

Abstract
A major obstacle in cancer gene therapy is the limited efficiency of in vivo gene transfer by replication-defective retrovirus vectors in current use. One strategy for circumventing this difficulty would be to use vectors capable of replication within tumor tissues. We have developed a replication-competent retrovirus (RCR) vector derived from murine leukemia virus (MuLV). This vector utilizes a unique design strategy in which an internal ribosome entry site–transgene cassette is positioned between the env gene and the 3′ long terminal repeat (LTR). The ability of this vector to replicate and transmit a transgene was examined in culture and in a solid tumor model in vivo. The RCR vector exhibited replication kinetics similar to those of wild-type MuLV and mediated efficient delivery of the transgene throughout an entire population of cells in culture after an initial inoculation with 1 plaque-forming unit (PFU) of vector per 2000 cells. After injection of 6 × 103 PFU of vector into established subcutaneous tumors, highly efficient spread of the transgene was observed over a period of 7 weeks, in some cases resulting in spread of the transgene throughout the entire tumor. MuLV-based RCR vectors show significant advantages over standard replication-defective vectors in efficiency of gene delivery both in culture and in vivo. This represents the first example of the use of an RCR vector in an adult mammalian host, and their first application to transduction of solid tumors.
OVERVIEW SUMMARY
Low efficiency of gene delivery by currently used viral vectors remains a significant barrier to the success of gene therapy of cancer. To examine the possibility of achieving superior transduction efficiency utilizing retroviral vectors that are capable of replication, we inserted an IRES–transgene cassette between the env gene and 3′ LTR of an infectious full-length murine leukemia virus (MuLV) clone. We found that the resulting vector replicated with kinetics similar to those of wild-type MuLV, and was stable through multiple serial passages in cultured cells. Injection of this vector into established subcutaneous tumors in mice resulted in highly efficient transmission of the transgene and, in some cases, transduction of entire tumor masses. These results demonstrate the potential utility of replication-competent retroviral vectors for cancer gene therapy.
INTRODUCTION
Advances in techniques for gene transfer and expression have made feasible the treatment of cancer at the genetic level by introduction of exogenous genes into tumor cells (Anderson, 1984). Clinical trials utilizing various gene therapy strategies are underway for a variety of malignancies. However, all of these strategies require efficient gene transfer and this step has been a major impediment (Robbins et al., 1998; Smith, 1999; Vile et al., 2000). Despite the use of viral vectors such as retroviruses, gene transfer efficiency in vivo has generally been inadequate for achieving significant therapeutic benefit.
Defective murine leukemia virus (MuLV)-based retroviral vectors, which have been the most commonly used gene delivery vehicles in clinical gene therapy protocols (Robbins et al., 1998), are incapable of secondary infection of adjacent cells because of the deletion of essential viral genes. More efficient transduction could be achieved if replication-competent retroviruses were used, as the virus would multiply after the initial infection event and each infected target cell would itself become a virus-producing cell. However, uncontrolled virus spread could result in adverse consequences, and the possibility of generating replication-competent retrovirus (RCR) during vector production has been a primary concern of gene therapy investigators (Cornetta et al., 1991a).
Nevertheless, replicating forms of other virus species, including adenovirus (Bischoff et al., 1996; Wildner et al., 1999; Alemany et al., 2000; Heise et al., 2000), paramyxoviruses (Lorence et al., 1988; Sinkovics and Horvath, 2000), herpesvirus (Walker et al., 1999; Pawlik et al., 2000), and reovirus (Coffey et al., 1998; Norman and Lee, 2000) have been exploited for cytolytic treatment of cancer. Replicative retroviral vectors have also been proposed for use in gene therapy (Russell, 1994; Vile et al., 1998), but there have been no previous studies reporting the development of RCR vectors for therapeutic applications. Although retroviruses are not cytolytic, the incorporation of a suicide transgene into an RCR vector could be used as a means to kill tumor cells, and would also serve as a safety mechanism to eliminate the vector after adequate levels of transduction are achieved. Furthermore, the initial rationale for use of defective retroviral vectors in cancer gene therapy would still hold true for RCR vectors, that is, MuLV-based vectors can transduce only cells that are actively dividing (Miller et al., 1990), and since the majority of normal cells are quiescent, transduction would be relatively selective for tumor cells.
All previously described replicating murine retroviral vectors have contained transgene inserts, ranging from 100 to 1200 bp, in the U3 region of the 3′ long terminal repeat (LTR) (Goff et al., 1981; Reik et al., 1985; Stuhlmann et al., 1989; Dillon et al., 1991). However, vectors based on this design have proven to be quite unstable, as most of these earlier constructs began losing their transgene inserts from the first infection cycle (Goff et al., 1981; Reik et al., 1985; Stuhlmann et al., 1989; Dillon et al., 1991), and even nonreplicating retroviral vectors containing U3 inserts show a high frequency of recombination and deletion events (Mavilio et al., 1984; Junker et al., 1995). Thus, none of these previous replicating MuLV vectors has been sufficiently stable to be useful for efficient and reliable gene delivery in vivo.
We therefore sought to develop a stable, nondefective retroviral vector capable of high-level transduction both in culture and within solid tumors. Employing a unique construct design, we have developed an MuLV-based RCR vector that contains an internal ribosome entry site (IRES)–transgene expression cassette inserted precisely at the env-3′ untranslated region (UTR) boundary in the MuLV genome. We have previously utilized this type of vector to achieve delivery of the cell cycle regulator p27(kip-1) to osteoblasts in vitro (Smith et al., 2000), demonstrating that this construct design produces a retroviral vector that can efficiently replicate and transduce mammalian cells in culture. Here we demonstrate that this vector is highly stable, being capable of replicating without observable deletions through multiple serial infection cycles in culture, and can achieve highly efficient gene delivery to solid tumors in vivo; hence this report represents the first use of such a nondefective retrovirus vector to achieve gene delivery in an adult mammalian host.
MATERIALS AND METHODS
Retroviral vector plasmid construction
An NheI fragment from plasmid pZAP (Shoemaker et al., 1981) (generously provided by J.A. Young, University of Wisconsin, Madison, WI) containing the wild-type ecotropic Moloney murine leukemia virus (MuLV) provirus was recloned to eliminate flanking rat genomic sequences, producing plasmid pZAP2. Overlap-extension polymerase chain reaction (PCR) (Horton et al., 1989) was used to fuse the 3′ end of the env gene to the encephalomyocarditis virus internal ribosome entry site (IRES) amplified from plasmid pEMCF, and the resulting plasmid was designated pZAPd. Insertion of the enhanced green fluorescent protein (GFP) cDNA (Cormack et al., 1996) from plasmid pEGFP-N1 (Clontech, Palo Alto, CA) into pZAPd, in frame with the authentic start codon of the IRES, resulted in the final construct pZAPd-GFP. All PCRs were performed with Pfu polymerase (Stratagene, La Jolla, CA), and the integrity of the pZAPd-GFP construct was verified by sequencing. The prefix p is omitted when referring to virus derived from plasmid pZAPd-GFP.
Cell culture and virus production
Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum, was used for culture for 293T (DuBridge et al., 1987) and NIH 3T3 (Jainchill et al., 1969) cell lines. NMU rat mammary carcinoma cells (Cohen, 1982) were grown in minimum essential medium supplemented with 10% fetal bovine serum. All cells were grown at 37°C under 5% CO2. The calcium phosphate precipitation method was used to transfect 293T cells with either pZAP2 or pZAPd-GFP for transient production of virus. The conditioned medium was harvested 48 hr posttransfection and filtered through 0.45-μm pore size syringe filters prior to use in transduction assays. All in vitro infections were carried out in the presence of Polybrene (4 μg/ml; Sigma, St. Louis, MO).
Retroviral assays
To determine the kinetics of virus spread in culture, NIH 3T3 cells were transfected with either pZAPd-GFP or pZAP2, using LipofectAMINE 2000 (Life Technologies, Rockville, MD). Reverse transcriptase assays were performed on the culture supernatants as described previously (Telesnitsky et al., 1995), followed by quantitation of reaction products with a Storm PhosphorImager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA). Vector titers were determined by the XC syncytial assay (Rowe et al., 1970) and are expressed as plaque-forming units (PFU) per milliliter.
Single cycle transduction with replicating vector
Virus vector stock at a multiplicity of infection (MOI) of 0.01 or 0.0005 was used to transduce NIH 3T3 cells at 20% confluence. On posttransduction days 3, 5, and 8, the target cells were examined by phase contrast and UV light microscopy using an Olympus IMT-2 inverted microscope (Olympus America, Melville, NY) with a 100-W mercury arc lamp and fluorescein filter cube. In some experiments, the transduced cells were trypsinized at each time point and one-fifth was replated, while the remainder was analyzed by flow cytometry.
Multiple cycle transductions with replicating vector
Virus vector stock at a dilution of 1:1000 was used to transduce NIH 3T3 cells at 20% confluence. On posttransduction day 2, the cells were trypsinized, one-fifth was replated, and the remainder was analyzed for GFP expression by fluorescence-activated cell sorting (FACS) as described below. On posttransduction day 4, the cells were again analyzed by FACS, and the conditioned medium was harvested, diluted 1:100, and used for the next round of transduction on a fresh plate of NIH 3T3 cells. This procedure was repeated for several cycles.
Flow cytometric analysis
NIH 3T3 cells were trypsinized, pelleted, and resuspended in phosphate-buffered saline (PBS). Flow cytometric analysis was performed on a FACScan (Becton Dickinson, Franklin Lakes, NJ), using the FL1 channel to quantitate GFP-expressing cells.
Southern blot analysis
After NheI digestion and agarose gel electrophoresis, 10 μg of genomic DNA from NIH 3T3 cells or NMU cell tumors was transferred onto Hybond-N+ filters (Amersham Pharmacia Biotech, Piscataway, NJ). The blots were hybridized with a random-prime [32P]dCTP-labeled probe for GFP or the 5′ LTR-gag region of MuLV, washed at high stringency, and analyzed by PhosphorImager.
Analysis of vector spread in solid tumors in vivo
Tumors were established by the subcutaneous injection of 2 × 106 NMU rat breast adenocarcinoma cells into the anterior flanks of 8-week-old nu/nu BALB/c mice (Simonsen Laboratories, Gilroy, CA). Four weeks later, the tumors had grown to 1–1.5 cm3, at which time they were injected with 6 × 103 PFU of vector. At regular intervals thereafter, subsets of the mice were humanely sacrificed, and their tumors were surgically removed. A portion of each tumor sample was immediately frozen in liquid nitrogen for later sectioning and immunohistochemical staining. The remaining portion of each tumor was used for FACS analysis, explantation, and isolation of genomic DNA for Southern hybridization. For immunohistochemistry, tumor sections were incubated overnight at 4°C with a 1:8000 dilution of GFP-specific monoclonal antibody (Clontech). Immunoreactivity was visualized with a Vectastain ABC kit (Vector Laboratories, Burlingame, CA) and diaminobenzidine as chromogen. All sections were counterstained with hematoxylin. For FACS analysis and explantation, cell suspensions from tumors were prepared by mincing of the tissue and incubation for 1–2 hr at 37°C on a rocking platform in Hanks’ balanced salt solution containing collagenase type III (100 U/ml) and 3 mM CaCl2. After removal of tissue fragments by passage through a cell strainer, dissociated cells were pelleted by low-speed centrifugation, resuspended in PBS, and either analyzed by FACS as described above or explanted into Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, and grown at 37°C under 5% CO2.
Detection of extratumoral spread of vector
Genomic DNA was extracted at autopsy from spleen, lung, kidney, liver, and heart tissue of ZAPd-GFP-injected animals. Six hundred nanograms of each DNA sample was used in a 50-μl PCR with PCR SuperMix (Life Technologies) and primers for the enhanced GFP. Five microliters of the reaction products was resolved on a 1% agarose gel and visualized by ethidium bromide staining. The detection sensitivity of this assay was determined by amplification of the GFP gene from serially diluted pZAPd-GFP plasmid in the presence of untransduced tissue genomic DNA. As an internal control for the amplification procedure, each DNA sample was used in PCR with primers that amplified a 500-bp target within the mouse β-casein gene. Tissues from the same organs were also dissociated and grown in explant cultures as described above.
RESULTS
Design and production of a unique MuLV-based RCR vector
As the U3–insert construct design utilized in all previously reported MuLV-derived replicative vectors apparently predisposes the vector to rapid loss of the inserted sequences, we hypothesized that insertion of the transgene into a less sensitive position might enhance the stability of the vector. To this end, we inserted a transgene expression cassette, consisting of the 0.6-kb internal ribosome entry site (IRES) from encephalomyocarditis virus (Jang et al., 1988) and the 0.7-kb green fluorescent protein (GFP) marker gene, precisely between the env stop codon and a region of the 3′ untranslated region (UTR) that binds reverse transcriptase during viral DNA synthesis (Wohrl et al., 1995); the resulting construct was designated pZAPd-GFP (Fig. 1A and B).
FIG. 1.

(A) Structure of replication-competent vector ZAPd-GFP, showing site of insertion of IRES–GFP cassette into the wild-type murine leukemia virus genome. (B) Nucleotide sequence of ZAPd-GFP at the junctions between env gene and IRES, IRES and GFP, and GFP and MuLV 3′ untranslated region. Start and stop codons for the env and GFP genes within the junctions are in boldface. (C) In vitro replication kinetics of ZAPd-GFP and wild-type MuLV in NIH 3T3 cells over a 14-day period after transfection with pZAPd-GFP or pZAP2. Reverse transcriptase assays were performed with supernatants collected on the indicated days after transfection. Values represent means obtained from two experiments, and are expressed in arbitrary units. A control transfection was performed without adding vector plasmid (mock).
Vector stocks were prepared by calcium phosphate transfection of 293T cells with pZAPd-GFP. The transfected cells were examined by UV light microscopy 48 hr later and found to brightly express GFP fluorescence (data not shown), confirming that the IRES–GFP transgene cassette was functional. Conditioned medium containing ZAPd-GFP vector particles was harvested from the 293T cells 48–72 hr after transfection. The initial vector stock titer as determined by XC assay ranged between 3 × 103 and 4 × 103 PFU/ml.
The ZAPd-GFP vector replicates efficiently in culture
To determine whether the insert-containing ZAPd-GFP vector was capable of efficient replication in culture, we transiently transfected NIH 3T3 cells with pZAPd-GFP or pZAP2. Every 2 days thereafter for a 2-week period, the transfected cell culture medium was sampled and assayed for reverse transcriptase activity. On the basis of the resulting reverse transcriptase activity profiles, the replication kinetics of the ZAPd-GFP vector appeared to be somewhat attenuated compared with that of wild-type MuLV (Fig. 1C). The titer of ZAPd-GFP on a fully transduced culture of NIH 3T3 cells reached 1.5–3.6 × 105 PFU/ml. In contrast, the titer of the parental wild-type MuLV when propagated on NIH 3T3 cells under the same conditions reached 2.0–3.4 × 106 PFU/ml. This suggests that the presence of the 1.3-kb IRES–transgene insert may have reduced virion production or impaired the replicative ability of the virus by approximately one order of magnitude.
The RCR vector mediates efficient in vitro expression and transmission of the GFP transgene
The reverse transcriptase activity profiles observed per se demonstrate that ZAPd-GFP can replicate but do not indicate whether the vector remained structurally intact and retained the IRES–GFP insert sequence through replication; nor does it indicate whether the transgene would be expressed from the virus. To answer these questions, we transduced NIH 3T3 cells with ZAPd-GFP at an MOI of 0.0005 and examined GFP expression in the culture by UV fluorescence microscopy at regular intervals thereafter (Fig. 2A). On day 3 postinoculation, GFP fluorescence was almost undetectable by microscopy; however, on day 5, GFP expression was detectable in about 10 to 20% of the cell population, and by day 9, almost the entire target cell population exhibited GFP fluorescence (Fig. 2B). Thus an initial inoculum of ZAPd-GFP on the order of 1 PFU per 2000 cells is sufficient to achieve highly efficient gene transfer to an entire cell population in culture.
FIG. 2.
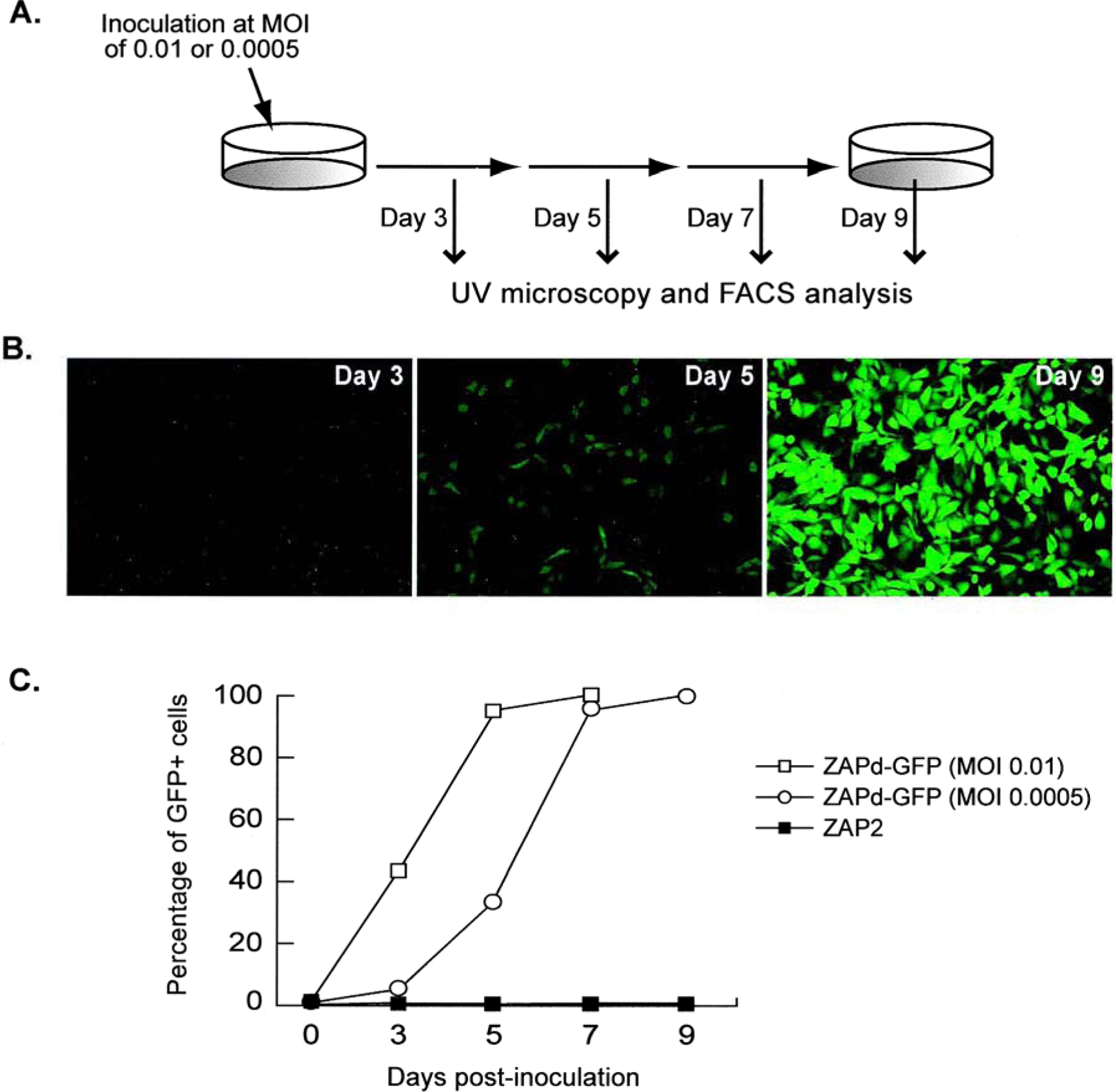
Transmission of GFP transgene through a single culture infected with ZAPd-GFP. (A) Schematic diagram of experimental procedure. Cells were inoculated with ZAPd-GFP at an MOI of 0.01 or 0.0005, and 3, 5, 7, and 9 days later the cells were examined by fluorescence microscopy, passaged, and an aliquot was analyzed by flow cytometry for GFP expression. (B) Appearance of cells at 95–100% confluence by fluorescence microscopy immediately prior to passage after initial inoculation at an MOI of 0.0005 (original magnification, ×100). (C) Results of flow cytometric analysis. y axis, means of three independent experiments; x axis, days postinoculation.
These experiments were repeated using MOIs of 0.01 as well as 0.0005 and the percentage of GFP-expressing cells was quantitated by FACS analysis (Fig. 2C). At an MOI of 0.0005, the FACS results demonstrate a classic lag phase over the first 2–3 days, followed by a logarithmic increase in GFP expression as the ZAPd-GFP vector replicates through the culture, reaching the plateau phase as the entire cell population is transduced (Fig. 2C). The average burst size from a retrovirus-infected cell has been calculated to be on the order of 100 infectious virions (Dimitrov et al., 1993), hence at an MOI of 0.01 (i.e., 1 PFU per 100 target cells) it is expected that a single replication cycle should be sufficient to achieve transduction of the entire culture. Indeed, at an MOI of 0.01, an almost immediate log-phase increase in GFP-positive cells is observed because of the higher initial input of vector particles, and the transduction kinetics are markedly accelerated as indicated by the transduction curve shifting to the left (Fig. 2C).
RCR vector-mediated GFP transmission is stable over multiple replication cycles
As the ability of the ZAPd-GFP vector to successfully transduce target cells during one or two rounds of replication had been established, we then sought to determine whether the RCR vector could stably retain and transmit the inserted transgene sequence through multiple replication cycles. Accordingly, we serially inoculated fresh populations of target cells and monitored spread of the GFP transgene through each culture. After initial inoculation of NIH 3T3 cells with ZAPd-GFP at an MOI of 0.001, the cells were examined by FACS on day 2, replated, and again examined by FACS on day 4 as described above. Prior to trypsinizing the cells for FACS analysis on day 4, the virus-containing culture medium was harvested, and a 100-fold dilution was used to inoculate a fresh population of NIH 3T3 cells. This procedure was repeated 6 additional times, using 100-fold dilutions of conditioned medium for each subsequent cycle, and GFP fluorescence was monitored by FACS on day 2 and day 4 of each cycle (Fig. 3A). Figure 3B shows the composite FACS results from three independent experiments. These results demonstrate that the percentage of GFP-positive cells increased between day 2 and day 4 during each individual replication cycle in a highly consistent and reproducible manner throughout, always reaching nearly 100% by day 4 of each cycle. This indicates that the ZAPd-GFP vector is highly stable in its ability to repeatedly transmit the GFP transgene throughout each fresh population of target cells.
FIG. 3.
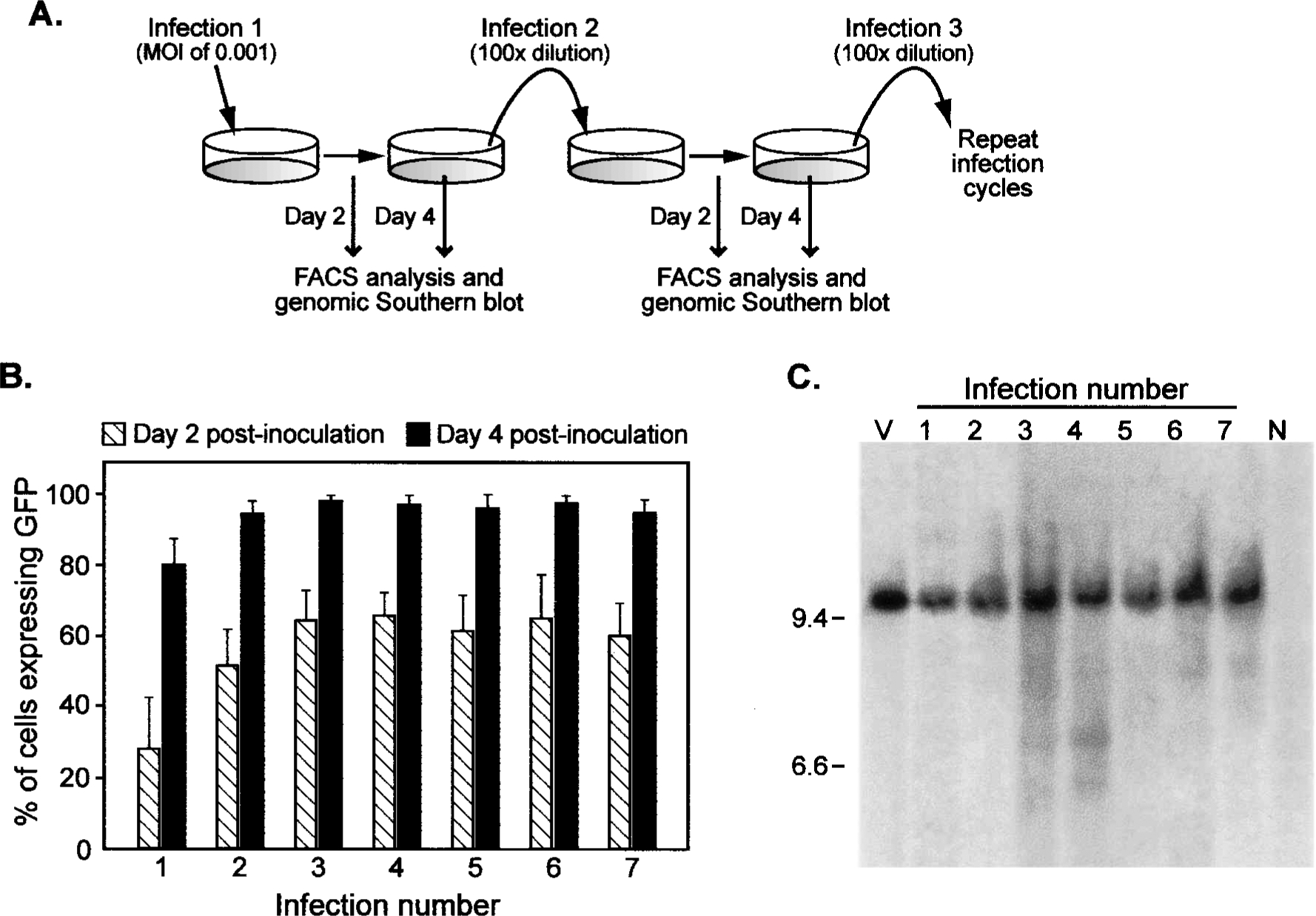
Spread of ZAPd-GFP over multiple serial infections. (A) Diagram depicting the experimental procedure. NIH 3T3 cells were infected with ZAPd-GFP at an MOI of 0.001 (infection number 1). Six subsequent serial infections were conducted in which a 100-fold dilution of supernatant from each infection cycle was used to inoculate the subsequent cell population. Each population of infected cells was examined by flow cytometry and Southern blot hybridization of genomic DNA. (B) Summary of results of flow cytometric analysis. Shown are the percentages of cells expressing GFP in each infected culture 2 and 4 days after the inoculation step of each infection cycle. (C) Southern hybridization analysis of DNA from each of the seven infected cultures. Each DNA sample was digested with NheI prior to blotting and hybridization with the GFP cDNA probe. NheI cuts once in each LTR of the ZAPd-GFP provirus, generating a 9.56-kb fragment containing the transgene. Lanes: V, 30 pg of pZAPd-GFP plasmid DNA; 1–7, genomic DNA from each of the seven infected cultures; N, DNA isolated from mock-infected NIH 3T3 cells.
The full-length RCR vector genome is retained during prolonged replication
To examine the structural stability of the ZAPd-GFP vector genome over the course of multiple replication cycles, and to rule out the possibility that the observed spread of the GFP transgene was primarily due to copackaging of a replication-defective vector retaining the GFP transgene and a replication-competent wild-type revertant lacking the transgene, we harvested genomic DNA from each serially infected NIH 3T3 cell population for Southern blot analysis. The genomic DNA samples were digested with NheI, which cuts once within each LTR and thereby releases the linear provirus, and probed with the GFP transgene sequence. Genomic DNA from all seven serial infection cycles demonstrated the presence of the full-length ZAPd-GFP provirus, predicted to be approximately 9.6 kb (equals approximately 8.3-kb wild-type MuLV genome plus 1.3-kb IRES–GFP transgene insert) (Fig. 3C). The presence of full-length ZAPd-GFP retaining the transgene, and the absence of deletion variants, was also observed by Southern blot analysis of unintegrated proviral DNA probed with the MuLV LTR-gag sequence (data not shown). Faint bands shorter than the full-length vector can be seen in some of the infection cycles, although it is unknown whether these represent deleted forms of the vector or nonspecific hybridization. The predominance of the full-length species in every infection cycle, however, suggests that serial GFP transmission was mediated primarily by the intact RCR vector.
The RCR vector spreads through solid tumors and achieves highly efficient gene transfer in vivo
The ability of ZAPd-GFP to transmit its transgene through multiple serially infected cultures indicated that this vector might be able to spread within solid tumors in vivo and mediate transfer of the transgene into large numbers of tumor cells. To examine the ability of ZAPd-GFP to achieve efficient gene delivery in tumors, we injected the vector into preestablished mammary cancer xenografts in nude mice. Tumors were established by subcutaneous injection of 2 × 106 NMU rat mammary carcinoma cells; after 4 weeks, the tumors had reached volumes of 1.0–1.5 cm3, and were injected with 6 × 103 PFU of ZAPd-GFP (Fig. 4A). Subsets of the mice were sacrificed and their tumors surgically removed 12, 22, 37, and 49 days after injection of the vector. After dissociation of the tumors into single-cell suspensions, the tumor cells were analyzed by flow cytometry for GFP expression (Fig. 4B). The first tumor harvest revealed minimal transduction in three out of four of the tumors examined. One of the four tumors examined, however, exhibited a moderate transduction level, with approximately 8% of its constituent cells expressing the GFP transgene. By day 22, the number of tumor cells infected with the virus had greatly increased. All four of the tumors removed from the mice at this time showed significant levels of infection, averaging approximately one-third of the cell population. Comparable increases in transduction levels occurred by the two subsequent tumor harvests, on the 37th and 49th days after injection of the vector. By the 49th day, the average transduction level was approximately 75%, with some tumors showing transduction levels approaching 100%. Explantation of dissociated tumor tissue and subsequent examination by fluorescence microscopy revealed that host-derived stromal cells within the tumor were also transduced by the vector (data not shown).
FIG. 4.
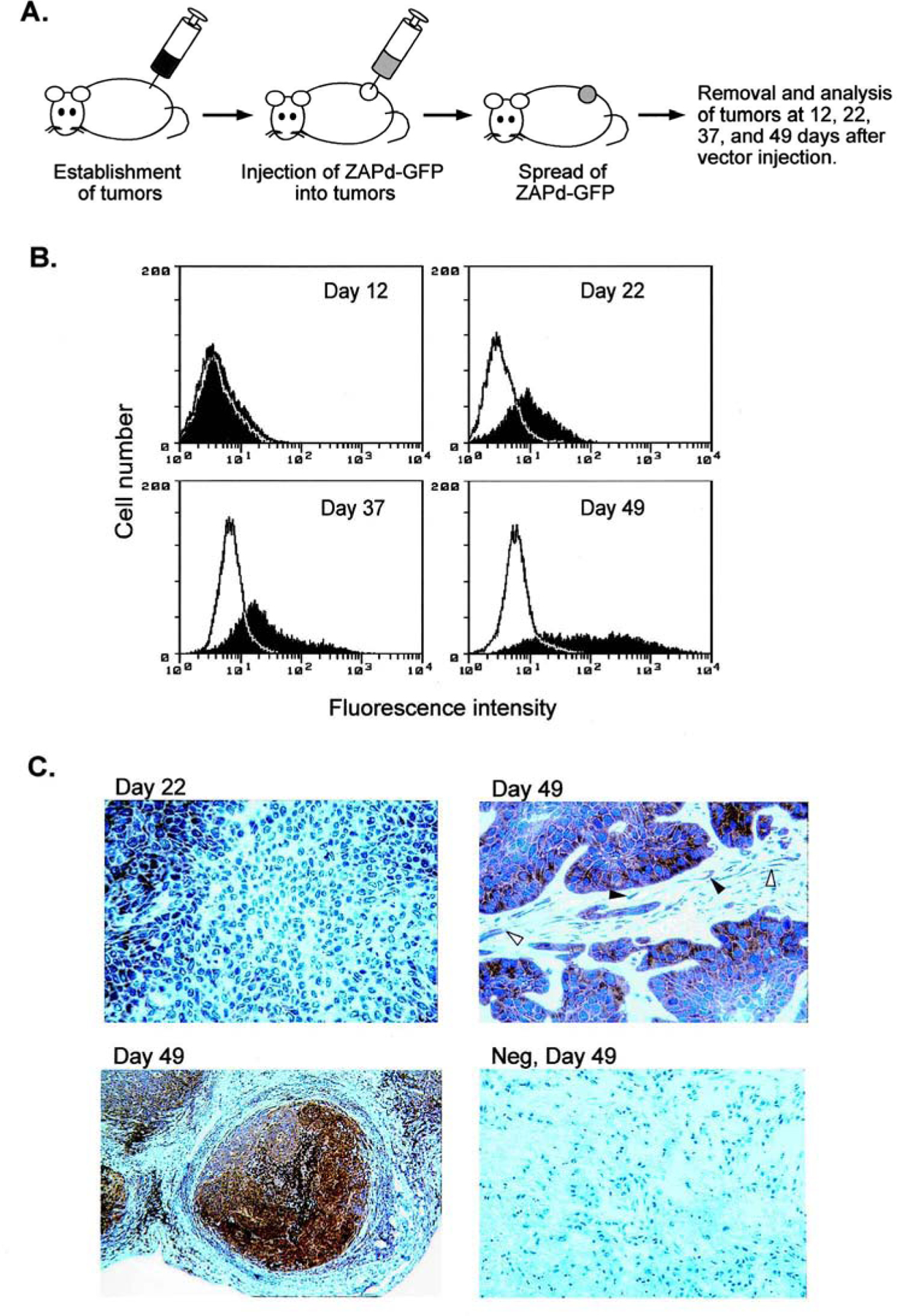
Spread of ZAPd-GFP through solid tumors in mice. (A) Subcutaneous tumors, 1 to 1.5 cm3 in volume, were injected with 6 × 103 PFU of ZAPd-GFP. Twelve, 22, 37, and 49 days after vector injection, tumors were removed from subsets of the mice and were analyzed for virus spread by flow cytometry and Southern blot hybridization. (B) Expression of GFP transgene in tumors. Tumor cells from each of the four time points were dissociated into single-cell suspensions and analyzed for GFP fluorescence by flow cytometry. Shaded histograms represent tumors injected with ZAPd-GFP, and open histograms represent untreated tumors. (C) Immunohistochemical staining of GFP in tumors injected with ZAPd-GFP. Tumors removed 22 and 49 days after vector injection were stained with a monoclonal antibody to GFP and counterstained with hematoxylin. Top left: Tumor removed 22 days after vector injection. Top right and bottom left: Tumors removed at 49 days. Open arrowheads indicate transduced fibroblasts and closed arrowheads indicate transduced endothelial cells. Bottom right: Negative control tumor removed 49 days after vector injection. Each panel represents a different mouse. Original magnification: Top left and bottom right, ×300; top right, ×400; bottom left, ×6.
Tumor tissue taken at the 22- and 49-day time points was also examined by immunohistochemistry with an anti-GFP antibody to confirm expression of GFP within the tumors. While tumors removed at the earlier time point revealed patchy staining for GFP, with clusters of transduced cells adjacent to clusters of untransduced cells (Fig. 4C, top left), low-magnification views of tumor tissue taken at the later time point demonstrate highly efficient transduction throughout the tumor mass (Fig. 4C, bottom left). The latter samples show intense staining in almost every tumor cell, as well as distinct staining in fibroblasts and some endothelial cells (Fig. 4C, top right).
The full-length RCR vector genome is retained during prolonged replication in vivo
To confirm that integrated vector provirus was present in the tumor cells and that the GFP transgene had been transmitted as part of the intact vector, we performed Southern analysis on genomic DNA from tumors removed 12 and 37 days after vector injection. The only bands detected by the GFP probe were the size expected for full-length ZAPd-GFP (Fig. 5A), demonstrating the presence of integrated vector and that the high-level transduction shown by FACS analysis and immunohistochemistry, was the result of the transmission of the GFP transgene by intact ZAPd-GFP. The MuLV LTR-gag probe also hybridized to the full-length ZAPd-GFP band, as well as to a smaller band that is likely to represent an endogenous provirus present in the NMU cell line or the BALB/c genome, as the same band is present in genomic DNA from the untransduced negative control tumors (Fig. 5B). These results suggest that the transmission of the GFP transgene through the tumors during the course of the 37 days was mediated primarily or exclusively by the intact vector, and that deletion variants, if present, occurred only at levels undetectable in our analysis.
FIG. 5.
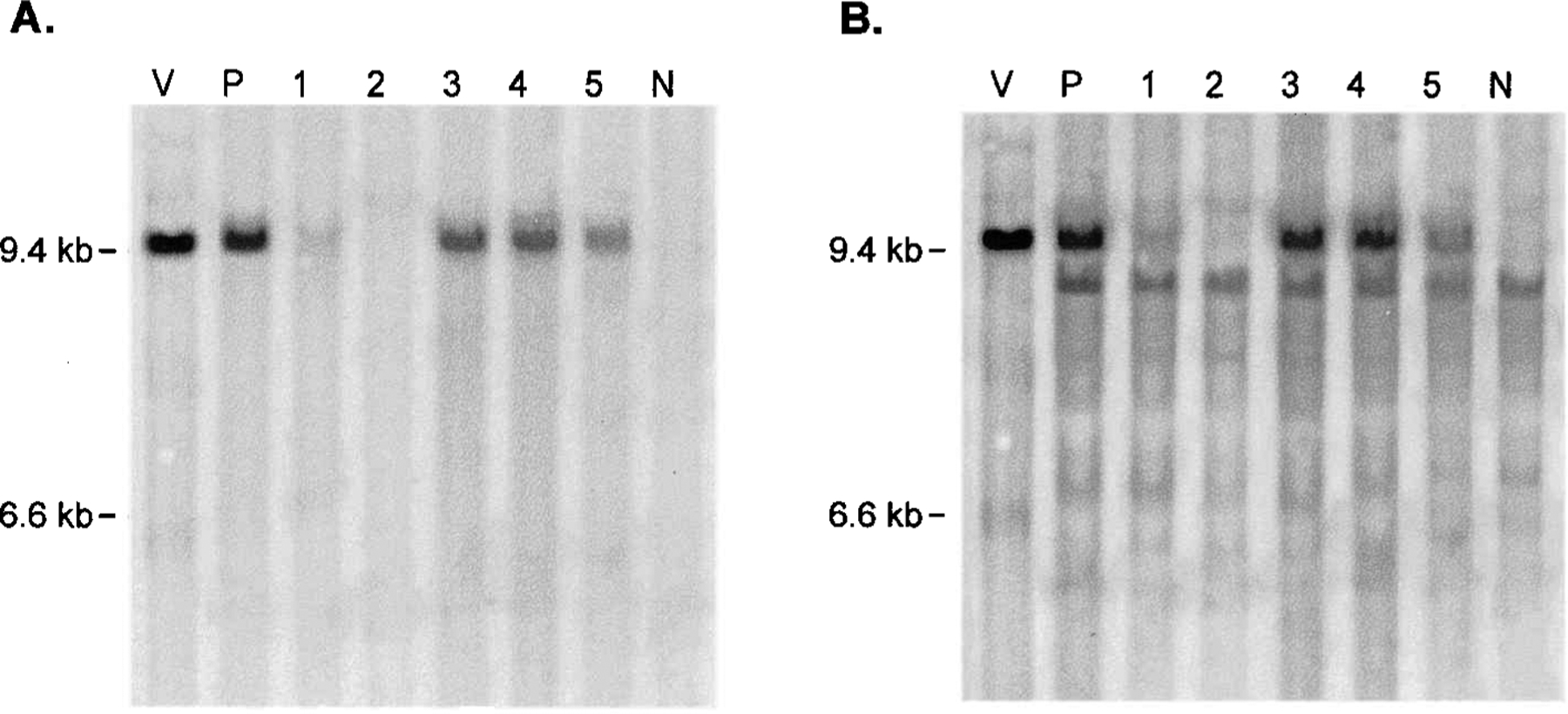
Southern blot analysis of genomic DNA from tumors injected with ZAPd-GFP. Ten micrograms of DNA from each tumor was digested to completion with NheI. After blotting onto a nylon membrane, the fragments were hybridized to a random-primed radiolabeled probe for the GFP transgene or for the MuLV LTR-gag sequence. Lanes: V, 30 pg of pZAPd-GFP plasmid DNA digested with NheI; P, DNA from tumor originating from cells that were infected with ZAPd-GFP prior to their injection into mouse; 1 and 2, DNA from tumors removed 12 days after vector injection; 3, 4, and 5, DNA from tumors removed 37 days after vector injection; N, negative control tumor injected with virus-free supernatant. (A) GFP-probed blot; (B) MuLV LTR-gag-probed blot. Top band, full-length ZAPd-GFP; bottom band, nonspecific hybridization signal (also present in negative control tumor).
Extratumoral RCR vector spread is not at detectable levels under present conditions
To detect any spread of ZAPd-GFP outside the tumors that might have occurred, a variety of extratumoral tissues including spleen, lung, kidney, liver, and heart were harvested at the time of autopsy. High molecular weight DNA was harvested from each of these tissues and was used, along with DNA extracted from tumors, in PCR with primers specific for the GFP transgene. Amplification of serial dilutions of pZAPd-GFP plasmid demonstrated that this assay could detect as few as 140 copies of GFP in a background of approximately 100,000 equivalents (600 ng) of untransduced genomic DNA, representing a transduction level of about 0.14%. Figure 6 shows the results of PCR using samples taken from mice sacrificed 49 days after vector injections. DNA from the tumor injected with vector revealed the presence of the full-length GFP transgene, but none of the nontumor tissues or the mock-treated tumor sample exhibited amplification. This unexpected result was also obtained on examination of tissues harvested from animals killed at earlier time points (data not shown). Flow cytometric analysis of the same tissues 1 week after explanting also revealed the presence of the GFP transgene only in tumors (data not shown). These results suggest that spread of the vector originating from a total initial inoculum of 6 × 103 PFU was minimal over the time course of these experiments.
FIG. 6.
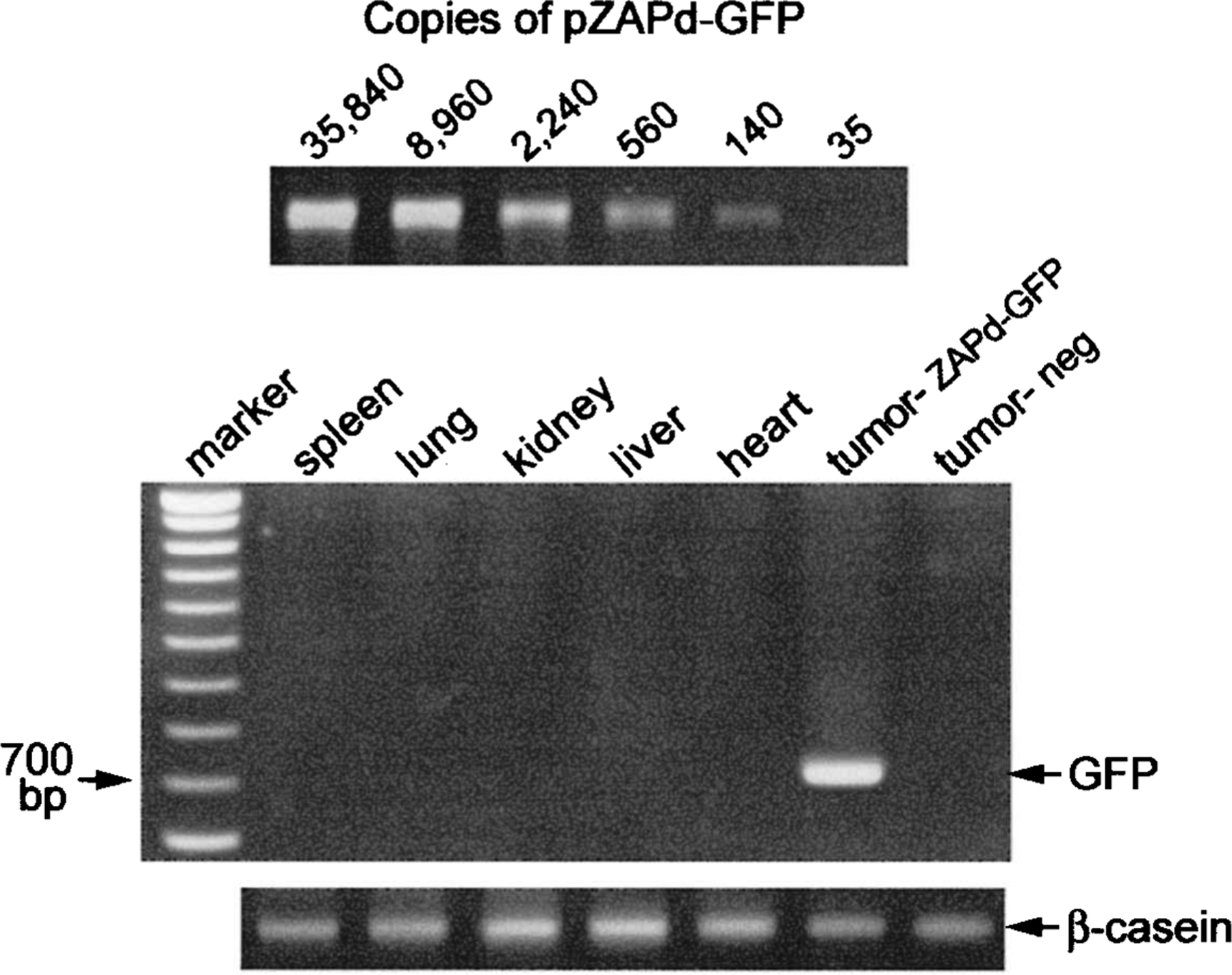
Analysis of extratumoral spread of ZAPd-GFP by PCR amplification of GFP transgene from genomic DNA. The sensitivity of the assay was determined by amplification, using 4-fold serial dilutions of pZAPd-GFP as template in the presence of untransduced genomic DNA (top). Six hundred nanograms of DNA, extracted from tumors and various extratumoral tissues at the time of autopsy, was used in PCR analysis for the GFP transgene (middle). Shown are results with tissues taken from mice 49 days after injection of tumors with vector. Expected size of full-length amplification product is 730 base pairs. A 500-bp region of the mouse β-casein gene was amplified from the same samples as an internal control (bottom).
DISCUSSION
This study demonstrates that the insertion of exogenous sequences precisely between the env gene and 3′ UTR of MuLV results in a fully replication-competent vector that exhibits a high degree of genomic stability, compared with previously reported MuLV-based RCR vectors that contained inserts within the U3 region of the LTR. We chose this particular insert position because (1) the packaging signal extends past the start codon of the gag gene, and thus positioning a transgene just upstream of the gag gene would likely impair packaging efficiency; (2) the gag and pol coding sequences are initially translated as a single polypeptide that is then cleaved, thus positioning a transgene between these coding sequences might interfere with proteolytic processing; (3) the 3′ end of pol overlaps with the 5′ end of env, precluding the insertion of a transgene between these genes; and (4) positioning the transgene, preceded by an IRES, just downstream of env would allow the transgene to be expressed from both the spliced env transcript as well as the unspliced genomic transcript. While the majority of previously reported U3–insert MuLV vectors, whose inserts were 100–1100 bp smaller than that of ZAPd-GFP, began losing their inserted sequences during the first replication cycle (Lobel et al., 1985; Reik et al., 1985; Stuhlmann et al., 1989; Coulombe et al., 1996), ZAPd-GFP was stable through continuous propagation over multiple replication cycles in culture, and furthermore, this vector efficiently replicated within solid tumors in vivo, leading to widespread delivery of the transgene at efficiencies significantly higher than those typically achieved using standard replication-defective retroviral vectors at much higher doses.
Most previous studies using standard replication-defective MuLV vectors for tumor transduction in vivo reported efficiencies of less than 10%, despite the use of large doses of vector or vector packaging cells, vector injection at multiple time points, or simultaneous injection of tumor and packaging cells (Short et al., 1990; Yang et al., 1996; Kruse et al., 1997; Ram et al., 1997; Smiley et al., 1997). The low in vivo transduction efficiency of defective retroviral vectors has been attributed to the inability of the vector particles to diffuse from the site of injection. The results of clinical trials of retroviral gene transfer into brain tumors, in which intratumoral injection of packaging cells resulted in transduction of cells only within a few cell diameters of the injection tracts, lend support to this notion (Ram et al., 1997; Rainov, 2000). Similar results were observed in studies involving direct intratumoral injection of defective adenovirus vectors (de Roos et al., 2000; Kurihara et al., 2000).
In contrast, the present study suggests that the capacity of replication-competent vectors for intercellular spread, which allows transduction of cells not initially exposed to the vector inoculum, largely circumvents such physical obstacles. Furthermore, since MuLV can infect only mitotically active cells, and the half-life of virion particles (5–8 hr) (Morgan et al., 1995; Chuck et al., 1996) is much shorter than the average cell cycle time of most human tumors (3–6 days), continuous release of replicating vector from initially infected cells also increases the likelihood that additional target cells will undergo division and thereby become infected.
The replication competence of the vector described here obviously raises questions about possible pathogenic effects resulting from spread of the vector in the host. Moloney MuLV is known to induce thymic lymphoma in newborn mice, and many other murine retroviruses are associated with characteristic malignancies. However, most of these viruses are not pathogenic in adult mice (Rosenberg and Jolicoeur, 1997). Nonpathogenic strains of MuLV have also been described, and these may be amenable for use in the construction of replicating vectors similar to those described here. Initial studies of the activity of amphotropic MuLV in rhesus monkeys could find no evidence of pathology in infected animals over a 3-year observation period, despite severe immune suppression at the time of infection and the administration of high doses (mean, 7.2 × 107 PFU) of replication-competent MuLV (Cornetta et al., 1990, 1991b). A later study, however, revealed that MuLV can be oncogenic in primates under certain conditions, on the basis of the observation that 3 of 10 rhesus monkey recipients of bone marrow cells infected with replication-competent MuLV developed T cell lymphoma (Donahue et al., 1992; Anderson et al., 1993). These results suggest that while MuLV has oncogenic potential in primates, the presence of a normally functioning immune system is sufficient to prevent the realization of this potential. As the mice used in the present study were athymic, the influence of a fully functional immune system on the spread of ZAPd-GFP has yet to be determined. The tumor microenvironment itself is known to be inherently immunosuppressive; therefore it is possible that RCR vector spread would be facilitated within the tumor even in immunocompetent hosts, while extratumoral spread would be restricted by the immune system.
The RCR vector was observed to transduce host-derived cells within the tumors, including fibroblasts and endothelial cells, apparently as efficiently as the tumor cells themselves. In some situations, for example, for angiogenesis inhibition strategies, achieving gene delivery to the nontumor cell compartment of tumors may be advantageous. These cells are more homogeneous in nature and lack the propensity of tumor cells to mutate rapidly; hence, such cells may be less likely to develop resistance to introduced therapeutic genes.
Nevertheless, the desirability of a mechanism to control RCR vector spread is underscored by the above observation that the vector efficiently transduced host-derived cells within the tumors. This was not unexpected, since Moloney MuLV is tropic for murine cells, and in its present form the vector employed in these studies is untargeted, except for its natural selectivity for proliferating cells. However, the finding that spread of this untargeted vector appeared to be confined to the tumor tissue was somewhat unexpected. Since the assay used to detect spread of the vector outside of the tumors, however, was based on amplification of the GFP transgene, the possibility remains that deletion variants of the vector lacking the transgene sequence were present in the tissues examined. Such deletion variants were not apparent by Southern blot analysis of DNA from the tumor tissue itself. The lack of detectable levels of extratumoral spread may be a consequence of the dilution of any vector that left the tumor, combined with the relative inability of MuLV to infect nonproliferating normal tissues.
As mentioned above, incorporating a suicide gene into RCR vectors would not only provide the means to kill transduced tumor cells, but would also serve as a self-limiting safety mechanism to terminate vector spread after adequate levels of transduction are achieved. An additional means of targeting spread of a replicating vector, and thereby minimizing risk to recipients, would be to engineer the virus to replicate only within particular cell subpopulations. One strategy that has been used to direct retroviral vector tropism is through the modification of the viral envelope protein to target the entry of vectors into cells expressing specific cell-surface proteins (Russell et al., 1993; Kasahara et al., 1994; Valsesia-Wittmann et al., 1994; Snitkovsky and Young, 1998). Tight control of retroviral tropism has also been achieved by replacement of the retroviral promoter/enhancer with cell type-specific transcriptional control elements, to target expression to particular tissues (Diaz et al., 1998; Jager et al., 1999). The development of tissue-targeted RCR vectors would represent a significant improvement in vector technology for gene therapy of cancer.
ACKNOWLEDGMENTS
We thank Paula Cannon for critical review of the manuscript, and Peng-Xuan Liu and Erlinda M. Gordon for technical assistance. This work was supported by GTI/Novartis (Gaithersburg, MD), the Department of Defense Breast Cancer Research Program grant BC980554, and NIH grant R01 CA85908. C.R.L. is the recipient of a Breast Cancer Research Project Fellowship through the Norris Comprehensive Cancer Center, and C.-K.T. is the recipient of a Susan G. Komen Breast Cancer Foundation predoctoral fellowship.
REFERENCES
- ALEMANY R, BALAGUE C, and CURIEL DT (2000). Replicative adenoviruses for cancer therapy. Nat. Biotechnol 18, 723–727. [DOI] [PubMed] [Google Scholar]
- ANDERSON WF (1984). Prospects for human gene therapy. Science 226, 401–409. [DOI] [PubMed] [Google Scholar]
- ANDERSON WF, MCGARRITY GJ, and MOEN RC (1993). Report to the NIH Recombinant DNA Advisory Committee on murine replication-competent retrovirus (RCR) assays (February 17, 1993). Hum. Gene Ther 4, 311–321. [DOI] [PubMed] [Google Scholar]
- BISCHOFF JR, KIRN DH, WILLIAMS A, HEISE C, HORN S, MUNA M, NG L, NYE JA, SAMPSON-JOHANNES A, FATTAEY A, and MCCORMICK F (1996). An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274, 373–376. [DOI] [PubMed] [Google Scholar]
- CHUCK AS, CLARKE MF, and PALSSON BO (1996). Retroviral infection is limited by Brownian motion. Hum. Gene Ther 7, 1527–1534. [DOI] [PubMed] [Google Scholar]
- COFFEY MC, STRONG JE, FORSYTH PA, and LEE PW (1998). Reovirus therapy of tumors with activated Ras pathway. Science 282, 1332–1334. [DOI] [PubMed] [Google Scholar]
- COHEN LA (1982). Isolation and characterization of a serially cultivated, neoplastic, epithelial cell line from the N-nitrosomethylurea induced rat mammary adenocarcinoma. In Vitro 18, 565–575. [DOI] [PubMed] [Google Scholar]
- CORMACK BP, VALDIVIA RH, and FALKOW S (1996). FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173, 33–38. [DOI] [PubMed] [Google Scholar]
- CORNETTA K, MOEN RC, CULVER K, MORGAN RA, MCLACHLIN JR, STURM S, SELEGUE J, LONDON W, BLAESE RM, and ANDERSON WF (1990). Amphotropic murine leukemia retrovirus is not an acute pathogen for primates. Hum. Gene Ther 1, 15–30. [DOI] [PubMed] [Google Scholar]
- CORNETTA K, MORGAN RA, and ANDERSON WF (1991a). Safety issues related to retroviral-mediated gene transfer in humans. Hum. Gene Ther 2, 5–14. [DOI] [PubMed] [Google Scholar]
- CORNETTA K, MORGAN RA, GILLIO A, STURM S, BALTRUCKI L, O’REILLY R, and ANDERSON WF (1991b). No retroviremia or pathology in long-term follow-up of monkeys exposed to a murine amphotropic retrovirus. Hum. Gene Ther 2, 215–219. [DOI] [PubMed] [Google Scholar]
- COULOMBE J, AVIS Y, and GRAY DA (1996). A replication-competent promoter-trap retrovirus. J. Virol 70, 6810–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE ROOS WK, DE WILT JH, VAN DER KAADEN ME, MANUSAMA ER, DE VRIES MW, BOUT A, TEN HAGEN TL, VALERIO D, and EGGERMONT AM (2000). Isolated limb perfusion for local gene delivery: Efficient and targeted adenovirus-mediated gene transfer into soft tissue sarcomas. Ann. Surg 232, 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIAZ RM, EISEN T, HART IR, and VILE RG (1998). Exchange of viral promoter/enhancer elements with heterologous regulatory sequences generates targeted hybrid long terminal repeat vectors for gene therapy of melanoma. J. Virol 72, 789–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DILLON PJ, LENZ J, and ROSEN CA (1991). Construction of a replication-competent murine retrovirus vector expressing the human immunodeficiency virus type 1 tat transactivator protein. J. Virol 65, 4490–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIMITROV DS, WILLEY RL, SATO H, CHANG LJ, BLUMENTHAL R, and MARTIN MA (1993). Quantitation of human immunodeficiency virus type 1 infection kinetics. J. Virol 67, 2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONAHUE RE, KESSLER SW, BODINE D, MCDONAGH K, DUNBAR C, GOODMAN S, AGRICOLA B, BYRNE E, RAFFELD M, MOEN R, BACHER J, ZSEBO KM, and NIENHUIS AN (1992). Helper virus induced T cell lymphoma in non-human primates after retroviral mediated gene transfer. J. Exp. Med 176, 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUBRIDGE RB, TANG P, HSIA HC, LEONG PM, MILLER JH, and CALOS MP (1987). Analysis of mutation in human cells by using an Epstein–Barr virus shuttle system. Mol. Cell. Biol 7, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOFF S, TRAKTMAN P, and BALTIMORE D (1981). Isolation and properties of Moloney murine leukemia virus mutants: Use of a rapid assay for release of virion reverse transcriptase. J. Virol 38, 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEISE C, HERMISTON T, JOHNSON L, BROOKS G, SAMPSON-JOHANNES A, WILLIAMS A, HAWKINS L, and KIRN D (2000). An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med 6, 1134–1139. [DOI] [PubMed] [Google Scholar]
- HORTON RM, HUNT HD, HO SN, PULLEN JK, and PEASE LR (1989). Engineering hybrid genes without the use of restriction enzymes: Gene splicing by overlap extension. Gene 77, 61–68. [DOI] [PubMed] [Google Scholar]
- JAGER U, ZHAO Y, and PORTER CD (1999). Endothelial cell-specific transcriptional targeting from a hybrid long terminal repeat retrovirus vector containing human prepro-endothelin-1 promoter sequences. J. Virol 73, 9702–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAINCHILL JL, AARONSON SA, and TODARO GJ (1969). Murine sarcoma and leukemia viruses: Assay using clonal lines of contact-inhibited mouse cells. J. Virol 4, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANG SK, KRAUSSLICH HG, NICKLIN MJ, DUKE GM, PALMENBERG AC, and WIMMER E (1988). A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol 62, 2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JUNKER U, BOHNLEIN E, and VERES G (1995). Genetic instability of a MoMLV-based antisense double-copy retroviral vector designed for HIV-1 gene therapy. Gene Ther. 2, 639–646. [PubMed] [Google Scholar]
- KASAHARA N, DOZY AM, and KAN YW (1994). Tissue-specific targeting of retroviral vectors through ligand–receptor interactions. Science 266, 1373–1376. [DOI] [PubMed] [Google Scholar]
- KRUSE CA, ROPER MD, KLEINSCHMIDT-DEMASTERS BK, BANUELOS SJ, SMILEY WR, ROBBINS JM, and BURROWS FJ (1997). Purified herpes simplex thymidine kinase Retrovector particles. I. In vitro characterization, in situ transduction efficiency, and histopathological analyses of gene therapy-treated brain tumors. Cancer Gene Ther. 4, 118–128. [PubMed] [Google Scholar]
- KURIHARA T, BROUGH DE, KOVESDI I, and KUFE DW (2000). Selectivity of a replication-competent adenovirus for human breast carcinoma cells expressing the MUC1 antigen. J. Clin. Invest 106, 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOBEL LI, PATEL M, KING W, NGUYEN-HUU MC, and GOFF SP (1985). Construction and recovery of viable retroviral genomes carrying a bacterial suppressor transfer RNA gene. Science 228, 329–332. [DOI] [PubMed] [Google Scholar]
- LORENCE RM, ROOD PA, and KELLEY KW (1988). New-castle disease virus as an antineoplastic agent: Induction of tumor necrosis factor-alpha and augmentation of its cytotoxicity. J. Natl. Cancer Inst 80, 1305–1312. [DOI] [PubMed] [Google Scholar]
- MAVILIO F, FERRARI G, ROSSINI S, NOBILI N, BONINI C, CASORATI G, TRAVERSARI C, and BORDIGNON C (1994). Peripheral blood lymphocytes as target cells of retroviral vector-mediated gene transfer. Blood 83, 1988–1997. [PubMed] [Google Scholar]
- MILLER DG, ADAM MA, and MILLER AD (1990). Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection [published erratum appears in Mol. Cell. Biol. 1992;12:433]. Mol. Cell. Biol 10, 4239–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORGAN JR, LEDOUX JM, SNOW RG, TOMPKINS RG, and YARMUSH ML (1995). Retrovirus infection: Effect of time and target cell number. J. Virol 69, 6994–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NORMAN KL, and LEE PW (2000). Reovirus as a novel oncolytic agent. J. Clin. Invest 105, 1035–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAWLIK TM, NAKAMURA H, YOON SS, MULLEN JT, CHANDRASEKHAR S, CHIOCCA EA, and TANABE KK (2000). Oncolysis of diffuse hepatocellular carcinoma by intravascular administration of a replication-competent, genetically engineered herpesvirus. Cancer Res. 60, 2790–2795. [PubMed] [Google Scholar]
- RAINOV NG (2000). A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther 11, 2389–2401. [DOI] [PubMed] [Google Scholar]
- RAM Z, CULVER KW, OSHIRO EM, VIOLA JJ, DEVROOM HL, OTTO E, LONG Z, CHIANG Y, MCGARRITY GJ, MUUL LM, KATZ D, BLAESE RM, and OLDFIELD EH (1997). Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat. Med 3, 1354–1361. [DOI] [PubMed] [Google Scholar]
- REIK W, WEIHER H, and JAENISCH R (1985). Replication-competent Moloney murine leukemia virus carrying a bacterial suppressor tRNA gene: Selective cloning of proviral and flanking host sequences. Proc. Natl. Acad. Sci. U.S.A 82, 1141–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBBINS PD, TAHARA H, and GHIVIZZANI SC (1998). Viral vectors for gene therapy. Trends Biotechnol. 16, 35–40. [DOI] [PubMed] [Google Scholar]
- ROSENBERG N, and JOLICOEUR P (1997). Retroviral pathogenesis. In Retroviruses. Coffin JM, Hughes SH, and Varmus H, eds. (Cold Spring Harbor Laboratory Press, Plainview, NY: ) pp. 475–585. [PubMed] [Google Scholar]
- ROWE WP, PUGH WE, and HARTLEY JW (1970). Plaque assay techniques for murine leukemia viruses. Virology 42, 1136–1139. [DOI] [PubMed] [Google Scholar]
- RUSSELL SJ (1994). Replicating vectors for gene therapy of cancer: Risks, limitations and prospects. Eur. J. Cancer 8, 1165–1171. [DOI] [PubMed] [Google Scholar]
- RUSSELL SJ, HAWKINS RE, and WINTER G (1993). Retroviral vectors displaying functional antibody fragments. Nucleic Acids Res. 21, 1081–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHOEMAKER C, HOFFMAN J, GOFF SP, and BALTIMORE D (1981). Intramolecular integration within Moloney murine leukemia virus DNA. J. Virol 40, 164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHORT MP, CHOI BC, LEE JK, MALICK A, BREAKEFIELD XO, and MARTUZA RL (1990). Gene delivery to glioma cells in rat brain by grafting of a retrovirus packaging cell line. J. Neurosci. Res 27, 427–439. [DOI] [PubMed] [Google Scholar]
- SINKOVICS JG, and HORVATH JC (2000). Newcastle disease virus (NDV): Brief history of its oncolytic strains. J. Clin. Virol 16, 1–15. [DOI] [PubMed] [Google Scholar]
- SMILEY WR, LAUBERT B, HOWARD BD, IBANEZ C, FONG TC, SUMMERS WS, and BURROWS FJ (1997). Establishment of parameters for optimal transduction efficiency and antitumor effects with purified high-titer HSV-TK retroviral vector in established solid tumors. Hum. Gene Ther 8, 965–977. [DOI] [PubMed] [Google Scholar]
- SMITH AE (1999). Gene therapy—where are we? Lancet 354(Suppl. 1), SI1–SI4. [PubMed] [Google Scholar]
- SMITH E, REDMAN RA, LOGG CR, COETZEE GA, KASAHARA N, and FRENKEL B (2000). Glucocorticoids inhibit developmental stage-specific osteoblast cell cycle. Dissociation of cyclin A-cyclin-dependent kinase 2 from E2F4–p130 complexes. J. Biol. Chem 275, 19992–20001. [DOI] [PubMed] [Google Scholar]
- SNITKOVSKY S, and YOUNG JA (1998). Cell-specific viral targeting mediated by a soluble retroviral receptor–ligand fusion protein. Proc. Natl. Acad. Sci. U.S.A 95, 7063–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STUHLMANN H, JAENISCH R, and MULLIGAN RC (1989). Construction and properties of replication-competent murine retroviral vectors encoding methotrexate resistance. Mol. Cell. Biol 9, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TELESNITSKY A, BLAIN S, and GOFF SP (1995). Assays for retroviral reverse transcriptase. Methods Enzymol. 262, 347–362. [DOI] [PubMed] [Google Scholar]
- VALSESIA-WITTMANN S, DRYNDA A, DELEAGE G, AUMAILLEY M, HEARD JM, DANOS O, VERDIER G, and COSSET FL (1994). Modifications in the binding domain of avian retrovirus envelope protein to redirect the host range of retroviral vectors. J. Virol 68, 4609–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VILE RG, SUNASSEE K, and DIAZ RM (1998). Strategies for achieving multiple layers of selectivity in gene therapy. Mol. Med. Today 4, 84–92. [DOI] [PubMed] [Google Scholar]
- VILE RG, RUSSELL SJ, and LEMOINE NR (2000). Cancer gene therapy: Hard lessons and new courses. Gene Ther. 7, 2–8. [DOI] [PubMed] [Google Scholar]
- WALKER JR, MCGEAGH KG, SUNDARESAN P, JORGENSEN TJ, RABKIN SD, and MARTUZA RL (1999). Local and systemic therapy of human prostate adenocarcinoma with the conditionally replicating herpes simplex virus vector G207. Hum. Gene Ther 10, 2237–2243. [DOI] [PubMed] [Google Scholar]
- WILDNER O, MORRIS JC, VAHANIAN NN, FORD H JR., RAMSEY WJ, and BLAESE RM (1999). Adenoviral vectors capable of replication improve the efficacy of HSVtk/GCV suicide gene therapy of cancer. Gene Ther. 6, 57–62. [DOI] [PubMed] [Google Scholar]
- WOHRL BM, GEORGIADIS MM, TELESNITSKY A, HENDRICKSON WA, and LE GRICE SF (1995). Footprint analysis of replicating murine leukemia virus reverse transcriptase. Science 267, 96–99. [DOI] [PubMed] [Google Scholar]
- YANG L, HWANG R, PANDIT L, GORDON EM, ANDERSON WF, and PAREKH D (1996). Gene therapy of metastatic pancreas cancer with intraperitoneal injections of concentrated retroviral herpes simplex thymidine kinase vector supernatant and ganciclovir. Ann. Surg 224, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]