

Abstract
The synthesis of diverse N-fused heterocycles, including the pyrido[1,2-a]indole scaffold, using an efficient pyrone remodeling strategy is described. The pyrido[1,2-a]indole core was demonstrated to be a versatile scaffold that can be site-selectively functionalized. The utility of this novel annulation strategy was showcased in a concise formal synthesis of three fascaplysin congeners.
The synthesis of diverse N-fused heterocycles, including the pyrido[1,2-a]indole scaffold, using an efficient pyrone remodeling strategy is described.
Introduction
The use of annulation reactions to construct complex structures remains a powerful strategy in chemical synthesis.1 For almost a century, 2-pyrones (A, Scheme 1a) have served as valuable heterocycles for annulations due to their versatile reactivity, which can be broadly categorized into two main paradigms: (1) pericyclic annulative processes and (2) regioselective opening via nucleophilic addition to unveil reactive intermediates poised for subsequent annulation. With respect to the first paradigm, pericyclic reactions, such as [4+2]-cycloadditions2 and 4π electrocyclizations,3 have been well documented to provide rapid access to bicycles such as B and C, which have been exploited in myriad ways.4,5 In contrast, there have been limited examples within the second paradigm. While nucleophilic 1,6-ring opening of 2-pyrones has proven to be a particularly effective strategy for orchestrating novel cyclization events via reactive intermediate D6 (our previous work6a,b), leveraging the dienolate functionality (E) accessible through 1,2-ring opening in annulation reactions remains underexplored.7
Scheme 1. Annulation strategies enabled by versatile reactivity of 2-pyrone derivatives.

We envisioned a strategy to N-fused bicycles in which a tethered reactive moiety (TRM) on 2-pyrone would engage an in situ generated dienolate (such as 1b) in an annulation reaction (Scheme 1b). The precursor N-heterocycle–pyrone adducts (e.g., 1) were anticipated to arise modularly by coupling N-heterocycle boronate esters and pyrones (e.g., 3-OTf pyrone)8via Suzuki coupling. The C2-borylated N-heterocycles were expected to arise directly from the precursor heterocycles by leveraging existing methods (e.g., C–H functionalization),9 thus enhancing the practicality of this approach. We hypothesized that opening 1 with a suitable nucleophile would first unveil dienolate 1a, which upon equilibration to 1b, would set the stage for annulation via direct capture of the aldehyde group by the TRM to provide N-fused heterocycle 2. Notably, varying the TRM would provide a general platform for diverse heterocycle synthesis.
To demonstrate the viability of this strategy, we initially focused on converting indole–pyrone adduct 3 to the pyrido[1,2-a]indole scaffold (3b, Scheme 2a)—a key structural motif present in a number of biologically active natural products including fascaplysin (4, Scheme 2b),10 goniomitine (5),11 and tronocarpine (6).12 While there exists numerous methods to access this biologically relevant scaffold,13–17 many of these tactics rely on reaction precursors with highly specific substitution patterns and, therefore, are unfortunately not general or modular. Specifically, we recognized that while heterocyclic–dienolate adducts (such as C3-substituted intermediate 3a) have proven to be effective precursors for benzannulation processes, strategies to install dienol/dienolate functionality at C2 of 1H-indoles lacking C3-substitution have remained elusive due to regioselectivity challenges.13b,18,19 Overall, we envisioned that our approach to coupling pyrone—a masked dienolate—to the C2-position of 1H-indole would provide a unique opportunity to address this longstanding regioselectivity challenge.
Scheme 2. Proposal to access pyrido[1,2-a]indole core.

Results and discussion
We commenced our investigations with indole–pyrone 7a (Table 1) and sodium methoxide as the nucleophile. Initially, we observed the formation of the desired pyrido[1,2-a]indole (8a) along with carbazole 9 and hemiaminal 10 as side products (entry 1). Changing the solvent from acetonitrile to 1,4-dioxane enhanced the formation of 9, which was generally more pronounced in relatively non-polar solvents.20 However, the use of polar solvents such as dimethylformamide resulted in complete decomposition of 7a (entry 3). The formation of hemiaminal 10 corroborates the proposed reaction mechanism illustrated in Scheme 1b and led us to investigate the use of polar protic solvents, such as methanol, to favor the conversion of 10 to 8a. We found, at this stage, that conducting the annulation in methanol furnished 8a in 45% yield (entry 4). Further investigation using co-solvents (entries 5–7) led to the identification of a dichloromethane/methanol solvent mixture as optimal, furnishing 8a in 61% yield (entry 7),21 presumably due to the increased solubility of 7a. Gratifyingly, the yield remained unaffected when the annulation was conducted both under open-flask conditions (entry 8) and on 1.3 g scale (entry 9). The structure of 8a was unambiguously confirmed by single-crystal X-ray analysis.
Reaction development and optimization.
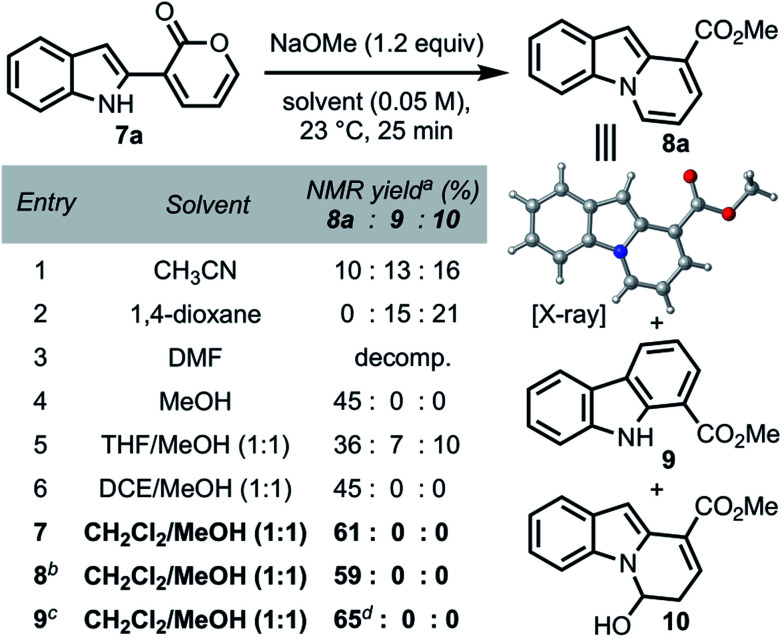
|
Determined by 1H NMR analysis using 1,2,3-trimethoxybenzene as an internal standard.
Open flask set-up under non-anhydrous solvent conditions.
Reaction conducted on 1.3 g scale.
Isolated yield.
With optimized conditions in hand, we investigated the scope of this operationally simple pyrido[1,2-a]indole synthesis (Scheme 3). Indole–pyrone substrates with varied substitution patterns were readily synthesized through Suzuki coupling of indole boronate esters9 with either 3-bromo-8a or 3-triflyloxy-2-pyrones.8b Indole substitution at both C3 and C7 had minimal influence on the ring-opening/annulation process, and the corresponding pyrido[1,2-a]indoles were isolated in comparable yields (8b–f, Scheme 3a). Interestingly, tetracyclic scaffolds such as lactam 8d and lactone 8e were accessed from indole–pyrones derived from tryptamine and tryptophol, respectively. Notably, 8d represents the core framework of tronocarpine (6). Next, we sought to investigate the tolerance of the overall transformation toward alterations of the electronics of the indole moiety. We observed that the presence of an electron-donating group, irrespective of the position, furnished the corresponding pyrido[1,2-a]indoles in high yields (8g–8i), whereas the product bearing an electron-withdrawing substituent (8j) was isolated in poor yield.22
Scheme 3. Scope of modular pyrido[1,2-a]indole synthesis. aIsolated both lactone and alcohol-ester precursor in a ratio of 2 : 1. bIsolated 8j along with the corresponding carbazole (29% yield). cOne-pot procedure: Suzuki coupling + ring-opening/annulation.

As shown in Scheme 3b, the established reaction conditions were also applicable to the efficient preparation of pyrido[1,2-a]indoles 8k–n bearing various substituents on the pyrone moiety. Unlike the electronic influence exerted by the substituents on the indole, C5-substitution on the pyrone moiety had little to no effect on the final reaction outcome with the sole exception being product 8k, which was isolated in diminished yield. Additionally, we investigated the effect of other alkoxide nucleophiles (Scheme 3c). With increasing basicity and sterics of the alkoxide, more forcing conditions were generally required, and the yield of the final products (8a, 8o–p) were also diminished.22
To further demonstrate the generality and versatility of our strategy, we next explored the synthesis of structurally diverse heterocyclic systems by subjecting various N-heterocyclic–pyrone adducts to the established reaction conditions (Scheme 4).23 Gratifyingly, upon coupling various TRMs, such as pyrrole, 7-aza-indole, pyrazole, and aniline moieties, to the C3 position of 2-pyrones, heterocycles such as indolizine 11, pyrido[3,2-b]indolizine 12, 3-aza-indolizine 13, and 1-naphthylamine 14 were isolated in moderate to high yields.
Scheme 4. Access to other novel heterocyclic cores. Conditions: NaOMe, CH2Cl2/MeOH, 23 or 55 °C, 10 min. aYield over two steps starting from SEM-protected 7-azaindole–pyrone substrate.

Each of the pyrone–heterocycle substrates described to this point contain a free N–H group, thus enabling cyclization directly from nitrogen to form a new N–C bond, with the sole exception being 1-naphthylamine 14.24 On the basis of the latter result and our initial hypothesis (Scheme 1b), we envisioned that employing N-protected substrates would direct the cyclization to the reactive carbon center, thus facilitating C–C bond formation25 and carbazole synthesis (Scheme 5). Interestingly, we found the annulation to be tolerant of various indole N-substituents, providing carbazoles 15a–c and 9 in high yields. Notably, unlike the pyrido[1,2-a]indole scope, the nature of the substituents—both on the indole and pyrone moieties—had little influence on the final reaction outcome, delivering the corresponding carbazoles (15d–g) in good yields.26
Scheme 5. Scope of modular carbazole synthesis. aSEM cleavage can also proceed in the same pot upon prolonged heating to furnish the free N–H carbazole 9.

We next sought to explore the subsequent reactivity of the C7-ester functionalized pyrido[1,2-a]indole products (Scheme 6). Friedel–Crafts acylation,27 copper-catalyzed carbenoid C–H insertion,28 Lewis acid-mediated epoxide opening/attendant lactonization,29 and chlorination30 all proceeded to provide the corresponding C10-functionalized pyrido[1,2-a]indoles 16–19. The structure of 18 and 19 were unambiguously confirmed by single-crystal X-ray analysis. Hydrogenation proceeded smoothly to furnish tetrahydro pyrido[1,2-a]indole 20. Treating 8a under Hartwig borylation conditions9,20 yielded boronate ester 21, resulting from borylation at the C7 position. Photo-mediated Heck coupling20,31 of 8a with iodobenzene gave biaryl compound 22, thus providing a platform to functionalize the C6 position as well, albeit at low conversion.32
Scheme 6. Derivatizations of pyrido[1,2-a]indoles. aSignificant portion of 8a (75%) remained unreacted.

With the generality of this strategy successfully established, we next turned our attention toward applying our pyrone remodeling strategy to access the fascaplysin family of natural products. As illustrated in Scheme 7, we began by hydrolyzing ester 8a to afford the intermediate carboxylic acid, which smoothly underwent Curtius rearrangement33 to furnish amine 23 in high yield.
Scheme 7. Formal synthesis of fascaplysin congeners.

Taking inspiration from methodology developed by Ackermann and co-workers,34 a palladium-catalyzed amination/C–H arylation domino coupling35 was employed to couple 23 and 1,2-dibromobenzene to furnish the pentacyclic core of the fascaplysin natural products (24), which possessed analytical data (1H and 13C NMR, HRMS, melting point, IR) in full agreement with those previously reported. The synthesis of 24 constitutes formal syntheses of fascaplysin (1) and homofascaplysins B and C (25 and 26), which can all be accessed independently in a single step from 24.36
Conclusions
In summary, we have developed a general, novel pyrone remodeling strategy, which capitalizes on the 1,2-ring opening of 2-pyrones, to construct diverse heterocyclic scaffolds. This transformation, which was initially validated through pyrido[1,2-a]indole synthesis, features a diverse substrate scope, with varied substitution patterns on both the indole and pyrone moieties. The scope was additionally extended to access carbazole cores and other N-fused heterocycles, thus, showcasing the generality of this strategy. The unusual reactivity of the pyrido[1,2-a]indole core was explored in several synthetic transformations, which enabled selective functionalization of three distinct carbon positions. Finally, the utility of this strategy was further demonstrated in a concise formal synthesis of three fascaplysin congeners. Studies to further expand the non-intuitive potential of 2-pyrone and its derivatives in the total synthesis of complex natural products are the focus of our current efforts.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
V. P. acknowledges TRDRP for a predoctoral fellowship. M. A. P. and K. E. G. thank the NSF for graduate research fellowships (DGE 1752814). Financial support for this research was provided to R. S. by the National Science Foundation (CHE-18566228). We thank Dr Hasan Celik and UC Berkeley's NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in CoC-NMR are supported in part by NIH S10OD024998. We are also grateful to Dr Nicholas Settineri (UC Berkeley) for single-crystal X-ray diffraction studies, and Dr Miao Zhang (UC Berkeley) for support with the acquisition of HRMS and IR data.
Electronic supplementary information (ESI) available. CCDC 2034052–2034054. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06317g
Notes and references
- (a) Wu L. Yu B. Li E.-Q. Recent advances in organocatalyst-mediated benzannulation reactions. Adv. Synth. Catal. 2020;362:4010. doi: 10.1002/adsc.202000608. [DOI] [Google Scholar]; (b) Li J. Ye Y. Zhang Y. Cycloaddition/annulation strategies for the construction of multisubstituted pyrrolidines and their applications in natural product synthesis. Org. Chem. Front. 2018;5:864. doi: 10.1039/C7QO01077J. [DOI] [Google Scholar]
- For selected examples of [4+2]-cycloadditions with 2-pyrones, see: ; (a) Cole C. J. F. Fuentes L. Snyder S. A. Asymmetric pyrone Diels–Alder reactions enabled by dienamine catalysis. Chem. Sci. 2020;11:2175. doi: 10.1039/C9SC05738B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liang X.-W. Zhao Y. Si X.-G. Xu M.-M. Tan J.-H. Zhang Z.-M. Zheng C.-G. Zheng C. Cai Q. Enantioselective synthesis of arene cis-dihydrodiols from 2-pyrones. Angew. Chem., Int. Ed. 2019;58:14562. doi: 10.1002/anie.201908284. [DOI] [PubMed] [Google Scholar]; (c) Wang Y. Li H. Wang Y.-Q. Liu Y. Foxman B. M. Deng L. Asymmetric Diels–Alder reactions of 2-pyrones with a bifunctional organic catalyst. J. Am. Chem. Soc. 2007;129:6364. doi: 10.1021/ja070859h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples of 4π electrocyclization of 2-pyrones, see: ; (a) Chapman O. L. McIntosh C. L. Pacansky J. Photochemical transformations. XLVIII. Cyclobutadiene. J. Am. Chem. Soc. 1973;95:614. doi: 10.1021/ja00783a066. [DOI] [Google Scholar]; (b) Pong R. G. S. Shirk J. S. Photochemistry of alpha-pyrone in solid argon. J. Am. Chem. Soc. 1973;95:248. doi: 10.1021/ja00782a049. [DOI] [Google Scholar]; (c) Pirkle W. H. McKendry L. H. Photochemical reactions of 2-pyrone and thermal reactions of the 2-pyrone photoproducts. J. Am. Chem. Soc. 1969;91:1179. doi: 10.1021/ja01033a025. [DOI] [Google Scholar]; (d) Corey E. J. Streith J. Internal photoaddition reactions of 2-pyrone and N-methyl-2-pyridone: a new synthetic approach to cyclobutadiene. J. Am. Chem. Soc. 1964;86:950. doi: 10.1021/ja01059a059. [DOI] [Google Scholar]
- Cai Q. The [4+2]-cycloaddition of 2-pyrone in total synthesis. Chin. J. Chem. 2019;37:946. doi: 10.1002/cjoc.201900048. [DOI] [Google Scholar]
- Coote S. C. 4-π-photocyclization: scope and synthetic applications. Eur. J. Org. Chem. 2020:1405. doi: 10.1002/ejoc.201901230. [DOI] [Google Scholar]
- For selected examples of annulation strategies involving 1,6-ring opening of 2-pyrones, see: ; (a) Palani V. Hugelshofer C. L. Sarpong R. A unified strategy for the enantiospecific total synthesis of delavatine A and formal synthesis of incarviatone A. J. Am. Chem. Soc. 2019;141:14421. doi: 10.1021/jacs.9b07693. [DOI] [PubMed] [Google Scholar]; (b) Palani V. Hugelshofer C. L. Kevlishvili I. Liu P. Sarpong R. A short synthesis of delavatine A unveils new insights into site-selective cross-coupling of 3,5-dibromo-2-pyrone. J. Am. Chem. Soc. 2019;141:2652. doi: 10.1021/jacs.8b13012. [DOI] [PubMed] [Google Scholar]; (c) Disadee W. Lekky A. Ruchirawat S. Metal-free, one-pot cascade annulation of 2-pyrones in water for the synthesis of peptidomimetics. J. Org. Chem. 2020;85:1802. doi: 10.1021/acs.joc.9b01856. [DOI] [PubMed] [Google Scholar]; (d) Maurya H. K. Vasudev P. G. Gupta A. A regioselective synthesis of 2,6-diarylpyridines. RSC Adv. 2013;3:12955. doi: 10.1039/C3RA41575A. [DOI] [Google Scholar]; (e) Usachev B. I. Usachev S. A. Röschenthaler G.-V. Sosnovskikh V. Y. A simple and convenient synthesis of 3-[5-(trifluoromethyl)-1,2,3-triazol-4-yl]cinnamic acids from 4-aryl-6-(trifluoromethyl)-2H-pyran-2-ones and sodium azide. Tetrahedron Lett. 2011;52:6723. doi: 10.1016/j.tetlet.2011.09.149. [DOI] [Google Scholar]; (f) Goel A. Verma D. Dixit M. Raghunandan R. Maulik P. R. Acetyltrimethylsilane: a novel reagent for the transformation of 2H-pyran-2-ones to unsymmetrical biaryls. J. Org. Chem. 2006;71:804. doi: 10.1021/jo052085y. [DOI] [PubMed] [Google Scholar]
- For known annulation strategies involving 1,2-ring opening of 2-pyrones, see: ; (a) Usachev S. A. Usachev B. I. Sosnovskikh V. Y. Synthesis of 6-hydroxy-5,6-dihydro-2-pyrones and pyridones by reaction of 4-aryl-6-trifluoromethyl-2-pyrones with water, hydrazine, and hydroxylamine. Chem. Heterocycl. Compd. 2017;53:1294. doi: 10.1007/s10593-018-2209-y. [DOI] [Google Scholar]; (b) Hansen C. A. Frost J. W. Deoxygenation of polyhydroxybenzenes: an alternate strategy for the benzene-free synthesis of aromatic chemicals. J. Am. Chem. Soc. 2002;124:5926. doi: 10.1021/ja0176346. [DOI] [PubMed] [Google Scholar]; (c) Tanyeli C. Tarhan O. Annulation reactions of 4-methoxy-2-pyrone with various active methyl compounds. Synth. Commun. 1989;19:2749. doi: 10.1080/00397918908053070. [DOI] [Google Scholar]
- For synthesis of 3,5-dibromo-2-pyrone and 3-triflyloxy-2-pyrone for cross-coupling, see: ; (a) Cho H.-K. Cho C.-G. Preparation of 3,5-dibromo-2-pyrone from coumalic acid. Org. Synth. 2015;92:148. doi: 10.15227/orgsyn.092.0148. [DOI] [Google Scholar]; (b) Frébault F. Oliveira M. T. Wöstefeld E. Maulide N. A concise access to 3-substituted 2-pyrones. J. Org. Chem. 2010;75:7962. doi: 10.1021/jo101843a. [DOI] [PubMed] [Google Scholar]
- The C–H borylation chemistry developed by Hartwig and co-workers can be employed to synthesize the N-heterocycle boronate ester precursors. For selected literature examples, see: ; (a) Larsen M. A. Hartwig J. F. Iridium-catalyzed C–H borylation of heteroarenes: scope, regioselectivity, application to late-stage functionalization, and mechanism. J. Am. Chem. Soc. 2014;136:4287. doi: 10.1021/ja412563e. [DOI] [PubMed] [Google Scholar]; (b) Ishiyama T. Nobuta Y. Hartwig J. F. Miyaura N. Room temperature borylation of arenes and heteroarenes using stoichiometric amounts of pinacolborane catalyzed by iridium complexes in an inert solvent. Chem. Commun. 2003:2924. doi: 10.1039/B311103B. [DOI] [PubMed] [Google Scholar]
- (a) Bharate S. B. Manda S. Mupparapu N. Battini N. Vishwakarma R. A. Chemistry and biology of fascaplysin, a potent marine-derived CDK-4 inhibitor. Mini-Rev. Med. Chem. 2012;12:650. doi: 10.2174/138955712800626719. [DOI] [PubMed] [Google Scholar]; (b) Segraves N. L. Robinson S. J. Garcia D. Said S. A. Fu X. Schmitz F. J. Pietraszkiewicz H. Valeriote F. A. Crews P. Comparison of fascaplysin and related alkaloids: a study of structures, cytotoxicities, and sources. J. Nat. Prod. 2004;67:783. doi: 10.1021/np049935+. [DOI] [PubMed] [Google Scholar]; (c) Segraves N. L. Lopez S. Johnson T. A. Said S. A. Fu X. Schmitz F. J. Pietraszkiewicz H. Valeriote F. A. Crews P. Structures and cytotoxicities of fascaplysin and related alkaloids from two marine phyla—Fascaplysinopsis sponges and Didemnum tunicates. Tetrahedron Lett. 2003;44:3471. doi: 10.1016/S0040-4039(03)00671-3. [DOI] [Google Scholar]
- (a) Bin H.-Y. Wang K. Yang D. Yang X.-H. Xie J.-H. Zhou Q.-L. Scalable enantioselective total synthesis of (−)-goniomitine. Angew. Chem., Int. Ed. 2019;58:1174. doi: 10.1002/anie.201812822. [DOI] [PubMed] [Google Scholar]; (b) Zhou S. Jia Y. Total synthesis of (−)-goniomitine. Org. Lett. 2014;16:3416. doi: 10.1021/ol501341b. [DOI] [PubMed] [Google Scholar]; (c) De Simone F. Gertsch J. Waser J. Catalytic selective cyclizations of aminocyclopropanes: formal synthesis of aspidospermidine and total synthesis of goniomitine. Angew. Chem., Int. Ed. 2010;49:5767. doi: 10.1002/anie.201001853. [DOI] [PubMed] [Google Scholar]; (d) Randriambola L. Quirion J.-C. Kan-Fan C. Husson H.-P. Structure of goniomitine, a new type of indole alkaloid. Tetrahedron Lett. 1987;28:2123. doi: 10.1016/S0040-4039(00)96059-3. [DOI] [Google Scholar]
- (a) Tan D.-X. Zhou J. Liu C.-Y. Han F.-S. Enantioselective total synthesis and absolute configuration assignment of (+)-tronocarpine enabled by an asymmetric Michael/aldol reaction. Angew. Chem., Int. Ed. 2020;59:3834. doi: 10.1002/anie.201914868. [DOI] [PubMed] [Google Scholar]; (b) Kam T.-S. Sim K.-M. Lim T.-M. Tronocarpine, a novel pentacyclic indole incorporating a seven-membered lactam moiety. Tetrahedron Lett. 2000;41:2733. doi: 10.1016/S0040-4039(00)00250-1. [DOI] [Google Scholar]
- For selected examples involving annulation strategy to access the pyrido[1,2-a]indole core, see: ; (a) Chuentragool P. Li Z. Randle K. Mahchi F. Ochir I. Assaf S. Gevorgyan V. General synthesis of pyrido[1,2-a]indoles via Pd-catalyzed cyclization of o-picolylbromoarenes. J. Organomet. Chem. 2018;867:273. doi: 10.1016/j.jorganchem.2018.03.003. [DOI] [Google Scholar]; (b) Dawande S. G. Lad B. S. Prajapati S. Katukojvala S. Rhodium-catalyzed pyridannulation of indoles with diazoenals: a direct approach to pyrido[1,2-a]indoles. Org. Biomol. Chem. 2016;14:5569. doi: 10.1039/C6OB00360E. [DOI] [PubMed] [Google Scholar]; (c) Jung Y. Kim I. Deformylative intramolecular hydroarylation: synthesis of benzo[e]pyrido[1,2-a]indoles. Org. Lett. 2015;17:4600. doi: 10.1021/acs.orglett.5b02331. [DOI] [PubMed] [Google Scholar]; (d) Karthikeyan I. Sekar G. Iron-catalyzed C–H bond functionalization for the exclusive synthesis of pyrido[1,2-a]indoles or triarylmethanols. Eur. J. Org. Chem. 2014:8055. doi: 10.1002/ejoc.201403233. [DOI] [Google Scholar]; (e) Sun L.-L. Liao Z.-Y. Tang R.-Y. Deng C.-L. Zhang X.-G. Palladium and copper cocatalyzed tandem N–H/C–H bond functionalization: synthesis of CF3-containing indolo- and pyrrolo[2,1-a]isoquinolines. J. Org. Chem. 2012;77:2850. doi: 10.1021/jo3000404. [DOI] [PubMed] [Google Scholar]; (f) Rogness D. C. Markina N. A. Waldo J. P. Larock R. C. Synthesis of pyrido[1,2-a]indole malonates and amines through aryne annulation. J. Org. Chem. 2012;77:2743. doi: 10.1021/jo2025543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples involving aza-Nazarov type cyclization to access the pyrido[1,2-a]indole core, see: ; (a) Karthikeyan I. Arunprasath D. Sekar G. An efficient synthesis of pyrido[1,2-a]indoles through aza-Nazarov type cyclization. Chem. Commun. 2015;51:1701. doi: 10.1039/C4CC08783F. [DOI] [PubMed] [Google Scholar]; (b) Naredla R. R. Zheng C. Lill S. O. N. Klumpp D. A. Charge delocalization and enhanced acidity in tricationic superelectrophiles. J. Am. Chem. Soc. 2011;133:13169. doi: 10.1021/ja2046364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For benzyne-mediated rearrangement to access the pyrido[1,2-a]indole core, see: ; Nikonov I. L. Kopchuk D. S. Kovalev I. S. Zyryanov G. V. Khasanov A. F. Slepukhin P. A. Rusinov V. L. Chupakhin O. N. Benzyne-mediated rearrangement of 3-(2-pyridyl)-1,2,4-triazines into 10-(1H-1,2,3-triazol-1-yl)pyrido[1,2-a]indoles. Tetrahedron Lett. 2013;54:6427. doi: 10.1016/j.tetlet.2013.09.042. [DOI] [Google Scholar]
- For cycloaddition strategy to access the pyrido[1,2-a]indole core, see: ; Beccalli E. M. Broggini G. Rosa C. L. Passarella D. Pilati T. Terraneo A. Zecchi G. Access to pyrrolo- and pyrido[1,2-a]indole derivatives by intramolecular nitrone cycloadditions. Effect of steric factors on the regioselective product formation. J. Org. Chem. 2000;65:8924. doi: 10.1021/jo000842g. [DOI] [PubMed] [Google Scholar]
- For multicomponent fragment coupling strategy to access the pyrido[1,2-a]indole core, see: ; (a) Zhu H. Stöckigt J. Yu Y. Zou H. “One-pot” multicomponent approach to indolizines and pyrido[1,2-a]indoles. Org. Lett. 2011;13:2792. doi: 10.1021/ol200883w. [DOI] [PubMed] [Google Scholar]; (b) Li T. Wang Z. Zhang M. Zhang H.-J. Wen T.-B. Rh/Cu-catalyzed multiple C–H, C–C, and C–N bon cleavage: facile synthesis of pyrido[2,1-a]indoles from 1-(pyridin-2-yl)-1H-indoles and γ-substituted propargyl alcohols. Chem. Commun. 2015;51:6777. doi: 10.1039/C5CC01412C. [DOI] [PubMed] [Google Scholar]
- Wu J.-Q. Yang Z. Zhang S.-S. Jiang C.-Y. Li Q. Huang Z.-S. Wang H. From indoles to carbazoles: tandem Cp*Rh(III)-catalyzed C–H activation/Brønsted acid-catalyzed cyclization reactions. ACS Catal. 2015;5:6453. doi: 10.1021/acscatal.5b01801. [DOI] [Google Scholar]
- Rathore K. S. Harode M. Katukojvala S. Regioselective π-extension of indoles with rhodium enalcarbenoids – synthesis of substituted carbazoles. Org. Biomol. Chem. 2014;12:8641. doi: 10.1039/C4OB01693A. [DOI] [PubMed] [Google Scholar]
- See the ESI† for detailed discussions
- Alternatively, pyrido[1,2-a]indole core can also be accessed from the Boc protected indole–pyrone precursor albeit in poor yield. See the ESI† for detailed experimental results
- Mechanistically, having an electron-withdrawing substituent on the indole moiety renders the free N–H of the precursor indole–pyrone more acidic, which upon exposure to sodium methoxide results in undesired deprotonation to yield the corresponding indole-1-ide, which is resistant toward the desired ring-opening/annulative process. For the same reason, increasing the basicity of the alkoxide source also has a negative effect on this overall transformation
- In general, N-heterocyclic–pyrone adducts with enhanced N–H acidity were more resistant toward the desired ring-opening/annulative process as mentioned in ref. 24. For instance, both the 7-aza-indole and pyrazole substrate required more forcing conditions to effect the desired transformation
- For the aniline substrate, the cyclization did not occur from the nitrogen center to provide the corresponding benzazepine core
- For the carbazole formation, addition of HCl was crucial to effect the C-addition to the unveiled aldehyde group
- As the precursors for carbazole synthesis lack a free N–H, the substituents on the indole fragment have little to no influence on the reaction outcome. This hypothesis supports the rationalization provided in ref. 22
- Ottoni O. Neder A. V. F. Dias A. K. B. Cruz R. P. A. Aquino L. B. Acylation of indole under Friedel–Crafts conditions – an improved method to obtain 3-acylindoles regioselectively. Org. Lett. 2001;3:1005. [PubMed] [Google Scholar]
- Maryanoff B. E. Reaction of dimethyl diazomalonate and ethyl-2-diazoacetoacetate with N-methylpyrrole. J. Org. Chem. 1982;47:3000. doi: 10.1021/jo00136a038. [DOI] [Google Scholar]
- Sueki S. Wang Z. Kuninobu Y. Manganese- and borane-mediated synthesis of isobenzofuranones from aromatic esters and oxiranes via C–H bond activation. Org. Lett. 2016;18:304. doi: 10.1021/acs.orglett.5b03474. [DOI] [PubMed] [Google Scholar]; . However, as reported in this reference, we did not observe the anticipated lactone formation
- Epstein W. W. Sweat F. W. Dimethyl sulfoxide oxidations. Chem. Rev. 1967;67:247. doi: 10.1021/cr60247a001. [DOI] [PubMed] [Google Scholar]
- (a) Chuentragool P. Kurandina D. Gevorgyan V. Catalysis with Palladium Complexes Photoexcited by Visible Light. Angew. Chem., Int. Ed. 2019;58:11586. doi: 10.1002/anie.201813523. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kurnadina D. Rivas M. Radzhabov M. Gevorgyan V. Heck Reaction of Electronically Diverse Tertiary Alkyl Halides. Org. Lett. 2018;20:357. doi: 10.1021/acs.orglett.7b03591. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Parasram M. Chuentragool P. Sarkar D. Gevorgyan V. Photoinduced Formation of Hybrid Aryl Pd-Radical Species Capable of 1,5-HAT: Selective Catalytic Oxidation of Silyl Ethers into Silyl Enol Ethers. J. Am. Chem. Soc. 2016;138:6340. doi: 10.1021/jacs.6b01628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Failed attempts to functionalize C6 include Lewis acid-mediated conjugate addition, nucleophilic radical addition, C–H insertion reactions, and [4+2]-cycloadditions
- Ghosh A. K. Sarkar A. Brindisi M. The Curtius rearrangement: mechanistic insight and recent applications in natural product syntheses. Org. Biomol. Chem. 2018;16:2006. doi: 10.1039/C8OB00138C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann L. Althammer A. Domino N–H/C–H bond activation: palladium-catalyzed synthesis of annulated heterocycles using dichloro(hetero)arenes. Angew. Chem., Int. Ed. 2007;46:1627. doi: 10.1002/anie.200603833. [DOI] [PubMed] [Google Scholar]
- After an extensive screening, a combination of Pd(OAc)2 and dppf in substoichiometric amounts have provided the best yields. See the ESI† for detailed optimization efforts
- For synthesis of fascaplysin congeners, see: ; (a) Waldmann H. Eberhardt L. Wittstein K. Kumar K. Silver catalyzed cascade synthesis of alkaloid ring systems: concise total synthesis of fascaplysin, homofascaplysin C and analogues. Chem. Commun. 2010;46:4622. doi: 10.1039/C001350A. [DOI] [PubMed] [Google Scholar]; (b) Gribble G. W. Pelcman B. Total syntheses of the marine sponge pigments fascaplysin and homofascaplysin B and C. J. Org. Chem. 1992;57:3636. doi: 10.1021/jo00039a024. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.