

Abstract
Mutations in the chromatin remodeller‐coding gene CHD7 cause CHARGE syndrome (CS). CS features include moderate to severe neurological and behavioural problems, clinically characterized by intellectual disability, attention‐deficit/hyperactivity disorder and autism spectrum disorder. To investigate the poorly characterized neurobiological role of CHD7, we here generate a zebrafish chd7 −/− model. chd7 −/− mutants have less GABAergic neurons and exhibit a hyperactivity behavioural phenotype. The GABAergic neuron defect is at least in part due to downregulation of the CHD7 direct target gene paqr3b, and subsequent upregulation of MAPK/ERK signalling, which is also dysregulated in CHD7 mutant human cells. Through a phenotype‐based screen in chd7 −/− zebrafish and Caenorhabditis elegans, we show that the small molecule ephedrine restores normal levels of MAPK/ERK signalling and improves both GABAergic defects and behavioural anomalies. We conclude that chd7 promotes paqr3b expression and that this is required for normal GABAergic network development. This work provides insight into the neuropathogenesis associated with CHD7 deficiency and identifies a promising compound for further preclinical studies.
Keywords: behaviour, CHD7, GABA, neurodevelopment, zebrafish
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Development & Differentiation; Neuroscience
Loss‐of‐function of chd7 causes defects in GABAergic neuron development and behavioural anomalies reminiscent of CHARGE syndrome, which are rescued by genetic and pharmacological interventions in zebrafish.
Introduction
Heterozygous loss‐of‐function mutations in CHD7 are the major cause of a rare congenital disorder termed CHARGE syndrome (CS), which stands for coloboma of the eye, heart defects, atresia choanae, retardation of growth and/or development, genital abnormalities and ear abnormalities (Pagon et al, 1981; Zentner et al, 2010; Janssen et al, 2012). The mutations are equally distributed along the coding region of CHD7, and the most prevalent types are nonsense mutations (44%), followed by frameshift deletions or insertions (34%) (Janssen et al, 2012). Although neurological abnormalities are not considered for clinical diagnosis of CS, many individuals with CS display moderate to severe neurological deficits, which include autism‐like behaviour, obsessive–compulsive disorder, attention‐deficit/hyperactivity disorder, anxiety, aggressivity and seizures (Hartshorne et al, 2005; Souriau et al, 2005; Johansson et al, 2006; Bergman et al, 2011; Hartshorne et al, 2017). Along these lines, CHD7 mutations have been identified in patients with autism spectrum disorder (ASD) (O'Roak et al, 2012; Takata et al, 2018). These reports strongly suggest an important role for CHD7 in the development and functioning of the central nervous system. However, the precise mechanisms underlying the neurological deficits in CS remain poorly understood. A recent study reported anxious‐ and aggressive‐like behaviours with increased expression of glycine transporters in adult chd7 heterozygous mutant zebrafish, leaving however unexplored the molecular and cellular mechanisms of brain circuitry (Liu & Liu, 2020). Also, noteworthy, there are no pharmacological and/or genetic treatments to ameliorate/rescue CS‐related neurological features. Current treatment options primarily focus on behavioural management as well as educational and physical therapies. Development of successful therapeutic strategies would benefit from the identification and targeting of causative factors.
Chd7 −/− mice die in utero around embryonic day 10.5 (Van Nostrand et al, 2014), a stage incompatible for studying the role of CHD7 in the neuropathogenesis of CS. Additionally, Chd7 +/− mice are viable and phenocopy a number of aspects of CS, but the full spectrum and severity of certain CS malformations are not seen (Payne et al, 2015). Yet, Chd7 +/− mice, Chd7 conditional knockout mice and other cellular and animal (Drosophila, zebrafish and Xenopus) models have provided insights on the general function of CHD7 (Schnetz et al, 2010; Patten et al, 2012; Ohta et al, 2016; Feng et al, 2017; Whittaker et al, 2017; Belanger et al, 2018). For instance, it has been shown that CHD7 is capable of both enhancing and inhibiting expression of embryonic stem cell genes (Schnetz et al, 2010). In that respect, CHD7 facilitates neural stem cell maintenance and proliferation in the developing brain (Ohta et al, 2016) and quiescence in the adult (Jones et al, 2015). It is also required for the formation of migratory neural crest cells (Bajpai et al, 2010; Okuno et al, 2017). CHD7 coordinates with the transcription factor SOX10 to regulate the initiation of myelinogenesis (He et al, 2016) and is required for oligodendrocyte precursor cell survival (Marie et al, 2018). Genetic inactivation of Chd7 in cerebellar granule neuron (GN) progenitors leads to cerebellar hypoplasia in mice, due to impairment of GN differentiation (Feng et al, 2017; Whittaker et al, 2017) but these cerebellar defects did not alter the social behaviour in these mice (Whittaker et al, 2017). Although these recent findings point to an important role of CHD7 in brain development, the precise neural substrates that may contribute to CS‐associated neurological deficits such as autistic traits and/or hyperactivity disorder remain poorly understood. Emerging evidence suggests that abnormalities in inhibitory GABAergic neurons development/function in the context of neurodevelopmental disorders are characterized by a shared symptomatology of ASD symptoms (Rubenstein & Merzenich, 2003; Coghlan et al, 2012). Whether such alterations in brain inhibitory neural networks underlie the neurological deficits in CHD7 mutation‐positive cases of CS is currently unknown.
The zebrafish is a powerful tool for studying neurological diseases including ASD (Stewart et al, 2014; Meshalkina et al, 2018). Here, we report the generation of a chd7 −/− mutant zebrafish line and show that these animals exhibit altered number and positioning of GABAergic neurons in the brain and display a hyperactive behaviour phenotype. Using genetic, pharmacological and biochemical approaches, we unravel the molecular mechanisms by which chd7 regulates GABAergic network development and behaviour in zebrafish. Finally, through a chemical‐genetic screen, we identified ephedrine that effectively ameliorates behavioural anomalies as well as the GABAergic defects in chd7 mutants. This study provides novel insights into the role of CHD7 in brain development and disease and has important translational implications.
Results
Zebrafish chd7 mutants display phenotypic characteristics of CHARGE syndrome
To investigate the neurobiological function of CHD7, we generated a chd7 knockout zebrafish line using CRISPR/Cas9 to target the helicase domain of the chd7 gene (17th exon) for disruption. A positive founder transmitting a single nucleotide insertion causing a frame‐shifting mutation was selected (Fig 1A). This mutation causes a premature stop codon 8 amino acids after the mutation site (Fig EV1A). To assess whether the mutant chd7 transcript underwent nonsense‐mediated decay upon that mutation, we performed qPCR. The relative abundance of chd7 mRNA in mutant zebrafish was significantly decreased, suggesting a loss of mutant transcript via nonsense‐mediated decay (Fig EV1B). While no major morphological differences were observed between wild‐type (chd7 +/+) and heterozygous (chd7 +/−) fish (Fig 1B), the survival rate of homozygous (chd7 −/−) larvae sharply declined after 10 days postfertilization (dpf) (Fig 1C). Remarkably, chd7 −/− zebrafish larvae displayed a small head phenotype (Fig 1D) compared with controls but nevertheless all the brain regions were fairly well‐preserved in mutant fish (Fig EV1C). Additionally, chd7 −/− zebrafish larvae exhibited a low frequency of pericardial oedema (20 %) (Fig EV1D), cranial cartilage malformations (Fig EV1E) and cranial nerve defects (Fig 1E). Notably, there were less arborizations of peripheral projections from the Vth cranial nerve in chd7 −/− fish. Precisely, chd7 −/− fish had reduced growth and branching of the peripheral axons, resulting in a significant decrease in the mean total length of the axon projections as compared to controls (Fig 1E). Strikingly, these phenotypic characteristics are hallmarks of CS (Hsu et al, 2014). Additionally, this new chd7 −/− mutant fish recapitulates other anomalies previously reported in chd7 morphants (Patten et al, 2012) and other chd7 mutant zebrafish lines (Cloney et al, 2018; Liu et al, 2018), but with less pronounced cardiac defects and no apparent blindness, thereby making it an ideal model to investigate the pathogenic mechanisms underlying CS‐associated neurological deficits. Behaviourally, chd7 −/− larvae were significantly hyperactive compared with wild‐type and chd7 +/− fish larvae at 5dpf (Fig 1F and G). This hyperactive phenotype was particularly prominent and persistent during the dark cycles (Fig 1G).
Figure 1. Generation of a zebrafish chd7 mutant using CRISPR/Cas9.
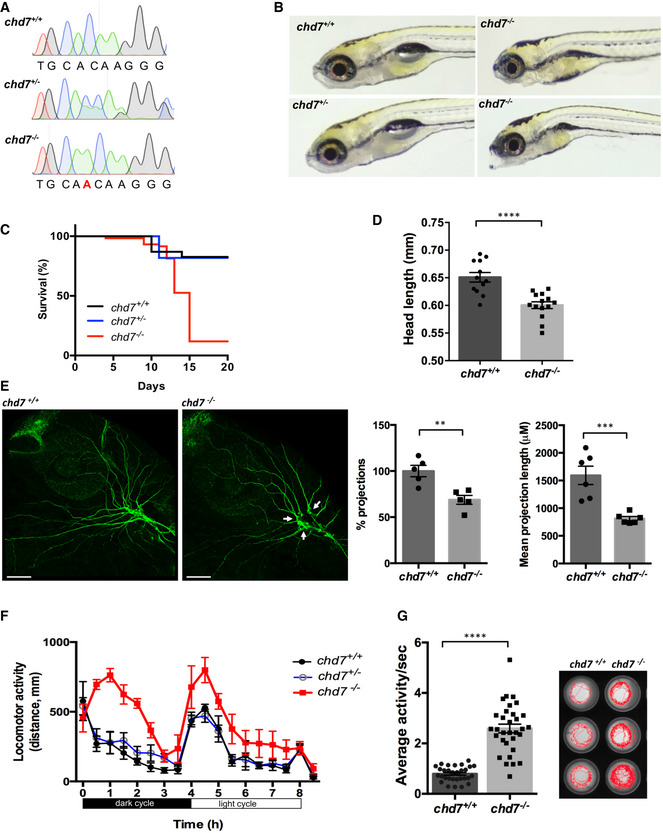
- Chromatograms showing the confirmation a 1‐nucleotide insertion mutation by Sanger sequencing.
- Gross morphological analyses of control (chd7 +/+; top left image), heterozygous (chd7 +/−; bottom left image) and knockout mutants (chd7 −/−; images in right panel).
- Kaplan–Meier survival plot showing low survival of chd7 mutants after 12 dpf (N = 5).
- Measurement of head size of control (n = 12) and mutants (n = 14) showing significantly smaller head size in chd7 −/− fish (****P < 0.0001, Student’s t‐test).
- Acetylated tubulin staining in 28 hpf controls (left) and mutants (right) showing severely affected outbranching of the trigeminal nerve (Vth cranial nerve). Notably, chd7 −/− display reduced branching of the Vth cranial nerve (arrows) and axonal arborization in the tectal area. Graphs showing quantitative analyses of percentage (n = 5) and mean total length of peripheral projections (n = 6) per zebrafish in controls and mutants (***P < 0.001; **P < 0.005, Student’s t‐test).
- Locomotor activity of control (black), heterozygous (blue) and mutants (red) showing significant hyperactivity of mutants in dark and light cycles (N = 3, n = 48).
- Average activity per second during dark cycle (left) is significantly increased in chd7 −/− mutants compared with chd7 +/+ (n = 32; ****P < 0.0001, Student’s t‐test). Representative swimming tracks during dark cycle of control and mutant fish (right). Mutant chd7 −/− larvae displayed hyperactive swimming.
Data information: ****P < 0.0001; ***P < 0.001; **P < 0.005, Student’s t‐test. Data are presented as mean ± SEM. Scale bar = 50 μm. n represents number of fish. N represents number of experimental repeats.
Figure EV1. Generation and characterization of chd7 mutants.
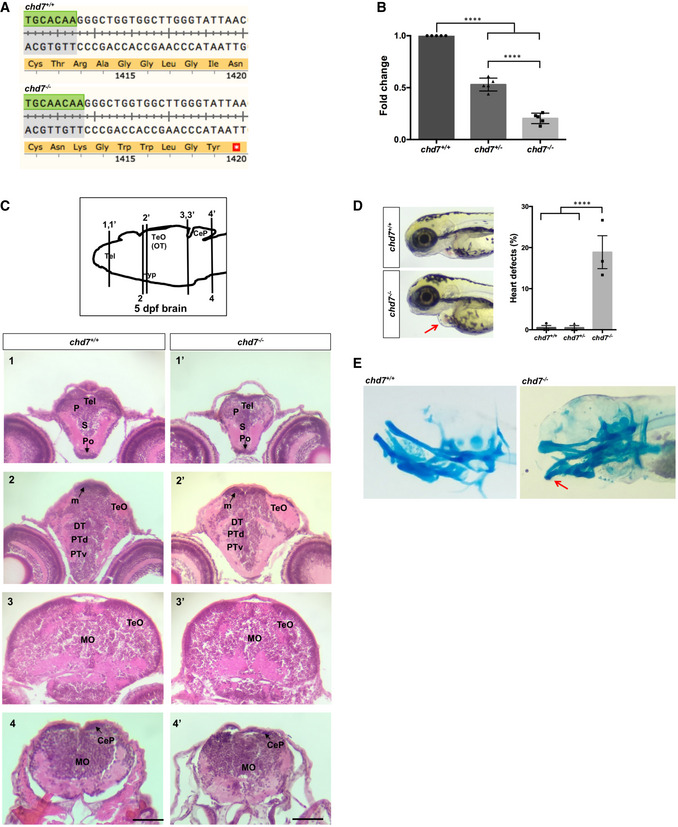
- Translation following genome editing resulted in a premature stop codon (*) in chd7 −/− fish.
- qPCR analysis of RNAs from 3dpf larvae shows a significant reduction of chd7 mRNA expression in both chd7 +/− and chd7 −/− compared with wild‐type (N = 5). ****P < 0.0001, one‐way ANOVA.
- Examination of brain tissues in chd7 +/+ (left images) and chd7 −/− (right images) by H&E staining at 5 dpf. Zebrafish brains were sectioned at telencephalic (1,1’), diencephalic (2, 2’), mesencephalic (3,3’) and rhombencephalic levels (4,4’). Levels of sections are indicated in the sketch of a sagittal view of a 5 dpf zebrafish brain (top image). The scale bar is 0.12 mm. P: pallium, S: subpallium, Po:preoptic region, Tel: Telencephalon; TeO: tectum opticum (or OT: optic tectum), m: medial tectal proliferating zone, DT: dorsal thalamus, PTd: dorsal part of posterior tuberculum, PTv: ventral part of posterior tuberculum, MO: medulla oblongata, Hyp: hypothalamus, CeP: cerebellar plate.
- chd7 −/− mutant fish displayed features of CS such as heart defects (red arrow) at a low penetrance (N = 3). ****P < 0.0001; Student’s t‐test.
- Alcian blue staining of 6dpf larvae showing craniofacial defects at Meckel’s cartilage (red arrow).
Data information: Data are presented as mean ± SEM. n is the number of fish used. N is the number of experimental repeats.
GABAergic neuron differentiation is defective in chd7−/− mutants
A hyperactivity behavioural phenotype in ASD mouse (Lee et al, 2018) and zebrafish (Hoffman et al, 2016) models has been reported to be due to alterations in GABAergic interneuron development. To test whether similar alterations in GABAergic neuron development occur in chd7 −/− mutants, we analysed the inhibitory GABAergic neuronal populations in wild‐type controls and chd7 −/− mutants during early brain development (Fig 2), using a transgenic line that labels GABAergic interneurons (Tg(dlx5a/6a:GFP)). Compared with controls, chd7 −/− larvae had a significant reduction in the density of GFP‐positive GABAergic cells in the brain at 5 dpf (Fig 2A and B). Particularly, we observed a highly significant decrease in the number of GABAergic neurons in the optic tectum (OT) (Fig 2C and D) and a near‐complete loss of GFP‐positive cells in the cerebellum (CB) compared with the controls. Reduced number and malpositioning of GABAergic cells were also observed in the hypothalamus (HYP; Fig 2E and F) and telencephalon (TEL; Fig 2G and H).
Figure 2. GABAergic neuron defects in zebrafish chd7 mutant brain.
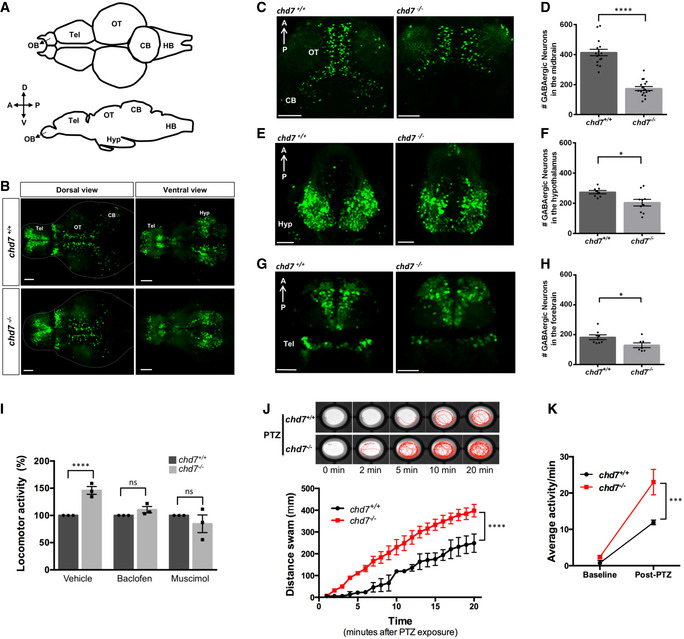
-
AStructural illustration of 5 dpf zebrafish brain from dorsal (top) and lateral (bottom) view (OB: Olfactory bulb, Tel: Telencephalon, OT: Optic tectum, CB: Cerebellum, HB: Hindbrain).
-
B5 dpf dlx5a/6a transgenic line showing GFP+ GABAergic neurons are reduced in chd7 −/− mutants (bottom) in comparison with controls (top) in both dorsal (left) and ventral (right) view.
-
C–HTotal number of GABAergic neurons (GFP+ cells) in (C, D) the optic tectum (OT) and cerebellum (CB) regions of 5 dpf wild‐type and chd7 mutant fish (n = 16; ****P < 0.0001; Student’s unpaired t‐test), (E, F) the hypothalamus (hyp) region (n = 10; *P = 0.0182; Student’s t‐test) and (G, H) the telencephalon (tel) (n = 7; *P = 0.0347; Student’s t‐test).
-
ITreatment of control (dark grey) and mutants (light grey) with GABA agonists Baclofen (N = 3, n = 24; ns, P = 0.1427; Student’s t‐test) and Muscimol (N = 3, n = 24; ns, P = 0.3987; Student’s t‐test) showing recovery of hyperactive locomotor activity in chd7 −/− mutants (vehicle: N = 3, n = 24, ****P < 0.0001; Student’s t‐test).
-
JFunctional analysis of GABAergic signalling shows increased responsiveness to GABA antagonist PTZ in both onset and overall locomotor activity (n = 24; ****P < 0.0001; one‐way ANOVA).
-
KAverage locomotor activity between 2 dpf controls (black) and chd7 −/− mutants (red) shows increased activity after 3 mM PTZ exposure (n = 24; ***P < 0.001; two‐way ANOVA).
Data information: Data are presented as mean ± SEM. Scale bar = 50 μm. n represents number of fish used. N represents number of experimental repeats.
We also examined the development of GABAergic neurons in chd7 −/− brain throughout major developmental phases between 1 and 5 dpf (Fig EV2A). The reduced number of GFP‐positive GABAergic neurons occurs very early in chd7 −/− embryos, with a striking decrease of GABAergic neurons posteriorly between 1 and 2 dpf (Fig EV2A). We next tested whether the reduced number of GABAergic neurons in chd7 −/− fish could be due to reduced proliferation, defects in neuronal differentiation and/or enhanced cell death. The zebrafish CNS proliferative profile is still very high at 2 dpf and is rapidly downregulated up to 5 dpf (Wullimann & Knipp, 2000). At 2 dpf, we did not observe a change in either the proliferation marker pH3 (Fig EV2B and C) or differences in the number of apoptotic cells (Fig EV2D). Additionally, we did not notice differences in the number of double‐positive cells in pH3 and NeuroD1 (neuronal progenitor marker) co‐staining (Fig EV2E). However, at 5 dpf, while no apoptosis was observed, a significant increase in pH3‐positive cells was detected (Fig EV2, EV3, EV4, EV5, EV6), suggesting a failure in differentiation of progenitor cells into GABAergic neurons.
Figure EV2. Proliferation and apoptosis analyses in wild‐type and chd7 mutant zebrafish.
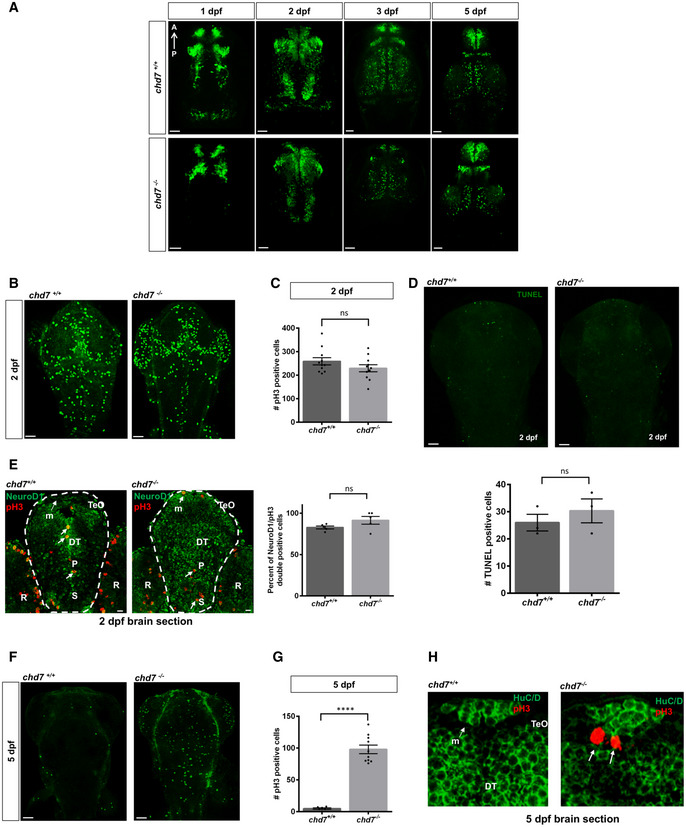
- Analysis of GABAergic neurons network development between 1 dpf and 5 dpf between control (top row) and chd7 mutants (bottom row) (N = 3).
- Proliferation analysis by pH3 staining at 2 dpf in control and chd7 mutants.
- Bar graph showing no difference in pH3‐positive cells in zebrafish brain at 2 dpf between control and mutants (n = 11; ns, P = 0.1816; Student’s t‐test).
- Cell death analysis by TUNEL assay in chd7 −/− and chd7 +/+ brains shows no change in apoptotic cells at 2 dpf (N = 3, n = 8; ns, P = 0.464; Student’s t‐test).
- Transverse sections of 2 dpf larvae after immunostaining with pH3 (red) and NeuroD1 (green). Bar graph showing no difference in pH3 and NeuroD1 double‐positive cells (arrows) in zebrafish brain at 2 dpf between control and mutants (N = 3, n = 5; ns, P = 0.124; Student’s t‐test). P: pallium, S: subpallium, TeO: tectum opticum, m: medial tectal proliferating zone, DT: dorsal thalamus R: retina.
- Proliferation analysis by pH3 staining at 5 dpf in control and chd7 mutants.
- An increase in pH3‐positive cells was noted in brains of mutant fish compared with controls at 5 dpf (N = 4, n = 10; ****P < 0.0001; Student’s t‐test).
- Transverse sections of 5 dpf larvae after immunostaining with pH3 (red) and HuC/D (green). pH3‐positive cells (arrows) were observed at 5 dpf in the medial tectal proliferating zone of mutant fish brains but none in controls (N = 3). P: pallium, S: subpallium, TeO: tectum opticum, m: medial tectal proliferating zone, DT: dorsal thalamus.
Data information: Data are presented as mean ± SEM. Scale bar = 50 μm and 10 μm for 2 dpf NeuroD1 and pH3 co‐stain.
Figure EV3. Aberrant GABAergic neuronal differentiation in chd7 mutant zebrafish.
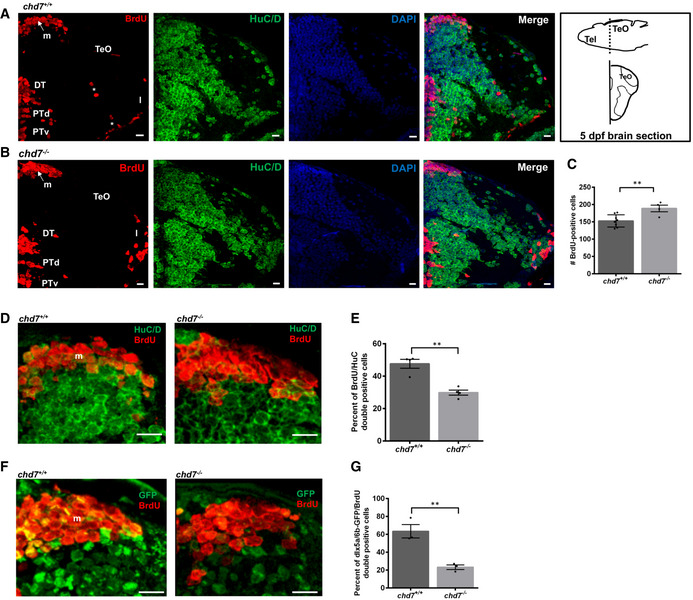
-
A, BImmunostaining with BrdU and HuC/D in brain sections of the zebrafish tectal region in chd7 +/+ (A) and chd7 −/− (B). Level of the sections is indicated in the sketch of a 5 dpf zebrafish brain (top right image in (A)). The scale bar is 10 μm. Tel: Telencephalon; TeO: tectum opticum, m: medial tectal proliferating zone, DT: dorsal thalamus, PTd: dorsal part of posterior tuberculum, PTv: ventral part of posterior tuberculum, l: lateral tectal proliferation zone. Asterisks (*) marks early migrated region of pretectum and proglomerular.
-
CThe number of BrdU‐positive cells in transverse sections of the zebrafish brain in chd7 +/+ and chd7 −/− (N = 3, chd7 +/+: n = 8; chd7 −/−: n = 4; **P < 0.05; Student’s t‐test).
-
DImmunostaining with BrdU and HuC/D in brain sections of the zebrafish medial tectal region. Scale bar = 10 μm. m: medial tectal.
-
EThe percentage of BrdU and HuC/D‐double positive cells among the BrdU‐positive cells in the medial tectal zone (N = 3, n = 4; **P < 0.05; Student’s t‐test).
-
FImmunostaining with BrdU and GFP (to label dlx5a/6a‐GFP + GABAergic neurons) in brain sections of the zebrafish medial tectal region. Scale bar = 10 μm. m: medial tectal.
-
GThe percentage of BrdU and dlx5a/6a‐GFP‐double positive cells among the BrdU‐positive cells in the medial tectal zone (N = 3, n = 3; **P < 0.05; Student’s t‐test).
Data information: Data are presented as mean ± SEM. n is the number of fish used. N is the number of experimental repeats.
Figure EV4. Impaired neurogenesis in chd7 mutant zebrafish.
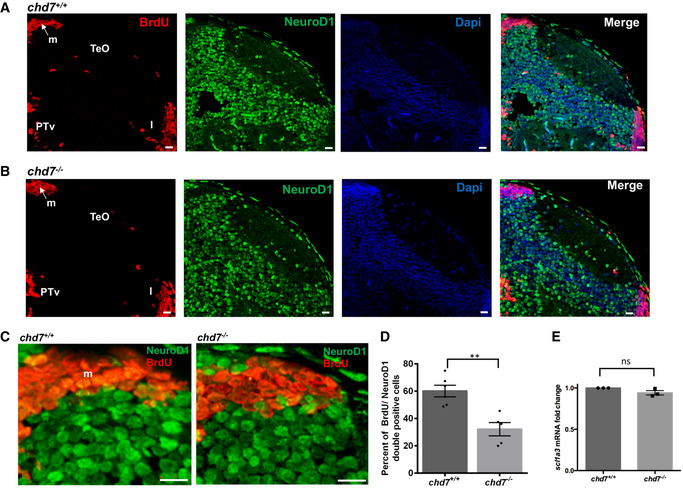
-
A, BImmunostaining with BrdU and NeuroD1 in brain sections of the zebrafish tectal region in chd7 +/+ (A) and chd7 −/− (B). The scale bar is 10 μm. Tel: Telencephalon; TeO: tectum opticum, m: medial tectal, DT: dorsal thalamus, PTd: dorsal part of posterior tuberculum, PTv: ventral part of posterior tuberculum, l: lateral tectal proliferation zone.
-
CImmunostaining with BrdU and NeuroD1 in brain sections of the zebrafish medial tectal region. Scale bar = 10 μm. m: medial tectal.
-
DThe percentage of BrdU and NeuroD1‐double positive cells among BrdU‐positive cells in the medial tectal zone (N = 3, chd7 +/+: n = 6; chd7 −/−: n = 6; **P < 0.05; Student’s t‐test).
-
EExpression level of scl1a3 mRNA in chd7 −/− relative to chd7 +/+ (N = 4). ns, not significant; Student’s t‐test.
Data information: Data are presented as mean ± SEM. n is the number of fish used. N is the number of experimental repeats.
Figure EV5. Expression of paqr3b in wild‐type and chd7 mutant zebrafish.
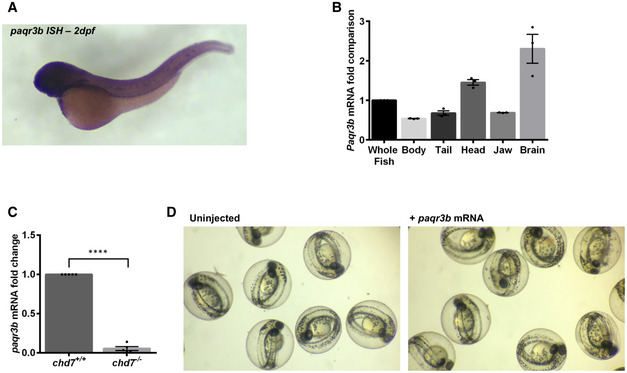
-
A, BExpression profile of paqr3b in whole‐mount zebrafish by in situ hybridization (A) and in tissues by qRT–PCR (B). N = 4.
-
CqRT–PCR validation of the downregulation of paqr3b (N = 4; ****P < 0.0001, Student’s t‐test).
-
DImages of gross morphology of 2 dpf zebrafish embryos with or without overexpression of paqr3b mRNA. Of note, neither abnormalities nor death were observed in zebrafish embryos upon overexpression of paqr3b mRNA.
Data information: Data are presented as mean ± SEM. n is the number of fish used. N is the number of experimental repeats.
Figure EV6. Pharmacological responses of chd7 mutants and amelioration of neuronal network development by ephedrine.
- PCR proof of a 700 bp deletion in the chd‐7 gene in chd‐7(gk290) mutant worms.
- Lifespan analyses of chd‐7(gk290) Caenorhabditis elegans mutants treated with ephedrine (green) compared with control DMSO (black). Log‐rank test was performed for statistical analyses. (N = 3, n = 50; *P < 0.05).
- Survival rate of chd7 −/− zebrafish mutants treated with ephedrine (blue) compared with untreated mutants (red). N = 3, n = 60.
- Acetylated tubulin staining in non‐treated and ephedrine‐treated chd7 −/− zebrafish mutants showing rescue of the severely affected outbranching structure of Vth cranial nerves. Graphs showing quantitative analyses of percentage and mean total length of peripheral projections per zebrafish in controls and mutants without and with ephedrine treatment (n = 5; ***P < 0.001; **P < 0.005; one‐way ANOVA).
Data information: Data are presented as mean ± SEM. Scale bar = 50 μm. n is the number of fish or worms used. N is the number of experimental repeats.
We, thus, next sought to evaluate further neurogenesis in chd7 mutants during brain development, with a focus on the midbrain region—the brain region where the reduced number of GABAergic neurons was more prominent in 5 dpf chd7 −/− fish. Zebrafish larvae (4 dpf) were exposed to BrdU‐containing media for 24 h and fixed. In both chd7 +/+ and chd7 −/− fish, BrdU‐labelled cells were noted in the medial tectal proliferation zone (m), dorsal thalamus (DT), posterior tuberculum (PT) and the lateral tectal proliferation zone (l) of the midbrain (Fig EV3A and B). Interestingly, compared with chd7 +/+, BrdU‐labelled cells in chd7 −/− did not migrate over long distances to reach the early migrated region of pretectum and proglomerular (Fig EV3A; asterisks), which are regions involved with visual and other sensorimotor circuits (ref). We also found an increased in the number of BrdU‐labelled cells in chd7 mutant fish compared with wild‐type controls (Fig EV3C).
In order to determine the phenotype of BrdU‐positive cells after the 24‐h incubation period, brain sections were double‐labelled to detect the colocalization of BrdU with HuC/D (a neuronal marker; Fig EV3A, B, D and E), dlx5a/6a‐GFP (a GABAergic neuron marker; Fig EV3F and G) or NeuroD1 (a neuronal progenitor marker; Fig EV4, EV5, EV6) in the midbrain area of the 5 dpf zebrafish larvae. The number of cells positive for both BrdU and HuC/D (Fig EV3E), BrdU and dlx5a/6a‐GFP (Fig EV3G) or BrdU and NeuroD1 (Fig EV4D) in the midbrain area, surrounding the proliferation zone m, of the 5 dpf larvae was significantly lower in the chd7 −/− larvae, indicating suppressed neurogenesis and impaired GABAergic neuronal differentiation in chd7 mutants. Noteworthy, we found no change in the expression of the early glial marker scl1a3 (Glast in mammals) between chd7 −/− and control fish (Fig EV4E), suggesting no alterations in gliogenesis upon loss of chd7.
GABAergic dysfunction is the underlying cause of behavioural defects
The altered number of GABAergic neurons in the chd7 −/− mutant zebrafish brain suggests that these fish may have abnormal neural network that subsequently gives rise to abnormal behavioural outputs. Indeed, a hyperactive behavioural phenotype during dark cycles in zebrafish has been closely linked to disturbances in GABAergic signalling (Hoffman et al, 2016). We thus attempted to rescue/ameliorate the night‐time hyperactive phenotype by targeting GABA receptors with two agonists, muscimol and baclofen (targeting GABA‐A and GABA‐B receptors, respectively). Both agonists rescued the constitutive hyperactivity observed in chd7 −/− mutants (Fig 2I). We further reasoned that if GABAergic signalling was perturbed in chd7 −/− mutants, these animals would be more susceptible to show signs of seizure upon treatment with pentylenetetrazol (PTZ), a GABA‐A receptor antagonist that induces seizures in rodents and zebrafish (Baraban et al, 2005; Watanabe et al, 2010). As expected, PTZ treatment induced more severe seizures in chd7 mutants compared with wild‐type controls, for both the time of onset and overall locomotor activity during the 20‐min test period (Fig 2J and K). Altogether, our findings provide strong evidence that neurodevelopmental GABAergic signalling deficits underlie behavioural defects in chd7 −/− mutants.
Brain gene expression profile is altered in chd7−/− zebrafish
CHD7 controls DNA accessibility by remodelling chromatin via translocating nucleosomes, thereby influencing gene transcription of many genes in both a positive and negative manner (Martin, 2010; Schnetz et al, 2010; Bouazoune & Kingston, 2012). To identify the molecular pathways underlying the neurodevelopmental defects in chd7 −/− mutants, we thus performed an unbiased transcriptomic (RNA‐seq) analysis on zebrafish chd7 −/− larval brains (5 dpf) compared with wild‐type controls. This age was chosen to perform an exhaustive analysis of the molecular phenotype because it corresponds to a stage when the behavioural phenotype is distinct. We identified 1,251 genes expressed in the chd7 −/− larval brain that were significantly up‐ (677 genes) or downregulated (574 genes; P < 0.05; Fig 3A, Dataset EV1). Gene ontology analysis revealed that several biological processes (Fig 3B) and molecular functions (Fig 3C) are enriched in the differentially expressed genes such as binding, signalling, catalytic activity, cellular process, metabolic process and biological adhesion. Using pathway analysis, the expression of the significantly dysregulated genes was mainly assigned to the following pathways: MAPK signalling, cell adhesion, calcium ion signalling, lipid transport, haem binding, tryptophan metabolism and sterol synthesis (Table EV1). Analysis of the chromatin immunoprecipitation‐sequencing (ChIP‐seq) datasets from the ENCODE Transcription Factor Targets project (Rouillard et al, 2016) revealed that many of the dysregulated genes within the above‐listed pathways are likely direct targets of CHD7 in murine and human cell lines (Table EV1).
Figure 3. chd7 regulates GABAergic neuron development via MAPK/ERK signalling.
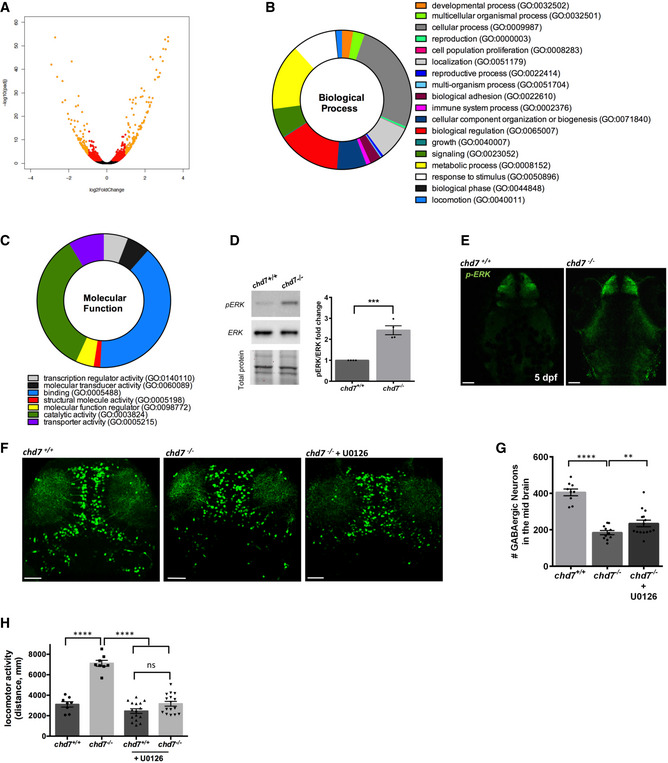
-
AVolcano plot showing each gene plotted according to its log2 fold change. All highly differentially expressed genes with P < 0.05 are in orange with fold change > 1.5.
-
B, CBiological processes (B) and molecular function (C) that are enriched in the differentially expressed genes.
-
DERK activation by phosphorylation is increased in mutants as shown by Western blot quantification (N = 4; ***P < 0.005; Student’s t‐test).
-
EpERK immunohistochemistry showing increased ERK activation in the mutant brain (N = 3, n = 8).
-
F, GTreatment with the ERK signalling inhibitor U0126 ameliorated the number of GABAergic neuron (n = 10–15; **P < 0.05; ****P < 0.0001, one‐way ANOVA).
-
HTreatment with the ERK signalling inhibitor U0126 rescues locomotor hyperactivity phenotype in chd7 mutant fish (n = 8 for chd7 +/+ and chd7 −/−; n = 16 for U0126‐treated fish; ****P < 0.0001; ns, not significant, one‐way ANOVA).
Data information: Data are presented as mean ± SEM. Scale bar = 50 μm. n represents number of fish used. N represents number of experimental repeats.
Downregulation of paqr3b in chd7−/− zebrafish contributes to GABAergic defects via MAPK/ERK signalling
Among the identified dysregulated pathways, MAPK signalling caught our attention the most based on its well‐known role in the pathogenesis of a wide range of neurodevelopmental disorders, including autism (Vithayathil et al, 2018). To complement our transcriptomic analysis, we thus decided to evaluate the status of MAPK/ERK signalling in chd7 −/− mutant brains and determine whether alteration of this pathway contributes to the observed GABAergic and behavioural defects. We detected a significant increase of phospho‐Erk1/2 (pERK) in 5 dpf chd7 mutant brains compared with wild‐type controls, using both Western blotting (Fig 3D) and immunostaining (Fig 3E). Additionally, treatment with a specific MEK/ERK inhibitor that prevents ERK phosphorylation (U0126) significantly increased the number of GABAergic neurons in chd7 −/− brains (Fig 3F and G) and reduced the hyperactive locomotor phenotype in chd7 −/− larvae (Fig 3H). Altogether, these data strongly suggest that CHD7 regulates GABAergic neuron development and behaviour via MAPK signalling.
Amongst the dysregulated genes in chd7 −/− brains that were assigned to the MAPK signalling pathway, we noted that 6 of them (paqr3b, flnb, nr4a1, dusp2, hspa8 and dusp16) were also identified as direct targets of CHD7 in murine and human cell lines tested in the ENCODE project (Table EV1). We further noted that paqr3b, an inhibitor of the MAPK/ERK signalling (Feng et al, 2007; Zhang et al, 2010), is normally highly expressed in the zebrafish brain (Fig EV5A and B), and the most dysregulated gene in our RNA‐seq dataset (Table EV1). Using RT–qPCR, we confirmed the strong downregulation of paqr3b in chd7 −/− mutant brains (Fig EV5C). Importantly, we also validated that this finding was relevant for human CS using previously described lymphoblastoid cell lines (LCLs) derived from a CHD7 mutation‐positive child and its unaffected parents (Bélanger et al, 2018). LCLs are especially useful for analysing molecular mechanisms relevant to CS (Bélanger et al, 2018). Accordingly, RT–qPCR analysis showed that PAQR3 gene expression was robustly decreased in CHD7 mutation‐positive LCLs compared with parental control LCLs (Fig 4A), while ChIP‐qPCR revealed that this decrease was associated with markedly reduced occupation of the PAQR3 proximal promoter by CHD7 (Fig 4B and C). These observations in human LCLs thus confirm that CHD7 directly regulates the expression of PAQR3.
Figure 4. CHD7 regulates PAQR3 expression in human cells.
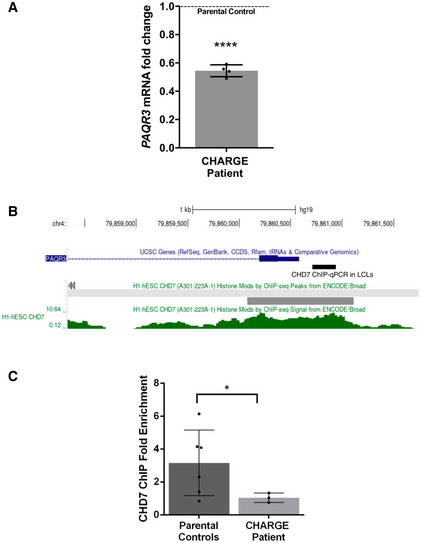
- qPCR analysis of PAQR3 expression in a CHD7 mutation‐positive patient compared with parental controls set (N = 4; ****P < 0.0001; Student’s t‐test). Fold change was calculated according to the method, using HPRT1 and RPS1 as housekeeping genes for normalization. All data were expressed as mean fold change ± SD across replicates, relative to control parents set to 1 (dotted line). N is the number of experimental repeats.
- Schematic view of PAQR3 exon 1 and proximal promoter on chromosome 4 (hg19 assembly), obtained with the UCSC genome browser (https://genome.ucsc.edu/) and showing the sequence amplified in ChIP‐qPCR assays in LCLs (thick black line) along with a previously described CHD7 ChIP‐seq peak (thick grey line) and signal (green) in H1‐hESC (ENCODE3).
- ChIP‐qPCR assays in LCLs showing decreased occupation of CHD7 on the PAQR3 proximal promoter in a CHD7 mutation‐positive patient (N = 3) compared with parental controls (N = 6); *P < 0.05; Student’s t‐test. All data were expressed as mean fold change ± SD. N is the number of experimental repeats.
Human PAQR3 is a regulator of ER (endoplasmic reticulum)‐to‐Golgi transport (Cao et al, 2018) that is essential for maintaining cellular and physiological homeostasis (Feng et al, 2007). To indirectly test this key role of PAQR3 in the context of GABAergic neuron development, we treated wild‐type zebrafish with the ER‐to‐Golgi transport blocker brefeldin A (BFA) (Donaldson et al, 1992). Using a low dose of BFA to affect ER‐to‐Golgi trafficking without inducing cell stress and death in wild‐type zebrafish (Le Corre et al, 2014), we found that BFA treatment was sufficient to recapitulate the decreased number and malpositioning of GABAergic neurons observed in chd7 −/− mutant brains (Fig 5A and B). Importantly, we found that overexpression of paqr3b mRNA in chd7 −/− mutants partially rescued the GABAergic neuron development defects at 3 dpf (Fig 5C and D) as well as restored pERK level to basal wild‐type level (Fig 5E). Of note, overexpression of paqr3b mRNA in zebrafish embryos did not alter their gross morphology and viability (Fig EV5D). Altogether, these findings strongly suggest that a CHD7‐PAQR3‐MAPK/ERK regulatory axis is especially important for proper development of GABAergic neurons.
Figure 5. Zebrafish paqr3b regulates GABAergic neuron development.
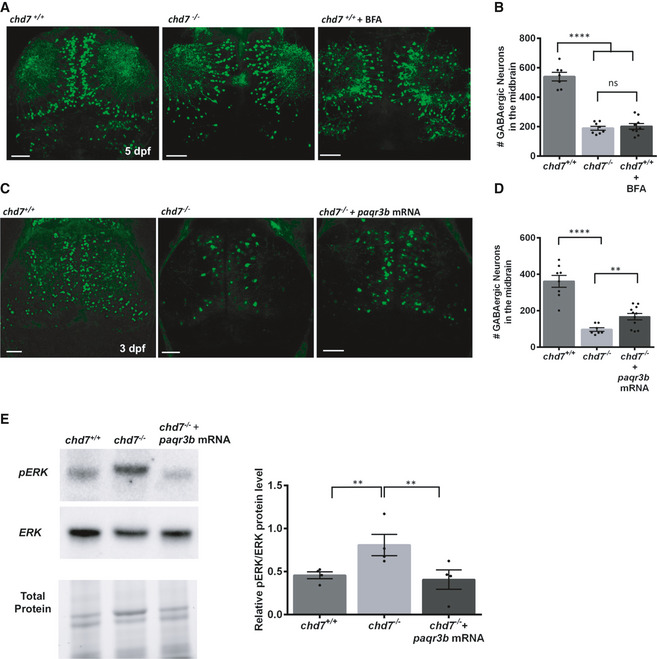
-
A, BGABAergic neuron defects in wild‐type fish treated with ER/Golgi traffic inhibitor BFA (n = 9; ****P < 0.0001; ns, not significant; one‐way ANOVA).
-
C, DOverexpression of paqr3b mRNA improve the number of GABAergic neurons in 3dpf chd7 mutant fish (n = 7 for chd7 −/− and n = 11 for chd7 −/− + paqr3b mRNA; ****P < 0.0001; **P < 0.05; One‐way ANOVA). Of note, the rescue experiment was performed at 3 dpf given the transient nature of mRNA.
-
EERK activation by phosphorylation in chd7 −/− mutants was restored to normal levels upon overexpression of paqr3b mRNA shown by Western blot quantification (N = 4; **P < 0.05; One‐way ANOVA, N is the number of experimental repeats).
Data information: Data are presented as mean ± SEM. Scale bar = 50 μm.
Ephedrine restores MAPK/ERK signalling and rescues GABAergic defects and associated behavioural anomalies
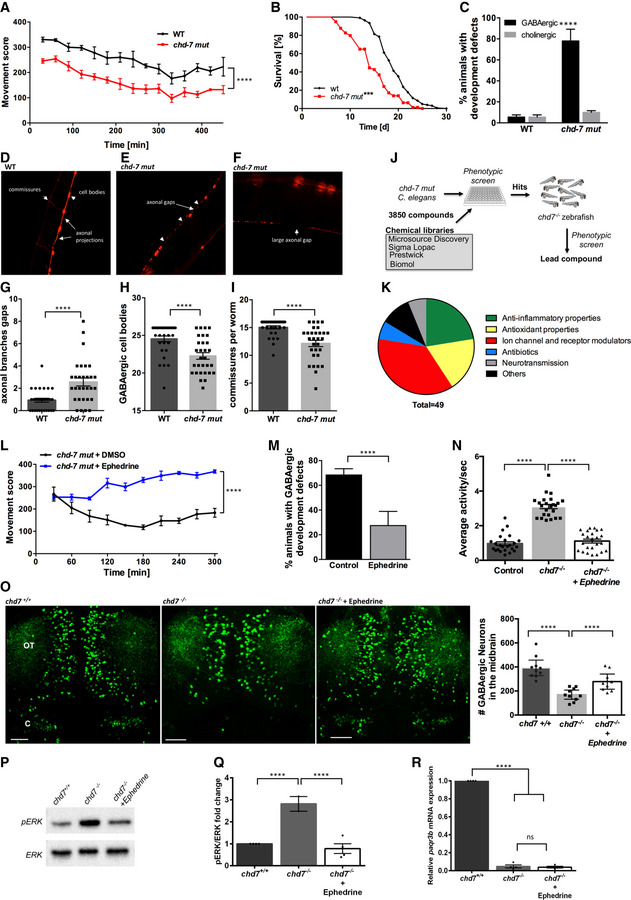
We recently demonstrated the power of combining simple genetic models such as worm (Caenorhabditis elegans) and zebrafish (Danio rerio) for identifying neuroprotective compounds that can rapidly be translated into preclinical and clinical testing (Patten et al, 2017). In C. elegans, loss‐of‐function of chd‐7 (Fig EV6A) leads to significant impairment of swimming locomotion when compared to wild‐type animals (Fig 6A), and reduced lifespan (Fig 6B). Locomotion of C. elegans is controlled by inhibitory GABAergic and excitatory cholinergic motor neurons (Zhen & Samuel, 2015). To visualize neurodevelopmental problems in these neurons, unc‐47p::mCherry and unc‐17p::GFP reporters for GABAergic and cholinergic motor neurons, respectively, were crossed into chd‐7 mutant animals (referred to chd‐7 mut hereafter). We found that GABAergic neurodevelopment is significantly impaired in chd‐7 mut when compared to wild‐type animals at the L4 stage, whereas the cholinergic motor system seems not to be affected by a mutation in the chd‐7 gene (Fig 6C). Particularly, a major neurodevelopmental problem in chd‐7 mut consists of interrupted connections in the GABAergic neuronal network, as indicated by significantly more axonal gaps and axonal loss/breaks larger than 50 µm (Fig 6D–G). The numbers of GABAergic cell bodies (Fig 6H) and commissures (Fig 6I) are slightly but significantly decreased to 22 and 12, respectively, compared with 26 and 16 in healthy worms. These observed phenotypes in chd‐7 mut worms are strikingly similar to those we previously reported in several C. elegans genetic models for ASD (Schmeisser et al, 2017).
Figure 6. GABAergic defects and behavioural anomalies are suppressed by ephedrine treatment in mut‐chd7 Caenorhabditis elegans .
-
AMovement scores of chd‐7(gk290) mutants (red) compared with N2 wildtype (WT; black). Two‐tailed t‐test was performed for statistical analyses, and movement score was considered different to WT (N = 3, n = 30; ****P < 0.0001, Student’s t‐test).
-
BLifespan analyses of chd‐7(gk290) mutants (n = 339; red) compared with WT (n = 444; black). Log‐rank test was performed for statistical analyses. ***P < 0.001.
-
CWT and chd‐7(gk290) animals with defects in the GABAergic (black) and cholinergic nervous system (grey) at the L4 stage (N = 4, n = 100; ****P < 0.0001; Student’s t‐test).
-
D–FExample pictures of the GABAergic nervous system cell bodies, commissures, axonal gaps and axonal breaks larger than 50 µm per worm in chd‐7(gk290) mutants expressing unc‐47p::mCherry.
-
G–IGABAergic neurodevelopmental defects of WT (black) and chd‐7(gk290) mutants (grey) at the L4 stage classified in axonal gaps per worm (G), number of GABAergic cell bodies (H) and number of commissures (I). N = 4, n = 100; ****P < 0.0001, Student’s t‐test.
-
JChemical libraries (3,850 compounds) were first screen in chd‐7(gk290) C. elegans mutants and positive hits were tested on chd7 zebrafish mutants.
-
K49 compounds that improved motility phenotypes in chd‐7(gk290) mutants were identified. These compounds were clustered in 6 main functional categories.
-
LMovement scores of chd‐7(gk290) mutants treated with ephedrine (blue) compared with the solvent DMSO (black). N = 3, n > 100; ****P < 0.0001, Student’s t‐test.
-
MDefects of the GABAergic nervous system at the L4 stage in chd‐7(gk290) mutants treated with ephedrine (grey) compared with DMSO (black) (N = 3, n > 100). ****P < 0.0001, Student’s t‐test.
-
NAverage activity per second is wild‐type and mutants treated with ephedrine compared with non‐treated mutant (wild‐type treated with ephedrine is referred to as Control). Ephedrine suppresses hyperactivity in chd7 mutant fish. n = 24; ****P < 0.0001, one‐way ANOVA.
-
ORepresentative images of GABAergic neurons and total number of GABAergic neurons (GFP+ cells) in the optic tectum (OT) and cerebellum (CB) regions of wild‐type, chd7 mutant and ephedrine‐treated fish (n = 9; ****P < 0.0001; One‐way ANOVA). Treatment with ephedrine ameliorated the number of GABAergic neurons.
-
P, QpERK is reduced in mutants following treatment with ephedrine as shown by Western blot quantification (N = 4). ****P < 0.0001, one‐way ANOVA.
-
RqPCR analysis of paqr3b expression in chd7 +/+ , chd7 −/− and chd7 −/− treated with ephedrine (N = 4; ****P < 0.0001; ns, not significant, one‐way ANOVA).
Data information: Data are presented as mean ± SEM. Scale bar = 50 μm. n is the number of fish or worms used. N is the number of experimental repeats.
We next exploited the impaired locomotion phenotype in chd‐7 mut worms to perform a comprehensive drug screen with 3,850 compounds (Fig 6J). We identified 49 compounds with beneficial effects that partially corrected the impaired locomotion of chd‐7 mut worms (Table EV2). Based on their suggested function, these compounds could be clustered into six main categories (Fig 6K). From all positive substances, three that improved swimming movement particularly well were chosen for further investigation: fisetin, meloxicam and ephedrine. The compounds were retested in worms and validated in our zebrafish model (Fig 6J) at various concentrations ranging from 1 to 50 μM. We confirmed two active compounds in zebrafish and identified ephedrine as the most potent lead compound. Ephedrine was found to significantly improve behavioural and GABAergic defects in both C. elegans (Fig 6L and M) and zebrafish (Fig 6N and O). Ephedrine also improved the Vth cranial nerve branching defects in zebrafish (Fig EV6).
To test whether ephedrine is exerting its beneficial effects by correcting the aberrant MAPK/ERK signalling in chd7 −/− mutants, we examined the level of pERK in these fish at 5dpf and found a significant reduction upon ephedrine treatment (Fig 6P and Q). Our findings thus support a model whereby development of GABAergic neurons in zebrafish is regulated by chd7 via MAPK/ERK signalling. However, ephedrine treatment did not affect the level of paqr3b expression in chd7 −/− mutants (Fig 6R), suggesting that ephedrine is acting downstream in the MAPK/ERK signalling cascade.
Discussion
Despite advances in genetic studies of CS, the underlying mechanisms for the neurological deficits in this disease remain poorly understood. In this study, we showed that loss‐of‐function of the chromatin remodeller‐coding gene chd7 disrupts normal number of the inhibitory GABAergic neurons in the zebrafish brain. Importantly, we also discovered that these GABAergic neuron defects result in behavioural anomalies that occur via an ERK‐dependent mechanism. Furthermore, through a phenotype‐based drug screening strategy, we identified a clinically approved drug, ephedrine, which proved to be very effective at correcting increased MAPK/ERK signalling, GABAergic defects and behaviour deficits caused by chd7 loss‐of‐function.
We observed a significant reduction in the number of GABAergic interneurons in the zebrafish chd7 −/− brain during development. GABAergic interneurons play an essential role in neural circuitry and behaviour. Proper differentiation of GABAergic neurons during brain development is important for establishing anatomical and functional circuitry. We found that the hyperactivity behavioural phenotype of chd7 −/− mutant fish could be suppressed by modulating GABAergic signalling with agonists. These findings suggest that the abnormal behaviour due to loss of chd7 in our mutants is likely in part due to defects in GABAergic network development. Interestingly, our observations are consistent with an ASD‐related phenotype recently described in zebrafish mutants of the ASD risk gene, CNTNAP2 (Hoffman et al, 2016). Patients with CHARGE syndrome frequently exhibit autism‐like behaviour (Hartshorne et al, 2005; Smith et al, 2005). Several studies have shown that GABAergic neurons and circuits are altered in ASD (Rubenstein & Merzenich, 2003; Coghlan et al, 2012) and the dysfunction of inhibitory GABAergic circuits has been proposed as a cause for ASD (Pizzarelli & Cherubini, 2011). Our work suggests that the overlapping symptom of autistic features in CS and ASD may share a common neurobiological pathway implicating improper GABAergic development and function.
Noteworthy, we did not observe any abnormalities in chd7 heterozygous mutants at the larval developmental stages that we studied. This is likely due to some teleost‐specific genetic compensation mechanisms occurring in heterozygous background. Such a lack of phenotype in the heterozygous mutant population has also been observed while modelling several autosomal dominant diseases in zebrafish such as Dravet syndrome, hyperekplexia and juvenile myoclonic epilepsy, where disease‐related phenotypes are only recapitulated in a homozygous mutant (−/−) background (Samarut et al, 2018; Samarut et al, 2019; Sourbron et al, 2019). It is, however, plausible that CHARGE‐related behavioural changes may occur in juvenile or adult chd7 heterozygous mutants and this warrants further studies.
PAQR3 negatively regulates Raf/MEK/ERK signalling (Feng et al, 2007; Zhang et al, 2010). The downregulation of paqr3b expression in chd7 −/− fish likely results in overactive Raf/MEK/ERK signalling, and hyperphosphorylation of ERK subsequently has detrimental consequences on neuronal network development. Xu et al (2016) showed PAQR3‐deleted mice display motor and behavioural abnormalities. It is thus possible that paqr3b is important for neural connectivity and motor function in vertebrates. Aberrant signalling through the MAPK/ERK pathway is involved in the pathogenesis of neurodevelopmental syndromes that involve autism, intellectual disability, neurodevelopmental delay and seizures (Vithayathil et al, 2018). Perturbation in MAPK/ERK activity in chd7 −/− mutants is consistent with findings observed in genetic models of ASD. Recent work by Holter et al (2021) revealed that hyperactive ERK signalling during mouse development affects the number of GABAergic interneurons. In our study, we similarly observed that with inhibition of ERK phosphorylation ameliorated the defective behaviour and number of GABAergic neurons. However, other mechanistic pathways under the control of CHD7 may also be important in GABAergic network development and this warrants further investigation. Indeed, we showed that the number of GABAergic neurons in chd7 −/− fish can be rescued by overexpressing paqr3b mRNA. PAQR3 is a key player in regulating ER‐to‐Golgi transport (Cao et al, 2018) and perturbing the secretory pathway may affect neurogenesis.
Ephedrine is both an α‐ and β‐adrenergic agonist (Vansal & Feller, 1999; Ma et al, 2007). Our findings showed that this adrenergic receptor agonist significantly reverses GABAergic defects as well as abnormal behaviour in our models. Adrenergic receptors (ARs), in particular α1‐AR, have also been shown to play a critical role in regulation of neurogenesis (Gupta et al, 2009). For instance, in neonates, α1‐ARs are important for the differentiation of neural progenitors into catecholaminergic neurons and GABAergic interneurons (Gupta et al, 2009). It has been suggested that α1‐AR is required in the neuronal maturation stages of neurogenesis by regulating the levels of the Dlx2, Mash1, NeuroD and bHlH mRNA (Gupta et al, 2009). Importantly, α1‐ARs colocalize with both GABAergic and NMDA receptor‐containing neurons and are likely to be involved in their regulation (Papay et al, 2006). As previously shown (Ferraro et al, 1993), α1‐ARs can modulate GABA release in the human cerebral cortex and they have also been shown to regulate CA1 GABAergic interneurons in the rat hippocampus (Bergles et al, 1996). Interestingly, AR agonist treatment can significantly increase the excitability of GABAergic interneurons (Bergles et al, 1996; Marek & Aghajanian, 1996; Kawaguchi & Shindou, 1998; Papay et al, 2006) while having a contrasting effect in pyramidal cells (Papay et al, 2006). An α1‐AR‐mediated facilitation of spontaneous GABA release from interneurons has been observed in several brain regions (Bergles et al, 1996; Kawaguchi & Shindou, 1998). It is possible that the increase in GABAergic signalling by ephedrine in our model is likely one of the underlying mechanisms in ameliorating the phenotypic anomalies in chd7 mutants. Ephedrine acts as both a direct and indirect sympathomimetic. Its primary mode of action is achieved indirectly, by inhibiting neuronal norepinephrine reuptake and by displacing more norepinephrine from storage vesicles (Wellman et al, 2003; Becker, 2012). In addition to norepinephrine, detection of dopamine has also been reported under ephedrine‐stimulated conditions in the CNS (Ruwe et al, 1985). Interestingly, dopaminergic signalling has been shown to regulate GABAergic neuron development and motor behaviour in zebrafish (Souza et al, 2011). The next step will be to unravel ephedrine’s exact target and mechanism of action in our chd7 models to increase the number of GABAergic neurons and to ameliorate behaviour.
In conclusion, we show for the first time that chd7 controls GABAergic network development in zebrafish brain via regulating paqr3b. We also provide novel insight on the pathogenic mechanisms—from molecular pathway to brain circuits and behaviour—associated with chd7 loss‐of‐function with relevance to CS and ASD. Five of nine members of the CHD family proteins have been implicated in neurodevelopmental disorders (Goodman & Bonni, 2019). Our findings may additionally be relevant to other neurodevelopmental diseases such as autism and epilepsy with which mutations in chromatin remodellers such as CHD8 and CHD2 have been associated. In this study, we also identify a clinically approved compound and its action on MAPK/ERK signalling that may be therapeutic avenues to be further explored in CHARGE syndrome. Overall, our work suggests that in vivo drug screening and experimental analysis of simple genetic models could prove extremely valuable in understanding and perhaps ultimately aid in developing treatments for certain neurological features associated with CHD7 deficiency.
Materials and Methods
Zebrafish
Fish husbandry
Adult zebrafish (D. rerio) were maintained at 28°C at a light/dark cycle of 12/12 h in accordance with Westerfield zebrafish book (Westerfield, 1993). Embryos were raised at 28.5°C, and collected and staged as previously described (Kimmel et al, 1995). The zebrafish lines used in this study were wild‐type, chd7 mutants, Tg(dlx5a/6a:GFP) which was obtained from the laboratory of Marc Ekker and used to generate Tg(dlx5a/6a:GFP;chd7 −/−) fish. All experiments were performed in compliance with the guidelines of the Canadian Council for Animal Care and the local ethics committee of INRS. For imaging studies, pigment formation was blocked by adding 0.003% phenylthiourea (PTU) dissolved in egg water at 24 h after fertilization (hpf).
CRISPR/Cas9‐generated mutagenesis
A guide RNA (gRNA) targeting the helicase domain of the chd7 gene was designed using the online tool CRISPRscan (TGTATTCCTGCTGTGCACAAGGG; PAM site underlined). Synthesis of gRNA and of Cas9 mRNA was performed as previously described (Swaminathan et al, 2018). Cas9 mRNA was synthesized using the mMESSAGE mMACHINE T3 kit from pT3TS‐nCas9n plasmid (Addgene #46757) linearized with Xba1. A volume of 1 nl containing a mix of 100 ng/µl Cas9 mRNA and 30 ng/µl gRNA was injected into one‐cell stage embryos using the Picospritzer III pressure ejector. Genotyping of chd7 +/+ (wild‐type), chd7 +/− (heterozygous) and chd7 −/− (homozygous) fish was performed by high‐resolution melting (HRM) analysis using genomic DNA extracted by boiling larva/clipped caudal fin in 50 mM NaOH for 10 min and then neutralized in 0.1 M Tris–HCl (pH8).
Rescue experiment was performed using paqr3b (NM_001030148.2) zebrafish open reading frame cloned into a pCS2+ expression vector. In vitro transcription was done using the SP6 message machine kit (Ambion), and 1 nl of paqr3b mRNA (40 ng/µl) was injected into the 1‐cell stage embryos.
Gross morphology and survival assessment
Larvae (chd7 +/+, chd7 +/− and chd7 −/−) were assessed for their survival rate and morphological phenotypes. The sample sizes for the different genotypes were as follows: three different batches (N = 5) each batch containing 30 larvae (n = 30) for chd7 +/+, chd7 −/− larvae and N = 3, n = 18–19 for chd7 +/− larvae. Gross morphology was observed under a stereomicroscope (Leica S6E). The head size at 3 dpf was measured using the software, ImageJ. Briefly, a straight line was drawn between the lowest point of the otolith and the anterior end of the brain, around the upper jaw area.
Neuronal network analysis
To visualize the axonal tracts, fluorescent immunohistochemistry was performed using the marker acetylated α‐tubulin (Sigma‐Aldrich; Cat# T7451). 28 hpf fish were fixed in Dent’s fixation (80% methanol and 20% DMSO) overnight (O/N) at 4°C. Samples were rehydrated in 75, 50 and 25% methanol in PBST for 30 min each. They were then washed 4 times in PBST for 30 min (twice under agitation and twice without followed by blocking in 10 % normal goat serum (NGS) and 2 % bovine serum albumin (BSA) in PBST for 1 h at room temperature (RT), under agitation). Primary acetyl‐tubulin monoclonal mouse antibody was added to the blocking (1:500) and incubated overnight at 4°C. After washing the primary antibody at least six times 30 min in PBST and blocking for 1 h, the secondary antibody Alexa 488 goat anti‐mouse (Sigma‐Aldrich; Cat# SAB4600387) was added O/N at a ratio of 1:1,000. Secondary antibody was washed and samples are mounted laterally for 28 hpf. Imaging was done on confocal microscope (Zeiss LSM780). The projection images were semi‐automatically traced with NIH ImageJ using the NeuronJ plug‐in. The total length of processes in each individual embryo was subsequently measured and analysed.
Craniofacial cartilage staining
To visualize the craniopharyngeal cartilage, Alcian blue staining was applied. Larvae were fixed in 4% paraformaldehyde (PFA) in PBST (one pellet in 200 ml dH2O and 0.1% Tween). Fish were fixed at 6 dpf for 5 h at RT. Then, the samples were dehydrated in methanol solution with increasing concentration; 25, 50 and 75% in PBST (10–15 min each) and stored at −20°C until use. Before the staining was started, the samples were rehydrated in the reverse order of methanol concentrations for 15 min each. After three quick washes in PBST, the samples were incubated in 0.1% Alcian blue solution with 70% ethanol (EtOH) and 0.37% HCl for 2 h, under agitation. Then, they were washed in EtOH and HCl solution; three quick washes and then twice 15 min. The larvae were digested in 10% trypsin in 30% saturated borax water which was pre‐warmed a 42°C. At the end, the samples were washed overnight at 4°C in 0.1% KOH in H2O. The following day, they were washed in 0.25% KOH for 1 h and stored in 80% glycerol and 0.25% KOH solution at 4°C until being imaged. Imaging was done on the Leica stereomicroscope (Leica S6E).
Behavioural analysis
Larvae (5 dpf) were separated into single wells of a 96‐well plate containing 200 µl of E3 media and habituated in the Daniovision® recording chamber (Noldus) for 1 h before start of experiment. Larval locomotor activity was monitored over light‐dark cycles using the Daniovision® apparatus. Analysis was performed using the EthoVision XT12 software (Noldus) to quantify the total swimming distance in given hours and the locomotor activity per second.
GABAergic neurons: confocal imaging and data analysis
To image and quantify GABAergic neurons (dlx5a/6a:GFP+ neurons), zebrafish larvae (1, 2, 3 and 5 dpf) of Tg(dlx5a/6a:GFP;chd7 +/+) and Tg(dlx5a/6a:GFP;chd7 −/−) were fixed in 4% PFA for 2 h. Zebrafish larvae (1, 2 and 3 dpf) were ventrally mounted for brain imaging, and in case of 5 dpf larvae, the brains were first dissected and then dorsally or ventrally mounted for imaging. Z‐stack images were taken using a Zeiss LSM780 confocal microscope (Carl Zeiss, Germany). GABAergic neurons (dlx5a/6a:GFP+ neurons) were counted in the brain regions of wild‐type and mutant larvae manually and blindly using the Cell Counter plug‐in for Fiji/Image J (NIH) Imaging software.
Bromodeoxyuridine (BrdU) labelling
Zebrafish larvae at 4 dpf were incubated in a 10 mM solution of BrdU (BD Biosciences; Cat# 550891) in fish water at a temperature of 28.5°C for 24 h. All of the larvae were then fixed at 5 dpf in 4% PFA O/N at 4°C and used to perform cryo‐sections of the brain.
Zebrafish brain cryo‐sections and double‐immunohistochemistry
Fixed larvae (5 dpf) were given serial sucrose treatment with 15 and 30% sucrose in 1X PBS, till the larvae sank to the bottom. The fish brains were then cryo‐sectioned (transverse section) in 10‐micron‐thick sections and dried at room temperature for 20 min, and frozen to store.
For the immunostaining, the 5 dpf zebrafish embryo brain sections were first post‐fixed in acetone at −20°C for 20 min. The sections were then washed with PBS for 15 min and processed for epitope retrieval with Tris–HCl (pH 8.2, 50 mM) at 85°C for 6‐min treatment (for HuC/D, NeuroD1) and additionally with HCl (4 N) at 37°C for 10 min followed by Sodium borate (0.1 M) washes for 20 min (for BrdU). Sections were then washed in 0.5% PBS‐Triton for 30 min, blocked in 10% NGS for 1 h at room temperature and then incubated in primary antibodies: HuC/D at 1:50 (Invitrogen; A21271); NeuroD1 at 1:500 (Abcam; ab60704), BrdU at 1:250 (Abcam; ab152095) and anti‐GFP at 1:250 (Invitrogen; GF28R) diluted in 5% NGS, 1% BSA in 0.1% PBS‐Triton, O/N at 4°C. The following day sections were washed in 0.3% PBS‐Triton and incubated with species‐specific secondary antibodies coupled to Alexa Fluor 488 or 555 (Invitrogen) diluted in 0.1% PBS‐Triton for 2–3 h at room temperature, followed by washes with 0.3% PBS‐Triton and mounted in DAPI glue (Invitrogen; Cat# P36941).
For the quantitative analyses, cells stained for BrdU, NeuroD1, GFP (for Dlx5/6) or HuC in the zebrafish midbrain area were taken under 40× magnification with an oil immersion lens using a Zeiss confocal microscope (LSM780; Carl Zeiss, Germany). The images were then processed with ZEN software (Carl Zeiss). Stained cells in consecutive sections from three brains per genotype (N = 3) were counted using ImageJ (NIH) and used to calculate the total number of double‐positive stained cells relative to BrdU‐positive cells in each larval brain section. Of note, the total number of sections (n) used varied between experiments. Neuroanatomical designations are taken from the Atlas of Early Zebrafish Brain Development (Mueller & Wullimann, 2015).
Haematoxylin and Eosin staining
For Haematoxylin and Eosin staining, brain sections were post‐fixed in 10% formol (Chaptec) for 5 min and rinsed with tap water. The sections were stained with Haematoxylin (StatLab) for 4 min, washed with alcohol‐acid, and were rinsed with tap water. The sections were then soaked in saturated Lithium Carbonate solution for 10 s and then rinsed it with tap water. Finally, staining was performed with Eosin Y (StatLab) for 2 min, and mounted under coverslip with permount mounting media. Neuroanatomical designations are taken from the Atlas of Early Zebrafish Brain Development (Mueller & Wullimann, 2015).
Drug treatment
Drugs were purchased from Sigma‐Aldrich, and stock solutions were prepared: Pentylenetetrazol (PTZ)—Muscimol‐30 mM, Baclofen‐15 mM, U0126‐10 mg/ml and Brefeldin A(BFA)—10 mg/ml. Embryos were treated from 8 hpf for GABA agonists Muscimol and Baclofen, and at 2 dpf for 20 min with 3 mM PTZ (Sigma‐Aldrich). For treatment with BFA, an established low dose concentration 3.56 μM (1 μg/ml; Sigma‐Aldrich) without inducing cell stress and death in zebrafish was used and added at 24 hpf (Le Corre et al, 2014). For treatment with U0126, a non‐toxic low dose of 4 μM (1.7 μg/ml; Sigma‐Aldrich) concentration was used and added at 8 hpf (Hawkins et al, 2008; Guo et al, 2015). The water was replaced every day with fresh water containing final concentration of the drug, until the activity measurement and/or brain imaging. For the chemical genetic screens, zebrafish embryos from 8 hpf were treated with fisetin, ephedrine and meloxicam (1–50 μM; all purchased from Sigma‐Aldrich) in E3 medium. The medium was replaced every day with fresh solution containing final concentration of the drug, until the locomotor activity measurement and/or imaging.
Western blotting
Larvae were collected at 5 dpf and placed in the dark for at least 1 hr, following which lysates were rapidly prepared by homogenization in high salt lysis buffer containing 150 mM NaCl, 50 mM Tris–HCl pH 7.5, 1% Triton, 0.1% SDS, 1% sodium deoxycolate and protease inhibitors cocktail (1:10, Sigma‐Aldrich). The lysates were centrifuged at 18,300 g for 10 min at 4°C. The supernatant was collected, and protein concentration was estimated using Bradford assay (Bio‐Rad). Western blotting was performed using 20 μg lysate per sample which were resolved on a 7.5% SDS‐polyacrylamide gel (Bio‐Rad). After electrophoresis, proteins on the gel were electrotransferred onto PVDF mini‐membranes (Bio‐Rad). The membranes were blocked with 5% non‐fat milk solution in 1X phosphate‐buffered saline or with 5% bovine serum albumin (Sigma) in 1X Tris‐buffered saline for immunoblotting with anti‐ERK (1: 2,000, Cell Signaling Technologies; Cat# 9102) and anti‐pERK (1: 2,000, Cell Signaling Technologies; Cat# 9101), respectively. Detection was performed using goat anti‐mouse and goat anti‐rabbit antibodies, respectively, conjugated with horseradish peroxidase. Bands were visualized with ECL and imaged using ChemiDoc (Bio‐Rad).
TUNEL assay
Whole‐mount TUNEL staining to determine apoptosis was performed on 2 and 5 dpf larvae as previously described (Jamadagni & Patten, 2019). Briefly, larvae were fixed in 4% PFA and then serially dehydrated and rehydrated with 25, 50 and 75% MeOH in PBST (0.1% Tween) and rinsed with PBST several times. The embryos were then digested with Proteinase K (10 μg/ml) for 20 min, followed by rinses with PBST and re‐fixed with 4% PFA for 20 min. This was followed by two quick washes and three long washes of 20 min. in PBST (1% Triton X), then were rinsed again with PBS and incubated in TUNEL reaction mix (as directed by the manufacturers; Roche/Sigma‐Aldrich) for 1 h at 37°C. The larvae were then mounted and imaged under a Zeiss LSM 780 confocal microscope.
Whole‐mount fluorescence immunohistochemistry
Whole‐mount fluorescence immunohistochemistry for proliferation (pH3 marker) was performed as previously described (Verduzco & Amatruda, 2011). Briefly, 2 and 5 dpf zebrafish larvae were fixed in 4% PFA overnight at 4°C. After fixation, the embryos were rinsed two times for 10 min each with PBST (0.1% Tween). They were then incubated with acetone (100%) at −20°C for 7 min. Following this, the larvae were rinsed with PBST (0.3% Triton X) and PBS‐DT (1% BSA, 1% DMSO, 1% Triton X) and further blocked in 5% NGS in PBS‐DT for 1 h. Primary antibody pAb Rabbit Anti‐phospho‐Histone H3 (Ser10) (1:250; Millipore, Cat# 06–570) was added to the blocking solution and incubated overnight at 4°C. The next day, larvae were rinsed with PBS‐DT and secondary antibody (Alexa Fluor 488, 1:1,000; Invitrogen) was then added and incubated overnight at 4°C. Larvae were rinsed and imaged with a Zeiss LSM 780 confocal microscope.
For pERK staining, zebrafish larvae (5 dpf) were placed in the dark for 1.5–2 h prior to rapid fixation in 4% PFA O/N. The larvae were then rinsed multiple times with PBST (0.1% Tween) followed by incubation in acetone (100%) at −20°C for 15 min. The larvae were then rinsed with PBST (0.3% Triton X) and PBS‐DT (1% BSA, 1% DMSO, 1% Triton X) and further blocked in 5% NGS in PBS‐DT for 1 h. Primary antibody anti‐phospho‐ERK (1:500, Cell Signaling Technologies; Cat# 9101) was added to the blocking solution and incubated overnight at 4°C. The following day, larvae were rinsed with PBS‐DT and incubated with secondary antibody (Alexa fluor 488, 1:1,000; Invitrogen) overnight at 4°C. Larvae were rinsed and imaged with a Zeiss LSM 780 confocal microscope.
Transcriptomic, differential expression and pathway analyses
Three independent batches of 5 dpf chd7+/+ and chd7 −/− larvae were dissected to extract the whole brain, by each of two experimenters, corresponding to experimental triplicates. Total RNA was extracted from these flash‐frozen brains using PicoPure RNA extraction kit (Thermo Fisher Scientific) following the manufacturer's standard protocol. For each sample, RNA extraction was made from five whole brains. The absence of contamination was assessed by Nanodrop using 260/280 and 260/230 ratios. Quality of total RNA was assessed with the BioAnalyzer Nano (Agilent), and all samples had a RIN above 8.3. Library preparation was performed using the TruSeq RNA (Illumina). Eight PCR cycles were required to amplify cDNA libraries. Libraries were quantified by Nanodrop and BioAnalyzer. All libraries were diluted to 10 nM and normalized with the Miseq SR50 v2. Libraries were pooled to equimolar concentration and multiplexed by six samples per lane. Sequencing was performed with the Illumina Hiseq2000 using the SBS Reagent Kit v3 (100 cycles, paired‐end) with 1.6 nM of the pooled library. Cluster density was targeted at around 800k clusters/mm2. Between 75 and 140 million reads were generated for each sample. Library preparation and sequencing was done at the genomics platform of the Institute for Research in Immunology and Cancer (University of Montreal). More than 93% of high‐quality reads were mappable onto the zv9 version of the zebrafish genome (ensemble release 77) using TopHat version 2.0.10. Differential gene expression analysis was assessed by DeSeq2 package using R software. Genes showing an absolute fold change >1.2 and an adjusted P value (false discovery rate) < 0.05 were considered to be significantly differentially expressed. Gene enrichment and pathway analysis were performed using PANTHER and DAVID bioinformatics resources (Huang da et al, 2009; Mi et al, 2019). The RNA‐seq has been deposited to the GEO database (GSE139623).
RT–qPCR
RT–qPCR was performed as previously described in Breuer et al, (2019). In short, RNA was isolated from ~ 30 embryos using TriReagent® (Sigma) according to manufacturer's protocol. 1 µg of RNA was used for cDNA synthesis by SuperScript®Vilo™ kit (Invitrogen). RT–qPCR was run with SYBR Green Master Mix (Bioline) using the LightCycler® 96 (Roche). ef1a was used as the reference gene for normalization and following primers were used for paqr3b: FW: 5' – CGCTGGCTTGCTCTGGATTA – 3'; RV:5' – CCTGCCTCCAAAAACTGTTGC – 3'.
Lymphoblastoid cell lines
Cell culture
Lymphoblastoid cell lines (LCLs) from a CHD7 mutation‐positive (c.5050+1G>T) CHARGE syndrome patient and its unaffected parents were maintained in RPMI medium as described previously (Belanger et al 2018). Families provided informed consent on studies approved by the respective institutional review board of the Baylor College of Medicine (experimental cohort for this study).
RT–qPCR (LCLs)
Relative transcript levels of PAQR3 mRNA in LCLs were analysed by RT–qPCR. The RT–qPCR analyses were performed as described above. The following primers were used for PAQR3: (Forward) 5′‐ CCAATTACCTCACGCAGCAA‐ 3′ and (Reverse) 5′‐GGAGCACCAATTCCTCCATT‐ 3′. Fold change was calculated according to the method, using HPRT1 and RPS1 as housekeeping genes for normalization. All data were expressed as mean fold change ± SD across replicates, relative to control parents set to 1.
ChIP assays
ChIP‐qPCR assays in 2 × 108 LCLs were performed as previously described (Belanger et al, 2018), using 2 µg of rabbit anti‐CHD7 antibody (Cell Signaling; Cat# 6505) and 2 µg of rabbit anti‐HA antibody (Abcam; Cat# ab9110) as negative control. Target sequence (196 bp) at the PAQR3 proximal promoter was determined based on a previously described ChIP‐seq peak of CHD7 in H1‐hESC (Sethi et al, 2020) that was downloaded from the ENCODE portal (Sloan et al, 2016) (https://www.encodeproject.org/); the relevant track (#ENCFF628RLE) was visualized on the UCSC genome browser using the GRCh37/hg19 human assembly (https://genome.ucsc.edu/). Primer sequences were: (Forward) 5′‐ GCT ACA GGC GAA TAC AAG TGG ‐ 3′ and (Reverse) 5′‐ CTG ACT TCA GCT TAG AAA TCC TC ‐ 3′). ChIP‐qPCR efficiency was calculated in % of CHD7 IP relative to input and expressed in fold enrichment relative to HA negative control.
Caenorhabditis elegans
Caenorhabditis elegans strains and maintenance
Caenorhabditis elegans was handled applying standard conditions (Stiernagle, 2006). Worms were kept on NGM agar with an Escherichia coli OP50 lawn at 15°C for maintenance and 20°C for assays. The mutant strain chd‐7(gk290) (VC606) was provided by the Caenorhabditis Genetics Center at the University of Minnesota and backcrossed four times to N2 wildtype (WT).
Caenorhabditis elegans locomotion assay
A worm tracking machine (Wmicrotracker, Phylum Tech) was used to track the swimming locomotion of C. elegans. Assays were performed in 96‐well‐microtiter plates with approximately 30 worms and 100 µl M9 buffer per well. Escherichia coli OP50 were added to the wells to prevent worms from starving. Each microtitre well was crossed by two infrared light rays from top to bottom and a detector determined interruptions of these light rays by worms moving in the well. From the signal, a movement score was calculated, which is defined as animal movement in a fixed time period. Movement was tracked for 10 h. The assay was performed in triplicates and movement scores of chd‐7(gk290) over time were compared with WT in three independent experiments using two‐tailed t‐test to determine significance.
Drug libraries
Natural and FDA‐approved compounds were provided by Sigma‐Aldrich (Sigma‐Aldrich's Library of Pharmacologically Active Compounds (Lopac) 1280 library, containing compounds as of July 2015, Oakville/Canada), Prestwick Chemicals (Prestwick Chemical Library; containing compounds as of March 2015, Illkirch/France), MicroSource (880 compounds, Gaylordsville, CT/USA), Enzo Life Science, Inc. (BML‐2865 Natural Products Library, containing compounds as of April 2015, Farmingdale, NY/USA). All compounds were dissolved in DMSO and tested at a concentration of 20 µM.
Drugs screen in C. elegans
Chd‐7(gk290) nematodes were exposed to the drugs from the libraries in microtitre wells at a concentration of 20 µM in M9 buffer with OP50 and movement was tracked for at least 4 h. The average movement score of C. elegans treated with a specific drug was compared with the DMSO control and the average movement score of the whole plate. If values for a drug were higher than the respective controls, a second screen to validate the increased movement due to the specific drug was performed. Compounds that increased locomotion of chd‐7(gk290) significantly according to a two‐tailed t‐test in the second screen were counted as positive. A complete list of all drugs tested can be found in reference (Schmeisser & Parker, 2017).
Assessment of neuronal integrity of GABAergic and cholinergic motor neurons in C. elegans
An unc‐47p::mCherry and unc‐17p::GFP reporter for GABAergic and cholinergic motor neurons was crossed into chd‐7(gk290) to analyse motor neurodevelopment at the L4 stage in vivo. Worms were put on microscopy slides with 2% agarose pads, paralysed with 5 mM levamisole and covered with a coverslip. Neuronal examinations were performed with a Zeiss Axio Imager M2 microscope (unc‐47p::mCherry) and a Leica DM6000 microscope (unc‐17p::GFP). About 100 worms were analysed in four independent experiments and compared with WT.
For a more detailed neuronal status in chd‐7(gk290), neuronal morphology was categorized into the following groups: gaps in the axonal branches per animal, number of axon commissures per animal, number of GABAergic and cholinergic cell bodies, and frequency of breaks > 50 µm in the nerve cords, which goes along with massive loss of neuronal tissue. About 30 chd‐7(gk290) worms with dysmorphic neuronal morphology were scored and compared with the neuronal morphology of WT worms.
Testing of beneficial compounds identified in the drug screen was performed as follows: NGM agar plates containing 20 µM of the respective substance were freshly prepared prior to each assay and seeded with OP50. Young adult C. elegans were allowed to lay eggs for about 4 h, and assays for neuronal morphology were performed in the progeny at the L4 stage as described above. Mean ± standard deviation of three independent experiments were calculated and two‐tailed t‐tests determined significance. Gaps in axonal branches, number of axon commissures, cell bodies and breaks > 50 µm in the nerve chords were determined in three independent experiments. Average and standard deviation of all tested animals were calculated and two‐tailed t‐tests were performed.
Caenorhabditis elegans lifespan assay
Lifespan assays were performed as previously described (Schmeisser et al, 2013). Briefly, worms were synchronized at the egg stage (day 0) and about 50 nematodes were transferred to each of three fresh lifespan plates per condition at the L4 stage. Worms were transferred on plates containing 10 µM FUDR (solved in water; applied on top of the grown bacteria lawn) after 24–48 h to prevent contamination with progeny generations. Nematodes that did not react to repeated gentle stimulation were scored as dead. Non‐natural deaths (bagging, protrusive vulvae) and lost animals were censored. JMP 11.0.0 (SAS Institute Inc.) was used for statistical analyses, and the log‐rang test determined P value and significance.
Statistical analysis
All zebrafish experiments were performed on at least three replicates (N) and each consisted of a sample size (n) of 8–30 fish. All C. elegans experiments have been performed for a minimum of three biological replicates. The number of samples was determined empirically. Data are presented as Mean ± SEM. Significance was determined using either Student’s t‐test, one‐way ANOVA or two‐way ANOVA using GraphPad PRISM software. All graphs were plotted using the GraphPad PRISM software.
Author contributions
SAP conceived this work. PJ designed, collected, analyzed and interpreted the results from studies related to GABAergic neurons and behaviour in zebrafish. ES, MB collected and analyzed the results of RNAseq. KS collected, analyzed and interpreted the results from studies related to C. elegans experiments. BK, SAP generated and characterized the CRISPR chd7 mutant line. PJ and BK performed the drug analyses in zebrafish. TC performed the ChIP analyses. PJ, MB, KS, JAP, ES, TC, NP and SAP interpreted the results. JAP, NP and SAP secured the research funding. PJ, KS, TC, NP and SAP drafted the manuscript. PJ and SAP with contributions from all authors prepared the final version of the manuscript. All authors read the final version of this manuscript.
Conflict of interest
The authors declare no competing interests. ES and JAP are co‐founders of Modelis Inc. The commercial affiliations did not play any role in this study; in particular, they did not have any additional role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Review Process File
Acknowledgements
This study was supported by the CHARGE Syndrome Foundation (KS, JAP and SP), Canada Foundation for Innovation (CFI; SP), the Natural Sciences and Engineering Research Council of Canada (NSERC; SP) and the Rare Disease Foundation (SP). SP holds an FRQS Junior 1 research scholar award and the Anna Sforza Djoukhadjian Research Chair. PJ is supported by a CERMO‐FC scholarship. KS was supported by a CIHR fellowship. JAP and NP are FRQS Senior research scholars, and NP is also the recipient of the UQAM Research Chair on rare genetic diseases. The authors thank Dr S. Lalani, Dr. J. W. Belmont and P. Hernandez (Baylor College of medicine) for generously providing the lymphoblastoid cell lines. We also thank Dr. Marie‐Claude Bélanger for critical reading and valuable comments on the manuscript; Claudia Maios for her help with the drug screens in C. elegans; and Valentin Lemoine, Alexandra Lissouba and Marc Allard for their help with zebrafish genotyping.
EMBO reports (2021) 22: e50958.
Data availability
The RNA‐seq has been deposited to the GEO database (GSE139623). (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139623).
References
- Bajpai R, Chen DA, Rada‐Iglesias A, Zhang J, Xiong Y, Helms J, Chang CP, Zhao Y, Swigut T, Wysocka J (2010) CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463: 958–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Taylor MR, Castro PA, Baier H (2005) Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c‐fos expression. Neuroscience 131: 759–768 [DOI] [PubMed] [Google Scholar]
- Becker DE (2012) Basic and clinical pharmacology of autonomic drugs. Anesth Prog 59: 159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger C, Berube‐Simard FA, Leduc E, Bernas G, Campeau PM, Lalani SR, Martin DM, Bielas S, Moccia A, Srivastava A et al (2018) Dysregulation of cotranscriptional alternative splicing underlies CHARGE syndrome. Proc Natl Acad Sci USA 115: E620–E629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Doze VA, Madison DV, Smith SJ (1996) Excitatory actions of norepinephrine on multiple classes of hippocampal CA1 interneurons. J Neurosci 16: 572–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij‐Arts CM (2011) CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet 48: 334–342 [DOI] [PubMed] [Google Scholar]
- Bouazoune K, Kingston RE (2012) Chromatin remodeling by the CHD7 protein is impaired by mutations that cause human developmental disorders. Proc Natl Acad Sci USA 109: 19238–19243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer M, Guglielmi L, Zielonka M, Hemberger V, Kolker S, Okun JG, Hoffmann GF, Carl M, Sauer SW, Opladen T (2019) QDPR homologues in Danio rerio regulate melanin synthesis, early gliogenesis, and glutamine homeostasis. PLoS One 14: e0215162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Wang Z, Wan H, Xu L, You X, Liao L, Chen Y (2018) PAQR3 regulates endoplasmic reticulum‐to‐golgi trafficking of COPII vesicle via interaction with Sec13/Sec31 Coat proteins. iScience 9: 382–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloney K, Steele SL, Stoyek MR, Croll RP, Smith FM, Prykhozhij SV, Brown MM, Midgen C, Blake K, Berman JN (2018) Etiology and functional validation of gastrointestinal motility dysfunction in a zebrafish model of CHARGE syndrome. FEBS J 285: 2125–2140 [DOI] [PubMed] [Google Scholar]
- Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ (2012) GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev 36: 2044–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD (1992) Brefeldin A inhibits Golgi membrane‐catalysed exchange of guanine nucleotide onto ARF protein. Nature 360: 350–352 [DOI] [PubMed] [Google Scholar]
- Feng L, Xie X, Ding Q, Luo X, He J, Fan F, Liu W, Wang Z, Chen Y (2007) Spatial regulation of Raf kinase signaling by RKTG. Proc Natl Acad Sci USA 104: 14348–14353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Kawauchi D, Korkel‐Qu H, Deng H, Serger E, Sieber L, Lieberman JA, Jimeno‐Gonzalez S, Lambo S, Hanna BS et al (2017) Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat Commun 8: 14758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro L, Tanganelli S, Calo G, Antonelli T, Fabrizi A, Acciarri N, Bianchi C, Beani L, Simonato M (1993) Noradrenergic modulation of gamma‐aminobutyric acid outflow from the human cerebral cortex. Brain Res 629: 103–108 [DOI] [PubMed] [Google Scholar]
- Goodman JV, Bonni A (2019) Regulation of neuronal connectivity in the mammalian brain by chromatin remodeling. Curr Opin Neurobiol 59: 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Wei H, Hu J, Sun S, Long J, Wang X (2015) U0126 inhibits pancreatic cancer progression via the KRAS signaling pathway in a zebrafish xenotransplantation model. Oncol Rep 34: 699–706 [DOI] [PubMed] [Google Scholar]
- Gupta MK, Papay RS, Jurgens CW, Gaivin RJ, Shi T, Doze VA, Perez DM (2009) alpha1‐Adrenergic receptors regulate neurogenesis and gliogenesis. Mol Pharmacol 76: 314–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne TS, Grialou TL, Parker KR (2005) Autistic‐like behavior in CHARGE syndrome. Am J Med Genet A 133A: 257–261 [DOI] [PubMed] [Google Scholar]
- Hartshorne TS, Stratton KK, Brown D, Madhavan‐Brown S, Schmittel MC (2017) Behavior in CHARGE syndrome. Am J Med Genet C Semin Med Genet 175: 431–438 [DOI] [PubMed] [Google Scholar]
- Hawkins TA, Cavodeassi F, Erdelyi F, Szabo G, Lele Z (2008) The small molecule Mek1/2 inhibitor U0126 disrupts the chordamesoderm to notochord transition in zebrafish. BMC Dev Biol 8: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D, Marie C, Zhao C, Kim B, Wang J, Deng Y, Clavairoly A, Frah M, Wang H, He X et al (2016) Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat Neurosci 19: 678–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EJ, Turner KJ, Fernandez JM, Cifuentes D, Ghosh M, Ijaz S, Jain RA, Kubo F, Bill BR, Baier H et al (2016) Estrogens suppress a behavioral phenotype in zebrafish mutants of the autism risk gene, CNTNAP2. Neuron 89: 725–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holter MC, Hewitt LT, Nishimura KJ, Knowles SJ, Bjorklund GR, Shah S, Fry NR, Rees KP, Gupta TA, Daniels CW et al (2021) Hyperactive MEK1 signaling in cortical GABAergic neurons promotes embryonic parvalbumin neuron loss and defects in behavioral inhibition. Cereb Cortex 10.1093/cercor/bhaa413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P, Ma A, Wilson M, Williams G, Curotta J, Munns CF, Mehr S (2014) CHARGE syndrome: a review. J Paediatr Child Health 50: 504–511 [DOI] [PubMed] [Google Scholar]
- da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Jamadagni P, Patten SA (2019) 25‐hydroxycholesterol impairs neuronal and muscular development in zebrafish. Neurotoxicology 75: 14–23 [DOI] [PubMed] [Google Scholar]
- Janssen N, Bergman JE, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, Hofstra RM, van Ravenswaaij‐Arts CM, Hoefsloot LH (2012) Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat 33: 1149–1160 [DOI] [PubMed] [Google Scholar]
- Johansson M, Rastam M, Billstedt E, Danielsson S, Stromland K, Miller M, Gillberg C (2006) Autism spectrum disorders and underlying brain pathology in CHARGE association. Dev Med Child Neurol 48: 40–50 [DOI] [PubMed] [Google Scholar]
- Jones KM, Saric N, Russell JP, Andoniadou CL, Scambler PJ, Basson MA (2015) CHD7 maintains neural stem cell quiescence and prevents premature stem cell depletion in the adult hippocampus. Stem Cells 33: 196–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Shindou T (1998) Noradrenergic excitation and inhibition of GABAergic cell types in rat frontal cortex. J Neurosci 18: 6963–6976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev Dynam 203: 253–310 [DOI] [PubMed] [Google Scholar]
- Le Corre S, Eyre D, Drummond IA (2014) Modulation of the secretory pathway rescues zebrafish polycystic kidney disease pathology. J Am Soc Nephrol 25: 1749–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee E, Kim R, Kim J, Lee S, Park H, Yang E, Kim H, Kim E (2018) Shank2 deletion in parvalbumin neurons leads to moderate hyperactivity, enhanced self‐grooming and suppressed seizure susceptibility in mice. Front Mol Neurosci 11: 209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZZ, Wang ZL, Choi TI, Huang WT, Wang HT, Han YY, Zhu LY, Kim HT, Choi JH, Lee JS et al (2018) Chd7 Is critical for early T‐cell development and thymus organogenesis in zebrafish. Am J Pathol 188: 1043–1058 [DOI] [PubMed] [Google Scholar]
- Liu H, Liu ZZ (2020) Aggressive‐like behavior and increased glycine transporters in a zebrafish model of CHARGE syndrome. Behav Brain Res 378: 112293 [DOI] [PubMed] [Google Scholar]
- Ma G, Bavadekar SA, Davis YM, Lalchandani SG, Nagmani R, Schaneberg BT, Khan IA, Feller DR (2007) Pharmacological effects of ephedrine alkaloids on human alpha(1)‐ and alpha(2)‐adrenergic receptor subtypes. J Pharmacol Exp Ther 322: 214–221 [DOI] [PubMed] [Google Scholar]
- Marek GJ, Aghajanian GK (1996) Alpha 1B‐adrenoceptor‐mediated excitation of piriform cortical interneurons. Eur J Pharmacol 305: 95–100 [DOI] [PubMed] [Google Scholar]
- Marie C, Clavairoly A, Frah M, Hmidan H, Yan J, Zhao C, Van Steenwinckel J, Daveau R, Zalc B, Hassan B et al (2018) Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc Natl Acad Sci USA 115: E8246–E8255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DM (2010) Chromatin remodeling in development and disease: focus on CHD7. PLoS Genet 6: e1001010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshalkina DA, N. Kizlyk M, V. Kysil E, Collier AD, Echevarria DJ, Abreu MS, Barcellos LJG, Song C, Warnick JE, Kyzar EJ et al (2018) Zebrafish models of autism spectrum disorder. Exp Neurol 299: 207–216 [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD (2019) PANTHER version 14: more genomes, a new PANTHER GO‐slim and improvements in enrichment analysis tools. Nucleic Acids Res 47: D419–D426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller T, Wullimann MF (2015) Atlas of Early zebrafish brain development: a tool for molecular neurogenetics, 2 nd edn, pp 258. Amsterdam: Elsevier; [Google Scholar]
- Ohta S, Yaguchi T, Okuno H, Chneiweiss H, Kawakami Y, Okano H (2016) CHD7 promotes proliferation of neural stem cells mediated by MIF. Mol Brain 9: 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno H, Renault Mihara F, Ohta S, Fukuda K, Kurosawa K, Akamatsu W, Sanosaka T, Kohyama J, Hayashi K, Nakajima K et al (2017) CHARGE syndrome modeling using patient‐iPSCs reveals defective migration of neural crest cells harboring CHD7 mutations. eLife 6: e21114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD et al (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagon RA, Graham JM Jr, Zonana J, Yong SL (1981) Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr 99: 223–227 [DOI] [PubMed] [Google Scholar]
- Papay R, Gaivin R, Jha A, McCune DF, McGrath JC, Rodrigo MC, Simpson PC, Doze VA, Perez DM (2006) Localization of the mouse alpha1A‐adrenergic receptor (AR) in the brain: alpha1AAR is expressed in neurons, GABAergic interneurons, and NG2 oligodendrocyte progenitors. J Comp Neurol 497: 209–222 [DOI] [PubMed] [Google Scholar]
- Patten SA, Jacobs‐McDaniels NL, Zaouter C, Drapeau P, Albertson RC, Moldovan F (2012) Role of Chd7 in zebrafish: a model for CHARGE syndrome. PLoS One 7: e31650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten SA, Aggad D, Martinez J, Tremblay E, Petrillo J, Armstrong GA, La Fontaine A, Maios C, Liao M, Ciura S et al (2017) Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight 2: e97152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne S, Burney MJ, McCue K, Popal N, Davidson SM, Anderson RH, Scambler PJ (2015) A critical role for the chromatin remodeller CHD7 in anterior mesoderm during cardiovascular development. Dev Biol 405: 82–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzarelli R, Cherubini E (2011) Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast 2011: 297153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma'ayan A (2016) The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016: baw100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM (2003) Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2: 255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruwe WD, Naylor AM, Bauce L, Veale WL (1985) Determination of the endogenous and evoked release of catecholamines from the hypothalamus and caudate nucleus of the conscious and unrestrained rat. Life Sci 37: 1749–1756 [DOI] [PubMed] [Google Scholar]
- Samarut E, Swaminathan A, Riche R, Liao M, Hassan‐Abdi R, Renault S, Allard M, Dufour L, Cossette P, Soussi‐Yanicostas N et al (2018) gamma‐Aminobutyric acid receptor alpha 1 subunit loss of function causes genetic generalized epilepsy by impairing inhibitory network neurodevelopment. Epilepsia 59: 2061–2074 [DOI] [PubMed] [Google Scholar]
- Samarut E, Chalopin D, Riche R, Allard M, Liao M, Drapeau P (2019) Individual knock out of glycine receptor alpha subunits identifies a specific requirement of glra1 for motor function in zebrafish. PLoS One 14: e0216159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser K, Mansfeld J, Kuhlow D, Weimer S, Priebe S, Heiland I, Birringer M, Groth M, Segref A, Kanfi Y et al (2013) Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat Chem Biol 9: 693–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser K, Fardghassemi Y, Parker JA (2017) A rapid chemical‐genetic screen utilizing impaired movement phenotypes in C. elegans: input into genetics of neurodevelopmental disorders. Exp Neurol 293: 101–114 [DOI] [PubMed] [Google Scholar]
- Schmeisser KFY, Parker JA (2017) A rapid chemical‐genetic screen utilizing impaired movement phenotypes in C. elegans: input into genetics of neurodevelopmental disorders. Exp Neurol 293: 101–114 [DOI] [PubMed] [Google Scholar]
- Schnetz MP, Handoko L, Akhtar‐Zaidi B, Bartels CF, Pereira CF, Fisher AG, Adams DJ, Flicek P, Crawford GE, Laframboise T et al (2010) CHD7 targets active gene enhancer elements to modulate ES cell‐specific gene expression. PLoS Genet 6: e1001023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi A, Gu M, Gumusgoz E, Chan L, Yan KK, Rozowsky J, Barozzi I, Afzal V, Akiyama JA, Plajzer‐Frick I et al (2020) Supervised enhancer prediction with epigenetic pattern recognition and targeted validation. Nat Methods 17: 807–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT et al (2016) ENCODE data at the ENCODE portal. Nucleic Acids Res 44: D726–D732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith IM, Nichols SL, Issekutz K, Blake K, Canadian Paediatric Surveillance P (2005) Behavioral profiles and symptoms of autism in CHARGE syndrome: preliminary Canadian epidemiological data. Am J Med Genet A 133A: 248–256 [DOI] [PubMed] [Google Scholar]
- Sourbron J, Partoens M, Scheldeman C, Zhang Y, Lagae L, de Witte P (2019) Drug repurposing for Dravet syndrome in scn1Lab(‐/‐) mutant zebrafish. Epilepsia 60: e8–e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souriau J, Gimenes M, Blouin C, Benbrik I, Benbrik E, Churakowskyi A, Churakowskyi B (2005) CHARGE syndrome: developmental and behavioral data. Am J Med Genet A 133A: 278–281 [DOI] [PubMed] [Google Scholar]
- Souza BR, Romano‐Silva MA, Tropepe V (2011) Dopamine D2 receptor activity modulates Akt signaling and alters GABAergic neuron development and motor behavior in zebrafish larvae. J Neurosc 31: 5512–5525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AM, Nguyen M, Wong K, Poudel MK, Kalueff AV (2014) Developing zebrafish models of autism spectrum disorder (ASD). Prog Neuropsychopharmacol Biol Psychiatry 50: 27–36 [DOI] [PubMed] [Google Scholar]
- Stiernagle T (2006) Maintenance of C. elegans . In WormBook, The C. elegans Research Community (ed) pp 1–11. Wormbook doi/10.1895/wormbook.1.101.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan A, Hassan‐Abdi R, Renault S, Siekierska A, Riche R, Liao M, de Witte PAM, Yanicostas C, Soussi‐Yanicostas N, Drapeau P et al (2018) Non‐canonical mTOR‐independent role of DEPDC5 in regulating GABAergic network development. Curr Biol 28: 1924–1937.e1925 [DOI] [PubMed] [Google Scholar]
- Takata A, Miyake N, Tsurusaki Y, Fukai R, Miyatake S, Koshimizu E, Kushima I, Okada T, Morikawa M, Uno Y et al (2018) Integrative analyses of de novo mutations provide deeper biological insights into autism spectrum disorder. Cell Rep 22: 734–747 [DOI] [PubMed] [Google Scholar]
- Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM, Lin CY, Lin CJ, Swiderski DL, Vogel H et al (2014) Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 514: 228–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vansal SS, Feller DR (1999) Direct effects of ephedrine isomers on human beta‐adrenergic receptor subtypes. Biochem Pharmacol 58: 807–810 [DOI] [PubMed] [Google Scholar]
- Verduzco D, Amatruda JF (2011) Analysis of cell proliferation, senescence, and cell death in zebrafish embryos. Methods Cell Biol 101: 19–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vithayathil J, Pucilowska J, Landreth GE (2018) ERK/MAPK signaling and autism spectrum disorders. Prog Brain Res 241: 63–112 [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Takechi K, Fujiwara A, Kamei C (2010) Effects of antiepileptics on behavioral and electroencephalographic seizure induced by pentetrazol in mice. J Pharmacol Sci 112: 282–289 [DOI] [PubMed] [Google Scholar]
- Wellman PJ, Miller DK, Ho DH (2003) Noradrenergic modulation of ephedrine‐induced hypophagia. Synapse 48: 18–24 [DOI] [PubMed] [Google Scholar]
- Westerfield M (1993) The zebrafish book : a guide for the laboratory use of zebrafish (Brachydanio rerio), Eugene, OR: M. Westerfield; [Google Scholar]
- Whittaker DE, Riegman KL, Kasah S, Mohan C, Yu T, Sala BP, Hebaishi H, Caruso A, Marques AC, Michetti C et al (2017) The chromatin remodeling factor CHD7 controls cerebellar development by regulating reelin expression. J Clin Investig 127: 874–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullimann MF, Knipp S (2000) Proliferation pattern changes in the zebrafish brain from embryonic through early postembryonic stages. Anat Embryol (Berl) 202: 385–400 [DOI] [PubMed] [Google Scholar]
- Xu DQ, Wang Z, Wang CY, Zhang DY, Wan HD, Zhao ZL, Gu J, Zhang YX, Li ZG, Man KY et al (2016) PAQR3 controls autophagy by integrating AMPK signaling to enhance ATG14L‐associated PI3K activity. The EMBO journal 35: 496–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Layman WS, Martin DM, Scacheri PC (2010) Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A 152A: 674–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jiang X, Qin X, Ye D, Yi Z, Liu M, Bai O, Liu W, Xie X, Wang Z et al (2010) RKTG inhibits angiogenesis by suppressing MAPK‐mediated autocrine VEGF signaling and is downregulated in clear‐cell renal cell carcinoma. Oncogene 29: 5404–5415 [DOI] [PubMed] [Google Scholar]
- Zhen M, Samuel AD (2015) C. elegans locomotion: small circuits, complex functions. Curr Opin Neurobiol 33: 117–126 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Review Process File
Data Availability Statement
The RNA‐seq has been deposited to the GEO database (GSE139623). (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139623).