

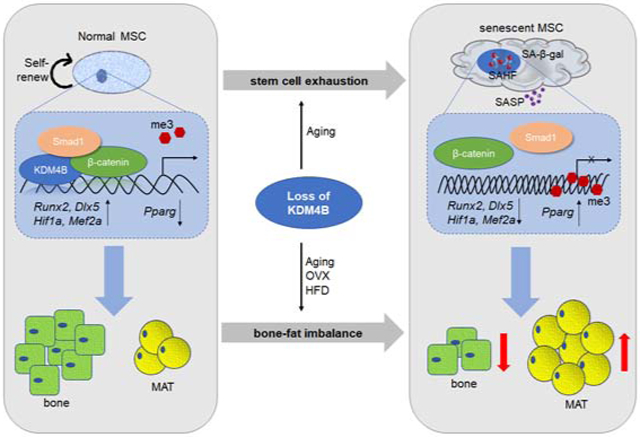
Summary
Skeletal aging is a complex process, characterized by a decrease in bone formation, an increase in marrow fat, and stem cell exhaustion. Loss of H3K9me3, a heterochromatin mark, has been proposed to be associated with aging. Here, we report that loss of KDM4B in mesenchymal stromal cells (MSCs) exacerbated skeletal aging and osteoporosis by reducing bone formation and increasing marrow adiposity via increasing H3K9me3. KDM4B epigenetically coordinated β-catenin/Smad1-mediated transcription by removing repressive H3K9me3. Importantly, KDM4B ablation impaired MSC self-renewal and promoted MSC exhaustion by inducing senescence-associated heterochromatin foci formation, providing a mechanistic explanation for stem cell exhaustion with aging. Moreover, while KDM4B was required for parathyroid hormone-mediated bone anabolism, KDM4B depletion accelerated bone loss and marrow adiposity induced by high fat diet. Our results suggest that the epigenetic rejuvenation and reversing bone-fat imbalance might be new strategies for preventing and treating skeletal aging and osteoporosis by activating KDM4B in MSCs.
Graphical Abstract
eTOC BLURB
Deng et al. show that loss of the epigenetic factor KDM4B in mesenchymal stem cells exacerbates skeletal aging and osteoporosis by reducing bone formation and increasing marrow adiposity via increasing H3K9me3, suggesting that targeting KDM4B might provide a new strategy for preventing and treating skeletal aging and osteoporosis.
Introduction
Aging causes significant changes to the skeleton, characterized by a decrease in bone formation and an increase in marrow adiposity (Fan et al., 2017; Kawai and Rosen, 2010; Zaidi et al., 2012). Age-related bone loss is a critical risk factor for osteoporosis that affects millions of patients worldwide. Osteoporosis is the most common metabolic bone disease and a leading cause of morbidity and mortality in the expanding aging population. In skeletal aging and osteoporosis, there is an inverse relationship between bone mass and marrow adiposity. Bone marrow adipose tissue (MAT) accumulation is increased at the expense of bone formation (Burge et al., 2007; Khosla and Hofbauer, 2017; Yu and Wang, 2016). Moreover, increased MAT accumulation and lower bone density are also hallmark symptoms of patients with radiation therapy and chemotherapy, or with type 1 diabetes (Naveiras et al., 2009; Yu and Wang, 2016). Therefore, it is important to understand the underlying mechanisms which control the switch between bone and fat in the bone marrow.
Bone marrow MSCs (also known as mesenchymal stem cells or skeletal stem cells) are believed to be the common progenitors for both osteoblasts and adipocytes in bone marrow, but commitment to these two lineages is mutually exclusive (Bianco et al., 2013; Zhou et al., 2014). Aging reduces bone marrow MSC number and its self-renewal, and inclines their differentiation into adipocytes at the expense of osteoblasts (Kawai and Rosen, 2010; Zaidi et al., 2012). Over the past decade, several transcription factors or signaling pathways have been identified in association with MSC fate commitment and differentiation (Chen et al., 2016; Crane and Cao, 2014; Deng et al., 2015; Fazeli et al., 2013; Wu et al., 2010). For example, the master osteogenic factor runt-related transcription factor 2 (RUNX2) was found to be a critical factor for MSC fate determination. Induction of RUNX2 inhibits adipogenesis while promoting osteogenesis (Chen et al., 2016; Deng et al., 2015; Fazeli et al., 2013). Parathyroid hormone (PTH) coordinates with the WNT/β-catenin and bone morphogenetic protein (BMP)/Smad signaling pathways to regulate MSC fate and differentiation (Crane and Cao, 2014; Wu et al., 2010). However, while these transcription factors and pathways have been found to govern switches between bone and fat, how epigenetic factors control skeletal aging remains poorly understood.
Epigenetic mechanisms play a critical role in aging, stem cell identity and fate. Stem cell exhaustion and epigenetic alteration are two of the hallmarks of aging (Sen et al., 2016). Both histone 3 lysine 9 (H3K9) hypermethylation and heterochromatin protein 1 (HP1) recruitment are hallmarks of heterochromatin formation. The loss of heterochromatin model of aging was proposed decades ago, and implicates that aging is associated with a loss of constitutive heterochromatin and cell senescence by decreasing H3K9me3 levels and delocalizing HP1 proteins (Nacarelli et al., 2017). In a premature aging disorder model of Werner syndrome, it was found that knock-in of catalytically inactive suppressor of variegation 3-9 homolog 1 (SUV39H1), a main H3K9me3 methyltransferase in MSCs, reduced H3K9me3 levels and accelerated MSC aging and replicative senescence in vitro (Zhang et al., 2015). On the contrary, some studies suggest that senescence-associated heterochromatin foci (SAHF; heterochromatin gain) are associated with cell aging, but most of these studies focus on oncogenic transformation (Narita et al., 2003). These two models are controversial in the field of aging, and there is very little genetic evidence which proves or disproves these models. We previously found that histone lysine demethylase 4B (KDM4B), a H3K9me3 demethylase, plays a critical role in osteogenic differentiation of MSCs, implying that erasing H3K9me3 may be required for bone formation in vivo (Ye et al., 2012). To explore the functional role of KDM4B in skeletal development and aging, we generated Kdm4b knockout mice.
Results
Exacerbating bone-fat imbalance in skeletal aging and osteoporosis by KDM4B loss
We generated conditional Kdm4b knockout mice in which the fifth exon containing the characteristic Jumonji domain (JMJD) was deleted in MSCs using Prx1Cre (Logan et al., 2002). Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) confirmed that Kdm4b was efficiently deleted in MSCs (Sca1+CD29+CD45−CD11b−) isolated from Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w littermates (Figure 1A). Unexpectedly, Prx1Cre;Kdm4bf/f mice were born normally and had no differences in size, weight or growth compared to Prx1Cre;Kdm4bw/w littermates. The whole-mount skeletal staining and histological analysis did not detect any developmental defects of craniofacial or long bones in neonatal Prx1Cre;Kdm4bf/f mice at P0 (Figures S1A-S1D). Recently, we showed that loss of Kdm4b in adipose tissues did not affect the adipocyte differentiation, but reduced the energy expenditure and caused obesity (Cheng et al., 2018). To further confirm our previous work on MSCs, we isolated MSCs from 3-month-old Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice and determined their lineage-specific differentiation potentials in vitro. Consistent with our previous findings, the deletion of Kdm4b in MSCs significantly impaired osteogenic differentiation (Figures S1E and S1F) as indicating by decreased alkaline phosphatase (ALP) activity, Alizarin Red S (ARS) staining and mRNA level of osteogenic makers and promoted adipogenic differentiation of MSCs in vitro by increased Oil Red O staining and mRNA level of adipogenic markers (Figures S1G and S1H). Colony forming units-osteoblast (CFU-OB) assay and colony forming units-adipocyte (CFU-AD) assay also confirmed loss of KDM4B promoted the MSC commitment to adipogenic lineage at the expense of osteogenic lineage (Figures S1I and S1J). To further confirm that loss of KDM4B altered lineage commitments of MSCs, Sca1+CD29+CD45−CD11b− MSCs from both Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice were sorted and seeded on 96-well plates to form individual clones. 16 to 20 individual clones from each mouse were picked after 2 weeks and then subjected them to osteogenic and adipogenic differentiation immediately. To minimize in vitro passages, half of the individual clones were utilized for ARS and Oil Red O staining, and the other half were used for gene expression studies. To rigidly test MSC lineage commitment, we induced osteogenic and adipogenic differentiation of MSCs with a mix of osteogenic and adipogenic induction medium (1: 1 ratio) as previously described by other groups (Fu et al., 2010; McBeath et al., 2004). ARS staining and Oil Red O staining demonstrated that all individual MSC clones from Kdm4b−/− mice exhibited a decrease in osteogenesis and an increase in adipogenesis compared with all clones from Kdm4b+/+ mice upon induction (Figures S1K-S1N). qRT-PCR found that, while the expression of osteogenic markers, Runx2 and Bglap, was significantly downregulated in Kdm4b−/− MSCs compared with Kdm4b+/+ MSCs, the expression of adipogenic markers, Pparg and Zfp423, was significantly upregulated in Kdm4b−/− MSCs (Figures S1O and S1P). Recently, Hif1a and Mef2a have been identified as key pro-osteogenic and anti-adipogenic transcription factors which control MSC fate (Rauch et al., 2019). We found that Hif1a and Mef2a were significantly reduced in Kdm4b−/− MSCs compared to Kdm4b+/+ MSCs (Figure S1Q).
Figure 1. Deletion of Kdm4b in mesenchymal cells using Prx1Cre exacerbates bone-fat imbalance in skeletal aging.

(A) Kdm4b mRNA in Sca1+CD29+CD45−CD11b− MSCs from 2-month-old Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice was assessed by qRT-PCR. n = 3.
(B) Representative μCT images of femurs from Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice at 3-, 12- and 18-month-old. Scale bar, 0.4 mm. n = 16.
(C) Quantitative measurements of BMD and BV/TV in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice by μCT. n = 16.
(D) Tb.N and Tb.Sp in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice by μCT. n = 16.
(E) Representative toluidine blue staining images of Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs. Blanks arrows indicate trabecular bones, and orange arrows indicate marrow adipose tissues. Scale bar, 0.4 mm. n = 16.
(F) Ob.N/BS and Ob.S/BS in 3-, 12- and 18-month-old Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice. n = 16.
(G) MAR and BFR as determined by dual labeling in Prx1Cre;Kdm4bw/wand Prx1Cre;Kdm4bf/f mice. n = 16.
(H) Representative μCT images of femoral cortex from Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice at 3-,12- and 18-month-old. Scale bar, 0.6 mm. n = 16.
(I) Quantitative measurements of cortical bone thickness (Cb.Th), cortex BMD (cBMD) and porosity by μCT. n = 16.
(J and K) Immunostaining of FABP4 and quantitative measurements of adipocytes in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice. Scale bar, 30 μm. n = 16.
(L and M) μCT analysis of MAT in tibiae of Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice. Scale bar, 1 mm. n = 16.
Data represent mean ± SD (error bars) from the pool of two independent experiments. The results were analyzed by two-way ANOVA with Holm-Sidak posthoc test except for (A) which was analyzed by Student’s t-test. *P < 0.05, **P < 0.01, between groups; #P < 0 .05; ##P < 0.01, age x genotype interaction.
Interestingly, in contrast to the lack of any defects in bone development, Prx1Cre;Kdm4bf/f mice displayed an accelerated age-related bone loss. With age advancing from 3 to 18 months, micro-computed tomography (μCT) analysis revealed a significantly greater decrease in both bone mineral density (BMD) and bone volume fraction (BV/TV) in Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w mice of mixed genders (Figures 1B and 1C). The 12- and 18-month-old Prx1Cre;Kdm4bf/f mice had significantly lower trabecular numbers (Tb.N) and greater trabecular spacing (Tb.Sp) than age-matched controls (Figure 1D). In addition, osteoblasts were also significantly reduced in 12- and 18-month-old Prx1Cre;Kdm4bf/f mice compared to age-matched Prx1Cre;Kdm4bw/w mice (Figures 1E and 1F). Furthermore, we performed a 7-day dynamic histomorphometric analysis using tetracycline labeling to determine if the accelerated bone loss was due to decreased bone formation. The mineral apposition rate (MAR) and bone formation rate (BFR) in 12- and 18-month -old Prx1Cre;Kdm4bf/f mice were significantly reduced compared to age-matched Prx1Cre;Kdm4bw/w mice (Figure 1G). Moreover, loss of Kdm4b also promoted cortical bone loss and increased porosity with age advancing from 3 to 18 months (Figures 1H and 1I). Prx1Cre is not expressed in the axial skeleton (Figures S2A and S2B) (Salazar et al., 2019), so to examine potential indirect effects of loss of Kdm4b in bone, we measured the bone mass in vertebrae (lumbar 4, L4) of Prx1Cre;Kdm4bf/f and Prx1Cre;Kdm4bw/w mice. We did not observe significant differences in vertebral bone mass and MAT between aged Prx1Cre;Kdm4bf/f and Prx1Cre;Kdm4bw/w mice (Figures S2C-S2F). In addition, osteoclasts were not affected by the deletion of KDM4B in MSCs (Figures S2G and S2H). The serum concentrations of procollagen type 1 N-terminal propeptide (P1NP), but not collagen type I c-telopeptide (CTX), were significantly lower in Prx1Cre;Kdm4bf/f mice than in Prx1Cre;Kdm4bw/w (Figure S2I). On the contrary, the number of adipocytes was significantly increased and MAT accumulation in the bone marrow was accelerated in aged Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w mice (Figures 1J and 1K). The distribution and regulation of in the bone marrow is region-specific, and is classified as constitutive MAT (cMAT) and regulated MAT (rMAT). While cMAT is relatively stable, rMAT responds to aging, hormone, and environmental cues. The proximal tibia and distal femur contain predominantly rMAT (Scheller et al., 2015). To further evaluate the region-specific effect of Kdm4b deletion on the formation of MAT, mouse tibial bone marrow lipids were stained with osmium-tetroxide as described previously (Scheller et al., 2014). μCT analysis revealed that there was a significant increase in rMAT in the proximal tibia of aged Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w littermates (Figures 1L and 1M). Moreover, the independent analysis of aged male and female mice also found that loss of KDM4B significantly promoted bone loss and MAT accumulation in both male and female mice (Figure S3).
To mimic the pathogenesis of osteoporotic bone loss, we performed ovariectomy (OVX) on female Prx1Cre;Kdm4bf/f and Prx1Cre;Kdm4bw/w mice to induce estrogen deficiency. As expected, μCT analysis found noticeable trabecular bone loss in Prx1Cre;Kdm4bw/w mice compared to sham controls 5 weeks after OVX. However, bone loss in Prx1Cre;Kdm4bf/f mice was significantly higher than in Prx1Cre;Kdm4bw/w mice (Figures 2A and 2B). Prx1Cre;Kdm4bf/f mice had more significantly decreased Tb.N and increased Tb.Sp compared to Prx1Cre;Kdm4bw/w mice following OVX (Figure 2C). Histomorphometric analysis showed that OVX-induced increase in osteoblast number and osteoblast surface were significantly suppressed in Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w mice (Figures 2D and 2E). Tetracycline labeling showed that the upregulated MAR and BFR, induced by OVX, were significantly blunted in Prx1Cre;Kdm4bf/f mice (Figure 2F), suggesting that loss of KDM4B suppressed the compensatory increase in bone formation after OVX. Osteoclast formation induced by OVX was not affected by loss of KDM4B in MSCs (Figures 2G). The serum concentration of P1NP and CTX further revealed that the increased net bone loss after OVX in Prx1Cre;Kdm4bf/f mice was caused by decelerating bone formation (Figure 2H). OVX significantly increased adipogenesis and MAT accumulation in Prx1Cre;Kdm4bw/w mice. However, adipocyte numbers and MAT accumulation were significantly greater in Prx1Cre;Kdm4bf/f mice than in Prx1Cre;Kdm4bw/w mice following OVX (Figures 2I and 2J).
Figure 2. Deletion of Kdm4b in mesenchymal cells using Prx1Cre exacerbated bone-fat imbalance in OVX-induced osteoporosis.

(A) Representative μCT images of femurs from Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice following sham and OVX. Scale bar, 0.4 mm. n = 8.
(B) Quantitative measurements of BMD and BV/TV in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice following sham and OVX by μCT. n = 8.
(C) Tb.N and Tb.Sp in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice following sham and OVX by μCT. n = 8.
(D) Representative toluidine blue staining images of Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following sham and OVX. Scale bar, 0.4 mm. n = 8.
(E) Ob.N/BS and Ob.S/BS in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following sham and OVX. n = 8.
(F) MAR and BFR as determined by dual labeling in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following sham and OVX. n = 8.
(G) Oc.N/BS and Oc.S/BS in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following sham and OVX. n = 8.
(H) ELISA of serum P1NP and CTX in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice following sham and OVX. n = 8.
(I and J) Representative immunostaining of FABP4 in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following sham and OVX. Scale bar, 30 μm. n = 8.
(K and L) Fat cell density and fat tissue fraction of FABP4-expressing tdTomato+ adipocytes in Prx1Cre;Kdm4bw/w;tdTomato and Prx1Cre;Kdm4bf/f;tdTomato mouse femurs following sham and OVX. Scale bar, 40 μm. n = 8.
(M and N) Representative immunostaining image of FABP4-expressing tdTomato− and tdTomato+ adipocytes in Prx1Cre/ERT2;Kdm4bw/w;tdTomato and Prx1Cre/ERT2;Kdm4bf/f;tdTomato mouse femurs following OVX, respectively. Scale bar, 40 μm. n = 6.
(O and P) μCT analysis of MAT in tibiae of Prx1Cre/ERT2;Kdm4bw/w;tdTomato and Prx1Cre/ERT2;Kdm4bf/f;tdTomato mice following sham and OVX. Scale bar, 1 mm. n = 6.
Data represent mean ± SD from the pool of two independent experiments. The results were analyzed by two-way ANOVA with Holm-Sidak posthoc test. *P < 0.05, **P < 0.01, between groups; #P < 0.05, ##P < 0.01 surgery x genotype interaction.
To further determine that KDM4B plays a key role in bone-fat switch in vivo, we generated Prx1Cre;Kdm4bf/f;tdTomato mice that expressed tdTomato in the Prx1Cre recombined cells, including osteoblasts and adipocytes (Madisen et al., 2010). OVX was then performed on both Prx1Cre;Kdm4bw/w;tdTomato and Prx1Cre;Kdm4bf/f;tdTomato mice. The exacerbation of adipocyte accumulation was validated by immunostaining for the adipocyte marker fatty acid-binding protein 4 (FABP4). All FABP4+ adipocytes were tdTomato-positive, confirming that adipocytes were differentiated from Prx1Cre-recombined MSCs. tdTomato+/FABP4+ adipocytes and MAT accumulation in Prx1Cre;Kdm4bf/f;tdTomato mice were significantly increased, compared to the Prx1Cre;Kdm4bw/w;tdTomato mice following OVX (Figures 2K and 2L). To further confirm our results, we generated Prx1Cre/ERT2;Kdm4bf/f;tdTomato and Prx1Cre/ERT2;Kdm4bw/w;tdTomato mice which allowed us to label new MSC-derived osteoblasts and adipocytes upon tamoxifen treatment. Immediately after OVX, tamoxifen was injected to induce tdTomato expression in Prx1-expressing MSCs. Similarly, the number of tdTomato+/FABP4+ adipocytes in Prx1Cre/ERT2;Kdm4bf/f;tdTomato mice was significantly greater compared to that in Prx1Cre/ERT2;Kdm4bw/w;tdTomato mice upon OVX. Of note, the small number of tdTomato−FABP4+ adipocytes in bone marrow were formed before tamoxifen injection and OVX, and their numbers did not differ between Prx1Cre/ERT2;Kdm4bf/f;tdTomato mice and Prx1Cre/ERT2;Kdm4bw/w;tdTomato mice (Figures 2M and 2N). μCT analysis of osmium-stained tibiae further confirmed the significant increase in rMAT in the proximal tibia of Prx1Cre/ERT2;Kdm4bf/f;tdTomato mice, compared to the littermates (Figures 2O and 2P).
MSCs in adult bone marrow specifically express the leptin receptor (LepR). In vivo lineage tracing has demonstrated that LepR+ MSCs are progenitors for both osteoblasts and adipocytes in adult mice (Yue et al., 2016; Zhou et al., 2014). To further confirm that loss of Kdm4b promoted bone loss and MAT accumulation, we generated LepRCre;Kdm4bf/f;tdTomato mice that expressed tdTomato in the LepRCre recombined cells, including osteoblasts and adipocytes. qRT-PCR confirmed that LepRCre efficiently deleted Kdm4b in MSCs (Figure S4A). OVX was then performed on both LepRCre;Kdm4bw/w;tdTomato mice and LepRCre;Kdm4bf/f;tdTomato mice. As expected, μCT analysis found that OVX-induced trabecular bone loss in LepRCre;Kdm4bw/w;tdTomato mice was significantly higher than in sham controls. However, bone loss in LepRCre;Kdm4bf/f;tdTomato mice was significantly exacerbated compared to LepRCre;Kdm4bw/w;tdTomato mice following OVX as determined by μCT and histological analysis (Figures S4B-S4G). Deletion of Kdm4b by LepRCre did not affect OVX-induced osteoclasts (Figures 4H). The merged immunofluorescent staining of FABP4 and tdTomato images confirmed that the FABP4-expressing adipocytes were differentiated from LepR+ MSCs. As predicted, OVX significantly induced adiposity as evidenced by an increase in adipocyte numbers and adipose tissue area in LepRCre;Kdm4bw/w;tdTomato mice. However, OVX-induced increase in adipocyte numbers and adipose tissue areas in LepRCre;Kdm4bf/f;tdTomato mice were significantly higher than in LepRCre;Kdm4bw/w;tdTomato mice (Figures S4I and S4J). μCT analysis revealed that there was a significant increase in rMAT accumulation in the proximal tibia of LepRCre;Kdm4bf/f;tdTomato mice compared to LepRCre;Kdm4bw/w;tdTomato following OVX (Figures S4K and S4L). In vitro differentiation assays also confirmed the deletion of Kdm4b by LepRCre promoted adipogenic differentiation of MSCs at the expense of osteogenic differentiation (Figures S4M-S4R).
Figure 4. KDM4B is required for PTH-mediated anabolic actions by controlling β-catenin/Smad1-mediated transcription.

(A) Representative μCT images of 12-month-old Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following PTH treatment. Scale bar, 0.4 mm. n = 8.
(B) Quantitative measurements of BMD and BV/TV in 12-month-old Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following PTH treatment by μCT. n = 8.
(C) Tb.N and Tb.Sp in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice following PTH treatment by μCT. n = 8.
(D) Representative H&E staining of Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following PTH treatment. Blanks arrows indicate trabecular bones, and orange arrows indicate marrow adipose tissues. Scale bar, 0.4 mm. n = 8.
(E) Ob.N/BS and Ob.S/BS in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following PTH treatment. n = 8.
(F) MAR and BFR as determined by dual labeling in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following PTH treatment. n = 8.
(G) Representative immunostaining of FABP4 in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following PTH treatment. Scale bar, 40 μm. n = 8.
(H) Fat cell density and fat tissue fraction in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs following PTH treatment. n = 8.
(I) qRT-PCR and Western blot showing that loss of KDM4B inhibited PTH-induced RUNX2 expression in mouse MSCs. n = 3.
(J-L) ChIP assays showing the enrichment of KDM4B (J), H3K9me3 (K) and β-catenin (L) at the Runx2 promoter treated with vehicle and PTH, respectively. n = 3.
(M and N) reChIP assays showing that β-catenin, Smad1 and KDM4B co-occupied on the Runx2 promoter. n = 3.
All data are presented as means ± SD. For (B), (C), (E), (F) and (H-L) the results were analyzed by two-way ANOVA with Holm-Sidak posthoc test. *P < 0.05, **P < 0.01, between groups; #P < 0.05, ##P < 0.01, treatment x genotype. For (M) and (N), *P < 0.05, **P < 0.01, Student’s t-test and one-way ANOVA with Tukey’s posthoc test.
Also see Figure S6
We further compared aging-associated bone loss in LepRCre;Kdm4bf/f;tdTomato mice with LepRCre;Kdm4bw/w;tdTomato mice. We found that trabecular bone loss in 12- and 18-month-old LepRCre;Kdm4bf/f;tdTomato mice was significantly greater than in age-matched LepRCre;Kdm4bw/w;tdTomato mice (Figures S5A-S5C). In contrast, rMAT was significantly elevated in 12- and 18-month-old LepRCre;Kdm4bf/f;tdTomato mice compared to age-matched LepRCre;Kdm4bw/w;tdTomato mice (Figures S5D and S5E). While KDM4B controls MSC fate decisions, it may also be required for osteoblast functions. To test these possibilities, we generated OcnCre;Kdm4bf/f mice in which Kdm4b was deleted in mature osteoblasts. No significant difference in bone loss was observed between OcnCre;Kdm4bf/f and OcnCre;Kdm4bw/w mice at 3-, 12- or 18-month-old ages (Figures S5F-S5M). The accumulation of MAT in aged OcnCre;Kdm4bf/f mice also did not differ significantly from that in aged control mice (Figures S5I and S5N).
Epigenetic control of gene transcription in MSC fate decision by KDM4B
To understand how loss of KDM4B accelerated skeletal aging and increased MAT, we isolated total RNA from both Kdm4b−/− and Kdm4b+/+ MSCs, respectively, and performed RNA-seq. Loss of KDM4B is expected to lead to decreased levels of gene expression due to elevated levels of H3K9 hypermethylation. In accordance gene ontology (GO) analysis revealed that downregulated genes (> 1.5 folds) by Kdm4b deficiency were enriched in skeletal system development (P = 1.7 x 10−20), protein catabolic process (P = 4.9 x 10−18), etc. (Figure 3A and Table S1) (Huang et al., 2008, 2009). Kyoto encyclopedia of genes and genomes (KEGG) analysis further showed that both Wnt signaling pathway (P = 5.8 x 10−8) and TGF-β signaling pathway (P = 8.6 x 10−7), including Runx2 and Dlx5, which are critical for osteogenesis, were affected by the deletion of Kdm4b in MSCs (Figure 3B, Tables S2 and S3). Gene set enrichment analysis (GSEA) revealed that the signature genes for Wnt/β-catenin and TGF-β signaling were downregulated in Kdm4b−/− MSCs (Figures 3C and 3D). In addition, GO analysis revealed the upregulated genes (> 1.5 folds) in Kdm4b−/− MSCs are associated with cell cycle (P = 2.0 x 10−21), cell division (P = 5.1 x 10−17) (Figure S6A). KEGG analysis showed these genes are associated with DNA replication (P = 2.4 x 10−10) and oxidative phosphorylation (P = 1.2 x 10−6) (Figure S6B). We also used GSEA to examine the signature genes associated with adipogenesis and adipocytokine signaling pathway in Kdm4b−/− and Kdm4b+/+ MSCs. The related genes might be upregulated with loss of KDM4B, however, the analysis did not reach statistical significance (Figure S6C).
Figure 3. KDM4B epigenetically controls Wnt/β-catenin- and BMP/Smad-mediated transcription by erasing H3K9me3 in mouse MSCs.

(A and B) GO (A) and KEGG (B) analysis of downregulated genes with Kdm4b deletion in MSCs.
(C and D) GSEA shows a significant decrease of Wnt/β-catenin (C) and TGF-β (D) gene signatures in Kdm4b−/− MSCs, respectively. Black bars represent individual genes in rank order. NES, normalized enrichment score; FDR, false discovery rate.
(E) Pie chart showing genome-wide binding profiles of KDM4B in mouse MSCs.
(F and G) The average bindings of KDM4B (F) and levels of H3K9me3 (G) for all refseq gene promoters, spanning ± 2 kb of the closest transcription start sites (TSSs). Red line, Kdm4b+/+ MSCs; green line, Kdm4b−/− MSCs.
(H) GO analysis of genes associated with KDM4B and over 2-fold H3K9me3 upregulation in Kdm4b−/− MSCs (±2kb of their TSSs). Bars represent −log10 of binomial raw p values.
(I) GREAT analysis of the nearest genes whose TSSs are within ±2kb of the unique KDM4B peaks in Kdm4b+/+ MSCs (upper) and unique H3K9me3 peaks in Kdm4b−/− MSCs (lower).
(J) Distribution of KDM4B and H3K9me3 at the ±5 kb regions centered at KDM4B peaks in Kdm4b+/+ and Kdm4b−/− MSCs (log2 scale).
(K and L) Gene tracks showing KDM4B binding and H3K9me3 levels at the Runx2 (K) and Ccnd1 (L) in Kdm4b+/+ and Kdm4b−/− MSCs. Bars above the gene tracks shows the significant peaks identified by using MACS2.
Also see Figure S6
To identify the direct targets of KDM4B, we performed chromatin immunoprecipitation sequencing (ChIP-seq) to determine the genome-wide occupancy of KDM4B. The cis-regulatory element annotation system (CEAS) analysis revealed that KDM4B showed very high relative enrichment in the promoter regions (±2 kb of the closest transcription start sites (TSS); 13.9%) compared to the genome background (2.0%) (Figure 3E). The enrichment of KDM4B at their promoters was dramatically decreased in Kdm4b−/− MSCs (Figure 3F), confirming the specificity of KDM4B binding peaks. H3K9me3 is a repressive mark of gene transcription. Notably, H3K9me3 enrichment at the promoter regions increased in Kdm4b−/− MSCs, corresponding to a dramatic decrease in KDM4B enrichment at the same region as compared to Kdm4b+/+ MSCs (Figure 3G). The genes that have both KDM4B enrichments in Kdm4b+/+ MCSs and H3K9me3 increases in Kdm4b−/− MSCs compared with Kdm4b+/+ MSCs at their promoters were chosen for GO analysis. We found that these genes were associated with transcription regulation (P = 2.7 x 10−25), skeletal system morphology (P = 3.3 x 10−4), Wnt/β-catenin (P = 3.9 x 10−3) and TGF-β signaling (P = 7.2 x 10−4) (Figure 3H). As revealed by the Genomic Region Annotation Tool (GREAT, version 4.0.4) (McLean et al., 2010), the nearest genes whose TSSs are within ±2 Kb of unique KDM4B peaks in Kdm4b+/+ MCSs and unique H3K9me3 peaks in Kdm4b−/− MSCs were enriched in aging (false discovery rate (FDR) value < 2.8 x 10−125), skeletal morphology (FDR value < 3.2 x 10−11) and body weight (FDR value < 3.9 x 10−10), respectively (Figure 3I). In addition, genome-wide analysis of KDM4B and H3K9me3 occupancy showed increased H3K9me3 enrichment after Kdm4b deletion (Figure 3J). Representative view of ChIP-seq results showed that KDM4B was present on the promoters of Wnt target genes, Runx2 and Ccnd1, and the deletion of Kdm4b significantly increased H3K9me3 levels on their promoters, respectively (Figures 3K and 3L). Individual chromatin Immunoprecipitation-quantitative PCR (ChIP-qPCR) also confirmed our ChIP-seq results (Figures S6D). Additionally, KDM4B was found occupied on the promoters of pro-osteogenic and anti-adipogenic transcription factors Hif1a and Mef2a (Rauch et al., 2019), and loss of KDM4B led to increased H3K9me3 levels on their promoters by ChIP-seq and Individual ChIP-qPCR assay (Figures S6E-S6H).
Epigenetic control of PTH-mediated anabolic metabolism by KDM4B
PTH promotes bone formation by orchestrating both Wnt/β-catenin and BMP/Smad signaling pathways (Wan et al., 2008). Since loss of KDM4B promoted osteoporotic bone loss and skeletal aging, we first examined whether PTH could regulate Kdm4b expression. PTH induces gene expression by activating protein kinase A (PKA) and Wnt/β-catenin signaling (Wang et al., 2006). Consistently, we found that the competitive PKA inhibitor H89 significantly inhibited PTH-induced expression of Kdm4b (Figure S6I). Similarly, siRNA-mediated depletion of β-catenin also suppressed Kdm4b expression induced by PTH (1-34) (Figure S6J). In order to further confirm whether PTH induces Kdm4b expression by PKA and Wnt/β-catenin signaling, we analyzed the promoter region of Kdm4b by using JASPAR profiles (Mathelier et al., 2016), and identified two potential cAMP-response element-binding protein (CREB) binding sites (−2973~−2966; −2178~−2171) next to T-cell factor/lymphoid enhancer factor 1 (TCF/LEF1) binding sites (−2957~−2947; −2093~−2083), respectively. ChIP assays revealed that both CREB and β-catenin were recruited to the promoter region of Kdm4b in response to PTH (1-34) treatment (Figures S6K and S6L).
Because PTH (1-34) induced Kdm4b expression, we hypothesized that KDM4B, as an epigenetic factor, might play a critical role in the PTH-mediated anabolic effect. To test our hypothesis, we treated 12-month-old Prx1Cre;Kdm4bf/f and Prx1Cre;Kdm4bw/w mice with PTH (1-34). PTH (1-34) administration significantly increased trabecular bone mass in Prx1Cre;Kdm4bw/w mice, but not in Prx1Cre;Kdm4bf/f mice, as determined by μCT analysis (Figures 4A and 4B). PTH (1-34) also significantly increased Tb.N and reduced Tb.Sp in Prx1Cre;Kdm4bw/w mice compared to Prx1Cre;Kdm4bf/f mice (Figure 4C). Histomorphologic analysis found that the increase in osteoblasts, MAR and BFR induced by PTH were significantly reduced in Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w mice (Figures 4D-4F). Analysis of serum concentration of P1NP confirmed that PTH-induced bone formation were blunted in Prx1Cre;Kdm4bf/f mice (Figure S6M). More importantly, while PTH (1-34) significantly reduced the number of adipocytes and MAT accumulation in Prx1Cre;Kdm4bw/w mice, it had a minimal inhibitory effect on the number of adipocytes and MAT accumulation in Prx1Cre;Kdm4bf/f mice (Figures 4G and 4H). μCT analysis further confirmed that KDM4B was required for PTH-mediated inhibition of MAT accumulation, while cMAT was not affected by PTH treatment (Figure S6N).
While the induction of Runx2 promotes osteogenic differentiation, it also inhibits adipogenic differentiation (Ge et al., 2016). qRT-PCR and Western blot demonstrated that the deletion of Kdm4b inhibited Runx2 expression induced by PTH (1-34) in MSCs (Figure 4I). ChIP-qPCR further confirmed that increased KDM4B was present on the promoter of Runx2 in Kdm4b+/+ MSCs upon PTH treatment (Figure 4J). Consistently, H3K9me3 levels were significantly reduced in Kdm4b+/+ MSCs upon PTH (1-34) stimulation (Figure 4K). In contrast, KDM4B could not be detected on the Runx2 promoter and H3K9me3 levels remained higher in Kdm4b−/− MSCs following PTH (1-34) treatment (Figures 4J and 4K). Our results suggest that PTH-induced KDM4B was critical for erasing repressive H3K9me3 marks on the Runx2 promoter in order to activate its transcription. Further, loss of KDM4B in MSCs abrogated the induction of β-catenin binding to the Runx2 promoter following PTH (1-34) treatment (Figure 4L). Because both β-catenin and Smad1 collaboratively induce Runx2 expression and are involved in PTH signaling (Ge et al., 2016; Rodríguez-Carballo et al., 2011), we further tested whether KDM4B coordinated with β-catenin and Smad1 to activate PTH (1-34)-induced gene transcription using re-ChIP assays. We treated MSCs with PTH (1-34) and isolated chromatin complexes. Fragmented chromatin complexes were first precipitated with anti-β-catenin and then subjected with anti-Smad1 or anti-KDM4B. Compared with IgG control, a significant amount of Smad1- and KDM4B-bound chromatin complexes were enriched from the initial β-catenin-associated chromatin complex, as determined by qPCR using Runx2-promoter specific primers (Figure 4M). The reverse re-ChIP-qPCR, first using anti-Smad1 and subsequently anti-KDM4B and anti-β-catenin, also revealed that both KDM4B and β-catenin on the Runx2 promoter were significantly enriched in Smad1-associaed complexes (Figure 4N).
Exacerbating bone-fat imbalance induced by HFD due to KDM4B loss
High fat diet (HFD) as an environmental factor has been reported to promote MAT expansion and bone loss in skeletal aging (Sen et al., 2016). To further demonstrate that KDM4B is an important epigenetic factor which regulates bone metabolism and bone-fat switch, we examined whether loss of KDM4B affected HFD-induced bone loss and MAT expansion. Previously, it has been shown that HFD significantly altered trabecular bone mass in 12 weeks (Tencerova et al., 2018). Thus, we tested whether deletion of Kdm4b could accelerate HFD-induced bone loss at 8 weeks. Male Prx1Cre;Kdm4bf/f and Prx1Cre;Kdm4bw/w mice were subjected to low-fat diet (LFD) and HFD for 8 weeks, respectively. Consistent with our recent findings (Cheng et al., 2018), the body weight gain induced by HFD for 8 weeks was enhanced in Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w mice probably due to the fact Prx1Cre also deleted Kdm4b in pre-adipocytes (Figure 5A). HFD significantly promoted trabecular bone loss in Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w mice (Figures 5B-5D). In contrast, HFD induced a significant increase in rMAT in Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w mice (Figures 5E and 5F). Histological analysis further confirmed that loss of KDM4B enhanced HFD-induced MAT expansion in bone marrow (Figures 5G-5I). However, vertebral bone loss was not significantly enhanced in Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w mice following HFD treatment, ruling out non-specific effects of body weight gain in Prx1Cre;Kdm4bf/f on femoral bone loss (Figures 5J and 5K).
Figure 5. Deletion of Kdm4b in MSCs exacerbates bone-fat imbalance in HFD-fed mice.

(A) Body weight growth curves of Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice fed a HFD or low fat diet (LFD) for 8 weeks. n = 7.
(B) Representative μCT images of femurs from Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice fed a HFD or LFD for 8 weeks. Scale bar, 0.4 mm. n = 7.
(C) Quantitative measurements of BMD and BV/TV in (B) by μCT. n = 7.
(D) Quantitative measurements of Tb.N and Tb.Sp in (B) by μCT. n = 7.
(E and F) μCT analysis of MAT in tibiae of Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice fed a HFD or LFD for 8 weeks. Scale bar, 1 mm. n = 7.
(G) Representative H&E staining images of Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs fed a HFD or LFD for 8 weeks. Scale bar, 0.4 mm. n = 7.
(H) Representative immunostaining of FABP4 in Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mouse femurs fed a HFD for 8 weeks. Scale bar, 50 μm. n = 7.
(I) Fat cell density and fat tissue fraction in femurs of Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice fed a HFD or LFD for 8 weeks. n = 7.
(J) Representative μCT images of the 4th lumbar vertebrae (L4) from Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice fed a HFD or LFD. Scale bar, 0.7 mm. n = 7.
(K) Quantitative measurements of BMD and BV/TV by μCT in L4 from Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice fed a HFD or LFD. n = 7.
Data represent mean ± SD (error bars) from the pool of two independent experiments and were analyzed by two-way ANOVA with Holm-Sidak posthoc test. *P<0.05, **P<0.01, between groups; #P<0.05, ##P<0.01, diet x genotype interaction.
Control of MSC self-renewal and SAHF in aging by KDM4B
Since loss of KDM4B exacerbated skeletal aging, we hypothesized that KDM4B might be required for MSC self-renewal and maintenance. MSCs were originally defined as nonhematopietic, plastic-adherent, colony-forming cells in culture, referred to as colony-forming unit-fibroblasts (CFU-Fs) (Bianco et al., 2013). We found that deletion of Kdm4b in MSCs of 18-month-old mice reduced the number of CFU-Fs, suggesting loss of KDM4B might reduce MSC pools (Figure 6A). Consistently, GSEA also revealed loss of KDM4B significantly decreased expression of stem cell signature genes in MSCs (Figure 6B).
Figure 6. Loss of KDM4B impairs MSC self-renewal.

(A) Representative images of CFU-Fs formed by cells from Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice. Scale bar, 8 mm. n = 5.
(B) GSEA shows a significant decrease of stem cell core gene signatures in Kdm4b−/− MSCs.
(C) Representative H&E staining images of the first transplants from Kdm4b+/+ and Kdm4b−/− MSCs. Scale bar, 150 μm. n = 8 for Kdm4b+/+ MSCs; n = 7 for Kdm4b−/− MSCs.
(D) Fluorescence images of bone tissue in the transplants from Kdm4b+/+ and Kdm4b−/− MSCs. Scale bar, 150 μm. n = 8 for Kdm4b+/+ MSCs; n = 7 for Kdm4b−/− MSCs.
(E) Representative images of FABP4-expressing tdTomato+ adipocytes in transplants from Kdm4b+/+ and Kdm4b−/− MSCs. Scale bar, 40 μm. n = 8 for Kdm4b+/+ MSCs; n = 7 for Kdm4b−/− MSCs.
(F and G) CFU-F (F) and CFU-OB (G) assays of cells from the first transplants. Scale bar, 8 mm. n = 3.
(H) Bone areas and fat areas in the secondary transplants. n = 7 for Kdm4b+/+ MSCs. n = 3 for Kdm4b−/− MSCs.
(I) CFU-F assays of cells from the secondary transplants. Scale bar, 8 mm. n = 3.
Data represent mean ± SD (error bars). For (A) the results were analyzed by two-way ANOVA with Holm-Sidak posthoc test. *P < 0.05, **P < 0.01, between groups; #P < 0.05, ##P < 0.01, age x genotype interaction. For (D-I), *P < 0.05, **P < 0.01, Student t’s test.
To further test the role of KDM4B in regulation of self-renewal in MSCs, we isolated MSCs from Prx1Cre;Kdm4bw/w;tdTomato and Prx1Cre;Kdm4bf/f;tdTomato mice, and assessed the self-renewal capacity of tdTomato+ MSCs by serial transplantation. For the primary transplantation, tdTomato+ Kdm4b+/+ and Kdm4b−/− MSCs were incubated with a gelfoam scaffold, and subcutaneously transplanted in the dorsal sites of nude mice. After 6 weeks, the mice were sacrificed and the transplants were collected for histology analysis. All Kdm4b+/+ MSCs (8 out of 8 transplants) and Kdm4b−/− MSCs (7 out of 8 transplants) gave rise to bone, fat, and stromal tissues in the transplants (Figure 6C). The cells in bone tissues expressed tdTomato, indicating that the bone was generated by the transplanted tdTomato+ MSCs (Figure 6D). Quantitative measurement of mineralized tissue areas revealed a 39% decrease in bone formation by Kdm4b−/− MSCs compared to Kdm4b+/+ MSCs (Figure 6D). The tdTomato+ adipocytes were further confirmed by immunostaining of FABP4 (Figure 6E). In contrast, Kdm4b−/− MSCs generated more adipose tissues compared to Kdm4b+/+ MSCs (Figure 6E). Importantly, the cells isolated from the transplants of Kdm4b−/− MSCs exhibited a significant decrease in CFU-F numbers (Figure 6F). In vitro CFU-osteoblasts (CFU-OB) assays also showed a significant decrease in alkaline phosphatase-positive colonies in Kdm4b−/− MSCs (Figure 6G). Subsequently, MSCs from the first transplantation were isolated and used for the secondary transplantation. After 6 weeks, while 7 out of 8 transplants of Kdm4b+/+ MSCs generated bone, fat, and stromal tissues, only 3 out of 7 secondary transplants of Kdm4b−/− MSCs formed bone, fat, and stromal tissues. The secondary transplants with Kdm4b−/− MSCs had a significant decrease in bone formation and a greater increase in adipose tissue formation compared to Kdm4b+/+ MSCs (Figure 6H). Furthermore, the cells from the secondary transplants of Kdm4b−/− MSCs were barely able to generate CFU-Fs (Figure 6I), so we could not perform the third transplantation, indicating a loss of self-renewal capacity.
Both H3K9 hypermethylation and heterochromatin protein 1 (HP1) recruitment are hallmarks of heterochromatin formation. The loss of heterochromatin model of aging was proposed decades ago and proposes that aging is associated with a loss of constitutive heterochromatin and cell senescence by decreasing H3K9me3 levels and delocalizing HP1 proteins (Nacarelli et al., 2017). Since KDM4B is a H3K9me3 demethylase, loss of KDM4B should lead to increased H3K9me3 in MSCs, and in turn help maintain constitutive heterochromatin which might be inconsistent with the loss of heterochromatin model of aging. To reconcile this, we isolated MSCs from 3-month-old Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice and examined their proliferation. Interestingly, EdU incorporation assays revealed that Kdm4b−/− MSCs significantly lost their proliferative potential compared to Kdm4b+/+ MSCs upon 5 passages in vitro (Figure 7A). Senescence-associated beta-galactosidase (SA-β-gal) staining revealed that deletion of Kdm4b promoted MSC replicative senescence in vitro (Figure 7B). The restoration of KDM4B, but not KDM4B(H189A), was able to rescue MSC senescence, indicating that the enzymatic activity of KDM4B is required for preventing senescence (Figure 7C). Interestingly, SA-β-gal staining found that MSCs from 20-month-old Prx1Cre;Kdm4bw/w mice had increased senescence at early passages (Figure 7D). SAHF is also proposed to be associated with aging, but most studies have focused on oncogene-induced senescence (Nacarelli et al., 2017). To explore whether loss of KDM4B promoted SAHF formation, we immunostained MSCs with 4’, 6-diamidino-2-phenylindole (DAPI), anti-H3K9me3 and anti-HP1α. While there was no difference in early passage of MSCs, the percentage of H3K9me3 and HP1α foci was significantly increased in MSCs from 3-month-old Prx1Cre;Kdm4bf/f;tdTomato mice compared to Prx1Cre;Kdm4bw/w;tdTomato mice after 5 passages (Figures S7A and S7B). Loss of KDM4B also significantly increased the nuclear size of MSCs after 5 passages in vitro (Figure S7B). Moreover, in vitro cultured early-passage MSCs from 20-month-old Prx1Cre;Kdm4bf/f;tdTomato mice had more SAHF and increased nuclear size compared to Prx1Cre;Kdm4bw/w;tdTomato mice (Figures S7C and S7D). MSCs from 20-month-old LepRCre;Kdm4bf/f;tdTomato mice also showed similar trends (Figures S7E and S7F).
Figure 7. Loss of KDM4B induces SAHF formation in MSCs and reduces MSC pool in aged mice.

(A) EdU incorporation assay of MSCs from Prx1Cre;Kdm4bw/w;tdTomato and Prx1Cre;Kdm4bf/f;tdTomato mice at P1 and P5, respectively. Scale bar, 40 μm. n = 4.
(B) SA-β-gal staining of MSCs from 3-month-old Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice at P1 and P5. Scale bar, 50 μm. n = 6.
(C) Restoration of KDM4B, but not KDM4B(H189A), recued the replicative senescence in Kdm4b−/− MSCs. Scale bar, 100 μm. n = 3.
(D) SA-β-gal staining of MSCs from 20-month-old Prx1Cre;Kdm4bw/w mice and Prx1Cre;Kdm4bf/f mice at passage 1. Scale bar, 50 μm. n = 4.
(E and F) Representative immunostaining of H3K9me3 (E) and HP1α (F) in Sca1+Tomato+CD45−CD11b− MSCs from 3- and 20-month-old Prx1Cre;Kdm4bw/w;tdTomato mice and Prx1Cre;Kdm4bf/f;tdTomato mice. Scale bars, 6 μm. n = 3.
(G) Measurements of SAHF positive cells and nuclear size in Sca1+Tomato+CD45−CD11b− MSCs from 3- and 20-month-old Prx1Cre;Kdm4bw/w;tdTomato mice and Prx1Cre;Kdm4bf/f;tdTomato mice. n = 3. 200 nuclei were analyzed in each group.
(H) Loss of KDM4B accelerated a decrease in Sca1+CD29+CD45−CD11b− MSCs in aged Prx1Cre;Kdm4bw/w mice and Prx1Cre;Kdm4bf/f mice. n = 5.
(I) CFU-Fs formed by the cells from 18-month-old Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice treated with vehicle and PTH, respectively. Scale bar, 8 mm. n = 3.
(J) Flow cytometry analysis for Sca1+CD29+CD45−CD11b− MSC population in bone marrow of 18-month-old Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice treated with vehicle and PTH, respectively. n = 3.
Data represent mean ± SD. For (A), (B), and (G-J), the results were analyzed by the two-way ANOVA with Holm-Sidak posthoc test. *P < 0.05, **P < 0.01, between groups; #P < 0.05 passage x genotype interaction, age x genotype interaction, or treatment x genotype interaction. For (C), *P < 0.05, one-way ANOVA with Tukey’s posthoc test; and for (D), **P < 0.01, the Student t’s test.
Also see Figure S7
To further confirm whether loss of KDM4B promoted SAHF formation in MSC in vivo, we modified our approaches and freshly sorted tdTomato+Sca1+CD45−CD11b− MSCs from 3- and 20-month-old Prx1Cre;Kdm4bw/w;tdTomato mice and Prx1Cre;Kdm4bf/f;tdTomato mice. After sorting, half of the MSCs were immediately used for total RNA isolation without culture in vitro, and the other half were plated on coverslips overnight to allow cell attachment for immunostaining with minimal interference in vitro. Immunostaining revealed that loss of KDM4B significantly increased the formation of both H3K9me3 and HP1α foci with advancing age from 3 months to 20 months (Figures 7E-7G). Moreover, loss of KDM4B also increased the nuclear size of MSCs in aged mice (Figure 7G). qRT-PCR showed that deletion of Kdm4b led to an increase in the expression of senescent genes, Cdkn2a (also known as p16) and Cdkn1a (also known as p21), in freshly isolated MSCs at aged mice (Figure S7G). Consistently, senescent MSCs in aged mice obtained the senescence-associated secretary phenotype (SASP). The expression of Il1a, Ccl2, Cxcl10 and tnf in freshly isolated MSCs was significantly elevated in aged Prx1Cre;Kdm4bf/f;tdTomato mice compared to Prx1Cre;Kdm4bw/w;tdTomato mice (Figure S7H). Consistently, FACS analysis revealed that Sca1+CD29+CD45−CD11b− MSC numbers were significantly reduced in the bone marrow of Prx1Cre;Kdm4bf/f mice compared to Prx1Cre;Kdm4bw/w mice with advancing age (Figures 7H). More importantly, PTH treatment increased the MSC pool in aged Prx1Cre;Kdm4bw/w mice, but not in Prx1Cre;Kdm4bf/f mice, as revealed by CFU-F assays (Figure 7I). Cell sorting for Sca1+CD29+CD45−CD11b− MSCs further revealed that PTH treatment increased the MSC pool in aged Prx1Cre;Kdm4bw/w mice, but not in Prx1Cre;Kdm4bf/f mice (Figures 7J).
Discussion
Our in vivo studies in the combination of genetic, epigenetic and pharmacologic approaches uncover that KDM4B is a critical epigenetic factor that controls skeletal aging and osteoporosis. Aging is the single largest risk factor for bone loss, reduced MSC pool and shifting of their fate commitment and differentiation potential (Farr et al, 2016; Geng et al., 2011). The loss of KDM4B increased MAT accumulation in aged mice and exacerbated osteoporotic bone loss. Interestingly, we found that KDM4B was both PTH-responsive and PTH-dependent. Deletion of Kdm4b in MSCs blunted the PTH-mediated increase in bone formation and inhibition of adiposity. In addition to bone-fat balance, we demonstrated that KDM4B controlled MSC self-renewal and maintained the MSC pool in vivo. Our results suggest that KDM4B might be an authentic epigenetic factor for controlling skeletal aging and osteoporosis.
The loss of heterochromatin with global H3K9me3 decrease has been identified in premature aging diseases such as the Hutchinson-Gilford Progeria Syndrome (HGPS) and Werner Syndrome (WS), implicating that the loss of heterochromatin is an important causal factor for aging (Nacarelli et al., 2017). The mutation of WRN is known to impair heterochromatin formation and lead to WS, also known as adult progeria. It has been shown that WRN-deficient MSCs globally lose H3K9me3 and heterochromatin, resulting in the replicative senescence and exhaustion of MSCs upon serial passaging in vitro (Zhang et al., 2015). However, it is unknown whether occurs in physiological aging. There may be differences in regulation of heterochromatin and aging between human and mouse. WRN knockout mice could survive to at least 2 years of age and did not show any histological signs of premature aging (Lombard et al, 2000). Our findings showed that loss of KDM4B promoted SAHF in vitro and in vivo and impaired MSC self-renewal by increasing H3K9me3. Moreover, contrary to the loss of heterochromatin model, the specific deletion of Kdm4b promoted skeletal aging and MAT accumulation in aged mice, but not in young adult mice. These divergent results might reflect mouse-human differences. Moreover, a delicate balance between H3K9 methylation and demethylation might be required for maintaining organ homeostasis or preventing aging. Excessive loss or gain of heterochromatin might target different genes or signaling pathways to lead to accelerated aging. Moreover, differences between premature and physiological aging are likely. It is possible that loss of heterochromatin could be mainly associated with premature aging in MSCs. Gaining H3K9me3 and SAHF might play an important role in physiological skeletal aging. While SAHF has been investigated in the context of oncogene-induced senescence, our results suggest that loss of Kdm4b can induce SAHF and promote skeletal aging. Supporting our studies, it has been shown that H3K9me3 inhibition improves spatial memory and increased spine formation in aged mice but not in young mice (Snigdha et al., 2016).
Estrogen replacement and intermittent PTH are currently FDA-approved treatment modalities for osteoporosis. In addition to their established and interconnected roles in bone turnover, both have been shown to regulate MSC lineage allocation (Fan et al., 2017; Okazaki et al., 2002), an important aspect of skeletal aging with poorly understood mechanisms. Our findings underline the importance of KDM4B in MSC lineage specification and MAT balance in various physiopathological conditions as an epigenetic switch. PTH has been found to attenuate osteoporosis and MAT by directing MSC fate decision (Fan et al., 2017). Importantly, our studies showed that the induction of KDM4B is required for PTH-mediated bone formation and MAT inhibition, providing insights into new epigenetic mechanism for PTH-mediated anabolic actions. Based on our RNA-seq and ChIP-seq results, we found that KDM4B promoted transcription in MSCs by erasing repressive H3K9me3 marks. While PTH induced KDM4B expression through β-catenin/CREB, loss of KDM4B also impaired the recruitment of β-catenin to chromatin induced by PTH. Intriguingly, since KDM4B forms a complex with β-catenin/Smad1 upon PTH stimulation, KDM4B might form a positive feedback loop to promote bone formation and inhibit MAT accumulation by enhancing β-catenin/Smad1-mediated transcription. Finally, we found that loss of KDM4B promoted bone loss and MAT induced by HFD, further supporting that KDM4B is an authentic epigenetic factor for regulating skeletal aging. Our results suggest that the epigenetic rejuvenation and reversing bone-fat imbalance might be new strategies for preventing or treating skeletal aging and osteoporosis by activating KDM4B in MSCs.
Limitations of the Study
Aging is trigged by multiple factors. While there are many similarities in epigenetic mechanisms of skeletal aging in both mouse and human, there are also some differences in regulation of heterochromatin and aging between mouse and human. We demonstrated that H3K9me3 levels are upregulated in Kdm4b−/− MSCs, especially at on the promoter regions. However, whether loss of KDM4B affects overall accessibility of the chromatin is unknown and needs to be further verified by ATAC-seq and DNase/MNase-seq in the future. The gain of function study needs to be performed to determine whether the induction of KDM4B could prevent skeletal aging by promoting overall accessibility of the chromatin in MSCs.
STAR★ METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Cun-Yu Wang (cwang@dentistry.ucla.edu).
Materials Availability
Plasmids used in this study are described in the Key Resource Table and available upon request. Mice are available upon request with the approved animal protocol. All requests need to execute a suitable Materials Transfer Agreement.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Polyclonal anti-KDM4B | Bethyl Laboratories | Cat#A301-478A RRID: AB_999594 |
| Rabbit monoclonal anti-KDM4B | Abcam | Cat#ab191434 RRID: AB_2721242 |
| Rabbit Polyclonal anti-H3K9me3 | Abcam | Cat#ab8898 RRID: AB_306848 |
| Rabbit Polyclonal anti-H3 | Cell Signaling Technology | Cat#2650 RRID: AB_2115124 |
| Rabbit Polyclonal anti-HP1α | Cell Signaling Technology | Cat#2616 RRID: AB_2070987 |
| Rabbit Polyclonal anti-FABP4 | Abcam | Cat#ab13979 RRID: AB_1951817 |
| Rabbit Polyclonal anti-Smad1 | Cell Signaling Technology | Cat#9743 RRID: AB_2107780 |
| Mouse monoclonal anti-β-catenin | BD Biosciences | Cat#610153 RRID: AB_397554 |
| Rabbit Polyclonal anti-RUNX2 | Abcam | Cat# ab23981 RRID: AB_777785 |
| Mouse monoclonal anti-α-tubulin | Sigma-Aldrich | Cat#T5168 RRID: AB_477579 |
| Mouse polyclonal anti-IgG isotype control | Abcam | Cat#ab37355 RRID: AB_2665484 |
| Rabbit polyclonal anti-IgG isotype control | Cell Signaling Technology | Cat#2729 RRID: AB_1031062 |
| Cy2 affiniPure donkey anti-rabbit IgG antibody | Jackson ImmunoResearch | Cat#711-225-152 RRID: AB_2340612 |
| FITC-conjugated anti-mouse Sca1 | Biolegend | Cat#108106 RRID: AB_313343 |
| PE-conjugated anti-mouse CD29 | Biolegend | Cat#102208 RRID: AB_312885 |
| APC-conjugated anti-mouse CD45 | Biolegend | Cat#103112 RRID: AB_312977 |
| PerCP-conjugated anti-mouse CD11b | Biolegend | Cat#101230 RRID: AB_2129374 |
| APC Rat IgG2b, κ Isotype Ctrl | Biolegend | Cat#400612 RRID: AB_326556 |
| PerCP Rat IgG2b, κ Isotype Ctrl | Biolegend | Cat#400630 RRID: AB_893676 |
| FITC Rat IgG2a, κ Isotype Ctrl | Biolegend | Cat#400506 RRID: AB_2736919 |
| PE Armenian Hamster IgG Isotype Ctrl | Biolegend | Cat#400908 RRID: AB_326593 |
| Bacterial and Virus Strains | ||
| pGreenFire1-TCF/LEF Lentivector | System Biosciences | Cat#TR013PA-1 |
| pCMV-VSV-G | Addgene | Cat#8454 |
| pCMV-dR8.2 dvpr | Addgene | Cat#8455 |
| pQCXIN control vector | Addgene | Cat#631514 |
| gag/pol | Addgene | Cat#14887 |
| pQCXIN-Flag-Kdm4b | This Paper | N/A |
| pQCXIN-Flag-Kdm4b (H189A) | This Paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Thermo Fisher Scientific | Cat#11995-065 |
| dexamethasone | Sigma-Aldrich | Cat#D4902 |
| β-Glycerophosphate | Sigma-Aldrich | Cat#G9422 |
| L-Ascorbic acid | Sigma-Aldrich | Cat#A4403 |
| 3-isobutyl-1-methylxanthine | Sigma-Aldrich | Cat#I7018 |
| Indomethacin | Sigma-Aldrich | Cat#I7378 |
| Insulin | Sigma-Aldrich | Cat#I0516 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat#10437028 |
| M-MuLV Reverse Transcriptase | New England Biolabs | Cat#M0253L |
| Random Hexamers | Thermo Fisher Scientific | Cat#N8080127 |
| dNTP | Thermo Fisher Scientific | Cat#18427013 |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | Cat#15140122 |
| Trypsin-EDTA | Thermo Fisher Scientific | Cat#R001100 |
| CelLytic M Cell Lysis Reagent | Sigma-Aldrich | Cat#C2978 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#15596026 |
| Tetracycline hydrochloride | Sigma-Aldrich | Cat#T3383 |
| Naphthol AS-TR phosphate | Sigma-Aldrich | Cat#N6125 |
| Fast Blue BB salt | Sigma-Aldrich | Cat#F0250 |
| ALP stabilizing buffer | Sigma-Aldrich | Cat#A9226 |
| pNPP liquid substrate | Sigma-Aldrich | Cat#P7998 |
| Alizarin red S | Sigma-Aldrich | Cat#A5533 |
| Cetylpyridinium chloride | Sigma-Aldrich | Cat#C0732 |
| Collagenase Type I | Worthington Biochemical | Cat#CLS-1 |
| Paraformaldehyde | Santa Cruz Biotechnology | Cat#sc-281692 |
| PTH 1-34, Recombinant Protein | Prospec | Cat#HOR-290 |
| H89 PKA inhibitor | Selleck Chemicals | Cat#S1582 |
| Gelatin gelfoam scaffolds | Ferrosan | Cat#1972 |
| Lipofectamine RNAiMAX reagent | Thermo Fisher Scientific | Cat#13778030 |
| Critical Commercial Assays | ||
| Oil-Red-O Staining Kit | Diagnostic Biosystems | Cat#KT025 |
| mouse P1NP ELISA kit | Immunodiagnostic Systems | Cat#AC-33F1 |
| Mouse CTX ELISA kit | Immunodiagnostic Systems | Cat#AC-06F1 |
| TRAP staining kit | Sigma-Aldrich | Cat#387A-1KT |
| QuantiTect SYBR Green PCR kit | Thermo Fisher Scientific | Cat#204143 |
| Envision Plus Kit | DAKO | Cat#K400811-2 |
| Luc-Screen kit | Tropix | Cat#T1035 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| Dynabeads mRNA Purification Kit | Thermo Fisher Scientific | Cat#61006 |
| Stranded RNA-Seq Library Preparation Kit | Kapa Biosystems | Cat#KK8400 |
| ChIP assay kit | Millipore | Cat#17-295 |
| KAPA Hyper Prep Kit | Kapa Biosystems | Cat#KK8502 |
| MesenCult™ Expansion Kit | Stem Cell technologies | Cat#05513 |
| Deposited Data | ||
| RNA-seq data | This paper | GEO: GSE104258 |
| ChIP-seq data | This paper | GEO: GSE104257 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Kdm4bf/f | Sanger Institute | N/A |
| Mouse: LepRCre (B6.129(Cg)-Leprtm2(cre)Rck/J) | Jackson Laboratory | JAX:008320 |
| Mouse: Prx1Cre (B6.Cg-Tg(Prrx1-cre)1Cjt/J) | Jackson Laboratory | JAX:005584 |
| Mouse: OcnCre (B6N.FVB-Tg(BGLAP-cre)1Clem/J) | Jackson Laboratory | JAX:019509 |
| Mouse: tdTomato (B6;129S6-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J) | Jackson Laboratory | JAX:007908 |
| Mouse: Prx1Cre/ERT2 (B6.Cg-Tg(Prrx1-cre/ERT2,-EGFP)1Smkm/J) | Jackson Laboratory | JAX:029011 |
| Oligonucleotides | ||
| siRNA targeting β-catenin (mouse) | Santa Cruz Biotechnology | Cat#sc-29210 |
| scrambled siRNA | Santa Cruz Biotechnology | Cat#sc-37007 |
| Primer for genotyping, qRT-PCR and ChIP-qPCR See Table S4. | This paper | N/A |
| Software and Algorithms | ||
| Cellsens | Olympus | http://www.olympus-lifescience.com/en/software/cellsens/ |
| SPOT4.0 Microscopy Imaging Software Advanced | Diagnostic Instruments | https://webstore.diaginc.com/SPOT-Imaging-Software-For-Microscopy-s/59.htm |
| Leica Application Suite X Version 3.7.0 | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| μCT Evaluation Program V4.4A and Ray V3.0 | Scanco Medical | http://www.scanco.ch/en/systems-solutions/software.html |
| NRecon Version 1.6 | Bruker | https://www.bruker.com/service/support-upgrades/software-downloads/micro-ct.html |
| Dataviewer Version 1.5.2.4 | Bruker | https://www.bruker.com/service/support-upgrades/software-downloads/micro-ct.html |
| CTan Version 1.15 | Bruker | https://www.bruker.com/service/support-upgrades/software-downloads/micro-ct.html |
| CTvox Version 3.2.0.0 | Bruker | https://www.bruker.com/service/support-upgrades/software-downloads/micro-ct.html |
| Cufflinks 2.1.1 | Open Source | http://cole-trapnell-lab.github.io/cufflinks/ |
| Tophat Version 2.0.9 | Open Source | http://ccb.jhu.edu/software/tophat/index.shtml |
| Genomic Region Annotation Tool (GREAT, version 4.0.4) | Open Source | http://great.stanford.edu/public/html/ |
| Bowtie2.2.1.0 | Langmead and Salzberg, 2012 | https://sourceforge.net/projects/bowtie-bio/files/bowtie2/ |
| Samtools Version 3.0 | Li et al., 2009 | http://www.htslib.org/ |
| MACS2 | Liu et al. 2014 | https://github.com/macs3-project/MACS |
| Integrated Genome Browser Version 9.1.0 | UNC Charlotte | https://bioviz.org/ |
| Cluster 3.0 | Open Source | http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm |
| DAVID Bioinformatics Resources 6.7&6.8 | National Institute of Allergy and Infectious Diseases, NIH | https://david.ncifcrf.gov/ |
| Gene Set Enrichment Analysis | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| EnrichR | Chen at al. 2013; Kuleshov et al. 2016 | https://amp.pharm.mssm.edu/Enrichr/ |
| JASPAR | Stormo et al. 2013; Wasserman et al. 2004 | http://jaspar.genereg.net/ |
Data and Code Availability
The authors declare that all relevant data are available within the article and its supplementary information files or from the corresponding author upon reasonable request. ChIP-seq datasets are under the accession number GSE104257. The RNA-seq datasets have been submitted to the NCBI database under the accession number GSE104258.
EXPERIMNTAL MODEL AND SUBJECT DETAILS
Mice.
Kdm4bf/f mice were generated as previously described (Cheng et al., 2018). We generated tissue-specific Kdm4b deficient mice by crossing Kdm4bf/f mice with Prx1Cre (Jackson Laboratory, Cat#005584), Prx1cre/ERT2 (Jackson Laboratory, Cat#029211), LepRCre (Jackson Laboratory, Cat#008320) and Loxp-tdTomato mice (Jackson Laboratory, Cat#007908). All mice were housed in pathogen-free facilities under 12-hr light and 12-hr dark cycle. All protocols (#2007-057, #2007-058, #2007-059, and #2013-002) were approved by the Division of Laboratory Animal Medicine of UCLA and were in accordance with US National Institute of Health guidelines. In aging studies, we utilized both male and female mice and randomly assigned to procedure groups at a sample size of ≥ 6 mice per group. For OVX, 8 mice per group were used. 7 mice per group of 8-week-old male mice were subjected to high fat diet (Research Diets, Cat#12492). PTH (1-34) (Prospec-Tany Technogene, Cat#HOR-290) was injected subcutaneously daily at a dose of 50 μg/kg body weight. However, the animal experiments were not conducted in a completely blinded fashion. The primer sequences for genotyping were listed in Table S4.
METHOD DETAILS
μCT analysis
Femurs and lumbar vertebrae were dissected, fixed in 10% neutral buffered formalin for 24~48 hours, and stored in 70% ethanol. The specimens were placed into a cylindrical holder (16.5 mm in diameter) with the long axis of the femur perpendicular to the X-ray source, and scanned using a Scanco μCT40 scanner (Scanco Medical AG, Basserdorf, Switzerland) at 55 kV/70 μA or using a Skyscan 1275 μCT imaging system (Skyscan, Kontich, Belgium) at 60 kV/166 μA following the guideline for assessment of bone microstructure in rodents (Chang et al., 2009). Volumetric reconstructions were performed using built-in software NRecon 1.6 and CTAn 1.15. For trabecular bone analysis, the regions of interest were defined as the areas between 0.3 mm and 0.4 mm proximal to the growth plate in the distal femurs. Cortical bone thickness was measured at the diaphysis (50 slices, midpoint of the bone length). For porosity measurements, slices from a point immediately distal to the third trochanter to a point immediately adjacent to the primary spongiosa were selected for analysis. To measure MAT in tibiae, samples were stained with osmium tetroxide (Polysciences, Cat#23311-10), and scanned as previously described (Scheller et al., 2014).
Histomorphometric analysis
To examine the rate of bone formation, mice were injected intraperitoneally with 5 mg/kg body mass tetracycline (Sigma-Aldrich, Cat#T3383) dissolved in physiological saline solution on day 0 and day 7. Samples were collected 3 days post the second injection, fixed in 10% neutral buffered formalin for 24 h, and sectioned without decalcification (8 μm) in Bone Histomorphometric Laboratory at UCLA. Mineral apposition rate (MAR) and bone formation rate (BFR) were measured as previously described (Chang et al., 2009; Egan et al., 2012; Yu et al., 2014). After toluidine blue staining, osteoblasts were identified as plump cuboidal cells with a perinuclear clear zone lining the osteoid surface.
Whole-mount skeletal staining
The whole-mount skeletons of pups at P0 was stained with Alizarin Red S and Alcian Blue as previously described (Rigueur and Lyons, 2014). Briefly, the samples were collected, skin and internal organs were removed, and the remaining bodies were fixed in 10 ml of 95% EtOH/per sample for 24 hours. Stain for cartilage was performed by placing enough Alcian blue stain (Alcian blue 8GX, Sigma Cat# A5268, 0.03% in 80% EtOH and 20% Acetic Acid) to cover the body for 3 days at room temperature. The samples were rinsed twice in 95% EtOH and de-stained in 95% EtOH overnight at room temperature. The samples were slightly cleared with 1% KOH for 1 hour and counterstained for bone in Alizarin Red stain (Alizarin Red S, Sigma Cat#A5533, 0.05% in 1% KOH) for 24 hours. The samples were placed in 1% KOH for 1 day and then in clearing solution (80:20 of 1% KOH to glycerol) for 1-3 days until the skeleton was clearly visible. The samples were stored indefinitely in 50:50 of 95% EtOH to 50% glycerol.
Immunohistochemistry and immunofluorescence staining
For paraffin sections, the specimens were decalcified in 10% ethylenediaminetetraacetic acid (EDTA, PH 7.4, Fisher, Cat#E478-500) for 2 weeks, and were sectioned (5 μm) for staining as previously described (Chang et al., 2009). The slides were incubated at 65 °C for 2 hrs, dewaxed with xylene (Fisher, Cat#X5-4), and rehydrated through gradient ethanol into water. Immunostaining was performed using a DAKO Envision Plus Kit (Cat#K400811-2) according to the manufacturers' instructions.
For tdTomato-expressing samples, bones were fixed in 4% paraformaldehyde (Santa Cruz Biotech, Cat#sc-281692), decalcified in 10% EDTA (PH 7.4) for 2 weeks, and dehydrated in 30% sucrose for at least 2 days in dark. After frozen section, slides were washed with PBS twice, blocked in PBS with 10% horse serum for 30 minutes, and then stained with rabbit anti-FABP4 (1:200, Abcam, Cat#ab13979) overnight at 4 °C. Cy2 affiniPure donkey anti-rabbit IgG antibody (Jackson ImmunoResearch, USA, Cat#711-225-152) was used as secondary antibodies (1:200). After being extensively washed with PBS, the slides were stained with 4’,6-diamidino-2-phenylindole (DAPI) and mounted with anti-fade mounting medium (Thermo Fisher Scientific, Cat#S36963). Images were taken and analyzed using an Olympus IX-51 microscope. To perform osteoclast quantification, TRAP (Tartrate-resistant acid phosphatase) staining was then done using a TRAP kit from Sigma-Aldrich based on manufacturers' instructions.
To detect SAHF, MSCs were seeded onto 8 chamber cell culture slides (Corning, USA, Cat#354118), and were allowed to attach overnight. The cells were then fixed in 4% paraformaldehyde for 15 min, and stained with DAPI, rabbit anti-H3K9me3 (1:100; Abcam, Cat#ab8898) and rabbit anti-HP1α (1:100; Cell Signaling Technology, Cat#2616S) as described. Images were taken and analyzed using an Olympus IX-51 microscope or Leica TCS SP8 confocal microscope.
Cell culture, ELISA, ALP, ARS, and Oil Red O staining
Intact bone marrow was isolated from the long bones and lumbar vertebrae by flushing and subjected to enzymatic digestion with Collagenase Type I (3 mg/mL; Worthington, Cat#CLS-1) and Dispase (4 mg/mL; Roche Diagnostics, Cat#4942078001) as described previously (Gulati et al., 2018; Suire et al., 2012). For CFU-F assays, the digested cells were plated in 6-well plates at a density of 1 x 106 cells/well, and cultured in a humidified 5% CO2 incubator at 37 °C in Dulbecco's Modified Eagle Medium (DMEM) (Gibco, Cat#11995-065) supplemented with 15% FBS (Gibco, Cat#26140079) and 1% penicillin/streptomycin (Gibco, Cat#15140122). CFU-F colonies were stained with 0.5% crystal violet (Sigma-Aldrich, Cat#C3886) in 4% Paraformaldehyde, and counted after 10 days of culture. For in vitro experiments, the digested cells were cultured as colonies for 2 weeks and passaged with trypsin at a ratio of 1:3 for expansion. Cells within 5 passages were used in this study. To induce osteogenic differentiation, cells were cultured in osteogenic induction medium containing 100 μM ascorbic acid (Sigma-Aldrich, Cat#A4403), 2 mM β-glycerophosphate (Sigma, Cat#G9422) and 10 nM dexamethasone (Sigma-Aldrich, Cat#D4902). To induce adipogenesis, cells were cultured in adipogenic induction medium containing 1 μM dexamethasone (Sigma-Aldrich, Cat#D4902), 10 μg/mL insulin (Sigma-Aldrich, Cat#I0516), 0.5 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich, Cat#I7018), and 0.2 mM indomethacin (Sigma, Cat#I7378). H89 PKA inhibitor (Selleck Chemicals, Cat#S1582) and PTH (1-34) (Prospec-Tany Technogene, Cat#HOR-290) and vehicle controls were added into the media as indicated. ELISAs were performed with a mouse P1NP ELISA kit (Cat#AC-33F1) and a CTX ELISA kit (Cat#AC-06F1, Immunodiagnostic Systems).
siRNA transfection was conducted using Lipofectamine RNAiMAX reagent (Invitrogen, Cat#13778030) according to manufacturer’s instructions. siRNA targeting mouse β-catenin was obtained from Santa Cruz Biotechnology (Cat#sc-29210). The scrambled siRNA (Santa Cruz Biotechnology, Cat#sc-37007) was used as the control.
For ALP staining, cells were fixed with 70% ethanol for 15 min and incubated with a mix of 0.25% naphthol AS-TR phosphate (Sigma-Aldrich, Cat#N6125) and 0.75% Fast Blue BB salt (Sigma-Aldrich, Cat#F0250) dissolved in 0.1 M Tris buffer (pH 9.6) at 37 °C. For quantitative determination of ALP activity, 20 μL cell extract was incubated with a mix of 50 μL ALP stabilizing buffer (Sigma-Aldrich, Cat#A9226) and 50 μL pNPP liquid substrate (Sigma-Aldrich, Cat#P7998) for 20 min at 37 °C. The absorbance was then measured at OD405 nm on a microplate reader (Bio-Rad). For ARS staining, after 2–3 weeks of osteogenic induction, the cells with fixed with 70% ethanol for 1 hour and stained with 1% ARS solution (Sigma-Aldrich, Cat#A5533). The stained cultures were then destained by 10% Cetylpyridinium chloride solution (Sigma-Aldrich, Cat#C0732), and absorbance of the solution was read at 562 nM. To detect the lipid droplet formation, Oil Red O staining was performed with a kit form Diagnostic Biosystems (Cat#KT025) according to the manufacturer’s instructions after 2-3 weeks of adipogenic differentiation.
To investigate the effect of KDM4B deletion on MSC proliferation, MSCs at passage 1 (P1) and passage 5 (P5) were seeded onto 8 chamber cell culture slides (Corning, Cat#354118), and were cultured with 10 μM 5-ethynyl-2-deoxyuridine (EdU) for 24 hours. Incorporated EdU was then detected by using click-it EdU alexa fluor 488 imaging kit according to the manufacturer's protocol (Thermo Fisher Scientific, Cat#C10337). SA-β-gal activity was analyzed in Kdm4b+/+ and Kdm4b−/− MSCs using SA-β-gal staining kit according to the manufacturer’s instruction (Cell Signaling Technology, Cat#9860).
Western blot analysis
Cells were collected and lysed in CelLytic™ MT Cell Lysis Reagent (Sigma-Aldrich, Cat#C3228) containing protease inhibitor cocktail (Roche, Cat#11836170001) on ice for 30 min, and centrifuged at 18,000 g for 15 min at 4 °C. Aliquots of the lysates were separated on a 7.5% - 12.5% sodium dodecyl sulfate-polyacrylamide gel, and then transferred onto Immun-Blot® PVDF Membrane (Bio-Rad, Cat#1620177). The membrane was incubated with primary antibodies at 4 °C for overnight, followed by horseradish peroxidase-conjugated secondary antibodies. Protein bands were detected with an enhanced chemiluminescence Western blotting detection kit (Thermo Fisher Scientific, Cat#34577). The primary antibodies were used as follows: mouse anti-α-tubulin (1:5000, Sigma-Aldrich, T5168), rabbit anti-RUNX2 (1:1000, Abcam, Cat#ab23981).
Flow cytometry, cell sorting and clonal expansion
Intact bone marrow was isolated from the long bones by flushing and was then triturated several times to break up clumps, and filtered through a 70-μm cell strainer (BD Biosciences, Cat#352350). The resulting cell suspension was treated with red blood cell lysis buffer (Biolegend, Cat#420301) for 5 min at room temperature, and washed with 2% FBS in PBS for 2 times. Cells were stained with directly conjugated antibodies for 30 min on ice in dark, washed with 2% FBS in PBS for 3 times, and resuspended in PBS. Samples were sorted on a BD FACS Aria III or analyzed on a BD LSR II analyzer at the UCLA Flow Cytometry Core. The gating was determined by comparison with unstained control, isotype negative controls and single stained controls. The following primary antibodies were used: FITC conjugated anti-Sca1 (Biolegend, Cat#122506), PE conjugated anti-CD29 (Biolegend, Cat#102208), APC conjugated anti-CD45 (Biolegend, Cat#103112), PerCP conjugated anti-CD11b (Biolegend, Cat#101230). All the FACS data were analyzed using Flowjo software (Version 10.5.3).
For comparing lineage commitment, Sca1+CD29+CD45−CD11b− MSCs were sorted from both Prx1Cre;Kdm4bw/w and Prx1Cre;Kdm4bf/f mice (n = 3) and seeded in 96-well plates for 2 weeks. 16 to 20 individual clones from each mouse (n = 3 for each genotype) were picked after 2 weeks and induced to osteogenic and adipogenic differentiation in a mix of osteogenic and adipogenic induction medium (1: 1 ratio) as previously described (Fu et al., 2010; McBeath et al., 2004). To minimize in vitro passages, a half of individual clones were utilized for ARS and Oil Red O staining, and another half of clones were used for gene expression studies. ARS and Oil Red O staining were performed after 2 ~ 3 weeks of osteogenic and adipogenic differentiation. Total RNA was isolated and examined by qRT-PCR after 1 week of osteogenic and adipogenic differentiation.
qRT-PCR and RNA-seq
For qRT-PCR, total RNA was extracted using Trizol reagents (Invitrogen, Cat#15596018) per the manufacturer’s instructions. 2 μg aliquots of RNAs were transcribed to cDNA with random hexamers (Invitrogen, Cat#N8080127) and reverse transcriptase (New England Biolabs, Cat#M0253L). qPCR then performed using the QuantiTect SYBR Green PCR kit (Qiagen, Cat#204143) and the CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad). The primer sequences for qRT-PCR were listed in Table S4.
For RNA-seq, RNA was isolated using RNeasy Mini Kit (Qiagen, Cat#74104) from three biological replicates of primary MSCs from Prx1Cre;Kdm4bf/f mice and control mice. Total RNA was then purified with Dynabeads™ mRNA Purification Kit (Invitrogen, Cat#61006). RNA-seq libraries were constructed using Stranded RNA-Seq Library Preparation Kit (Kapa Biosystems, Cat#KK8400), and sequenced using an illumina HiSeq 3000 sequencer at the Technology Center for Genomics & Bioinformatics (TCGB) core at UCLA.
ChIP-qPCR and ChIP-seq
ChIP assays were performed using a ChIP assay kit (Millipore, USA, Cat#17-295) per the manufacturer's instructions. Briefly, cells were treated with 3'-dithiobispropionimidate solution (10 mM; Thermo Fisher Scientific, Cat#20665) for 10 min at room temperature and then incubated in 1% formaldehyde for 15 min in a 37 °C water bath for cross-linking. Nuclei were extracted and sonicated for an hour to generate 200–500 bp genomic DNA fragments with a sonicator. Chromatin complexes were immunoprecipitated with the related antibodies and Dynabeads Protein A/G (Thermo Fisher Scientific, Cat#10002D/10004D), and the precipitated DNA was quantified by qPCR. Data was expressed as the percentage of input DNA. The following antibodies were used: mouse anti-β-catenin (1: 200, BD Biosciences, Cat#610153), rabbit anti-Smad1 (1:100, Cell Signaling Technology, Cat#9743S), rabbit anti-KDM4B (1:400, Bethyl Laboratories, USA, Cat#A301-478A), rabbit anti-KDM4B (1:400, Abcam, Cat#ab191434), rabbit anti-H3K9me3 (1:500, Abcam, Cat#ab8898), rabbit anti-H3 (1:200, Cell Signaling Technology, Cat#2650), anti-mouse IgG (1:500 Abcam, Cat#ab197767), and anti-rabbit IgG (1:200, Cell Signaling Technology, Cat#2729). The primer sequences for ChIP-qPCR were listed in Table S4.
For ChIP-seq, yeast chromatin was added as a spike-in control for H3K9me3 ChIP-seq as described previously (Orlando et al., 2014). Libraries were prepared using KAPA Hyper Prep Kit (Kapa Biosystems, KK8502) per the manufacturer’s protocol. The sequencing was completed using HiSeq 3000 system (Illumina) or Nextseq 500 at the Technology Center for Genomics & Bioinformatics (TCGB) of UCLA.
In vivo serial transplantation
In the initial transplantation, MSCs from bone marrow of Prx1Cre;Kdm4bw/w;tdTomato and Prx1Cre;Kdm4bf/f;tdTomato mice by mechanical and enzymatic dissociation as stated above, respectively. After expansion, 100 μl of cell suspension with approximately 1 x 106 cells were incubated with Gelatin gelfoam scaffolds (Ferrosan, Soeborg, Denmark, Cat#1972) at 37 °C for 6 hrs, and then transplanted subcutaneously at the dorsal sites of nude mice (n = 8). 6 weeks after transplantation, the scaffolds from nude mice were explanted and digested to give rise to cells for secondary transplantation. Again, 6 weeks after secondary transplantation, the scaffolds were fixed with 4% paraformaldehyde, decalcified by 10% ethylenediaminetetraacetic acid (EDTA) (PH 7.4), and were subjected to frozen section. Representative sections were stained with H&E. The area of mineralized tissue and adipose tissue versus total area were measured using SPOT 4.0 software (Diagnostic Instruments, Michigan, USA). Expression of tdTomato was detected and considered as an evidence that the newly formed bone tissue and adipose tissue were generated by MSCs from bone marrow of Prx1Cre;Kdm4bw/w;tdTomato and Prx1Cre;Kdm4bf/f;tdTomato mice during the serial transplantation.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq analysis
The reads were aligned to the mm9 build of the mouse genome using Tophat. Expression of the transcript was determined using Cufflinks with Refseq mRNAs to guide assembly. GSEA was performed to generate the list of the most differentially expressed genes, including Wnt and TGF-β signaling pathway signatures and stem cell signatures (Subramanian et al., 2005). Transcripts that were downregulated in Kdm4b−/− MSCs from greater than 1.5-fold were used in GO term and KEGG pathway analysis to assess the enriched functional annotations. The RNA-seq datasets have been submitted to the NCBI database under the accession number GSE104258.
ChIP-seq analysis
All sequencing reads were mapped to human genome (GRCh37/hg19), mouse genome (NCBI37/mm9) and yeast genome (UCSC version sacCer3) using the software Bowtie (version 1.2.2), respectively. Only reads that aligned to a unique genomic position with no more than two mismatches were retained for the above analysis. When multiple reads mapped to the same position in the genome, only one was counted. The mapped reads were subjected to custom ChIP-seq peak-calling algorithm implemented in MACS2 (Zhang et al., 2008). KDM4B and total H3 was normalized to input, and H3K9me3 was normalized to total H3 (Stroud et al., 2017). The peak calling parameters for KDM4B are: macs2 callpeak -t KDM4B_sample.bam -c input.bam -g mm -q 0.001 -B -f BAM (FDR < 0.001); and the peak calling parameters for H3K9me3 are: macs2 callpeak --nomodel --nolambda -t H3K9me3_sample.bam -c H3_sample.bam -g mm -B -f BAM -q 0.05 (FDR < 0.05). Bedtools was used to get the unique KDM4B peaks in Kdm4b+/+ MSCs, and the parameters are “bedtools intersect -v -a KDM4B_WT.bed -b KDM4B_KO.bed > unique_KDM4B_peaks.bed”. Similarly, the unique H3K9me3 peaks in Kdm4b−/− MSCs was obtained by “bedtools intersect -v -a H3K9me3_KO.bed -b H3K9me3_WT.bed > unique_H3K9me3_peaks.bed”. GREAT (version 4.0.4) was used to analyze the nearest genes whose TSSs are within ±2 Kb of the unique KDM4B and H3K9me3 peaks. For H3K9me3 ChIP-seq, 19055 reads were mapped to the yeast genome in Kdm4b+/+ sample, and 12603 reads were mapped to the yeast genome in Kdm4b−/− sample. Representative ChIP-seq enriched regions were visualized in the Integrated Genome Browser (version 9.1.0) with spike-in normalization. Gene ontology (GO) analysis was utilized to determine signaling pathways associated with genes in which KDM4B was enriched at their promoters (over 1000 reads within ±2 Kb of TSSs) and H3K9me3 levels were upregulated by at least 2 folds in KDM4B-depleted MSCs. The binding heatmaps were generated with CEAS package and Cluster 3.0.
Statistical analysis
All values are expressed as mean ± SD. For single comparisons, an unpaired two-tailed Student’s t-test was used. And the statistical significance of differences among more than two groups was calculated using one-way analysis of variance (ANOVA) with Tukey’s posthoc test. For comparison between groups involving two genotypes of mice along with surgery, PTH treatment, HFD treatment, or aging conditions, the differences were assessed using two-way ANOVA with Holm-Sidak posthoc test. P value < 0.05 was considered to be statistically significant.
Supplementary Material
Highlights.
Loss of KDM4B exacerbates bone-fat imbalance in skeletal aging and osteoporosis
Loss of KDM4B impairs MSC self-renewal and promotes MSC exhaustion
Loss of KDM4B induces senescence-associated heterochromatin foci formation
KDM4B is required for the anabolic effects of parathyroid hormone
Acknowledgments
This work was supported by NIH grants R01DE028260, R01AR063089 and R01DE16513, a Core Voucher Award from UCLA Clinical and Translational Science Institute and the Hsien family chartable funds.
Footnotes
Declaration of Interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung U-I, Kubota N, Terauchi Y, Harada Y, Azuma Y, Nakamura K et al. (2004). PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest 113, 846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, and Wang C-Y (2013). The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med 19, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, and Tosteson A (2007). Incidence and Economic Burden of Osteoporosis-Related Fractures in the United States, 2005–2025. J Bone Miner Res 22, 465–475. [DOI] [PubMed] [Google Scholar]
- Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, Guan K, Krebsbach PH, and Wang CY (2009). Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med 15, 682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Shou P, Zheng C, Jiang M, Cao G, Yang Q, Cao J, Xie N, Velletri T, Zhang X, et al. (2016). Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell Death Differ 23, 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Yuan Q, Vergnes L, Rong X, Youn JY, Li J, Yu Y, Liu W, Cai H, Lin JD, et al. (2018). KDM4B protects against obesity and metabolic dysfunction. Proc Natl Acad Sci U S A 115, E5566–E5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JL, and Cao X (2014). Bone marrow mesenchymal stem cells and TGF-β signaling in bone remodeling. J Clin Invest 124, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng P, Chen Q-M, Hong C, and Wang C-Y (2015). Histone methyltransferases and demethylases: regulators in balancing osteogenic and adipogenic differentiation of mesenchymal stem cells. Int J Oral Sci 7, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan KP, Brennan TA, and Pignolo RJ (2012). Bone histomorphometry using free and commonly available software. Histopathology 61, 1168–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Hanai J-I, Le PT, Bi R, Maridas D, DeMambro V, Figueroa CA, Kir S, Zhou X, Mannstadt M et al. (2017). Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab 25, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, Drake MT, Tchkonia T, LeBrasseur NK, Kirkland JL, et al. (2016). Identification of Senescent Cells in the Bone Microenvironment. J Bone Miner Res 31, 1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazeli PK, Horowitz MC, MacDougald OA, Scheller EL, Rodeheffer MS, Rosen CJ, and Klibanski A (2013). Marrow fat and bone--new perspectives. J Clin Endocrinol Metab 98, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Wang Y-K, Yang MT, Desai RA, Yu X, Liu Z, and Chen CS (2010). Mechanical regulation of cell function with geometrically modulated elastomeric substrates. Nat Methods 7, 733–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C, Cawthorn WP, Li Y, Zhao G, Macdougald OA, and Franceschi RT (2016). Reciprocal Control of Osteogenic and Adipogenic Differentiation by ERK/MAP Kinase Phosphorylation of Runx2 and PPARγ Transcription Factors. J Cell Physiol 231, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng S, Zhou S, and Glowacki J (2011). Age-related decline in osteoblastogenesis and 1α-hydroxylase/CYP27B1 in human mesenchymal stem cells: stimulation by parathyroid hormone. Aging Cell 10, 962–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati GS, Murphy MP, Marecic O, Lopez M, Brewer RE, Koepke LS, Manjunath A, Ransom RC, Salhotra A, Weissman IL, et al. (2018). Isolation and functional assessment of mouse skeletal stem cell lineage. Nature protocols 13, 1294–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2008). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai M, and Rosen CJ (2010). PPARγ: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol 6, 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla S, and Hofbauer LC (2017). Osteoporosis treatment: recent developments and ongoing challenges. Lancet Diabetes Endocrinol 5, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan M, Martin JF, Nagy A, Lobe C, Olson EN, and Tabin CJ (2002). Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. genesis 33, 77–80. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Fornes O, Arenillas DJ, Chen C-Y, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R, et al. (2016). JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 44, D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBeath R, Pirone DM, Nelson CM, Bhadriraju K, and Chen CS (2004). Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Developmental cell 6, 483–495. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat biotech 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacarelli T, Liu P, and Zhang R (2017). Epigenetic Basis of Cellular Senescence and Its Implications in Aging. Genes (Basel) 8, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Nuñez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, and Lowe SW (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716. [DOI] [PubMed] [Google Scholar]
- Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, and Daley GQ (2009). Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 460, 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki R, Inoue D, Shibata M, Saika M, Kido S, Ooka H, Tomiyama H, Sakamoto Y, and Matsumoto T (2002). Estrogen promotes early osteoblast differentiation and inhibits adipocyte differentiation in mouse bone marrow stromal cell lines that express estrogen receptor (ER) alpha or beta. Endocrinology 143, 2349–2356. [DOI] [PubMed] [Google Scholar]
- Orlando David A., Chen Mei W., Brown Victoria E., Solanki S, Choi Yoon J., Olson Eric R., Fritz Christian C., Bradner James E., and Guenther Matthew G. (2014). Quantitative ChIP-Seq Normalization Reveals Global Modulation of the Epigenome. Cell Rep 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Rauch A, Haakonsson AK, Madsen JGS, Larsen M, Forss I, Madsen MR, Van Hauwaert EL, Wiwie C, Jespersen NZ, Tencerova M, et al. (2019). Osteogenesis depends on commissioning of a network of stem cell transcription factors that act as repressors of adipogenesis. Nat Genet 51, 716–727. [DOI] [PubMed] [Google Scholar]
- Rigueur D, and Lyons KM (2014). Whole-Mount Skeletal Staining. In Skeletal Development and Repair: Methods and Protocols, Hilton MJ, ed. (Totowa, NJ: Humana Press; ), pp. 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Carballo E, Ulsamer A, Susperregui AR, Manzanares-Céspedes C, Sánchez-García E, Bartrons R, Rosa JL, and Ventura F (2011). Conserved regulatory motifs in osteogenic gene promoters integrate cooperative effects of canonical Wnt and BMP pathways. J Bone Miner Res 26, 718–729. [DOI] [PubMed] [Google Scholar]
- Salazar VS, Capelo LP, Cantù C, Zimmerli D, Gosalia N, Pregizer S, Cox K, Ohte S, Feigenson M, Gamer L, et al. (2019). Reactivation of a developmental Bmp2 signaling center is required for therapeutic control of the murine periosteal niche. eLife 8, e42386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller EL, Doucette CR, Learman BS, Cawthorn WP, Khandaker S, Schell B, Wu B, Ding S-Y, Bredella MA, Fazeli PK, et al. (2015). Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat Commun 6, 7808–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller EL, Troiano N, Vanhoutan JN, Bouxsein MA, Fretz JA, Xi Y, Nelson T, Katz G, Berry R, Church CD, et al. (2014). Use of osmium tetroxide staining with microcomputerized tomography to visualize and quantify bone marrow adipose tissue in vivo. Methods Enzymol 537, 123–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Shah PP, Nativio R, and Berger SL (2016). Epigenetic Mechanisms of Longevity and Aging. Cell 166, 822–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snigdha S, Prieto GA, Petrosyan A, Loertscher BM, Dieskau AP, Overman LE, and Cotman CW (2016). H3K9me3 Inhibition Improves Memory, Promotes Spine Formation, and Increases BDNF Levels in the Aged Hippocampus. J Neurosci 36, 3611–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Su SC, Hrvatin S, Greben AW, Renthal W, Boxer LD, Nagy MA, Hochbaum DR, Kinde B, Gabel HW, et al. (2017). Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 171, 1151–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suire C, Brouard N, Hirschi K, and Simmons PJ (2012). Isolation of the stromal-vascular fraction of mouse bone marrow markedly enhances the yield of clonogenic stromal progenitors. Blood 119, e86–e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tencerova M, Figeac F, Ditzel N, Taipaleenmaki H, Nielsen TK, and Kassem M (2018). High-Fat Diet-Induced Obesity Promotes Expansion of Bone Marrow Adipose Tissue and Impairs Skeletal Stem Cell Functions in Mice. J Bone Miner Res 33, 1154–1165. [DOI] [PubMed] [Google Scholar]
- Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, and Cao X (2008). Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev 22, 2968–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BL, Dai CL, Quan JX, Zhu ZF, Zheng F, Zhang HX, Guo SY, Guo G, Zhang JY, and Qiu MC (2006). Parathyroid hormone regulates osterix and Runx2 mRNA expression predominantly through protein kinase A signaling in osteoblast-like cells. J Endocrinol Investig 29, 101–108. [DOI] [PubMed] [Google Scholar]
- Wu X, Pang L, Lei W, Lu W, Li J, Li Z, Frassica FJ, Chen X, Wan M, and Cao X (2010). Inhibition of Sca-1-positive skeletal stem cell recruitment by alendronate blunts the anabolic effects of parathyroid hormone on bone remodeling. Cell stem cell 7, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Fan Z, Yu B, Chang J, Al Hezaimi K, Zhou X, Park N-H, and Wang C-Y (2012). Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell stem cell 11, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Chang J, Liu Y, Li J, Kevork K, Al-Hezaimi K, Graves DT, Park N-H, and Wang C-Y (2014). Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor-κB. Nat Med 20, 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, and Wang C-Y (2016). Osteoporosis: the result of an 'aged' bone microenvironment. Trends Mol Med 22, 641–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue R, Zhou Bo O., Shimada Issei S., Zhao Z, and Morrison Sean J. (2016). Leptin Receptor Promotes Adipogenesis and Reduces Osteogenesis by Regulating Mesenchymal Stromal Cells in Adult Bone Marrow. Cell Stem Cell 18, 782–796. [DOI] [PubMed] [Google Scholar]
- Zaidi M, Buettner C, Sun L, and Iqbal J (2012). Minireview: The link between fat and bone: does mass beget mass? Endocrinology 153, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Li J, Suzuki K, Qu J, Wang P, Zhou J, Liu X, Ren R, Xu X, Ocampo A, et al. (2015). Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348, 1160–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BO, Yue R, Murphy MM, Peyer JG, and Morrison SJ (2014). Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell stem cell 15, 154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all relevant data are available within the article and its supplementary information files or from the corresponding author upon reasonable request. ChIP-seq datasets are under the accession number GSE104257. The RNA-seq datasets have been submitted to the NCBI database under the accession number GSE104258.