

Abstract
Natural products remain a critical source of medicines and drug leads. One of the most rapidly growing superclasses of natural products is RiPPs: ribosomally synthesized and posttranslationally modified peptides. RiPPs have rich and diverse bioactivities. This review highlights examples of the molecular mechanisms of action that underly those bioactivities. Particular emphasis is placed on RiPP/target interactions for which there is structural information. This detailed mechanism of action work is critical toward the development of RiPPs as therapeutics and can also be used to prioritize hits in RiPP genome mining studies.
Keywords: RiPPs, Natural products, Mechanism of action
Introduction
Much of our current pharmaceutical arsenal owes its existence to natural products, molecules produced by bacteria, fungi, and plants (Li & Vederas, 2009). In addition to their utility in medicine, natural products have also captured the interest of scientists from a variety of disciplines. For decades, synthetic chemists have focused on recapitulating the architectural and stereochemical complexity of natural products in the laboratory while enzymologists have studied the intricate steps of natural product biosynthesis. More recently, with the exponential rise of sequencing data in the 21st century, the field of bioinformatics has played an increasingly prominent role in natural products discovery. Great strides have been made in predicting the structure of a natural product based on the gene cluster that encodes its biosynthesis (Blin, Pascal Andreu, et al., 2019; Blin, Shaw, et al., 2019). One superfamily of natural products that has particularly benefited from “genome mining” approaches is the RiPPs: ribosomally synthesized and posttranslationally modified peptides (Arnison et al., 2013). The biosynthesis of RiPPs starts with a gene-encoded precursor peptide, and the gene encoding this precursor is often colocalized with genes encoding the enzymes that mature it into the final natural product. Many powerful software packages have been developed to identify RiPP biosynthetic gene clusters (BGCs) from genomic sequence data (Russell & Truman, 2020). This has led to the rapid discovery of novel RiPP chemistry and biosynthesis, subjects that have been extensively and authoritatively covered in a recent review paper (Montalbán-López et al., 2021). Thus, we provide only basic information about RiPP structure and biosynthesis below. Li and Rebuffat have also recently reviewed the roles of RiPPs in physiology and ecology (Li & Rebuffat, 2020). Here, we review the molecular mechanisms of action of select RiPPs with a particular focus on RiPPs with either antimicrobial or cytotoxic activity. RiPPs exert these activities by targeting key cellular processes such as DNA replication, transcription, and translation (Travin et al., 2020). RiPPs also interact extensively with the cell envelope in both Gram-negative and Gram-positive bacteria (Fig. 1). Rather than organizing this review by RiPP families, we have organized it into sections based on the targets of the RiPPs. In this way, both commonalities and diversity in RiPP mechanisms of action can be appreciated.
Fig. 1.

The varied targets of bioactive RiPPs. Members of the RiPPs superfamily exert antibiotic and/or cytotoxic activity via inhibition of nearly all major cellular processes. Many RiPPs are membrane-active, compromising the permeability barrier of the cytoplasmic membrane in both Gram-negative and Gram-positive bacteria (only a Gram-negative cell is shown for simplicity's sake). Other RiPPs active at the cell envelope disrupt outer membrane biogenesis in Gram-negative bacteria and peptidoglycan (cell wall) biosynthesis. RiPPs also engage cytoplasmic targets disrupting the central processes of DNA replication, transcription, and translation. OM: outer membrane, IM: inner membrane.
RiPPs Targeting the Cell Envelope
Lanthipeptides that Attack the Bacterial Cell Envelope
A major target of RiPPs is the bacterial cell envelope, an intricate, multilayered structure that surrounds and protects bacterial cells from their environment (Silhavy et al., 2010). The integrity of the cell envelope is central to bacterial viability and a breach in this structure oftentimes leads to cell death. As such, drugs that cripple cell envelope biogenesis are highly sought-after because the pathway is essential and unique to bacteria and many cell envelope targets are accessible from outside the cell. Broadly, there are two types of bacterial cell envelope, those belonging to Gram-negative versus Gram-positive organisms. The Gram-negative cell envelope comprises an inner and outer membrane with a thin layer of peptidoglycan sandwiched between the membranes (Fig. 2). Gram-positive bacteria lack an outer membrane and instead are encased in a thick layer of peptidoglycan that sits above the cytoplasmic membrane. Many cell envelope-targeting RiPPs are thought to be pore-forming peptides that penetrate the cytoplasmic membrane. An important step in engagement of these RiPPs with the cell membrane is oftentimes recognition of key intermediates in the biosynthesis of the peptidoglycan cell wall.
Fig. 2.

The bacterial cell envelope is a common target for RiPPs. (A) Cartoon and chemical structure of lipid II, the precursor to peptidoglycan. The stem peptides of lipid II from different bacteria vary at the positions indicated by R1, R2, and R3. GlcNAc: N-acetylglucosamine, MurNAc: N-acetylmuramic acid. (B) Schematic of cell envelope biosynthesis processes disrupted by RiPPs. Both lanthipeptides and the lasso peptide siamycin-I bind to lipid II, disrupting peptidoglycan biosynthesis. The recently discovered RiPP darobactin inhibits BamA, disrupting the assembly of outer membrane proteins in Gram-negative bacteria. BAM: β-barrel assembly machinery.
Peptidoglycan is a crosslinked glycopolymer of repeating disaccharide-peptide units that is assembled from a basic building block called lipid II (Fig. 2A) (Vollmer et al., 2008). Despite the diversity of bacteria, the lipid II molecules that make up their cell wall are chemically conserved. Lipid II contains a disaccharide of N-acetylmuramic acid (MurNAc) and N-acetylglucosamine (GlcNAc). An undecaprenyl pyrophosphate lipid tail is linked to the anomeric carbon of MurNAc and a short peptide side chain is covalently attached to the MurNAc C3 carbon. The identity of the amino acids that comprise the peptide side chain can vary in different bacteria, but for the most part is similar across many species. Lipid II is synthesized inside the cell and the fully assembled molecule is then translocated across the cytoplasmic membrane to the outer leaflet, where peptidoglycan glycosyltransferase enzymes polymerize the units into glycan strands (Fig. 2B) (Bouhss et al., 2008; Egan et al., 2020; Typas et al., 2012; van Heijenoort, 2007). To reinforce the cell wall matrix, enzymes with peptidoglycan transpeptidase function form crosslinks between the peptide side chains of the strands (Sauvage et al., 2008). The conserved chemistry and synthesis of cell wall intermediates make them highly effective antibiotic targets.
In 1928, around the time that penicillin was discovered, one of the first antimicrobial RiPPs was also found (Rogers, 1928), though the fact that it was ribosomally synthesized was not confirmed until much later. This RiPP, called nisin, is a lanthipeptide from Lactococcus lactis that is present in fermented milk. Nisin has been used as a food preservative with broad-spectrum antimicrobial activity for decades, but only recently have we begun to understand its mechanism of action (McAuliffe et al., 2001). Early studies reported that nisin caused membrane leakage similar to a detergent and inhibited cell wall synthesis (Breukink et al., 1999; Linnett & Strominger, 1973; Van Heusden et al., 2002; Wiedemann et al., 2004). Advances in structural biology tools and access to defined cell wall substrates now have enabled studies clarifying that nisin docks on lipid II and thereafter permeabilizes the cytoplasmic membrane, leading to changes in the membrane potential and cell death (Breukink et al., 2003; Hsu et al., 2004; ‘t Hart et al., 2016).
Nisin is synthesized as a precursor peptide that is processed into the 34-residue active form through the actions of a protease and modification enzymes (Buchman et al., 1988; Gross & Morell, 1971; Ra et al., 1999). A defining structural feature of nisin is the five intramolecular (methyl)lanthionine rings that are posttranslationally installed along the elongated peptide backbone (Fig. 3A) (Gross & Morell, 1971). A solution NMR structure of nisin in complex with lipid II revealed that the N-terminal (methyl)lanthionine rings in particular are important for nisin bioactivity as they mediate binding to lipid II (Fig. 3B) (Hsu et al., 2004). To determine the structure, a variant of lipid II with a triprenyl tail was used as the substrate to maximize solubility. The shortened tail was not expected to adversely affect nisin binding, and indeed, the nisin-lipid II structure showed that nisin recognizes MurNAc and only the first isoprene unit in the lipid tail as well as the pyrophosphate group linking the sugar and lipid. Upon binding to the substrate, nisin forms a pocket comprised of its first two residues, Ile1 and Dhb2, the N-terminal lanthionine ring A, and methyllanthionine ring B that encloses these regions of lipid II. Notably, hydrogen bonding between the pyrophosphate group of lipid II and the backbone amides, rather than specific side chains, of the nisin pocket drives the interaction.
Fig. 3.

Structure and mechanism of action of the lanthipeptide nisin. (A) Representation of nisin structure. Dhb: dehydrobutyrine, Dha: dehydroalanine, Abu: aminobutyric acid. The thioether linkage between Ala and Ala is referred to as a lanthionine moiety while the linkage between Abu and Ala is a methyllanthionine linkage. (B) NMR structure (PDB file 1WCO) of nisin bound to a lipid II analog. Nisin is shown as a space-filling model and the lipid II analog is shown as sticks. The N-terminal (methyl)lanthionine rings (space-filling tan) of nisin envelop the pyrophosphate moiety (magenta and cyan sticks) of lipid II.
The second step in the mode of action of nisin is generation of membrane pores. A structure of membrane-embedded nisin is unavailable, but the nisin-lipid II structure together with other in vitro and modeling studies suggest a mechanism for pore formation (Van Heusden et al., 2002; Wiedemann et al., 2004). Whereas the N-terminus of nisin binds to lipid II, it is the C-terminus of nisin that penetrates the cytoplasmic membrane. The N-and C-terminal regions are separated by a few residues that do not contribute to the rigid (methyl)lanthionine rings. These linear residues act as a flexible hinge (Fig. 3A) around which the C-terminus of nisin can pivot and insert perpendicularly into the phospholipid bilayer (Van Heusden et al., 2002). Therefore, lipid II positions nisin at the membrane and also appears to help stabilize the membrane-spanning nisin pores that form (‘t Hart et al., 2016). Interactions with membrane phospholipids may contribute to nisin bioactivity (Prince et al., 2016), but it is evident that lipid II plays a central role in the mode of action of nisin. A mechanism that relies on binding with the chemically conserved lipid II moiety explains how nisin is able to kill diverse Gram-positive pathogens. At the same time, this mechanism ensures targeted activity as nisin does not simply disrupt any lipid environment.
Other Gram-positive lanthipeptide antibiotics that inhibit bacterial cell wall synthesis appear to bind lipid II as well for bioactivity (McAuliffe et al., 2001). Two examples are provided by the lanthipeptides epidermin (Allgaier et al., 1985) and mutacin 1140 (Hillman et al., 1998), which are elongated nisin-like molecules that contain four thioether rings. Using purified cell wall substrates, epidermin was shown to form a complex with lipid II (Brötz et al., 1998). The interaction with lipid II is critical for the pore-forming activity of epidermin, as the peptide was only able to permeabilize synthetic liposomes when lipid II was incorporated in the liposomes (Brötz et al., 1998). Similar use of lipid-II containing liposomes support the binding interaction of mutacin 1140 to lipid II (Smith et al., 2008). Mersacidin is a small Bacillus-derived lanthipeptide that also contains four thioether rings (Chatterjee et al., 1992). However, unlike nisin, epidermin, and mutacin 1140, mersacidin adopts a more compact, globular shape (Bierbaum et al., 1995; Prasch et al., 1997). Early studies using macromolecular synthesis inhibition assays pinpointed peptidoglycan synthesis as the pathway that mersacidin targets (Brötz et al., 1995). Reconstitution of peptidoglycan synthesis with a cell-free system then showed that mersacidin inhibits peptidoglycan polymerization and leads to accumulation of lipid II (Brötz et al., 1997). In line with these observations, purified lipid II added in excess was found to antagonize mersacidin activity, increasing the minimal inhibitory concentration (MIC) of mersacidin against an indicator strain (Brötz et al., 1998). Consequently, it was proposed that mersacidin sequesters lipid II to inhibit peptidoglycan synthesis. A solution NMR study in membrane-mimetic dodecylphosphocholine (DPC) micelles later provided evidence of lipid II binding that is driven by electrostatic interactions with the E17 acidic side chain in mersacidin (Hsu et al., 2003). The GlcNAc sugar of lipid II may be involved in the binding interaction, as mersacidin has a much higher affinity for lipid II than lipid I, an intermediate that lacks GlcNAc (Fig. 2B) (Brötz et al., 1997, 1998).
Similar mechanistic and structural studies of actagardine, microbisporicin, and the two-component lacticin 3147 system show that these lanthipeptides likewise inhibit peptidoglycan synthesis. Some of these lanthipeptides also disrupt the cytoplasmic membrane. In the case of lacticin 3147, two peptides act synergistically to effect cell death (McAuliffe et al., 1998; Ryan et al., 1996). The first peptide, lacticin A1, has four (methyl)lanthionine rings and structurally resembles mersacidin. Lacticin A1 contains an equal number of acidic (D10 and E24) and basic (H23 and K30) residues. The second peptide, lacticin A2, has three (methyl)lanthionine rings and a more elongated structure (Martin et al., 2004). Lacticin A2 is cationic due to the presence of K24 and R27 at the C-terminus. It has been proposed that these two peptides recapitulate the two-step mechanism of nisin: lacticin A1 binds lipid II to associate with the cytoplasmic membrane and recruits lacticin A2, which inserts into the membrane (Wiedemann et al., 2006). Membrane insertion leads to formation of pores that release potassium ions, dissipating the membrane potential (Wiedemann et al., 2006). The absence of either peptide compromises lacticin activity (Wiedemann et al., 2006). Also important for lacticin bioactivity are its charged residues, in particular E24 of lacticin A1, which may mediate interaction with the cell membrane (Deegan et al., 2010). Interestingly, the location of charged residues in lacticin 3147 is quite conserved in other two-component lanthipeptides (Deegan et al., 2010). Actagardine (Arioli et al., 1976; Boakes et al., 2009; Coronelli et al., 1976; Parenti et al., 1976; Somma et al., 1977; Zimmermann & Jung, 1997) and microbisporicin (Castiglione et al., 2007, 2008; Münch et al., 2014; Vasile et al., 2012) are similar to lacticin in that they also have mersacidin-like domains that presumably underlie their peptidoglycan synthesis inhibiting activity. Microbisporicin, which is commercially known as NAI-107, appears to form a 1:1 or 2:1 complex with lipid II through a binding event that involves its N-terminal thioether rings and the lipid II pyrophosphate group as seen with nisin (Castiglione et al., 2007, 2008; Münch et al., 2014; Vasile et al., 2012). However, unlike nisin, microbisporicin treatment does not detectably induce the formation of membrane pores (Münch et al., 2014). A different lanthipeptide that has membrane-disrupting activity is bovicin HJ50 (Xiao et al., 2004). There is some evidence that bovicin HJ50 interacts with lipid II in the process of pore formation because the N-terminal fragment of nisin, which is known to bind lipid II, competitively inhibits bovicin activity (Zhang et al., 2014). Direct demonstration of this interaction is still needed.
Lipid II binding is evidently a key step in the action of many antibacterial lanthipeptides, but other lipid molecules can also serve as docking substrates. A well-known example is illustrated by the 19-residue cyclic lanthipeptide cinnamycin (Takemoto et al., 1984), also known as Ro09–0198, which binds the membrane phospholipid phosphatidylethanolamine (Choung, Kobayashi, Inoue, et al., 1988; Choung, Kobayashi, Takemoto, et al., 1988). Cinnamycin forms a compact structure as a result of three internal thioether crosslinks and an additional crosslink between the sidechains of the sixth and last residues, A6 and K19 (Fredenhagen et al., 1990; Kaletta et al., 1991; Kessler et al., 1987). Early investigations on the bioactivity of cinnamycin revealed that it is hemolytic against mammalian erythrocytes (Choung, Kobayashi, Inoue, et al., 1988), but its activity is not confined to targeting solely eukaryotic cell membranes. Cinnamycin also attacks the bacterial cell membrane through a conserved mechanism that relies on recognition of phosphatidylethanolamine lipids (O'Rourke et al., 2017; Widdick et al., 2003). Structurally, phosphatidylethanolamine is built on a glycerol backbone to which two fatty acids and an ethanolamine-phosphate head group are esterified. Binding studies with different types of lipids suggested that the identity of the polar head group—the presence of a free amino end and phosphate moiety—dictated whether cinnamycin could bind the lipid and permeabilize the membrane (Choung, Kobayashi, Inoue, et al., 1988; Choung, Kobayashi, Takemoto, et al., 1988). Subsequent NMR analysis and molecular dynamics simulation of the cinnamycin-phosphatidylethanolamine complex revealed key contacts underlying the interaction and explained how the peptide displays such selectivity toward the lipid (Hosoda et al., 1996; Vestergaard et al., 2019; Wakamatsu et al., 1990). Cinnamycin forms a snug pocket that surrounds the phosphatidylethanolamine polar head group in a 1:1 complex, with the acyl chains sticking out from the peptide (Hosoda et al., 1996; Vestergaard et al., 2019; Wakamatsu et al., 1990). The primary amine of the lipid head group contacts the backbone carbonyls of cinnamycin and the hydroxylated D15 residue through hydrogen bonding. A network of hydrogen bonds between the backbone of cinnamycin residues F10-Abu11-F12-V13 and the phosphate group of phosphatidylethanolamine contributes to further stabilization of the complex (Vestergaard et al., 2019). How cinnamycin engages the lipid presumably affects its capacity to permeabilize the membrane (Vestergaard et al., 2019).
These lanthipeptide studies point to key features of this class of RiPPs. Many lanthipeptides are pore-forming agents that seemingly have evolved to appropriate lipid II as an anchor for attachment to the cell membrane (McAuliffe et al., 2001). Peptide binding to lipid II would also adversely impact cell wall synthesis because the enzymes that make new peptidoglycan material can no longer physically access this key substrate. The steady-state levels of Lipid II are after all normally low in cells (van Heijenoort, 2007; Bouhss et al., 2008). It is worth noting that the lanthipeptides discussed herein can recognize a different region of lipid II from glycopeptides such as vancomycin, which instead binds the terminal d-Ala-d-Ala residues of the lipid II stem peptide (Perkins, 1969). In fact, competitive lipid II binding assays helped reveal that the mode of action of mersacidin, for example, is distinct from those of glycopeptides (Brötz et al., 1998). Such unique mechanisms may be informative for developing combinatorial antibiotics. Another theme that emerges from comparison of these lanthipeptides is the structural conservation of domains that bind lipid II and how homology-based searches can hone in on mechanisms of action.
Polytheonamide Toxins that form Membrane Channels
Some of the largest and most heavily modified RiPPs discovered to date are the polytheonamides, a class of RiPPs that exhibits ion channel activity. Polytheonamides are marine sponge-derived founding members of the proteusin class of RiPPs, a moniker fittingly inspired by the Greek sea god Proteus (Freeman et al., 2012). Polytheonamides are potent cytotoxic agents against mammalian cells whose mode of action involves ion channel formation (Iwamoto et al., 2010). In 1994, Hamada et al. isolated two polytheonamides, A and B, from the marine sponge Theonella swinhoei and preliminarily analyzed their primary sequences (Hamada et al., 1994a; Hamada et al., 1994b). A revised characterization of the sequences revealed that the polytheonamides are 49-residue linear peptides that extensively contain unusual amino acids in the d-configuration (Hamada et al., 2005). The A and B peptides differ only in the stereochemistry of the sulfoxide modification at residue 44 (Hamada et al., 2005). Given the high prevalence of nonproteinogenic amino acids, for a long time the polytheonamides were thought to be nonribosomal peptides (Hamada et al., 2005). However, the enzymatic machinery responsible for their synthesis remained elusive for almost two decades. In 2012, a seminal report unveiled the polytheonamide BGC (Freeman et al., 2012). The relatively few modification enzymes encoded within the polytheonamide gene cluster could account for most of the 40+ posttranslational modifications observed (Freeman et al., 2017). Identification of the gene cluster answered a longstanding question. That is, by finding the structural gene which specifies the precursor peptides, the authors demonstrated that polytheonamides A and B were in fact ribosomal peptides. Moreover, the operon-like organization of the gene cluster suggested a bacterial origin. The polytheonamide producer was subsequently identified as an uncultivated filamentous bacterial symbiont from the genus Entotheonella that lives in marine sponges (Wilson et al., 2014).
That the polytheonamides are capable of forming ion channels which induce cytotoxicity can be attributed to their three-dimensional structure. Not only are the polytheonamides replete of unusual amino acids, many of the amino acids are hydrophobic in nature. The N-terminus of the peptides is also blocked by a tert-butylated N-acyl group (Hamada et al., 2005). The overall hydrophobicity of the peptide implies membrane localization. Another striking feature of polytheonamides is the alternating pattern of l- and d-amino acids along the length of the peptides (Hamada et al., 2005). This pattern is a signature of the nonribosomal gramicidin peptides, which form membrane ion channels that disturb the cellular balance of ions (Kelkar & Chattopadhyay, 2007). Polytheonamide B was likewise found to structurally organize as a β-helix resembling a spring or corkscrew (Hamada et al., 2010). The solution NMR structure in membrane-mimetic methanol: chloroform (1:1) solvent indicated that the polytheonamide B helix is approximately 45 Å in length and the diameter of the pore is 4 Å (Fig. 4). These physical dimensions have two important functional implications. The average thickness of the plasma membrane has been estimated to be 30–40 Å (Andersen & Koeppe, 2007; Mitra et al., 2004; van Meer et al., 2008); as such, a single polytheonamide helix can span the entire phospholipid bilayer to form an ion channel (Hamada et al., 2010). Second, the ionic radius of a potassium ion—a demonstrated cargo of polytheonamide channels (Hamada et al., 2010; Iwamoto et al., 2014)—is 1.33 Å (Huang et al., 1978), which means that the ions can readily be conveyed through the interior of the helix assuming the absence of competing cargo. Besides potassium, other monovalent cations also demonstrate the ability to traverse the polytheonamide channel with the experimentally tested selectivity of H+ > Cs+ > Rb+ > K+ > Na+ (Hamada et al., 2010; Oiki et al., 1997). As expected for an ion channel, the polytheonamide B helix has a hydrophilic core (Hamada et al., 2010). Another feature of the peptide is that its N-terminus is more hydrophobic than its C-terminus, which led to the hypothesis that the N-terminus initially punctures the membrane (Hamada et al., 2010; Shinohara et al., 2012). In addition to forming plasma membrane ion channels, polytheonamides may exert secondary activity in other cellular compartments. According to a recent report, polytheonamide B dissipates the pH gradient of lysosomes purportedly through similar pore-forming activity (Hayata et al., 2018). Mechanisms to maintain ion gradients across the cell membrane are essential to life. Protein channels ferry ions across the impermeable cell membrane, and importantly, are subject to stringent regulation. By dysregulating the flow of ions, polytheonamide toxins effectively disrupt cellular ion homeostasis and membrane potential.
Fig. 4.

The β-spiral structure of polytheonamide B in organic solvent to mimic the membrane environment (PDB file 2RQO). Left: View showing the length of polytheonamide B to be roughly the same thickness as a plasma membrane. Note the density of polar residues at the C-terminus of the structure. Right: The interior of polytheonamide B is polar, allowing for the passage of cations such as potassium. Polytheonamide B is a potent cytotoxin, functioning as an ion channel.
Membrane-Active Bacteriocin RiPPs
Another widespread class of bacterial ribosomal peptides with membrane-targeting activity is the bacteriocins. Bacteriocins are not classified according to a unique posttranslational modification, but rather are broadly defined as antimicrobial proteinaceous substances. As such, bacteriocins come in many different varieties, harboring diverse chemical modifications and having a broad spectrum of activity. Nisin and the other lanthipeptides discussed above constitute a subset of bacteriocins. Like nisin, many bacteriocins are used as natural, safe preservatives to kill foodborne pathogens and minimize food spoilage (Drider et al., 2006). Some bacteriocins, such as lactococcin G (Kjos et al., 2014; Nissen-Meyer et al., 1992) and plantaricin JK (Anderssen et al., 1998; Oppegård et al., 2016), consist of two peptide components and lack posttranslational modifications. Over the past century, activity-driven assays have unearthed hundreds of bacteriocins. For instance, lactic acid bacteria alone harbor more than 230 putative bacteriocin BGCs and are prolific producers of these RiPPs (Alvarez-Sieiro et al., 2016). The biosynthesis and activities of bacteriocins have been extensively reviewed (Arnison et al., 2013; Li & Rebuffat, 2020). This section will survey representative peptides from a few different classes of membrane-active, posttranslationally modified bacteriocins for which structural data exist. The mechanistic studies that revealed how peptide form dictates function will be the subject of focus.
Pediocin-like bacteriocins are defined by a conserved, minimally modified tripartite structure consisting of an N-terminal antiparallel β-sheet, an intervening α-helix, and an unstructured C-terminal tail (Balandin et al., 2019; Fimland et al., 2005; Ríos Colombo et al., 2018). A prototypical example of this RiPP class is pediocin PA-1, also known as pediocin AcH (Bhunia et al., 1988; Gonzalez & Kunka, 1987; Henderson et al., 1992; Motlagh et al., 1992). Pediocin PA-1 has only a single type of posttranslational modification in the form of two disulfide bridges linking C9–C14 at the N-terminus and C24–C44 at the C-terminus (Fig. 5) (Henderson et al., 1992). The N-terminal disulfide bridge acts as a staple to stabilize the β-sheet and is found in almost all pediocin-like bacteriocins (Bédard et al., 2018). Another conserved N-terminal motif found in pediocin PA-1 and similar peptides is the pediocin box, which describes the sequence YGNG(V/L) that when mutated leads to reduction of pediocin antimicrobial activity (Miller et al., 1998). Mutations of the disulfide bond-forming cysteines similarly reduces pediocin activity (Bédard et al., 2018; Miller et al., 1998). Pediocin PA-1 was found to adsorb to Gram-positive cells in a process that likely involved electrostatic interactions, providing initial evidence of membrane-targeting activity (Bhunia et al., 1991). Curiously, pediocin PA-1 could adsorb to both pediocin-resistant and susceptible Gram-positive strains. This observation led to the proposal that a cell-surface receptor that is the specific docking site of pediocin PA-1 is present only in susceptible cells. Adsorption of pediocin PA-1 to susceptible cells caused cells to lyse or cease growing; release of potassium ions indicative of membrane potential collapse was also observed. Efflux and transport assays, transmembrane potential measurements, and binding studies provided additional evidence that pediocin PA-1 permeabilizes the bacterial cytoplasmic membrane (Chen, Ludescher, et al., 1997; Chen, Shapira, et al., 1997; Chikindas et al., 1993). Specifically, the cationic N-terminus of pediocin PA-1 alone can bind membrane vesicles but the more hydrophobic C-terminus is required for pore formation (Chen, Ludescher, et al., 1997). The C-terminus also predominantly determines target cell specificity (Johnsen et al., 2005). However, whether pediocin PA-1 required a membrane protein receptor for activity was debatable because it could release carboxyfluorescein from vesicles composed entirely of synthetic phospholipids (Chen, Shapira, et al., 1997). The σ54-regulated mannose phosphotransferase (Man-PTS) system has been proposed as the membrane receptor for pediocin-like bacteriocins (Dalet et al., 2000, 2001; Ramnath et al., 2000; Robichon et al., 1997). Yet, the relative contribution of this putative receptor toward peptide-membrane interaction is unclear. How the different regions of the multisubunit Man-PTS system would engage with the peptide is also poorly understood. Interestingly, the Man-PTS system is apparently required for the bioactivity of other structurally distinct bacteriocin RiPPs and may serve as a membrane receptor in these cases as well. The nonpediocin-like garvicin A/B/C peptides that target a Man-PTS subunit provide one such example (Tymoszewska et al., 2018).
Fig. 5.
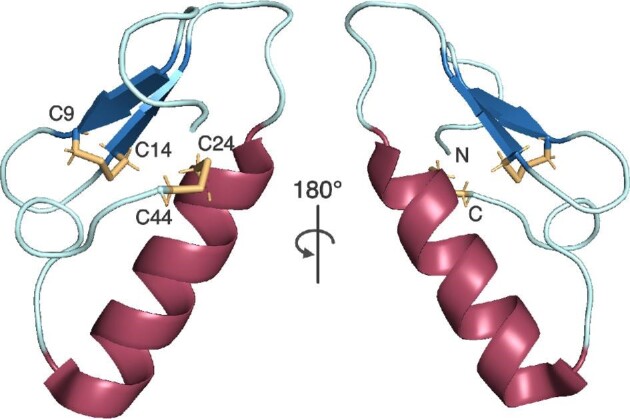
Structure of a synthetic analog of pediocin PA-1, a membrane-active bacteriocin. This compact structure is formed via two disulfide bonds, which are highlighted in orange. The N-terminal region of pediocin PA-1 is cationic, allowing for electrostatic interactions with bacterial membrane lipid head groups, while its C-terminal region is hydrophobic, allowing for pore formation in membranes. Figure drawn from PDB file 5UKZ.
The head-to-tail circularized bacteriocins are another group of membrane-active RiPPs that lack extensive posttranslational modifications. The founding member of this group is enterocin AS-48, a 70-residue peptide whose N- and C-termini are connected through a peptide bond (Burgos et al., 2014; Gálvez et al., 1986, 1989; Martinez-Bueno et al., 1994; Samyn et al., 1994). Enterocin AS-48 has broad-spectrum antimicrobial activity against several Gram-positive pathogens and to a lesser extent some Gram-negative organisms (Gálvez et al., 1986). Treatment of sensitive cells with enterocin AS-48 led to collapse of cytoplasmic membrane potential and formation of pores through which solutes could leak (Gálvez et al., 1991). As enterocin AS-48 can permeabilize artificial lipid vesicles, its interaction with the membrane may occur independently of a protein receptor (Gálvez et al., 1991). Solution NMR and crystal structures of enterocin AS-48 provided a model for how the peptide acts on the cell membrane (González et al., 2000; Sánchez-Barrena et al., 2003). The peptide folds into a compact globular shape that consists of five α-helices arranged in such a way that their hydrophobic side chains are directed toward the core of the 3D structure (González et al., 2000). The α-helices and the intervening loops are stabilized by a network of hydrogen bonds. Due to the high number of lysines present in the sequence, enterocin AS-48 has a net positive charge. However, the charged residues cluster within helices α4 and α5, the latter of which contains the head-to-tail junction of the cyclized peptide (Fig. 6A). One side of the enterocin AS-48 three-dimensional surface is more cationic as a consequence of this asymmetrical distribution of charged residues. The initial stages of peptide-membrane engagement may rely on electrostatic interactions between this cationic surface and the anionic head groups of acidic phospholipids that are enriched in bacterial cell membranes (González et al., 2000). Once enterocin AS-48 attaches with the membrane, rearrangement of the peptide to expose hydrophobic residues may facilitate membrane insertion (Sánchez-Barrena et al., 2003). Crystal structures of enterocin AS-48 in two different oligomeric conformations differing in the relative positions of the hydrophobic and hydrophilic α-helices support this rearrangement-dependent mechanism (Fig. 6B, C) (Sánchez-Barrena et al., 2003).
Fig. 6.

Structure of the membrane-active bacteriocin enterocin AS-48. (A) Enterocin AS-48 is cyclized between its N- and C-termini and highly helical. Sidechains of negatively and positively charged amino acids are clustered together in helices 4 and 5 and are shown as sticks (PDB file 1O82). (B and C) Two distinct dimeric forms of enterocin AS-48 are found within the crystal structure. Dimeric form I (DF-I) shows the hydrophobic side chains of helices 1 and 2 in gray. Dimeric form II (DF-II) includes a sulfate ion at the protomer–protomer interface.
Gram-negative bacteria also produce modified bacteriocin RiPPs that target the cytoplasmic membrane of closely related species. An example of a membrane-active, Gram-negative bacteriocin is microcin E492, which was isolated from Klebsiella pneumoniae strain RYC492 and showed broad activity against members of the Enterobacteriaceae family (de Lorenzo, 1984; Lagos et al., 2009). Microcin E492 is a low-molecular weight peptide as is typical for the microcin family of RiPPs, but its production is specified by a BGC that resides on the chromosome instead of a plasmid as for many other microcins. Identification (Wilkens et al., 1997) and sequencing (Lagos et al., 2008) of the BGC revealed several potential modification enzymes, whose roles were later revealed to be installation of a C-glucosylated linear trimer of N-(2,3 dihydroxybenzoyl)-l-serine (DHBS) to the C-terminus of the precursor peptide (Vassiliadis et al., 2007). Microcin E492 exists in both the modified and unmodified form, but it is the modified form that exhibits more pronounced antimicrobial activity (Thomas et al., 2004). The observed difference in activity has been attributed to the relative affinities of the two peptide forms for the outer membrane transporter that delivers microcin E492 to its site of action at the cytoplasmic membrane (Destoumieux-Garzón et al., 2006; Thomas et al., 2004). In fact, the C-glucosylated DHBS unit of the modified peptide structurally resembles the catecholate siderophore salmochelin (Thomas et al., 2004; Vassiliadis et al., 2007). Siderophore uptake occurs through TonB-dependent transporters (TBDTs) (Noinaj et al., 2010). Therefore, it has been proposed that the siderophore-like appendage of microcin E492 directs peptide transport across the outer membrane through a TBDT. Consistent with this role, it was shown that TonB and the FepA, Fiu, and Cir outer membrane receptors of target cells are required for microcin E492 bioactivity (Patzer et al., 2003; Thomas et al., 2004). Perhaps also consistent with TonB-dependent transport of microcin E492, another catecholate-type siderophore, enterochelin, was found to inhibit bioactivity possibly through competition for the same transporter (Orellana & Lagos, 1996). Mechanistic and structural information on microcin E492-receptor interaction is still unknown. Once it has crossed the outer membrane barrier, microcin E492 is thought to permeabilize the cytoplasmic membrane (De Lorenzo & Pugsley, 1985). Membrane insertion releases membrane potential-sensitive probes from preloaded cells, indicating depolarization of the membrane (De Lorenzo & Pugsley, 1985; Destoumieux-Garzón et al., 2003). In addition to antibacterial activity, microcin E492 has been reported to induce apoptosis of some human cancer cell lines, although whether this secondary activity is related to its pore-forming ability is unclear (Hetz et al., 2002). Going forward, how microcin E492 engages with the cytoplasmic membrane and how this process may involve inner membrane proteins remain to be seen (Bieler et al., 2006). It has also been reported that microcin E492 forms amyloid-like fibrils that preclude activity (Bieler et al., 2005). What domains of microcin E492 are necessary and sufficient for bioactivity and the route of conversion to a different oligomeric state are potential future areas of investigation.
Sactipeptides are a growing class of bacteriocin RiPPs that are defined by unusual intramolecular covalent linkages between the side chain sulfur of cysteine residues and the α-carbon of other residues. The most well-characterized sactipeptide is sutilosin A, which was first isolated from the common laboratory strain Bacillus subtilis 168 (Babasaki et al., 1985) and subsequently from other Bacillus species (Sutyak et al., 2008; Zheng et al., 1999). Subtilosin A has bactericidal activity against Gram-positive pathogens (Babasaki et al., 1985) and reportedly spermicidal activity as well (Silkin et al., 2008). Four intramolecular linkages contribute to the semirigid, cyclic nature of subtilosin A (Babasaki et al., 1985; Kawulka et al., 2003, 2004; Marx et al., 2001). In addition to three thioether bridges that connect C4 with F31, C7 with T28, and C13 with F22, an amide bond between N1 and G35 links the N- and C-terminus of the peptide (Fig. 7A) (Kawulka et al., 2003, 2004). A solution NMR structure of subtilosin A resembled a bent hairpin that forms a shallow basin (Fig. 7B) (Kawulka et al., 2003). The two sides of the hairpin are held close together by the thioether linkages and the side chains of the backbone point outward from the center of the hairpin loop (Kawulka et al., 2003, 2004). Subtilosin A has many hydrophobic residues, but there are also three acidic residues that are concentrated on the loop end of the folded peptide and a basic residue present on the opposite end. This separation of charge is believed to impact how subtilosin A targets the cytoplasmic membrane of sensitive cells (Thennarasu et al., 2005). Subtilosin A bound and permeabilized unilamellar vesicles composed of synthetic phospholipids (Thennarasu et al., 2005). The model proposed for binding involves insertion of the loop-distal end of subtilosin A into the lipid bilayer first (Thennarasu et al., 2005). This end contains the single basic residue, K2, which may interact with the negatively charged phosphate headgroup and a large, hydrophobic tryptophan residue, W34, which may perturb the lipid bilayer (Thennarasu et al., 2005). Release of membrane potential- and pH-sensitive probes from preloaded cells suggest that partial insertion of subtilosin A leads to ATP efflux and depletion of the transmembrane ion gradient (Noll et al., 2011).
Fig. 7.

The structure of sactipeptide subtilosin A. (A) The 35 aa subtilosin A is cyclized via a head-to-tail peptide bond and three unusual thioether linkages between the side chain of cysteine and the α-carbon of a distal amino acid. (B) The solution structure of subtilosin A in methanol (PDB file 1PXQ) shows a compact hairpin-like structure. Acidic (D16, D21, E23) and basic (K2) residues are concentrated at different ends of the folded peptide. Residues participating in thioether linkages are shown as gold sticks.
Similarly, the two-component sactipeptide thuricin CD, which has nanomolar potency against Clostridium species, also attacks the bacterial cytoplasmic membrane (Mathur et al., 2017; Rea et al., 2010). Thuricin CD is composed of two structurally related peptides, α and β, that share 39.6 per cent sequence identity and act synergistically (Rea et al., 2010). The residues at positions 21, 25, and 28 in both peptides are modified through α-carbon-cysteine sulfur linkages. Neither peptide has the N-to-C amide linkage present in subtilosin A, but thuricin-α and thuricin-β each forms a folded hairpin structure with the backbone side chains pointed outward similar to subtilosin A (Sit et al., 2011). The solution NMR structure of thuricin CD also showed that the surfaces of the constituent peptides are highly hydrophobic (Sit et al., 2011). Upon thuricin CD challenge, sensitive cells undergo membrane depolarization and cell lysis (Mathur et al., 2017). A more recent example of a sactipeptide with proposed membrane-targeting activity is thuricin Z (Mo et al., 2019). Thuricin Z has four α-carbon to cysteine sulfur linkages installed at residues D26-C16, T30-C12, Y34-C8, and H38-C4, highlighting a degree of periodicity in the modified sites (Mo et al., 2019). To our knowledge, the three-dimensional structure of thuricin Z has not been solved. However, like subtilosin A, thuricin Z also permeabilizes bacterial membrane-mimicking unilamellar vesicles (Mo et al., 2019; Thennarasu et al., 2005).
Bacteriocin RiPPs are extremely diverse in structure and tailoring modifications, but a striking number appear to act on the same target, the bacterial cytoplasmic membrane. Binding studies with purified peptides and model membrane vesicles have provided insights into the chemical requirements for interaction and pore formation. Electrostatic interactions for one appear to play a strong role. Bacterial cell membranes are rich in anionic phospholipids (Sohlenkamp & Geiger, 2016). Different bacteria have distinctive membrane lipid content and even within the same organism, the membrane lipid content can adapt depending on the growth environment (Sohlenkamp & Geiger, 2016). The precise molecular interactions between bacteriocin RiPPs and diverse membrane structures remain to be dissected. Furthermore, it is worth considering how in reality the crowded physiological environment of a cell membrane would impact peptide binding. Sometimes, a concentration of bacteriocin higher than its MIC was required to observe membrane permeabilization in vitro (Thennarasu et al., 2005). This difference may reflect the mode by which the peptide engages the membrane. Even if a peptide can bind synthetic lipid-only vesicles in vitro, one of the many membrane-embedded proteins may promote binding in cellulo. Understanding these interactions will explain and enable prediction of spectrum of activity.
Two Recently Discovered Cell Envelope-Targeting RiPPs
Aside from lanthipeptides, a member of the lasso peptide class of RiPPs has been reported to also inhibit bacterial cell wall synthesis by targeting lipid II. Siamycin-I is a 21-residue peptide that folds into a three-dimensional lasso structure containing a ring, loop, and tail regions (Detlefsen et al., 1995). An isopeptide bond between the N-terminal cysteine and side chain of D9 produces a nine-membered ring through which the remaining residues are threaded. Two disulfide bridges connecting the ring with the loop above and tail below further rigidify the lasso fold (Maksimov et al., 2012). The lipid II-targeting activity of siamycin-I was only recently reported (Daniel-Ivad et al., 2017; Tan et al., 2019), but siamycin-I had been shown to possess other bioactivities since its initial discovery more than two decades ago (Chokekijchai et al., 1995; Tsunakawa et al., 1995). Siamycin-I inhibits the human immunodeficiency virus (Chokekijchai et al., 1995; Lin et al., 1996; Tsunakawa et al., 1995), myosin light chain kinase involved in smooth muscle contraction (Yano et al., 1996), and ATP-dependent enzymes (Ma et al., 2011). Siamycin-I was rediscovered in a screen for cryptic secondary metabolites that are regulated by a two-component system (Daniel-Ivad et al., 2017). Expressing a constitutively active pleiotropic regulator triggered the production of this lasso peptide in multiple Streptomyces species (Daniel-Ivad et al., 2017). Several lines of evidence thereafter suggested siamycin-I inhibits bacterial cell wall synthesis. Treatment of sensitive cells with siamycin-I phenocopied treatment with the cell wall-targeting antibiotics vancomycin, bacitracin, and nisin (Daniel-Ivad et al., 2017). Mutations that conferred siamycin-I resistance mapped to the WalKR genes, which is a signal transduction pathway that controls cell wall metabolism (Tan et al., 2019). Finally, biochemical reconstitution with defined cell wall substrates demonstrated that siamycin-I blocks the conversion of polyprenyl pyrophosphate-linked substrates (Tan et al., 2019). Thus, it was concluded that siamycin-I binds lipid II through recognition of the conserved pyrophosphate motif similar to nisin (Tan et al., 2019). Unlike nisin however, siamycin-I treatment does not lead to formation of membrane pores (Tan et al., 2019). Siamycin-I is active against Gram-positive but not Gram-negative bacteria possibly due to the presence of an outer membrane barrier in the latter group (Daniel-Ivad et al., 2017; Tsunakawa et al., 1995). Nonetheless, the extent to which siamycin-I inhibits different Gram-positive bacteria does vary (Daniel-Ivad et al., 2017).
Darobactin is the founding member of a new class of RiPPs that are defined by the presence of strained cyclophane rings (Imai et al., 2019; Nguyen et al., 2020). Darobactin is a modified heptapeptide that contains two macrocycles formed through an aromatic-aliphatic ether linkage between the W1 side chain and W3 β-carbon and a carbon-carbon bond between the W3 side chain and K5 β-carbon (Fig. 8) (Imai et al., 2019). Imai et al. isolated darobactin by screening concentrated extracts from bacterial cultures of nematode gut symbionts for antimicrobial activity (Imai et al., 2019). Darobactin has high therapeutic potential because it showed broad-spectrum activity against Gram-negative bacteria and low cytotoxicity against human cell lines and commensals (Imai et al., 2019). An early clue in determining the mechanism of action was the observation that darobactin treatment decreased the abundance of outer membrane proteins while increasing the abundance of periplasmic chaperones (Imai et al., 2019). Spontaneous suppressor mutants that were resistant to darobactin mapped to BamA (Imai et al., 2019). The BamABCDE complex, or β-barrel assembly machinery (BAM), is an essential outer membrane complex that folds and inserts proteins into the outer membrane (Fig. 2B) (Bakelar et al., 2016; Rigel & Silhavy, 2012; Tomasek et al., 2020; Wu et al., 2005). The BamA subunit is a β-barrel protein that templates the folding of nascent β-barrels (Tomasek et al., 2020). BamA exists in different conformational states and switches from the lateral gate-closed to gate-opened state to accommodate entry of the unfolded substrate for folding (Hartmann et al., 2018; Tomasek et al., 2020). In vitro reconstitution of BAM in lipid nanodiscs showed that darobactin directly inhibited BAM activity and that activity was dependent on the modified peptide scaffold as a linear variant was inactive (Imai et al., 2019). Isothermal titration calorimetry and solution NMR experiments confirmed that darobactin bound BamA and stabilized its gate-closed conformation, thereby preventing opening of the lateral gate for substrate entry and folding (Imai et al., 2019). A closed BamA gate would lead to accumulation of unfolded proteins in the periplasm, which explains the observed increase in periplasmic chaperones (Imai et al., 2019). High-priority ESKAPE pathogens, many of which are Gram-negatives, have developed resistance to clinically used antibiotics (CDC, 2019). Antibiotics that are effective against Gram-negative pathogens are thus increasingly needed. A major impediment in developing these antibiotics is that the outer membrane provides a barrier that prevents accessibility to druggable targets. Cell-surface components, and in particular outer membrane components such as BamA, are suitable targets that circumvent the issue of outer membrane permeability. Notably, mutations in outer membrane components that confer resistance to a drug in vitro may simultaneously impair virulence in vivo. For instance, darobactin-resistant E. coli mutants that have acquired mutations in BamA have attenuated virulence in mice (Imai et al., 2019). This duality means that darobactin would be even more effective as a drug because resistant strains that arise are likely to also be less virulent.
Fig. 8.
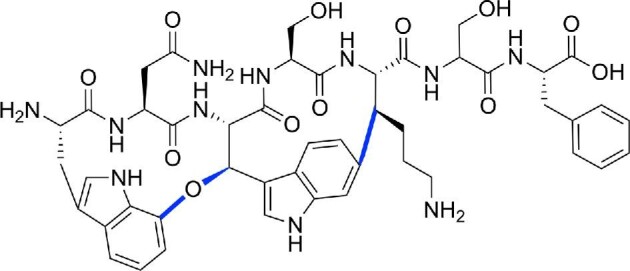
Chemical structure of the heptapeptide RiPP darobactin. Linkages between sidechains are colored in blue.
Transcription-Inhibiting RiPPs
Transcription is an essential step of gene expression where a specific coding sequence of DNA is converted to a corresponding messenger RNA by the enzyme RNA polymerase. The essentiality of this process makes RNA polymerase a promising drug target. In fact, various RiPPs, including many members in the family of lasso peptides and the cyclic peptide α-amanitin, have been unveiled to be RNA polymerase inhibitors through either crystallography or in vitro inhibition assays (Braffman et al., 2019; Cheung-Lee, Parry, Cartagena, et al., 2019; Cheung-Lee, Parry, Zong, et al., 2020; Mukhopadhyay et al., 2004; Solbiati et al., 1999).
Lasso peptides are characterized by their [1] rotaxane structure, where an isopeptide bond joins the N-terminus of the core peptide to the side chain of a D or E residue, templating a 7–9 aa macrocyclic ring (Cheung-Lee & Link, 2019; Hegemann et al., 2015; Maksimov et al., 2012; Maksimov & Link, 2014). Since the discovery of the first isolated lasso peptide, microcin J25 (MccJ25), lasso peptides have been a hot topic of research due to both their unique rotaxane structure and diverse bioactivities (Salomón & Farías, 1992). Lasso peptides that inhibit RNA polymerases have all been isolated from Gram-negative proteobacteria, including MccJ25 (Salomón & Farías, 1992; Wilson et al., 2003), capistruin (Knappe et al., 2008; Kuznedelov et al., 2011), citrocin (Cheung-Lee, Parry, Cartagena, et al., 2019), ubonodin (Cheung-Lee, Parry, Zong, et al., 2020), acinetodin (Metelev, Arseniev, Bushin, et al., 2017), and klebsidin (Metelev, Arseniev, Bushin, et al., 2017) (Fig. 9A). Braffman et al. recently reported the crystal structures of microcin J25 and capistruin bound to E. coli RNA polymerase (Braffman et al., 2019). MccJ25 binds deep within the secondary channel in a way that is expected to interfere with nucleoside triphosphate (NTP) substrate binding (Fig. 9B, C), in agreement with the partial competitive mechanism of inhibition regarding NTPs reported by Mukhopadhyay et al. (2004). The Y9 residue of MccJ25 is found to be strictly essential for RNA polymerase inhibition (Pavlova et al., 2008), and this residue makes the most extensive set of contacts with RNA polymerase. Additionally, the side chain hydroxyl groups of Y9 and Y20 as well as the C-terminus of G21 of MccJ25 form hydrogen bonds with the RNA polymerase, suggesting their importance in the mode of action of MccJ25. It is worth noting that the Y-YG motif, where the first Y is the first residue of the loop, the second Y is the lower lock residue, and the G is at the C-terminus, is conserved in MccJ25, citrocin, and ubonodin (Cheung-Lee, Parry, Cartagena, et al., 2019; Cheung-Lee, Parry, Zong, et al., 2020; Salomón & Farías, 1992). The C-terminal YG motif is also conserved in the case of acinetodin and klebsidin, where the first residue of the loop is T and F, respectively (Metelev, Arseniev, Bushin, et al., 2017). The importance of this motif will be beneficial in discovering novel antimicrobial lasso peptides targeting RNA polymerase through a precursor-centric genome mining approach (Maksimov et al., 2012). In the case of citrocin and ubonodin, abortive transcription initiation assays with E. coli RNA polymerase were carried out to confirm both peptides inhibit transcription initiation (Cheung-Lee, Parry, Cartagena, et al., 2019; Cheung-Lee, Parry, Zong, et al., 2020). On the other hand, in the case of acinetodin and klebsidin, single molecule acoustic force spectroscopy experiment was
Fig. 9.

RiPP inhibitors of RNA polymerase (RNAP). (A) Schematics of microcin J25 (MccJ25), capistruin, and α-amanitin. ILX: 4,5-dihydroxyisoleucine, TRX: 6-hydroxytryptophan, CSX: S-oxy cysteine, HYP: HYP: 4-hydroxyproline. Note the tryptathionine linkage between 6-hydroxytryptophan and S-oxy cysteine. (B) From left to right, crystal structures of E. coli RNAP showing the binding site of MccJ25, E. coli RNAP showing the binding site of capistruin, and yeast RNA polymerase II (Pol II) showing the binding site for α-amanitin. Residues in RNAP or Pol II that directly contact these RiPPs are colored red. (C) Zoomed-in view of MccJ25 (gold/blue) bound to RNAP (cyan). Key amino acids for this interaction are labeled. Dashed lines are H-bonds (yellow). (D) As in part C, but for the RNAP–capistruin interaction. (E) Zoomed-in view of the α-amanitin-Pol II binding site with key interacting residues highlighted. HYP: hydroxyproline, ILX: dihydroxyisoleucine, TRX: hydroxytryptophan. Drawn from PDB files 6N60 (MccJ25), 6N61 (capistruin), and 3CQZ (α-amanitin).
carried out to measure the speed at which the nucleotides are getting transcribed, and showed both peptides slowed transcription (Metelev, Arseniev, Bushin, et al., 2017). In contrast, capistruin, lacking the Y-YG motif, binds further from the RNA polymerase active site and does not sterically hinder NTP binding (Fig. 9B, D) (Braffman et al., 2019). This observation is also supported by the fact that capistruin inhibition is partially noncompetitive with respect to NTPs (Braffman et al., 2019). It is also worth noting that the uptake of MccJ25 by its susceptible Escherichia and Salmonella strains is one of the best studied examples of peptide transport in the RiPPs field, which will be discussed further in the Cellular Uptake of RiPPs section (Mathavan et al., 2014; Salomón & Farías, 1993, 1995).
α-amanitin is one of the deadliest amatoxins found in species of the mushroom genus Amanita. Structurally, it is a bicyclic octapeptide with C-to-N (head to tail) condensation of the peptide backbone and a cross-bridge between 6-hydroxytryptophan and S-oxy cysteine (Fig. 9A) (Hallen et al., 2007; Luo et al., 2012). It is highly specific for RNA polymerase II, causing cytolysis of hepatocytes (Magdalan et al., 2009). Bushnell et al. first solved the cocrystal structure of α-amanitin and RNA polymerase II (Pol II) from Saccharomyces cerevisiae, in which α-amanitin interacts with the bridge helix in RNA polymerase II (Fig. 9B, E) (Bushnell et al., 2002). Binding of α-amanitin allows nucleotide entry to the active site and RNA synthesis but prevents the translocation of DNA and RNA needed to enable the next round of synthesis (Bushnell et al., 2002). Brueckner and coworkers, as well as Keplan et al., have subsequently solved two additional cocrystal structures of α-amanitin with S. cerevisiae Pol II. Both structures suggested α-amanitin traps the trigger loop (TL) and shifts the bridge helix (Brueckner & Cramer, 2008; Kaplan et al., 2008). The importance of the hydroxyl group of the hydroxyproline residue 2 is highlighted by Brueckner and coworkers, which binds not only to the bridge helix residue E822, but also interacts with the TL residue H1085, and a possible contact with N1082, which binds the bridge helix residue E826 (Brueckner & Cramer, 2008). This observation also agrees with previous analysis on α-amanitin variants, where the hydroxyl group of the hydroxyproline residue 2 has been shown to be crucial for its activity (Wienland & Faulstich, 1991; Zanotti et al., 2009).
Other than targeting the RNA polymerase, linear azo(lin)e peptide (LAP) family member microcin B17 (MccB17) targets DNA gyrase, which can indirectly affect RNA transcription (Booker et al., 2010; Higgins et al., 1988; Peter et al., 2004; Rovinskiy et al., 2012). The mechanism of action of MccB17 will be further detailed in the RiPPs with Other Mechanisms of Action section.
Translation-Inhibiting RiPPs
Following the production of mRNA, translation is the process during which the ribosome synthesizes proteins by decoding the mRNA. The complementary tRNA anticodon sequences bind to the mRNA codon and the amino acids carried by the individual tRNAs are linked together to form the elongated polypeptides. Diverse classes of RiPPs, including LAPs, bottromycins, thiopeptides, microcin C (McC), have been shown to target various machinery required for this essential process, including the ribosome (Harms et al., 2008; Metelev, Osterman, Ghilarov, et al., 2017; Otaka & Kaji, 1976, 1981, 1983; Travin et al., 2019), the elongation factor Tu (EF-Tu) (Heffron & Jurnak, 2000; Morris et al., 2009), and the aspartyl-tRNA synthetase (AspRS) (Kazakov et al., 2008; Metlitskaya et al., 2006; Piskunova et al., 2017; Ran et al., 2017).
RiPPs that inhibit the ribosome include LAPs, thiopeptides, and bottromycins, including klebsazolicin (Metelev, Osterman, Ghilarov, et al., 2017), phazolicin (Travin et al., 2019), thiostrepton (Bagley et al., 2005; Kelly et al., 2009; Liao et al., 2009), nosiheptide (Shoji et al., 1976) micrococcin (Cundliffe & Thompson, 1981), and bottromycin A2 (Otaka & Kaji, 1976, 1981, 1983). Other than bottromycin A2, crystal structures of these peptides bound to ribosome have been solved (Harms et al., 2008; Metelev, Osterman, & Ghilarov, et al., 2017; Travin et al., 2019). Klebsazolicin and phazolicin belong to LAPs, a family of RiPPs defined by the presence of heterocycles derived from C, S, and T residues in the precursor peptide (Cox et al., 2015). Metelev et al. have crystallized the Thermus thermophilus 70S ribosome in the presence of klebsazolicin, mRNA, and A-, P-, and E-site tRNAs, where klebsazolicin is curled to a compacted form in the peptide exit channel on the large ribosomal subunit (Fig. 10B, C) and serves as a compelling example of how flexible RiPPs can be when bound to their molecular targets (Metelev, Osterman, Ghilarov, et al., 2017). Klebsazolicin forms extensive contacts with 23S rRNA of the ribosome; the first and the second thiazole rings (Thz7 and Thz10) stack over C2586-C1782 and A2062-m2A2503 base pairs, respectively, and the hydroxyl groups of the S1 and S9 side chain form H-bonds with U2585 and U2506, respectively (Fig. 10C) (Metelev, Osterman, & Ghilarov, et al., 2017). On the other hand, Travin et al. crystallized phazolicin bound to E. coli 70S ribosome, after numerous attempts with 70S ribosome from T. thermophilus, mRNA, and tRNAs failed (Travin et al., 2019). Similar to klebsazolicin, phazolicin forms a compact globule that obstructs the nascent peptide exit tunnel of the ribosome, and binds further away than klebsazolicin from the peptidyl transferase center of the ribosome (Fig. 10A, D). One striking feature of this cocrystal structure is its complex intramolecular interaction involving the π–π interactions of thiazole12 (Thz12), oxazole21 (Oxz21), Thz6, Oxz18, along with the nucleotide U2609 from the 23S rRNA. Moreover, multiple intermolecular interactions were observed; Thz3, Oxz15, and Oxz18 are involved in π–π stacking with nucleotides A751, C2611, and U2609, respectively, and positively charged side chains of R5 and R11, along with hydrophilic side chains of D7 and S8, form additional H-bonds with the nucleobases and backbone phosphate groups of the 23S RNA.
Fig. 10.

Examples of RiPPs that inhibit translation. (A) Crystal structures of bacterial ribosomes with thiopeptide thiostrepton binding site (PDB file 3CF5) colored in blue and linear azo(lin)e peptide (LAP) phazolicin binding site colored in red (PDB file 6U48). (B) Chemical structure of the LAP klebsazolicin. (C) Key interactions between klebsazolicin (gold) and 23S rRNA (blue), drawn from PDB file 5W4K. Dashed lines are H-bonds (yellow), and double-sided arrows represent π stacking. (D): As in part C, but for the phazolicin:ribosome interaction, drawn from PDB file 6U48. (E) Key interactions between thiostrepton (gold), 23S rRNA (blue), and ribosomal protein L11 (cyan) drawn from PDB file 3CF5. (F) As in part E, but for the nosiheptide:ribosome interaction, drawn from PDB file 2ZJP. THZ: thiazole, OXZ: oxazole, QA: quinaldic acid.
Thiopeptides are distinguished by their six-membered nitrogen heterocycles, dehydrated amino acids, and thiazole rings formed from dehydrated S/T and cyclized cysteine residues (Bagley et al., 2005; Shen et al., 2019). In contrast to klebsazolicin and phazolicin, thiopeptides that target the ribosome, including thiostrepton, nosiheptide, and micrococcin, target the GTPase-associated center of the ribosome, which is responsible for binding and stimulation of the GTPase activity of translation factors from all phases of translation (Fig. 10A) (Harms et al., 2008; Wilson & Nierhaus, 2005). Harms et al. crystallized the Deinococcus radiodurans 50S large ribosomal subunit in complex with thiostrepton, nosiheptide, and micrococcin. Both thiostrepton and nosiheptide were observed in a cleft between the N-terminal domain of ribosomal protein L11 and the tips of H43 and H44 of the 23S rRNA (Fig. 10E, F) (Harms et al., 2008). In particular, Thz6 and Thz14 of thiostrepton stack on P22 and P26 of L11, respectively, and Thz1 forms stacking interactions with A1095 of the 23S rRNA. The binding site of thiostrepton and nosiheptide sterically clashes with the elongation factor-G (EF-G), and both peptides also lock the N-terminus of L11 in a position that will prevent stable interaction between EF-G and the stalk base. In contrast, micrococcin does not bind in the same position as thiostrepton and nosiheptide, with the macrocyclic loop of micrococcin being displaced by ∼6 Å compared to that of thiostrepton. Unlike thiostrepton and nosiheptide, micrococcin was shown to stimulate uncoupled EF-G-dependent GTPase activity (Cameron et al., 2002; Harms et al., 2008; Lentzen et al., 2003). However, due to resolution limitations, the precise molecular interactions of micrococcin with the ribosome were not reported (Harms et al., 2008). Other than inhibiting the ribosome, Bird et al. have recently reported that thiostrepton, nosiheptide, and micrococcin P1, along with thiopeptides thiocillin III, GE37468, and berninamycin, can induce cellular mitophagy (Bird et al., 2020).
Bottromycins are unique peptide antibiotics that contain a macrolactamidine, several nonproteinogenenic amino acids and a thiazole (Gomez-Escribano et al., 2012). Similar to polytheonamides, botthromycins had long been thought to be synthesized through nonribosomal peptide synthetases pathways since their first discovery in 1957 (Waisvisz et al., 1957). In 2012, four studies sequentially confirmed they are indeed RiPPs (Crone et al., 2012; Gomez-Escribano et al., 2012; Hou et al., 2012; Huo et al., 2012). Otaka and coworkers demonstrated that bottromycin A2 targeted the A site of the ribosome by weakening the affinity of peptidyl or aminoacyl-tRNA for the A site (Otaka & Kaji, 1976, 1981, 1983). However, a cocrystal structure of bottromycin with ribosome has yet to be solved.
The elongation factor thermo unstable (EF-Tu), a guanine nucleotide-binding protein that binds and directs aminoacyl-tRNA to the A-site of the ribosome (Harvey et al., 2019), has also shown to be the molecular target for a group of thiopeptides, including GE2270A (Möhrle et al., 1997; Zuurmond et al., 2000), thiomuracin A (Morris et al., 2009), and amythiamicin A (Shimanaka et al., 1994, 1995). Notably, Heffron and coworkers solved the crystal structure of an EF-Tu complex with GE2270A (Fig. 11A) (Heffron & Jurnak, 2000). In the solved structure, GE2270A binds to a pocket in the second domain of EF-Tu-GDP. The strongest interactions are localized to residues including D216, G222, R223, T256, G257, E259, F261, R262, N273, V274, G275, and L277 (Fig. 11B) (Heffron & Jurnak, 2000). In particular, GE2270A appears to be locked into the binding site by a salt bridge between R223 and E259 of EF-Tu, and five H-bond interactions were observed between GE2270A and EF-Tu (Fig. 11B) (Heffron & Jurnak, 2000).
Fig. 11.

Interaction of thiopeptide GE2270A with elongation factor EF-Tu. (A) Crystal structure of the EF-Tu: GE2270A complex, drawn from PDB file 1D8T. (B) Close up view of the EF-Tu (cyan):GE2270A (magenta) complex. Amino acids from EF-Tu making direct contact with the thiopeptide are labeled. Atoms from GE2270A making direct contact are labeled. Dashed lines are H-bonds (yellow) or a salt bridge (blue) between R223 and E259 that accounts for the high stability of the GE2270A:EF-Tu complex.
In addition, the AspRS has been shown to be targeted by McC, a MRTGNAD heptapeptide with an adenosine monophosphate (AMP) analog attached to the C-terminus through an N-acyl phosphoramidate linkage and N-terminal formylation (Severinov & Nair, 2012). Once in the susceptible cells, McC is first deformylated, and then digested to form the toxic processed McC, a nonhydrolyzable analogue of aspartyl-adenylate (Kazakov et al., 2008; Metlitskaya et al., 2006). This mimic inhibits AspRS, which therefore blocks protein synthesis. In vitro luciferase gene transcription-translation reaction was carried out to illustrate the mode of action of McC (Metlitskaya et al., 2006). In the presence of processed McC, the exogenously added aminoacylated tRNAAsp was quickly used up, and the second addition of aminoacylated tRNAAsp led to a further increase in luciferase production (Metlitskaya et al., 2006). In the meantime, an orthogonal experiment showed that E. coli cells overproducing D. radiodurans AspRS were resistant to McC (Metlitskaya et al., 2006). Metlitskaya et al. further illustrated that the processed McC with the aminopropyl group was shown to be approximately 10-fold more potent than the variant without this modification (Metlitskaya et al., 2009).
RiPPs with Other Mechanisms of Action
Other than the aforementioned mechanisms of action by various RiPPs, there are some notable exceptions that do not fit these categories. The lasso peptide lassomycin is active against multidrug resistant Mycobacterium tuberculosis by targeting the Hsp100 family chaperone ClpC1 (Gavrish et al., 2014). ClpC1 complexes with the proteases ClpP1 and ClpP2 to form a protein degradation machine similar to the proteasome. Six resistance mutants of M. tuberculosis were raised, and all of them showed mutations in the clpC1 gene, which encodes the subunit of the hexameric ATPase complex, ClpC1 (Gavrish et al., 2014). In the presence of low concentrations of lassomycin, ATP hydrolysis by ClpC1 increased up to 7- to 10-fold, and this activation by lassomycin showed an apparent Kd of 0.41 μM, which resembles its MIC value against M. tuberculosis (Gavrish et al., 2014). However, lassomycin completely eliminated the ability of ClpC1P1P2 to degrade casein (Gavrish et al., 2014).
Microcin B17 (MccB17) is an E. coli antibiotic that inhibits DNA gyrase, an essential bacterial enzyme that catalyzes the ATP-dependent negative supercoiling of double-stranded closed circular DNA (Reece & Maxwell, 1991). Inhibition of DNA gyrase would result in changes in the average supercoil density, which would alter the efficiency and phenotype of proteins involved in transcription (Booker et al., 2010; Higgins et al., 1988; Peter et al., 2004; Rovinskiy et al., 2012), DNA replication (Pang et al., 2005), chromosome segregation (Champion & Higgins, 2007; Ogura et al., 1990), and transposition (Sternglanz et al., 1981). MccB17 binds to the gyrase(A2B2)–DNA complex in a way that interrupts, but does not completely inhibit the strand-passage reaction, and stabilizes the conformation of the enzyme where the DNA is cleaved and covalently bound to the enzyme (Collin & Maxwell, 2019; Zamble et al., 2001). Although a structure of MccB17 bound to DNA gyrase has yet to be solved, MccB17 has been shown to inhibit DNA supercoiling while still allowing DNA cleavage in vitro (Zamble et al., 2001). MccB17 variant studies suggest that the thiazole-oxazole motif at position 39 and oxazole-thiazole motif at position 54 are crucial for its antimicrobial activity (Collin et al., 2013; Collin & Maxwell, 2019).
Sporulation killing factor (SKF) is a 26-residue sactipeptide produced by Bacillus subtilius Py79 and B. subtilis 168, which is characterized by its thioether bond between C4 and the α-carbon of M12 (Flühe et al., 2013; Gonzalez-Pastor, 2003; Grell et al., 2018). When a B. subtilis population is deprived of nutrition, the regulatory protein Spo0A activates the expression of the skf and sdp operons, which results in the production of SKF and the sporulation delay protein, respectively. Both products are exported, and Spo0A-inactive cells are lysed due to the extracellular SKF (Engelberg-Kulka, 2003; Gonzalez-Pastor, 2003; Liu et al., 2010). This process releases additional nutrients for Spo0A-active B. subtilis subpopulation, enabling them to delay the main sporulation event (Flühe et al., 2013; Gonzalez-Pastor, 2003).
Thiopeptide siomycin A targets oncogenic transcription factor Forkhead box M1 (FoxM1), a therapeutic target expressed in a majority of human tumors (Bhat et al., 2009; Laoukili et al., 2007; Radhakrishnan et al., 2006). Siomycin A inhibits expression and transcriptional activity of FoxM1, but not other members of the Forkhead box family. Bhat et al. have shown that siomycin A inhibits the growth and induces potent apoptosis in leukemia CEM, HL60, U937 and liver Hep-3B, Huh7, and SK-Hep cancer cells (Bhat et al., 2009). The apoptosis induced by siomycin A correlates with the suppression of FoxM1 expression, while overexpression of FoxM1 partially protected cancer cells from siomycin-mediated death (Bhat et al., 2009). It is worth noting that thiostrepton, a structurally similar thiopeptide that has shown to inhibit ribosome, also shows similar activity as siomycin A against FoxM1 (Bhat et al., 2009).
Cellular Uptake of RiPPs
The bioactivity of RiPPs is a product of their ability to first access and then bind their target. We have focused thus far in this review on direct interactions between RiPPs and their cellular targets, but the mechanisms by which the RiPPs are delivered to their targets equally impact bioactivity. Consider the lasso peptides that inhibit RNA polymerase and the thiopeptides that inhibit the ribosome. In both scenarios, the target is cytoplasmic, which means that the exogenous peptides must cross the cytoplasmic membrane and, in the case of a Gram-negative target cell, the outer membrane as well before reaching the intracellular target. Any disruption in this path would curtail bioactivity. In fact, that some RiPPs display a narrow spectrum of activity despite having highly conserved targets has been attributed to whether they can be internalized. However, very little is known about the cellular uptake mechanisms of RiPPs and much of the present knowledge has been confined to the microcin family of RiPPs. Even from these limited studies, a common theme that has emerged is the promiscuous role that endogenous nutrient transporters play in RiPP transport (Mathavan & Beis, 2012).
Genetic screens laid the foundation for uncovering the cellular uptake mechanisms of RiPPs and implicated a small number of proteins as putative membrane transporters. FhuA-mediated uptake of MccJ25 is perhaps the best-understood example of RiPP transport (Mathavan et al., 2014; Salomón & Farías, 1993), but uptake pathways have also been proposed for microcin E492 (Destoumieux-Garzón et al., 2003, 2006; Patzer et al., 2003; Pugsley et al., 1986; Thomas et al., 2004) and microcin B17 (Laviña et al., 1986). As previously discussed, microcin E492 harbors a siderophore-like modification that directs the peptide through TonB-dependent receptors (Destoumieux-Garzón et al., 2003, 2006; Patzer et al., 2003; Pugsley et al., 1986; Thomas et al., 2004). Another factor that is potentially involved in microcin E492 uptake is semA, which encodes a protein of unknown function; semA mutants were resistant to microcin E492 through an as yet undetermined mechanism (Pugsley et al., 1986). Selection and mapping of microcin B17-resistant mutants similarly revealed that this peptide co-opts a protein native to susceptible cells to cross the outer membrane barrier (Laviña et al., 1986). Based on the genetic evidence, microcin B17 may enter cells through the general porin OmpF, a channel that is normally used for the passive diffusion of small hydrophilic molecules (Laviña et al., 1986). After entering the periplasm, microcin B17 undergoes a second transport step across the inner membrane before delivery to its cytoplasmic target. The polytopic inner membrane protein SbmA has been proposed as the transporter that catalyzes this step (Laviña et al., 1986). The native function of SbmA remains unknown, although SbmA-null mutants are resistant to many antimicrobial peptides including MccJ25 (Salomón & Farías, 1995), which will be discussed shortly, and citrocin (Cheung-Lee, Parry, Cartagena, et al., 2019). Still, it would be counterintuitive for bacteria to evolve a factor that simply serves to sensitize them to harmful agents.
Cellular uptake of MccJ25 involves the outer membrane receptor FhuA (Mathavan et al., 2014; Salomón & Farías, 1993) and the inner membrane protein SbmA (Salomón & Farías, 1995). Mutagenesis studies helped to identify the residues that are dispensable for inhibition of RNA polymerase but essential for uptake (Pavlova et al., 2008). The histidine at position 5 within the ring of MccJ25 is important for transport through SbmA (De Cristóbal et al., 2006). The binding of MccJ25 to FhuA is known to involve many more residues, owing to the availability of structural information. FhuA is a TBDT that primarily functions in acquisition of the essential nutrient iron (Braun, 2009; Noinaj et al., 2010). In the environment, iron often exists in the insoluble, oxidized ferric form, that is, iron(III), that bacteria cannot readily internalize. Instead, bacteria produce high-affinity iron-chelating agents called siderophores that bind and carry iron through outer membrane transporters such as FhuA. Transport is coupled to the proton motive force via the TonB-ExbB-ExbD inner membrane complex, which spans the periplasm and directly contacts the transporter (Fig. 12A). FhuA is a monomeric β-barrel protein comprising 22 antiparallel β-strands that wrap around to form a central pore (Ferguson et al., 1998; Locher et al., 1998). The β-strands are connected by short turns on the periplasmic face of the barrel and by long loops on the extracellular face. The central pore is normally gated because the N-terminus of FhuA forms a plug or cork-like domain that inserts from the periplasmic side and occludes the cavity. Binding of a ligand to FhuA displaces the plug and signals TonB to drive transport across the now-continuous channel (Pawelek et al., 2006).
Fig. 12.

Cellular uptake of lasso peptide microcin J25 (MccJ25) is mediated by the TonB-dependent transporter FhuA. (A) The canonical role of FhuA, an outer membrane (OM) receptor, is transport of iron-siderophore complexes across the OM into the periplasm (P). This energy-dependent process is mediated by the TonB/ExbB/ExbD inner membrane (IM) protein complex and driven by the proton motive force (PMF). (B) Crystal structure of MccJ25 bound to the extracellular face of FhuA. (C) Zoomed-in view of the FhuA:MccJ25 complex showing close contacts between the cork domain (gold) and β-barrel (cyan) of FhuA with MccJ25 (magenta). Dashed lines are H-bonds (yellow). All structure figures drawn from PDB file 4CU4.
The natural cargo of FhuA is the hydroxamate siderophore ferrichrome, but FhuA also moonlights as a receptor for many foreign agents including bacteriophages and colicin toxins (Braun, 2009). MccJ25 likewise sneaks into susceptible cells through FhuA. MccJ25 docks on the exposed extracellular loops of FhuA but also forms contacts with the β-barrel and cork domain, effectively obstructing the pore near the space that ferrichrome normally occupies (Fig. 12B). This interaction involves the MccJ25 loop region, which is oriented toward the extracellular space (Destoumieux-Garzón et al., 2005; Mathavan et al., 2014; Socias et al., 2009). Upon FhuA binding, the MccJ25 loop is remodeled into a more open configuration relative to its structure in solution (Mathavan et al., 2014). The plane of the MccJ25 8-membered ring lies vertical to the membrane surface and does not undergo major conformational change in the presence of FhuA (Mathavan et al., 2014) The 2.3-Å FhuA-MccJ25 cocrystal structure showed that in addition to abundant hydrophobic contacts across the length of the peptide, three key hydrogen bonds are formed between the MccJ25 ring and the FhuA cork domain (Fig. 12C) (Mathavan et al., 2014). The histidine at position 5 within the MccJ25 ring contributes two of the three hydrogen bonds; it is bonded to the FhuA cork domain residues Y116 and F115 (Mathavan et al., 2014). Moreover, the side chain of this H5 residue is buried in the FhuA pore and is critical for FhuA recognition because an MccJ25H5A mutant has reduced binding affinity (Mathavan et al., 2014). These specific contacts may explain the narrow spectrum activity of MccJ25, as only certain FhuA homologs are capable of MccJ25 uptake (Vincent et al., 2004).
Nature has evolved RiPPs with diverse modes of action. It is remarkable that RiPPs have also evolved to exploit the native receptors of target cells for entry. To repurpose RiPPs into potent antibiotics, one must know the range of bioactivity and a first step toward this goal is to understand cellular uptake. How can we predict what the transporters are in target cells? Early studies on the production and uptake of MccJ25 may provide some clues. Iron deprivation was shown to increase MccJ25 production (Salomón & Fariás, 1994) and MccJ25 uses an iron transporter to enter cells (Mathavan et al., 2014; Salomón & Farías, 1993). This connection may not be a mere coincidence and perhaps suggests that finding the signals which regulate biosynthesis of RiPPs in producer cells can lead us to discovering their transporters in target cells.
Conclusion
In this review, we have described the molecular mechanisms of action of a variety of different RiPPs for which detailed structural or mechanistic information is available. RiPPs have several defining structural features. First, RiPPs tend to be larger in size than natural products from other classes such as polyketides and nonribosomal peptides. Second, many (though not all) RiPPs exhibit some form of macrocyclization, which results in the RiPP having a more compact structure than a comparable linear peptide. Third, by definition, many RiPPs include chemical moieties not found in the canonical set of amino acid building blocks. All of these features of RiPPs contribute to their mechanisms of activity.
With regards to the size of RiPPs, lasso peptides like capistruin and MccJ25 are sufficiently bulky to occlude most of the secondary channel of RNAP (Fig. 9). The large size of klebsazolicin allows for it to coil up and occupy a large pocket within the 23S rRNA (Fig. 10). Similarly, the large size of polytheonamide B is just the right size for a single molecule of this peptide to span the plasma membrane (Fig. 4). Many RiPPs are too large to passively cross the permeability barriers of bacteria and other cells. While some RiPPs have deduced ways to hijack energy-coupled transport systems (Fig. 12), many other RiPPs simply exert their bioactivity via interactions at the cell envelope instead.
The macrocyclization of RiPPs likely plays a role in generating the right pharmacophore for target engagement. In the lasso peptide MccJ25, the Y9 and Y20 residues make crucial contacts with RNAP (Fig. 9A). These amino acids are in close proximity in the threaded lasso form of MccJ25, but are not near each other in either a linear or unthreaded peptide. Indeed, macrocyclization via the mechanical bond in MccJ25 is critical for the antimicrobial activity of this peptide (Wilson et al., 2003). Head-to-tail cyclization of the bacteriocins (Figs 6 and 7) likely pre-pays some of the entropic penalty associated with membrane binding and pore formation. Another likely role of macrocyclization in RiPPs has more to do with the complex, protease-laden environment into which RiPPs are exported. The organisms that produce RiPPs live in a vast variety of different environments, including dense, complex microbiomes such as soil and the human gut. The macrocyclization of RiPPs allows for enhanced stability of the RiPP relative to a linear peptide in these hostile environments.
Finally, the posttranslational modifications inherent to RiPPs play specific roles in their mechanism of action. The alternating l- and d-amino acids of polytheonamide B give rise to the unusual β-spiral structure that allows it to function as an ion channel (Fig. 4). The thioether rings of nisin help form a cage that binds the pyrophosphate moiety of lipid II (Fig. 3). The aromatic thiazoles and oxazoles in klebsazolicin and phazolicin mediate numerous π–π stacking interactions with ribosomal RNA (Fig. 10).
Structurally diverse RiPPs with novel mechanisms of action will continue to be discovered for decades to come. In the past, it took many years and often the effort of multiple groups to reach an understanding of the mechanism of action following initial isolation of the natural product. As mentioned in the introduction, bioinformatic genome mining has quickened the pace of RiPP discovery. However, these genome mining approaches do not come with a guarantee of bioactivity. Even with bioactivity, determination of the molecular mechanism of action of a RiPP can be arduous work, requiring expertise in genetics, biochemistry, and structural biology. This work is important, however, if RiPPs are to progress to the clinic as antibiotics or cytotoxic payloads. Going forward, we propose that integration of detailed mechanism of action information with genome mining is a way to prioritize the hits from the avalanche of new genome mining data to improve the chances of isolating bioactive RiPPs.
Contributor Information
Li Cao, Department of Chemical and Biological Engineering, Princeton University, Princeton, NJ 08544, USA.
Truc Do, Department of Chemical and Biological Engineering, Princeton University, Princeton, NJ 08544, USA.
A James Link, Department of Chemical and Biological Engineering, Princeton University, Princeton, NJ 08544, USA; Department of Chemistry, Princeton University, Princeton, NJ 08544, USA; Department of Molecular Biology, Princeton University, Princeton, NJ 08544, USA.
Funding
Work in the Link laboratory on RiPPs was funded by National Institutes of Health grant (GM107036) and a grant from Princeton University School of Engineering and Applied Sciences (Focused Research Team on Precision Antibiotics). L.C. was supported by an National Science Foundation Graduate Research Fellowship (DGE-1656466).
Conflict of Interest
The authors declare no conflict of interest.
References
- Allgaier H., Jung G., Werner R. G., Schneider U., Zähner H. (1985). Elucidation of the structure of epidermin, a ribosomally synthesized, tetracyclic heterodetic polypeptide antibiotic. Angewandte Chemie (International Ed. in English), 24, 1051–1053. 10.1002/anie.198510511. [DOI] [Google Scholar]
- Alvarez-Sieiro P., Montalbán-López M., Mu D., Kuipers O. P. (2016). Bacteriocins of lactic acid bacteria: Extending the family. Applied Microbiology and Biotechnology, 100, 2939–2951. 10.1007/s00253-016-7343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen O. S., Koeppe R. E. (2007). Bilayer thickness and membrane protein function: An energetic perspective. Annual Review of Biophysics and Biomolecular Structure, 36, 107–130. 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- Anderssen E. L., Diep D. B., Nes I. F., Eijsink V. G. H., Nissen-Meyer J. (1998). Antagonistic activity of Lactobacillus plantarum C11: Two new two- peptide bacteriocins, plantaricins EF and JK, and the induction factor plantaricin A. Applied and Environmental Microbiology, 64, 2269–2272. 10.1128/aem.64.6.2269-2272.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arioli V., Berti M., Silvestri L. G. (1976). Gardimycin, a new antibiotic from Actionoplanes. III. Biological properties. Journal of Antibiotics (Tokyo), 29, 511–515. 10.7164/antibiotics.29.511. [DOI] [PubMed] [Google Scholar]
- Arnison P. G., Bibb M. J., Bierbaum G., Bowers A. A., Bugni T. S., Bulaj G., Camarero J. A., Campopiano D. J., Challis G. L., Clardy J., Cotter P. D., Craik D. J., Dawson M., Dittmann E., Donadio S., Dorrestein P. C., Entian K. D., Fischbach M. A., Garavelli J. S., van der Donk W. A. (2013). Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Natural Product Reports, 30, 108–160. 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babasaki K., Takao T., Shimonishi Y., Kurahashi K. (1985). Subtilosin A, a new antibiotic peptide produced by Bacillus subtilis 168: Isolation, structural analysis, and biogenesis. Journal of Biochemistry, 98, 585–603. 10.1093/oxfordjournals.jbchem.a135315. [DOI] [PubMed] [Google Scholar]
- Bagley M. C., Dale J. W., Merritt E. A., Xiong X. (2005). Thiopeptide antibiotics. Chemical Reviews., 105, 685–714. 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- Bakelar J., Buchanan S. K., Noinaj N. (2016). The structure of the barrel assembly machinery complex. Science, 351, 180–186. 10.1126/science.aad3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balandin S. V., Sheremeteva E. V., Ovchinnikova T. V. (2019). Pediocin-like antimicrobial peptides of bacteria. Biochemistry, 84, 464–478. 10.1134/S000629791905002X. [DOI] [PubMed] [Google Scholar]
- Bédard F., Hammami R., Zirah S., Rebuffat S., Fliss I., Biron E. (2018). Synthesis, antimicrobial activity and conformational analysis of the class IIa bacteriocin pediocin PA-1 and analogs thereof. Scientific Reports, 8, 9029. 10.1038/s41598-018-27225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat U. G., Halasi M., Gartel A. L. (2009). Thiazole antibiotics target FoxM1 and induce apoptosis in human cancer cells. PLoS One, 4, 1–7. 10.1371/journal.pone.0005592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhunia A. K., Johnson M. C., Ray B. (1988). Purification, characterization and antimicrobial spectrum of a bacteriocin produced by Pediococcus acidilactici. Journal of Applied Bacteriology, 65, 261–268. 10.1111/j.1365-2672.1988.tb01893.x. [DOI] [PubMed] [Google Scholar]
- Bhunia A. K., Johnson M. C., Ray B., Kalchayanand N. (1991). Mode of action of pediocin AcH from Pediococcus acidilactici H on sensitive bacterial strains. Journal of Applied Bacteriology, 70, 25–33. 10.1111/j.1365-2672.1991.tb03782.x. [DOI] [Google Scholar]
- Bieler S., Estrada L., Lagos R., Baeza M., Castilla J., Soto C. (2005). Amyloid formation modulates the biological activity of a bacterial protein. Journal of Biological Chemistry, 280, 26880–26885. 10.1074/jbc.M502031200. [DOI] [PubMed] [Google Scholar]
- Bieler S., Silva F., Soto C., Belin D. (2006). Bactericidal activity of both secreted and nonsecreted microcin E492 requires the mannose permease. Journal of Bacteriology, 188, 7049–7061. 10.1128/JB.00688-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierbaum G., Brötz H., Koller K. P., Sahl H. G. (1995). Cloning, sequencing and production of the lantibiotic mersacidin. FEMS Microbiology Letters, 127, 121–126. 10.1016/0378-1097(95)00048-A. [DOI] [PubMed] [Google Scholar]
- Bird K. E., Xander C., Murcia S., Schmalstig A. A., Wang X., Emanuele M. J., Braunstein M., Bowers A. A. (2020). Thiopeptides induce proteasome-independent activation of cellular mitophagy. ACS Chemical Biology, 15, 2164–2174. 10.1021/acschembio.0c00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K., Pascal Andreu V., de los Santos E. L. C., Del Carratore F., Lee S. Y., Medema M. H., Weber T. (2019). The antiSMASH database version 2: A comprehensive resource on secondary metabolite biosynthetic gene clusters. Nucleic Acids Research, 47, D625–D630. 10.1093/nar/gky1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S. Y., Medema M. H, Weber T. (2019). antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Research, 47, W81–W87. 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boakes S., Cortés J., Appleyard A. N., Rudd B. A. M., Dawson M. J. (2009). Organization of the genes encoding the biosynthesis of actagardine and engineering of a variant generation system. Molecular Microbiology, 72, 1126–1136. 10.1111/j.1365-2958.2009.06708.x. [DOI] [PubMed] [Google Scholar]
- Booker B. M., Deng S., Higgins N. P. (2010). DNA topology of highly transcribed operons in Salmonella enterica serovar Typhimurium. Molecular Microbiology, 78, 1348–1364. 10.1111/j.1365-2958.2010.07394.x. [DOI] [PubMed] [Google Scholar]
- Bouhss A., Trunkfield A. E., Bugg T. D. H., Mengin-Lecreulx D. (2008). The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiology Reviews, 32, 208–233. 10.1111/j.1574-6976.2007.00089.x. [DOI] [PubMed] [Google Scholar]
- Braffman N. R., Piscotta F. J., Hauver J., Campbell E. A., Link A. J., Darst S. A. (2019). Structural mechanism of transcription inhibition by lasso peptides microcin J25 and capistruin. Proceedings of the National Academy of Sciences of the USA, 116, 1273–1278. 10.1073/pnas.1817352116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun V. (2009). FhuA (TonA), the career of a protein. Journal of Bacteriology, 191, 3431–3436. 10.1128/JB.00106-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breukink E., Van Heusden H. E., Vollmerhaus P. J., Swiezewska E., Brunner L., Walker S., Heck A. J. R., De Kruijff B. (2003). Lipid II is an intrinsic component of the pore induced by nisin in bacterial membranes. Journal of Biological Chemistry, 278, 19898–19903. 10.1074/jbc.M301463200. [DOI] [PubMed] [Google Scholar]
- Breukink E., Wiedemann I., Van Kraaij C., Kuipers O. P., Sahl H. G., De Kruijff B. (1999). Use of the cell wail precursor lipid II by a pore-forming peptide antibiotic. Science, 286, 2361–2364. 10.1126/science.286.5448.2361. [DOI] [PubMed] [Google Scholar]
- Brötz H., Bierbaum G., Leopold K., Reynolds P. E., Sahl H. G. (1998). The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrobial Agents and Chemotherapy, 42, 154–160. 10.1128/aac.42.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brötz H., Bierbaum G., Markus A., Molitor E., Sahl H. G. (1995). Mode of action of the lantibiotic mersacidin: Inhibition of peptidoglycan biosynthesis via a novel mechanism? Antimicrobial Agents and Chemotherapy, 39, 714–719. 10.1128/AAC.39.3.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brötz H., Bierbaum G., Reynolds P. E., Sahl H. G. (1997). The lantibiotic mersacidin inhibits peptidoglycan biosynthesis at the level of transglycosylation. European Journal of Biochemistry FEBS, 246, 193–199. 10.1111/j.1432-1033.1997.t01-1-00193.x. [DOI] [PubMed] [Google Scholar]
- Brötz H., Josten M., Wiedemann I., Schneider U., Götz F., Bierbaum G., Sahl H. G. (1998). Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Molecular Microbiology, 30, 317–327. 10.1046/j.1365-2958.1998.01065.x. [DOI] [PubMed] [Google Scholar]
- Brueckner F., Cramer P. (2008). Structural basis of transcription inhibition by α-amanitin and implications for RNA polymerase II translocation. Nature Structural & Molecular Biology, 15, 811–818. 10.1038/nsmb.1458. [DOI] [PubMed] [Google Scholar]
- Buchman G. W., Banerjee S., Hansen J. N. (1988). Structure, expression, and evolution of a gene encoding the precursor of nisin, a small protein antibiotic. Journal of Biological Chemistry, 263, 16260–16266. [PubMed] [Google Scholar]
- Burgos M. J. G., Pulido R. P., del Carmen López Aguayo M., Gálvez A., Lucas R. (2014). The cyclic antibacterial peptide enterocin AS-48: Isolation, mode of action, and possible food applications. International Journal of Molecular Science, 15, 22706–22727. 10.3390/ijms151222706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell D. A., Cramer P., Kornberg R. D. (2002). Structural basis of transcription: α-amanitin-RNA polymerase II cocrystal at 2.8 Å resolution. Proceedings of the National Academy of Sciences of the USA, 99, 1218–1222. 10.1073/pnas.251664698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron D. M., Thompson J., March P. E., Dahlberg A. E. (2002). Initiation factor IF2, thiostrepton and micrococcin prevent the binding of elongation factor G to the Escherichia coli ribosome. Journal of Molecular Biology, 319, 27–35. 10.1016/S0022-2836(02)00235-8. [DOI] [PubMed] [Google Scholar]
- Castiglione F., Cavaletti L., Losi D., Lazzarini A., Carrano L., Feroggio M., Ciciliato I., Corti E., Candiani G., Marinelli F., Selva E. (2007). A novel lantibiotic acting on bacterial cell wall synthesis produced by the uncommon actinomycete Planomonospora sp. Biochemistry, 46, 5884–5895. 10.1021/bi700131x. [DOI] [PubMed] [Google Scholar]
- Castiglione F., Lazzarini A., Carrano L., Corti E., Ciciliato I., Gastaldo L., Candiani P., Losi D., Marinelli F., Selva E., Parenti F. (2008). Determining the structure and mode of action of microbisporicin, a potent lantibiotic active against multiresistant pathogens. Chemistry & Biology, 15, 22–31. 10.1016/j.chembiol.2007.11.009. [DOI] [PubMed] [Google Scholar]
- CDC . (2019). Antibiotic resistance threats in the United States, 2019. U.S. Department of Health and Human Services, CDC. [Google Scholar]
- Champion K., Higgins N. P. (2007). Growth rate toxicity phenotypes and homeostatic supercoil control differentiate Escherichia coli from Salmonella enterica serovar typhimurium. Journal of Bacteriology, 189, 5839–5849. 10.1128/JB.00083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S., Chatterjee D. K., Jani R. H., Blumbach J., Ganguli B. N., Klesel N., Limbert M., Seibert G. (1992). Mersacidin, a new antibiotic from Bacillus in vitro and in vivo antibacterial activity. Journal of Antibiotics (Tokyo), 45, 839–845. 10.7164/antibiotics.45.839. [DOI] [PubMed] [Google Scholar]
- Chen Y., Ludescher R. D., Montville T. J. (1997). Electrostatic interactions, but not the YGNGV consensus motif, govern the binding of pediocin PA-1 and its fragments to phospholipid vesicles. Applied and Environmental Microbiology, 63, 4770–4777. 10.1128/aem.63.12.4770-4777.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Shapira R., Eisenstein M., Montville T. J. (1997). Functional characterization of pediocin PA-1 binding to liposomes in the absence of a protein receptor and its relationship to a predicted tertiary structure. Applied and Environmental Microbiology, 63, 524–531. 10.1128/AEM.63.2.524-531.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung-Lee W. L., Link A. J. (2019). Genome mining for lasso peptides: Past, present, and future. Journal of Industrial Microbiology & Biotechnology, 46, 1371–1379. 10.1007/s10295-019-02197-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung-Lee W. L., Parry M. E., Cartagena A. J., Darst S. A., Link A. J. (2019). Discovery and structure of the antimicrobial lasso peptide citrocin. Journal of Biological Chemistry, 294, 6822–6830. 10.1074/jbc.RA118.006494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung-Lee W. L., Parry M. E., Zong C., Jaramillo Cartagena A., Darst S. A., Connell N. D., Russo R., Link A. J. (2020). Discovery of ubonodin, an antimicrobial lasso peptide active against members of the Burkholderia cepacia complex. ChemBioChem, 21, 1335–1340. 10.1002/cbic.201900707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikindas M. L., García-Garcerá M. J., Driessen A. J. M., Ledeboer A. M., Nissen- Meyer J., Nes I. F., Abee T., Konings W. N., Venema G. (1993). Pediocin PA-1, a bacteriocin from Pediococcus acidilactici PAC1.0, forms hydrophilic pores in the cytoplasmic membrane of target cells. Applied and Environmental Microbiology, 59, 3577–3584. 10.1128/aem.59.11.3577-3584.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chokekijchai S., Kojima E., Anderson S., Nomizu M., Tanaka M., Machida M., Date T., Toyota K., Ishida S., Watanabe K., Yoshioka H., Roller P. P., Murakami K., Mitsuya H. (1995). NP-06: A novel anti-human immunodeficiency virus polypeptide produced by a Streptomyces species. Antimicrobial Agents and Chemotherapy, 39, 2345–2347. 10.1128/AAC.39.10.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choung S.-Y., Kobayashi T., Inoue J., Takemoto K., Ishitsuka H., Inoue K. (1988). Hemolytic activity of a cyclic peptide Ro09-0198 isolated from Streptoverticillium. Biochimica et Biophysica Acta – Biomembranes, 940, 171–179. 10.1016/0005-2736(88)90192-7. [DOI] [PubMed] [Google Scholar]
- Choung S.-Y., Kobayashi T., Takemoto K., Ishitsuka H., Inoue K. (1988). Interaction of a cyclic peptide, Ro09-0198, with phosphatidylethanolamine in liposomal membranes. Biochimica et Biophysica Acta – Biomembranes, 940, 180–187. 10.1016/0005-2736(88)90193-9. [DOI] [PubMed] [Google Scholar]
- Collin F., Maxwell A. (2019). The microbial toxin microcin B17: Prospects for the development of new antibacterial agents. Journal of Molecular Biology, 431, 3400–3426. 10.1016/j.jmb.2019.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin F., Thompson R. E., Jolliffe K. A., Payne R. J., Maxwell A. (2013). Fragments of the bacterial toxin microcin B17 as gyrase poisons. PLoS One, 8, 1–9. 10.1371/journal.pone.0061459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronelli C., Tamoni G., Lancini G. C. (1976). Gardimycin, a new antibiotic from actinoplanes. II. Isolation and preliminary characterization. Journal of Antibiotics (Tokyo), 29, 507–510. 10.7164/antibiotics.29.507. [DOI] [PubMed] [Google Scholar]
- Cox C. L., Doroghazi J. R., Mitchell D. A. (2015). The genomic landscape of ribosomal peptides containing thiazole and oxazole heterocycles. BMC Genomics, 16, 1–16. 10.1186/s12864-015-2008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone W. J. K., Leeper F. J., Truman A. W. (2012). Identification and characterisation of the gene cluster for the anti-MRSA antibiotic bottromycin: Expanding the biosynthetic diversity of ribosomal peptides. Chemical Science, 3, 3516–3521. 10.1039/c2sc21190d. [DOI] [Google Scholar]
- Cundliffe E., Thompson J. (1981). Concerning the mode of action of micrococcin upon bacterial protein synthesis. European Journal of Biochemistry FEBS, 118, 47–52. 10.1111/j.1432-1033.1981.tb05484.x. [DOI] [PubMed] [Google Scholar]
- Dalet K., Briand C., Cenatiempo Y., Héchard Y. (2000). The rpoN gene of Enterococcus faecalis directs sensitivity to subclass IIa bacteriocins. Current Microbiology, 41, 441–443. 10.1007/s002840010164. [DOI] [PubMed] [Google Scholar]
- Dalet K., Cenatiempo Y., Cossart P., Héchard Y. (2001). A σ54-dependent PTS permease of the mannose family is responsible for sensitivity of Listeria monocytogenes to mesentericin Y105. Microbiology (Reading, England), 147, 3263–3269. 10.1099/00221287-147-12-3263. [DOI] [PubMed] [Google Scholar]
- Daniel-Ivad M., Hameed N., Tan S., Dhanjal R., Socko D., Pak P., Gverzdys T., Elliot M. A., Nodwell J. R. (2017). An engineered allele of afsQ1 facilitates the discovery and investigation of cryptic natural products. ACS Chemical Biology, 12, 628–634. 10.1021/acschembio.6b01002. [DOI] [PubMed] [Google Scholar]
- De Cristóbal R. E., Solbiati J. O., Zenoff A. M., Vincent P. A., Salomón R. A., Yuzenkova J., Severinov K., Farías R. N. (2006). Microcin J25 uptake: His5 of the MccJ25 lariat ring is involved in interaction with the inner membrane MccJ25 transporter protein SbmA. Journal of Bacteriology, 188, 3324–3328. 10.1128/JB.188.9.3324-3328.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deegan L. H., Suda S., Lawton E. M., Draper L. A., Hugenholtz F., Peschel A., Hill C., Cotter P. D., Ross R. P. (2010). Manipulation of charged residues within the two-peptide lantibiotic lacticin 3147. Microbial Biotechnology, 3, 222–234. 10.1111/j.1751-7915.2009.00145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lorenzo V. (1984). Isolation and characterization of microcin E 492 from Klebsiella pneumoniae. Archives of Microbiology, 139, 72–75. 10.1007/BF00692715. [DOI] [PubMed] [Google Scholar]
- De Lorenzo V., Pugsley A. P. (1985). Microcin E492, a low-molecular-weight peptide antibiotic which causes depolarization of the Escherichia coli cytoplasmic membrane. Antimicrobial Agents and Chemotherapy, 27, 666–669. 10.1128/AAC.27.4.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destoumieux-Garzón D., Duquesne S., Peduzzi J., Goulard C., Desmadril M., Letellier L., Rebuffat S., Boulanger P. (2005). The iron–siderophore transporter FhuA is the receptor for the antimicrobial peptide microcin J25: Role of the microcin Val11–Pro16 β-hairpin region in the recognition mechanism. Biochemical Journal, 389, 869–876. 10.1042/BJ20042107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destoumieux-Garzón D., Peduzzi J., Thomas X., Djediat C., Rebuffat S. (2006). Parasitism of iron-siderophore receptors of Escherichia coli by the siderophore-peptide microcin E492m and its unmodified counterpart. Biometals, 19, 181–191. 10.1007/s10534-005-4452-9. [DOI] [PubMed] [Google Scholar]
- Destoumieux-Garzón D., Thomas X., Santamaria M., Goulard C., Barthélémy M., Boscher B., Bessin Y., Molle G., Pons A. M., Letellier L., Peduzzi J., Rebuffat S. (2003). Microcin E492 antibacterial activity: Evidence for a TonB-dependent inner membrane permeabilization on Escherichia coli. Molecular Microbiology, 49, 1031–1041. 10.1046/j.1365-2958.2003.03610.x. [DOI] [PubMed] [Google Scholar]
- Detlefsen D. J., Hill S. E., Volk K. J., Klohr S. E., Tsunakawa M., Furumai T., Lin P. F., Oki T., Nishio M., Kawano K., Lee M. S. (1995). Siamycins I and II, new anti-HIV-1 peptides: II. Sequence analysis and structure determination of siamycin I. Journal of Antibiotics (Tokyo), 48, 1515–1517. 10.7164/antibiotics.48.1515. [DOI] [PubMed] [Google Scholar]
- Drider D., Fimland G., Héchard Y., McMullen L. M., Prévost H. (2006). The continuing story of class IIa Bacteriocins. Microbiology and Molecular Biology Reviews, 70, 564–582. 10.1128/mmbr.00016-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan A. J. F., Errington J., Vollmer W. (2020). Regulation of peptidoglycan synthesis and remodelling. Nature Reviews Microbiology, 18, 446–460. 10.1038/s41579-020-0366-3. [DOI] [PubMed] [Google Scholar]
- Engelberg-Kulka H. (2003). Cannibals Defy starvation and avoid sporulation. Science, 301, 467–468. 10.1126/science.1088051. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D., Hofmann E., Coulton J. W., Diederichs K., Welte W. (1998). Siderophore-mediated iron transport: Crystal structure of FhuA with bound lipopolysaccharide. Science, 282, 2215–2220. 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- Fimland G., Johnsen L., Dalhus B., Nissen-Meyer J. (2005). Pediocin-like antimicrobial peptides (class IIa bacteriocins) and their immunity proteins: Biosynthesis, structure, and mode of action. Journal of Peptide Science, 11, 688–696. 10.1002/psc.699. [DOI] [PubMed] [Google Scholar]
- Flühe L., Burghaus O., Wieckowski B. M., Giessen T. W., Linne U., Marahiel M. A. (2013). Two [4Fe-4S] clusters containing radical SAM enzyme SkfB catalyze thioether bond formation during the maturation of the sporulation killing factor. Journal of the American Chemical Society, 135, 959–962. 10.1021/ja310542g. [DOI] [PubMed] [Google Scholar]
- Fredenhagen A., Fendrich G., Märki F., Märki W., Gruner J., Raschdorf F., Peter H. H. (1990). Duramycins B and C, two new lanthionine containing antibiotics as inhibitors of phospholipase A2. Structural revision of duramycin and cinnamycin. Journal of Antibiotics (Tokyo), 43, 1403–1412. 10.7164/antibiotics.43.1403. [DOI] [PubMed] [Google Scholar]
- Freeman M. F., Gurgui C., Helf M. J., Morinaka B. I., Uria A. R., Oldham N. J., Sahl H.-G., Matsunaga S., Piel J. (2012). Metagenome mining reveals polytheonamides as posttranslationally modified ribosomal peptides. Science, 338, 387–390. 10.1126/science.1226121. [DOI] [PubMed] [Google Scholar]
- Freeman M. F., Helf M. J., Bhushan A., Morinaka B. I., Piel J. (2017). Seven enzymes create extraordinary molecular complexity in an uncultivated bacterium. Nature Chemistry, 9, 387–395. 10.1038/nchem.2666. [DOI] [PubMed] [Google Scholar]
- Gálvez A., Giménez-Gallego G., Maqueda M., Valdivia E. (1989). Purification and amino acid composition of peptide antibiotic AS-48 produced by Streptococcus (Enterococcus) faecalis subsp. liquefaciens S-48. Antimicrobial Agents and Chemotherapy, 33, 437–441. 10.1128/AAC.33.4.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gálvez A., Maqueda M., Martínez-Bueno M., Valdivia E. (1991). Permeation of bacterial cells, permeation of cytoplasmic and artificial membrane vesicles, and channel formation on lipid bilayers by peptide antibiotic AS-48. Journal of Bacteriology, 173, 886–892. 10.1128/jb.173.2.886-892.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gálvez A., Maqueda M., Valdivia E., Quesada A., Montoya E. (1986). Characterization and partial purification of a broad spectrum antibiotic AS-48 produced by Streptococcus faecalis. Canadian Journal of Microbiology, 32, 765–771. 10.1139/m86-141. [DOI] [PubMed] [Google Scholar]
- Gavrish E., Sit C. S., Cao S., Kandror O., Spoering A., Peoples A., Ling L., Fetterman A., Hughes D., Bissell A., Torrey H., Akopian T., Mueller A., Epstein S., Goldberg A., Clardy J., Lewis K. (2014). Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chemistry & Biology, 21, 509–518. 10.1016/j.chembiol.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Escribano J. P., Song L., Bibb M. J., Challis G. L. (2012). Posttranslational β-methylation and macrolactamidination in the biosynthesis of the bottromycin complex of ribosomal peptide antibiotics. Chemical Science, 3, 3522–3525. 10.1039/c2sc21183a. [DOI] [Google Scholar]
- Gonzalez C. F., Kunka B. S. (1987). Plasmid-associated bacteriocin production and sucrose fermentation in Pediococcus acidilactici. Applied and Environmental Microbiology, 53, 2534–2538. 10.1128/aem.53.10.2534-2538.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González C., Langdon G. M., Bruix M., Gálvez A., Valdivia E., Maqueda M., Rico M. (2000). Bacteriocin AS-48, a microbial cyclic polypeptide structurally and functionally related to mammalian NK-lysin. Proceedings of the National Academy of Sciences of the USA, 97, 11221–11226. 10.1073/pnas.210301097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Pastor J. E. (2003). Cannibalism by sporulating bacteria. Science, 301, 510–513. 10.1126/science.1086462. [DOI] [PubMed] [Google Scholar]
- Grell T. A. J., Kincannon W. M., Bruender N. A., Blaesi E. J., Krebs C., Bandarian V., Drennan C. L. (2018). Structural and spectroscopic analyses of the sporulation killing factor biosynthetic enzyme SkfB, a bacterial AdoMet radical sactisynthase. Journal of Biological Chemistry, 293, 17349–17361. 10.1074/jbc.RA118.005369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross E., Morell J. L. (1971). Structure of nisin. Journal of the American Chemical Society, 93, 4634–4635. 10.1021/ja00747a073. [DOI] [PubMed] [Google Scholar]
- Hallen H. E., Luo H., Scott-Craig J. S., Walton J. D. (2007). Gene family encoding the major toxins of lethal Amanita mushrooms. Proceedings of the National Academy of Sciences of the USA,104, 19097–19101. 10.1073/pnas.0707340104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada T., Matsunaga S., Fujiwara M., Fujita K., Hirota H., Schmucki R., Güntert P., Fusetani N. (2010). Solution structure of polytheonamide B, a highly cytotoxic nonribosomal polypeptide from marine sponge. Journal of the American Chemical Society, 132, 12941–12945. 10.1021/ja104616z. [DOI] [PubMed] [Google Scholar]
- Hamada T., Matsunaga S., Yano G., Fusetani N. (2005). Polytheonamides A and B, highly cytotoxic, linear polypeptides with unprecedented structural features, from the marine sponge, Theonella swinhoei. Journal of the American Chemical Society, 127, 110–118. 10.1021/ja045749e. [DOI] [PubMed] [Google Scholar]
- Hamada T., Sugawara T., Matsunaga S., Fusetani N. (1994a). Polytheonamides, unprecedented highly cytotoxic polypeptides from the marine sponge Theonella swinhoei: Structure elucidation. Tetrahedron Letters, 35, 609–612. 10.1016/S0040-4039(00)75851-5. [DOI] [Google Scholar]
- Hamada T., Sugawara T., Matsunaga S., Fusetani N. (1994b). Polytheonamides, unprecedented highly cytotoxic polypeptides, from the marine sponge Theonella swinhoei: Isolation and component amino acids. Tetrahedron Letters, 35, 719–720. 10.1016/S0040-4039(00)75799-6. [DOI] [Google Scholar]
- Harms J. M., Wilson D. N., Schluenzen F., Connell S. R., Stachelhaus T., Zaborowska Z., Spahn C. M. T., Fucini P. (2008). Translational regulation via L11: Molecular switches on the ribosome turned on and off by thiostrepton and micrococcin. Molecular Cell, 30, 26–38. 10.1016/j.molcel.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Hartmann J. B., Zahn M., Burmann I. M., Bibow S., Hiller S. (2018). Sequence-specific solution NMR assignments of the β-barrel insertase BamA to monitor its conformational ensemble at the atomic level. Journal of the American Chemical Society, 140, 11252–11260. 10.1021/jacs.8b03220. [DOI] [PubMed] [Google Scholar]
- Harvey K. L., Jarocki V. M., Charles I. G., Djordjevic S. P. (2019). The diverse functional roles of elongation factor tu (EF-Tu) in microbial pathogenesis. Frontiers in Microbiology, 10, 1–19. 10.3389/fmicb.2019.02351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayata A., Itoh H., Inoue M. (2018). Solid-phase total synthesis and dual mechanism of action of the channel-forming 48-mer peptide polytheonamide B. Journal of the American Chemical Society, 140, 10602–10611. 10.1021/jacs.8b06755. [DOI] [PubMed] [Google Scholar]
- Heffron S. E., Jurnak F. (2000). Structure of an EF-Tu complex with a thiazolyl peptide antibiotic determined at 2.35 Å resolution: Atomic basis for GE2270A inhibition of EF- Tu. Biochemistry, 39, 37–45. 10.1021/bi9913597. [DOI] [PubMed] [Google Scholar]
- Hegemann J. D., Zimmermann M., Xie X., Marahiel M. A. (2015). Lasso peptides: An intriguing class of bacterial natural products. Accounts of Chemical Research, 48, 1909–1919. 10.1021/acs.accounts.5b00156. [DOI] [PubMed] [Google Scholar]
- Henderson J. T., Chopko A. L., van Wassenaar P. D. (1992). Purification and primary structure of pediocin PA-1 produced by Pediococcus acidilactici PAC-1.0. Archives of Biochemistry and Biophysics, 295, 5–12. 10.1016/0003-9861(92)90480-K. [DOI] [PubMed] [Google Scholar]
- Hetz C., Bono M. R., Barros L. F., Lagos R. (2002). Microcin E492, a channel-forming bacteriocin from Klebsiella pneumoniae, induces apoptosis in some human cell lines. Proceedings of the National Academy of Sciences of the USA, 99, 2696–2701. 10.1073/pnas.052709699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins C. F., Dorman C. J., Stirling D. A., Waddell L., Booth I. R., May G., Bremer E. (1988). A physiological role for DNA supercoiling in the osmotic regulation of gene expression in S. typhimurium and E. coli. Cell, 52, 569–584. 10.1016/0092-8674(88)90470-9. [DOI] [PubMed] [Google Scholar]
- Hillman J. D., Novák J., Sagura E., Gutierrez J. A., Brooks T. A., Crowley P. J., Hess M., Azizi A., Leung K.-P., Cvitkovitch D., Bleiweis A. S. (1998). Genetic and biochemical analysis of mutacin 1140, a lantibiotic from Streptococcus mutans. Infection and Immunity, 66, 2743–2749. 10.1128/IAI.66.6.2743-2749.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda K., Ohya M., Kohno T., Maeda T., Endo S., Wakamatsu K. (1996). Structure determination of an immunopotentiator peptide, cinnamycin, complexed with lysophosphatidylethanolamine by 1H-NMR. Journal of Biochemistry, 119, 226–230. 10.1093/oxfordjournals.jbchem.a021226. [DOI] [PubMed] [Google Scholar]
- Hou Y., Tianero M. D. B., Kwan J. C., Wyche T. P., Michel C. R., Ellis G. A., Vazquez-Rivera E., Braun D. R., Rose W. E., Schmidt E. W., Bugni T. S. (2012). Structure and biosynthesis of the antibiotic bottromycin D. Organic Letters, 14, 5050–5053. 10.1021/ol3022758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S. T. D., Breukink E., Bierbaum G., Sahl H. G., De Kruijff B., Kaptein R., Van Nuland N. A. J., Bonvin A. (2003). NMR study of mersacidin and lipid II interaction in dodecylphosphocholine micelles: Conformational changes are a key to antimicrobial activity. Journal of Biological Chemistry, 278, 13110–13117. 10.1074/jbc.M211144200. [DOI] [PubMed] [Google Scholar]
- Hsu S. T. D., Breukink E., Tischenko E., Lutters M. A. G., De Kruijff B., Kaptein R., Bonvin A., Van Nuland N. A. J. (2004). The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nature Structural & Molecular Biology, 11, 963–967. 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- Huang L. Y., Catterall W. A., Ehrenstein G. (1978). Selectivity of cations and nonelectrolytes for acetylcholine-activated channels in cultured muscle cells. Journal of General Physiology, 71, 397–410. 10.1085/jgp.71.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L., Rachid S., Stadler M., Wenzel S. C., Müller R. (2012). Synthetic biotechnology to study and engineer ribosomal bottromycin biosynthesis. Chemistry & Biology, 19, 1278–1287. 10.1016/j.chembiol.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Imai Y., Meyer K. J., Iinishi A., Favre-Godal Q., Green R., Manuse S., Caboni M., Mori M., Niles S., Ghiglieri M., Honrao C., Ma X., Guo J. J., Makriyannis A., Linares-Otoya L., Böhringer N., Wuisan Z. G., Kaur H., Wu R., Lewis K. (2019). A new antibiotic selectively kills Gram-negative pathogens. Nature, 576, 459–464. 10.1038/s41586-019-1791-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M., Matsunaga S., Oiki S. (2014). Paradoxical one-ion pore behavior of the long β-helical peptide of marine cytotoxic polytheonamide B Scitific Reports, 4, 1–7. 10.1038/srep03636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M., Shimizu H., Muramatsu I., Oiki S. (2010). A cytotoxic peptide from a marine sponge exhibits ion channel activity through vectorial-insertion into the membrane. FEBS Letters, 584, 3995–3999. 10.1016/j.febslet.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Johnsen L., Fimland G., Nissen-Meyer J. (2005). The C-terminal domain of pediocin-like antimicrobial peptides (class IIa bacteriocins) is involved in specific recognition of the C-terminal part of cognate immunity proteins and in determining the antimicrobial spectrum. Journal of Biological Chemistry, 280, 9243–9250. 10.1074/jbc.M412712200. [DOI] [PubMed] [Google Scholar]
- Kaletta C., Entian K.-D., Jung G. (1991). Prepeptide sequence of cinnamycin (Ro 09-0198): The first structural gene of a duramycin-type lantibiotic. European Journal of Biochemistry FEBS, 199, 411–415. 10.1111/j.1432-1033.1991.tb16138.x. [DOI] [PubMed] [Google Scholar]
- Kaplan C. D., Larsson K. M., Kornberg R. D. (2008). The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by α-amanitin. Molecular Cell, 30, 547–556. 10.1016/j.molcel.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawulka K. E., Sprules T., Diaper C. M., Whittal R. M., McKay R. T., Mercier P., Zuber P., Vederas J. C. (2004). Structure of subtilosin A, a cyclic antimicrobial peptide from Bacillus subtilis with unusual sulfur to α-carbon cross-links: Formation and reduction of α-thio-α-amino acid derivatives. Biochemistry, 43, 3385–3395. 10.1021/bi0359527. [DOI] [PubMed] [Google Scholar]
- Kawulka K. E, Sprules T., McKay R. T., Mercier P., Diaper C. M., Zuber P., Vederas J. C. (2003). Structure of subtilosin A, an antimicrobial peptide from Bacillus subtilis with unusual posttranslational modifications linking cysteine sulfurs to α-carbons of phenylalanine and threonine. Journal of the American Chemical Society, 125, 4726–4727. 10.1021/ja029654t. [DOI] [PubMed] [Google Scholar]
- Kazakov T., Vondenhoff G. H., Datsenko K. A., Novikova M., Metlitskaya A., Wanner B. L., Severinov K. (2008). Escherichia coli peptidase A, B, or N can process translation inhibitor microcin C. Journal of Bacteriology, 190, 2607–2610. 10.1128/JB.01956-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelkar D. A., Chattopadhyay A. (2007). The gramicidin ion channel: A model membrane protein. Biochimica et Biophysica Acta – Biomembranes, 1768, 2011–2025. 10.1016/j.bbamem.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Kelly W. L., Pan L., Li C. (2009). Thiostrepton biosynthesis: Prototype for a new family of bacteriocins. Journal of the American Chemical Society, 131, 4327–4334. 10.1021/ja807890a. [DOI] [PubMed] [Google Scholar]
- Kessler H., Steuernagel S., Gillessen D., Kamiyama T. (1987). Complete sequence determination and localisation of one imino and three sulfide bridges of the nonadecapeptide Ro 09-0198 by homonuclear 2D-NMR spectroscopy. The DQF-RELAYED-NOESY-experiment. Helvetica Chimica Acta, 70, 726–741. 10.1002/hlca.19870700322. [DOI] [Google Scholar]
- Kjos M., Oppegård C., Diep D. B., Nes I. F., Veening J.-W., Nissen-Meyer J., Kristensen T. (2014). Sensitivity to the two-peptide bacteriocin lactococcin G is dependent on UppP, an enzyme involved in cell-wall synthesis. Molecular Microbiology, 92, 1177–1187. 10.1111/mmi.12632. [DOI] [PubMed] [Google Scholar]
- Knappe T. A., Linne U., Zirah S., Rebuffat S., Xie X., Marahiel M. A. (2008). Isolation and structural characterization of capistruin, a lasso peptide predicted from the genome sequence of Burkholderia thailandensis E264. Journal of the American Chemical Society, 130, 11446–11454. 10.1021/ja802966g. [DOI] [PubMed] [Google Scholar]
- Kuznedelov K., Semenova E., Knappe T. A., Mukhamedyarov D., Srivastava A., Chatterjee S., Ebright R. H., Marahiel M. A., Severinov K. (2011). The antibacterial threaded-lasso peptide capistruin inhibits bacterial RNA polymerase. Journal of Molecular Biology, 412, 842–848. 10.1016/j.jmb.2011.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos R., Baeza M., Corsini G., Hetz C., Strahsburger E., Castillo J. A., Vergara C., Monasterio O. (2001). Structure, organization and characterization of the gene cluster involved in the production of microcin E492, a channel-forming bacteriocin. Molecular Microbiology, 42, 229–243. 10.1046/j.1365-2958.2001.02630.x. [DOI] [PubMed] [Google Scholar]
- Lagos R., Tello M., Mercado G., García V., Monasterio O. (2009). Antibacterial and antitumorigenic properties of microcin E492, a pore-forming bacteriocin. Current Pharmaceutical Biotechnology, 10, 74–85. 10.2174/138920109787048643. [DOI] [PubMed] [Google Scholar]
- Laoukili J., Stahl M., Medema R. H. (2007). FoxM1: At the crossroads of ageing and cancer. Biochimica et Biophysica Acta – Reviews on Cancer, 1775, 92–102. 10.1016/j.bbcan.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Laviña M., Pugsley A. P., Moreno F. (1986). Identification, mapping, cloning and characterization of a gene (sbm A) required for microcin B17 action on Escherichia coli K12. Journal of General Microbiology, 132, 1685–1693. 10.1099/00221287-132-6-1685. [DOI] [PubMed] [Google Scholar]
- Lentzen G., Klinck R., Matassova N., Aboul-ela F., Murchie A. I. H. (2003). Structural basis for contrasting activities of ribosome binding thiazole antibiotics. Chemistry & Biology, 10, 769–778. 10.1016/S1074-5521(03)00173-X. [DOI] [PubMed] [Google Scholar]
- Li J. W. H., Vederas J. C. (2009). Drug discovery and natural products: End of an era or an endless frontier? Science, 325, 161–165. 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- Li Y., Rebuffat S. (2020). The manifold roles of microbial ribosomal peptide-based natural products in physiology and ecology. Journal of Biological Chemistry, 295, 34–54. 10.1074/jbc.REV119.006545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao R., Duan L., Lei C., Pan H., Ding Y., Zhang Q., Chen D., Shen B., Yu Y., Liu W. (2009). Thiopeptide biosynthesis featuring ribosomally synthesized precursor peptides and conserved posttranslational modifications. Chemistry & Biology, 16, 141–147. 10.1016/j.chembiol.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P. F., Samanta H., Bechtold C. M., Deminie C. A., Patick A. K., Alam M., Riccardi K., Rose R. E., White R. J., Colonno R. J. (1996). Characterization of siamycin I, a human immunodeficiency virus fusion inhibitor. Antimicrobial Agents and Chemotherapy, 40, 133–138. 10.1128/aac.40.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnett P. E., Strominger J. L. (1973). Additional antibiotic inhibitors of peptidoglycan synthesis. Antimicrobial Agents and Chemotherapy, 4, 231–236. 10.1128/AAC.4.3.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. T., Yang Y. L., Xu Y., Lamsa A., Haste N. M., Yang J. Y., Ng J., Gonzalez D., Ellermeier C. D., Straight P. D., Pevzner P. A., Pogliano J., Nizet V., Pogliano K., Dorrestein P. C. (2010). Imaging mass spectrometry of intraspecies metabolic exchange revealed the cannibalistic factors of Bacillus subtilis. Proceedings of the National Academy of Sciences of the USA, 107, 16286–16290. 10.1073/pnas.1008368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher K. P., Rees B., Koebnik R., Mitschler A., Moulinier L., Rosenbusch J. P., Moras D. (1998). Transmembrane signaling across the ligand-gated FhuA receptor: Crystal structures of free and ferrichrome-bound states reveal allosteric changes. Cell, 95, 771–778. 10.1016/S0092-8674(00)81700-6. [DOI] [PubMed] [Google Scholar]
- Luo H., Hallen-Adams H. E., Scott-Craig J. S., Walton J. D. (2012). Ribosomal biosynthesis of α-amanitin in Galerina marginata. Fungal Genetics and Biology, 49, 123–129. 10.1016/j.fgb.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma P., Nishiguchi K., Yuille H. M., Davis L. M., Nakayama J., Phillips-Jones M. K. (2011). Anti-HIV siamycin i directly inhibits autophosphorylation activity of the bacterial FsrC quorum sensor and other ATP-dependent enzyme activities. FEBS Letters, 585, 2660–2664. 10.1016/j.febslet.2011.07.026. [DOI] [PubMed] [Google Scholar]
- Magdalan J., Ostrowska A., Podhorska-Okołów M., Piotrowska A., Iżykowska I., Nowak M., Dolińska-Krajewska B., Zabel M., Szeląg A., Dzięgiel P. (2009). Early morphological and functional alterations in canine hepatocytes due to α-amanitin, a major toxin of Amanita phalloides. Archives of Toxicology, 83, 55–60. 10.1007/s00204-008-0376-9. [DOI] [PubMed] [Google Scholar]
- Maksimov M. O., Link A. J. (2014). Prospecting genomes for lasso peptides. Journal of Industrial Microbiology & Biotechnology, 41, 333–344. 10.1007/s10295-013-1357-4. [DOI] [PubMed] [Google Scholar]
- Maksimov M. O., Pan S. J., Link A. J. (2012). Lasso peptides: Structure, function, biosynthesis, and engineering. Natural Product Reports, 29, 996–1006. 10.1039/c2np20070h. [DOI] [PubMed] [Google Scholar]
- Maksimov M. O., Pelczer I., Link A. J. (2012). Precursor-centric genome-mining approach for lasso peptide discovery. Proceedings of the National Academy of Sciences of the USA, 109, 15223–15228. 10.1073/pnas.1208978109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N. I., Sprules T., Carpenter M. R., Cotter P. D., Hill C., Ross R. P., Vederas J. C. (2004). Structural characterization of lacticin 3147, a two-peptide lantibiotic with synergistic activity. Biochemistry, 43, 3049–3056. 10.1021/bi0362065. [DOI] [PubMed] [Google Scholar]
- Martinez-Bueno M., Maqueda M., Gálvez A., Samyn B., Van Beeumen J., Coyette J., Valdivia E. (1994). Determination of the gene sequence and the molecular structure of the enterococcal peptide antibiotic AS-48. Journal of Bacteriology, 176, 6334–6339. 10.1128/jb.176.20.6334-6339.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx R., Stein T., Entian K. D., Glaser S. J. (2001). Structure of the Bacillus subtilis peptide antibiotic subtilosin A determined by 1H-NMR and matrix assisted laser desorption/ionization time-of-flight mass spectrometry. Journal of Protein Chemistry, 20, 501–506. 10.1023/A:1012562631268. [DOI] [PubMed] [Google Scholar]
- Mathavan I., Beis K. (2012). The role of bacterial membrane proteins in the internalization of microcin MccJ25 and MccB17. Biochemical Society Transactions, 40, 1539–1543. 10.1042/BST20120176. [DOI] [PubMed] [Google Scholar]
- Mathavan I., Zirah S., Mehmood S., Choudhury H. G., Goulard C., Li Y., Robinson C. V., Rebuffat S., Beis K. (2014). Structural basis for hijacking siderophore receptors by antimicrobial lasso peptides. Nature Chemical Biology, 10, 340–342. 10.1038/nchembio.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur H., Fallico V., O'Connor P. M., Rea M. C., Cotter P. D., Hill C., Ross R. P. (2017). Insights into the mode of action of the sactibiotic thuricin CD. Frontiers in Microbiology, 8, 1–14. 10.3389/fmicb.2017.00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuliffe O., Ross R. P., Hill C. (2001). Lantibiotics: Structure, biosynthesis and mode of action. FEMS Microbiology Reviews, 25, 285–308. 10.1111/j.1574-6976.2001.tb00579.x. [DOI] [PubMed] [Google Scholar]
- McAuliffe O., Ryan M. P., Ross R. P., Hill C., Breeuwer P., Abee T. (1998). Lacticin 3147, a broad-spectrum bacteriocin which selectively dissipates the membrane potential. Applied and Environmental Microbiology, 64, 439–445. 10.1128/aem.64.2.439-445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metelev M., Arseniev A., Bushin L. B., Kuznedelov K., Artamonova T. O., Kondratenko R., Khodorkovskii M., Seyedsayamdost M. R., Severinov K. (2017). Acinetodin and klebsidin, RNA polymerase targeting lasso peptides produced by human isolates of Acinetobacter gyllenbergii and Klebsiella pneumoniae. ACS Chemical Biology, 12, 814–824. 10.1021/acschembio.6b01154. [DOI] [PubMed] [Google Scholar]
- Metelev M., Osterman I. A., Ghilarov D., Khabibullina N. F., Yakimov A., Shabalin K., Utkina I., Travin D. Y., Komarova E. S., Serebryakova M., Artamonova T., Khodorkovskii M., Konevega A. L., Sergiev P. V., Severinov K., Polikanov Y. S. (2017). Klebsazolicin inhibits 70S ribosome by obstructing the peptide exit tunnel. Nature Chemical Biology, 13, 1129–1136. 10.1038/nchembio.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metlitskaya A., Kazakov T., Kommer A., Pavlova O., Praetorius-Ibba M., Ibba M., Krasheninnikov I., Kolb V., Khmel I., Severinov K. (2006). Aspartyl-tRNA synthetase is the target of peptide nucleotide antibiotic microcin C. Journal of Biological Chemistry, 281, 18033–18042. 10.1074/jbc.M513174200. [DOI] [PubMed] [Google Scholar]
- Metlitskaya A., Kazakov T., Vondenhoff G. H., Novikova M., Shashkov A., Zatsepin T., Semenova E., Zaitseva N., Ramensky V., Aerschot A. V., Severinov K. (2009). Maturation of the translation inhibitor microcin. Journal of Bacteriology, 191, 2380–2387. 10.1128/JB.00999-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller K. W., Schamber R., Osmanagaoglu O., Ray B. (1998). Isolation and characterization of pediocin AcH chimeric protein mutants with altered bactericidal activity. Applied and Environmental Microbiology, 64, 1997–2005. 10.1128/aem.64.6.1997-2005.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K., Ubarretxena-Belandia I., Taguchi T., Warren G., Engelman D. M. (2004). Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proceedings of the National Academy of Sciences, 101, 4083–4088. 10.1073/pnas.0307332101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch D., Müller A., Schneider T., Kohl B., Wenzel M., Bandow J. E., Maffioli S., Sosio M., Donadio S., Wimmer R., Sahl H. G. (2014). The lantibiotic NAI-107 binds to bactoprenol-bound cell wall precursors and impairs membrane functions. Journal of Biological Chemistry, 289, 12063–12076. 10.1074/jbc.M113.537449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo T., Ji X., Yuan W., Mandalapu D., Wang F., Zhong Y., Li F., Chen Q., Ding W., Deng Z., Yu S., Zhang Q. (2019). Thuricin Z: A narrow-spectrum sactibiotic that targets the cell membrane. Angewandte Chemie, International Edition, 58, 18793–18797. 10.1002/anie.201908490. [DOI] [PubMed] [Google Scholar]
- Möhrle V. G., Tieleman L. N., Kraal B. (1997). Elongation factor Tu1 of the antibiotic GE2270A producer Planobispora rosea Has an unexpected resistance profile against EF-Tu targeted antibiotics. Biochemical and Biophysical Research Communications, 230, 320–326. 10.1006/bbrc.1996.5947. [DOI] [PubMed] [Google Scholar]
- Montalbán-López M., Scott T. A., Ramesh S., Rahman I. R., van Heel A. J., Viel J. H., Bandarian V., Dittmann E., Genilloud O., Goto Y., Grande Burgos M. J., Hill C., Kim S., Koehnke J., Latham J. A., Link A. J., Martínez B., Nair S. K., Nicolet Y., van der Donk W. A. (2021). New developments in RiPP discovery, enzymology and engineering. Natural Product Reports, 28, 130–239. 10.1039/D0NP00027B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. P., Leeds J. A., Naegeli H. U., Oberer L., Memmert K., Weber E., LaMarche M. J., Parker C. N., Burrer N., Esterow S., Hein A. E., Schmitt E. K., Krastel P. (2009). Ribosomally synthesized thiopeptide antibiotics targeting Elongation Factor Tu. Journal of the American Chemical Society, 131, 5946–5955. 10.1021/ja900488a. [DOI] [PubMed] [Google Scholar]
- Motlagh A. M., Bhunia A. K., Szostek F., Hansen T. R., Johnson M. C., Ray B. (1992). Nucleotide and amino acid sequence of pap-gene (pediocin AcH production) in Pediococcus acidilactici H. Letters in Applied Microbiology, 15, 45–48. 10.1111/j.1472-765X.1992.tb00721.x. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay J., Sineva E., Knight J., Levy R. M., Ebright R. H. (2004). Antibacterial peptide microcin J25 inhibits transcription by binding within and obstructing the RNA polymerase secondary channel. Molecular Cell, 14, 739–751. 10.1016/j.molcel.2004.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. Q. N., Tooh Y. W., Sugiyama R., Nguyen T. P. D., Purushothaman M., Leow L. C., Hanif K., Yong R. H. S., Agatha I., Winnerdy F. R., Gugger M., Phan A. T., Morinaka B. I. (2020). Post-translational formation of strained cyclophanes in bacteria. Nature Chemistry, 12, 1042–1053. 10.1038/s41557-020-0519-z. [DOI] [PubMed] [Google Scholar]
- Nissen-Meyer J., Holo H., Håvarstein L. S., Sletten K., Nes I. F. (1992). A novel lactococcal bacteriocin whose activity depends on the complementary action of two peptides. Journal of Bacteriology, 174, 5686–5692. 10.1128/jb.174.17.5686-5692.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N., Guillier M., Barnard T. J., Buchanan S. K. (2010). TonB-dependent transporters: Regulation, structure, and function. Annual Review of Microbiology, 64, 43–60. 10.1146/annurev.micro.112408.134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll K. S., Sinko P. J., Chikindas M. L. (2011). Elucidation of the molecular mechanisms of action of the natural antimicrobial peptide subtilosin against the bacterial vaginosis-associated pathogen Gardnerella vaginalis. Probiotics & Antimicrobial Proteins, 3, 41–47. 10.1007/s12602-010-9061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T., Niki H., Mori H., Morita M., Hasegawa M., Ichinose C., Hiraga S. (1990). Identification and characterization of gyrB mutants of Escherichia coli that are defective in partitioning of mini-F plasmids. Journal of Bacteriology, 172, 1562–1568. 10.1128/jb.172.3.1562-1568.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oiki S., Muramatsu I., Matsunaga S., Fusetani N. (1997). A channel-forming peptide toxin: Polytheonamide from marine sponge (Theonella swinhoei). Folia Pharmacologica Japonica, 110, 195–198. 10.1254/fpj.110.supplement_195. [DOI] [PubMed] [Google Scholar]
- Oppegård C., Kjos M., Veening J.-W., Nissen-Meyer J., Kristensen T. (2016). A putative amino acid transporter determines sensitivity to the two-peptide bacteriocin plantaricin JK. Microbiologyopen, 5, 700–708. 10.1002/mbo3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana C., Lagos R. (1996). The activity of microcin E492 from Klebsiella pneumoniae is regulated by a microcin antagonist. FEMS Microbiology Letters, 136, 297–303. 10.1016/0378-1097(96)00025-0. [DOI] [PubMed] [Google Scholar]
- O'Rourke S., Widdick D., Bibb M. (2017). A novel mechanism of immunity controls the onset of cinnamycin biosynthesis in Streptomyces cinnamoneus DSM 40646. Journal of Industrial Microbiology & Biotechnology, 44, 563–572. 10.1007/s10295-016-1869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otaka T., Kaji A. (1976). Mode of action of bottromycin A2. Release of aminoacyl- or peptidyl-tRNA from ribosomes. Journal of Biological Chemistry, 251, 2299–306. [PubMed] [Google Scholar]
- Otaka T., Kaji A. (1981). Mode of action of bottromycin A2: Effect on peptide bond formation. FEBS Letters, 123, 173–176. 10.1016/0014-5793(81)80280-3. [DOI] [PubMed] [Google Scholar]
- Otaka T., Kaji A. (1983). Mode of action of bottromycin A2: Effect of bottromycin A2 on polysomes. FEBS Letters, 153, 53–59. 10.1016/0014-5793(83)80118-5. [DOI] [PubMed] [Google Scholar]
- Pang Z., Chen R., Manna D., Higgins N. P. (2005). A gyrase mutant with low activity disrupts supercoiling at the replication terminus. Journal of Bacteriology, 187, 7773–7783. 10.1128/JB.187.22.7773-7783.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti F., Pagani H., Beretta G. (1976). Gardimycin, a new antibiotic from actionoplanes. I. Description of the producer strain and fermentation studies. Journal of Antibiotics (Tokyo), 29, 501–506. 10.7164/antibiotics.29.501. [DOI] [PubMed] [Google Scholar]
- Patzer S. I., Baquero M. R., Bravo D., Moreno F., Hantke K. (2003). The colicin G, H and X determinants encode microcins M and H47, which might utilize the catecholate siderophore receptors FepA, Cir, Fiu and IronN. Microbiology (Reading, England), 149, 2557–2570. 10.1099/mic.0.26396-0. [DOI] [PubMed] [Google Scholar]
- Pavlova O., Mukhopadhyay J., Sineva E., Ebright R. H., Severinov K. (2008). Systematic structure–activity analysis of microcin J25. Journal of Biological Chemistry, 283, 25589–25595. 10.1074/jbc.M803995200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelek P. D., Croteau N., Ng-Thow-Hing C., Khursigara C. M., Moiseeva N., Allaire M., Coulton J. W. (2006). Structure of TonB in complex with FhuA, E. coli outer membrane receptor. Science, 312, 1399–1402. 10.1126/science.1128057. [DOI] [PubMed] [Google Scholar]
- Perkins H. R. (1969). Specificity of combination between mucopeptide precursors and vancomycin or ristocetin. Biochemical Journal, 111, 195–205. 10.1042/bj1110195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter B. J., Arsuaga J., Breier A. M., Khodursky A. B., Brown P. O., Cozzarelli N. R. (2004). Genomic transcriptional response to loss of chromosomal supercoiling in Escherichia coli. Genome Biology, 5, 1–16. 10.1186/gb-2004-5-11-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskunova J., Maisonneuve E., Germain E., Gerdes K., Severinov K. (2017). Peptide-nucleotide antibiotic microcin C is a potent inducer of stringent response and persistence in both sensitive and producing cells. Molecular Microbiology, 104, 463–471. 10.1111/mmi.13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasch T., Naumann T., Marker R. L. M., Sattler M., Schubert W., Schaal S., Bauch M., Kogler H., Griesinger C. (1997). Constitution and solution conformation of the antibiotic mersacidin determined by NMR and molecular dynamics. European Journal of Biochemistry FEBS, 244, 501–512. 10.1111/j.1432-1033.1997.00501.x. [DOI] [PubMed] [Google Scholar]
- Prince A., Sandhu P., Kumar P., Dash E., Sharma S., Arakha M., Jha S., Akhter Y., Saleem M. (2016). Lipid-II independent antimicrobial mechanism of nisin depends on its crowding and degree of oligomerization. Scientific Reports, 6, 1–14. 10.1038/srep37908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugsley A. P., Moreno F., De Lorenzo V. (1986). Microcin-E492-insensitive mutants of Escherichia coli K12. Microbiology (Reading, England), 132, 3253–3259. 10.1099/00221287-132-12-3253. [DOI] [PubMed] [Google Scholar]
- Ra R., Beerthuyzen M. M., De Vos W. M., Saris P. E. J., Kuipers O. P. (1999). Effects of gene disruptions in the nisin gene cluster of Lactococcus lactis on nisin production and producer immunity. Microbiology (Reading, England), 145, 1227–1233. 10.1099/13500872-145-5-1227. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan S. K., Bhat U. G., Hughes D. E., Wang I. C., Costa R. H., Gartel A. L. (2006). Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Research, 66, 9731–9735. 10.1158/0008-5472.CAN-06-1576. [DOI] [PubMed] [Google Scholar]
- Ramnath M., Beukes M., Tamura K., Hastings J. W. (2000). Absence of a putative mannose-specific phosphotransferase system enzyme IIAB component in a leucocin a-resistant strain of Listeria monocytogenes, as shown by two-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Applied and Environmental Microbiology, 66, 3098–3101. 10.1128/AEM.66.7.3098-3101.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran R., Zeng H., Zhao D., Liu R., Xu X. (2017). The novel property of heptapeptide of microcin C7 in affecting the cell growth of Escherichia coli. Molecules (Basel, Switzerland), 22, 1–13. 10.3390/molecules22030432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea M. C., Sit C. S., Clayton E., O'Connor P. M., Whittal R. M., Zheng J., Vederas J. C., Ross R. P., Hill C. (2010). Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proceedings of the National Academy of Sciences of the USA, 107, 9352–9357. 10.1073/pnas.0913554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece R. J., Maxwell A. (1991). DNA gyrase: Structure and function. Critical Reviews in Biochemistry and Molecular Biology, 26, 335–375. 10.3109/10409239109114072. [DOI] [PubMed] [Google Scholar]
- Rigel N. W., Silhavy T. J. (2012). Making a beta-barrel: Assembly of outer membrane proteins in Gram-negative bacteria. Current Opinion in Microbiology, 15, 189–193. 10.1016/j.mib.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos Colombo N. S., Chalón M. C., Navarro S. A., Bellomio A. (2018). Pediocin-like bacteriocins: New perspectives on mechanism of action and immunity. Current Genetics, 64, 345–351. 10.1007/s00294-017-0757-9. [DOI] [PubMed] [Google Scholar]
- Robichon D., Gouin E., Débarbouillé M., Cossart P., Cenatiempo Y., Héchard Y. (1997). The rpoN (σ54) gene from Listeria monocytogenes is involved in resistance to mesentericin Y105, an antibacterial peptide from Leuconostoc mesenteroides. Journal of Bacteriology, 179, 7591–7594. 10.1128/jb.179.23.7591-7594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers L. A. (1928). The inhibiting effect of Streptococcus lactis on Lactobacillus bulgaricus. Journal of Bacteriology, 16, 321–5. 10.1128/JB.16.5.321-325.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovinskiy N., Agbleke A. A., Chesnokova O., Pang Z., Higgins N. P. (2012). Rates of gyrase supercoiling and transcription elongation control supercoil density in a bacterial chromosome. PLoS Genetics, 8, e1002845. 10.1371/journal.pgen.1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell A. H., Truman A. W. (2020). Genome mining strategies for ribosomally synthesised and post-translationally modified peptides. Computational and Structural Biotechnology Journal, 18, 1838–1851. 10.1016/j.csbj.2020.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M. P., Rea M. C., Hill C., Ross R. P. (1996). An application in cheddar cheese manufacture for a strain of Lactococcus lactis producing a novel broad-spectrum bacteriocin, lacticin 3147. Applied and Environmental Microbiology, 62, 612–619. 10.1128/aem.62.2.612-619.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomón R. A., Farías R. N. (1992). Microcin 25, a novel antimicrobial peptide produced by Escherichia coli. Journal of Bacteriology, 174, 7428–7435. 10.1128/jb.174.22.7428-7435.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomón R. A., Farías R. N. (1993). The fhuA protein is involved in microcin 25 uptake. Journal of Bacteriology, 175, 7741–7742. 10.1128/jb.175.23.7741-7742.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomón R. A., Fariás R. N. (1994). Influence of iron on microcin 25 production. FEMS Microbiology Letters, 121, 275–280. 10.1016/0378-1097(94)90303-4. [DOI] [PubMed] [Google Scholar]
- Salomón R. A., Farías R. N. (1995). The peptide antibiotic microcin 25 is imported through the tonB pathway and the sbmA protein. Journal of Bacteriology, 177, 3323–3325. 10.1128/jb.177.11.3323-3325.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samyn B., Martinez-Bueno M., Devreese B., Maqueda M., Gálvez A., Valdivia E., Coyette J., Van Beeumen J. (1994). The cyclic structure of the enterococcal peptide antibiotic AS-48. FEBS Letters, 352, 87–90. 10.1016/0014-5793(94)00925-2. [DOI] [PubMed] [Google Scholar]
- Sánchez-Barrena M. J., Martínez-Ripoll M., Gálvez A., Valdivia E., Maqueda M., Cruz V., Albert A. (2003). Structure of bacteriocin AS-48: From soluble state to membrane bound state. Journal of Molecular Biology, 334, 541–549. 10.1016/j.jmb.2003.09.060. [DOI] [PubMed] [Google Scholar]
- Sauvage E., Kerff F., Terrak M., Ayala J. A., Charlier P. (2008). The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiology Reviews, 32, 234–258. 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- Severinov K., Nair S. K. (2012). Microcin C: Biosynthesis and mechanisms of bacterial resistance. Future Microbiology, 7, 281–289. 10.2217/fmb.11.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X., Mustafa M., Chen Y., Cao Y., Gao J. (2019). Natural thiopeptides as a privileged scaffold for drug discovery and therapeutic development. Medicinal Chemistry Research, 28, 1063–1098. 10.1007/s00044-019-02361-1. [DOI] [Google Scholar]
- Shimanaka K., Iinuma H., Hamada M., Ikeno S., Tsuchiya K. S., Arita M., Hori M. (1995). Novel antibiotics, amythiamicins IV. A Mutation in the elongation factor Tu gene in a resistant mutant of B. subtilis. Journal of Antibiotics (Tokyo), 48, 182–184. 10.7164/antibiotics.48.182. [DOI] [PubMed] [Google Scholar]
- Shimanaka K., Takahashi Y., Iinuma H., Naganawa H., Takeuchi T. (1994). Novel antibiotic, amythiamicins. III. Structure elucidations of amythiamicins A, B and C. Journal of Antibiotics (Tokyo), 47, 1153–1159. 10.7164/antibiotics.47.1153. [DOI] [PubMed] [Google Scholar]
- Shinohara N., Itoh H., Matsuoka S., Inoue M. (2012). Selective modification of the N-terminal structure of Polytheonamide B significantly changes its cytotoxicity and activity as an ion channel. Chemmedchem, 7, 1770–1773. 10.1002/cmdc.201200142. [DOI] [PubMed] [Google Scholar]
- Shoji J., Hinoo H., Wakisaka Y., Koizumi K., Mayama M., Matsuura S., Matsumoto K. (1976). Studies on antibiotics from the genus Bacillus. VIII. Isolation of three new antibiotics, thiocillins I, II and III, related to micrococcin P. Journal of Antibiotics, 29, 366–374. 10.7164/antibiotics.29.366. [DOI] [PubMed] [Google Scholar]
- Silhavy T. J., Kahne D., Walker S. (2010). The bacterial cell envelope. Cold Spring Harbor Perspectives in Biology, 2, a000414. 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkin L., Hamza S., Kaufman S., Cobb S. L., Vederas J. C. (2008). Spermicidal bacteriocins: Lacticin 3147 and subtilosin A. Bioorganic & Medicinal Chemistry Letters, 18, 3103–3106. 10.1016/j.bmcl.2007.11.024. [DOI] [PubMed] [Google Scholar]
- Sit C. S., McKay R. T., Hill C., Ross R. P., Vederas J. C. (2011). The 3D structure of Thuricin CD, a two-component bacteriocin with cysteine sulfur to α-carbon cross-links. Journal of the American Chemical Society, 133, 7680–7683. 10.1021/ja201802f. [DOI] [PubMed] [Google Scholar]
- Smith L., Hasper H., Breukink E., Novak J., Ćerkasov J., Hillman J. D., Wilson-Stanford S., Orugunty R. S. (2008). Elucidation of the antimicrobial mechanism of mutacin 1140. Biochemistry, 47, 3308–3314. 10.1021/bi701262z. [DOI] [PubMed] [Google Scholar]
- Socias S. B., Severinov K., Salomon R. A. (2009). The Ile13 residue of microcin J25 is essential for recognition by the receptor FhuA, but not by the inner membrane transporter SbmA. FEMS Microbiology Letters, 301, 124–129. 10.1111/j.1574-6968.2009.01805.x. [DOI] [PubMed] [Google Scholar]
- Sohlenkamp C., Geiger O. (2015). Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiology Reviews, 40, 133–159. 10.1093/femsre/fuv008. [DOI] [PubMed] [Google Scholar]
- Solbiati J. O., Ciaccio M., Farías R. N., González-Pastor J. E., Moreno F., Salomón R. A. (1999). Sequence analysis of the four plasmid genes required to produce the circular peptide antibiotic microcin J25. Journal of Bacteriology, 181, 2659–2562. 10.1128/JB.181.8.2659-2662.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somma S., Merati W., Parenti F. (1977). Gardimycin, a new antibiotic inhibiting peptidoglycan synthesis. Antimicrobial Agents and Chemotherapy, 11, 396–401. 10.1128/AAC.11.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternglanz R., DiNardo S., Voelkel K. A., Nishimura Y., Hirota Y., Becherer K., Zumstein L., Wang J. C. (1981). Mutations in the gene coding for Escherichia coli DNA topoisomerase I affect transcription and transposition. Proceedings of the National Academy of Sciences of the USA, 78, 2747–2751. 10.1073/pnas.78.5.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutyak K. E., Wirawan R. E., Aroutcheva A. A., Chikindas M. L. (2008). Isolation of the Bacillus subtilis antimicrobial peptide subtilosin from the dairy product-derived Bacillus amyloliquefaciens. Journal of Applied Microbiology, 104, 1067–1074. 10.1111/j.1365-2672.2007.03626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto K., Miyasaka Y., Ishitsuka H., Suhara Y., Maruyama H. (1984). Pentadecapeptide. U.S. Patent No. 4452782A.
- Tan S., Ludwig K. C., Müller A., Schneider T., Nodwell J. R. (2019). The lasso peptide siamycin-I targets lipid II at the gram-positive cell surface. ACS Chemical Biology, 14, 966–974. 10.1021/acschembio.9b00157. [DOI] [PubMed] [Google Scholar]
- ‘t Hart P., Oppedijk S. F., Breukink E., Martin N. I. (2016). New insights into nisin's antibacterial mechanism revealed by binding studies with synthetic lipid II analogues. Biochemistry, 55, 232–237. 10.1021/acs.biochem.5b01173. [DOI] [PubMed] [Google Scholar]
- Thennarasu S., Lee D.-K., Poon A., Kawulka K. E., Vederas J. C., Ramamoorthy A. (2005). Membrane permeabilization, orientation, and antimicrobial mechanism of subtilosin A. Chemistry and Physics of Lipids, 137, 38–51. 10.1016/j.chemphyslip.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Thomas X., Destoumieux-Garzón D., Peduzzi J., Afonso C., Blond A., Birlirakis N., Goulard C., Dubost L., Thai R., Tabet J. C., Rebuffat S. (2004). Siderophore peptide, a new type of post-translationally modified antibacterial peptide with potent activity. Journal of Biological Chemistry, 279, 28233–28242. 10.1074/jbc.M400228200. [DOI] [PubMed] [Google Scholar]
- Tomasek D., Rawson S., Lee J., Wzorek J. S., Harrison S. C., Li Z., Kahne D. (2020). Structure of a nascent membrane protein as it folds on the BAM complex. Nature, 583, 473–478. 10.1038/s41586-020-2370-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travin D. Y., Bikmetov D., Severinov K. (2020). Translation-targeting RiPPs and where to find them. Frontiers in Genetics, 11, 1–16. 10.3389/fgene.2020.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travin D. Y., Watson Z. L., Metelev M., Ward F. R., Osterman I. A., Khven I. M., Khabibullina N. F., Serebryakova M., Mergaert P., Polikanov Y. S., Cate J. H. D., Severinov K. (2019). Structure of ribosome-bound azole-modified peptide phazolicin rationalizes its species-specific mode of bacterial translation inhibition. Nature communications, 10, 4563. 10.1038/s41467-019-12589-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunakawa M., Hu S.-L., Hoshino Y., Detlefson D. J., Hill S. E., Furumai T., White R. J., Nishio M., Kawano K., Yyamamoto S., Fukagawa Y., Oki T. (1995). Siamycins I and II, new anti-HIV peptides. I. Fermentation, isolation, biological activity and initial characterization. Journal of Antibiotics (Tokyo), 48, 433–434. 10.7164/antibiotics.48.433. [DOI] [PubMed] [Google Scholar]
- Tymoszewska A., Diep D. B., Aleksandrzak-Piekarczyk T. (2018). The extracellular loop of Man-PTS subunit IID is responsible for the sensitivity of Lactococcus garvieae to garvicins A, B and C. Scientific Reports, 8, 1–15. 10.1038/s41598-018-34087-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A., Banzhaf M., Gross C. A., Vollmer W. (2012). From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nature Reviews Microbiology, 10, 123–136. 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heijenoort J. (2007). Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiology and Molecular Biology Reviews, 71, 620–635. 10.1128/MMBR.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Heusden H. E., De Kruijff B., Breukink E. (2002). Lipid II induces a transmembrane orientation of the pore-forming peptide lantibiotic nisin. Biochemistry, 41, 12171–12178. 10.1021/bi026090x. [DOI] [PubMed] [Google Scholar]
- van Meer G., Voelker D. R., Feigenson G. W. (2008). Membrane lipids: Where they are and how they behave. Nature Reviews Molecular Cell Biology, 9, 112–124. 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasile F., Potenza D., Marsiglia B., Maffioli S., Donadio S. (2012). Solution structure by nuclear magnetic resonance of the two lantibiotics 97518 and NAI-107. Journal of Peptide Science, 18, 129–134. 10.1002/psc.1425. [DOI] [PubMed] [Google Scholar]
- Vassiliadis G., Peduzzi J., Zirah S., Thomas X., Rebuffat S., Destoumieux-Garzón D. (2007). Insight into siderophore-carrying peptide biosynthesis: Enterobactin is a precursor for microcin E492 posttranslational modification. Antimicrob Agents Chemother, 51, 3546–3553. 10.1128/AAC.00261-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard M., Berglund N. A., Hsu P.-C., Song C., Koldsø H., Schiøtt B., Sansom M. S. P. (2019). Structure and dynamics of cinnamycin–lipid complexes: Mechanisms of selectivity for phosphatidylethanolamine lipids. ACS Omega, 4, 18889–18899. 10.1021/acsomega.9b02949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent P. A., Delgado M. A., Farías R. N., Salomón R. A. (2004). Inhibition of Salmonella enterica serovars by microcin J25. FEMS Microbiology Letters, 236, 103–107. 10.1016/j.femsle.2004.05.027. [DOI] [PubMed] [Google Scholar]
- Vollmer W., Blanot D., De Pedro M. A. (2008). Peptidoglycan structure and architecture. FEMS Microbiology Reviews, 32, 149–167. 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- Waisvisz J. M., van der Hoeven M. G., van Peppen J., Zwennis W. C. M. (1957). Bottromycin. I. A new sulfur-containing antibiotic. Journal of the American Chemical Society, 79, 4520–4521. 10.1021/ja01573a072. [DOI] [Google Scholar]
- Wakamatsu K., Choung S.-Y., Kobayashi T., Inoue K., Higashijima T., Miyazawa T. (1990). Complex formation of peptide antibiotic Ro09-0198 with lysophosphatidylethanolamine: 1H NMR analyses in dimethyl sulfoxide solution. Biochemistry, 29, 113–118. 10.1021/bi00453a013. [DOI] [PubMed] [Google Scholar]
- Widdick D. A., Dodd H. M., Barraille P., White J., Stein T. H., Chater K. F., Gasson M. J., Bibb M. J. (2003). Cloning and engineering of the cinnamycin biosynthetic gene cluster from Streptomyces cinnamoneus DSM 40005. Proceedings of the National Academy of Sciences of the USA, 100, 4316–4321. 10.1073/pnas.0230516100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann I., Benz R., Sahl H. G. (2004). Lipid II-mediated pore formation by the peptide antibiotic nisin: A black lipid membrane study. Journal of Bacteriology, 186, 3259–3261. 10.1128/JB.186.10.3259-3261.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann I., Böttiger T., Bonelli R. R., Wiese A., Hagge S. O., Gutsmann T., Seydel U., Deegan L., Hill C., Ross P., Sahl H. G. (2006). The mode of action of the lantibiotic lacticin 3147 – A complex mechanism involving specific interaction of two peptides and the cell wall precursor lipid II. Molecular Microbiology, 61, 285–296. 10.1111/j.1365-2958.2006.05223.x. [DOI] [PubMed] [Google Scholar]
- Wienland T., Faulstich H. (1991). Fifty years of amanitin. Experientia, 47, 1186–1193. 10.1007/BF01918382. [DOI] [PubMed] [Google Scholar]
- Wilkens M., Villanueva J. E., Cofré J., Chnaiderman J., Lagos R. (1997). Cloning and expression in Escherichia coli of genetic determinants for production of and immunity to microcin E492 from Klebsiella pneumoniae. Journal of Bacteriology, 179, 4789–4794. 10.1128/JB.179.15.4789-4794.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D. N., Nierhaus K. H. (2005). Ribosomal proteins in the spotlight. Critical Reviews in Biochemistry and Molecular Biology, 40, 243–267. 10.1080/10409230500256523. [DOI] [PubMed] [Google Scholar]
- Wilson K. A., Kalkum M., Ottesen J., Yuzenkova J., Chait B. T., Landick R., Muir T., Severinov K., Darst S. A. (2003). Structure of microcin J25, a peptide inhibitor of bacterial RNA polymerase, is a lassoed tail. Journal of the American Chemical Society, 125, 12475–12483. 10.1021/ja036756q. [DOI] [PubMed] [Google Scholar]
- Wilson M. C., Mori T., Rückert C., Uria A. R., Helf M. J., Takada K., Gernert C., Steffens U. A. E., Heycke N., Schmitt S., Rinke C., Helfrich E. J. N., Brachmann A. O., Gurgui C., Wakimoto T., Kracht M., Crüsemann M., Hentschel U., Abe I., Piel J. (2014). An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature, 506, 58–62. 10.1038/nature12959. [DOI] [PubMed] [Google Scholar]
- Wu T., Malinverni J., Ruiz N., Kim S., Silhavy T. J., Kahne D. (2005). Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell, 121, 235–245. 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Xiao H., Chen X., Chen M., Tang S., Zhao X., Huan L. (2004). Bovicin HJ50, a novel lantibiotic produced by Streptococcus bovis HJ50. Microbiology (Reading, England), 150, 103–108. 10.1099/mic.0.26437-0. [DOI] [PubMed] [Google Scholar]
- Yano K., Toki S., Nakanishi S., Ochiai K., Ando K., Yoshida M., Matsuda Y., Yamasaki M. (1996). MS-271, a novel inhibitor of calmodulin-activated myosin light chain kinase from Streptomyces sp.—I. isolation, structural determination and biological properties of MS-271. Bioorganic & Medicinal Chemistry, 4, 115–120. 10.1016/0968-0896(95)00175-1. [DOI] [PubMed] [Google Scholar]
- Zamble D. B., Miller D. A., Heddle J. G., Maxwell A., Walsh C. T., Hollfelder F. (2001). In vitro characterization of DNA gyrase inhibition by microcin B17 analogs with altered bisheterocyclic sites. Proceedings of the National Academy of Sciences of the USA, 98, 7712–7717. 10.1073/pnas.141225698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti G., Petersen G., Wieland T. (2009). Structure–toxicity relationships in the amatoxin series. International Journal of Peptide and Protein Research, 40, 551–558. 10.1111/j.1399-3011.1992.tb00440.x. [DOI] [PubMed] [Google Scholar]
- Zhang J., Feng Y., Teng K., Lin Y., Gao Y., Wang J., Zhong J. (2014). Type AII lantibiotic bovicin HJ50 with a rare disulfide bond: Structure, structure–activity relationships and mode of action. Biochemical Journal, 461, 497–508. 10.1042/BJ20131524. [DOI] [PubMed] [Google Scholar]
- Zheng G., Yan L. Z., Vederas J. C., Zuber P. (1999). Genes of the sbo-alb locus of Bacillus subtilis are required for production of the antilisterial bacteriocin subtilosin. Journal of Bacteriology, 181, 7346–7355. 10.1128/jb.181.23.7346-7355.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann N., Jung G. (1997). The three-dimensional solution structure of the lantibiotic murein-biosynthesis-inhibitor actagardine determined by NMR. European Journal of Biochemistry FEBS, 246, 809–819. 10.1111/j.1432-1033.1997.00809.x. [DOI] [PubMed] [Google Scholar]
- Zuurmond A. M., De Graaf J. M., Olsthoorn-Tieleman L. N., Van Duyl B. Y., Mörhle V. G., Jurnak F., Mesters J. R., Hilgenfeld R., Kraal B. (2000). Ge2270A-resistant mutations in elongation factor Tu allow productive aminoacyl-tRNA binding to EF-TU·GTP·GE2270A complexes. Journal of Molecular Biology, 304, 995–1005. 10.1006/jmbi.2000.4260. [DOI] [PubMed] [Google Scholar]