

ABSTRACT
While treatments for colorectal cancer continue to improve, some 50% of patients succumb within 5 years, pointing to the need for additional therapeutic options. We have developed a modified non-replicating adenoviral vector for gene transfer, called AdRGD-PG, which offers improved levels of transduction and transgene expression. Here, we employ the p53-responsive PG promoter to drive expression of p53 or human interferon-β (hIFNβ) in human colorectal cancer cell lines HCT116wt (wtp53), HCT116−/- (p53 deficient) and HT29 (mutant p53). The HCT116 cell lines were both easily killed with p53 gene transfer, while combined p53 and hIFNβ cooperated for the induction of HT29 cell death and emission of immunogenic cell death (ICD) markers. Elevated annexinV staining and caspase 3/7 activity point to cell death by a mechanism consistent with apoptosis. P53 gene transfer alone or in combination with hIFNβ sensitized all cell lines to chemotherapy, permitting the application of low drug doses while still achieving significant loss of viability. While endogenous p53 status was not sufficient to predict response to treatment, combined p53 and hIFNβ provided an additive effect in HT29 cells. We propose that this approach may prove effective for the treatment of colorectal cancer, permitting the use of limited drug doses.
KEYWORDS: Adenovirus, apoptosis, immunogenic cell death, gene therapy, chemotherapy
Introduction
Colorectal malignancies remain the second leading cause of death due to cancer1 and it is expected that globally, cases will increase by about 60% to more than 2.2 million new cases with an estimated 1.1 million deaths by 2030.2 While current treatments for colorectal cancer (CRC) can be effective, new approaches are needed since the expected 5-year survival rate is 50%, leaving many patients with limited options for therapy. Mutations in the adenomatous polyposis coli (APC), KRAS and TP53 genes are described as the main events driving the malignant progression of adenoma lesions.3,4 Among the genetic drivers, p53, known as the guardian of genome, is mutated in almost half of the sporadic cases of CRC.5 Additionally, the presence of p53 mutations is closely associated with lymphatic invasion and advanced stages, and tumors from proximal colon present a poorer response to chemotherapy when harboring mutated p53.5 Considering the importance of wild-type p53 in limiting tumor progression, it is not surprising that some of the early gene therapy approaches for cancer aimed to overexpress p53.6
Targeting the p53 pathway as part of a therapeutic approach is expected to capitalize on the variety of anti-neoplastic functions coordinated by p53. For example, p53 acts as a transcriptional regulator of genes involved in cell death via the mitochondrial pathway, activating pro-apoptotic genes such as BAX, NOXA (PMAIP1) and PUMA (BBC3), and down regulating members of the BCL2 family, which in turn destabilizes the mitochondrial membrane, initiating the caspase cascade and apoptosis.7 Moreover, p53 can promote the expression of cell death receptors, such as DR5, Fas and PIDD, supporting the extrinsic apoptosis pathway.7 However, mutations in the p53 DNA binding domain lead to the loss of most of these functions, thus contributing to resistance to cell death, including upon cancer treatment.8 For these reasons, compounds have been designed to change the conformation of mutated p53 in order to restore its normal transcriptional activity.9 p53 activity may also be restored by interrupting its interaction with MDM2 or, as mentioned above, upon p53 gene transfer.10
The immune system also plays a major role in malignant transformation and can also be leveraged as part of cancer treatment regimens. During progression, tumor cells evade the immune system through a process called cancer immunoediting.11 The type I interferon family, originally described as a component of the antiviral response, is a key mediator of anti-tumor immunity and dysfunction in this pathway contributes to escape from immunosurveillance.12,13 Support for this notion was demonstrated when interferon alpha/beta receptor 1 (Ifnar1) knockout mice was shown to be more prone to develop tumors when exposed to the carcinogen methylcholanthrene. Moreover, tumors derived from type I IFN deficient mice were more immunogenic, meaning that the lack of this pathway allowed the development of a broad range of poorly immunoedited cells that could be recognized in mice with a normal type I IFN pathway.12 Both IFNα and IFNβ bind to IFNAR1/IFNAR2 receptors, however IFNβ binds more stably which leads to improved signaling as compared to other family members.13 Once bound to its receptor, type I interferon triggers a cascade that recruits the JAK/STAT pathway, culminating in expression of cell death mediators such as FAS and TRAIL, immune regulators including MHC-1 and CXCL10, and, among others, genes linked to anti-viral response.13 The type I interferon pathway is an interesting target for cancer treatment, through the use of recombinant protein is confounded by toxic side effects.13 Alternatively, gene transfer of type I interferon may provide high, but localized, protein levels.
Our group has developed specialized adenoviral vectors for gene transfer targeting the p53 and interferon pathways. For example, the combination of vectors encoding p19Arf (an inhibitor of MDM2) and mouse IFNβ (mIFNβ) induced immunogenic cell death and an anti-tumor immune response in mouse models of melanoma and lung cancer.14–16 Moreover, transcription of these genes is controlled by a p53-responsive promoter, termed PG, which confers high-level expression of the transferred genes through a positive feedback mechanism.17,18 Use of these vectors for the transfer of p53 was shown to improve killing of prostate cancer cells in vitro as well as cooperate with cabazitaxel, resulting in essentially complete tumor elimination in vivo.19,20
Here we explore the cellular responses to adenovirus carrying p53 and human IFNβ (hIFNβ) under the control of the PG promoter in colorectal cancer cell lines HCT116wt (p53 wt), HCT116−/- (p53−/-) and HT29 (p53 R273H, mutated in the DNA-binding domain). This represents a departure from our previous studies involving IFNβ since we are focusing on human cDNAs transferred to human cells that differ in their p53 status. In addition, the combination of p53 plus hIFNβ, predicted to cooperate for cell killing,21 has not yet been examined in a gene transfer setting in CRC. In addition, we examine the association of gene transfer with chemotherapies that are relevant for CRC. Our data show that endogenous p53 status did not clearly influence response to treatment among the cell lines tested. Gene transfer sensitized the cells to the action of the chemotherapies. Combined gene transfer of p53 and hIFNβ cooperated to provide higher levels of cell killing in certain situations.
Materials and methods
Cell culture:
HCT116wt and HCT116−/- were kindly provided by Mari Sogayar (NUCEL, FMUSP) and HT29 was provided by Ramon Kaneno (UNESP-Botucatu). All cell lines were authenticated (STR profile) and monitored for mycoplasma. Cells were maintained in RPMI medium, 10% fetal calf serum (FCS) and 1x antibiotic-antimycotic (all reagents from Thermo Fisher Scientific, Waltham, MA, USA) at 37°C, 5% CO2.
Vectors and virus production:
The AdRGD-CMV-LacZ vector was kindly provided by Hiroyuki Mizuguchi (Osaka University, Japan). The AdRGD-PG-eGFP and AdRGD-PG-p53 vectors have been described previously.19,22 For AdRGD-PG-hIFNβ, the human IFNβ cDNA was isolated from pLG104R (ATCC 31092) and inserted in pEntr-PG and followed by site-specific recombination in pAdRGD-promoterless-Dest, as described previously.22,23 Vector maps are provided in Figure S1. Virus production was performed as per Peng et al.,24 where cell lysate containing virus is purified in an iodixanol gradient before desalting. Purified virus was then aliquoted and stored at −80°C. Titration was performed using the Adeno X Rapid Titration kit (Takara, Palo Alto, CA, USA). The resulting biological titer was used in calculating the multiplicity of infection (MOI) used for transduction.
Transduction/treatment regimen:
When applied alone, transduction with the control vector was performed using a MOI of 25 for HCT116 cell lines or 100 for HT29, while co-transductions were done using a MOI of 12.5 for each vector in HCT116 or 50 for each vector for HT29, thus the total MOI was constant across all conditions for each cell line. Cells were plated and transduced the following day using virus diluted to the desired MOI in a minimal volume of medium containing 2% FCS, applied to cells and incubated for 6 to 8 hours before completing with medium containing 10% FCS. When necessary, drug treatments were initiated the following day by removing medium and replacing it with fresh medium containing doxorubicin (Doxorubicin hydrochloride, Sigma, Darmstadt, Germany), 5-fluorouracil (5-FU, SIGMA, Darmstadt, Germany), cisplatin (SIGMA, Darmstadt, Germany) or nutlin-3 (Sigma-Aldrich, St. Louis, MO, USA) or the excipient, DMSO (Sigma-Aldrich). Then cells were incubated as indicated for each experiment.
Transgene expression:
After 48 h incubation, proteins were extracted, quantified via Bradford method, separated by 12% SDS-PAGE, transferred to nitrocellulose membrane, incubated with rabbit anti-p53 (SC6243, Santa Cruz Biotechnology, Santa Cruz, CA, USA). The Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences, Marlborough, MA, USA) was used for chemiluminescent detection in the Image Quant LAS 4000 (GE Healthcare Life Sciences). hIFNβ was detected using cell culture supernatant after 48 h incubation, using Verikine human IFN beta ELISA kit (PBL Assay Science, Piscataway, NJ, USA). GFP expression was evaluated 48 h after transduction, cells were harvested, kept on ice, and fluorescence was detected via flow cytometry and analyzed using manufacturer’s software (Attune, Thermo Fisher Scientific).
qPCR: As per our previous work22 total RNA was extracted using TRIzol™ reagent (Thermo Fisher Scientific) as recommended by the supplier. Quantitative PCR reactions were performed by using GoTaq® 1-Step RT-qPCR System (Promega, Madison, WI, USA) and 7500 Real-Time PCR System (Thermo Fisher Scientific) as recommended by the supplier. Expression levels were analyzed using the ΔΔCt method. GAPDH mRNA level was used as an internal control. Oligonucleotide sequences are listed in Table S1.
Cell number alteration, colony formation and MTT
In order to evaluate cell number, 72 h after transduction, cells were gently washed with PBS, harvested with trypsin, stained with trypan blue and counted excluding stained cells. For the clonogenic assay, 24 h after transduction, 200 cells were seeded on six well plates. After six days, cells were gently washed with PBS, fixed with methanol and acetic acid and stained with trypan blue 20%. For MTT, 5.103 cells were seeded on 96 well plate and treated with vectors on the same day. After 24 h, the media was completed with chemotherapeutics at indicated concentrations. MTT standard test was evaluated as previously described.18
Sub-G1, AnnexinV and caspase 3/7
Cell cycle and Annexin-V/PI methods were described in previous studies.18,25 Briefly, adherent and floating cells were harvested. For cell cycle analysis, a portion of these cells was fixed with 70% ethanol, followed by RNAse treatment and staining with propidium iodide (PI). Annexin-V/PI staining was performed immediately after harvest using the remaining cells without fixation, staining with AnnexinV-Alexa 488 (Thermo Fisher Scientific) and PI. For caspase activity, a portion of the cells were incubated with CellEvent caspase 3/7 detection reagent (Thermo Fisher Scientific) following manufacturer’s instruction. In either case, cells were observed by flow cytometry and analysis was performed using the manufacturer’s software (Attune, Thermo Fisher Scientific).
ICD: Immunogenic cell death markers were analyzed as previously described.26 Seventy-two hours after transduction, cells and supernatant were collected. Cells were incubated with a rabbit anti-calreticulin (#NB300-545, Novus Biologicals, Littleton, CO, USA), washed and incubated with secondary Alexa488-conjugated anti-rabbit secondary antibody (Thermo Fisher Scientific) and analyzed by flow cytometry (Attune, Thermo Fisher Scientific). ATP in the supernatant collected at 72 h was measured via ELISA using the ENLITEN ATP Assay System (Promega, Madison, WI, USA) and Bioluminescence detection was made with microplate reader Victor (Perkin-Elmer, Waltham, MA, USA).
HMGB1 was measured by Western blot where cell culture media from treated cells was harvested and centrifuged 1 min at 12000x g and supplemented with protease inhibitor cocktail (Thermo Fisher Scientific). From this, 180 μl of media were concentrated (Concentrator Plus – Eppendorf, Hamburg, Germany) and resolved by SDS-Page and electroblotted onto nitrocellulose membranes (Millipore, Burlington, MA, USA). Blots were incubated with anti-HMGB1 (1:1000, Abcam ab79823, Cambridge, UK) antibody resuspended in 5% nonfat dry milk (Sigma-Aldrich, St. Louis, MO, USA) in PBS plus 0.1% Tween-20 overnight at 4°C. Detection was achieved using a horseradish peroxidase-conjugate secondary antibody (1:10000, Sigma Aldrich), in 5% nonfat dry milk in PBS plus 0.1% Tween-20, visualized with ECL (GE Healthcare, Chicago, IL, USA) and images were recorded by using an ImageQuant LAS4000 imaging platform (GE Healthcare) and analyzed by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Results
The colorectal carcinoma cell lines HCT116wt (wild type p53), HCT116−/- (biallelic p53 knockout27) or HT29 (mutant p53) were first evaluated for their susceptibility to adenoviral transduction and transgene expression from the p53-responsive PG promoter. Upon transduction with either AdRGD-PG-eGFP or AdRGD-CMV-LacZ, we found that the two HCT116 cells tolerated a multiplicity of infection (MOI) of 25 without toxicity, while for HT29 a MOI of 100 was not deleterious (data not shown). Expression of exogenous p53 or human interferon-β (hIFNβ) upon transduction with the AdRGD-PG-p53 or AdRGD-PG-hIFNβ vectors was readily detected in all cell lines (Figure 1a and B). Transduction with AdRGD-PG-eGFP resulted in approximately 60% GFP-positive HCT116wt or HCT116−/-, while about 30% of HT29 cells presented GFP activity (Figure 1c). Co-transduction of the p53-responsive GFP vector along with the virus encoding p53 led to a significantly increased intensity of GFP fluorescence (Figure 1d), while hIFNβ had no impact on PG promoter activity. Transgene activity was further confirmed by qPCR detection of transcripts from p53 target genes as well as mediators of the type I interferon response (Figures S2, S3 and S4), showing the expected activation of these pathways.
Figure 1.
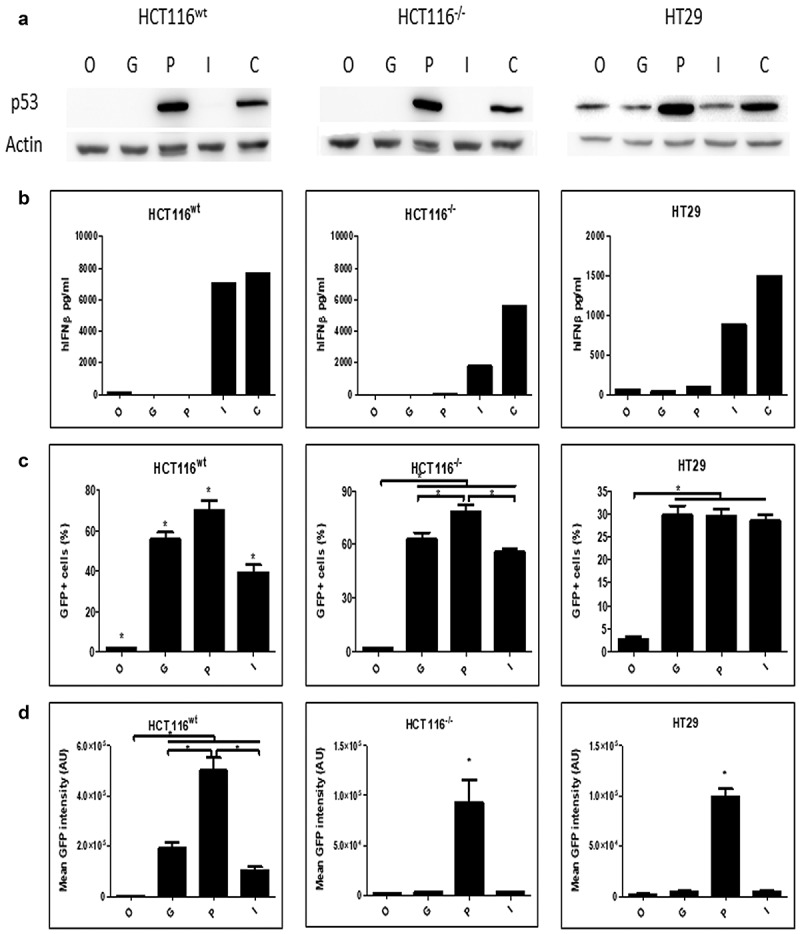
Colorectal cell lines are efficiently transduced and express the transgenes
HCT116wt, HCT116−/- and HT29 (p53 R273H) cells were seeded in 24 wells plate (5.104 cells per well). After 24 h, cell number was verified and then transduced using AdRGD-PG-eGFP (G), AdRGD-PG-p53 + AdRGD-PG-eGFP (P), AdRGD-PG-hIFNβ + AdRGD-PG-eGFP (I) AdRGD-PG-p53 + AdRGD-PG-hIFNβ (C) or no vector (O). Cells and supernatant were harvested 48 h after transduction and analyzed by the respective methodology. A. Western blot showing p53 expression. B. IFN-β measurement in cell supernatant via ELISA. These data are qualitative and included to confirm expression, thus do not conclusively indicate relative expression levels. C. Percentage of positive cells for GFP detected by flow cytometry. D. Intensity of GFP fluorescence (AU, arbitrary units). * p < .05 One Way Anova; Tukey posttest. In panels C and D, results represent the average and standard deviation of three independent experiments.
Next, the impact of transgene expression on cell proliferation, clonogenicity and mitochondrial activity was assessed. The cell lines were transduced, and viable cells were counted after 72 hours incubation, revealing that p53 or hIFNβ when applied alone or in combination, significantly reduced the viable cell population, yet for HCT116wt, combined gene transfer had an additive impact (Figure 2a). In a clonogenic assay, cells were transduced, counted, replated and observed for colony formation, revealing significantly reduced colonies for either p53 or hIFNβ, yet their combination was especially effective in HTC116wt and HT29 (Figure 2b). Mitochondrial activity, as revealed in a standard MTT assay, was also reduced by either transgene, though combined gene transfer did not further reduce MTT staining (Figure 2c). These assays show that either p53 or hIFNβ can impede cellular functions and the combination, in some cases, provides an additive effect.
Figure 2.
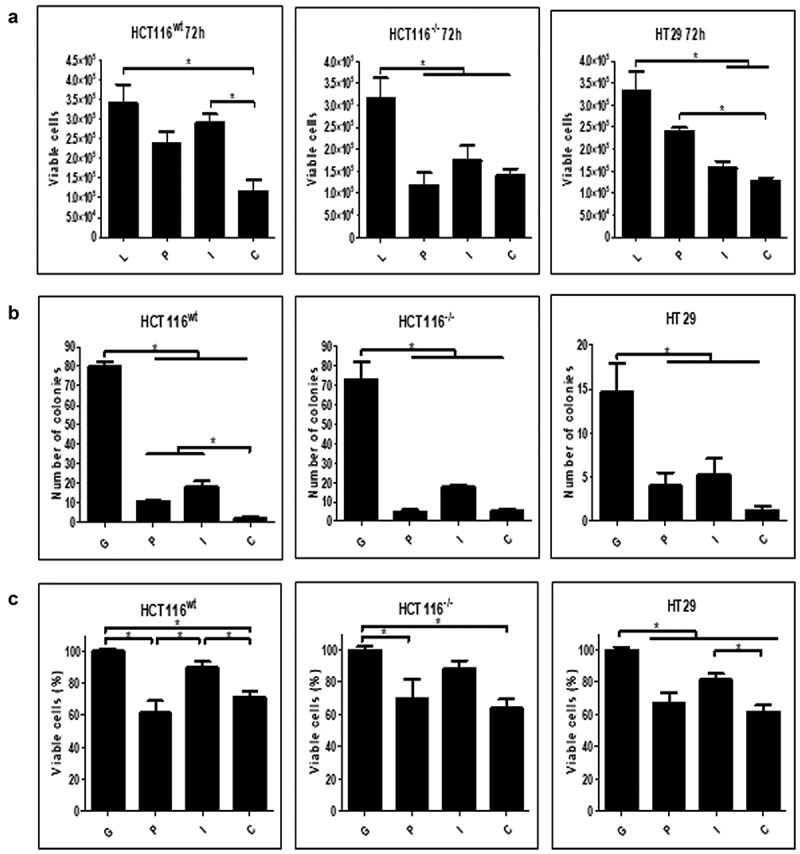
Cellular response to transgene activity
HCT116wt, HCT116−/- and HT29 (p53 R273H) cells were seeded in 24 wells plate (5.104 cells per well). After 24 h, cell number was verified and then transduced using AdRGD-CMV-LacZ (L), AdRGD-PG-eGFP (G), AdRGD-PG-p53 + AdRGD-PG-eGFP (P), AdRGD-PG-hIFNβ + AdRGD-PG-eGFP (I) or the combination AdRGD-PG-p53 + AdRGD-PG-hIFNβ (C). Cells were harvested at indicated times and analyzed using the respective methodology. A. Viable cells counted 72 h post-transduction. B. Number of colonies formed. C. Viability is measured via MTT 96 h post-transduction. * p < .05 One Way Anova; Tukey posttest. Results represent the average and standard deviation of three independent experiments.
Evaluation of cell death upon gene transfer revealed significant accumulation of sub-G1 (hypodiploid) populations (Figure 3a), indicating a predominant role for p53 in the HCT116 cell lines and a clear additive effect of p53 plus hIFNβ in HT29. A similar pattern was seen when treated cells were stained with AnnexinV/PI (Figure 3b). Caspase 3/7 activity was also observed upon gene transfer, especially when p53 and hIFNβ were combined (Figure 3c). These results point to cell death by a mechanism consistent with apoptosis that, in some cases, was observed at elevated levels upon combined p53 and hIFNβ treatment.
Figure 3.
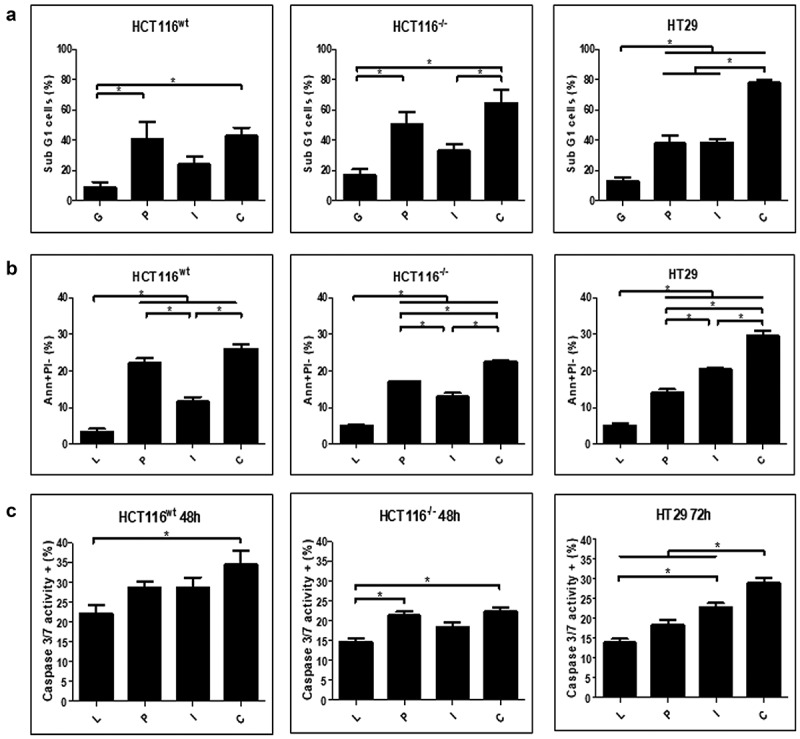
Induction of cell death markers upon treatment with adenovirus
HCT116wt, HCT116−/- and HT29 (p53 R273H) cells were seeded in 24 wells plate (5.104 cells per well). After 24 h, cell number was verified and then transduced using AdRGD-CMV-LacZ (L), AdRGD-PG-eGFP (G), AdRGD-PG-p53 + AdRGD-PG-eGFP (P), AdRGD-PG-hIFNβ + AdRGD-PG-eGFP (I) or the combination AdRGD-PG-p53 + AdRGD-PG-hIFNβ (C). Cells were harvested at the indicated times and analyzed using the respective methodology. A. Percentage of sub G1 (hypodiploid) cells was detected by PI staining and flow cytometry 72 h post-transduction. B. Percentage of cells positive for Annexin V staining and negative for PI staining at 72 h as detected by flow cytometry. C. Percentage of cells positive for active caspase 3 and 7. * p < .05 One Way Anova; Tukey posttest. Results represent the average and standard deviation of three independent experiments.
We also investigated markers of immunogenic cell death (ICD) upon gene transfer. The detection of calreticulin on the cell surface was elevated in the presence of p53 gene transfer, yet was further enhanced upon combined p53 and hIFNβ treatment (Figure 4a), a pattern that closely matched annexinV/PI staining and accumulation of sub-G1 cells. Liberation of ATP was associated with p53 gene transfer in the HCT116 cell lines yet was significantly induced by hIFNβ alone or in combination with p53 in HT29 cells (Figure 4b). Similarly, extracellular HMGB1 was detected upon p53 gene transfer in the HCT116 cell lines or in HT29 upon hIFNβ gene transfer alone or in combination with p53 (Figure 4c). As shown here, p53 gene transfer is sufficient to induce emission of ICD markers in HCT116 cells, whereas hIFNβ alone or in combination with p53 promoted these markers in the HT29 cell line.
Figure 4.
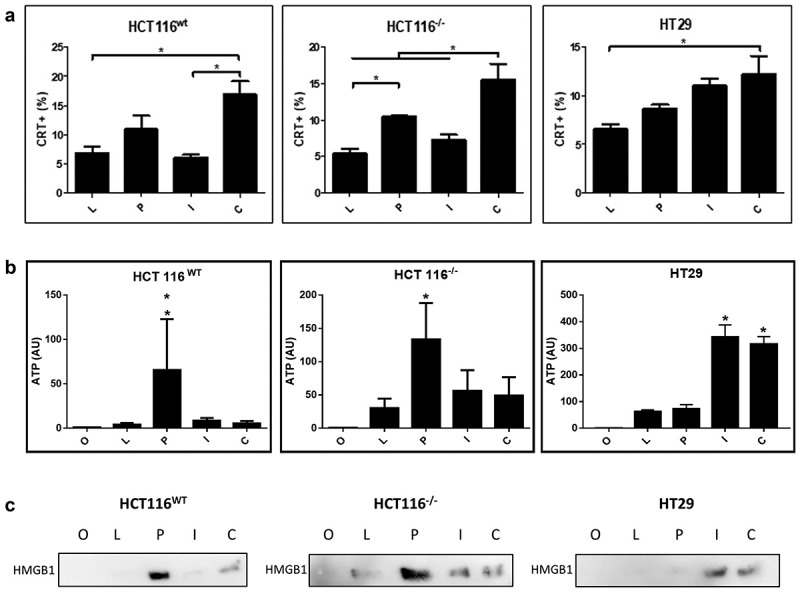
Emission of immunogenic cell death markers in response do gene transfer
HCT116wt, HCT116−/- and HT29 (p53 R273H) cells were seeded in 24 wells plate (5.104 cells per well). After 24 h, cell number was verified and then transduced using AdRGD-PG-Luc (L), AdRGD-PG-p53 + AdRGD-PG-eGFP (P), AdRGD-PG-hIFNβ + AdRGD-PG-eGFP (I) AdRGD-PG-p53 + AdRGD-PG-hIFNβ (C) or no vector (O). Cells and supernatant were harvested at 72 hours post transduction and analyzed using the respective methodology. A. Percentage of cells with calreticulin (CRT+) exposure on the cell surface as observed by flow cytometry. (B) Detection of ATP measured as intensity of luciferase activity (AU, arbitrary units) and (C) release of HMGB1 measured by Western blot using equal quantities of conditioned medium. * p < .05 One Way Anova; Tukey posttest. Results represent the average and standard deviation of three independent experiments.
Having demonstrated cell death upon gene transfer, we next explored whether p53 and/or hIFNβ would sensitize the colorectal carcinoma cell lines to chemotherapeutic agents (Figure 5). For both the HCT116wt and HCT116−/- cell lines, even the lowest concentration of doxorubicin resulted in significant inhibition of MTT staining in the presence of p53 alone or in combination with hIFNβ. Similarly, p53 alone or in combination with hIFNβ increased sensitivity to cisplatin and nutlin-3. Yet p53, hIFNβ or combined gene transfer conferred sensitivity to 5-FU. Use of these isogenic cell lines reveals that presence of endogenous p53wt did not strongly influence these results. Whereas p53 alone or together with hIFNβ was especially effective for sensitizing HT29 cells to cisplatin. Gene transfer generally facilitated the cellular response of HT29 to doxorubicin, 5-FU and nutlin-3, seen as decreased MTT staining.
Figure 5.
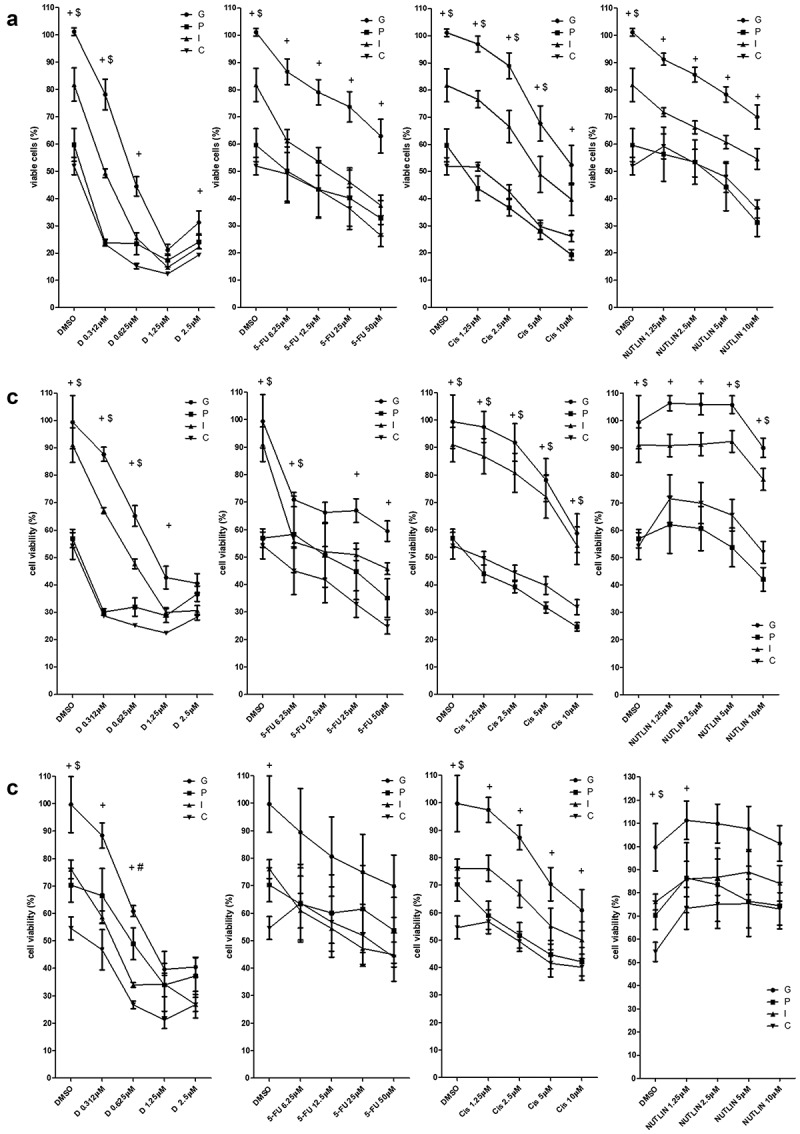
Association of chemotherapy with gene transfer
Cell lines were seeded (5000 cells per well) and transduced using AdRGD-PGeGFP (G) AdRGD-PGp53 + AdRGD-PG-eGFP (P), AdRGD-PG-hIFNβ + AdRGD-PG-eGFP (I) AdRGD-PG-p53 + AdRGD-PG-hIFNβ (C). After 24 hours incubation, doxorrubicin (D) cisplatina (Cis), 5-fluorouracil (5-FU), Nutlin-3 (NUTLIN) or the diluent (DMSO) were added at the concentrations indicated. Then, after 72 hours incubation, MTT assay was performed and viability normalized against the GFP control. (A) HCT116wt, (B) HCT116−/-and (C) HT29. + (C ≠ G); # (C ≠ P); $ (C ≠ I); p < .05 Two Way ANOVA, Bonferroni posttest. Results represent the average and standard deviation of four independent experiments.
Discussion
Here we explore the potential therapeutic effect of adenovirus carrying p53 and hIFNβ under control of the p53-responsive PG promoter, in CRC cell lines. As approximately 50% of CRC cases present mutations in p53, we evaluated the proposed therapy in three cell lines, HCT116wt (p53 wild type), HCT116−/- (p53 null) and HT29 (mutant p53 – R273H), serving as representatives of the three scenarios of p53 status. While transgene expression was observed in all cell lines, the magnitude of expression was correlated with functional p53. Even so, both of the HCT116 cell lines were especially sensitive to p53, while HT29 responded better to combined gene transfer, in terms of inhibiting proliferation or inducing cell death. P53 gene transfer was especially effective for sensitizing cells to the effects of chemotherapy regardless of the endogenous p53 status. In this model we show that the association between gene transfer and chemotherapy was more effective than either treatment alone and that, in some situations, combined p53 and hIFNβ gene transfer led to higher levels of cell death than seen for treatment with a single gene.
The AdRGD-PG vectors provided reliable transduction and transgene expression. As expected, exogenous p53 expression was readily detected and was not altered by endogenous p53 status. However, the HCT116−/- or HT29 cells showed lower secretion of hIFNβ or GFP activity, yet in either case expression was elevated in the presence of wild-type p53. Thus the p53-responsive character of the PG promoter was confirmed, as has been demonstrated in previous publications.14,16,25,28
As seen in the qPCR assays, response to the p53 and hIFNβ transgenes was confirmed. These assays also revealed the expected activation of the PG promoter in the presence of exogenous p53 since hIFNβ transcripts were elevated in the presence of the AdRGD-PG-p53 vector. Even so, both lower and higher levels of hIFNβ transcript were correlated with the expression of interferon responsive genes at the similar intensities. These assays also revealed subtle differences between the response of the HCT116 cell lines as compared to HT29. For example, induction of BAX was seen in HT29 when treated with the combination of p53 and hIFNβ, but this did not occur in the HTC116 cell lines.
Considering the biological effects resulting from gene transfer, either p53 or IFNβ reduced cell number, viability or colony formation. As revealed by annexinV staining and caspase 3/7 activity, cell death was consistent with apoptosis. For the two HCT116 cell lines, p53 gene transfer alone was sufficient to bring about cell death and its combination with IFNβ did not seem to provide added benefit, at least as measured here. While previous studies indicate that HCT116 cells do not die upon p53 gene transfer,29,30 we did not encounter such resistance using our modified vectors, suggesting that the improvements in transduction and expression may overcome the barriers to p53 activity in these cells. Interestingly, combined gene transfer provided a cooperative effect in HT29 cells. Previous results from our group showed that combining 19Arf and mIFNβ conferred higher levels of cell death in mouse cell lines with wild-type endogenous p53,14,25,26 thus the influence of p53 deficiencies has not yet been studied. In the case of HT29, we have no direct evidence that the mutant p53 participated in the cellular response. Even so, crosstalk between mutant p53 and IFNβ has been reported in the literature. While mutant p53 could inhibit IFNβ signaling, IFNβ released by cancer-associated fibroblasts downregulated the expression of mutant p53.31 In our assays, expression of IFNβ did not seem to impact expression of endogenous mutant p53 (Figure 1). Moreover, exogenous p53 levels were much higher than endogenous mutant p53, suggesting that wild-type p53 may have overcome this potential barrier to its function.
While the main objective of this study was to explore cell death in response to p53 plus hIFNβ gene transfer, one of our goals includes the induction of an antitumor immune response. Our previous studies demonstrated that the combination p19Arf and mIFNβ induces immunogenic cell death and immune activation.15,26 Here we take the first steps to reveal if the p53 plus hIFNβ combination would provide a similar effect. For the HT29 cells, the combined gene transfer led to emission of three ICD markers, calreticulin, ATP and HMGB1. Since hIFNβ is also crucial for ICD, we can conclude that treatment of HT29 cells resulted in the release of four critical ICD factors. Further testing will be necessary to determine bona fide ICD. For the HCT116 cell lines, p53 gene transfer was sufficient to promote the emission of ATP and HMBG1 and an additive effect with hIFNβ was seen only for calreticulin. It is possible that timing could be critical for the detection of these markers. Even so, while previous studies indicate the resistance of HCT116 to the effects of p53 gene transfer, the use of our modified vector, AdRGD-PG-p53, yielded cell death by including emission of ICD markers.
Gene transfer, especially p53, was associated with increased sensitivity to chemotherapeutic agents. The presence of exogenous hIFNβ did not confer additional benefit as measured by the MTT assay. Even so, reduced drug concentrations could bring about significant reductions in cell viability in the presence of p53 alone or in combination with hIFNβ. As seen in our previous studies, this may provide an opportunity for effective tumor inhibition while avoiding toxic side effects.20 One important question that also remains is whether the association of gene transfer with chemotherapeutics will lead to ICD and consequent immune activation.
As shown here, the strategy of combining p53 and hIFNβ gene transfer resulted in the induction of cell death compatible with apoptosis and sensitization of CRC cell lines to chemotherapy. Future studies will delve more deeply into the mechanism of cell death, including ICD. Of particular importance would be to use clinically relevant models to demonstrate therapeutic potential, including assays in vivo and in organotypic systems.
Supplementary Material
Acknowledgments
We are grateful to Dr. Roger Chammas for continued support and encouragement.
Funding Statement
This work was supported by the Sao Paulo Reseach Foundation (Fundação de Amparo à Pesquisa do Estado de São Paulo, FAPESP), #2015/26580-9.
Author contributions:
PRDV: Conceived the project, performed experiments, interpreted data, drafted and edited the manuscript.
SAM: Performed experiments and edited the manuscript.
AH: Performed experiments and edited the manuscript.
RET: Performed experiments and edited the manuscript.
DBZ: Performed experiments and edited the manuscript.
FA: Performed experiments and edited the manuscript.
BES: Conceived the project, provided funding, interpreted data, drafted and edited the manuscript.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Organization WH . Cancer fact sheet [Internet]. In: Centre GWM, ed., 2018.
- 2.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. doi: 10.1002/ijc.29210 [DOI] [PubMed] [Google Scholar]
- 3.Fumagalli A, Drost J, Suijkerbuijk SJ, Van Boxtel R, De Ligt J, Offerhaus GJ, Begthel H, Beerling E, Tan EH, Sansom OJ, et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc Natl Acad Sci U S A. 2017;114(12):E2357–E64. doi: 10.1073/pnas.1701219114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drost J, Van Jaarsveld RH, Ponsioen B, Zimberlin C, Van Boxtel R, Buijs A, Sachs N, Overmeer RM, Offerhaus GJ, Begthel H, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521(7550):43–47. doi: 10.1038/nature14415 [DOI] [PubMed] [Google Scholar]
- 5.Iacopetta B, Russo A, Bazan V, Dardanoni G, Gebbia N, Soussi T, Kerr D, Elsaleh H, Soong R, Kandioler D. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. J Clin Oncol. 2005;23:7518–7528. [DOI] [PubMed] [Google Scholar]
- 6.Zhang WW, Li L, Li D, Liu J, Li X, Li W, Xu X, Zhang MJ, Chandler LA, Lin H. The first approved gene therapy product for cancer Ad-p53 (Gendicine): 12 years in the clinic. Hum Gene Ther. 2018;29:160–179. [DOI] [PubMed] [Google Scholar]
- 7.Beckerman R, Prives C.. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935. doi: 10.1101/cshperspect.a000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li XL, Zhou J, Chen ZR, Chng WJ. p53 mutations in colorectal cancer- molecular pathogenesis and pharmacological reactivation. World J Gastroenterol. 2015;21(1):84–93. doi: 10.3748/wjg.v21.i1.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheok CF, Lane DP. Exploiting the p53 pathway for therapy. Cold Spring Harb Perspect Med. 2017;7(3):7. doi: 10.1101/cshperspect.a026310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–998. doi: 10.1038/ni1102-991 [DOI] [PubMed] [Google Scholar]
- 12.Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6(7):722–729. doi: 10.1038/ni1213 [DOI] [PubMed] [Google Scholar]
- 13.Medrano RFV, Hunger A, Mendonca SA, Barbuto JAM, Strauss BE. Immunomodulatory and antitumor effects of type I interferons and their application in cancer therapy. Oncotarget. 2017;8(41):71249–71284. doi: 10.18632/oncotarget.19531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catani JPP, Medrano RFV, Hunger A, Del Valle P, Adjemian S, Zanatta DB, Kroemer G, Costanzi-Strauss E, Strauss BE. Intratumoral immunization by p19Arf and interferon-β gene transfer in a heterotopic mouse model of lung carcinoma. Transl Oncol. 2016;9(6):565–574. doi: 10.1016/j.tranon.2016.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medrano RF, Catani JP, Ribeiro AH, Tomaz SL, Merkel CA, Costanzi-Strauss E, Strauss BE. Vaccination using melanoma cells treated with p19arf and interferon beta gene transfer in a mouse model: a novel combination for cancer immunotherapy. Cancer Immunol Immunother. 2016;65(4):371–382. doi: 10.1007/s00262-016-1807-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunger A, Medrano RF, Strauss BE. Harnessing combined p19Arf and interferon-beta gene transfer as an inducer of immunogenic cell death and mediator of cancer immunotherapy. Cell Death Dis. 2017;8(5):e2784. doi: 10.1038/cddis.2017.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bajgelman MC, Strauss BE. Development of an adenoviral vector with robust expression driven by p53. Virology. 2008;371(1):8–13. doi: 10.1016/j.virol.2007.11.015 [DOI] [PubMed] [Google Scholar]
- 18.Tamura RE, Da Silva Soares RB, Costanzi-Strauss E, Strauss BE. Autoregulated expression of p53 from an adenoviral vector confers superior tumor inhibition in a model of prostate carcinoma gene therapy. Cancer Biol Ther. 2016;17(12):1221–1230. doi: 10.1080/15384047.2016.1235655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamura RE, Hunger A, Fernandes DC, Laurindo FR, Costanzi-Strauss E, Strauss BE. Induction of oxidants distinguishes susceptibility of prostate carcinoma cell lines to p53 gene transfer mediated by an improved adenoviral vector. Hum Gene Ther. 2017;28(8):639–653. doi: 10.1089/hum.2016.139 [DOI] [PubMed] [Google Scholar]
- 20.Tamura RE, Lana MG, Costanzi-Strauss E, Strauss BE. Combination of cabazitaxel and p53 gene therapy abolishes prostate carcinoma tumor growth. Gene Ther. 2019;27:15–26. [DOI] [PubMed] [Google Scholar]
- 21.Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424(6948):516–523. doi: 10.1038/nature01850 [DOI] [PubMed] [Google Scholar]
- 22.Hunger A, Medrano RF, Zanatta DB, Del Valle PR, Merkel CA, Salles TA, Ferrari DG, Furuya TK, Bustos SO, De Freitas Saito R, et al. Reestablishment of p53/Arf and interferon-beta pathways mediated by a novel adenoviral vector potentiates antiviral response and immunogenic cell death. Cell Death Discov. 2017;3(1):17017. doi: 10.1038/cddiscovery.2017.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.David TIP, Cerqueira OLD, Lana MG, Medrano RFV, Hunger A, Strauss BE. Response of human melanoma cell lines to interferon-beta gene transfer mediated by a modified adenoviral vector. Sci Rep. 2020;10(1):17893. doi: 10.1038/s41598-020-74826-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng HH, Wu S, Davis JJ, Wang L, Roth JA, Marini FC 3rd, Fang B. A rapid and efficient method for purification of recombinant adenovirus with arginine-glycine-aspartic acid-modified fibers. Anal Biochem. 2006;354(1):140–147. doi: 10.1016/j.ab.2006.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merkel CA, Medrano RF, Barauna VG, Strauss BE. Combined p19Arf and interferon-beta gene transfer enhances cell death of B16 melanoma in vitro and in vivo. Cancer Gene Ther. 2013;20(5):317–325. doi: 10.1038/cgt.2013.23 [DOI] [PubMed] [Google Scholar]
- 26.Hunger A, Medrano RF, Zanatta DB, Del Valle PR, Merkel CA, Salles TA, Ferrari DG, Furuya TK, Bustos SO, De Freitas Saito R, et al. Reestablishment of p53/Arf and interferon-. Cell Death Discov. 2017;3(1):17017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282(5393):1497–1501. doi: 10.1126/science.282.5393.1497 [DOI] [PubMed] [Google Scholar]
- 28.Strauss BE, Costanzi-Strauss E. pCLPG: a p53-driven retroviral system. Virology. 2004;321(2):165–172. doi: 10.1016/j.virol.2003.12.021 [DOI] [PubMed] [Google Scholar]
- 29.Polyak K, Waldman T, He TC, Kinzler KW, Vogelstein B. Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev. 1996;10(15):1945–1952. doi: 10.1101/gad.10.15.1945 [DOI] [PubMed] [Google Scholar]
- 30.Sasaki Y, Morimoto I, Ishida S, Yamashita T, Imai K, Tokino T. Adenovirus-mediated transfer of the p53 family genes, p73 and p51/p63 induces cell cycle arrest and apoptosis in colorectal cancer cell lines: potential application to gene therapy of colorectal cancer. Gene Ther. 2001;8(18):1401–1408. doi: 10.1038/sj.gt.3301538 [DOI] [PubMed] [Google Scholar]
- 31.Madar S, Harel E, Goldstein I, Stein Y, Kogan-Sakin I, Kamer I, Solomon H, Dekel E, Tal P, Goldfinger N, et al. Mutant p53 attenuates the anti-tumorigenic activity of fibroblasts-secreted interferon beta. PLoS One. 2013;8(4):e61353. doi: 10.1371/journal.pone.0061353 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.