

Abstract
Identification of tumor-specific mutations, called neoantigens, offers new exciting opportunities for personalized cancer immunotherapy. However, it remains challenging to achieve robust induction of neoantigen-specific T cells and drive their infiltration into the tumor microenvironment (TME). Here, we have developed a novel polyethyleneimine (PEI)-based personalized vaccine platform carrying neoantigen peptides and CpG adjuvants in a compact nanoparticle (NP) for their spatio-temporally concerted delivery. The NP vaccine significantly enhanced activation and antigen cross-presentation of dendritic cells, resulting in strong priming of neoantigen-specific CD8+ T cells with the frequency in the systemic circulation reaching as high as 23 ± 7% after a single subcutaneous administration. However, activated CD8+ T cells in circulation exhibited limited tumor infiltration, leading to poor anti-tumor efficacy. Notably, local administration of stimulator of interferon genes (STING) agonist promoted tumor infiltration of vaccine-primed CD8+ T cells, thereby overcoming one of the major challenges in achieving strong anti-tumor efficacy with cancer vaccination. The NP vaccination combined with STING agonist therapy eliminated tumors in murine models of MC-38 colon carcinoma and B16F10 melanoma and established long-term immunological memory. Our approach provides a novel therapeutic strategy based on combination nano-immunotherapy for personalized cancer immunotherapy.
Keywords: cancer vaccine, neoantigen, STING, tumor infiltrating lymphocytes
1. Introduction
Genomic sequencing of malignant cells has revealed the presence of mutations unique to cancer cells [1–3]. Some of these mutational genomic sequences are transcribed and translated into proteins that expose otherwise immunologically stealth cancer cells to the immune system. These proteins or peptides, called neoantigens, have recently garnered significant research interest, and many clinical studies are underway worldwide to examine the therapeutic potential of neoantigen-based cancer vaccination [4–6]. Exome sequencing of tumor genome, followed by algorithmic selection, enables the identification of neoantigen candidates, which are then constructed as peptides or mRNAs for personalized cancer vaccination [4, 7]. Also, with the recent progresses in nanotechnology, nanoparticle-based vaccines have been employed to elicit neoantigen-specific T cell responses [8–10]. However, many neoantigen vaccine delivery platforms induce suboptimal immune activation [11], and even if vaccination generates sufficient anti-tumor CD8+ T cells in the systemic circulation, intratumoral trafficking of CD8+ T cells is often limited [12], thus resulting in poor anti-tumor effects. Thus, new approaches are needed to promote the induction of neoantigen-specific T cells and increase their tumor infiltration.
Notably, tumor infiltration of CD8+ T cells is correlated with patient prognosis, as shown by recent retrospective analyses of clinical cases [13–16]. Also, tumors with sparse T cell infiltration are less responsive to immune checkpoint blockade (ICB) therapies designed to reinvigorate tumor-infiltrating lymphocytes [17]. Such immunologically “cold” tumors have various immunosuppressive mechanisms to exclude or reprogram immune cells, thus shaping the tumor microenvironment (TME) to evade the immune surveillance [18, 19]. While chemotherapy [20, 21], oncolytic virus [22], and anti-angiogenic agents [23] have been applied to reverse immunosuppression and convert non-immunogenic “cold” tumors into T cell-inflamed “hot” tumors, it remains to be seen how to combine them effectively with cancer vaccination.
Here, to overcome these urgent challenges, we have developed a facile personalized nanoparticle vaccine using polyethyleneimine (PEI) that offers versatile functionality for modular incorporation of neoantigens and simple electrostatic assembly of CpG adjuvant. The facile conjugation chemistry and self-assembly allows the incorporation of a variety of antigen peptides into compact nano-sized particles together with CpG for efficient co-delivery of neoantigens and adjuvants while preserving their immunological activities. As a result, the nanoparticle (NP) vaccine significantly enhanced cellular uptake of antigens and adjuvant molecules to promote activation and antigen cross-presentation of dendritic cells (DCs), resulting in robust priming of neoantigen-specific CD8+ T cell. However, despite high frequency of anti-tumor CD8+ T cells in the systemic circulation, we observed weak anti-tumor effect due to limited intratumoral trafficking of activated CD8+ T cells. STING is a pattern recognition receptor (PRR) that triggers NF-κB and IRF3 pathways, leading to strong type 1 IFN responses [24], and recent studies have reported promising therapeutic efficacy of STING agonists for cancer treatment [25, 26]. Here, we have demonstrated that intratumoral administration of STING agonist induced robust secretion of cytokines and chemokines that promoted T cell trafficking into the TME. When combined with NP vaccination, treatment with STING agonist led to strong recruitment of vaccine-primed, neoantigen-specific CD8+ T cells into the TME, leading to significantly improved anti-tumor efficacy, compared to either treatment alone. Overall, we have developed a PEI-based nanovaccine platform that elicits robust neoantigen-specific cytotoxic T cells with simple and facile incorporation of neoantigens and adjuvants and demonstrated a novel combination therapy using STING agonist for promoting tumor infiltration of neoantigen-specific cytotoxic T cells. These studies present a new combination nano-immunotherapy for inducing neoantigen-specific T cells and their tumor infiltration for achieving potent anti-tumor efficacy.
2. Materials and Methods
Synthesis of PEI conjugates and NP Vacc.
Conjugated polymer between PEI and PEG (PEI-PEG) was prepared using branched PEI (Sigma-Aldrich, molecular weight ~25,000) and methoxy poly(ethyleneglycol) propionic acid N-hydroxysuccinimide (Nanocs, molecular weight 5000) as reported previously [27]. Antigen peptides employed in this study include SIINFEKL, CSSSIINFEKL, ASMTNMELM (Adpgk), CSSASMTNMELM (CSS-Adpgk), and LCPGNKYEM (M27). All antigen peptides were obtained from Genemed Synthesis. For PEI conjugates (PEI-PEG/CSS-antigen or PEI-PEG/M27), PEI-PEG was dissolved in DMSO and added with 3-(2-Pyridyldithio)propionic acid N-hydroxysuccinimide ester (SPDP, Sigma-Aldrich) to create thiol-reactive disulfide bond. After stirring for 3h, the mixture was reacted with CSS-antigen overnight, followed by dialysis-purification using Amicon ultra 10 kDa MW cutoff centrifugal filters. The resulting PEI conjugate was vigorously mixed with CpG (CpG 1826, Integrated DNA Technology) in PBS at varying PEI conjugate/CpG weight ratio of 0.5, 1, 2, or 3 to form respective NP Vacc by CpG nano-complexation.
Animal studies.
Animals were cared for following federal, state and local guidelines. All experiments performed on animals were in accordance with and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan, Ann Arbor. 5–6 weeks old female C57BL/6 mice were purchased from Jackson Laboratories. Tumor cells (6×105 MC-38 cells per mouse (1.2×106 for re-challenge study) and 3×105 B16F10 cells per mouse) were injected s.c. on the right flank of each mouse. Vaccines were administered s.c. at the tail-base on indicated days. Four days after the vaccination, CDA (dissolved in PBS; 0.5 μg for the MC-38 model and 5 μg for the B16F10 model) was injected into tumors on the indicated days. Tumor sizes were measured using a caliper.
In vitro BMDC studies.
BMDCs were collected from hind femurs of C57BL/6 mice, cultured with media supplemented with GM-CSF for 8–10 days as previously reported [28]. For the analysis of cytokine secretion and proliferation by NP Vacc, immature BMDCs were treated with the free PEI conjugate containing CSS-modified Adpgk (3 μg/ml), free CpG (1 μg/ml), or NP Vacc formulated at different weight ratios of PEI conjugate/CpG (1 μg/ml CpG and the respective amount of PEI conjugate). Cells were washed after 2 h, supplemented with fresh media, and further incubated for 24 h for the detection of IL-12p70 in cell culture media using ELISA, while BMDC proliferation was measured using a cell counting kit (CCK-8; Dojindo Molecular Technologies, Inc.).
For studying CpG uptake by BMDCs, 5’ phosphate group of CpG was tethered with ethylenediamine and subsequently labeled with Alexa Fluor® 647 NHS Ester (AF647-NHS, Invitrogen) as described before [27]. In parallel, PEI conjugates were labeled with Alexa Fluor® 488 NHS Ester (AF488-NHS, Invitrogen). BMDCs were then incubated with soluble formulation or NP Vacc at dose of 10 μg/ml CpG-AF467 and 20 μg/ml PEI conjugates-AF488 (equivalent of 10 μg/ml for free Adpgk). At the indicated time points, cells were collected, washed with FACS buffer (1% BSA in PBS), and then subjected to flow cytometry for measuring fluorescence signals. To visualize cellular localization, BMDCs were grown onto 12 mm glass coverslips in 24 well plates at a density of 5 × 105 cells/well and treated with samples as above for 24 h. Cells were further incubated with Hoechst 33342 (5 g/ml, Invitrogen) and Lysotracker Red DND-99 (100 nM, Invitrogen) for 30 min for the staining of nuclei and endolysosomes, respectively. Then, cells were fixed with 4% formaldehyde in PBS and mounted on slide glass using ProLong™ Diamond Antifade Mountant (Invitrogen) for confocal microscopy (Nikon A1Rsi).
For assessing activation and antigen cross-presentation, NP Vacc were constructed using PEI conjugate containing CSS-modified SIINFEKL and CpG. BMDCs were incubated for 24 h with either soluble or NP vaccine formulation containing SIINFEKL and CpG (2 μg/ml SIINFEKL and 1 μg/ml CpG). Cells were then stained with antibody-fluorophore conjugates including CD40-APC (Invitrogen), CD86-PE/Cy7 (BD Biosciences), and SIINFEKL/H-2kb-PE (Invitrogen) for 30 min at room temperature. Cells were washed with FACS buffer and analyzed using flow cytometry.
For the analysis of cytokine secretion by BMDCs in response to CDA treatment, BMDCs were seeded at a density of 105 cells/well in a 96-well tissue culture plate and incubated overnight at 37 °C with 5% CO2. Cells were washed with PBS twice, and then added with 5 μg of CDA in 200 μl of fresh culture media. Culture media were retrieved after 6 hrs for ELISA assay.
Analyses of CD8+ T cells and cytokines in blood and tumor.
Blood was collected at the indicated time points by submandibular bleeding, treated with ACS lysing buffer (Gibco), and washed with PBS to obtain PBMCs. For TME analysis, tumor tissues were excised from tumor-bearing mice, cut into small pieces (1–2 mm) with scissors, and treated with 1 mg/ml of collagenase A (Sigma-Aldrich) and 100 IU of DNase I (Sigma-Aldrich) for 30 min at 37 °C with continuous shaking. Samples were placed on top of 40 μm strainers and mashed through with a plunger, followed by centrifugation at 1000 g for 5 min. The cell pellet was resuspended and washed twice with PBS by centrifuging at 1500 g for 3 min. PBMCs and tumor cells were stained with a live/dead staining dye (eBioscience) and fluorophore-labeled antibodies including CD3-FITC (Biolegend), CD8-APC (BD Biosciences), and Adpgk tetramer-PE (NIH Tetramer Core Facility), fixed with 2% formaldehyde, and then suspended in FACS buffer for flow cytometry. Blood sera and tumor tissue supernatants were separately collected for detection of cytokines using ELISA. For immunohistochemistry of tumor tissues, portions of tumor tissues were embedded in optimal cutting temperature compound (OCT compound; Tissue Tek) and frozen in −80 °C. Tissue sections were prepared using a cryostat (Thermo Scientific Cryostar™ NX50). The sections were placed on surface-treated glass slides (Superfrost Plus; Fisher), dried for 30 min in room temperature, fixed with acetone at −20 °C for 10 min, and washed and hydrated with Tris buffered saline (TBS). Samples were then treated with blocking buffer (TBS containing 10% goat serum and 1% bovine serum albumin) for 1 hr at room temperature, followed by staining with rat anti-mouse CD8 antibody for overnight at 4 °C. After washing the samples twice with TBS, secondary antibody (goat anti-rat IgG-AF647) was added and incubated for 1 hr at room temperature. Next, the samples were washed twice with TBS and stained with DAPI for 10 min at room temperature. Coverslips were mounted on top of the samples using a mount medium (Prolong Diamond Antifade; Fisher Scientific). Samples analyzed with a confocal microscope.
Statistical analysis.
Statistical analysis was performed with Prism 7.0e (GraphPad Software). Statistical comparisons were performed using one-way ANOVA followed by Tukey’s HSD multiple comparison post hoc test or two-way ANOVA followed by Sidak’s multiple comparisons test, as indicated in the figure legends. Animal survival was analyzed by the log-rank (Mantel–Cox) test. Statistical significances are indicated as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
3. Results and Discussion
Synthesis and characterization of neoantigen vaccine nanoparticles (NP Vacc)
Specifically, we prepared a personalized NP vaccine based on PEI by taking advantage of its versatile functionality for chemical conjugation and electrostatic complexation. PEI was grafted with PEG for enhancing colloidal stability and biocompatibility [29–31], followed by conjugation with CSS-modified neoantigen peptides via reduction sensitive disulfide bond. The conjugate formed by conjugation between PEI, PEG, and CSS-modified neoantigen is hereafter denoted as “PEI conjugate”. Because of the strong cationic characteristic of PEI, the simple mixture of PEI conjugates with anionic CpG formed a nanoscale condensate by electrostatic complexation. (Figure 1a). CpG was employed as an adjuvant not only because of its strong anionic property but also for its potent immunostimulatory property [32].
Figure 1. Synthesis and characterization of NP Vacc.
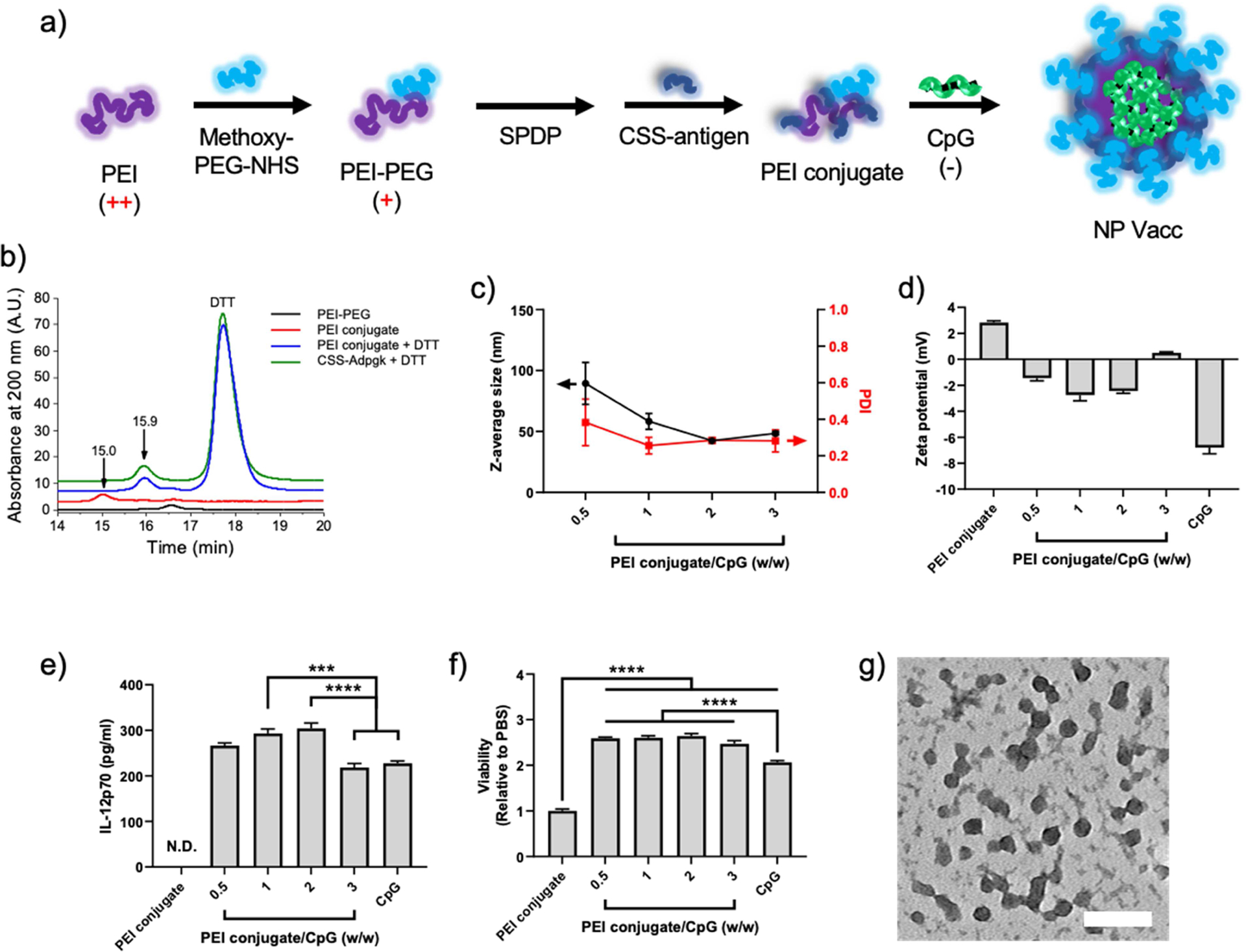
a) Schematic drawing of PEI nanoparticle vaccine formation. PEI-PEG was conjugated with CSS-antigen via disulfide linkage (PEI conjugate) and then condensated with CpG by electrostatic complexation to form NP Vacc. b) GPC chromatograms, indicating the formation of PEI conjugate. c) Hydrodynamic size and d) surface charge of NP Vacc synthesized in different PEI conjugate:CpG weight ratios, as measured by DLS and zeta potential. e) IL-12p70 secretion and f) relative proliferation of BMDCs after incubation with PEI conjugate, CpG, or PEI NPs. g) NP Vacc composed of a PEI conjugate:CpG weight ratio of 2:1 were imaged using TEM after 2% uranyl acetate staining. Scale bar = 100 nm. Data are presented as mean ± SEM. ***p < 0.001, ****p < 0.0001, analyzed by one-way ANOVA, followed by Tukey’s HSD multiple comparison post hoc test.
PEG was conjugated to PEI by the reaction between methoxy poly(ethyleneglycol) propionic acid N-hydroxysuccinimide (methoxy-PEG-NHS) with the primary amine of PEI, resulting in a conjugated polymer between PEI and PEG (PEI-PEG). The stoichiometry PEI:PEG was controlled to 1:15, which was reported to diminish cytotoxicity of PEI [29, 30]. The conjugation was confirmed by quantification of free amine groups using 2,4,6-trinitrobenzene sulfonic acid (data not shown). To prepare a personalized NP vaccine, we employed Adpgk, a neo-epitope identified in MC-38 mouse colon adenocarcinoma [33]. PEI-PEG was incubated with 3-(2-pyridyldithio)propionic acid N-hydroxysuccinimide ester (SPDP) at room temperature (RT) for 3 hr for amine-to-sulfhydryl cross-linking, followed by overnight incubation with CSS-Adpgk to form the PEI conjugate via a reducible disulfide bond. The resulting PEI conjugate was analyzed by gel permeation chromatography (GPC). As shown in Figure 1B, PEI conjugate had a major elution peak at 15 min; however, this particular peak was absent in PEI-PEG, suggesting successful conjugation of Adpgk onto PEI-PEG. A brief treatment (~5 min) with dithiothreitol (DTT) delayed the elution of the PEI conjugate by ~0.9 min to be overlapped with the peak of free CSS-Adpgk + DTT sample (Figure 1B), indicating efficient release of CSS-Adpgk from the PEI conjugate in a reduction sensitive manner.
Incubation of the PEI conjugate with CpG led to the formation of nanoparticles (hereafter denoted as “NP Vacc”). Dynamic light scattering (DLS) measurement showed that the hydrodynamic size of NP Vacc decreased with an increasing feed amount of CpG, resulting in 40 – 50 nm Z-average size and 0.2 – 0.3 polydispersity index with the PEI conjugate/CpG weight ratio of > 1 (Figure 1c). In addition, the zeta potential values of NP Vacc were between those of free PEI conjugates and free CpG with almost neutral charges (Figure 1d), thus suggesting PEI/CpG charge compensation and PEG surface passivation. To assess the impact of complexation on immune activation by CpG, bone marrow-derived dendritic cells (BMDCs) were incubated with NP Vacc, and IL-12p70 secretion and cellular viability were measured. In particular, IL-12p70, a biologically active form of IL-12, is an important pro-inflammatory cytokine that plays an essential role in CD4 T helper-1 response and peptide antigen priming of naïve CD8+ T cells, which is critical for immunological performance and anti-tumor efficacy of cancer vaccine [34, 35]. NP Vacc formulated at the PEI-conjugate/CpG weight ratio of 1 or 2 promoted higher amount of IL-12p70 and proliferation of BMDCs, compared to PBS and free CpG (Figure 1e,f). In contrast, PEI conjugate alone did not induce the secretion of IL-12p70 nor proliferation of BMDCs, which indicated the crucial role of CpG in immune activation by NP Vacc [36]. NP Vacc formulated at the PEI conjugate/CpG weight ratio of 2 exhibited relatively uniform size of 20 – 30 nm under transmission electron microscope (Figure 1g), which correlated well with the DLS measurements. Based on these results, NP Vacc formulated at the PEI conjugate/CpG weight ratio of 2 was chosen for the subsequent studies.
NP Vacc enhance DC activation and antigen presentation
Next, we investigated the impact of NP Vacc on DC uptake and subsequent activation and antigen presentation. Fluorophore-labeled CpG was formulated into NP Vacc for fluorescence-based analysis of NP uptake by BMDCs. NP Vacc promoted the uptake of CpG as early as after 2 h of incubation and showed > 30-fold increase over 72 h, compared to free CpG (Figure 2a). The presence of Adpgk or CSS-Adpgk peptide admixed with CpG did not affect the uptake of CpG, showing comparable levels with free CpG treatment alone. We further examined cellular uptake using confocal microscopy and confirmed that BMDCs incubated with NP Vacc displayed brighter CpG fluorescence within the endo-lysosomal compartments, compared with BMDCs incubated with free CpG (Figure 2b). Enhanced endo-lysosomal CpG delivery can potentiate engagement and activation of TLR9 receptors, which are located within the endo-lysosomes [37]. We also investigated maturation and antigen presentation of BMDCs by employing SIINFEKL peptide, a MHC-I-restricted epitope of ovalbumin protein. BMDCs were incubated with NP Vacc consisting of PEI-PEG/CSS-SIINFEKL conjugates or control soluble formulations, followed by flow cytometric analysis. NP Vacc significantly increased the expression of co-stimulatory markers, including CD40 and CD86, on BMDCs (Figure 2c). Next, cells were stained with 25-D1.16 monoclonal antibody that recognizes SIINFEKL peptide complexed with H-2Kb MHC-I molecule (H-2Kb-SIINFEKL). Admixture of soluble SIINFEKL + CpG increased the expression of H-2Kb-SIINFEKL within 8 h, which rapidly declined to the baseline level after 24 h (Figure 2d). In contrast, NP Vacc significantly increased antigen presentation after 24 h, as shown by highly elevated levels of H-2Kb-SIINFEKL, compared to SIINFEKL + CpG (p < 0.0001, Figure 2d). Thus, despite the slow kinetics, NP Vacc significantly increased the cumulative extent of antigen presentation, compared with soluble formulations. SIINFEKL + CpG and CSS-SIINFEKL + CpG induced comparable stimulation and antigen presentation of BMDCs, indicating that N-terminal CSS modification did not alter the immunological property of SIINFEKL peptide. Taken together, these results show that NP Vacc promote cellular uptake of antigen and CpG, leading to enhanced DC activation, maturation, and antigen presentation, compared with their free admixture.
Figure 2. NP Vacc promotes cellular uptake of antigen and CpG by DCs and improves DC maturation and antigen cross-presentation.
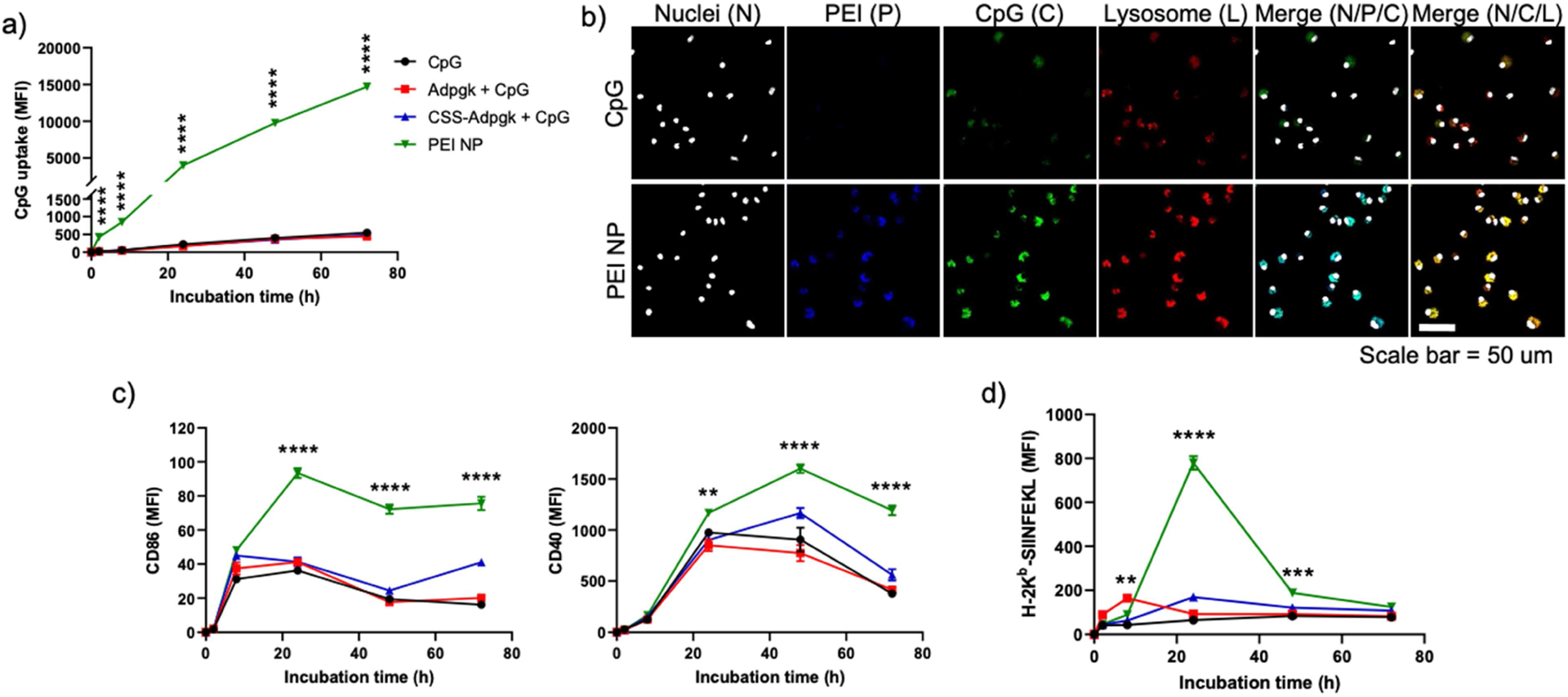
a-b) BMDCs were incubated in vitro with fluorophore-labeled CpG in the formulations of CpG, Adpgk + CpG, CSS-Adpgk + CpG, or corresponding NP VACC, and fluorescence signals were measured using a) flow cytometry and b) confocal microscopy (scale bar = 50 μm). c-d) BMDCs were incubated with CpG, SIINFEKL + CpG, CSS-SIINFEKL + CpG, or NP Vacc, and c) DC activation and d) antigen presentation were measured by staining cells with c) anti-CD86 and anti-CD40 antibodies or d) anti-H-2Kb-SIINFEKL antibody, respectively, followed by flow cytometry. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, analyzed by two-way ANOVA, followed by Tukey’s HSD multiple comparison post hoc test. Asterisks represent comparison between NP Vacc vs. Adpgk + CpG in a), and between NP Vacc vs. SIINFEKL + CpG in c) and d).
NP Vacc elicit systemic CD8+ T cell responses but fail to control tumor growth
Next, we examined the immunogenicity and anti-tumor efficacy of NP Vacc in vivo. C57BL/6 mice were injected subcutaneously (s.c.) with 6×105 MC-38 mouse colon carcinoma cells on the right flank on day 0. On day 7 when tumors were palpable, mice were vaccinated with various doses of NP Vacc composed of PEI-PEG/CSS-Adpgk conjugates and CpG (NP Vacc) via s.c. injection at tail base (Figure 3a). We set 10 μg Adpgk peptide and 15 μg CpG as 1x dose according to our previous report [8] and conducted a dose-sparing study using lower doses of 0.1x and 0.5x. Soluble vaccines composed of free Adpgk and CpG (Sol Vacc) at the corresponding doses were used as control groups. To assess the priming of antigen-specific CD8+ T cells, peripheral blood mononuclear cells (PBMCs) were collected 7 days after the vaccination, and the frequencies of Adpgk-specific CD8+ T cells were measured by the tetramer assay. NP Vacc induced a dose-dependent increase in the frequency of Adpgk-specific CD8+ T cells in peripheral blood (Figure 3b). Notably, a single vaccination with NP Vacc at 1x dose (containing 10 μg of Adpgk peptide + 15 μg CpG) promoted clonal expansion of Adpgk-specific CD8+ cells as high as 23 ± 6.9% among all CD8+ T cells in PBMCs, representing 3-fold enhancement compared with 1x dose of Sol Vacc (p < 0.05, Figure 3b). In addition, whereas Sol Vacc increased the systemic levels of IL-6, IL-12p70, and TNF-α in serum and caused acute body weight loss, NP Vaccine group did not show any sign of systemic toxicity (Fig. S1), potentially by reducing the systemic exposure of free CpG. However, despite this significant expansion of neoantigen-specific CD8+ T cells in the systemic circulation, we did not observe meaningful anti-tumor effect for any of the vaccine groups (Figure 3c). Since successful cancer immunotherapy requires sufficient infiltration of anti-tumor T cells into the TME [38], we analyzed the TME for the frequency of CD8+ T cells. The total number of tumor-infiltrating CD8+ T cells in the TME was similar for all the treatment groups (Figure 3d). Although NP Vacc induced a slight increase in the number of Adpgk-specific CD8+ T cells, compared with PBS (Figure 3e), there was no statistical difference between the NP Vacc and Sol Vacc groups. Taken together, NP Vacc effectively expanded tumor-specific CD8+ T cells in the systemic compartment, but their anti-tumor efficacy was limited, potentially due to poor tumor infiltration of activated T cells.
Figure 3. NP Vacc elicits tumor-specific CD8+ T cell responses in the systemic compartment but fails to inhibit MC-38 tumor growth.
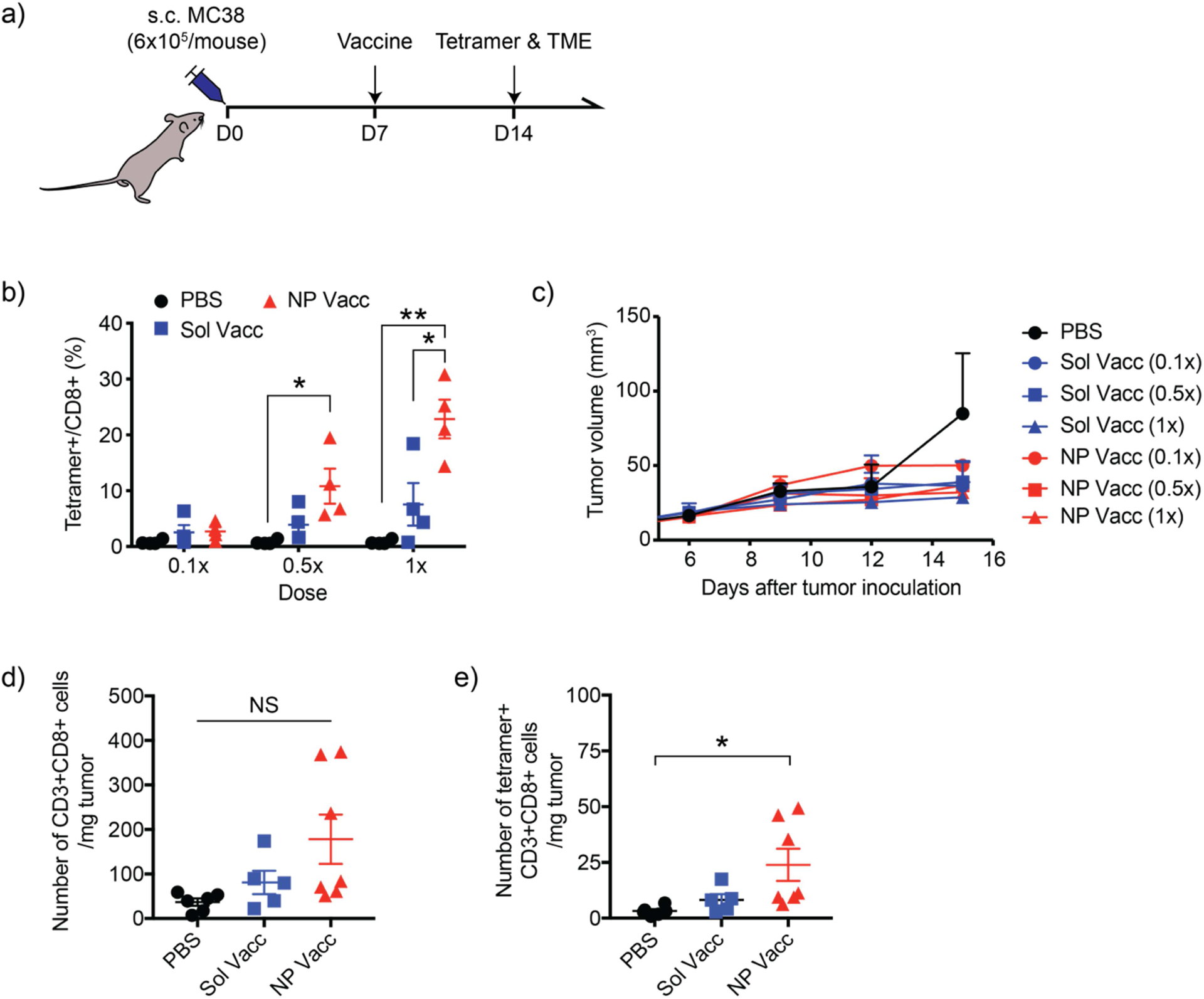
a) Timeline of experiment. b) C57BL/6 mice were inoculated at s.c. flank with 6×105 MC-38 colon carcinoma cells on day 0 and vaccinated at s.c. tail base on day 7 with increasing doses of vaccines. Neoantigen-specific CD8+ T cell levels in blood circulation were quantified on day 14 by tetramer staining and flow cytometry. c) Shown are the average MC-38 tumor growth curves. d-e) Numbers of tumor-infiltrating d) CD3+CD8+ T cells and e) tetramer+CD3+CD8+ T cells were measured on day 14. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ****p < 0.0001, analyzed by one-way ANOVA, followed by Tukey’s HSD multiple comparison post hoc test.
Combination of NP Vacc and STING agonist recruits T cells into tumors and eliminates MC-38 tumors.
STING has been reported to promote tumor infiltration of peripheral T cells through the type I interferon (IFN) pathway [39, 40]. Based on these studies, we investigated whether STING agonist can recruit vaccine-primed circulating CD8+ T cells into tumors. First, we examined whether cyclic-di-adenosine monophosphate (CDA), a potent STING agonist, can induce secretion of chemokines essential for recruitment of T cells. BMDCs treated with CDA in vitro produced increased levels of CXCL10, CCL2, CCL3, and CCL5 (Fig. S2). When CDA was administered directly into MC-38 tumors in vivo, this led to increased serum concentrations of IFN-β, CXCL10, CCL2, and CCL5 in a dose-dependent manner (Fig. S3a). Intratumoral (i.t.) administration of CDA at the dose range of 1–20 μg was well tolerated, and mice did not show any abnormal change in body weight (Fig. S3b).
Next, we investigated the effects of i.t. administration of CDA on vaccine-primed CD8+ T cells. C57BL/6 mice were inoculated with 6×105 MC-38 cells at s.c. flank. On day 7 when tumors were palpable, mice were s.c. vaccinated with NP Vacc, Sol Vacc, or PBS, followed by i.t. injection of CDA on day 11 at the priming phase of CD8+ cells [41] (Figure 4a). Analyses of PBMCs on day 14 for the frequency of tumor-specific CD8+ T cells indicated that CDA monotherapy induced weak anti-tumor CD8+ T cells in the systemic circulation (Figure 4b). Additional i.t. administration of CDA followed by s.c. NP Vacc or Sol Vacc slightly increased the mean frequency of vaccine-primed tumor-specific CD8+ T cells in circulation, but the increases were not statistically significant (Figure 4b). In stark contrast, NP Vacc + CDA potently increased the number of CD3+CD8+ T cells in the TME (Figure 4c) and achieved 10.9-fold (p < 0.0001), 3.6-fold (p < 0.001), and 3.7-fold (p < 0.001) higher numbers of intratumoral neoantigen-specific CD8+ T cells, compared with CDA alone, NP Vacc alone, or Sol Vacc + CDA, respectively (Figure 4d). On the other hand, Sol Vacc + CDA had a minor effect on the level of CD8+ T cells in the TME, inducing only slight increase from the PBS control (Figure 4c–d), probably due to weaker priming of CD8+ T cells by Sol Vacc. Immunohistochemistry also showed an increased number of CD8+ T cells infiltrating MC38 tumors for the NP Vacc + CDA treatment group (Figure 4e). Moreover, NP Vacc + CDA significantly elevated the intratumoral concentration of IFN-γ (Figure 4f), which is a pleiotropic cytokine involved in cellular migration and effector functions of T cells during tumor rejection [42–46]. On the other hand, the numbers of CD4+ T cells and B cells within the tumors were not significantly different among the treatment groups (Fig. S4), which indicate that NP Vacc + CDA combination treatment induced a CD8+ T cell-driven immune response. We also monitored the effect of Vacc + CDA combination therapy on tumor growth and survival of MC-38-bearing mice. MC-38 tumor bearing mice were vaccinated with either NP Vacc or Sol Vacc on day 7, followed by i.t. administration of CDA on days 11, 14, and 17 (Figure 4g). Sol Vacc + CDA slowed the average tumor growth, but the majority of animals succumbed to tumor growth by day 50 with 32% survival rate (Figure 4h,i). Importantly, NP Vacc + CDA exhibited remarkable anti-tumor efficacy, leading to robust tumor regression and complete tumor elimination in ~70% of animals (Figure 4h,i). In contrast, NP Vacc or CDA administered as a single agent showed only minor impact on the tumor growth suppression and survival rate. In addition, 100% of the survivors from the NP Vacc + CDA group were resistant to tumor re-challenge with 1.2×106 MC-38 cells injected in the contralateral s.c. flank on day 90 (Figure 4j), showing long-term immunological memory response. Overall, these results demonstrate that CDA promotes tumor trafficking of antigen-specific CD8+ T cells primed by NP Vacc, leading to potent anti-tumor efficacy and long-term memory response against tumor relapse.
Figure 4. Combination of NP Vacc and STING agonist elicits tumor-specific CD8+ T cells in peripheral blood and the TME, leading to robust anti-tumor efficacy.
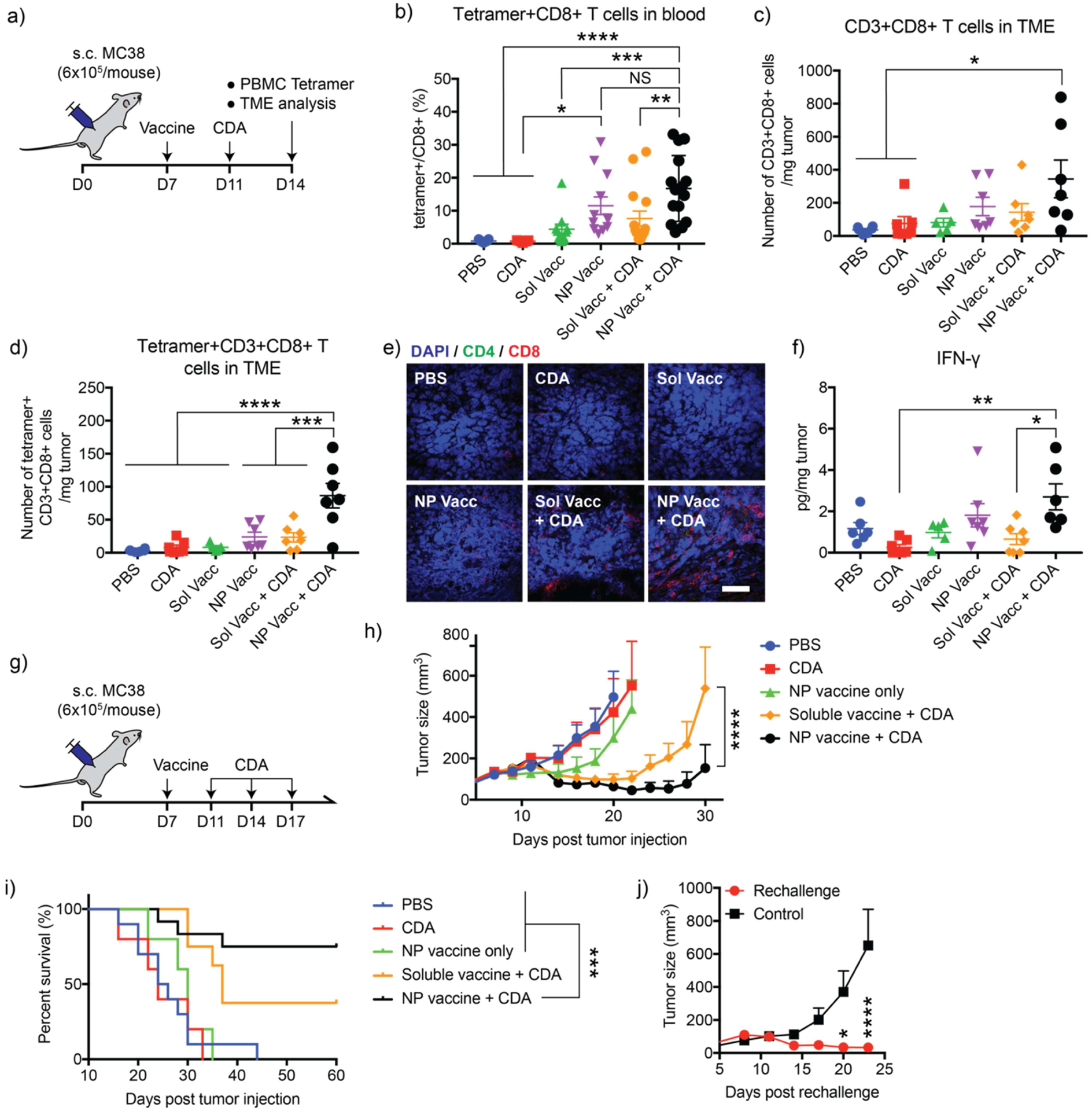
a) Timeline of experiment. C57BL/6 mice were inoculated at s.c. flank with 6×105 MC-38 colon carcinoma cells on day 0 and vaccinated at s.c. tail base on day 7 with the indicated vaccines. A subset of animals also received intratumoral administration of 0.5 g CDA on day 11. On day 14, animals were analyzed for b) Adpgk-tetramer+CD8+ T cells within PBMCs, c) tumor-infiltrating CD3+CD8+ T cells, and d) tumor-infiltrating Adpgk-tetramer+CD3+CD8+ T cells by flow cytometry. e) Immunohistochemistry images showing T cells infiltrating MC38 tumors. Scale bar = 50 μm. f) Concentrations of IFN-γ were measured in tumors using ELISA. g) Timeline of experiment. C57BL/6 mice inoculated at s.c. flank with 6×105 MC-38 colon carcinoma cells on day 0 were vaccinated on day 7 via s.c. tail base. These mice received intratumoral administration of 0.5 g CDA on days 11, 14, and 17. Shown are h) the average tumor growth curves and i) animal survival. j) Tumor growth curve after re-challenging survivors with MC-38 cells. Mice were re-challenged with 1.2×106 MC-38 cells by subcutaneous injection on the left-side flanks 90 days after the initial tumor inoculation. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, analyzed by b,c,d,f) one-way ANOVA followed by Tukey’s HSD multiple comparison post hoc test or h,j) two-way ANOVA followed by Sidak’s multiple comparisons test. i) The survival curves were analyzed by the log-rank (Mantel–Cox) test.
Combination of NP Vacc and STING agonist eliminates poorly immunogenic B16F10 tumors.
Lastly, we sought to demonstrate the therapeutic potential and generality of NP Vacc + CDA combination therapy using B16F10 melanoma model, which is highly aggressive and resistant to conventional immunotherapies [47, 48]. M27 peptide, a MHC class I-restricted neoantigen identified in B16F10 cells [49], was utilized to prepare PEI-PEG/M27 conjugates and subsequently formulated into NP Vacc with CpG as described earlier. C57BL/6 mice were inoculated at s.c. flank with 3 × 105 B16F10 tumor cells and vaccinated at s.c. tail base with NP Vacc on day 4, followed by i.t. administration of CDA on days 8, 11, and 14 (Figure 5a). Proliferation and activation of M27-specific CD8+ T cells were analyzed by IFN-γ ELISPOT assay performed on day 11 using splenocytes. NP Vacc elicited robust M27-specific IFN-γ+CD8+ T cells, generating 57-fold higher ELISPOT responses than CDA mono-therapy (p < 0.0001, Figure 5b). Whereas combination CDA treatment slightly decreased the frequency of M27-specific IFN-γ+ T cells in spleen (Figure 5b), it significantly increased the concentrations of IFN-β, CCL5, and CXCL10 in tumor (Figure 5c), compared with NP Vacc alone. Moreover, NP Vacc + CDA combination treatment promoted robust tumor-infiltration of CD8+ T cells as shown by flow cytometry and immunohistochemistry (Figure 5d,e). In contrast, we did not observe any significance difference among the frequency of CD4+ T cells or B cells within the TME (Fig. S5), indicating a CD8+ T cell-driven immune response promoted by the combination treatment. In parallel, we monitored animals for tumor growth and survival (Figure 5d,e). As B16F10 melanoma is a poorly immunogenic [47, 48], NP Vacc alone failed to slow the tumor growth or extend the animal survival (Figure 5d,e). While CDA mono-therapy slightly delayed the tumor growth, all mice succumbed to tumor growth within 35 days. On the other hand, NP Vacc + CDA exhibited a remarkable anti-tumor efficacy, eradicating B16F10 tumors in 100% of treated mice (Figure 5d,e). Overall, these results showed that NP Vacc + CDA combination therapy exerts potent anti-tumor efficacy against even poorly immunogenic tumors.
Figure 5. Combination treatment of NP Vacc and CDA regresses poorly immunogenic B16F10 melanoma.
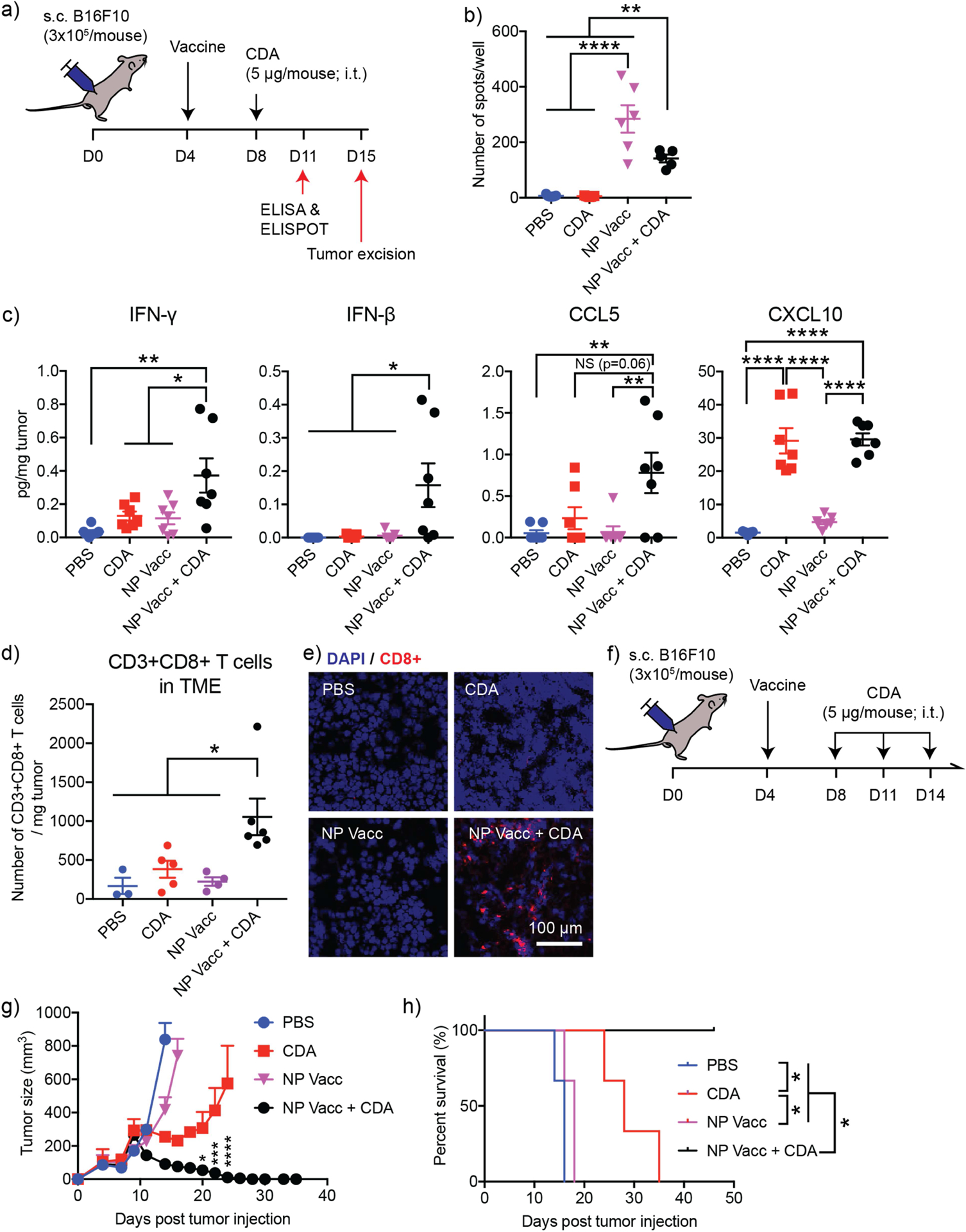
a) Timeline of experiment. B16F10-bearing mice were vaccinated on day 4, followed by CDA injection on day 8. Mice were sacrificed on day 11 for ELISPOT and ELISA analyses using spleens and tumor samples, respectively. A subset of mice was sacrificed on day 15 for the analysis of TME. b) Neoantigen peptide (M27)-specific cells within splenocytes were detected with IFN-γ ELISPOT assay. c) Cytokine concentrations in tumor measured by ELISA. Tumor-infiltrating CD8+ T cells were analyzed by d) flow cytometry and e) immunohistochemistry. Scale bar = 100 μm. f) Timeline of survival study. B16F10-bearing mice were vaccinated on day 4, followed by intratumoral administration of 5 μg CDA on days 8, 11, and 14. g) Tumor growth and h) survival curves of B16F10 tumor-bearing mice. Asterisks in g) indicate statistical comparison between CDA and NP Vacc + CDA. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001, analyzed by b,c,d) one-way ANOVA followed by Tukey’s HSD multiple comparison post hoc test or g) two-way ANOVA followed by Sidak’s multiple comparisons test. h) The survival curves were analyzed by the log-rank (Mantel–Cox) test.
4. Conclusions
In conclusion, we have developed PEI-based NPs co-delivering CpG adjuvants and tumor-specific neoantigen peptides for personalized cancer immunotherapy and showed that they elicit potent anti-tumor CD8+ T cell responses in the systemic circulation; however, this was not sufficient to inhibit tumor growth, in part due to poor tumor infiltration of activated CD8+ T cells. We demonstrated for the first time that local administration of STING agonist can significantly enhance infiltration of NP vaccine-primed CD8+ T cells into the TME by promoting secretion of T cell-attracting chemokines and cytokines, leading to robust tumor regression and long-term immunological memory. Our strategy of utilizing STING-based immunotherapy for potentiating NP-based cancer vaccines present a new combination nano-immunotherapy that is be widely applicable for combination cancer immunotherapy.
Supplementary Material
Supplementary Figure 1. Changes in serum cytokine levels and body weight after vaccination.
Supplementary Figure 2. Chemokine secretion by mouse BMDCs in vitro after CDA treatment.
Supplementary Figure 3. Changes in serum chemokines and cytokines and body weight after intratumoral injection of CDA in MC-38 tumor-bearing mice.
Supplementary Figure 4. CD4+ T cells and B cells infiltrating MC38 TME.
Supplementary Figure 5. CD4+ T cells and B cells infiltrating B16F10 TME.
Acknowledgement
This work was supported in part by NIH (R01AI127070, R01CA210273, R01DK125087, and U01CA210152) and through the University of Michigan Rogel Cancer Center Support Grant (P30CA46592). J.J.M. is supported by NSF CAREER Award (1553831). K.S.P. acknowledges financial support from the UM TEAM Training Program (DE007057 from NIDCR). We acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for the provision of MHC-I tetramers.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Potential of Conflicts of Interest
Authors declare no conflict of interest.
5. References
- [1].Schumacher TN, Schreiber RD, Neoantigens in cancer immunotherapy, Science 348(6230) (2015) 69–74. [DOI] [PubMed] [Google Scholar]
- [2].Castle JC, Kreiter S, Diekmann J, Löwer M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, Koslowski M, Kuhn AN, Britten CM, Huber C, Türeci O, Sahin U, Exploiting the mutanome for tumor vaccination, Cancer Res 72(5) (2012) 1081–91. [DOI] [PubMed] [Google Scholar]
- [3].Heemskerk B, Kvistborg P, Schumacher TN, The cancer antigenome, EMBO J 32(2) (2013) 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ, An immunogenic personal neoantigen vaccine for patients with melanoma, Nature 547(7662) (2017) 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, Shukla SA, Hu Z, Li L, Le PM, Allesøe RL, Richman AR, Kowalczyk MS, Abdelrahman S, Geduldig JE, Charbonneau S, Pelton K, Iorgulescu JB, Elagina L, Zhang W, Olive O, McCluskey C, Olsen LR, Stevens J, Lane WJ, Salazar AM, Daley H, Wen PY, Chiocca EA, Harden M, Lennon NJ, Gabriel S, Getz G, Lander ES, Regev A, Ritz J, Neuberg D, Rodig SJ, Ligon KL, Suvà ML, Wucherpfennig KW, Hacohen N, Fritsch EF, Livak KJ, Ott PA, Wu CJ, Reardon DA, Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial, Nature 565(7738) (2019) 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fang Y, Mo F, Shou J, Wang H, Luo K, Zhang S, Han N, Li H, Ye S, Zhou Z, Chen R, Chen L, Liu L, Pan H, Chen S, A pan-cancer clinical study of personalized neoantigen vaccine monotherapy in treating patients with various types of advanced solid tumors, Clin Cancer Res (2020). [DOI] [PubMed] [Google Scholar]
- [7].Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Müller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Brück AK, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Höller C, Utikal J, Huber C, Loquai C, Türeci Ö, Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer, Nature 547(7662) (2017) 222–226. [DOI] [PubMed] [Google Scholar]
- [8].Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ, Designer vaccine nanodiscs for personalized cancer immunotherapy, Nat Mater 16(4) (2017) 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Scheetz L, Park KS, Li Q, Lowenstein PR, Castro MG, Schwendeman A, Moon JJ, Engineering patient-specific cancer immunotherapies, Nat Biomed Eng 3(10) (2019) 768–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ni Q, Zhang F, Liu Y, Wang Z, Yu G, Liang B, Niu G, Su T, Zhu G, Lu G, Zhang L, Chen X, A bi-adjuvant nanovaccine that potentiates immunogenicity of neoantigen for combination immunotherapy of colorectal cancer, Sci Adv 6(12) (2020) eaaw6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Blass E, Ott PA, Advances in the development of personalized neoantigen-based therapeutic cancer vaccines, Nat Rev Clin Oncol (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Anderson KG, Stromnes IM, Greenberg PD, Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies, Cancer Cell 31(3) (2017) 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Barnes TA, Amir E, HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer, Br J Cancer 117(4) (2017) 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shimizu S, Hiratsuka H, Koike K, Tsuchihashi K, Sonoda T, Ogi K, Miyakawa A, Kobayashi J, Kaneko T, Igarashi T, Hasegawa T, Miyazaki A, Tumor-infiltrating CD8+ T-cell density is an independent prognostic marker for oral squamous cell carcinoma, Cancer Med 8(1) (2019) 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Idos GE, Kwok J, Bonthala N, Kysh L, Gruber SB, Qu C, The Prognostic Implications of Tumor Infiltrating Lymphocytes in Colorectal Cancer: A Systematic Review and Meta-Analysis, Sci Rep 10(1) (2020) 3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pagès F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, Lugli A, Zlobec I, Rau TT, Berger MD, Nagtegaal ID, Vink-Börger E, Hartmann A, Geppert C, Kolwelter J, Merkel S, Grützmann R, Van den Eynde M, Jouret-Mourin A, Kartheuser A, Léonard D, Remue C, Wang JY, Bavi P, Roehrl MHA, Ohashi PS, Nguyen LT, Han S, MacGregor HL, Hafezi-Bakhtiari S, Wouters BG, Masucci GV, Andersson EK, Zavadova E, Vocka M, Spacek J, Petruzelka L, Konopasek B, Dundr P, Skalova H, Nemejcova K, Botti G, Tatangelo F, Delrio P, Ciliberto G, Maio M, Laghi L, Grizzi F, Fredriksen T, Buttard B, Angelova M, Vasaturo A, Maby P, Church SE, Angell HK, Lafontaine L, Bruni D, El Sissy C, Haicheur N, Kirilovsky A, Berger A, Lagorce C, Meyers JP, Paustian C, Feng Z, Ballesteros-Merino C, Dijkstra J, van de Water C, van Lent-van Vliet S, Knijn N, Mușină AM, Scripcariu DV, Popivanova B, Xu M, Fujita T, Hazama S, Suzuki N, Nagano H, Okuno K, Torigoe T, Sato N, Furuhata T, Takemasa I, Itoh K, Patel PS, Vora HH, Shah B, Patel JB, Rajvik KN, Pandya SJ, Shukla SN, Wang Y, Zhang G, Kawakami Y, Marincola FM, Ascierto PA, Sargent DJ, Fox BA, Galon J, International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study, Lancet 391(10135) (2018) 2128–2139. [DOI] [PubMed] [Google Scholar]
- [17].Petitprez F, Meylan M, de Reyniès A, Sautès-Fridman C, Fridman WH, The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies, Front Immunol 11 (2020) 784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, Caux C, Depil S, Cold Tumors: A Therapeutic Challenge for Immunotherapy, Front Immunol 10 (2019) 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Duan Q, Zhang H, Zheng J, Zhang L, Turning Cold into Hot: Firing up the Tumor Microenvironment, Trends Cancer 6(7) (2020) 605–618. [DOI] [PubMed] [Google Scholar]
- [20].Demaria S, Volm MD, Shapiro RL, Yee HT, Oratz R, Formenti SC, Muggia F, Symmans WF, Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy, Clin Cancer Res 7(10) (2001) 3025–30. [PubMed] [Google Scholar]
- [21].Heeren AM, van Luijk IF, Lakeman J, Pocorni N, Kole J, de Menezes RX, Kenter GG, Bosse T, de Kroon CD, Jordanova ES, Neoadjuvant cisplatin and paclitaxel modulate tumor-infiltrating T cells in patients with cervical cancer, Cancer Immunol Immunother 68(11) (2019) 1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Achard C, Surendran A, Wedge ME, Ungerechts G, Bell J, Ilkow CS, Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy, EBioMedicine 31 (2018) 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lanitis E, Irving M, Coukos G, Targeting the tumor vasculature to enhance T cell activity, Curr Opin Immunol 33 (2015) 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Corrales L, Gajewski TF, Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of Cancer, Clin Cancer Res 21(21) (2015) 4774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW, Gajewski TF, Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity, Cell Rep 11(7) (2015) 1018–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, Hudson TE, Vu UT, Francica BJ, Banda T, Katibah GE, Kanne DB, Leong JJ, Metchette K, Bruml JR, Ndubaku CO, McKenna JM, Feng Y, Zheng L, Bender SL, Cho CY, Leong ML, van Elsas A, Dubensky TW, McWhirter SM, Magnitude of Therapeutic STING Activation Determines CD8+ T Cell-Mediated Anti-tumor Immunity, Cell Rep 25(11) (2018) 3074–3085.e5. [DOI] [PubMed] [Google Scholar]
- [27].Nam J, Son S, Moon JJ, Adjuvant-Loaded Spiky Gold Nanoparticles for Activation of Innate Immune Cells, Cell Mol Bioeng 10(5) (2017) 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ochyl LJ, Bazzill JD, Park C, Xu Y, Kuai R, Moon JJ, PEGylated tumor cell membrane vesicles as a new vaccine platform for cancer immunotherapy, Biomaterials 182 (2018) 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Petersen H, Fechner PM, Martin AL, Kunath K, Stolnik S, Roberts CJ, Fischer D, Davies MC, Kissel T, Polyethylenimine-graft-Poly(ethylene glycol) Copolymers: Influence of Copolymer Block Structure on DNA Complexation and Biological Activities as Gene Delivery System, Bioconjugate Chemistry 13(4) (2002) 845–854. [DOI] [PubMed] [Google Scholar]
- [30].Petersen H, Fechner PM, Fischer D, Kissel T, Synthesis, Characterization, and Biocompatibility of Polyethylenimine-graft-poly(ethylene glycol) Block Copolymers, Macromolecules 35(18) (2002) 6867–6874. [Google Scholar]
- [31].Nam J, Son S, Park KS, Moon JJ, Modularly Programmable Nanoparticle Vaccine Based on Polyethyleneimine for Personalized Cancer Immunotherapy, Advanced Science (2198–3844) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vollmer J, Krieg AM, Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists, Advanced Drug Delivery Reviews 61(3) (2009) 195–204. [DOI] [PubMed] [Google Scholar]
- [33].Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, Modrusan Z, Mellman I, Lill JR, Delamarre L, Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing, Nature 515(7528) (2014) 572–6. [DOI] [PubMed] [Google Scholar]
- [34].Colombo MP, Trinchieri G, Interleukin-12 in anti-tumor immunity and immunotherapy, Cytokine Growth Factor Rev 13(2) (2002) 155–68. [DOI] [PubMed] [Google Scholar]
- [35].Schmidt CS, Mescher MF, Peptide antigen priming of naive, but not memory, CD8 T cells requires a third signal that can be provided by IL-12, J Immunol 168(11) (2002) 5521–9. [DOI] [PubMed] [Google Scholar]
- [36].Li H, Li Y, Wang X, Hou Y, Hong X, Gong T, Zhang Z, Sun X, Rational design of Polymeric Hybrid Micelles to Overcome Lymphatic and Intracellular Delivery Barriers in Cancer Immunotherapy, Theranostics 7(18) (2017) 4383–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kawai T, Akira S, Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity, Immunity 34(5) (2011) 637–650. [DOI] [PubMed] [Google Scholar]
- [38].Chen Daniel S., I. Mellman, Oncology Meets Immunology: The Cancer-Immunity Cycle, Immunity 39(1) (2013) 1–10. [DOI] [PubMed] [Google Scholar]
- [39].Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre ML, Gajewski TF, STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors, Immunity 41(5) (2014) 830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Vatner RE, Janssen EM, STING, DCs and the link between innate and adaptive tumor immunity, Mol Immunol 110 (2019) 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, Fong L, Nolan GP, Engleman EG, Systemic Immunity Is Required for Effective Cancer Immunotherapy, Cell 168(3) (2017) 487–502.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nakajima C, Uekusa Y, Iwasaki M, Yamaguchi N, Mukai T, Gao P, Tomura M, Ono S, Tsujimura T, Fujiwara H, Hamaoka T, A role of interferon-gamma (IFN-gamma) in tumor immunity: T cells with the capacity to reject tumor cells are generated but fail to migrate to tumor sites in IFN-gamma-deficient mice, Cancer Res 61(8) (2001) 3399–405. [PubMed] [Google Scholar]
- [43].Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ, Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion, Front Immunol 9 (2018) 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Melero I, Rouzaut A, Motz GT, Coukos G, T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy, Cancer Discov 4(5) (2014) 522–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bhat P, Leggatt G, Matthaei KI, Frazer IH, The kinematics of cytotoxic lymphocytes influence their ability to kill target cells, PLoS One 9(5) (2014) e95248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bhat P, Leggatt G, Waterhouse N, Frazer IH, Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity, Cell Death Dis 8(6) (2017) e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Curran MA, Montalvo W, Yagita H, Allison JP, PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors, Proc Natl Acad Sci U S A 107(9) (2010) 4275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, Douglas M, Tibbitts T, Sharma S, Proctor J, Kosmider N, White K, Stern H, Soglia J, Adams J, Palombella VJ, McGovern K, Kutok JL, Wolchok JD, Merghoub T, Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells, Nature 539(7629) (2016) 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kuai R, Sun X, Yuan W, Xu Y, Schwendeman A, Moon JJ, Subcutaneous Nanodisc Vaccination with Neoantigens for Combination Cancer Immunotherapy, Bioconjug Chem 29(3) (2018) 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Changes in serum cytokine levels and body weight after vaccination.
Supplementary Figure 2. Chemokine secretion by mouse BMDCs in vitro after CDA treatment.
Supplementary Figure 3. Changes in serum chemokines and cytokines and body weight after intratumoral injection of CDA in MC-38 tumor-bearing mice.
Supplementary Figure 4. CD4+ T cells and B cells infiltrating MC38 TME.
Supplementary Figure 5. CD4+ T cells and B cells infiltrating B16F10 TME.