, Frumm SM, et al. Targeting MTHFD2 in acute myeloid leukemia. The Journal of Experimental Medicine 2016;213:1285–306 doi 10.1084/jem.20151574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell 2004;13(2):169–78 doi 10.1016/s1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- 56.Case D, Babin V, Berryman J, Betz R, Cai Q, Cerutti D, et al. Amber 2014. 2014. [Google Scholar]
- 57.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 2018;27(1):14–25 doi 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tasian SK, Hurtz C, Wertheim GB, Bailey NG, Lim MS, Harvey RC, et al. High incidence of Philadelphia chromosome-like acute lymphoblastic leukemia in older adults with B-ALL. Leukemia 2017;31(4):981–4 doi 10.1038/leu.2016.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen IM, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood 2017;129(2):177–87 doi 10.1182/blood-2016-05-707653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hurtz C, Wertheim GB, Loftus JP, Blumenthal D, Lehman A, Li Y, et al. Oncogene-independent BCR-like signaling adaptation confers drug resistance in Ph-like ALL. J Clin Invest 2020. doi 10.1172/JCI134424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.