

Summary
Repair pathway “choice” at stalled mammalian replication forks is an important determinant of genome stability; however, the underlying mechanisms are poorly understood. FANCM encodes a multi-domain scaffolding and motor protein that interacts with several distinct repair protein complexes at stalled forks. Here we use defined mutations engineered within endogenous Fancm in mouse embryonic stem cells to study how Fancm regulates stalled fork repair. We find that distinct FANCM repair functions are enacted by molecularly separable scaffolding domains. These findings define FANCM as a key mediator of repair pathway choice at stalled replication forks and reveal its molecular mechanism. Notably, mutations that inactivate FANCM ATPase function disable all its repair functions and “trap” FANCM at stalled forks. We find that Brca1 hypomorphic mutants are synthetic lethal with Fancm null or Fancm ATPase-defective mutants. The ATPase function of FANCM may therefore represent a promising “druggable” target for therapy of BRCA1-linked cancer.
Keywords: FANCM, BRCA1, Fanconi anemia, Bloom’s syndrome helicase, genomic instability, homologous recombination, replication restart, break-induced replication, tandem duplication, synthetic lethality
Graphical Abstract
eTOC blurb
FANCM has both scaffolding and motor functions at stalled replication forks. Panday et al. use Fancm separation-of-function alleles to link specific FANCM scaffolding functions to distinct pathways of stalled fork repair, implicating FANCM in stalled fork repair pathway choice. FANCM-mediated ATP hydrolysis is required for all Fancm-dependent stalled fork repair functions. ATP hydrolysis-defective Fancm mutation is synthetic lethal with Brca1 mutation in mouse embryonic stem cells, identifying FANCM ATPase activity as a candidate therapeutic target in BRCA1-linked cancer.
Introduction
Genomic instability underlies many human diseases, ranging from rare hereditary developmental syndromes to common diseases such as cancer (Ciccia and Elledge, 2010; Taylor et al., 2019). A major threat to genome stability arises when replication forks stall at sites of DNA damage or abnormal DNA structure (Cortez, 2019; Zeman and Cimprich, 2014). Eukaryotic replication fork stalling activates S-phase checkpoint signaling, triggers fork remodeling and exposes the stalled fork to a range of possible repair outcomes (Neelsen and Lopes, 2015; Quinet et al., 2017; Rickman and Smogorzewska, 2019). Conservative repair of stalled forks requires the homologous recombination (HR) pathway, which mediates protective fork-remodeling in addition to canonical repair (Duxin and Walter, 2015; Schlacher et al., 2011; Scully et al., 2019). Alternative, error-prone repair pathways include aberrant DNA end joining and replication restart mechanisms (Adamo et al., 2010; Pace et al., 2010; Willis et al., 2017). The molecular basis of repair pathway “choice” at stalled mammalian forks remains poorly understood.
Stalled fork repair is defective in Fanconi anemia (FA)—a rare genetic disorder associated with childhood anemia and increased cancer incidence (Niraj et al., 2019). Cells from FA patients reveal spontaneous genomic instability and are hypersensitive to DNA interstrand crosslinking agents (ICLs), such as mitomycin C (MMC), as well as to endogenous aldehydes, which may also induce DNA-protein crosslinks (Hodskinson et al., 2020; Langevin et al., 2011; Rosado et al., 2011). Twenty-two distinct FA genes have been identified to date, defining a pathway that includes the hereditary breast and ovarian cancer (HBOC) predisposition genes, BRCA1 and BRCA2, as well as other general HR genes such as RAD51 (Niraj et al., 2019; Prakash et al., 2015). Conversely, a small proportion of HBOC risk is accounted for by monoallelic germline mutation of FA genes other than BRCA1 and BRCA2. These genes include Rad51C/FANCO and FANCM (Bogliolo et al., 2018; Castéra et al., 2018; Catucci et al., 2018; Figlioli et al., 2020; Neidhardt et al., 2017; Peterlongo et al., 2015).
ICL processing by the FA pathway is activated by bidirectional replication fork stalling at the ICL, followed by replisome disassembly and asymmetrical fork reversal, with recruitment of the FANCD2/FANCI heterodimer (Amunugama et al., 2018; Deans and West, 2011; Knipscheer et al., 2009; Long et al., 2011; Raschle et al., 2008). FANCD2/FANCI is monoubiquitinated by the FA “core” complex, comprised of FANC A, B, C, E, F, G and L—FANCL being the E3 ubiquitin ligase (Kim and D'Andrea, 2012; Meetei et al., 2003). Downstream targets include the SLX4 nuclease scaffold, which mediates dual incisions of one sister chromatid to generate an “unhooked” ICL on one sister and a two-ended DSB on the other (Hodskinson et al., 2014; Klein Douwel et al., 2014; Zhang and Walter, 2014).
Mammalian FANCM encodes a large scaffolding and motor protein that is part of a family of conserved DEAH-type DNA-dependent ATPases and dsDNA translocases related to the Archaeal Hef protein (Meetei et al., 2005; Nandi and Whitby, 2012; Prakash et al., 2005; Sun et al., 2008; Whitby, 2010; Xue et al., 2015). FANCM homologs mediate branch migration, D loop dissociation, replication fork reversal and 3’-5’ DNA helicase activity, the last of these being absent from the mammalian protein (Gari et al., 2008a; Gari et al., 2008b; Meetei et al., 2005; Sun et al., 2008; Whitby, 2010; Xue et al., 2015; Zheng et al., 2011). At the cellular level, FANCM homologs contribute to the non-crossover “synthesis-dependent strand annealing” (SDSA) pathway of HR (Paques and Haber, 1999; Prakash et al., 2009; Sun et al., 2008), suppressing both crossing over and sister chromatid exchange (SCE) (Bakker et al., 2009; Deans and West, 2009; Rosado et al., 2009). FANCM ATP hydrolysis mutants are defective for ICL resistance, suggesting that FANCM motor function contributes to stalled fork repair (Nandi and Whitby, 2012; Xue et al., 2008).
FANCM binds FAAP24 and two Major Histone Fold proteins MHF1 and MHF2 (Ciccia et al., 2007; Singh et al., 2010; Tao et al., 2012; Yan et al., 2010). This complex facilitates FANCM recruitment to stalled forks and ATR-dependent S phase checkpoint responses (Collis et al., 2008). The FANCM MM1 domain binds the FA core complex via direct interaction with FANCF, while its MM2 domain binds the Bloom's syndrome helicase (BLM)-TOP3A-RMI1-RMI2 (BTR) complex via direct interaction with RMI1 (Deans and West, 2009) (Figure 1A). Both FANCM ΔMM1 and ΔMM2 mutants were found to be defective for ICL-resistance and for SCE suppression (Deans and West, 2009). It is unknown how individual biochemical functions of FANCM are connected to specific pathways of stalled fork repair. All known FANCM mutations in HBOC are predicted to truncate and destabilize the gene product, the early truncating mutant FANCM:p.Arg658* conferring the highest risk of breast cancer (Figlioli et al., 2019).
Figure 1. FANCM is recruited to Tus/Ter-stalled mammalian replication forks.

See also Figure S1. A. Cartoon of FANCM protein and gene structure. DEAH: helicase domain. B. 6xTer-HR reporter and repair products of Tus/Ter-induced fork stalling. Grey boxes: mutant GFP alleles. Orange triangle: 6xTer array. Blue line: I-SceI restriction site. Ovals A and B: artificial 5’ and 3’ RFP exons. Red ovals: wild type RFP-coding exons. STGC/LTGC: short/long tract gene conversion. TD: tandem duplication. Red zig-zag: non-homologous TD breakpoint. C. FancmΔ85 allele, showing frame-shift product with premature stop codon (*). D. Left panel: Immunoblot of chromatin-extracted FANCM in Fancm+/+ and FancmΔ85/Δ clones. H3: Histone H3 loading control. Right panel: loss of FANCM band in siFANCM-treated samples. siLuc: control siRNA to Luciferase. *: background band. E. Immunoblot showing FANCD2 ubiquitination in Fancm+/+ and FancmΔ85/Δ clones in presence or absence of MMC. β-tubulin: loading control. F. Proliferative competition assay in MMC, measuring enrichment of GFP+ Fancm+/+ vs. GFP− FancmΔ85/Δ cells. Data shows mean ± standard deviation (SD), n=3. Here and all subsequent growth assays, data normalized to 0 μg/mL MMC. G. ChIP analysis of FANCM at Tus/Ter. Cartoon shows qPCR primer positions for ChIP (red half-arrows; GFP sequence not shown). Numbers indicate distance in bp from outer primer to nearest edge of 6xTer array. Orange triangles: Ter sites. Blue line: I-SceI restriction site. Lower panel: FANCM ChIP 24 hours after transfection with empty vector (EV; gold) or Tus-F140A (purple). Data here and in all subsequent ChIP figures shows mean of 2−ΔΔCT values, normalized to EV and β-Actin control locus (see STAR methods). Data shows mean ± SD. Analysis by one-way ANOVA (n=3). In this and all subsequent figures: *: P < 0.05: **: P < 0.01; ***: P < 0.001; ****: P < 0.0001; ns: not significant. H. ChIP analysis of FANCM spreading at Tus/Ter or at I-SceI-induced DSB. Data shows mean ± SD (n=3).
The Saccharomyces cerevisiae FANCM and BLM homologs, MPH1 and SGS1, delay the onset of break-induced replication (BIR) (Jain et al., 2016) and MPH1 mediates template-switching during the onset of BIR (Stafa et al., 2014), suggesting that FANCM and BLM can disassemble BIR intermediates in vivo. Mammalian FANCM also mediates “traverse” of an ICL by the replicative helicase (Huang et al., 2013; Huang et al., 2019). FANCM and BLM have both collaborative and distinct roles in the Alternative Lengthening of Telomeres (ALT) pathway, acting concertedly to suppress ALT initiation at stalled forks; BLM additionally supports ALTrelated telomere synthesis (Lu et al., 2019; Pan et al., 2017; Silva et al., 2019).
To study stalled fork repair in mammalian cells, we adapted the Escherichia coli Tus/Ter replication fork barrier (RFB) to induce site-specific, bidirectional replication fork stalling and HR at a defined chromosomal locus in mammalian cells (Willis et al., 2014). By placing an array of six 23 bp Ter sites within an HR reporter adjacent to a target site for the rare-cutting homing endonuclease I-SceI, we quantify stalled fork repair and, in parallel, conventional DSB repair. We identified three distinct pathways of stalled fork repair (Figure 1B): “short tract” gene conversion (STGC), “long tract” gene conversion (LTGC) and the formation of non-homologous tandem duplications (TDs) (Willis et al., 2017).
STGC is a conservative HR outcome, in which limited gene conversion between two mutant heteroalleles of GFP (encoding enhanced GFP) converts the cell to GFP+. Tus/Ter-induced STGC is a product of two-ended HR, implicating bidirectional fork stalling in its mechanism, and is mediated by the canonical BRCA-Rad51 HR pathway (Willis et al., 2014; Willis et al., 2017). In contrast to DSB-induced HR, which competes with the second major DSB repair pathway, classical non-homologous end joining (cNHEJ), Tus/Ter-induced STGC is unaffected by the status of cNHEJ (Scully et al., 2019; Willis et al., 2018).
LTGC is an error-prone replicative response, likely related to BIR in yeast (Chandramouly et al., 2013; Saini et al., 2013; Willis et al., 2015). LTGC, which accounts for a minority of Tus/Ter-induced HR events in wild type cells, results in an expansion of the repaired sister chromatid and converts the cell to GFP+RFP+ (Figure 1B). Unlike its DSB-induced counterpart, Tus/Ter-induced LTGC is Rad51-independent and is increased in the absence of BRCA1, suggesting a non-classical mechanism of initiation (Willis et al., 2014).
Non-homologous TDs of ~3-6 kb—scored as GFP−RFP+ products (Figure 1B)—can be observed during the stalled fork response but not during conventional DSB repair (Willis et al., 2017). Tus/Ter-induced TD formation is suppressed by BRCA1 and its binding partners BARD1 and CtIP, but is unaffected by BRCA2 or Rad51. Remarkably, the genomes of human breast and ovarian cancers lacking BRCA1, but not those lacking BRCA2, contain abundant small ~10 kb non-homologous TDs (termed “Group 1” TDs), which drive BRCA1-linked cancer primarily by disrupting tumor suppressor genes (Menghi et al., 2018; Menghi et al., 2016; Nik-Zainal et al., 2016). Thus, the Tus/Ter system recapitulates Group 1 TD formation in BRCA1-linked cancer. Tus/Ter-induced TDs arise by an aberrant replication fork restart/replication bypass mechanism and are completed by an end joining step (Willis et al., 2017). Notably, although depletion of either FANCM or BLM has little impact on TD frequencies in wild type cells, depletion of either protein in Brca1 mutants boosts Tus/Ter-induced TDs >10-fold (Willis et al., 2017).
Precisely how FANCM, BLM and BRCA1 interact in stalled fork repair, including in the suppression of Group 1 TDs, is unknown. Are the distinct stalled fork repair outcomes noted above reflections of the function or dysfunction of a single overarching activity, or are they independently regulated? In this study, we answer this question and reveal a key role for FANCM in repair pathway choice at stalled forks. We also identify how genetic interactions between Fancm and Brca1 impact repair pathway choice, and reveal unexpected synthetic lethal interactions between loss-of-function mutations of Fancm and Brca1.
Results
FANCM is recruited to Tus/Ter-stalled mammalian replication forks.
To study FANCM in stalled fork repair, we engineered endogenous Fancm mutations in a mouse embryonic stem (ES) cell line that carries a conditional allele of Brca1, described later (Willis et al., 2014; Willis et al., 2017; Xu et al., 1999), and contains a single copy of a 6xTer-HR reporter targeted to Rosa26 on chromosome 6 (Figure 1B) (Willis et al., 2014; Willis et al., 2017). We used CRISPR/Cas9 with dual sgRNA incisions to engineer an 85 bp deletion within exon 2, introducing a frame-shift early in the Fancm open reading frame (ORF). We derived multiple independent clones with potentially biallelic FancmΔ85 mutations, as well as multiple independent parallel Cas9/sgRNA-exposed clones that retained wild type Fancm—(here termed Fancm+/+ cells; Figures 1C, S1A and S1B).
CRISPR/Cas9-engineered FancmΔ85/Δ clones might contain one precise 85 bp deletion and one larger deletion affecting the second Fancm allele (Kosicki et al., 2018). We used whole genome sequencing to determine the genotype of FancmΔ85/Δ Ter-HR reporter clone #39 (Figure S1C). In addition to the expected Brca1 conditional genotype, we observed heterozygous deletions in Fancm—the planned 85bp deletion and a deletion of 2544 bp sharing its 5’ end with the Δ85 allele but extending into the 2nd intron. This deletion disrupts the Fancm ORF as effectively as the Δ85 bp deletion. Three independent FancmΔ85/Δ clones revealed markedly reduced Fancm mRNA levels compared to Fancm+/+ clones, suggestive of nonsense-mediated decay (Figure S1D). Full-length FANCM protein was undetectable in all three FancmΔ85/Δ clones (Figure 1D), as was FANCD2 monoubiquitination, even following treatment with MMC (Figure 1E). FancmΔ85/Δ cells revealed a mild cell cycle phenotype, with increase in the G2/M fraction (Figure S1E). To test the sensitivity of FancmΔ85/Δ cells to MMC, we exposed a mixed culture of ~90% unmarked (GFP−) FancmΔ85/Δ clone #39 cells and ~10% GFP+-marked wild type cells to titrated doses of MMC for 72 hours (Figure 1F). GFP+ cells were progressively enriched at higher doses of MMC, indicating a relative fitness advantage of the GFP+ wild type cells over the GFP− FancmΔ85/Δ cells. No equivalent enrichment was observed when ~10% GFP+ wild type cells were mixed with ~90% GFP− Fancm+/+ cells. Thus, FancmΔ85/Δ cells are sensitive to MMC, consistent with previous studies of FANCM mutants (Deans and West, 2009; Rosado et al., 2009).
We used chromatin-immunoprecipitation (ChIP) to determine whether FANCM accumulates at the Tus/Ter RFB. We observed strong enrichment of FANCM at the 6xTer array in Tustransfected Fancm+/+ cells but none in FancmΔ85/Δ cells (Figure 1G). To study FANCM distribution near Tus/Ter, we generated FancmΔ85/Δ and control Fancm+/+ derivatives of conditional Brca1 ES cells that contain a Rosa26-targeted 6x Ter and I-SceI construct that lacks GFP repeat sequences (Willis et al., 2018). Interestingly, the FANCM signals at both the Tus/Ter RFB and an I-SceI-induced DSB were restricted to <1 kb either side of the inducing lesion (Figure 1H). Thus, FANCM localizes specifically both to stalled fork structures and in close proximity to a site-specific DSB.
FANCM regulates three distinct pathways of stalled fork repair
To study how Fancm loss affects stalled fork repair, we measured Tus/Ter-induced repair in 3 independent FancmΔ85/Δ Ter-HR reporter clones (#39, 44 and 49) and 3 independent Fancm+/+ clones (#46, 47 and 48). We measured repair 72 hours following transfection with Tus, I-SceIexpression vectors or empty vector (EV; see STAR methods). All FancmΔ85/Δ clones revealed a ~4-fold reduction in Tus/Ter-induced STGC and a ~2-fold increase in LTGC, compared to Fancm+/+ controls; consequently, loss of Fancm skewed the proportion of LTGC:Total HR products at Tus/Ter from ~10% to ~70% (Figure 2A). Neither Fancm+/+ nor FancmΔ85/Δ cells revealed Tus/Ter-induced TDs (GFP−RFP+ products) in the absence of BRCA1 depletion. FancmΔ85/Δ cells revealed a modest reduction in I-SceI-induced STGC but no change in I-SceI-induced LTGC (Figure 2B). Thus, FancmΔ85/Δ cells reveal HR defects that are specific to stalled fork repair, as revealed by severe impairment of STGC and elevated LTGC frequencies.
Figure 2. FANCM regulates three distinct pathways of stalled fork repair.

See also Figure S2. A. Tus/Ter-induced repair in Fancm+/+ (white) clones vs. FancmΔ85/Δ (gray) clones. Data shows mean ± SEM. Analysis by one-way ANOVA (n=4). B. I-SceI-induced HR measured in same experiment. C. Representative raw FACS data (uncorrected for transfection efficiency) for Fancm+/+ and FancmΔ85/Δ cells co-transfected with empty vector (EV), I-SceI or Tus and siRNAs as shown. FACS plots pooled from n=4. Numbers show percentages. D. Tus/Ter-induced repair in Fancm+/+ clone #48 and FancmΔ85/Δ85 clone #39 co-transfected with Tus and siRNAs as shown (see STAR Methods). Data shows mean ± SEM. Analysis by Student’s t-test (n=4). E. I-SceI-induced repair measured in parallel in same experiment.
We used siRNA to deplete BRCA1 in FancmΔ85/Δ or Fancm+/+ Ter-HR reporter cells co-transfected with Tus, I-SceI or control empty vectors (see STAR methods and Figure S2A). siRNA to luciferase (siLUC) served as a control for siBRCA1. Figure 2C shows representative raw data, uncorrected for transfection efficiency. Depletion of BRCA1 in Fancm+/+ Ter-HR reporter cells produced the expected reduction in Tus/Ter-induced STGC and increase in LTGC (Figure 2D) (Willis et al., 2014). BRCA1 depletion did not further reduce Tus/Ter-induced STGC in FancmΔ85/Δ cells, but it further increased Tus/Ter-induced LTGC. FancmΔ85/Δ cells depleted of BRCA1 revealed strong induction of Tus/Ter-induced TDs (Figures 2C and 2D), recapitulating the previously noted relationship between FANCM and BRCA1 in TD suppression (Willis et al., 2017). Defects in I-SceI-induced STGC caused by loss of Fancm and BRCA1 depletion appeared to be additive, suggesting possible differences in the relationships between FANCM and BRCA1 in STGC regulation at stalled forks and at DSBs (Figure 2E). We noted no significant induction of I-SceI-induced GFP−RFP+ products in FancmΔ85/Δ cells depleted of BRCA1, confirming specificity of TD induction for the stalled fork response. Indeed, our unpublished work shows that I-SceI-induced GFP−RFP+ products are not TDs.
Fancm hemizygotes phenocopy wild type cells in stalled fork repair
To study the contribution of individual FANCM domains to stalled fork repair, we engineered in-frame mutations in endogenous Fancm in Brca1 conditional Ter-HR reporter cells. This method has the advantage of preserving physiological Fancm gene expression regulation. We first generated Brca1 conditional Ter-HR reporter cells that are strictly hemizygous for Fancm. We used CRISPR/Cas9 with dual sgRNA targeting of exons 2 and 23 to delete the entire 53.2 kb Fancm ORF (deleted allele termed Fancm−; Figures S2B and S2C). Sequencing of a selected Fancm+/− clone revealed no Cas9-induced indels at the sgRNA target sites of the wild type Fancm allele, and Fancm expression matched that of Fancm+/+ cells, implying transcriptional compensation for hemizygosity (Figures S2C and S2D). The Fancm+/− Brca1 conditional Ter-HR reporter clone revealed wild type repair frequencies (Figure S2E).
The FANCM-FA core complex interaction specifically mediates Tus/Ter-induced STGC.
The FANCM MM1 domain interacts with FANCF and is implicated in recruitment of the FA core complex to stalled forks (Deans and West, 2009). We used Cas9-dual sgRNA incisions to generate an in-frame 366 bp deletion of the MM1 coding region of exon 14 in Fancm+/− Ter-HR reporter cells (Figures 3A, S3A and S3B). FancmΔMM1/− cells showed a normal cell cycle distribution (Figure S3C). We studied four independent clones of FancmΔMM1/− (#13, 19, 47 and 77) and equivalently Cas9/sgRNA-exposed Fancm+/− cells (#01, 07, 14 and 16); each clone’s genotype was confirmed by sequencing. In FancmΔMM1/− clones, MM1-encoding mRNA was undetectable, whereas MM2-encoding mRNA was detected at normal levels, as was the FANCM protein (Figures 3A and 3B). However, we detected no FANCD2 monoubiquitination in FancmΔMM1/− cells, even following MMC treatment (Figure 3C). Consistent with a previous study, FancmΔMM1/− cells revealed increased sensitivity to MMC (Deans and West, 2009) (Figure 3D). ChIP revealed FANCM ΔMM1 at normal levels at Tus/Ter (Figure 3E). We detected robust Tus-dependent recruitment of FANCA and FANCL to Tus/Ter in Fancm+/− cells, but no recruitment in FancmΔMM1/− cells, while BLM recruitment was unaltered (Figure 3F). This data shows that the FANCM MM1 domain is required for stable interaction of the FA core complex with Tus/Ter-stalled forks, but is not required for recruitment of BLM or FANCM itself.
Figure 3. The FANCM-FA core complex interaction specifically mediates Tus/Ter-induced STGC.

See also Figure S3. A. FancmΔMM1 allele and RT qPCR analysis of MM1 and MM2 encoding mRNA in Fancm+/− and FancmΔMM1/− cells. Red half-arrows: RT qPCR primers. Here and in all subsequent expression analyses, data normalized to Gapdh mRNA using the 2−ΔCT method (see STAR methods). Data shows mean ± SD. Analysis by Student’s t-test (n=3). B. Immunoblot of chromatin-extracted FANCM in Fancm+/− and FancmΔMM1/− clones. *: non-specific band. C. Immunoblot showing FANCD2 ubiquitination in Fancm+/− and FancmΔMM1/− clones. D. Proliferative competition assay in MMC, measuring enrichment of GFP+ Fancm+/− vs. GFP− FancmΔMM1/− cells. Data shows mean ± SD (n=3). E and F. ChIP analysis of FANCM (E) and FANCA, FANCL and BLM (F) at Tus/Ter in Fancm+/− and FancmΔMM1/− cells. Data shows mean ± SD. Analysis by one-way ANOVA (n=3). G. Tus/Ter-induced repair in Fancm+/− (white) clones vs. FancmΔMM1/− (gray) clones. Data shows mean ± SEM. Analysis by one-way ANOVA (n=5). H. Tus/Ter-induced repair in Fancm+/− vs. FancmΔMM1/− clones co-transfected with Tus and siRNAs as shown. Data shows mean ± SEM. Analysis by Student’s t-test (n=6).
We analyzed stalled fork and DSB repair functions of each of the four FancmΔMM1/− and isogenic Fancm+/− clones. Tus/Ter-induced STGC was reduced in FancmΔMM1/− cells, albeit less markedly than in FancmΔ85/Δ cells (Figure 3G; compare with Figure 2A). Tus/Ter-induced LTGC was unaltered by deletion of MM1, producing a compensatory increase in the LTGC/Total HR ratio. No TD induction was noted. Depletion of BRCA1 in FancmΔMM1/− cells exacerbated the defect in Tus/Ter-induced STGC and elevated LTGC (Figure 3H). Therefore, BRCA1 controls Tus/Ter-induced HR independently of the FANCM MM1 domain. In contrast to FancmΔ85/Δ cells, FancmΔMM1/− cells revealed no induction of Tus/Ter-induced TDs following BRCA1 depletion (Figure 3H). We noted modest, proportionate reductions in I-SceI-induced STGC and LTGC in FancmΔMM1/− cells, but no other repair defects (Figures S3D and S3E).
Given the failure of FA core complex recruitment to stalled forks in FancmΔMM1/− cells, we asked whether FancmΔMM1 is epistatic with FA core complex in Tus/Ter-induced STGC. Indeed, depletion of either FANCA or FANCF diminished Tus/Ter-induced STGC in Fancm+/− cells to levels equivalent to those of FancmΔMM1/− cells, while having no impact on LTGC (Figures S3F and G). Importantly, in the same experiment, FANCA or FANCF depletion had no impact on STGC in FancmΔMM1/− cells. This result suggests that the STGC function of the FANCM MM1 domain is mediated by its interaction with the FA core complex. The lack of LTGC dysregulation in FancmΔMM1/− cells and the absence of Tus/Ter-induced TDs following BRCA1-depletion identify FancmΔMM1 as a separation-of-function allele that discriminates control of STGC from other FANCM-mediated stalled fork repair functions.
The FANCM-BLM interaction suppresses LTGC and TD formation at stalled forks
The FANCM MM2 domain interacts with the BTR complex via direct interactions with RMI1 (Figure 1A) (Deans and West, 2009). We used Cas9/sgRNA incisions to generate an in-frame 114 bp deletion of the MM2-encoding region of exon 14 in Fancm+/− Ter-HR reporter cells (Figures 4A, S4A and S4B). FancmΔMM2/− cells revealed only modest MMC sensitivity and no cell cycle phenotype (Figures S4C and S4D). We studied three independent clones of FancmΔMM2/− (#03, 06 and 09) and three equivalently Cas9/sgRNA-exposed Fancm+/− clones (#22, 26, 27); each clone’s genotype was confirmed by sequencing. The FANCM ΔMM2 protein was present at wild type levels and FANCD2 monoubiquitination was normal at basal levels but slightly blunted in FancmΔMM2/− cells following MMC (Figures 4B and 4C). ChIP revealed reduced recruitment of BLM to Tus/Ter in FancmΔMM2/− cells; in contrast, the BLM ChIP signal at Tus/Ter was abolished in FancmΔ85/Δ cells (Figure 4D). This shows that FANCM MM2 domain is required for efficient BLM interaction with stalled forks, but that additional FANCM elements also contribute to BLM recruitment. This data is consistent with previous reports (Deans and West, 2009; Ling et al., 2016). The recruitment of FANCM itself, FANCA and FANCL were all modestly reduced in FancmΔMM2/− cells or in cells depleted of BLM with siRNA (Figures 4E and S4E). Thus, FANCM and BLM recruitment to stalled forks is, at least in part, co-dependent, as was reported previously (Ling et al., 2016).
Figure 4. The FANCM-BLM interaction suppresses LTGC and TD formation at stalled forks.

See also Figure S4. A. FancmΔMM2 allele and RT qPCR analysis of MM1 and MM2 encoding mRNA in Fancm+/− and FancmΔMM2/−clones. Data shows mean ± SD. Analysis by Student’s t-test (n=3). B. Immunoblot of chromatin-extracted FANCM in Fancm+/− and FancmΔMM2/− clones. C. Immunoblot showing FANCD2 ubiquitination in Fancm+/− and FancmΔMM2/− cells. D and E. ChIP analysis of BLM (D), FANCM, FANCA, and FANCL (E) at Tus/Ter in Fancm+/− and FancmΔMM2/− cells. Analysis by one-way ANOVA (n=3). F. Tus/Ter-induced HR in Fancm+/− (white) clones vs. FancmΔMM2/− (gray) clones. Data shows mean ± SEM. Analysis by one-way ANOVA (n=4). G. Tus/Ter-induced repair in Fancm+/− vs. FancmΔMM2/− clones co-transfected with Tus and siRNAs as shown. Data shows mean ± SEM. Analysis by Student’s t-test (n=5).
FancmΔMM2/− cells revealed unaltered Tus/Ter-induced STGC and no Tus/Ter-induced TDs in the absence of BRCA1 depletion; however, LTGC was elevated ~1.5-fold (Figure 4F). Following depletion of BRCA1, Tus/Ter-induced STGC was reduced and LTGC further increased, indicating that BRCA1 functions independently of the FANCM MM2 domain in stalled fork HR. Notably, BRCA1 depletion boosted Tus/Ter-induced TDs in FancmΔMM2/− cells, albeit to frequencies lower than those observed in BRCA1-depleted FancmΔ85/Δ cells (Figure 4G; compare with Figure 2D). No significant alterations in I-SceI-induced HR were noted in FancmΔMM2/− cells (Figures S4F and S4G). These findings show that MM2 specifically suppresses Tus/Ter-induced LTGC and TD formation (the latter in cells lacking BRCA1), but has no role in STGC. Despite the lower levels of FA core recruitment in ChIP experiments, the FA pathway remains active in FancmΔMM2/− cells. Fancm1ΔMM2 is therefore a separation-of-function allele that discriminates suppression of LTGC and TD formation from STGC control in stalled fork repair.
FANCM ATP hydrolysis mutants are defective for FANCM-mediated stalled fork repair
To study the role of FANCM ATPase function in stalled fork repair, we used Cas9/sgRNA incisions to engineer an in-frame 66 bp deletion within the FANCM ATPase domain-encoding region of exon 2 of Fancm+/− Ter-HR reporter cells (Figures 5A, S5A and S5B). The resulting FancmΔDEAH product lacks 22 amino acid residues spanning the DEAH motif of the Walker B box and is predicted to be defective for ATP hydrolysis. We studied three independent clones of FancmΔDEAH/− (#16, 55 and 67) and three equivalently Cas9/sgRNA-exposed Fancm+/− clones (#12, 13 and 14); each clone’s genotype was confirmed by sequencing. The FANCM ΔDEAH protein was present at wild type levels and supported FANCD2 monoubiquitination, although the MMC response of FANCD2 was slightly blunted (Figures 5B and 5C). There was no cell cycle phenotype (Figure S5C). We detected normal levels of BLM at Tus/Ter (Figure 5D). However, the FANCM ΔDEAH mutant protein accumulated at Tus/Ter to higher levels than wild type FANCM (Figure 5E). FANCA and FANCL were recruited to Tus/Ter at normal levels (Figures 5F and 5G). Thus, the motor function of FANCM is dispensable for recruitment of the FA core complex and BLM to Tus/Ter-stalled forks, albeit with some alteration in stoichiometry of binding partners. In contrast, ATP hydrolysis by FANCM appears to be required for the timely release of FANCM from the stalled fork. Despite this evidence of FANCD2 activation and BLM recruitment to Tus/Ter, FancmΔDEAH/− cells were MMC sensitive (Figure 5H), raising the possibility that steps of the FA mechanism downstream of FANCD2 monoubiquitination are impaired in FANCM ATP hydrolysis mutants.
Figure 5. FANCM ATP hydrolysis mutants are defective for FANCM-mediated stalled fork repair.

See also Figures S5 and S6. A. FancmΔDEAH allele and RT qPCR analysis of MM2 and DEAH encoding mRNA in Fancm+/− and FancmΔDEAH/− clones. Data shows mean ± SD. Analysis by Student’s t-test (n=3). B. Immunoblot of chromatin-extracted FANCM in Fancm+/− and FancmΔDEAH/− clones. C. Immunoblot showing FANCD2 ubiquitination in Fancm+/− and FancmΔDEAH clones. D-G. ChIP analysis of BLM (D), FANCM (E), FANCA (F) and FANCL (G) at Tus/Ter in Fancm+/− and FancmΔDEAH/− cells. Data shows mean ± SD. Analysis by one-way ANOVA (n=3). H. Proliferative competition assay in presence of MMC, measuring enrichment of GFP+ Fancm+/− vs. GFP− FancmΔDEAH/−cells (n=3). Error bars: SD. I. Tus/Ter-induced HR in Fancm+/− (white) clones vs. FancmΔDEAH/− (gray) clones. Data shows mean ± SEM. Analysis by one-way ANOVA (n=5). J. Tus/Ter-induced repair in Fancm+/− vs. FancmΔDEAH/− clones co-transfected with Tus and siRNAs as shown. Data shows mean ± SEM. Analysis by Student’s t-test (n=5).
Notably, FancmΔDEAH/− clones revealed a ~3-fold reduction in Tus/Ter-induced STGC, a ~3-fold increase in LTGC (GFP+RFP+ products), and a corresponding skewing of the proportion of LTGC:Total HR products at Tus/Ter from ~10% in Fancm+/− cells to ~50% in FancmΔDEAH/− cells (Figure 5I). FancmΔDEAH/− cells revealed no Tus/Ter-induced TDs in the absence of BRCA1 depletion. BRCA1 depletion exacerbated the defect in Tus/Ter-induced STGC and increased LTGC in FancmΔDEAH/− cells, showing that BRCA1 acts independently of FANCM ATPase function in stalled fork repair (Figure 5J). Tus/Ter-induced TDs were elevated in FancmΔDEAH/− cells depleted of BRCA1, to levels reminiscent of FancmΔ85/Δ cells (compare Figures 5J and 2D). We noted a modest reduction in I-SceI-induced STGC, but no change in I-SceI-induced LTGC (Figures S5D and S5E). In summary, FancmΔDEAH/− cells phenocopy FancmΔ85/Δ cells functionally, with the exception that Tus/Ter-induced STGC is further diminished by BRCA1 depletion in FancmΔDEAH/− cells. This difference raises the possibility that a scaffolding function of FANCM contributes to BRCA1-mediated STGC at stalled forks. Thus, FancmΔDEAH/− cells reveal striking repair defects, despite retaining the ability to recruit the FA core complex and BLM to stalled forks.
Loss of 22 amino acids within the motor domain of the ΔDEAH mutant might have deleterious effects on protein folding. We therefore engineered a FancmD202A/− line, in which FANCM ATPase activity is ablated by the mutation of D202A (Figure S5F). This line phenocopied FancmΔDEAH/− cells biochemically and functionally, including the finding of an increased ChIP signal of FANCM D202A at Tus/Ter (Figures S5G-K). These results suggest that the phenotype of FancmΔDEAH/− cells is a specific product of its inability to hydrolyze ATP.
BLM can act independently of Fancm in stalled fork repair
The complex relationships between FANCM and BLM raise the question: to what extent are BLM stalled fork repair functions dependent on Fancm? To address this, we developed Blm+/− cells and targeted the 3’ end of the residual wild type Blm ORF with a dual auxin-inducible (Natsume et al., 2016; Nishimura et al., 2020) and SMASh (Chung et al., 2015) degron that also introduced an 8x HA epitope tag C-terminal to the AID domain but N-terminal to the SMASh protease target site, to generate Blmdeg/− reporter cells (Figure S6A and STAR methods). In cells transfected with Oryza sativa TIR1 F74A (Nishimura et al., 2020), activation of each degron alone depleted BLM (detected by anti-HA western blotting). Combined degron activation achieved the most effective depletion and markedly diminished BLM-HA recruitment to Tus/Ter (Figures S6A and S6B). We generated FancmΔ85/Δ and control Fancm+/+ derivatives of Blmdeg/− cells and used these to determine whether BLM can act independently of Fancm in stalled fork repair.
siRNA-mediated BLM depletion modestly elevates Tus/Ter-induced STGC and LTGC (Willis et al., 2017). Degron-mediated BLM depletion in either Fancm+/+ or FancmΔ85/Δ cells reproduced these effects (Figures S6C and S6D). Notably, degron activation triggered, or further increased, Tus/Ter-induced TD formation in BRCA1-depleted Blmdeg/− cells on both Fancm+/+ and FancmΔ85/Δ backgrounds. Similarly, siRNA-mediated BLM depletion (on a Blm wild type background) further increased TD frequencies in BRCA1-depleted FancmΔMM2/−, FancmΔ85/Δ and FancmΔDEAH/− cells, in comparison to controls with intact BLM levels (Figures S6E-G). The impact of BLM depletion on TD frequencies was approximately additive with the Fancm mutant TD phenotype. These results suggest that, although the bulk recruitment of BLM to stalled forks requires Fancm, BLM regulates stalled fork repair even in presumptively null FancmΔ85/Δ cells, where BLM is not detected at Tus/Ter by ChIP. These Fancm-independent repair functions of BLM may involve transient stalled fork-BLM interactions that are not readily detectable by ChIP.
Synthetic lethal interaction between Brca1 and Fancm mutations
To study relationships between loss-of function mutations in Fancm and Brca1, we sought to conditionally delete wild type Brca1 in Fancm mutant cells. The conditional allele, Brca1fl, contains loxP sites flanking a large in-frame central exon, commonly termed “exon 11” (although it is in fact the 10th exon of Brca1) (Miki et al., 1994; Willis et al., 2014; Willis et al., 2017; Xu et al., 1999). Cre-mediated deletion of exon 11 generates Brca1Δ, a hypomorphic allele (Figure 6A). Expression of the second Brca1 allele, Brca111, is disrupted by a hygromycin resistance gene in place of the 3’ half of exon 11. Brca1fl/11 ES cells and their Cre-treated Brca1Δ/11 derivatives grow at equivalent rates in vitro, but Brca1Δ/11 cells reveal HR defects and are defective for TD suppression (Willis et al., 2014; Willis et al., 2017).
Figure 6. Synthetic lethal interaction between Brca1 and Fancm mutations.

See also Figure S7. A. Left panel: wild type BRCA1 product of Brca1fl allele. Exon 11-encoded region shown. Cre converts Brca1fl to Brca1Δ, with in-frame deletion of exon 11. Right panel: Brca1 exons 10-12 in Brca1fl allele with PCR primers indicated (red half-arrows). Black triangles: loxP elements. B. Brca1fl/11 and Brca1Δ/11 PCR products using primers from panel A. C. RT qPCR analysis of Brca1 Exon 11-encoded mRNA in Brca1fl/11 and Brca1Δ/11 clones. Brca1 gene expression level was normalized to Gapdh using the 2−ΔCT method. Data shows mean ± SEM (n=3). D. Brca1fl/11 and Brca1Δ/11 allele recovery in unselected clones following Cre transduction of Brca1fl\11 cells carrying the Fancm genotypes shown.
We exposed cultures of Brca1fl/11 Ter-HR reporter ES cells of different Fancm genotypes to adenovirus encoding the Cre recombinase and screened clones for loss or retention of wild type Brca1 (Figures 6B-D; see STAR methods). In the first experiment, we retrieved 14/48 (30%) Cre-treated Fancm+/+ Brca1Δ/11 clones, the remainder being of the genotype Fancm+/+ Brca1fl/11. In parallel samples of FancmΔ85/Δ Brca1fl/11 cells exposed to the same titer of adeno-Cre in the same experiment, we retrieved only one FancmΔ85/Δ Brca1Δ/11 clone out of 48 tested, the remaining 47 clones being FancmΔ85/Δ Brca1fl/11 (Figure 6D). The solitary FancmΔ85/Δ Brca1Δ/11 clone (#68) was slow-growing compared to its FancmΔ85/Δ Brca1fl/11 siblings. We used whole genome sequencing to confirm its genotype (Figures S7A and S7B).
FancmΔ85/Δ Brca1Δ/11 clone #68 revealed levels of Tus/Ter-induced STGC and LTGC equivalent to an isogenic FancmΔ85/Δ Brca1fl/11 clone, suggesting that some compensatory changes had occurred in the clone (Figure S7C). In contrast, clone #68 revealed higher frequencies of Tus/Ter-induced TDs than its FancmΔ85/Δ Brca1fl/11 sibling, either in the presence or absence of BRCA1 co-depletion (Figure S7C). Clone #68 also revealed lower levels of I-SceI-induced STGC than the isogenic FancmΔ85/Δ Brca1fl/11 clone (Figure S7D).
We transduced each different Fancm genotype of Brca1fl/11 Ter-HR reporter ES cells with adeno-Cre, using appropriate isogenic control clones, and analyzed the resulting colonies. Brca1Δ/11 clones were recovered at normal frequencies in FancmΔMM1/− and FancmΔMM2/− cells (Figure 6D). In contrast, we recovered no FancmΔDEAH/− Brca1Δ/11 clones, despite retrieving 15/48 (31%) Brca1Δ/11 clones from parallel control Fancm+/− Brca1fl/11 cultures in the same experiment. Thus, a Fancm ATP hydrolysis-defective mutant is synthetic lethal with Brca1Δ/11 in mouse ES cells.
Discussion
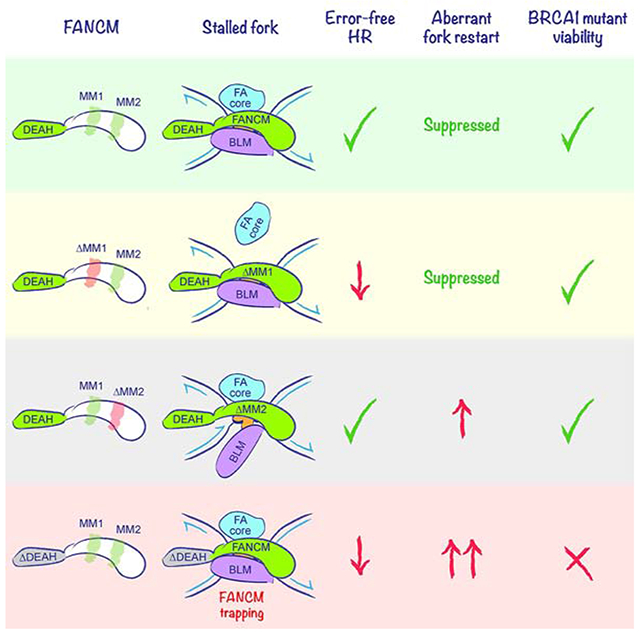
Work described here identifies separation-of-function mutants of Fancm in mammalian stalled fork repair and identifies a synthetic lethal interaction between mutations of Fancm and Brca1. We show that FANCM positively regulates error-free STGC in response to a Tus/Ter RFB, while suppressing error-prone replicative responses of LTGC and TD formation. Prior to this study, the regulatory relationships between error-free and error-prone repair at stalled forks were unclear. Our identification of separation-of-function Fancm mutants shows that STGC support and LTGC/TD suppression at stalled forks are regulated in a mutually independent fashion, and that FANCM critically determines the balance of flux through these pathways. Underpinning all FANCM-mediated repair is a requirement for its motor protein function, as revealed by the phenotype of the ATPase-defective FancmΔDEAH and FancmD202A mutants. Notably, the FANCM ΔDEAH and D202A proteins accumulates to unusually high levels at stalled forks, implicating ATP hydrolysis in the timely displacement of FANCM from the stalled fork. “Trapping” of the FANCM ATPase-defective mutant may be an important corollary of FancmΔDEAH mutant repair defects. Finally, we identify synthetic lethal interactions between Fancm null or ATPase mutants and Brca1 mutation. Collectively, these findings identify the FANCM ATPase as a potentially “druggable” target for therapy of BRCA1-linked cancers.
FANCM in STGC at stalled forks: the Fanconi anemia pathway
The interaction of FANCM MM1 domain with the FA core complex specifically supports error-free HR (i.e., STGC) at stalled forks, while having no role in regulating the aberrant replicative stalled fork repair responses of LTGC and TD formation. These findings draw an explicit connection between Tus/Ter-induced STGC and the FA pathway of replication-coupled ICL repair (Figure 7A). Notably, the FA pathway is required for cellular viability in the presence of aldehydes, where the major fork-stalling lesions may include DNA-protein crosslinks (Langevin et al., 2011; Rosado et al., 2011). Perhaps Tus/Ter will serve as a useful model of how the FA pathway processes such lesions. Similar to the FA pathway of ICL repair, Tus/Ter-induced STGC entails conservative two-ended HR, implicating bidirectional fork arrest as an intermediate (Knipscheer et al., 2009; Raschle et al., 2008; Willis et al., 2014; Zhang and Walter, 2014). The incision step of replication-coupled ICL repair is mediated by the DNA structurespecific endonuclease-binding scaffold SLX4/FANCP (Hodskinson et al., 2014; Klein Douwel et al., 2014). If similar FA-mediated incisions normally occur at Tus/Ter, they might be impaired in FANCM ΔMM1 mutants, thereby disabling STGC (Figure 7B). It will be interesting to determine the role of SLX4 and other FA genes in Tus/Ter-induced repair.
Figure 7. Mechanisms of FANCM in stalled fork repair.

A. FANCM mediates error-free HR (i.e., STGC) at forks bidirectionally arrested at Tus/Ter (orange triangles). FANCM recruits FA core complex, promoting FANCD2 ubiquitination (D2-Ub) and SLX4-mediated incisions (red triangles). FANCM motor function (green dashed arrows) promotes fork remodeling and timely release of FANCM from stalled fork. B. Upper panel: FANCM ΔMM1 mutant retains motor functions but is defective for FA core complex recruitment and FANCD2 ubiquitination, resulting in reduced STGC and increased MMC sensitivity. Lower panel: FANCM ΔDEAH mutant retains FA core complex recruitment and FANCD2 ubiquitination. Defective fork remodeling, possibly combined with FANCM trapping, results in reduced STGC and increased MMC sensitivity. C. FANCM and BLM act in a concerted fashion to suppress aberrant replication fork restart. Hypothetical mechanism of D-loop formation at stalled fork in the absence of a strand exchange step. BLM can dissolve post-replicative double Holliday junction (dHJ). Alternative processing by HJ resolution (red triangles) generates D-loop at stalled fork, with accompanying sister chromatid exchange. Dashed light blue lines: resected nascent lagging strands at stalled fork. FANCM/BLM-mediated branch migration (green and purple dashed arrow) dissociates D-loop, preventing aberrant fork restart. D. Defects in FANCM/BLM interaction allow D-loop to persist, favoring resumption of nascent leading strand synthesis (red half-arrow) with displacement of Tus from Ter (empty orange triangles). Engagement of unknown helicase(s) (red dashed arrow) extends aberrant replication restart by bubble migration. This mechanism is BIR-like but, as shown in C, might not be break-induced. Leftward normal fork duplicates genomic segment ‘a’ bounded, at one end, by the site of fork stalling and, at the other end, by the site of displacement of the restarted leading strand (red). BRCA1 loss impairs DNA end resection, preventing collapse of TD back to single copy by single strand annealing. By default, tandem duplication forms by end joining.
In addition to the MM1 domain, we find that ATP hydrolysis by FANCM is required for efficient Tus/Ter-induced STGC. Notably, FANCM ΔDEAH and D202A mutants uncouple the correlation between FANCD2 monoubiquitination competence and MMC resistance that is evident in other Fancm mutants. This finding suggests that ATP hydrolysis by FANCM is required for the FA mechanism “downstream” of FANCD2 monoubiquitination—potentially, by supporting a fork remodeling/reversal step (Amunugama et al., 2018; Gari et al., 2008a; Gari et al., 2008b) (Figure 7A). In addition, our finding that ATPase-defective FANCM mutants accumulate to abnormally high levels at Tus/Ter suggests that timely release of FANCM may be important for STGC. Conversely, “trapping” of ATPase-defective FANCM at the stalled fork might interfere with the STGC mechanism (Figure 7B).
We find that BLM modestly suppresses Tus/Ter-induced STGC and, thus, opposes FANCM in this function. Further, the FANCM ΔMM2 mutant, which is impaired in the ability to interact with BLM, exhibits no defects in STGC. These observations suggest that FANCM regulates STGC independently of BLM (Figure 7A).
FANCM in LTGC and TD suppression: aberrant replication restart
FancmΔMM2 mutants reveal increased Tus/Ter-induced LTGC and TD formation (the latter in the absence of BRCA1). Thus, in contrast to its mechanism of action in STGC, FANCM must interact with the BTR complex for optimal suppression of LTGC and TDs. The mild MMC sensitivity of FancmΔMM2/− cells suggests that the primary impact of dysregulated LTGC and TD suppression in mouse ES cells may be on mutagenesis/structural variation, rather than on cell viability in response to MMC. In contrast, in HEK 293 cells, the FANCM ΔMM2 mutant revealed MMC sensitivity similar to that of the ΔMM1 mutant, suggesting that the impact of MM2 on MMC sensitivity varies between different cell types (Deans and West, 2009).
In our ChIP studies, we found that the bulk recruitment of BLM to Tus/Ter is dependent on Fancm. However, even in FancmΔ85/Δ cells, which are likely Fancm null and reveal no FANCM or BLM ChIP signal at Tus/Ter, degron- or siRNA-mediated BLM depletion nonetheless influenced stalled fork repair, increasing the frequencies of LTGC and TD formation in BRCA1-depleted cells. The simplest explanation of this apparent contradiction is that BLM can access stalled fork intermediates in FancmΔ85/Δ cells, but is present at levels below the limits of detection by ChIP.
In the experiments reported here, the ability of a Fancm mutant to suppress Tus/Ter-induced LTGC predicted its ability to suppress TDs. This suggests that Tus/Ter-induced LTGC and TD formation—both Rad51-independent processes—may arise by a common mechanism (Willis et al., 2014; Willis et al., 2017). Tus/Ter-induced TDs are products of aberrant replication fork restart (Willis et al., 2017). In this regard, replication fork restart in S. pombe occurs by a BIR-like mechanism (Jalan et al., 2019; Nguyen et al., 2015). Taken together, these observations suggest that FANCM/BLM may suppress Tus/Ter-induced LTGC and TD formation by disallowing BIR-type replication restart at Tus/Ter (Figure 7C). A critical step in this restart mechanism would be formation of a stable D-loop at the site of stalling. Given the Rad51-independence of the restart mechanism, it will be relevant to determine the role of RAD52 in this process. However, in principle, a D-loop could be established at a stalled fork without the need for a strand exchange or annealing step, through the processing of post-replicative Holliday junctions in the vicinity of the fork (Figure 7C). Both FANCM and BLM mediate branch migration of Holliday junctions in vitro, and the BTR complex additionally mediates dissolution of double Holliday junctions (Gari et al., 2008b; Karow et al., 2000; Willis et al., 2014; Wu and Hickson, 2003; Xue et al., 2015). Thus, BLM and, possibly, FANCM might act on post-replicative HJs to reduce the likelihood of their conversion to D-loops at stalled forks. Indeed, the Bloom’s homolog SGS1 plays a role in post-replication repair at a Tus/Ter RFB in S. cerevisiae (Larsen et al., 2017).
Whatever its mechanism of formation, the established D-loop would also be a target for direct dissociation by FANCM/BLM (Bachrati et al., 2006; Gari et al., 2008a; Sun et al., 2008; Xue et al., 2015) (Figure 7C). In the context of FANCM/BLM dysfunction, failure to dissociate D-loops in a timely fashion might permit resumption of DNA synthesis and the establishment of a BIR-like bubble migration mechanism (Figure 7D). This model is supported by in vivo studies in yeast: ScMPH1 (the FANCM homolog) promotes template switching in preference to processive BIR; in addition, ScMPH1 and SGS1 (the BLM homolog) confer a delay in the onset of BIR (Jain et al., 2016; Stafa et al., 2014).
The dramatic synergistic induction of TDs in cells lacking both FANCM and BRCA1 suggests that the two proteins act at different steps to suppress TD formation. If FANCM acts at the stalled fork to disallow BIR-type fork restart, BRCA1 may act upon a later step to suppress TD formation. We have suggested that BRCA1 suppresses TDs by promoting collapse of the nascent TD back to single copy status, through BRCA1’s role as a mediator of single strand annealing (SSA) (Scully et al., 2019). In the absence of BRCA1, the failure of DNA end resection and SSA permit the TD to form by end joining (Figure 7D).
Phenotype of ATPase-defective FANCM: synthetic lethality with Brca1 mutation
We find that the ATPase (motor) function of FANCM is essential for all aspects of FANCM-mediated repair. This might reflect a role for a FANCM-mediated fork remodeling in these repair pathways. An additional clue came from our ChIP analysis of FANCM at the Tus/Ter RFB. Strikingly, the FANCM ΔDEAH and D202A proteins accumulate at Tus/Ter to levels ~2-fold higher than the wild type protein. Since the cellular abundance of FANCM ΔDEAH and FANCM D202A proteins were equivalent to wild type FANCM, the increased signal at Tus/Ter suggests that the residence time of ATPase-defective FANCM at the stalled fork is abnormally prolonged. ATP hydrolysis by FANCM may therefore contribute to the timely displacement of FANCM from the stalled fork. Conversely, a defect in ATP hydrolysis might “trap” FANCM at the stalled fork, with potential for neomorphic or antimorphic functions. Interestingly, small molecule inhibitors of poly(ADP ribose) polymerase (PARP), which are used in the treatment of BRCA-linked cancers, are thought to act, in part, by trapping PARP1 at sites of DNA damage (Murai et al., 2012).
Unexpectedly, we observed synthetic lethality between certain Fancm mutations and Brca1 mutation. FancmΔ85/Δ (presumptively Fancm null) and FancmΔDEAH cells were synthetic lethal with Brca1 exon 11 deletion, while FancmΔMM1 and FancmΔMM2 cells were viable on this Brca1 mutant background. These findings show that synthetic lethal interactions between BRCA1 and FANCM are not restricted to ALT cells (Pan et al., 2017). It will be important to identify the mechanisms underlying synthetic lethality. Thus far, we have not observed homozygous FANCM loss in BRCA1-linked breast or ovarian cancer. However, homozygous FANCM mutations are rare in cancer and it may be necessary to explore larger datasets to reach a conclusion about the relationship between FANCM and BRCA1 mutations in cancer. If loss of FANCM ATPase function were lethal in BRCA1 mutant cancers—as it is in Brca1 mutant ES cells—this interaction could be exploited for cancer therapy, since the ATPase function of FANCM should, in principle, be “druggable”. Small molecule FANCM ATPase inhibitors might selectively kill BRCA1 mutant cancer cells, while leaving surrounding BRCA1+/− cells intact.
Limitations
We have restricted our study to the genetically tractable mouse ES cell system. The extent to which our findings are generalizable to other cell types and other organisms is an important open question. Although our work explicitly connects Tus/Ter-induced STGC to the FA pathway, this hypothesis should be tested further by studying additional FA genes, such as FANCD2 and SLX4. Another limitation is evident in the model of fork restart in FANCM/BLM-defective cells (Figure 7D). Although we invoke a BIR-like bubble migration mechanism, current evidence in support of this mechanism is indirect.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents and resources should be directed and will be fulfilled by the Lead Contact, Ralph Scully (rscully@bidmc.harvard.edu).
Materials Availability
Plasmids and cell lines generated in the study are available upon request from the lead contact.
Data and code availability
Unprocessed blots and gels are available at: https://data.mendeley.com/datasets/gwjs4kp9mg/draft?a=8c165752-21e7-4b4d-a17a-8426b21a9ca0
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
mES cells used in this study were cultured under humidified conditions at 37°C 5% CO2 in DMEM supplemented with 15% fetal bovine Serum (FBS), recombinant LIF, and additional factors as described previously (Willis and Scully, 2021). Mouse embryonic fibroblasts were grown in DMEM supplemented with 10% FBS and feeders prepared as described previously (Willis and Scully, 2021). Plasmid, Cas9/sgRNA RNP, siRNA, and ultramer oligo transfections were performed using Lipofectamine 2000 (ThermoFisher Scientific, 11668019).
METHOD DETAILS
Molecular biology, siRNA and sgRNA oligos
C-terminal HA tagged Tus-F140 expression plasmid (pcDNA3β-MYC-NLS-Tus-F140A-3xHA) was derived from the parental vector (Willis et al., 2014; Willis et al., 2017) and 6xTer HR reporters were engineered by conventional cloning methods as previously described (Willis et al., 2014; Willis et al., 2017). All plasmids used for transfection were prepared by endotoxin-free maxiprep (Qiagen, 12362). The Blm exon22 targeting construct was generated specifically using large synthetic double-strand DNA Gblocks (Integrated DNA Technologies). All primers used for genotyping, RT-qPCR, sgRNA synthesis, and chromatin immunoprecipitation qPCR were purchased from ThermoFisher Scientific. siRNA SMARTpools were purchased from Horizon Scientific/Dharmacon. Cas9-sgRNA target sites in mouse genome sequence were identified using the Heidelberg CCtop tool: https://cctop.cos.uni-heidelberg.de. sgRNAencoding DNA oligos for in vitro transcription were purchased from ThermoFisher Scientific. sgRNA in vitro synthesis was performed using the Engen sgRNA Synthesis Kit (New England Biolabs, E3322S) and RNA product purified using the Clean and Concentrate Kit (Zymo Research, R1017). sgRNA product quality was assessed by denaturing gel electrophoresis using Novex 10% TBE-Urea (ThermoFisher Scientific, EC6875BOX).
Cell culture and the generation of mouse Fancm mutant cell lines
Fancm mutant cell lines were derived from a conditional Brca1fl/exon11 founder cell line carrying one Brca1 allele in which much of exon 11 is partially replaced by the hygromycin resistance cassette, and one functionally wild type Brca1 allele in which exon 11 is flanked by loxp elements and may be excised Cre recombinase. The founder cell line contains a single copy of the 6X Ter-HR reporter cassette, targeted to the rosa26 locus and verified by southern blot (Chandramouly et al., 2013; Willis et al., 2014; Willis et al., 2017). mES cells are routinely thawed onto plates coated with mouse embryonic fibroblast (MEF) feeders, maintained in ES medium on gelatinized plates, and regularly tested for mycoplasma infection by Myco-Alert assay (Lonza, LT07-318). Fancm mutant cell lines were generated using CRISPR/Cas9 mediated mutation of the Fancm locus. Cas9-sgRNA RNP was pre-assembled in vitro in OptiMEM by mixing Spy NLS Cas9 (New England Biolabs, M0646T) and purified sgRNA. Cells were co-transfected with 0.45 μg Cas9(1.1) expression plasmid and 0.05 μg of each of two purified sgRNA using Lipofectamine 2000 (ThermoFisher Scientific, 11668019). After 72 hr, transfected cells were plated onto 6-cm dishes containing feeder MEFs without selection. Individual clones were picked for expansion between 9 and 14 days later and Fancm mutant clones were identified by PCR and confirmed by sequencing.
Generation of mouse Fancm hemizygous and in-frame mutant cell lines
Fancm domain and ATPase mutants were derived from a hemizygous (Fancm+/−) clone. To generate Fancm+/− clones in Brca1fl/exon11 6X Ter-HR reporter cells, Fancm exons 2-23 were deleted by co-transfection with 30 nM preassembled Cas9-sgRNA RNP. RNP was preassembled in vitro in OptiMEM mixing Spy NLS Cas9 and purified sgRNA. Fancm+/− clones which retained one wild type allele were identified by PCR and confirmed by PCR product sequencing. To generate MM1 (FancmΔMM1/−), MM2 (FancmΔMM2/−), DEAH (FancmΔDEAH/−), the Fancm+/− mutant cell line was subjected to a second round of Cas9-sgRNA RNP-mediated targeted gene modification. Clonal candidates harboring the expected in-frame domain deletion were identified by PCR and confirmed by sequencing. For sgRNA targeted DNA sequences including PAM targeting Fancm and primers used for PCR screening and sequencing, see Supplemental Table S1.
Generation of mouse Fancm ATPase/translocase point mutant D202A cell line
The D202A point mutation was generated by using a single-strand oligodeoxynucleotide (ssODN) targeted to exon 2 using Cas9-sgRNA RNP. The ssODN donor (Integrated DNA Technologies) used encodes an aspartate to alanine substitution at amino acid position 202 with silent mutations to the sgRNA targeted PAM sequence to prevent Cas9/sgRNA RNP cutting of the ssODN flanked by 100 bp homology. The targeting sgRNA within exon 2 supports expected Cas9 nuclease incisions 20 bp from the mutation site. To generate this mutant, Brca1fl/exon11 founder line cells were co-transfected with 30 nM preassembled Cas9-sgRNA RNP supplemented with 100ng ssODN using Lipofectamine 2000. After 72 hr, transfected cells were plated onto 6-cm dishes containing feeder MEFs without selection. Individual clones were picked for screening between 9 and 14 days later. Clones harboring the FancmD202A/+ point mutation were identified by PCR and confirmed by sequencing. Subsequently, the remaining wildtype allele was mutated to generate FancmD202A/Δ85 using as described for generating the Δ85 allele. FancmD202A/Δ85 clones were identified by PCR and confirmed by sequencing.
Generation of mouse Blm hemizygous, degron tagged mutant cell lines
Cas9-sgRNA mediated Blm locus deletion was accomplished by Lipofectamine 2000-mediated transfection: 1.6 x 105 cells were co-transfected in suspension with 30nM RNP preassembled in OptiMEM using Spy NLS Cas9 and purified in vitro transcribed sgRNA. Three days later, four technical duplicates containing a total of 2M cells were pooled and plated on MEF feeder coated 6cm dishes without selection and individual clones picked one week later. Lines retaining wild type or mutated sequence were identified by conventional PCR and sequenced. Subsequently, BLM C-terminal tagging was accomplished by RNP co-transfection: 1.6 x 105 Blm−/+ cells were co-transfected as above with the addition of 90 ng Blm exon22 targeting vector complementary to the BLM C-terminus encoding sequence containing elements encoding an 8xHA-SMASh-AiD-neomycin resistance cassette. Four hours after transfection, four technical duplicates containing a total of 800,000 cells were pooled and plated onto neomycinresistant MEF feeder coated 10cm dishes. 24 hours after transfection, media was supplemented with 800 μg/mL G418 and emergent individual clones picked one week later. Cell lines harboring Blm-targeted cassette sequence were identified by PCR and the region of interest and integrated cassette verified by sequencing. For sgRNA targeted DNA sequences including PAM targeting Blm locus (expected 80,122 bp deletion), Blm 5’ locus breaksite, Blm 3’ locus breaksite, and Blm exon22 for C-terminal targeting, and primers used for PCR screening and sequencing, see Table S2.
Degron-tagged BLM depletion for Immunoblotting
5 μM 5-adamantyl-indoleacetic acid (5-IAA) (TCI Chemicals, A3390) and 1μM Asunaprevir (MedChemExpress, BMS-650032) stocks were each prepared in DMSO and kept frozen until use. For Western blotting, 24 technical transfection replicates of 2.0 x 105 cells were performed in suspension with 0.4 μg empty vector and 0.1 μg Tir1-F74A expression plasmid. Cells were treated with 5 nM 5-IAA and 1nM Asunaprevir beginning 6 hours after transfection and protein extracted 18 hours after addition of drug(s). The chromatin fraction from transfected cell lysates was prepared as described (Gillotin et al., 2018); see also “Isolation of Chromatin-bound Proteins for FANCM and BLM Immunoblotting” section below. Protein was resolved by 4-12 % Bis-Tris SDS-PAGE (ThermoFisher Scientific, NW04122BOX) and analyzed by immunoblotting using an antibody against HA tag (Abcam, ab9110, 1:1000).
Recombination assays
For measurement of HR repair frequencies, 1.6 × 105 cells were co-transfected in suspension with 0.35 μg empty vector, pcDNA3β -myc NLS-I-SceI, or pcDNA3β -myc NLS-Tus and 20 pmol ON Targetplus siRNA SMARTpool using Lipofectamine 2000 (ThermoFisher Scientific, 11668019). For functional assays involving the degron-tagged Blm−/+ derivatives, cells were co-transfected with 0.35 μg empty vector, Tus, or I-SceI expression plasmids, 0.1 μg Tir1-F74A expression plasmid, and 10 pmol siRNA. Cells were treated with 5 nM 5-IAA and 1nM Asunaprevir beginning 6 hours after transfection with replenishment with fresh drug 24 hours after transfection. For all functional assays, GFP+RFP−, GFP+RFP+ and GFP−RFP+ frequencies were measured in duplicate 72 hr after transfection by using flow cytometry (Beckman Coulter CytoFlex LX). The total number of events that were scored for each sample was 3X105–6X105. Repair frequencies showed are corrected for background events and for transfection efficiency (55–85%). To measure transfection efficiency, parallel transfection was done with 0.05 μg wild-type GFP expression vector, 0.30 μg control vector and 20 pmol siRNA. To deplete two gene targets, 10 pmol of each siRNA was used, while single depletion controls received 10 pmol of the target siRNA and 10 pmol of control luciferase siRNA. Data presented represent mean and error bars represent the s.e.m. of between 3 (n=3) and 8 (n=8) independent experiments (n values given in figure legends).
Competition assays
To assess the mitomycin C (Sigma-Aldrich, M4287-2mg) sensitivity of Fancm mutant clones, competition experiments were performed. 1.6 x 105 cells were co-transfected in suspension with 0.45 μ9 empty vector and either 50 ng empty vector for each Fancm mutant clone or 50 ng GFP-expression plasmid for the parental wild type clone, using Lipofectamine 2000. 18 hours after transfection, cells were counted, and each Fancm mutant clone mixed and plates 1:5 with the GFP-marked parental wild type clone. 6 hours after cell plating, growth medium was replaced with media containing titrated doses of mitomycin C. Two days later, GFP+ frequencies were scored on a Beckman Coulter CytoFlex LX. Fold enrichment of cultures transiently co-transfected with GFP-expression plasmid normalized to 0 μg/mL mitomycin C control. Plots represent the mean of triplicate samples from three independent experiments (n=3).
Isolation of Chromatin-bound Proteins for FANCM and BLM Immunoblotting
Freshly harvested cell pellets were washed with 1x PBS and lysed in 5 volumes of chilled A1 buffer (50 mM HEPES,140 mM NaCl,1 mMEDTA,10% glycerol,0.5% NP-40, 0.25% TritonX-100,1 mM DTT, and 1X protease inhibitor). Each lysate was centrifuged at 1,100 x g at 4 °C for 2 min and the supernatant was discarded. Pellets were resuspended by gentle pipetting in A1 buffer and samples were incubated for 10 minutes on ice. Each suspension was centrifuged at 1,100 x g at 4 °C for 2 min and the supernatant again discarded. Pellets were resuspended by gentle pipetting in 2 volumes of ice cold E2 buffer (10 mM Tris-HCl pH-8, 200 mM NaCl, 1 mM EDTA and 0.5 mM EGTA, and 1X protease inhibitor). Each suspension was centrifuged at 1,100 x g at 4 °C for 2 min and the supernatant discarded. Pellets were resuspended by gentle pipetting with A1 buffer and incubated for 10 minutes on ice. Each suspension was centrifuged at 1,100 x g at 4 °C for 2 min and the supernatant discarded. Pellets were resuspended by gentle pipetting in 2 volumes of ice cold E3 buffer (500 mM Tris-HCl pH-6.8, 500 mM NaCl and 1x protease inhibitor. Each sample was sonicated in a water bath using Diagenode Bioruptor 300 with attached 4°C chiller cycling 30 sec on and 30 sec off on high setting for 5 min. Sonicated samples were centrifuged at 16,000xg for 15 min at 4°C. The supernatant corresponding to the chromatin fraction was transferred and subjected for western blot analysis using following antibodies; FANCM (Abcam, ab95014, 1:1000), Histone H3 (Abcam, ab1791,1:1000).
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation was performed as described previously (Willis et al., 2018). Transfection of 2.0 x 105 cells containing a single copy of a minimal cassette lacking redundant sequence harboring a single GFP allele interrupted by 6xTer-I-SceI elements targeted to the Rosa26 locus were performed with 0.5 μg empty vector, pcDNA3β-MYC NLS-I-SceI or pcDNA3β-MYC NLS-Tus-F140A-3xHA using Lipofectamine 2000. 10 million cells were collected 24 hours after transfection and fixed in 1% formaldehyde supplemented serum free media. Fixation was quenched by addition of glycine to 125 mM. Cells were lysed in lysis buffer (0.1% SDS, 20 mM EDTA, 50 mM Tris pH 8.1) containing 1x protease inhibitor (Sigma-Aldrich). Chromatin shearing to ~500 bp was accomplished using a Diagenode Bioruptor 300 with attached 4°C chiller for 20 cycles, 15 seconds on and 30 seconds off set to medium setting. To avoid non-specific binding to protein A/G beads, 100 μl lysates for each ChIP sample were precleared by the addition of 10 μl activated Magna ChIP magnetic beads (Millipore Sigma, 16-663) in ChIP dilution buffer (1% Triton-X-100, 2 mM EDTA, 150 mM NaCI, 20 mM Tris pH 8.1) and incubation for 1 hr at 4°C with gentle mixing. After removal of beads by magnet, for each immunoprecipitation, 3 μl of anti-FANCM (Abcam, ab95014), 3 μl of anti-FANCA (Abcam, ab97578), 3 μl of anti FANCL (Abcam, ab94458), or 5 μl of anti- Blm (Bethyl Labs, A300-110A) was added and mixed for 12 hours at 4°C followed by addition of 10 μl activated Magna ChIP beads and mixing for 16 hours at 4°C. Beads were washed six times in ice-cold ChIP RIPA buffer (50 mM HEPES pH 7.6, 1 mM EDTA, 7 mg/mL sodium deoxycholate, 1% NP-40) followed by two washes in ice-cold TE (10 mM Tris pH8.0, 1 mM EDTA). Crosslinks were reversed and DNA was eluted by incubation in 100 uL Elution buffer (1% SDS, 200 mM sodium bicarbonate, 5.6 μg/mL RNAse A) overnight at 65°C. Protein was removed by proteinase K digest for 30 min at 55°C. Released DNA was purified by PCR Purification column (Qiagen, 28106) and analyzed by qPCR on an ABI Prism 7300 or QuantStudio3 using SYBR Green (Applied Biosystems, 4368702). For primers used for qPCR see Table S3. Data are presented as the mean calculated from three independent experiments (n = 3) normalized against untreated controls (empty vector) and control locus (beta-actin) using the 2−ΔΔCT method (Schmittgen and Livak, 2008).
RT qPCR analysis
RNA extraction was performed using RNeasy Mini Kit (Qiagen, 74106). All analyses of Gapdh and genes of interest were performed using an Applied Biosystems 7300 Real time PCR System or QuantStudio 3 using Power SYBR Green RNA-to CT ™ 1-Step Kit (Applied Biosystems, 4389986). For primer sequences for RT qPCR see Table S4. mRNA was extracted and measured in biological triplicates. Target gene expression level was normalized to Gapdh using the 2−ΔCT method (Schmittgen and Livak, 2008).
Cell Cycle analysis
Mouse ES cells were seeded on 6-well plates until 70% confluency. Cells were incubated with 10 μM BrdU for 15 min and immediately disaggregated to single cells by trypsinization and quenching with standard ES cell media. Cells were washed with chilled 1x PBS pH 7.0 and pelleted by centrifugation at 1000rpm for 5 mins. The cell pellet was gently resuspended in 50μl 1x PBS and suspended, clump-free cells fixed by the dropwise addition of 5mL chilled 70% ethanol with gentle vortexing. Fixed cells are stored at 4°C in the dark until further processed.
For BrdU immunostaining, ethanol was removed and cells subjected to DNA denaturation by addition of 0.5 mL of 2 M HCl, 0.5 % Triton-X-100 solution with gentle vortexing, Cells were incubated under denaturing conditions for 30 mins at RT with intermittent vortexing every 10 mins. Cells were pelleted, denaturant aspirated and cells neutralized in 1 mL 0.1 M Sodium Borate Decahydrate. Cells were immediately pelleted and resuspended in 50-100μl of 1x PBS 1% BSA. BrdU was stained by incubation with anti-BrdU antibody (Abcam, ab8039, 1:100) at RT for 30 mins in the dark in 1x PBS 1% BSA 0.5% tween-20. Cells were washed with 5 mL chilled 1x PBS 20 mM HEPES pH 7.4, 0.5 % tween-20 and resuspended in 50-100μl of 1x PBS 1% BSA 0.5% tween-20 containing the secondary FITC-conjugated rabbit anti-mouse antibody (1:50). Cells were incubated at RT for 30 mins in the dark. Cells were washed with 5 mL chilled 1x PBS 20 mM HEPES pH 7.4, 0.5 % tween-20 and resuspended in 300 μl 38 mM sodium citrate, 69 μM propidium iodide, 5 μl/mL RNaseA. Samples were incubated in the dark for 15 mins at 37 °C before analysis. Approximately 104 events were acquired using Cytoflex LX and the results were analyzed using FloJo software.
FANCD2 Ubiquitination Western blot
Mouse ES cells were grown until they reached 70% confluency followed by incubation with 0.08 μg/ml mitomycin C for 12 hours. After drug treatment cells were lysed in RIPA buffer (50mM Tris-HCl, pH 8.0, 250 mM NaCl, 0.1% sodium dodecyl sulfate, 1% NP-40 containing PMSF, and Sigma-Aldrich complete protease inhibitor tablet) and protein resolved by 8% SDS-PAGE at 50V for 18hr at 4°C. Protein abundance was analyzed by immunoblotting using the FANCD2 antibody(Abcam ab108928, 1:1,000).
Whole Genome Sequencing
Genomic DNA libraries of 450 bp insert size were derived for clones #68 and 39, using a KAPA Hyper Prep Kit according to manufacturer guidelines and 150 bp paired-end sequence reads were generated using the NovaSeq 6000 system and aligned to the mouse genome (Mm10). The Fancm and Brca1 loci were visualized and manually inspected using the Integrative Genomics Viewer (IGV) tool (Robinson et al., 2017). WGS data are available from the Sequence Read Archive database (www.ncbi.nlm.nih.gov/sra), SRA: Pending.
QUANTITATION AND STATISTICAL ANALYSIS
All statistics were performed using GraphPad Prism v7.0e software. Data shown represents the arithmetic mean and error bars represent the standard error of the mean (s.e.m.) of between three (n=3) and nine (n=9) independent experiments (n values given in figure legends). Each Figure Legend reports the number of independent experiments (n) that were performed to generate the data presented. Data points for each independent experiment were typically collected as the mean of technical duplicates. This mean value was taken as the solitary data point for that individual experiment. The arithmetic mean of samples collected for groups of independent experiments for repair frequency statistical analysis, was calculated and data points for each independent experiment used to calculate the mean and s.e.m., calculated as standard deviation/ n, (n indicates the number of independent experiments). Differences between sample pairs repair frequencies and fold enrichment for ChIP were analyzed by Student's two-tailed unpaired t-test, assuming unequal variance. One-way ANOVA statistical analysis of greater than three samples was performed when indicated P-values are indicated in the figure and or figure legends. No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-HA tag antibody | Abcam | Cat#ab9110, RRID:AB_307019 |
| Rabbit anti-BLM Antibody | Bethyl labs | Cat#A300-110A, RRID:AB_2064794 |
| Rabbit anti-FANCL antibody | Abcam | Cat#ab94458, RRID:AB_10675676 |
| Rabbit anti-FANCM antibody | Abcam | Cat#ab95014, RRID:AB_10675719 |
| Rabbit anti-Histone H3 antibody | Abcam | Cat#ab1791, RRID:AB_302613 |
| Rabbit anti-FANCA antibody | Abcam | Cat#ab97578, RRID:AB_10680297 |
| Rabbit anti-FANCD2 [EPR2302] antibody | Abcam | Cat#ab108928, RRID:AB_10862535 |
| mouse anti-BrdU antibody | Abcam | Cat#ab8039 RRID:AB_306213 |
| Goat anti-mouse FITC conjugated IgG | Jackson Immuno Research | Cat#115-095-146 RRID:AB_2338599 |
| Bacterial and Virus Strains | ||
| One Shot Stbl3 Chemically Competent E. coli | ThermoFisher Scientific | Cat#C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-adamantyl-indoleacetic acid | TCI Chemicals | Cat#A3390 |
| Asunaprevir | MedChem Express | Cat#BMS-650032 |
| Spy NLS Cas9 | New England Biolabs | Cat#M0646T |
| 5mL 4mg/mL RNaseA | Qiagen | Cat#158924 |
| proteinase K | Fisher Scientific | Cat#BP1700500 |
| sodium bicarbonate | Fisher Scientific | Cat#S233500 |
| formaldehyde, ACS reagent, 37% | Sigma-Aldrich | Cat#252549-25ML |
| Complete Protease Inhibitor Cocktail Tablets | Sigma-Aldrich | Cat#4693132001 |
| Mitomycin C from Streptomyces caespitosus | Millipore Sigma | Cat#475820-10MG |
| Lipofectamine 2000 | ThermoFisher Scientific | Cat#11668019 |
| Novex TBE-Urea gels, 10%, 10 well | ThermoFisher Scientific | EC6875BOX |
| Bolt 4-12% Bis-Tris 1 mm Mini Protein Gel | ThermoFisher Scientific | NW04122BOX |
| Magna ChIP Protein A+G magnetic beads | Fisher Scientific | Cat#16-663 |
| UltraPure Salmon Sperm DNA Solution | ThermoFisher Scientific | Cat#15632011 |
| 10mg/mL BSA, Molecular Grade | New England Biolabs | Cat#B9000S |
| Propidium Iodide | Sigma-Aldrich | P4170-100MG |
| Critical Commercial Assays | ||
| EnGen®sgRNA Synthesis Kit, S. pyogenes | New England Biolabs | Cat#E3322S |
| PCR Purification Kit | Qiagen | Cat#28106 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| Endo-free Maxiprep kit | Qiagen | Cat#12362 |
| 2x Power SYBR Green | Applied Biosystems | Cat#4368702 |
| Power SYBR Green RNA-to CT 1-Step Kit | Applied Biosystems | Cat#4389986 |
| Taq DNA Polymerase Kit (250 U) | Qiagen | Cat#201203 |
| MycoAlert Mycoplasma Detection Kit | Lonza | Cat#LT07-318 |
| RNA Clean and Concentrator Kit | Zymo Research | Cat#R1017 |
| Cell line | ||
| Brca1 exon 11 conditional mouse ES cells | Dr. Chuxia Deng (Xu et al., 1999) | N/A |
| Oligonucleotides | ||
| See Supplemental Table S1. | ||
| Recombinant DNA | ||
| Plasmid: pHIV-Tir1-74A-8xMYC-IRES-NAT-T2A-hCD52 | This study and (Willis et al., 2018) | N/A |
| Plasmid: pcDNA-1xAID-8xHA-SMASH-3xHA-T2A-neo (mBlm C-term targeting) | This study | N/A |
| Plasmid: pcDNA3β-MYC-NLS-Tus-F140A-3xHA | This study | N/A |
| Plasmid: pX330-U6-Chimeric_BB-CBh-hSpCas9(1.1) | This study | Addgene #71814 |
| Plasmid: pcDNA3β -myc NLS-I-SceI | (Puget et al., 2005) | N/A |
| Plasmid: pcDNA3β | (Puget et al., 2005) | N/A |
| Plasmid: pcDNA3β -myc NLS-Tus | (Willis et al., 2014) | N/A |
| Software and Algorithms | ||
| Prism 7.0e for Mac | GraphPad Software | https://www.graphpad.com/scientific-software/prism/, RRID:SCR_000306 |
| CCTop-CRISPR/Cas9 target gene predictor | (Stemmer et al., 2015) | https://cctop.cos.uni-heidelberg.de, RRID:SCR_016890 |
| Other | ||
| Diagenode Bioruptor Plus UCD-300 TM | Diagenode | Cat#B01020001 |
| Eppendorf Thermomixer C (REF 5382000023) | Fisher Scientific | Cat#05-412-503 |
| Smartblock Thermoblock (REF 5367000025) 24x 1.5-2.0mL | Fisher Scientific | Cat#05-412-510 |
| Diagenode Bioruptor water cooler | Diagenode | Cat#B02010003 |
| Medmark Technologies Nutator mixer | Fisher Scientific | Cat#NC0597936 |
| Dynal MPC-S Magnetic Rack #A13346 | Fisher Scientific | Cat#501148229 |
| Deposited data | ||
| Unprocessed blots and gels | This paper | https://data.mendeley.com/datasets/gwis4kp9mg |
| Whole genome sequencing data | This paper | SRA BioProject ID: PRJNA704440 |
Highlights.
Separation-of-function Fancm mutants in stalled fork repair pathway choice.
Bloom’s syndrome helicase can act independently of Fancm in stalled fork repair.
ATP hydrolysis by FANCM required for all its stalled fork repair functions.
Synthetic lethality between FANCM ATPase-dead mutant and Brca1 mutation.
Acknowledgements
We thank Drs. Andrew Deans, Hilda Pickett, Angelos Constantinou and Johannes Walter for helpful discussions. This work was supported by AACR fellowship 19-40-12-PAND (to A.P.), CDMRP grant BC160172 (to E.T.L. and R.S.) and NIH grants R01CA095175 and R01CA217991 (to R.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, and La Volpe A (2010). Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell 39, 25–35. [DOI] [PubMed] [Google Scholar]
- Amunugama R, Willcox S, Wu RA, Abdullah UB, El-Sagheer AH, Brown T, McHugh PJ, Griffith JD, and Walter JC (2018). Replication Fork Reversal during DNA Interstrand Crosslink Repair Requires CMG Unloading. Cell reports 23, 3419–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachrati CZ, Borts RH, and Hickson ID (2006). Mobile D-loops are a preferred substrate for the Bloom's syndrome helicase. Nucleic Acids Res 34, 2269–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, Delzenne-Goette E, van der Wal A, van der Valk M, Joenje H, te Riele H, et al. (2009). Fancmdeficient mice reveal unique features of Fanconi anemia complementation group M. Human molecular genetics 18, 3484–3495. [DOI] [PubMed] [Google Scholar]
- Bogliolo M, Bluteau D, Lespinasse J, Pujol R, Vasquez N, d'Enghien CD, Stoppa-Lyonnet D, Leblanc T, Soulier J, and Surrallés J (2018). Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genetics in medicine : official journal of the American College of Medical Genetics 20, 458–463. [DOI] [PubMed] [Google Scholar]
- Castera L, Harter V, Muller E, Krieger S, Goardon N, Ricou A, Rousselin A, Paimparay G, Legros A, Bruet O, et al. (2018). Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genetics in medicine : official journal of the American College of Medical Genetics 20, 1677–1686. [DOI] [PubMed] [Google Scholar]
- Catucci I, Osorio A, Arver B, Neidhardt G, Bogliolo M, Zanardi F, Riboni M, Minardi S, Pujol R, Azzollini J, et al. (2018). Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genetics in medicine : official journal of the American College of Medical Genetics 20, 452–457. [DOI] [PubMed] [Google Scholar]
- Chandramouly G, Kwok A, Huang B, Willis NA, Xie A, and Scully R (2013). BRCA1 and CtIP suppress long-tract gene conversion between sister chromatids. Nat Commun 4, 2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Jacobs CL, Huo Y, Yang J, Krumm SA, Plemper RK, Tsien RY, and Lin MZ (2015). Tunable and reversible drug control of protein production via a self-excising degron. Nat Chem Biol 11, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani el H, Joenje H, McDonald N, de Winter JP, et al. (2007). Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell 25, 331–343. [DOI] [PubMed] [Google Scholar]
- Collis SJ, Ciccia A, Deans AJ, Horejsi Z, Martin JS, Maslen SL, Skehel JM, Elledge SJ, West SC, and Boulton SJ (2008). FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell 32, 313–324. [DOI] [PubMed] [Google Scholar]
- Cortez D (2019). Replication-Coupled DNA Repair. Mol Cell 74, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans AJ, and West SC (2009). FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi Anemia. Mol Cell 36, 943–953. [DOI] [PubMed] [Google Scholar]
- Deans AJ, and West SC (2011). DNA interstrand crosslink repair and cancer. Nature reviews 11, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, and Walter JC (2015). What is the DNA repair defect underlying Fanconi anemia? Current opinion in cell biology 37, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlioli G, Bogliolo M, Catucci I, Caleca L, Lasheras SV, Pujol R, Kiiski JI, Muranen TA, Barnes DR, Dennis J, et al. (2019). The FANCM:p.Arg658* truncating variant is associated with risk of triple-negative breast cancer. NPJ breast cancer 5, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlioli G, Kvist A, Tham E, Soukupova J, Kleiblova P, Muranen TA, Andrieu N, Azzollini J, Balmaña J, Barroso A, et al. (2020). The Spectrum of FANCM Protein Truncating Variants in European Breast Cancer Cases. Cancers 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Decaillet C, Delannoy M, Wu L, and Constantinou A (2008a). Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci U S A 105, 16107–16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Decaillet C, Stasiak AZ, Stasiak A, and Constantinou A (2008b). The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol Cell 29, 141–148. [DOI] [PubMed] [Google Scholar]
- Gillotin S, Davies JD, and Philpott A (2018). Subcellular localisation modulates ubiquitylation and degradation of Ascl1. Scientific reports 8, 4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodskinson MR, Bolner A, Sato K, Kamimae-Lanning AN, Rooijers K, Witte M, Mahesh M, Silhan J, Petek M, Williams DM, et al. (2020). Alcohol-derived DNA crosslinks are repaired by two distinct mechanisms. Nature 579, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodskinson MR, Silhan J, Crossan GP, Garaycoechea JI, Mukherjee S, Johnson CM, Scharer OD, and Patel KJ (2014). Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol Cell 54, 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, and Seidman MM (2013). The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell 52, 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang J, Bellani MA, Pokharel D, Gichimu J, James RC, Gali H, Ling C, Yan Z, Xu D, et al. (2019). Remodeling of Interstrand Crosslink Proximal Replisomes Is Dependent on ATR, FANCM, and FANCD2. Cell reports 27, 1794–1808.e1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Sugawara N, Mehta A, Ryu T, and Haber JE (2016). Sgs1 and Mph1 Helicases Enforce the Recombination Execution Checkpoint During DNA Double-Strand Break Repair in Saccharomyces cerevisiae. Genetics 203, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalan M, Oehler J, Morrow CA, Osman F, and Whitby MC (2019). Factors affecting template switch recombination associated with restarted DNA replication. eLife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karow JK, Constantinou A, Li JL, West SC, and Hickson ID (2000). The Bloom's syndrome gene product promotes branch migration of holliday junctions. Proc Natl Acad Sci U S A 97, 6504–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, and D'Andrea AD (2012). Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 26, 1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Raschle M, Walter JC, and Knipscheer P (2014). XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 54, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, and Walter JC (2009). The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326, 1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M, Tomberg K, and Bradley A (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nature biotechnology 36, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, and Patel KJ (2011). Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58. [DOI] [PubMed] [Google Scholar]
- Larsen NB, Liberti SE, Vogel I, Jorgensen SW, Hickson ID, and Mankouri HW (2017). Stalled replication forks generate a distinct mutational signature in yeast. Proc Natl Acad Sci U S A 114, 9665–9670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C, Huang J, Yan Z, Li Y, Ohzeki M, Ishiai M, Xu D, Takata M, Seidman M, and Wang W (2016). Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell discovery 2, 16047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Raschle M, Joukov V, and Walter JC (2011). Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, O'Rourke JJ, Sobinoff AP, Allen JAM, Nelson CB, Tomlinson CG, Lee M, Reddel RR, Deans AJ, and Pickett HA (2019). The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat Commun 10, 2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, et al. (2003). A novel ubiquitin ligase is deficient in Fanconi anemia. Nature genetics 35, 165–170. [DOI] [PubMed] [Google Scholar]
- Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, et al. (2005). A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nature genetics 37, 958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menghi F, Barthel FP, Yadav V, Tang M, Ji B, Tang Z, Carter GW, Ruan Y, Scully R, Verhaak RGW, et al. (2018). The Tandem Duplicator Phenotype Is a Prevalent Genome-Wide Cancer Configuration Driven by Distinct Gene Mutations. Cancer Cell 34, 197–210 e195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menghi F, Inaki K, Woo X, Kumar PA, Grzeda KR, Malhotra A, Yadav V, Kim H, Marquez EJ, Ucar D, et al. (2016). The tandem duplicator phenotype as a distinct genomic configuration in cancer. Proc Natl Acad Sci U S A 113, E2373–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. (1994). A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71. [DOI] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, and Pommier Y (2012). Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi S, and Whitby MC (2012). The ATPase activity of Fml1 is essential for its roles in homologous recombination and DNA repair. Nucleic Acids Res 40, 9584–9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Kiyomitsu T, Saga Y, and Kanemaki MT (2016). Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors. Cell reports 15, 210–218. [DOI] [PubMed] [Google Scholar]
- Neelsen KJ, and Lopes M (2015). Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16, 207–220. [DOI] [PubMed] [Google Scholar]
- Neidhardt G, Hauke J, Ramser J, Groβ E, Gehrig A, Müller CR, Kahlert AK, Hackmann K, Honisch E, Niederacher D, et al. (2017). Association Between Loss-of-Function Mutations Within the FANCM Gene and Early-Onset Familial Breast Cancer. JAMA oncology 3, 1245–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MO, Jalan M, Morrow CA, Osman F, and Whitby MC (2015). Recombination occurs within minutes of replication blockage by RTS1 producing restarted forks that are prone to collapse. eLife 4, e04539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. (2016). Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niraj J, Färkkilä A, and D'Andrea AD (2019). The Fanconi Anemia Pathway in Cancer. Annual review of cancer biology 3, 457–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Yamada R, Hagihara S, Iwasaki R, Uchida N, Kamura T, Takahashi K, Torii KU, and Fukagawa T (2020). A super-sensitive auxin-inducible degron system with an engineered auxin-TIR1 pair. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, and Patel KJ (2010). Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 329, 219–223. [DOI] [PubMed] [Google Scholar]
- Pan X, Drosopoulos WC, Sethi L, Madireddy A, Schildkraut CL, and Zhang D (2017). FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc Natl Acad Sci U S A 114, E5940–e5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, and Haber JE (1999). Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiology and molecular biology reviews : MMBR 63, 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlongo P, Catucci I, Colombo M, Caleca L, Mucaki E, Bogliolo M, Marin M, Damiola F, Bernard L, Pensotti V, et al. (2015). FANCM c.5791C>T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity and is a familial breast cancer risk factor. Human molecular genetics 24, 5345–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Krejci L, Van Komen S, Anke Schurer K, Kramer W, and Sung P (2005). Saccharomyces cerevisiae MPH1 gene, required for homologous recombination-mediated mutation avoidance, encodes a 3' to 5' DNA helicase. J Biol Chem 280, 7854–7860. [DOI] [PubMed] [Google Scholar]
- Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P, et al. (2009). Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev 23, 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Zhang Y, Feng W, and Jasin M (2015). Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7, a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puget N, Knowlton M, and Scully R (2005). Molecular analysis of sister chromatid recombination in mammalian cells. DNA Repair (Amst) 4, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Lemaçon D, and Vindigni A (2017). Replication Fork Reversal: Players and Guardians. Mol Cell 68, 830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, and Walter JC (2008). Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134, 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman K, and Smogorzewska A (2019). Advances in understanding DNA processing and protection at stalled replication forks. The Journal of cell biology 218, 1096–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, and Mesirov JP (2017). Variant Review with the Integrative Genomics Viewer. Cancer research 77, e31–e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado IV, Langevin F, Crossan GP, Takata M, and Patel KJ (2011). Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nature structural & molecular biology 18, 1432–1434. [DOI] [PubMed] [Google Scholar]
- Rosado IV, Niedzwiedz W, Alpi AF, and Patel KJ (2009). The Walker B motif in avian FANCM is required to limit sister chromatid exchanges but is dispensable for DNA crosslink repair. Nucleic Acids Res 37, 4360–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, and Malkova A (2013). Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502, 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, and Livak KJ (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Scully R, Panday A, Elango R, and Willis NA (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva B, Pentz R, Figueira AM, Arora R, Lee YW, Hodson C, Wischnewski H, Deans AJ, and Azzalin CM (2019). FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat Commun 10, 2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh TR, Saro D, Ali AM, Zheng XF, Du CH, Killen MW, Sachpatzidis A, Wahengbam K, Pierce AJ, Xiong Y, et al. (2010). MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol Cell 37, 879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafa A, Donnianni RA, Timashev LA, Lam AF, and Symington LS (2014). Template switching during break-induced replication is promoted by the Mph1 helicase in Saccharomyces cerevisiae. Genetics 196, 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer M, Thumberger T, Del Sol Keyer M, Wittbrodt J, and Mateo JL (2015). CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PloS one 10, e0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Nandi S, Osman F, Ahn JS, Jakovleska J, Lorenz A, and Whitby MC (2008). The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol Cell 32, 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Jin C, Li X, Qi S, Chu L, Niu L, Yao X, and Teng M (2012). The structure of the FANCM-MHF complex reveals physical features for functional assembly. Nat Commun 3, 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AMR, Rothblum-Oviatt C, Ellis NA, Hickson ID, Meyer S, Crawford TO, Smogorzewska A, Pietrucha B, Weemaes C, and Stewart GS (2019). Chromosome instability syndromes. Nature reviews Disease primers 5, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby MC (2010). The FANCM family of DNA helicases/translocases. DNA Repair (Amst) 9, 224–236. [DOI] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, and Scully R (2014). BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Frock RL, Menghi F, Duffey EE, Panday A, Camacho V, Hasty EP, Liu ET, Alt FW, and Scully R (2017). Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature 551, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Panday A, Duffey EE, and Scully R (2018). Rad51 recruitment and exclusion of non-homologous end joining during homologous recombination at a Tus/Ter mammalian replication fork barrier. PLoS genetics 14, e1007486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Rass E, and Scully R (2015). Deciphering the Code of the Cancer Genome: Mechanisms of Chromosome Rearrangement. Trends Cancer 1, 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, and Scully R (2021). Measurement of Homologous Recombination at Stalled Mammalian Replication Forks. Methods Mol Biol 2153, 329–353. [DOI] [PubMed] [Google Scholar]
- Wu L, and Hickson ID (2003). The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874. [DOI] [PubMed] [Google Scholar]
- Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, and Deng CX (1999). Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nature genetics 22, 37–43. [DOI] [PubMed] [Google Scholar]
- Xue X, Sung P, and Zhao X (2015). Functions and regulation of the multitasking FANCM family of DNA motor proteins. Genes Dev 29, 1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Li Y, Guo R, Ling C, and Wang W (2008). FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Human molecular genetics 17, 1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Delannoy M, Ling C, Daee D, Osman F, Muniandy PA, Shen X, Oostra AB, Du H, Steltenpool J, et al. (2010). A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol Cell 37, 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Walter JC (2014). Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst) 19, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XF, Prakash R, Saro D, Longerich S, Niu H, and Sung P (2011). Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae Mph1 protein. DNA Repair (Amst) 10, 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed blots and gels are available at: https://data.mendeley.com/datasets/gwjs4kp9mg/draft?a=8c165752-21e7-4b4d-a17a-8426b21a9ca0