

Abstract
Cholangiocarcinoma (CCA) is a highly aggressive malignancy with extremely poor prognoses. The oncogenic role and prognostic value of c-Myc in CCA is not well elucidated. WD repeat domain 5 (WDR5) is a critical regulatory factor directly interacting with c-Myc to regulate c-Myc recruitment at chromosomal locations, but the interaction of WDR5 and c-Myc in CCA was uncovered. In our study, we detected WDR5 and c-Myc expression in all CCA types, including intrahepatic (iCCA), perihilar (pCCA), and distal (dCCA) CCA, and evaluated their prognostic significance. Consequently, we demonstrated that WDR5 was significantly correlated with poor prognosis of CCA and that WDR5 and c-Myc co-expression was a more sensitive prognostic factor. With in vitro and in vivo experiments and bioinformatics, we showed that WDR5 interacted with the Myc box IIIb (MBIIIb) motif of c-Myc and facilitated Myc-induced HIF1A transcription, thereby promoting the epithelial-mesenchymal transition (EMT), invasion, and metastasis of CCA. Moreover, WDR5 enhanced hypoxia-inducible factor 1 subunit α (HIF-1α) accumulation by binding with histone deacetylase 2 (HDAC2) and increasing histone 3 lysine 4 acetylation (H3K4ac) deacetylation of the prolyl hydroxylase domain protein 2 (PHD2) promoter, resulting in the attenuation of chromatin opening and PHD2 expression, and eventually leading to HIF-1α stabilization and accumulation. In conclusion, WDR5 facilitated EMT and metastasis of CCA by increasing HIF-1α accumulation in a Myc-dependent pathway to promote HIF-1α transcription and a Myc-independent pathway to stabilize HIF-1α.
Keywords: WDR5, c-Myc, CCA, EMT, HIF-1α, PHD2
Graphical abstract
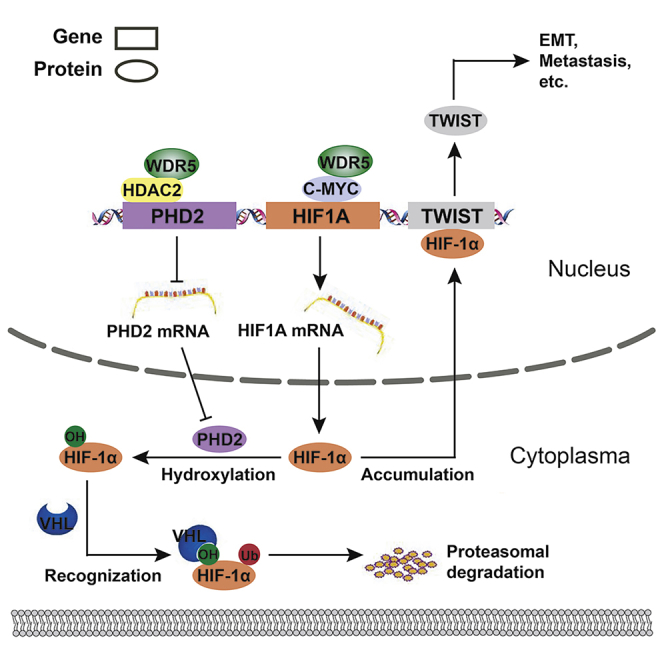
Chen et al. identified WDR5 as a prognostic biomarker of cholangiocarcinoma and depicted two distinct mechanisms of WDR5-enhanced HIF-1α accumulation: (1) WDR5 promoted HIF-1α mRNA synthesis via direct interaction with c-Myc, and (2) WDR5 inhibited HIF-1α protein hydroxylation and degradation by suppressing PHD2 expression, which relied on interacting with HDAC2 and deacetylizing H3K4ac.
Introduction
Cholangiocarcinoma (CCA) is a type of highly aggressive malignancy originating from the biliary system with a rising incidence worldwide.1 The prognosis of CCA is extremely poor due to the difficulties of early diagnosis and the resistance to chemotherapy and radiotherapy.2 According to the anatomical location, CCA is further classified into three subtypes, including intrahepatic (iCCA), perihilar (pCCA), and distal (dCCA) CCA.3 pCCA is the most prevalent type of CCA, accounting for about 50%–60% of total CCA cases, while dCCA and iCCA accounted for 20%–30% and 10%, respectively.4 Surgical resection is the only curative option for CCA patients. Unfortunately, most patients present in an advanced stage and lose the surgical opportunity.4 The 5-year overall survival (OS) rate of CCA is very unsatisfactory, remaining less than 30% after radical resection.5 The dismal outcome of CCA can be partially attributed to the insensitivity to systemic treatment such as chemotherapy.6 Moreover, there are very few well-accepted target drugs of CCA, and the effects of most current targeted drugs are unsatisfactory. Therefore, new biomarkers and targeted therapies are urgently needed to advance the survival of CCA patients.
Unfortunately, the discovery of CCA effective biomarkers to guide precise treatment or predict promising drug targets remains stagnant compared with that of common cancer types, such as lung cancer. Many reasons have led to this result, including the low radical surgical rate and the difficulty of obtaining specimens and establishing cohorts.7 Emerging evidence has indicated that each CCA subtype has not only different morbidities, clinical characteristics, treatment strategies, and prognoses, but also molecular features and biological behaviors,1,8 so these three CCA subtypes should be investigated separately, including the biomarker studies. However, this issue did not get enough attention before the Seventh Edition of the American Joint Committee on Cancer (AJCC)/International Union Against Cancer (UICC) classification defined CCA into iCCA, pCCA, and dCCA in 2007.
In the results of CCA high-throughput sequencing, c-Myc is a promising target and is considered as an indicator for the proliferation subtype of extrahepatic CCA by high-throughput sequencing.9 As an oncoprotein and a transcription factor, c-Myc is overexpressed in multiple malignant tumors.10 c-Myc drives tumor initiation, development, and maintenance mainly by activating the transcriptional program of downstream gene networks, and it is regarded as a valuable target for cancer therapy.11,12 Overexpression and ectopic activation of c-Myc were previously reported to promote cholangiocarcinogenesis and CCA progression.13 However, the prognostic significance of c-Myc in CCA has not received a consensus. In our previous study, we demonstrated that c-Myc activation promoted CCA progression, but the expression of c-Myc was not a strong prognostic predictor of pCCA.14 In summary, in vitro and in vivo experiments suggested the significant oncogenic function of c-Myc in cancer progression, but the prognostic significance of c-Myc is usually not remarkable in clinical studies. This discrepancy is extraordinarily prominent in CCA, and we suspect that there are other effectors participating in Myc-involved progression.
WD repeat domain 5 (WDR5) is an important component of the histone methyltransferase complex SET1/MLL, which mainly catalyzes histone 3 lysine 4 methylation (H3K4me).15 It is upregulated in prostate cancer, bladder cancer, breast cancer, and colorectal cancer, and it is associated with tumor progression and poor outcome.16 Recent studies revealed that WDR5 enhances c-Myc recruitment to chromosomal locations via direct interaction with c-Myc.17 WDR5 is responsible for Myc-induced gene transcription by binding a conserved motif of c-Myc, which is identified as Myc box IIIb (MBIIIb) (residues 258–267 of human c-Myc). This interaction of c-Myc and WDR5 creates an avidity-based chromatin recognition mechanism, making c-Myc able to select its target genes in response to different upstream stimulators.18 The mutations of this motif (I262/V264/V265 to E, named as WBM) can destroy Myc-WDR5 interaction at about 80% of the chromosomal locations of c-Myc, eventually disabling the ability of c-Myc to facilitate tumorigenesis, indicating a critical role of WDR5 in Myc-driven tumor progression.17,19
Besides functioning as a chaperon of c-Myc for its recognition and interaction with chromatin, WDR5 itself is able to regulate gene transcription by regulating histone modification. These histone modifications, such as phosphorylation, acetylation, methylation, and ubiquitination, change the local charge on the nucleosome, hence altering the nucleosome structure and stability, which affects the openness of the chromatin fiber (also called chromatin accessibility) and subsequent gene transcription.20,21 As an essential component of the histone methyltransferase complex, WDR5 participates in H3K4me and promotes transcription by generating an open chromatin structure and enabling better access to the transcriptional machinery.15 Moreover, WDR5 interacts with histone deacetylases (HDACs) to promote histone deacetylation, which induces the compact state of the nucleosome, and thus represses DNA transcription.22, 23, 24 However, the modulation of WDR5 on histone methylation and acetylation is tissue- and context-specific, and this function has not yet been elucidated in CCA.
Hypoxia-inducible factor 1 (HIF1), consisting of an unstable α-subunit (HIF-1α) and a stable β-subunit (HIF-1β), is able to regulate the cellular response to hypoxia and also to promote tumor progression.25 HIF-1α translocates into the nucleus to combine with HIF-1β and promotes transcription in hypoxic conditions, but it quickly degrades under normoxic conditions.26,27 Prolyl hydroxylase domain protein (PHD) is a key oxygen sensor and also a HIF-1α suppressor by directly controlling HIF-1α stability.28 Under normoxic conditions, HIF-1α is hydroxylated by PHDs at prolines 564 and 402, recognized by von Hippel-Lindau (VHL), and eventually degraded through the E3 ubiquitin ligase-proteasome pathway. However, PHD activity is diminished by hypoxia, resulting in the stabilization and accumulation of HIF-1α.29, 30, 31 HIF-1α is involved in a variety of tumor biological behaviors, including cell-cycle regulation, angiogenesis, and epithelial-mesenchymal transition (EMT) by inducing transcription of oncogenes such as CCND1, VEGFA, and TWIST1.32, 33, 34, 35 However, the function of HIF-1α in the tumorigenesis and progression of CCA is still obscure.
In this study, we investigated the expressions of c-Myc and WDR5 in a large CCA cohort consisting of 78 iCCAs, 141 pCCAs, and 88 dCCAs, and we found that co-expression of c-Myc and WDR5 predicted the worst prognosis. Results from in vitro and in vivo experiments confirmed the critical role of WDR5 in the invasion, metastasis, and EMT of CCA. Meanwhile, we investigated the role of WDR5 in Myc-induced CCA metastasis and elucidated the molecular mechanism. During the above processes, we found that there existed other effectors responsible for WDR5-induced metastasis apart from c-Myc, and we identified HIF-1α as the molecule by RNA sequencing (RNA-seq) and bioinformatics. Most importantly, we depicted two distinct mechanisms of how WDR5 enhanced HIF-1α accumulation: (1) WDR5 promoted HIF-1α messenger RNA (mRNA) synthesis via direct interaction with c-Myc, and (2) WDR5 inhibited HIF-1α protein hydroxylation, ubiquitination, and degradation through suppressing PHD2 expression, which relied on interacting with HDAC2 and deacetylizing histone 3 lysine 4 acetylation (H3K4ac).
Results
Co-expression of c-Myc and WDR5 correlated with the most unfavorable prognosis of CCA
c-Myc is a well-known oncogene in numerous cancer types, so we detected its function in CCA progression. iCCA cell line RBE and pCCA cell line QBC939 were used for Cell Counting Kit-8 (CCK-8) and transwell assays after regulating c-Myc expression (Figures S1A and 1B). The results suggested that c-Myc knockdown impaired the proliferation and invasion of CCA cells (Figures 1A and 1B; Figure S1C). The expression of c-Myc was further detected with immunohistochemistry (IHC) in CCA tissues, including 78 iCCAs, 141 pCCAs, and 88 dCCAs (Figure 1C). With the Kaplan-Meier method, we showed that c-Myc was associated with prognosis in iCCA and dCCA, but the statistical significance in pCCA was not remarkable (p = 0.127) (Figure 1D). This result was inconsistent with our previous study that c-Myc alone was not sufficient to predict prognosis of pCCA.14 This discrepancy between experimental results and clinical observation drove us to investigate whether there are other factors cooperating with c-Myc to lead to tumor progression and poor prognosis of CCA.
Figure 1.

Co-expression of high c-Myc and WDR5 was correlated with the poorest prognosis of CCA
(A and B) Knockdown of c-Myc inhibited cell proliferation (A) and invasion (B) of the iCCA cell line RBE and the pCCA cell line QBC939. (C) Representative IHC staining of c-Myc in iCCA, pCCA, and dCCA. Scale bars, 50 μm. (D) Overall survival curves of iCCA, pCCA, and dCCA patients were stratified by c-Myc expression. (E) TCGA data of WDR5 mRNA level in 9 normal tissues and 36 CCA tissues. The expression of WDR5 mRNA was quantified using log2(TPM + 1). (F) mRNA levels of WDR5 in 18 iCCA, 16 pCCA, and 14 dCCA tissues as well as their corresponding normal tissues. (G) Representative IHC staining of WDR5 expression in iCCA, pCCA, and dCCA tissues. (H) IHC scores of WDR5 in iCCA, pCCA, and corresponding normal tissues. (I) Overall survival curves of iCCA, pCCA, and dCCA patients were stratified by WDR5 expression. (J) Survival curves were further stratified into subgroups with the co-expression, single expression, and low expression of c-Myc and WDR5. ∗p < 0.05, ∗∗p < 0.01. In (A), data were calculated by two-way ANOVA. In (B) and (E), data were calculated by one-way ANOVA. In (F) and (H), data were calculated by a paired t test. In (D), (I), and (J), data were calculated by a log-rank test, and the C-index was calculated to evaluate the accuracy of the prognostic model.
Recent research has identified WDR5 as a critical determinant for the recruitment of c-Myc.17,19 We first queried The Cancer Genome Atlas (TCGA) for CCA data (including 36 CCAs and 9 normal tissues) and found that WDR5 mRNA in CCAs was substantially higher than that in normal bile duct tissues (Figure 1E). In iCCAs, pCCAs, dCCAs, and their corresponding normal tissues, we detected WDR5 mRNA by quantitative real-time PCR and demonstrated that WDR5 was upregulated in CCAs (Figure 1F). WDR5 expression was further investigated in tissue microarrays of iCCAs, pCCAs, and dCCAs (Figure 1G). The IHC scores of WDR5 in iCCA and pCCA were higher than those in tumor-adjacent tissues (Figure 1H; Figure S1D). All of these results suggested the potential role of WDR5 in CCA tumorigenesis and progression. Moreover, we evaluated the prognostic significance of WDR5 in CCAs and confirmed that WDR5 was a significant prognostic biomarker of iCCA, pCCA, and dCCA (Figure 1I).
Moreover, the CCA patients were categorized into three subgroups based on their expression pattern: co-expression of WDR5 and c-Myc (double-positive), single overexpression of WDR5 or c-Myc (single-positive), and double low expression of WDR5 and c-Myc (double-negative). The concordance index (C-index) was introduced to calculate the accuracy of biomarkers to predict prognosis.36,37 In iCCA, pCCA, and dCCA, three molecular subsets based on both WDR5 and c-Myc expression had a lower p value and higher C-index than did those considering WDR5 or c-Myc separately, which suggested that the combination of WDR5 and c-Myc was able to predict prognosis of CCA more precisely (Figure 1J). These results demonstrated that patients with co-expression of c-Myc and WDR5 had significantly poorer prognosis than did those with only c-Myc or WDR5 overexpression, and they also indicated that WDR5 could facilitate Myc-involved progression and prognosis of CCA.
Prognostic significance of c-Myc and WDR5 in CCA
Univariate analysis with a log-rank test was first performed to analyze the prognostic value of WDR5, c-Myc, and other clinicopathological factors (Table S5). In iCCA patients, advanced age (p = 0.012), advanced T stage (p = 0.003), positive lymph node metastasis (p < 0.001), distant metastasis (p = 0.030), and advanced tumor-lymph-node-metastasis (TNM) stage (p = 0.001) showed a strong correlation with poor prognosis (Figure S1E). In pCCA patients, advanced age (p = 0.010), positive lymph node metastasis (p = 0.002), distant metastasis (p < 0.001), and advanced TNM stage (p < 0.001) were all associated with lower OS rate (Figure S1F). In dCCA patients, positive lymph node metastasis (p = 0.022), distant metastasis (p < 0.001), and advanced TNM stage (p = 0.020) predicted the worse outcome (Figure S1G).
Multivariate analysis was also performed to identify the independent prognostic factors using the Cox regression model (Table S6). All factors with a p value ≤0.10 in univariate analysis were enrolled into multivariate analysis, except TNM stage, because of its natural interaction with the T, N, or M stage. We found that WDR5 was an independent prognostic factor in pCCA (p = 0.030) and dCCA (p = 0.048) rather than in iCCA (p = 0.477). Patients with pCCA and dCCA had a 1.67- and 1.82-fold higher risk of cancer-caused death than did those with lower WDR5.
WDR5 facilitated CCA cell metastasis in vitro and in vivo
In our study, WDR5 expression was detected in different human CCA cell lines and normal bile epithelium cell human intrahepatic biliary epithelial cells (HIBEpiCs). WDR5 was detectable in all cells and had a lower level in HIBEpiCs (Figures 2A and 2B; Figure S2A). After overexpressing WDR5 in RBE cells and silencing WDR5 in QBC939 cells (Figures 2C–2E), the proliferation, migration, and invasion were detected with CCK-8, wound-healing, and transwell assays. WDR5 had no obvious effect on CCA proliferation (Figure S2B), but it was required in CCA migration and invasion (Figures 2F and 2G; Figures S2C and 2SD). Previous studies reported that WDR5 was able to activate EMT in several malignancies, which can confer tumor cells more aggressive invasion.16,38,39 Therefore, we investigated the regulation of WDR5 on EMT-specific proteins, including E-cadherin, N-cadherin, and vimentin. WDR5 knockdown increased E-cadherin expression and decreased expression of N-cadherin and vimentin, while WDR5 overexpression had opposite changes on the expression of EMT biomarkers (Figure 2H; Figure S2E), indicating that WDR5 was able to promote EMT.
Figure 2.

WDR5 facilitated the migration, invasion, and metastasis of CCA
(A and B) The expression of WDR5 in different CCA cell lines and in a normal biliary epithelium cell line (HIBEpiCs) was detected with qPCR (A) and western blot (B). (C) WDR5 was overexpressed in RBE cells with lentivirus encoding LV5-WDR5. (D and E) WDR5 was knocked down in QBC939 cells with two different shRNAs in different vectors, namely LV3-shWDR5-1 (D) and pGPU6-shWDR5-2 (E). (F and G) Invasion and migration of RBE (F) and QBC939 (G) cells were detected with wound-healing (left panel) and transwell assays (right panel) after overexpressing or silencing WDR5. (H) EMT biomarkers were detected after WDR5 overexpression in RBE cells and knockdown in QBC939 cells. (I) Stable WDR5-silenced cells and control cells were injected via the tail vein of BALB/c nude mice. The body weights of mice injected with scramble (LV3-scr.) and shWDR5 (LV3-sh1) cells were measured weekly. (J) Livers were weighed to assess the tumor burden of liver metastatic foci. (K) Number of metastasis nodules in livers and lungs of mice. (L) Representative gross specimens and H&E staining of metastasis lesions in liver and lung. Arrows indicate the metastasis lesions. Scale bars, 50 μm. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗ p < 0.001. In (C)–(K), data were analyzed with a one-way ANOVA. In (I), data were analyzed with a two-way ANOVA.
Experiments in vivo were performed to verify the function of WDR5 in metastasis. Stable WDR5-silenced or control QBC939 cells were injected into BALB/c nude mice via the tail vein. The mice injected with WDR5-silenced QBC939 cells had heavier body weight, lower liver weight, and fewer metastasis nodules in livers and lungs (Figures 2I–2L). H&E staining of the metastasis nodules on livers and lungs was further performed to confirm that the nodules were formed by CCA cells (Figure 2J). All of these results suggested the potency of WDR5 to promote CCA metastasis.
WDR5 promoted CCA metastasis via interaction with c-Myc
c-Myc can bind directly with WDR5 through the Myc-MBIIIb motif, and WBM mutation of MBIIIb can efficiently disrupt the interaction between c-Myc and WDR517,19 (Figure 3A). In RBE and QBC939 cells, we performed a coIP assay after overexpressing Myc-wild-type (WT) or Myc-WBM. Consequently, WBM mutation overexpression dramatically decreased the interaction between WDR5 and c-Myc, showing that WBM mutation was dominantly negative for the Myc-WDR5 interface (Figure 3B; Figure S3A). Moreover, WBM mutation also impaired the invasion and EMT of RBE and QBC939 cells (Figures 3C and 3D; Figure S3B and S3C). In vivo experiments were performed to evaluate the influence of Myc-WBM on metastasis. The mice with stable QBC939 cells with Myc-WBM overexpression had higher body weight, lower liver weight, and fewer metastasis nodules compared with those with Myc-WT-overexpressed cells (Figures 3E–3H). These results suggested that the MBIIIb motif of c-Myc was essential in WDR5-induced EMT, invasion, and metastasis of CCA.
Figure 3.

WDR5 promoted CCA metastasis via the interaction with c-Myc
(A) Schematic diagram of human wild-type (WT) and WBM mutant c-Myc protein. The MBIIIb motif refers to amino acids from 258 to 268. WBM represents the I262E/V264E/V265E mutation of the MBIIIb motif. (B) Co-immunoprecipitation (coIP) of WT and WBM mutant c-Myc with endogenous WDR5 in RBE (left) and QBC939 (right) cells. (C) The invasion of RBE and QBC939 cells transfected with Myc-WT or Myc-WBM was detected with a transwell assay. (D) In RBE and QBC939 cells stably expressing Myc-WT or Myc-WBM, the expression levels of EMT-specific proteins were detected with western blot. (E) Weekly body weight of BALB/c mice injected with QBC939 cells overexpressing Myc-WT or Myc-WBM. (F) Livers were weighed to assess the tumor burden of liver metastatic foci. (G) Number of metastasis nodules in livers and lungs of mice. (H) Representative H&E staining of metastasis lesions in liver and lung. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, calculated with a two-way ANOVA (E) or with a one-way ANOVA (C, F, and G).
HIF-1α was a downstream target of c-Myc and WDR5
In above experiments, we proved that the MBIIIb motif of c-Myc was essential in the WDR5-facilitated EMT, invasion, and tumor metastasis, so we further screened the downstream effectors that were responsible for CCA progression. First, the datasets of chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) from HEK293 cells were re-analyzed (Gene Expression Omnibus [GEO]: GSE60897). Myc-WT and Myc-WBM interacted with the promoter of 1,501 and 374 genes, respectively (Figure 4A), and a total of 298 genes were overlapped (Figure 4B). Our target genes should be in the subset of 1,203 genes whose promoter interacted with Myc-WT but not Myc-WBM (Figure 4B). In ChIP-seq of HEK293 cells (GEO: GSE60897), 10,095 genes promoters that may interact with WDR5 were screened out (Figure 4C), and 1,113 of them were overlapped with the genes binding with Myc-WT, accounting for about 80% of all Myc-interacting genes but only 11% of WDR5-binding promoters (Figure 4D). To further assess WDR5 effects on Myc-induced transcription, we retrieved a published mRNA sequencing (mRNA-seq) dataset of lymphoma Ramos cells (GEO: GSE126207).19 Based on the criteria as a log2 fold change ≥0.75 and a p value <0.05, 3,593 downregulated genes and 6,533 upregulated genes were selected in the differentially expressed genes (DEGs) between Myc-WT and Myc-WBM overexpression (Figure 4E). Moreover, we performed mRNA-seq in QBC939 cells transfected with scrambled short hairpin RNA (shRNA) and shWDR5 and screened out 3,877 upregulated and 3,360 downregulated genes (GEO: GSE149536) (Figure 4E). The expressions of 1,203 genes in Figure 4B were verified in mRNA-seq results of QBC939 (Figures S4A and S4B) and Ramos (Figures S4C and S4D) cells. The DEGs of these 1,203 genes were detailed in Tables S7 and S8.
Figure 4.

HIF-1α was the primary direct target of c-Myc and WDR5
(A) Re-analysis of the ChIP-seq datasets regarding Myc-WT and Myc-WBM in HEK293 cells (GEO: GSE60897). Pie chart and histogram show the percentage and number of different binding regions mapped for WT and WBM of c-Myc. (B) Venn diagram shows the overlap of genes binding with Myc-WT and Myc-WBM. (C) ChIP-seq datasets show the number and binding sites of genes interacting with WDR5 in HEK293 cells (GEO: GSE60897). (D) Overlapped genes interacting with the promoter-TSS region of both WDR5 and Myc-WT are shown in the Venn diagram. (E) Left panel: mutual regulated mRNAs in Ramos cells, a lymphoma cell line, with overexpression of Myc-WT and Myc-WBM (GEO: GSE126207). Right panel: WDR5 in QBC939 cells was silenced by shRNA, and mRNA sequencing was performed to screen the upregulated or downregulated genes (GEO: GSE149536). (F) Flowchart of screening downstream genes of c-Myc and WDR5 by the bioinformatics method. Upper panel: a total of 30 overlapped genes were screened out by the results in the ChIP-seq datasets and RNA-seq, which are shown in (A)–(E). Bottom panel: the overlapped 30 genes had only 2 common genes with EMT-involved genes in GO analysis, which were HIF1A and LEF1. (G) In QBC939 cells, mRNAs of HIF1A and LEF1 were detected with qPCR after silencing WDR5 (upper two panels) or overexpressing Myc-WT/WBM (bottom panel). (H) HIF-1α expression was detected with western blot after silencing WDR5 (upper two panels) or overexpressing Myc-WT/WBM (bottom panel) in QBC939 cells. (I) ChIP assay revealed that WDR5 knockdown (upper two panels) or Myc-WBM mutation (bottom panel) inhibited the recruitment of c-Myc on the HIF1A promoter. ∗p < 0.05, ∗∗p < 0.01, by one-way ANOVA. (J and K) mRNA correlations between HIF-1α and WDR5 (J) or c-Myc (K) were analyzed with a Pearson correlation test. (L and M) Protein correlations between HIF-1α and WDR5 (L) or c-Myc (M) were analyzed with a Pearson correlation test. (N) Protein levels of HIF-1α in CCA patients with double-positive, single-positive, and double-negative expression of c-Myc and WDR5. ∗∗∗p < 0.001, by one-way ANOVA.
By overlapping the four gene sets shown in Figures 4A, 4C, and 4E, 30 genes were selected as candidates that may be regulated by both WDR5 and Myc-WT, but not by Myc-WBM. Furthermore, only HIF1A and lymphoid enhancer-binding factor 1 (LEF1) were selected out when we combined these 30 genes with EMT-related genes in Gene Ontology (GO) analysis (Figure 4F). In addition, we silenced WDR5 expression or overexpressed c-Myc in QBC939 cells and showed that both HIF1A and LEF1 mRNA were decreased after WDR5 knockdown, but only HIF1A mRNA in cells with Myc-WBM overexpression was significantly lower than that in cells with Myc overexpression (Figure 4G). Moreover, the read count of LEF1 was very low in our results of mRNA-seq (Figure S4E). These results suggested that HIF-1α was the most promising effector of WDR5/Myc-induced EMT and metastasis. To confirm the results from bioinformatics analysis, we detected HIF-1α expression and HIF1A promoter interaction with c-Myc by quantitative real-time PCR and ChIP, respectively, after regulating the expression of WDR5 and c-Myc. HIF-1α expression was extensively reduced when WDR5 was knocked down, and it increased when Myc-WT, but not Myc-WBM, was overexpressed (Figure 4H; Figure S4F). Moreover, the interaction of the HIF1A promoter with c-Myc was also significantly impaired by WDR5 knockdown or Myc-WBM overexpression (Figure 4I). These results suggested that interaction of WDR5 and c-Myc synergistically promoted the HIF1A transcription and expression, and the MBIIIb motif was required in that process. Moreover, we detected HIF-1α expression in 18 fresh iCCAs, 16 pCCAs, and 14 dCCAs by quantitative real-time PCR, and in CCA tissue microarrays by IHC (Figure S4G). In fresh CCAs, we found that HIF-1α expression was positively associated with WDR5 and c-Myc (Figures 4J and 4K). In CCA tissue microarrays, we analyzed the correlations between HIF-1α and c-Myc or WDR5 with a Pearson test (Figures 4L and 4M) and Chi-square test (Figure S4H), which further verified the significant correlations between HIF-1α and c-Myc or WDR5. In addition, patients with co-expression of WDR5 and c-Myc had a remarkably higher IHC score of HIF-1α than did those with low WDR5/c-Myc (Figure 4L). Taken together, these results supported that HIF-1α expression was increased by WDR5 and c-Myc.
HIF-1α was an important effector in WDR5-induced CCA metastasis
HIF-1α has been reported to participate in several malignant biological behaviors, including tumor metastasis and EMT.40,41 HIF-1α was proved as a prognostic biomarker of iCCA in a small-sample-size cohort,42 but the functions of HIF-1α in CCA progression were rarely reported. TCGA CCA data and IHC scores of CCAs pairs confirmed the upregulation of HIF-1α in CCA tissues (Figure 5A; Figure S5A). Twist1 was a well-known effector of HIF-1α and was also a well-known biomarker of EMT. In QBC939 cells, Twist1 expression was suppressed by two independent HIF-1α inhibitors, LW6 and PX-478 (Figures 5B and 5C; Figure S5B). In survival analyses, HIF-1α was a prognostic biomarker of iCCA, pCCA, and dCCA (Figure 5D). In mRNA-seq of WDR5-silenced QBC939 cells, proteins involved in the HIF-1α pathway were significantly downregulated (Figure 5E), so we further investigated the role of HIF-1α in WDR5-induced progression. LW6 or PX-478 substantially attenuated invasion and EMT of QBC939 cells (Figures 5F and 5G; Figure S5C and S5D), indicating a critical role of HIF-1α in the WDR5-induced invasion and EMT. In vivo experiments were also conducted to confirm the role of HIF-1α in WDR5-induced metastasis. Stable WDR5-overexpressed or control QBC939 cells were injected into BALB/c nude mice via the tail vein, and LW6 or PX-478 was used to inhibit HIF-1α expression through oral gavage or intraperitoneal injection. An in vivo imaging system (IVIS) was used to assess the tumor burden. The mice injected with WDR5-overexpressed QBC939 cells showed higher tumor burden, liver weight, and more metastasis nodules in livers and lungs, and LW6 or PX-478 reversed this effect (Figures 5H–5L; Figure S5E). Moreover, the prognostic significance of the co-expression of HIF-1α and WDR5 was further estimated to evaluate the facilitating role of HIF-1α in WDR5-induced progression. Co-expression of HIF-1α and WDR5 was a more sensitive factor compared with WDR5 alone in iCCA, pCCA, and dCCA (Figure 5M). These results further underscored the crucial role of HIF-1α in WDR5-involved CCA progression and prognosis.
Figure 5.

HIF-1α was an important effector in WDR5-induced CCA metastasis
(A) HIF1A mRNA in CCAs was significantly higher than that in normal tissues. (B and C) Quantitative real-time PCR (B) and western blot (C) show that the HIF-1α inhibitor LW6 (20 μM) or PX-478 (25 μM) decreased TWIST1 expression. (D) Survival curves of iCCA, pCCA, and dCCA patients were stratified by HIF-1α expression. (E) Genes in the HIF1A signaling pathway were changed along with WDR5 knockdown. (F and G) Invasive ability (F) and expression of EMT-specific proteins (G) in WDR5-overexpressed QBC939 cells in the presence or absence of LW6 (20 μM) or PX-478 (25 μM). (H) Metastatic models were established by tail vein injection of WDR5-overexpressed QBC939 cells in the treatment of HIF-1α inhibitor. Two HIF-1α inhibitors, LW6 (10 mg/kg p.o.) and PX-478 (100 mg/kg i.p.), were used to inhibit HIF-1α expression in vivo. The tumor metastases were monitored by a live imaging system (n = 6). (I) Radiant efficiency of in vivo fluorescence in (H) was measured to quantify the tumor burden of mice. (J) Livers of mice in (H) were weighed to assess the tumor burden of liver metastatic foci. (K and L) The numbers of metastasis nodules in livers (J) and lungs (K) of mice in (H) were counted. (M) The survival curves were further stratified into subgroups with the co-expression, single expression, and double low expression of WDR5 and HIF-1α in iCCA, pCCA and dCCA. In (B), (F), and (I)–(L), ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, calculated by one-way ANOVA. In (D) and (M), data were calculated by a log-rank test, and the C-index was calculated to evaluate the accuracy of the prognostic model.
WDR5 increased HIF-1α accumulation via suppressing the PHD2-mediated degradation pathway independently of c-Myc
Since c-Myc knockout mice were embryo lethal,43 we established stable Myc-silenced QBC939 cells and treated them with 40 μM c-Myc inhibitor 10058-F4 to eliminate c-Myc function as much as possible (sh+F4 group). In this c-Myc loss-of-function model, the ChIP assay showed that the interaction between c-Myc and the HIF1A promoter was almost blocked (Figure 6A). Moreover, the combination of sh-Myc and 10058-F4 abolished the WDR5-induced upregulation of HIF1A mRNA (Figure 6B), but it could not totally wipe out the change of HIF-1α expression (Figure 6C; Figure S6A). These results demonstrated that abrogating c-Myc function can suppress HIF1A transcription, but there were other post-transcriptional mechanisms responsible for WDR5-regulated HIF-1α accumulation, which was independent of c-Myc.
Figure 6.

WDR5 promoted HIF-1α accumulation via suppressing the PHD2-mediated degradation pathway
(A) ChIP assay proved that the combination of shMyc (sh) and c-Myc inhibitor 10058-F4 (40 μM) almost completely suppressed c-Myc recruitment on the HIF1A promoter. (B and C) In QBC939 cells treated with shMyc and inhibitor 10058-F4, quantitative real-time PCR (B) and western blot (C) showed that WDR5 overexpression had little effect on HIF1A mRNA, but it increased HIF-1α accumulation. (D) Half-life analysis of HIF-1α protein. Cells were treated with 10 μg/mL cycloheximide (CHX) for the indicated times. The gray scale values of western blot bands were quantified with ImageJ software. (E) Effect of ubiquitination inhibitor MG-132 (10 μM) on control and WDR5-overexpressed QBC939 cells. MG-132 attenuated the HIF-1α elevation induced by WDR5 overexpression in QBC939 cells treated with shMyc and inhibitor 10058-F4. (F) In QBC939 cells treated with shMyc and inhibitor 10058-F4, WDR5 overexpression decreased hydroxylation of HIF-1α. (G and H) The mRNA levels of the PHD family and VHL in QBC939 cells transfected with scramble and WDR5 knockdown were detected by RNA-seq (G) and verified by quantitative real-time PCR (H). (I) The expression levels of the PHD family in QBC939 cells transfected with control, vector, and LV5-WDR5 were detected by western blot in the treatment with shMyc and inhibitor 10058-F4. (J) Effect of PHD1–PHD3 knockdown on the expression of total HIF-1α and hydroxy-HIF-1α in QBC939 cells treated with shMyc and inhibitor 10058-F4. (K) Survival curves of CCA were stratified with PHD1–PHD3 in TGCA database. (L) The correlations between WDR5 and PHD2 mRNA in iCCA, pCCA, and dCCA were analyzed with a Pearson correlation test. (M) Expression levels of HIF-1α in WDR5-silenced and/or PHD2-silenced QBC939 cells after treatment with shMyc and inhibitor 10058-F4. PHD2 knockdown attenuated the influence of WDR5 knockdown on HIF-1α reduction. Data were analyzed with one-way ANOVA in (A), (B), and (H). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. n.s., not significant.
In our study, cycloheximide (CHX) was introduced to inhibit protein synthesis for half-life analysis of HIF-1α protein. As expected, with the treatment of shMyc+F4, HIF-1α protein had a shorter half-life in control cells compared with that in WDR5-overexpressed QBC939 cells (Figure 6D; Figure S6B). These data implicated that overexpression of WDR5 stabilized HIF-1α protein. Ubiquitination is the major pathway of protein degradation including HIF-1α, so we used the ubiquitination inhibitor MG132 and detected HIF-1α stabilization. In sh+F4 cells, MG132 diminished the HIF-1α increase that was caused by WDR5 overexpression (Figure 6E; Figure S6C), showing that WDR5-induced HIF-1α accumulation was dependent on HIF-1α ubiquitination.
Ubiquitination of HIF-1α requires hydroxylation on prolines 564 and 402 by PHDs and the recognition of VHL for the degradation through the E3 ubiquitin ligase/proteasome pathway.29, 30, 31 Therefore, we evaluated the hydroxylation of HIF-1α in sh+F4 QBC939 cells and found that WDR5 overexpression suppressed HIF-1α hydroxylation (Figure 6F; Figure S6D). Furthermore, mRNA levels of the proteins that were related with HIF-1α hydroxylation were investigated, including PHD1–PHD3 and VHL. Our RNA-seq data showed that PHD2 and PHD3 were increased in WDR5-silenced QBC939 cells (Figure 6G). Quantitative real-time PCR and western blot (WB) analysis revealed that PHD2 and PHD3 expression levels were increased after WDR5 knockdown, and they were decreased when WDR5 was overexpressed (Figures 6H and 6I; Figure S6E). Moreover, PHD1–PHD3 were silenced, respectively, and HIF-1α was detected in sh+F4 QBC939 cells (Figure S6F). As a result, only PHD2 exhibited a significant influence on HIF-1α stabilization in CCA (Figure 6J; Figures S6G and 6H). In TGCA CCA data, PHD2 was the only PHD member with prognostic value of CCA (Figure 6K). With quantitative real-time PCR, we showed that PHD2 was negatively correlated with WDR5 in CCA tissues (Figure 6L). Finally, we knocked down PHD2 in WDR5-silenced QBC939 cells treated with sh+F4 and confirmed that PHD2 was essentially required in WDR5-induced HIF-1α stabilization (Figure 6M; Figure S6I).
WDR5 suppressed PHD2 expression through HDAC2-induced H3K4 deacetylation
To further investigate the mechanism of how WDR5 suppressed PHD2 expression in CCA, we used actinomycin D (Act D) to inhibit mRNA synthesis and detected the half-life of PHD2 mRNA in WDR5-silenced QBC939 cells. No significant difference of PHD2 mRNA half-life was observed in WDR5-silenced and control cells (Figure 7A), indicating that the inhibitory effect of WDR5 on PHD2 mRNA was the result of mRNA synthesis impairment rather than mRNA degradation. Given that WDR5 was involved in multiple histone modifications, we then analyzed the chromatin accessibility of the PHD2 promoter region and found that WDR5 knockdown remarkably enhanced the chromatin accessibility of the PHD2 promoter (Figure 7B). This result was in conflict with the main function of WDR5, which was promoting H3K4me, opening chromatin structure, and enabling the transcriptional machinery.15 Previous studies reported that WDR5 interacted with HDACs to promote histone deacetylation, which induced the compacted nucleosome and thus repressed DNA transcription,22, 23, 24 so we further performed ChIP assays to detect the H3K4ac of histone interacting with the promoter region of PHD2. The results showed that WDR5 knockdown enhanced the H3K4ac levels of histone interacting with the PHD2 gene promoter, and they indicated that WDR5-induced PHD2 transcriptional repression might correlate with H3K4 deacetylation (Figure 7C). To further validate this result and provide a global view of WDR5-involved H3K4ac, we applied a cleavage under targets and tagmentation (CUT&Tag) assay, a newly developed assay to investigate protein-DNA interaction,44,45 with QBC939 cells (GEO: GSE162968). EGLN1, the gene encoding PHD2, was the gene with the most obvious H3K4ac increase after WDR5 knockdown (Figures 7D–7F; Table S9).
Figure 7.

WDR5 suppressed PHD2 expression through HDAC2-induced H3K4 deacetylation
(A) Half-life detection of PHD2 mRNA was performed using 10 μM actinomycin D (Act D) for the indicated times. (B) Chromatin accessibility of PHD2 promoters was evaluated by quantitative real-time PCR with DNase I-pretreated nucleus of control, scramble, and WDR5-silenced QBC939 cells. Fold change was analyzed using 2−ΔΔCt. (C) ChIP assay showed that WDR5-silenced QBC939 cells had elevated H3K4ac modification at the PHD2 promoter regions. H3K4ac antibody was used for immunoprecipitation (IP) after WDR5 knockdown, and quantitative real-time PCR of the PHD2 promoter in output was performed. (D) A CUT&Tag assay was performed to confirm that WDR5 knockdown promoted H3K4ac modification at PHD2 promoter regions. WDR5 was knocked down by shWDR5 in QBC939 cells, and H3K4Ac antibody was used to interact with the H3K4-acetylized protein. Log2(RPM + 1) was used for quantification. (E) Overlay of H3K4ac at EGLN1 (PHD2) loci in scramble (purple) and WDR5 knockdown (green) QBC939 cells. Significantly increased regions are underscored (red box). (F) List of 10 DEGs with most significant change in CUT&Tag data. EGLN1 (PHD2) had the highest fold change after WDR5 knockdown. (G) CoIP of WDR5 with HDAC1–HDAC3 in QBC939 cells. (H) Effect of HDAC1–HDAC3 knockdown on the expression of PHD2 protein in QBC939 cells. (I) ChIP assays show that H3K4ac on PHD2 promoters in HDAC2 silenced QBC939 cells. H3K4ac antibody was used for IP after HDAC2 knockdown, and qPCR of the PHD2 promoter in output was performed. (J) ChIP assay revealed that WDR5 directly interacts with the promoter region of PHD2 gene. WDR5 antibody was used for IP, and qPCR of the PHD2 promoter in output was performed. (K) ChIP assay revealed that WDR5 knockdown attenuated HDAC2 binding to the PHD2 promoter. HDAC2 antibody was used for IP after WDR5 knockdown. (L) ChIP assay revealed that WDR5 overexpression disrupted H3K4ac at PHD2 promoter regions and knockdown or inhibition of HDAC2 reversed it. H3K4ac antibody was used for IP, and quantitative real-time PCR of PHD2 promoter in output was performed. (M) Expression of PHD2 in WDR5-overexpressed QBC939 cells that were treated with 50 μM SCA for 48 h or transfected with siHDAC2. Data were analyzed with two-way ANOVA in (A), or with one-way ANOVA in (B), (C), and (I)–(L). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. n.s., not significant.
H3K4ac is deacetylated by the HDAC family, including HDAC1–HDAC10. In the BioGRID protein interactions database, the affinity-capture mass spectrometry(ACMS) results indicated that WDR5 could bind directly with HDAC1, HDAC2, and HDAC3.46 Therefore, we performed co-immunoprecipitation (coIP) in QBC939 cells to confirm the interaction of WDR5 to HDAC1–HDAC3 (Figure 7G). Furthermore, we knocked down HDAC1–HDAC3 separately by small interfering RNA (siRNA) and detected PHD2 expression. HDAC2 knockdown caused the most significant enhancement of PHD2 expression in QBC939 cells (Figure 7H). ChIP assays confirmed that HDAC2 knockdown extensively increased H3K4ac of histone, which interacted with the PHD2 promoter (Figure 7I), and that WDR5 could interact with the PHD2 promoter (Figure 7J). Moreover, WDR5 knockdown reduced HDAC2 binding to the PHD2 promoter (Figure 7K). In addition, WDR5-overexpressed QBC939 cells were transfected with siHDAC2, or incubated in 50 μM Santacruzamate A (SCA), a specific inhibitor of HDAC2, for 48 h. ChIP and western blot results showed that HDAC2 inhibition or knockdown increased the H3K4ac of histone interacting with the PHD2 promoter, and thus promoted PHD2 expression, which was an inverse effect of WDR5 overexpression (Figures 7L and 7M). All of the above data demonstrated that WDR5 suppressed PHD2 expression through HDAC2-induced H3K4 deacetylation.
Overall, we found that WDR5 interacted with the MBIIIb motif of c-Myc in CCA and facilitated Myc-induced transcription of HIF1A. Additionally, WDR5 interacted with HDAC2 and enhanced HDAC2-induced H3K4 deacetylation, and therefore decreased PHD2 transcription. Reduction of PHD2 expression attenuated HIF-1α hydroxylation and thus stabilized HIF-1α. The accumulated HIF-1α could eventually promote CCA invasion, metastasis, and EMT via inducing TWIST1 expression (Figure 8).
Figure 8.

Schematic depiction of the mechanisms underlying WDR5 facilitating CCA metastasis and EMT by increasing HIF-1α accumulation
Two distinct mechanisms of how WDR5 enhanced HIF-1α accumulation: (1) WDR5 promoted HIF-1α mRNA synthesis via directly interacting with c-Myc and (2) WDR5 inhibited HIF-1α protein hydroxylation, ubiquitination, and degradation through suppressing PHD2 expression, which relied on interacting with HDAC2 and deacetylizing H3K4ac.
Discussion
c-Myc, a well-known tumor driver oncoprotein, is overexpressed in more than half of human malignancies. It was estimated that up to one-third of all cancer deaths were attributed to aberrant c-Myc expression or activity.47 However, the clinical and prognostic significance of c-Myc in CCA has not reached a consensus. Our previous study supported that c-Myc was not a good prognostic biomarker of pCCA.14 In the present study, we systemically investigated the expression of c-Myc and WDR5 in all CCA subtypes and demonstrated that co-expression of c-Myc and WDR5 was able to predict poor prognosis with more sensitivity. Our results suggested that WDR5-Myc interaction and their oncogenic functions were important and common molecular features of CCAs, although iCCA, pCCA, and dCCA were generally considered to have different molecular profiles. Moreover, this would be a significant breakthrough in CCA biomarker study if our results were verified in larger cohorts and multiple medical centers, because our results indicated that detection of c-Myc and WDR5 could screen the high-risk patients and guide the individual treatment more precisely.
The extensive involvement of c-Myc in cancer provided the possibility of Myc-targeted therapy for human malignancies. Currently, several strategies have been developed to decrease MYC expression or to interfere with downstream signaling triggered by MYC.48,49 However, the approaches to directly inhibit c-Myc activity, disrupt c-Myc interaction with the promoters of target genes, and suppress its transcription activity are still not amenable11,19 because of the inaccessibility of the c-Myc-MAX interface for small-molecule inhibitors. Moreover, suppressing all kinds of the transcription activity of Myc may be deadly to normal cells. The promising approach is to suppress the oncogenic activity of c-Myc but preserve its normal function.17 The result that WDR5 could enhance the recruitment of c-Myc to its target genes is a milestone in the studies of c-Myc oncogenic function.50, 51, 52 These studies showed that WDR5 was required for only a part of Myc-induced gene transcription, and that destroying WDR5-Myc interaction can disable c-Myc ability for facilitating tumorigenesis in vivo.17,19 Therefore, an inhibitor suppressing the WDR5-Myc interaction is regarded as a promising method to separate the oncogenic role from the normal function of Myc.18,53 The small-molecule inhibitor to block WDR5-Myc interaction has just been screened out in a recent study,53 which may confer our study of greater clinical significance. For the first time, we demonstrated that WDR5 was essential in Myc-induced progression of CCA by binding with the MBIIIb motif, and we identified the WDR5/Myc downstream effector as HIF-1α. This is an important supplement to the discovery of new drug targets for CCA, especially at this time when the effect of current CCA-targeted therapies is usually limited.
In this study, we identified HIF1A as a target gene of c-Myc and WDR5 by RNA-seq and published ChIP-seq datasets.17,19 As a well-known hypoxia-responsive transcription factor, HIF-1α regulates the transcription of genes that are involved in cell stress to hypoxia, including angiogenesis, apoptosis, and malignant biological behaviors such as tumor metastasis and EMT.40,41 In the present study, we demonstrate that HIF-1α was a direct effector of WDR5/Myc and was an indicator of CCA prognosis. HIF-1α could be a viable drug target of CCA, since there was a variety of HIF-1α-specific inhibitors, and several of them have been demonstrated to suppress tumor progression with in vitro and in vivo experiments.54
Recent studies revealed that HIF-1α also exerted sustained oncogenicity under normoxic conditions.55 However, the intracellular expression of HIF-1α in normoxic condition was normally low. Aberrant accumulation of HIF-1α protein was associated with tumorigenesis and malignant progression.40,41 We demonstrated that WDR5 increased HIF-1α accumulation in two independent pathways: (1) WDR5 facilitated Myc-induced transcription of HIF1A and (2) WDR5 bound with HDAC2 and increased the deacetylation of H3K4ac of the histone interacting with the PHD2 promoter, resulting in the attenuation of chromatin opening and PHD2 expression, which eventually led to HIF-1α stabilization and accumulation. Both of the above-noted regulating pathways of HIF-1α are reported in our study for the first time. The pleiotropic effects of WDR5 on HIF-1α accumulation provided new insights to the oncogenic function of WDR5 and HIF-1α regulation. The correlation between WDR5 and metastasis has been hinted at in breast cancer, glioma, and colon cancer, by participating in several signaling pathways such as transforming growth factor (TGF)-β signaling and the phosphatidylinositol 3-kinase (PI3K)/AKT pathway.16,56,57 For the first time, we linked WDR5 with HIF-1α and its downstream TWIST1 and suggested that WDR5 resulted in metastasis by promoting HIF-1α-induced EMT. This conclusion enhanced current understanding of WDR5 as an oncoprotein, especially in CCA.
In conclusion, we demonstrated that WDR5 interacted with the MBIIIb motif of c-Myc and facilitated Myc-induced transcription of downstream genes. Co-expression of WDR5 and c-Myc was a sensitive factor predicting poor prognosis of all CCA types, including iCCA, pCCA, and dCCA. WDR5 had pleiotropic effects on HIF-1α accumulation by promoting Myc-induced HIF1A transcription or decreasing PHD2 expression via HDAC2-involved H3K4 deacetylation. HIF-1α was the main effector of WDR5-induced EMT, invasion, and metastasis of CCA. Our results suggested that the detection of WDR5, c-Myc, and HIF-1α can help stratify the high-risk CCA patients more precisely and guide the individual treatment. WDR5 and HIF-1α are promising drug targets of CCA, and blocking the WDR5-Myc interface and HIF-1α accumulation would be a potential approach to treat CCA.
Materials and methods
Patients and ethics
The primary cohort was comprised of patients who underwent surgery for iCCA (258 cases), pCCA (412 cases), and dCCA (260 cases) in Qilu Hospital of Shandong University from 2012 to 2018 (Table S1). The validation cohort consisting of 78 iCCA patients, 141 pCCA patients, and 88 dCCA patients was further selected using the following criteria: (1) patients who underwent radical resection with a clear surgical margin; (2) patients with available formalin-fixed tumor tissues, follow-up information, and complete medical records; (3) patients with a postsurgical survival time of more than 1 month; and (4) patients with no history of other malignancies. The tumors were classified and staged according to the eighth edition of the AJCC/UICC TNM classification system. Informed consent was obtained from all patients. All experiments were approved and supervised by the Ethics Committee of Qilu Hospital of Shandong University.
Cells and agents
Human pCCA cell lines QBC939 and FRH-0201, iCCA cell lines RBE and HCCC-9810, and HIBEpiCs were obtained by the Cell Bank of the Chinese Academy of Sciences. RBE cells, HCCC-9810 cells, and HIBEpiCs were cultured in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA). QBC939 and FRH-0201 cells were cultured in DMEM (Thermo Fisher Scientific). The media for the cell lines were supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), 100 U/ml penicillin, and 100 μg/mL streptomycin at 37°C under 95% air and 5% CO2. All cell lines were authenticated using short tandem repeat (STR) analysis, and the databases of the Chinese Academy of Sciences and American Type Culture Collection were used as references.
The HIF-1α inhibitors LW6 and PX-478, the c-Myc inhibitor 10058-F4, the proteasome inhibitor MG132, and the HDAC2 inhibitor SCA were all purchased from MedChemExpress (Monmouth, NJ, USA). Other agents without special instruction were purchased from Sigma-Aldrich (St. Louis, MO, USA). The following primary antibodies were used: WDR5 (Abcam, ab178410), c-Myc (Santa Cruz Biotechnology, sc-40), E-cadherin (Cell Signaling Technology, #3195), N-cadherin (Cell Signaling Technology, #13116), vimentin (Cell Signaling Technology, #5741), Twist1 (Santa Cruz Biotechnology, sc-81417), HIF-1α (Abcam, ab2185), hydroxy-HIF-1α (Pro564) (Cell Signaling Technology, #3434), PHD1 (Abcam, ab108980), PHD2 (Santa Cruz Biotechnology, sc271835), PHD3 (Proteintech, 18325-1-AP), VHL (GeneTex, GTX101087), HDAC1 (Cell Signaling Technology, #5356), HDAC2 (Cell Signaling Technology, #5113), HDAC3 (Abcam, ab7030), and β-actin (Cell Signaling Technology, #4970).
IHC
IHC was performed as described.7 EDTA (pH 9.0) was used for antigen retrieval. Primary anti-WDR5 antibody (R&D Systems, AF5810, 1:50), anti-c-Myc antibody (Abcam, ab32072, 1:200), or anti-HIF-1α (Abcam, ab2185, 1:100) was applied and incubated at 4°C overnight. Goat anti-rabbit and rabbit anti-goat antibodies labeled with biotin (Zsbio, Beijing, China) were used for 30 min at room temperature. The peroxidase reaction was performed with 3,3′-diaminobenzidine (DAB) solution (Zsbio).
The IHC score was evaluated by Quant Center software, which calculated the staining intensity and the area of each staining. IHC score = (percentage of cells of weak intensity × 1) + (percentage of cells of moderate intensity × 2) + (percentage of cells of strong intensity × 3), according to previous studies.14 The cohorts were divided into subgroups with high and low expression by the cutoff of the IHC score, which was confirmed as the point with the highest sum of specificity and sensitivity in the receiver operating characteristic (ROC) curves according to a previous study.58
Western blotting
Total protein was extracted using radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime) with 1% PMSF (Beyotime) and 1% phosphatase inhibitor (Solarbio). Protein concentration was determined using a bicinchoninic acid (BCA) assay kit (Beyotime). An equal amount of protein was separated with 10% SDS-PAGE after denaturation and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were incubated overnight in the primary antibody at 4°C and then incubated for 1 h in the second antibody. Enhanced chemiluminescence (Millipore) was used for horseradish peroxidase (HRP) detection. Quantitative analysis of western blotting was performed using ImageJ.
Quantitative real-time PCR
Quantitative real-time PCR was performed as described.7 Briefly, TRIzol reagent (Thermo Fisher Scientific) was used for extraction of total RNA in cells or tissues. A reverse transcriptase kit (Toyobo) was used for cDNA synthesis according to the manufacturer’s recommendations. SYBR Green master mix (Roche) was used for real-time PCR, and the Ct values were calculated by a Roche LightCycler 480 PCR instrument. The 2−ΔΔCt method was performed for comparison between groups, and GAPDH was used as an internal control. All primers were designed using Primer Express version 5.0 software (Applied Biosystems). The primers used for qPCR are listed in Table S2.
Transfection and stable cell lines
The full-length sequence and shRNA of WDR5 were constructed into the lentivirus vector (GeneChem, GenePharma). The processes of transfection and the establishment of stable cell lines were performed as described.7 The efficiency levels of knockdown and overexpression were verified by western blotting and quantitative real-time PCR. The sequences of siRNAs and shRNAs are listed in Table S3.
CCK-8 assays
CCK-8 assays were performed as described.7,59 Briefly, cells transfected with siRNA/shRNA sequences or corresponding scramble oligonucleotides were plated into 96-well plates (3 × 103 cells/well) and incubated for 1–4 days. Then, 10 μL of CCK-8 reagent (Dojindo, Kumamoto, Japan) was added and incubated at 37°C for 1 h. The optical density at 450 nm was detected using a spectrophotometer (Molecular Devices).
Transwell assays
Transwell assays were performed using 24-well plates with transwell chambers (8.0-μm pore diameter; Corning Life Sciences). 5 × 104 RBE cells or 10 × 104 QBC939 cells were seeded into the upper chambers with or without Matrigel coating (diluted at 1:6 with RPMI 1640 or DMEM; Corning Life Sciences). Approximately 600 μL of complete medium was added into the lower chamber. After 36 h of incubation, the cells attached to the bottoms of chambers were fixed with methanol and stained with 0.5% crystal violet (Beyotime) for 30 min. Cell numbers were counted by ImageJ in five random visual fields of microscopy at ×200 magnification.
In vivo experiments
For the in vivo metastasis assay, 4 × 105 QBC939 cells with Myc overexpression or WDR5 knockdown or overexpression were injected into the caudal vein of each nude mouse in the presence or absence of HIF-1α inhibitor LW6 (10 mg/kg per os [p.o.]) or PX-478 (100 mg/kg intraperitoneally [i.p.]). The tumor metastases were monitored by a live imaging system (IVIS Spectrum). Radiant efficiency was measured to quantify the tumor burden of mice. Mouse weights were measured every week, and the weights of livers were measured to assess the actual tumor burden. The number of nodules on the livers and lungs was confirmed by H&E staining and counted. All animal experiments were approved by the Medical Ethics Committee of Shandong University.
CoIP
Total protein was isolated from cells using Nonidet P-40 (NP-40) lysis buffer (Beyotime) with 1% PMSF (Beyotime). The protein lysate was incubated with primary antibody at 4°C overnight. Protein A/G beads (Santa Cruz Biotechnology) were added and incubated at 4°C for 2 h. Immunocomplexes were collected by centrifugation (12,000 rpm, 1 min, 4°C) and washed with NP-40 lysis buffer for the next western blotting analysis.
Plasmid construction and site-directed mutagenesis
Full-length c-Myc was amplified by PCR and subcloned into pcDNA3.1. WBM mutation of c-Myc was introduced through PCR-mediated site-directed mutagenesis by simultaneously mutating I262/V264/V265 to glutamic acid. The sequences were verified via DNA sequencing.
Bioinformation analysis
The ChIP-seq datasets of Myc-WT, Myc-WBM, and WDR5 from the previous study were downloaded from the GEO database (GEO: GSE60897). HOMER software (Hypergeometric Optimization of Motif Enrichment) was used to annotate the three sets of ChIP-seq peaks.
RNA-seq datasets regarding Myc-WT and Myc-WBM Ramos cells were downloaded from the GEO database (GEO: GSE126207). Read counts were normalized, and the differential expression analysis of two groups was performed using DESeq2 of the R package.
RNA isolation and RNA-seq
Total mRNA of QBC939 cells was isolated using TRIzol reagent (Thermo Fisher Scientific). After undergoing quality control, three scramble and three shWDR5 samples were sequenced by Illumina HiSeq 4000 (LC-Bio Technologies). The sequence reads were mapped to the reference genome using HISAT2 software. The transcript abundances (read counts) were estimated with StringTie. Normalization and differential expression analysis of mRNAs was conducted using DESeq2 of the R package. The heatmap was created by the pheatmap R package.
ChIP
ChIP was conducted using an enzymatic ChIP kit (Cell Signaling Technology, #9003) according to the manufacturer’s instructions. Chromatin samples were incubated with anti-c-Myc (Santa Cruz Biotechnology, sc-40), anti-HDAC2 (Abcam, ab7029), and anti-H3K4Ac (Active Motif, #39381) antibodies. Mouse immunoglobulin G (IgG) (Santa Cruz Biotechnology, sc-2025) and rabbit IgG (Cell Signaling Technology, #2729) were used as negative controls. A non-immunoprecipitated sample (2%) was used as the input control. The purified DNA was then detected by qPCR. The primers used for ChIP-qPCR are listed in Table S4.
mRNA half-life determination
Cells were planted into six-well plates and treated with 10 μM Act D (MCE) to inhibit the activity of RNA polymerase II. Total RNA was extracted with TRIzol at 0, 1, 2, and 4 h after Act D addition. Quantitative real-time PCR was performed to measure the amount of mRNA, and mRNA half-life was calculated using linear regression.
Protein half-life determination
CHX (MedChemExpress) was a translational inhibitor that was generally used for protein half-life determination. Cells were cultured with 10 μg/mL CHX and collected at the indicated time points (0, 20, 40, and 60 min). Protein levels were measured by western blotting and quantified using ImageJ.
Chromatin accessibility
In chromatin accessibility analysis, nuclei were first treated with DNase I (Thermo Fisher Scientific) at 37°C. After 30 min, the reaction was halted by 50 mM EDTA. Genomic DNA was purified using phenol-chloroform extraction, and the products were used for qPCR. The fold change was analyzed using 2−ΔΔCt.
CUT&Tag assay
CUT&Tag-IT for Illumina was purchased from Vazyme (TD901). The assay was performed according to the protocol. Briefly, 1 × 105 QBC939 cells were washed twice with wash buffer and incubated with concanavalin A (Con A)-coated magnetic beads at room temperature for 15 min. Bead-bound cells were collected by a magnet stand and resuspended in 50 μL of antibody buffer. 1 μg of anti-H3K4ac antibody (Active Motif, #39381) was added and incubated at room temperature for 2 h with slow rotation. 1 μg of secondary antibody (goat anti-rabbit IgG H&L, Vazyme, Ab206) was diluted in 50 μL of digitonin (Dig) wash buffer, and the cells were incubated at room temperature for 50 min. The hyperactive pG-Tn5 transposon was diluted 1:100 in Dig-300 buffer and incubated with cells at room temperature for 1 h. Cells were then resuspended in tagmentation buffer for DNA fragmentation. After 1 h, EDTA, SDS, and Proteinase K were added to terminate tagmentation. DNA was then purified using phenol-chloroform-isoamyl alcohol extraction and ethanol precipitation.
Library amplification was also performed according to the protocol. 24 μL of DNA was mixed with 1 μL of Tris-acetate-EDTA (TAE), 10 μL of 5× TruePrep Amplify Buffer (TAB), 5 μL of double-distilled H2O (ddH2O), as well as 5 μL of i5 and i7 primers from TruePrep index kit v2 for Illumina (Vazyme, TD202). The PCR program was set according to the protocol. VAHTS DNA clean beads (Vazyme) were used for DNA purification. Libraries were sequenced on NovaSeq (Illumina, USA) at Novogene Science and Technology (Beijing, China), and 150-bp paired-end reads were generated. Fastp software was used to remove adaptor and low-quality reads. All reads produced by CUT&Tag of H3K4ac were aligned to the hg19 human genome using BWA (Burrows-Wheeler Aligner). MACS2 was used for peak calling. Genes with a peak within 3 kb of the transcription start site (TSS) were considered target genes. An area of 6 kb surrounding each TSS was selected to get CUT&Tag profiles of H3K4ac using deepTools software. Annotation of peaks was performed using ChIPseeker.
Statistical analysis
SPSS 17.0 and GraphPad Prism 5.0 software were used for statistical analysis and chart generation. The correlation between WDR5 and clinicopathologic features was assessed by a χ2 test. The survival curves were plotted using the Kaplan-Meier method, and the log-rank test was conducted to determine the statistical significance. The Harrell’s C-index, which measures the proportion of pairs for which the predicted and observed outcomes are concordant, was calculated using “survival” of the R package by RStudio software. The independent prognostic factors were analyzed in a Cox proportional hazards regression model. A paired t test was used for the comparison of two paired groups, and one-way or two-way ANOVA was performed to analyze the difference of multiple groups. p values <0.05 were considered statistically significant.
Acknowledgments
We thank Dr. Xiaoqing Yang for evaluating the IHC score of CCA. This work was supported by the National Natural Science Foundation of China (grant no. 82072676); the Shandong University Multidisciplinary Research and Innovation Team of Young Scholars (grant no. 2020QNQT002); the Shandong Province Major Research and Design Program (grant no. 2018GSF118169); the Natural Science Foundation of Shandong Province (grant no. ZR2019MH008); the Jinan City Science and Technology Development Program (grant nos. 201805017 and 201805013); the Clinical Research Innovation Fund Project (grant no. CXPJJH11800001-2018240); and by the Hengrui Hepatobiliary and Pancreatic Foundation (grant no. Y-2017-144).
Author contributions
T.C., K.L., R.S., and Z. Li carried out research. Y.X. designed all of the experiments. Z.Z., T.C., R.S., J.L., X.Z., G.R., and Z. Liu collected the specimens and performed the follow-up. Z.Z. collected the funding. Y.X. and T.C. participated in data analysis and interpretation. Y.X. and T.C. wrote the manuscript. All of the authors have read and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.02.017.
Contributor Information
Yunfei Xu, Email: xuyunfei1988@126.com.
Zongli Zhang, Email: zzlzzl1900@163.com.
Supplemental information
References
- 1.Rizvi S., Khan S.A., Hallemeier C.L., Kelley R.K., Gores G.J. Cholangiocarcinoma—evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018;15:95–111. doi: 10.1038/nrclinonc.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pitt H.A., Nakeeb A., Abrams R.A., Coleman J., Piantadosi S., Yeo C.J., Lillemore K.D., Cameron J.L. Perihilar cholangiocarcinoma. Postoperative radiotherapy does not improve survival. Ann. Surg. 1995;221:788–797. doi: 10.1097/00000658-199506000-00017. discussion 797–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu Y.F., Yang X.Q., Lu X.F., Guo S., Liu Y., Iqbal M., Ning S.L., Yang H., Suo N., Chen Y.X. Fibroblast growth factor receptor 4 promotes progression and correlates to poor prognosis in cholangiocarcinoma. Biochem. Biophys. Res. Commun. 2014;446:54–60. doi: 10.1016/j.bbrc.2014.02.050. [DOI] [PubMed] [Google Scholar]
- 4.Blechacz B. Cholangiocarcinoma: current knowledge and new developments. Gut Liver. 2017;11:13–26. doi: 10.5009/gnl15568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeOliveira M.L., Cunningham S.C., Cameron J.L., Kamangar F., Winter J.M., Lillemoe K.D., Choti M.A., Yeo C.J., Schulick R.D. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann. Surg. 2007;245:755–762. doi: 10.1097/01.sla.0000251366.62632.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu Y.F., Liu Z.L., Pan C., Yang X.Q., Ning S.L., Liu H.D., Guo S., Yu J.M., Zhang Z.L. HMGB1 correlates with angiogenesis and poor prognosis of perihilar cholangiocarcinoma via elevating VEGFR2 of vessel endothelium. Oncogene. 2019;38:868–880. doi: 10.1038/s41388-018-0485-8. [DOI] [PubMed] [Google Scholar]
- 7.Qiu B., Chen T., Sun R., Liu Z., Zhang X., Li Z., Xu Y., Zhang Z. Sprouty4 correlates with favorable prognosis in perihilar cholangiocarcinoma by blocking the FGFR-ERK signaling pathway and arresting the cell cycle. EBioMedicine. 2019;50:166–177. doi: 10.1016/j.ebiom.2019.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Razumilava N., Gores G.J. Cholangiocarcinoma. Lancet. 2014;383:2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montal R., Sia D., Montironi C., Leow W.Q., Esteban-Fabró R., Pinyol R., Torres-Martin M., Bassaganyas L., Moeini A., Peix J. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 2020;73:315–327. doi: 10.1016/j.jhep.2020.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stine Z.E., Walton Z.E., Altman B.J., Hsieh A.L., Dang C.V. MYC, metabolism, and cancer. Cancer Discov. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dang C.V., O’Donnell K.A., Zeller K.I., Nguyen T., Osthus R.C., Li F. The c-Myc target gene network. Semin. Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Dang C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013;3:a014217. doi: 10.1101/cshperspect.a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berasain C., Fernández-Barrena M.G., Avila M.A. New molecular interactions of c-Myc in cholangiocarcinoma may open new therapeutic opportunities. Hepatology. 2016;64:336–339. doi: 10.1002/hep.28607. [DOI] [PubMed] [Google Scholar]
- 14.Liu Z., Sun R., Zhang X., Qiu B., Chen T., Li Z., Xu Y., Zhang Z. Transcription factor 7 promotes the progression of perihilar cholangiocarcinoma by inducing the transcription of c-Myc and FOS-like antigen 1. EBioMedicine. 2019;45:181–191. doi: 10.1016/j.ebiom.2019.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guarnaccia A.D., Tansey W.P. Moonlighting with WDR5: a cellular multitasker. J. Clin. Med. 2018;7:21. doi: 10.3390/jcm7020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan X., Chen S., Wu J., Lin J., Pan C., Ying X., Pan Z., Qiu L., Liu R., Geng R., Huang W. PI3K/AKT-mediated upregulation of WDR5 promotes colorectal cancer metastasis by directly targeting ZNF407. Cell Death Dis. 2017;8:e2686. doi: 10.1038/cddis.2017.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas L.R., Wang Q., Grieb B.C., Phan J., Foshage A.M., Sun Q., Olejniczak E.T., Clark T., Dey S., Lorey S. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell. 2015;58:440–452. doi: 10.1016/j.molcel.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas L.R., Foshage A.M., Weissmiller A.M., Tansey W.P. The MYC-WDR5 nexus and cancer. Cancer Res. 2015;75:4012–4015. doi: 10.1158/0008-5472.CAN-15-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas L.R., Adams C.M., Wang J., Weissmiller A.M., Creighton J., Lorey S.L., Liu Q., Fesik S.W., Eischen C.M., Tansey W.P. Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad. Sci. USA. 2019;116:25260–25268. doi: 10.1073/pnas.1910391116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bannister A.J., Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goll M.G., Bestor T.H. Histone modification and replacement in chromatin activation. Genes Dev. 2002;16:1739–1742. doi: 10.1101/gad.1013902. [DOI] [PubMed] [Google Scholar]
- 22.Vilhais-Neto G.C., Fournier M., Plassat J.L., Sardiu M.E., Saraf A., Garnier J.M., Maruhashi M., Florens L., Washburn M.P., Pourquié O. The WHHERE coactivator complex is required for retinoic acid-dependent regulation of embryonic symmetry. Nat. Commun. 2017;8:728. doi: 10.1038/s41467-017-00593-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Z., He Y., Ju J., Rank G., Cerruti L., Ma C., Simpson R.J., Moritz R.L., Jane S.M., Zhao Q. The role of WDR5 in silencing human fetal globin gene expression. Haematologica. 2012;97:1632–1640. doi: 10.3324/haematol.2012.061937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu M.Z., Tsai Y.P., Yang M.H., Huang C.H., Chang S.Y., Chang C.C., Teng S.C., Wu K.J. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol. Cell. 2011;43:811–822. doi: 10.1016/j.molcel.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 25.Semenza G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang G.L., Jiang B.H., Rue E.A., Semenza G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kallio P.J., Okamoto K., O’Brien S., Carrero P., Makino Y., Tanaka H., Poellinger L. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1α. EMBO J. 1998;17:6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Appelhoff R.J., Tian Y.M., Raval R.R., Turley H., Harris A.L., Pugh C.W., Ratcliffe P.J., Gleadle J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 29.Kaelin W.G., Jr., Ratcliffe P.J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 30.Cockman M.E., Masson N., Mole D.R., Jaakkola P., Chang G.W., Clifford S.C., Maher E.R., Pugh C.W., Ratcliffe P.J., Maxwell P.H. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000;275:25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 31.Epstein A.C., Gleadle J.M., McNeill L.A., Hewitson K.S., O’Rourke J., Mole D.R., Mukherji M., Metzen E., Wilson M.I., Dhanda A. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 32.Schodel J., Bardella C., Sciesielski L.K., Brown J.M., Pugh C.W., Buckle V., Tomlinson I.P., Ratcliffe P.J., Mole D.R. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat. Genet. 2012;44:420–425. doi: 10.1038/ng.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma S., Lu C.C., Yang L.Y., Wang J.J., Wang B.S., Cai H.Q., Hao J.J., Xu X., Cai Y., Zhang Y., Wang M.R. ANXA2 promotes esophageal cancer progression by activating MYC-HIF1A-VEGF axis. J. Exp. Clin. Cancer Res. 2018;37:183. doi: 10.1186/s13046-018-0851-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun S., Ning X., Zhang Y., Lu Y., Nie Y., Han S., Liu L., Du R., Xia L., He L., Fan D. Hypoxia-inducible factor-1α induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 2009;75:1278–1287. doi: 10.1038/ki.2009.62. [DOI] [PubMed] [Google Scholar]
- 35.Semenza G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 36.Weiss A., Chavez-MacGregor M., Lichtensztajn D.Y., Yi M., Tadros A., Hortobagyi G.N., Giordano S.H., Hunt K.K., Mittendorf E.A. Validation study of the American Joint Committee on Cancer eighth edition prognostic stage compared with the anatomic stage in breast cancer. JAMA Oncol. 2018;4:203–209. doi: 10.1001/jamaoncol.2017.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Callegaro D., Miceli R., Bonvalot S., Ferguson P., Strauss D.C., Levy A., Griffin A., Hayes A.J., Stacchiotti S., Pechoux C.L. Development and external validation of two nomograms to predict overall survival and occurrence of distant metastases in adults after surgical resection of localised soft-tissue sarcomas of the extremities: a retrospective analysis. Lancet Oncol. 2016;17:671–680. doi: 10.1016/S1470-2045(16)00010-3. [DOI] [PubMed] [Google Scholar]
- 38.He W., Zhong G., Jiang N., Wang B., Fan X., Chen C., Chen X., Huang J., Lin T. Long noncoding RNA BLACAT2 promotes bladder cancer-associated lymphangiogenesis and lymphatic metastasis. J. Clin. Invest. 2018;128:861–875. doi: 10.1172/JCI96218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malek R., Gajula R.P., Williams R.D., Nghiem B., Simons B.W., Nugent K., Wang H., Taparra K., Lemtiri-Chlieh G., Yoon A.R. TWIST1-WDR5-Hottip regulates Hoxa9 chromatin to facilitate prostate cancer metastasis. Cancer Res. 2017;77:3181–3193. doi: 10.1158/0008-5472.CAN-16-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rankin E.B., Giaccia A.J. Hypoxic control of metastasis. Science. 2016;352:175–180. doi: 10.1126/science.aaf4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lai H.H., Li J.N., Wang M.Y., Huang H.Y., Croce C.M., Sun H.L., Lyu Y.J., Kang J.W., Chiu C.F., Hung M.C. HIF-1α promotes autophagic proteolysis of Dicer and enhances tumor metastasis. J. Clin. Invest. 2018;128:625–643. doi: 10.1172/JCI89212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morine Y., Shimada M., Utsunomiya T., Imura S., Ikemoto T., Mori H., Hanaoka J., Kanamoto M., Iwahashi S., Miyake H. Hypoxia inducible factor expression in intrahepatic cholangiocarcinoma. Hepatogastroenterology. 2011;58:1439–1444. doi: 10.5754/hge11156. [DOI] [PubMed] [Google Scholar]
- 43.Davis A.C., Wims M., Spotts G.D., Hann S.R., Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7:671–682. doi: 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- 44.Kaya-Okur H.S., Wu S.J., Codomo C.A., Pledger E.S., Bryson T.D., Henikoff J.G., Ahmad K., Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019;10:1930. doi: 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaya-Okur H.S., Janssens D.H., Henikoff J.G., Ahmad K., Henikoff S. Efficient low-cost chromatin profiling with CUT&Tag. Nat. Protoc. 2020;15:3264–3283. doi: 10.1038/s41596-020-0373-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oughtred R., Stark C., Breitkreutz B.J., Rust J., Boucher L., Chang C., Kolas N., O’Donnell L., Leung G., McAdam R. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019;47(D1):D529–D541. doi: 10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tansey W.P. Mammalian MYC proteins and cancer. New J. Sci. 2014;2014:757534. [Google Scholar]
- 48.Delmore J.E., Issa G.C., Lemieux M.E., Rahl P.B., Shi J., Jacobs H.M., Kastritis E., Gilpatrick T., Paranal R.M., Qi J. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dang C.V. Therapeutic targeting of Myc-reprogrammed cancer cell metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011;76:369–374. doi: 10.1101/sqb.2011.76.011296. [DOI] [PubMed] [Google Scholar]
- 50.Carugo A., Genovese G., Seth S., Nezi L., Rose J.L., Bossi D., Cicalese A., Shah P.K., Viale A., Pettazzoni P.F. In vivo functional platform targeting patient-derived xenografts identifies WDR5-Myc association as a critical determinant of pancreatic cancer. Cell Rep. 2016;16:133–147. doi: 10.1016/j.celrep.2016.05.063. [DOI] [PubMed] [Google Scholar]
- 51.Richart L., Carrillo-de Santa Pau E., Río-Machín A., de Andrés M.P., Cigudosa J.C., Lobo V.J.S., Real F.X. BPTF is required for c-MYC transcriptional activity and in vivo tumorigenesis. Nat. Commun. 2016;7:10153. doi: 10.1038/ncomms10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerlach J.M., Furrer M., Gallant M., Birkel D., Baluapuri A., Wolf E., Gallant P. PAF1 complex component Leo1 helps recruit Drosophila Myc to promoters. Proc. Natl. Acad. Sci. USA. 2017;114:E9224–E9232. doi: 10.1073/pnas.1705816114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chacón Simon S., Wang F., Thomas L.R., Phan J., Zhao B., Olejniczak E.T., Macdonald J.D., Shaw J.G., Schlund C., Payne W. Discovery of WD repeat-containing protein 5 (WDR5)-MYC inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2020;63:4315–4333. doi: 10.1021/acs.jmedchem.0c00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fallah J., Rini B.I. HIF inhibitors: status of current clinical development. Curr. Oncol. Rep. 2019;21:6. doi: 10.1007/s11912-019-0752-z. [DOI] [PubMed] [Google Scholar]
- 55.Dong Z.R., Sun D., Yang Y.F., Zhou W., Wu R., Wang X.W., Shi K., Yan Y.C., Yan L.J., Yao C.Y. TMPRSS4 drives angiogenesis in hepatocellular carcinoma by promoting HB-EGF expression and proteolytic cleavage. Hepatology. 2020;72:923–939. doi: 10.1002/hep.31076. [DOI] [PubMed] [Google Scholar]
- 56.Punzi S., Balestrieri C., D’Alesio C., Bossi D., Dellino G.I., Gatti E., Pruneri G., Criscitiello C., Lovati G., Meliksetyan M. WDR5 inhibition halts metastasis dissemination by repressing the mesenchymal phenotype of breast cancer cells. Breast Cancer Res. 2019;21:123. doi: 10.1186/s13058-019-1216-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dai B., Xiao Z., Zhu G., Mao B., Huang H., Guan F., Lin Z., Peng W., Liang X., Zhang B., Hu Z. WD repeat domain 5 promotes invasion, metastasis and tumor growth in glioma through up-regulated zinc finger e-box binding homeobox 1 expression. Cancer Manag. Res. 2020;12:3223–3235. doi: 10.2147/CMAR.S237582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Y.F., Liu H.D., Liu Z.L., Pan C., Yang X.Q., Ning S.L., Zhang Z.L., Guo S., Yu J.M. Sprouty2 suppresses progression and correlates to favourable prognosis of intrahepatic cholangiocarcinoma via antagonizing FGFR2 signalling. J. Cell. Mol. Med. 2018;22:5596–5606. doi: 10.1111/jcmm.13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun R., Liu Z., Qiu B., Chen T., Li Z., Zhang X., Xu Y., Zhang Z. Annexin10 promotes extrahepatic cholangiocarcinoma metastasis by facilitating EMT via PLA2G4A/PGE2/STAT3 pathway. EBioMedicine. 2019;47:142–155. doi: 10.1016/j.ebiom.2019.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.