

Abstract
Accurate microbial identification and abundance estimation are crucial for metagenomics analysis. Various methods for classifying metagenomic data and estimating taxonomic profiles, broadly referred to as metagenomic profilers, have been developed. Yet, benchmarking metagenomic profilers remains challenging because some tools are designed to report relative sequence abundance while others report relative taxonomic abundance. Here, we show how misleading conclusions can be drawn by neglecting this distinction between relative abundance types when benchmarking metagenomic profilers. Moreover, we show compelling evidence that interchanging sequence abundance and taxonomic abundance will influence both per-sample summary statistics and cross-sample comparisons. We suggest that the microbiome research community should pay attention to potentially misleading biological conclusions arising from this issue when benchmarking metagenomic profilers, by carefully considering the type of abundance data that was analyzed and interpreted, and clearly stating the strategy used for metagenomic profiling.
By directly interrogating the community composition in an unbiased and culture-independent manner, metagenomic sequencing is transforming microbiology by enabling more rapid species detection and discovery1. This has a wide range of applications in environmental and clinical microbiology. Various computational methods have been developed to identify species contained in the samples by classifying sequencing reads and quantifying their relative abundances1–5. Those computational methods are broadly referred to as metagenomic profilers.
Following a previous benchmarking study3, metagenomic profilers can be categorized based on their reference database type (Fig.1a): (1) DNA-to-DNA methods (e.g., Kraken6, 7, Bracken8, and PathSeq9), which compare sequence reads with comprehensive metagenome databases; (2) DNA-to-Protein methods (e.g., Kaiju10 and Diamond11), which compare sequence reads with genomic databases of protein-coding sequences; or (3) DNA-to-Marker methods (e.g., MetaPhlAn12, 13 and mOTUs5, 14), which only include specific gene families in their reference databases. Note that those metagenomic profilers all rely on reference databases. They should not be confused with de novo assembly-based methods that do not use any reference databases15, 16. Those reference-free binning methods cannot taxonomically classify sequences15, 16 and are not directly comparable with the metagenomic profilers evaluated here.
Figure 1. Comparison of profiling results.
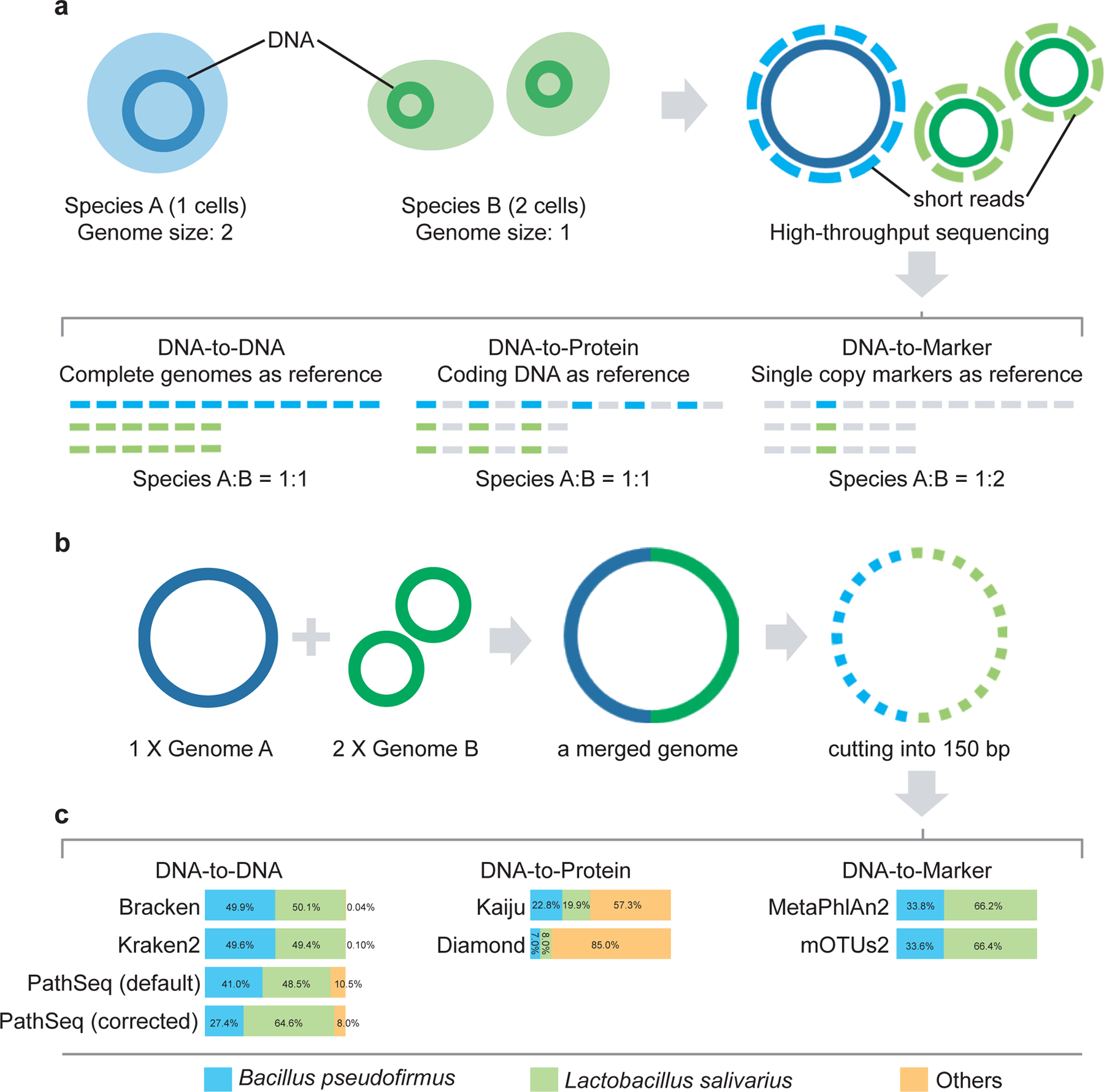
a, Illustration of the reference databases and the default output abundance type for DNA-to-DNA, DNA-to-Protein and DNA-to-Marker profilers on a mixture of two species A (1 cell) and B (2 cells). b, A simulated microbial community with only two genomes: Bacillus pseudofirmus (genome size 4.2MB) and Lactobacillus salivarius (genome size 2.1MB). We merged one copy of Bacillus pseudofirmus genome (genome A) with two copies of Lactobacillus salivarius genome (genome B) sequences into one metagenome file. Then we sheared the merged metagenomic sequences into 150bp to simulate a typical metagenomic dataset. c, Profiling results (default output) of different profilers for the simulated microbial community. The bar plots show the estimated relative abundance of the two microbial members A and B using different metagenomics profilers. PathSeq (default) represents the profiling result generated by the default setting of PathSeq (without genome-length correction). PathSeq (corrected) represents the profiling result of PathSeq with the parameter “--divide-by-genome-length” (i.e., genome-length correction) enabled.
Many studies have benchmarked metagenomic profilers2, 17–20, finding that the abundance-estimation performance of different profilers varies considerably even on the same benchmark datasets. For example, in a recent benchmarking study2, the abundance-estimation performance of 20 metagenomic profilers were evaluated based on the L2 distance between the observed and true relative abundance profiles. It was found that DNA-to-DNA methods were among the best-scoring methods, with typical average L2 distance < 0.1, while DNA-to-Marker methods had much higher L2 distance, indicating less favorable performance.
Here we show that this apparently high performance-variation largely arises because the profilers report one of two fundamentally different types of relative abundances: sequence abundance or taxonomic abundance. For example, the raw output of DNA-to-DNA methods is the relative abundance of a given taxon calculated as the proportion of sequences assigned to it out of the total number of sequences, i.e., the sequence abundance. For DNA-to-Protein methods, the output type is the relative sequence abundance of protein-coding sequences10, 11. By contrast, DNA-to-Marker methods directly output the relative abundance of each taxon calculated as the number of genomes of that taxon relative to the total number of genomes detected, i.e., the taxonomic abundance. Unfortunately, the distinction between the two types of relative abundances has rarely been carefully considered in previous benchmarking studies2, 18, 19, 21.
In this paper, we show that the two types of relative abundances are not related by any universal algebraic relation. Moreover, interchanging them leads to very misleading performance assessments of metagenomic profilers. These results imply that many benchmarking results presented in the literature require re-examination. Beyond re-evaluating previous benchmark results in light of the confounded use of relative abundance types, we further point out the serious issues in microbiome data analysis based on sequence abundances, which are typically produced by DNA-to-DNA methods and have been applied in thousands of published microbiome studies (e.g., Kraken: 1,438 citations; Kraken2: 204 citations; Bracken: 202 citations, by March 2021, according to their official websites). We find that microbiome data analysis based on sequence abundance will underestimate (or overestimate) the relative abundances of microbes with smaller (or larger) genome sizes, respectively. This will fundamentally affect differential abundance analyses and other analytical methods that rely on accurate taxon counts in their input contingency matrix. Without careful consideration, these issues could impede cross-study comparisons of differentially abundant taxa identified from different methods and hence warrant more attention from the entire microbiome research community.
Results
Illustration of the caveat in benchmarking metagenomic profilers.
To illustrate the caveat of confusing sequence abundance and taxonomic abundance in benchmarking metagenomic profilers, we simulated a simple microbial community with only two genomes (see Fig.1b), where genome A (Bacillus pseudofirmus, GCF_000005825.2, size: 4.2MB) is twice the size of genome B (Lactobacillus salivarius, GCF_000008925.1, size: 2.1MB). In this simulated community, the sequence abundance ratio of genome A : genome B = 1:1, while the taxonomic abundance ratio of genome A : genome B = 1:2.
As shown in Fig.1c, the DNA-to-DNA profiler Bracken (or Kraken2) reported the abundance ratio between Bacillus pseudofirmus and Lactobacillus salivarius as 49.9% : 50.1% ≈ 1 : 1.004 (or 49.6% : 49.4% ≈ 1 : 0.996, respectively), which is very close to the ground truth of sequence abundance ratio (1:1). Moreover, Bracken (or Kraken2) identified only one false-positive species --- Lactobacillus plantarum with very low sequence abundance 0.04% (or 0.1%), respectively. Notably, for another DNA-to-DNA profiler PathSeq, when the genome-length correction is disabled (by default), it reported the abundance ratio between Bacillus pseudofirmus and Lactobacillus salivarius as 40.1% : 48.5% ≈ 1 : 1.209, which is quite different to the ground truth of sequence abundance ratio (1:1). With enabled genome length correction, PathSeq reported the abundance ratio as 27.4% : 64.6% ≈ 1 : 2.358, which certainly doesn’t represent the sequence abundance ratio (1:1), and is still quite different from the ground truth of taxonomic abundance ratio (1:2). Hence, the genome-length correction in PathSeq does not work as well as we expected. We suspect that this is largely due to its very high false-positive rate. Indeed, 786 of the 788 species identified by PathSeq are false-positive, with accumulated sequence abundance ~10.5% and taxonomic abundance ~8.0%. This simple example clearly demonstrates that neither the raw sequence abundance profile nor the taxonomic profile obtained after genome-length correction produced by DNA-to-DNA profilers represents the true taxonomic profile of a microbiome sample.
DNA-to-Protein profiler Kaiju (or Diamond) reported the abundance ratio between Bacillus pseudofirmus and Lactobacillus salivarius as 22.8% : 19.9% ≈ 1 : 0.873 (or 7.0% : 8.0% ≈ 1 : 1.143), respectively (Fig.1c). The ratios are close to 1:1, indicating the methods are indeed reporting sequence abundance. However, due to the conservation of protein sequence22, these two methods reported a very large number of false-positive species: 330 for Kaiju and 152 for Diamond, with accumulated abundance 57.3% and 85.0%, respectively.
DNA-to-Markers profiler MetaPhlAn2 (or mOTUs2) reported the abundance ratio between Bacillus pseudofirmus and Lactobacillus salivarius as 33.8% : 66.2% ≈ 1 : 1.959 (or 33.6% : 66.4% ≈ 1 : 1.976), respectively, without any false-positive species (Fig.1c). The ratio between the relative abundances of the two species is roughly 1:2, indicating the methods are indeed reporting taxonomic abundances.
To avoid the potential impact of false positives on benchmarking metagenomic profilers, going forward we will focus on DNA-to-DNA and DNA-to-Markers methods. And for DNA-to-DNA methods, we will focus on Bracken and Kraken2.
No universal algebraic relation between the two types of relative abundances.
We emphasize that mathematically there is no universal (i.e., sample-independent) algebraic relation between the two types of relative abundances, even in the ideal case (when all genomes/taxa are known). To demonstrate this, let’s denote Ri as the number of metagenomic reads assigned to the genome of a microbial taxon i with genome size Li and ploidy Pi (i.e., the number of copies of the genome in one cell of taxon i). The number of microbial cells classified as taxon i is then given by Ci = Ri/(LiPi). Let n be the number of identified taxa in the sample. Then the sequence abundance of taxon i is given by
| [1] |
and its taxonomic abundance is given by
| [2] |
From Eqs.[1–2], we have Si/Ti ∝ LiPi, but the coefficient is complicated and sample-specific. Hence, as long as Li and Pi vary across different taxa, Si and Ti are not connected by any universal or sample-independent algebraic relation.
The variation of genome size Li of different taxa can be very large. Indeed, in the recently updated microbial genome database (NCBI RefSeq, 2020 Nov 6th), the sizes of fully sequenced and assembled microbial genomes vary considerably (Fig.2a). For example, just within the bacteria kingdom, the genome size variation can be more than 100-fold, e.g., Candidatus Nasuia deltocephalinicola (GCF_000442605.1) with 112,091 bp vs. Sorangium cellulosum (GCF_000418325.1) with 14,782,125 bp. Therefore, microbial genome sizes could vary radically within a single microbiome sample, especially when viruses (which tend to have shorter genomes, Fig.2a) are analyzed together with bacteria in shotgun metagenomics.
Figure 2. Correlation between sequence abundance and taxonomic abundance in synthetic profiles based on different kingdoms.
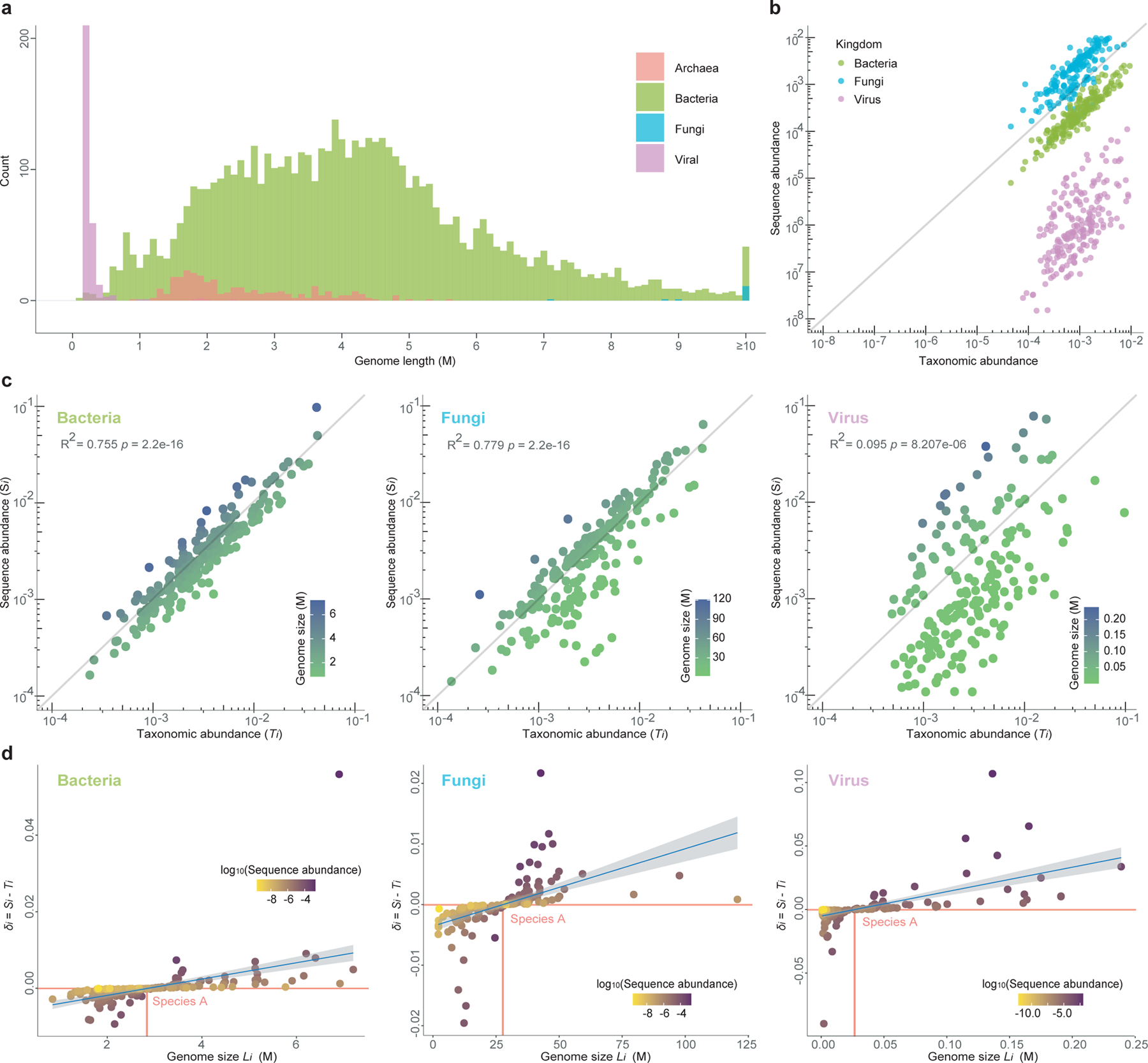
a, Genome size distribution of microorganisms calculated from the microbial genome database (NCBI RefSeq 2020 Nov 6th) that includes 171,927 bacteria, 293 fungi, 945 archaea, and 9,362 viruses. b, The scatter plot shows the correlation between taxonomic abundance (x axis) and sequence abundance (y axis) of 600 randomly selected species in a simulated profile (n=1) which includes bacteria (species number=200), fungi (species number=200) and virus (species number=200). c, Correlation between taxonomic abundance (x-axis) and sequence abundance (y-axis) of 200 randomly selected species in a simulated microbial community within each of the three kingdoms: bacteria (n=1 simulated profile), fungi (n=1), and virus (n=1). d, Relationship between the genome length of a species (Li) and the difference between its sequence and taxonomic abundances (denoted as δi). Each point represents a species and is colored by its log10-transformed sequence abundance in the simulated microbial communities shown in c. The “reference” species A has the minimum |δi|. In those figures, the minimum |δi|’s is close to 0, indicating that species A has almost identical sequence and taxonomic abundances in those communities. The confidence interval (CI) is 0.95.
Regrading ploidy Pi, although prokaryotes are usually thought to contain one copy of a circular chromosome, previous studies have demonstrated that many species of archaea and bacteria are polyploid and can contain more than ten copies of their chromosome23. In fact, extreme polyploidy has been observed in a large bacterium Epulopiscium, which contains tens of thousands of copies of its genome24.
The variations in Li and Pi drive the theoretical distinction between sequence abundance and taxonomic abundance. To further illustrate the difference between the two relative abundance types, we generated synthetic microbial communities based on the NCBI RefSeq database. As shown in Fig.2b, where we investigate a complex microbial community consisting microbes from all different kingdoms (fungi, bacteria, and virus), Si tends to overestimate Ti of species with larger genome sizes (e.g., fungi) and underestimate Ti of species with smaller genome sizes (e.g., viruses). This is true even if we investigate a community consisting of microbes from the same kingdom (Fig.2c). For synthetic communities, the over- and underestimation of taxonomic abundances can be quantified as follows. For a given community, let’s denote the difference between taxon-i’s sequence abundance Si and taxonomic abundance Ti as δi ≡ Si − Ti. First, we identify a “reference” species A that has the minimum |δi|. (In the ideal case, δA = 0, implying that species A has identical Si and Ti in the community. Then for those species that have a larger (or smaller) genome sizes Li than LA, their Ti will tend to be overestimated (or underestimated), i.e., their δi will tend to be positive (or negative), respectively. Indeed, for the simulated bacterial, fungal, and viral communities analyzed in Fig.2c, we found that, δi > 0 (or < 0) for those taxa with genome sizes Li > LA (or < LA), respectively (Fig.2d). But we emphasize there is so simple relationship between δi and (Li – LA). In fact, we found some “outlier species”, which have genome sizes much larger or smaller than LA, and yet their δi values are close to 0. Those outlier species typically have very low abundances. Note that here, for the sake of simplicity, in our simulations we did not consider the variation of ploidy, but only focused on the variation of genome sizes. Hence, the difference between sequence abundance and taxonomic abundance demonstrated in Fig.2c,d is very conservative.
In reality, many factors will further complicate the relation between Si and Ti, and hence affect the benchmarking of metagenomic profilers. First of all, most metagenomic profilers do not consider ploidy in their abundance estimation, because the ploidy information is still lacking for many genomes. Second, the presence of unknown genomes/taxa renders the conversion from sequence abundance to taxonomic abundance extremely challenging, and significantly affects the benchmarking based on real data. This is because different profilers report different types of relative abundance and handle unknown genomes/taxa differently. Finally, instead of converting Si to Ti through Li and Pi correction, DNA-to-Marker methods directly calculate Ti as the ratio of sequence coverage of single-copy marker genes of each taxon to that of all taxa, which naturally avoids the genome-size and copy-number corrections. This also affects the direct comparability of metagenomic profilers, because the taxonomic abundances produced by DNA-to-Marker methods are meant to reflect relative cell abundances, which can be achieved by classifying only those metagenomics reads that map against taxonomic marker genes, rather than sequences abundances produced by DNA-to-DNA methods, which are based on classifying all metagenomic reads in a given sample. In addition, transforming taxonomic abundance to sequence abundance may introduce systematic error since it requires accurate genome size information.
Benchmarking results depend on the abundance type.
To further illustrate the problem of mixing sequence abundance and taxonomic abundance in benchmarking metagenomic profilers, we simulated metagenomic sequencing reads for 25 communities from distinct habitats (e.g., gut, oral, skin, vagina and building, five communities for each habitat, see Methods). To avoid reference database biases of different metagenomic profilers, the genomes used to generate simulated communities were selected from the intersection among the reference databases of MetaPhlAn2, mOTUs2, and Kraken2. (Bracken and Kraken2 use the same reference database.) Then we calculated the distance between the ground truth abundance profiles and the estimated ones from different profilers. We notice that in previous benchmarking studies, typically L120 or L22 distance was used. Yet, just like other popular distance/dissimilarity measures used in microbiome data analysis, e.g., Bray-Curtis dissimilarity (BC) and root Jensen-Shannon divergence (rJSD), those measures (L1, L2, BC and rJSD) are not compositionally aware25. This prompted us to ask if the compositional unawareness of those measures will affect the benchmarking result. It is well known that the classical Aitchison distance (based on centered log-ratio transform) is a compositionally aware distance measure26. However, it suffers from the inflated zero counts in microbiome data because log-transform of zero counts is undefined unless arbitrary pseudocounts are added to each taxon. Fortunately, a recently developed distance measure --- the robust Aitchison distance (rAD)27 does not involve any pseudocounts, and hence naturally avoids the issue of dealing with sparse zero counts using the classical Aitchison distance. Therefore, in this work, to systematically study the potential impact of compositional-unawareness of dissimilarity/distance measures on the benchmarking result, we used the following five measures: BC, rJSD, L1, L2, and rAD.
As shown in Fig.3a,b, we found that for BC, rJSD, L1, and L2, if the sequence abundance is used as the ground truth, Bracken and Kraken2 outperform MetaPhlAn2 and mOTUs2; while if the taxonomic abundance is used as the ground truth, MetaPhlAn2 and mOTUs2 outperform Bracken and Kraken2.
Figure 3. Quantitative and qualitative benchmarking results of four representative metagenomic profilers using 25 simulated communities.
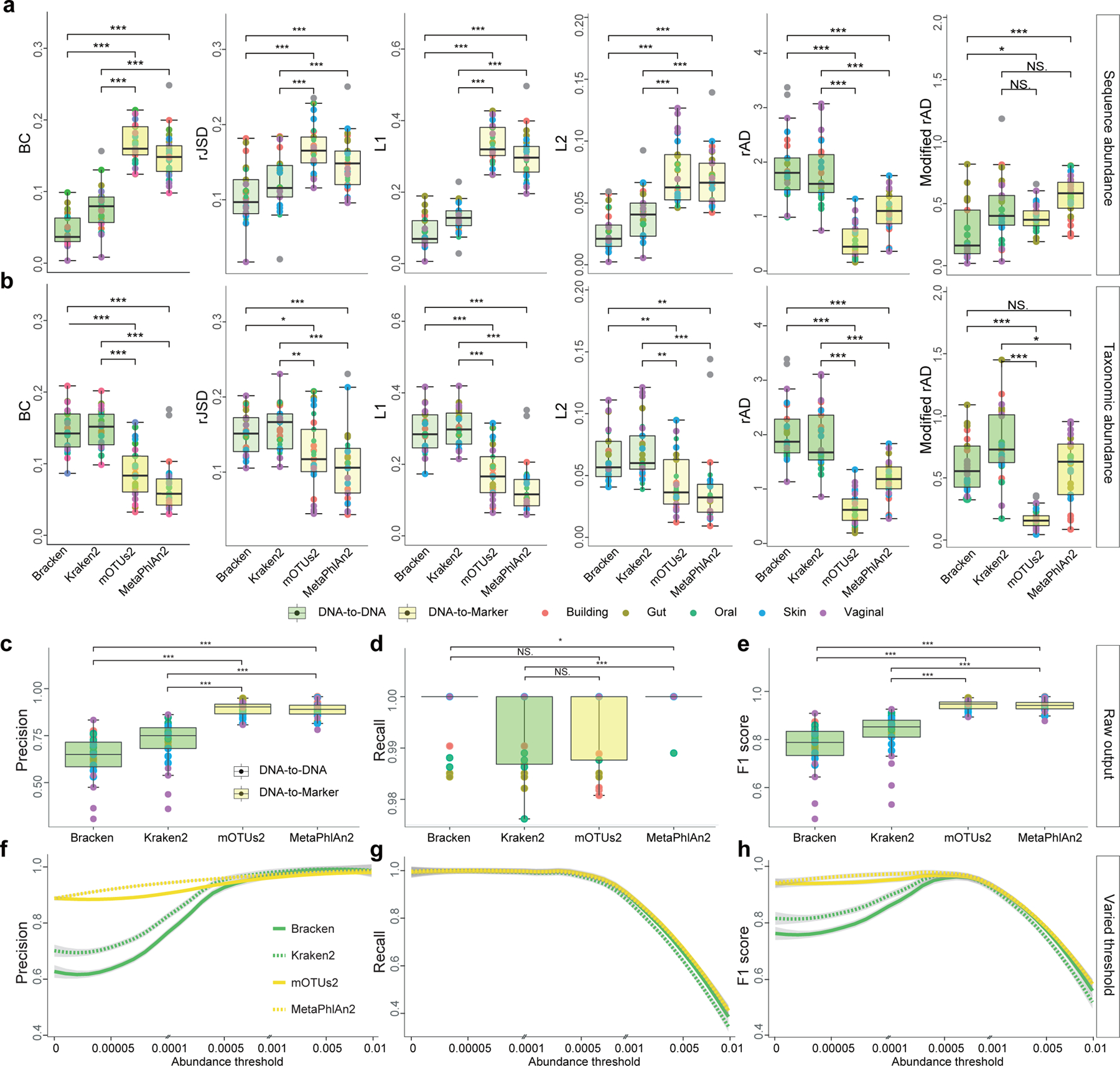
a-b: Differential benchmarking results of four representative metagenomics profilers using two types of relative abundance as ground truth: sequence abundance (a) and taxonomic abundance (b). The boxplots indicate the dissimilarities based on L1, L2, root Jensen-Shannon divergence (rJSD), Bray-Curtis (BC), and robust Aitchison distance (rAD) between the ground-truth profiles and the profiles predicted by different metagenomics profilers (Bracken, Kraken2, mOTUs2, and MetaPhlAn2) at the species level. For each metagenomic profiler, we performed the dissimilarity calculations based on 25 simulated microbial communities from five representative environmental habitats (gut, oral, skin, vagina and building) separately. Note that for each profiler based on any evaluation metric, its performance variation across different synthetic communities is due to microbiome complexity difference (e.g., species composition and richness). c-d: Precision-recall analysis. c-e: Boxplots indicate the precision (c), recall (d), and F1 score (e) based on the default profiling results of four metagenomic profilers (without any abundance thresholding) using either sequence abundance (green) or taxonomic abundance (yellow) as the ground truth. f-h: The change of the precision (f), recall (g), and F1 score (h) with abundance threshold tuned from 0 to 0.01. Each dot represents the microbial profile of a simulated community, n = 25 simulated datasets. Significance levels: p-value<0.05 (*), <0.01 (**), <0.001 (***), NS (non-significance); two-sided Wilcoxon signed-rank test. Exact p-values are provided in the Source Data File. The lower and upper hinges correspond to the first and third quartiles, and the center refers to the median value. The upper (or lower) whisker extends from the hinge to the largest (smallest) value no further (at most) than 1.5 * IQR from the hinge. Data beyond the end of the whiskers are plotted individually.
Interestingly, with rAD as the evaluation metric, regardless of the ground truth being sequence or taxonomic abundance, mOTUs2 and MetaPhlan2 always outperform Bracken and Kraken. This could be due to the fact that, as a compositionally aware distance measure, rAD weighs low-abundance taxa more than the other measures. To test this idea, we sought to rule out the bias introduced by false positives and calculated rAD based on taxonomic profilers where false positives have been removed (Methods). This is denoted as modified rAD in Fig.3a,b. We found that, after removal of averagely 27 ± 10 species from default profiling results in Kraken2 (with accumulated abundance 29.26% ± 12.13%), 40 ± 14 in Bracken (36.91% ± 12.11%), 8 ± 4 in mOTUs2 (11.47% ± 4.62%), and 9 ± 4 in MetaPhlAn2 (11.29% ± 4.19%), the benchmarking result based on rAD is the same as that of using BC, rJSD, L1, and L2, or their modified versions (Fig.S1). (Note that ± represents standard deviation throughout the paper.) We always found the same pattern: if the sequence abundance is used as the ground truth, Bracken and Kraken2 outperform MetaPhlAn2 and mOTUs2; while if the taxonomic abundance is used as the ground truth, MetaPhlAn2 and mOTUs2 outperform Bracken and Kraken2. This result strongly indicates that the benchmarking result of metagenomic profilers depends on the selected abundance type.
Moreover, we emphasize that even though the five distance/dissimilarity measures (BC, rJSD, L1, L2, and rAD) all showed the similar results in the performance evaluation (after the removal of false positives), L2 was not designed for compositional data analysis. To investigate whether the discriminating power of these distance measures for the two sequence types persists with varied microbial diversity, we simulated a set of abundance tables (for both taxonomic abundance and sequence abundance) with different species counts ranging from 10 to 500 (see Methods). We then calculated the distance or dissimilarity between the sequence abundance and taxonomic abundance profiles (Fig.S2). We found that with an increasing number of species, the discriminative power of L2 keeps decreasing, while BC, rJSD, L1 and rAD can still distinguish the two abundance types. This result suggests that L2 distance cannot discriminate the two types of relative abundances in microbiome samples of high species richness. This might be due to the fact that L2 distance is not appropriate for compositional data analysis at all26, 28.
Precision-recall analysis of different metagenomic profilers.
We emphasize that the above contradicting performance evaluations due to different abundance types cannot be detected by the Precision-Recall analysis. This is because Precision and Recall only concern the difference of presence/absence patterns in the ground truth and predicted abundance profiles, and by definition the ground truth sequence and taxonomic abundance profiles share exactly the same presence/absence pattern. We also want to emphasize that the evaluation of Precision and Recall is largely impacted by the reference database used by different profilers29. To avoid the bias introduced by the database differences, we selected genomes from the intersection among the reference databases of MetaPhlAn2, mOTUs2, and Kraken2/Bracken. This enables us to evaluate the Precision and Recall of different profilers in an unbiased manner (see Supplementary Note, Fig.3c–h and Figs.S3 for details).
Impact of abundance type on the alpha diversity calculation.
Interchanging sequence abundance and taxonomic abundance strongly influences per-sample summary statistics. To demonstrate this issue, we simulated 500 abundance profiles representing microbiota from distinct habitats (gut, oral, skin, vagina, and building, 100 profiles for each, see Methods) with known sequence abundance and taxonomic abundance profiles. Note that here we didn’t use profiling results generated by different metagenomic profilers. This is mainly because, in the empirical study, we found it is technically challenging to accurately convert sequence abundance to taxonomic abundance for all detected taxa, which can heavily impact the alpha diversity calculation. For simulated abundance profiles, the species richness will not be affected by using sequence abundance or taxonomic abundance as ground truth, because they share the same absence/presence pattern. However, we found that statistically the Shannon index, Simpson index, and Pielou’s evenness index calculated from taxonomic abundances are significantly higher than those calculated from sequence abundances (p-value<0.001, two sided Wilcoxon signed-rank test) regardless of the habitat (Fig.4a,c,e). Interestingly, when ranking the samples according to their alpha diversity measures calculated from sequence or taxonomic abundance, the rankings are not fully concordant with each other (Spearman correlation of the rank vectors is 0.929 ± 0.020 for Shannon index, 0.835 ± 0.042 for Simpson index, and 0.808±0.045 for Pielou’s evenness index). In fact, in the histograms of the differences between those indices calculated from sequence and taxonomic abundances (denoted as ΔShannon, ΔSimpson, ΔPielou), we found both negative and positive parts, despite the mean is always negative (Fig.4b,d,f). These results suggest that alpha diversity calculations and comparisons can be strongly affected by the type of relative abundance used.
Figure 4. Alpha diversity based on sequence abundance and taxonomic abundance.
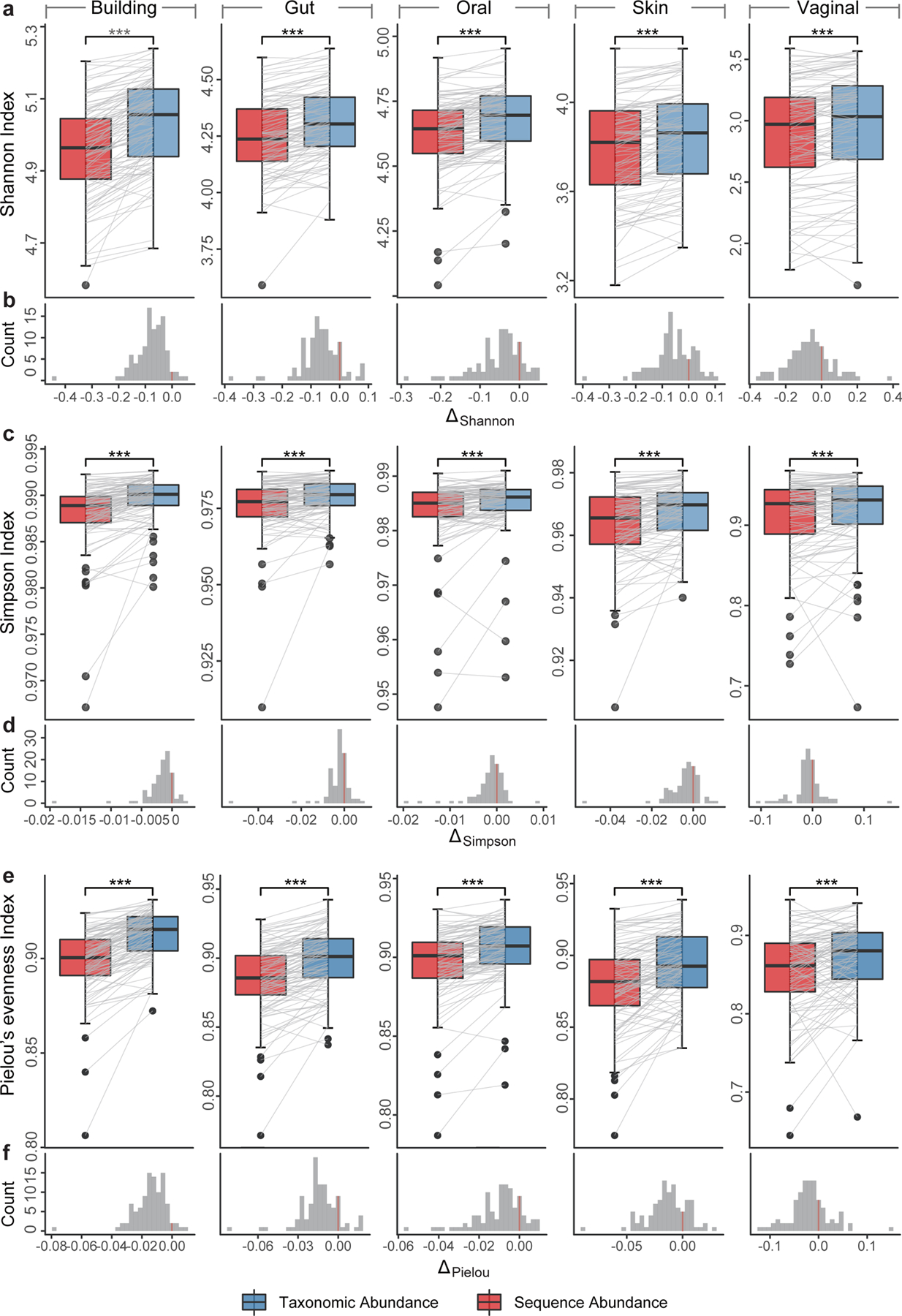
Alpha diversity (a-b: Shannon index; c-d: Simpson index; and e-f: Pielou’s evenness index) based on ground truth of simulated data from different habitats, and the histogram of the alpha-diversity difference calculated from two different abundance types. For each sample, the indices calculated from two abundance types were connected by a gray line to illustrate their difference. Significance levels: p-value<0.05 (*), <0.01 (**), <0.001 (***), NS (non-significance); two-sided Wilcoxon signed-rank test. Exact p-values are provided in the Source Data File.
Impact of abundance types on the beta diversity and ordination analyses.
To check if mixing sequence abundance and taxonomic abundance will also influence between-sample attributes such as beta diversity and ordination analyses, we re-analyzed the 500 samples generated for Fig.4. In order to quantify the impact of influence on beta diversity introduced by abundance type, we performed Mantel test30, 31 to compare the beta-diversity (in terms of BC, rJSD, L1, L2 and rAD) calculated from the taxonomic abundance and sequence abundance profiles of those samples (see Methods). Interestingly, regardless of the species richness in the habitats, the abundance type has some influence on the cross-sample comparisons based on the BC, rJSD and L1 measures (Spearman coefficient r = 0.944 ± 0.006, 0.947 ± 0.009, 0.944 ± 0.006, respectively; p-value =10−4 for all), but affects L2 and rAD more strongly (r = 0.844 ± 0.026, 0.519 ± 0.137, respectively; p-value=10−4 for both). These results demonstrate the inconsistent relative relationships between samples introduced by different abundance types in beta diversity calculation.
We then performed ordination analyses using four different methods: Non-metric Multidimensional Scaling (NMDS)32, Principal Coordinates Analysis (PCoA)33, t-distributed stochastic neighbor embedding (t-SNE)34, and Uniform Manifold Approximation and Projection (UMAP)35. We found that, regardless of the distance/dissimilarity measures used (e.g. rJSD, BC and rAD), taxonomic abundance and sequence abundance profiles are drastically different in all the four ordination results (Fig.5, Figs.S4–S5). We then performed Procrustes analysis36, 37 to analyze the congruence of two-dimensional shapes produced from superimposition of ordination analyses from two abundance types. We found very low similarity between the ordination results calculated from sequence and taxonomic abundances (Fig.5, Figs.S4–S5, Monte Carlo p-value<0.05). These results indicate that both beta diversity (especially for L2 and rAD) and ordination analyses can be heavily affected by the relative abundance type used.
Figure 5. . Ordination analyses of simulated profiles based on rJSD.
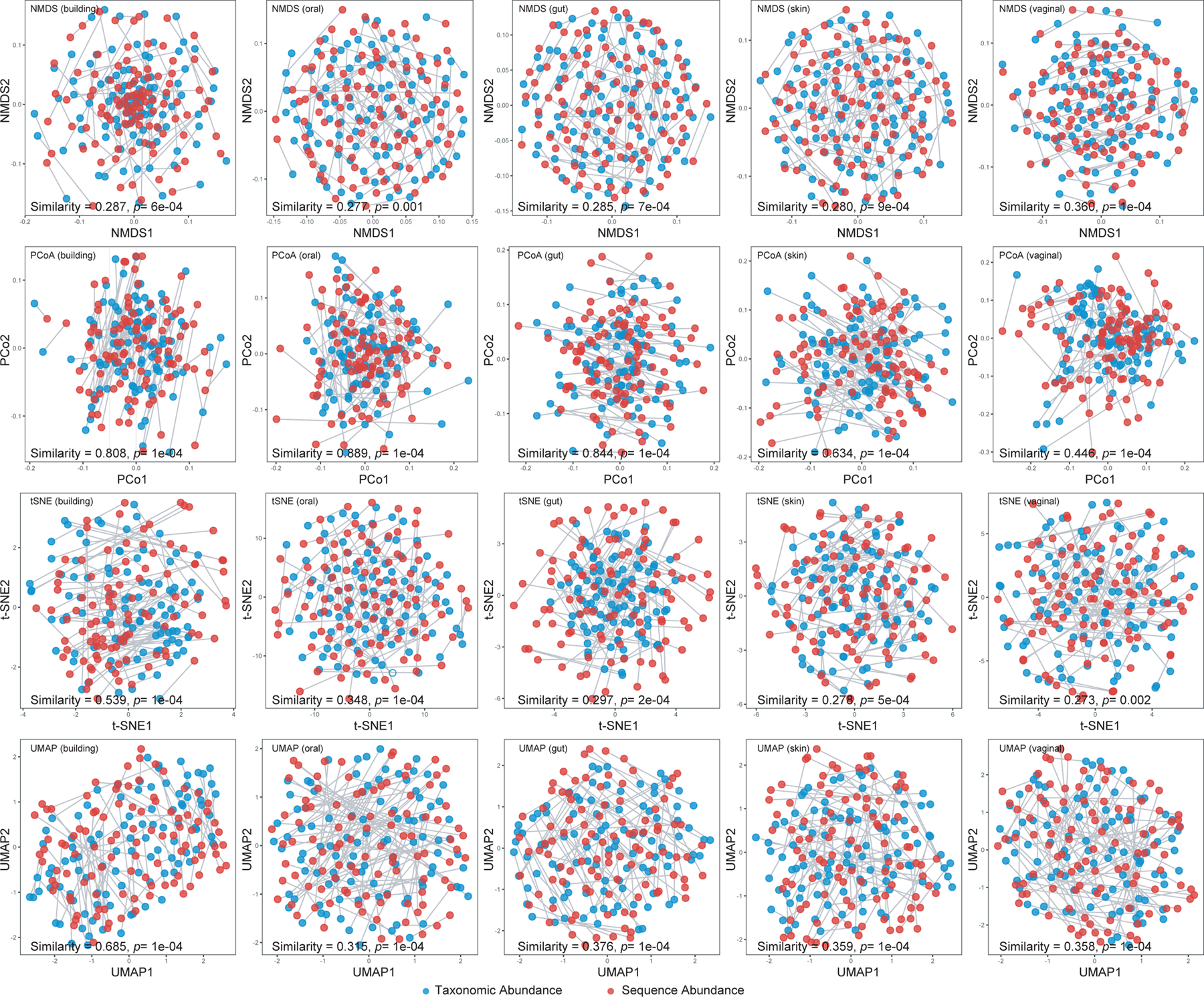
Scatter plots of NMDS, PCoA, t-SNE and UMAP illustrate the dissimilarities between the sequence abundance (red dots) and taxonomic abundance (blue dots), which are the ground truth of the simulated 100 gut profiles. Root Jensen-Shannon divergence (rJSD) was used in the ordination analyses. The plots of the ordination analyses based on sequence abundance and taxonomic abundance were adjusted to overlap with each other first, then the similarity was calculated by the Monte-Carlo test. For each simulated profile, the two dots (corresponding to two abundance types) were connected by a grey line to demonstrate the difference of their positions in the ordination plot.
Discussion
Taken together, we emphasize the importance of differentiating between sequence abundance and taxonomic abundance in metagenomic profiling. Ignoring this distinction can underestimate (or overestimate) the relative abundances of organisms with small (or large) genome sizes, respectively. Sequence abundances are typically produced by DNA-to-DNA or DNA-to-Protein methods, which rely on microbial genomes or genes as the reference database, report relative sequence abundance, i.e. the fraction of sequence reads assigned to each entity in the database. By contrast, DNA-to-Marker methods output relative taxonomic abundance representing the fraction of each detected taxon.
Our results demonstrate that misleading performance assessment of metagenomic profilers and spurious alpha and beta diversity patterns can arise from interchanging sequence abundance with taxonomic abundance. For alpha diversity measures (Shannon index, Simpson index, and Pielou’s evenness index), statistically they are higher based on taxonomic abundance than that based on sequence abundance. Yet, the relative rankings of those measures calculated from taxonomic or sequence abundances are not fully concordant with each other. Indeed, their differences (ΔShannon, ΔSimpson, ΔPielou) can be either negative or positive. Dramatic changes in the relative position between samples are also shown in the ordination analysis. Therefore, interchanging abundance types could have a deleterious effect on the interpretation of alpha and beta diversity analyses and meta-analyses.
The distinction between the two types of relative abundances was known to the field of microbiome research (at least to the developers of various metagenomic profilers), and has been conceptually considered in some benchmark studies (e.g., CAMI19). However, the consequences of ignoring this distinction for benchmarking metagenomic classifiers, per-sample summary statistics, and cross-sample comparisons have not been quantitatively studied or clearly illustrated so far. In particular, the vast majority of end users of those metagenomic profilers should be clearly aware of the distinction between sequence abundance and taxonomic abundance, and of the consequences of ignoring this distinction in selecting metagenomics tools, data interpretation, and cross-study comparison of differentially abundant taxa identified by different tools.
Theoretically, sequencing abundance can be converted to taxonomic abundance through genome-size and ploidy corrections, as shown in Eq. [2]. Yet, in reality, unknown microbial genomes/taxa, missing ploidy information, and misclassification of reads from conserved regions across different species render the conversion very challenging, if not impossible. It is not our intention to imply that one should convert sequence abundance to taxonomic abundance. In some cases, the conversion is actually not needed. For example, instead of converting sequence abundance to taxonomic abundance, DNA-to-Marker methods directly calculate taxonomic abundance as the ratio of sequence coverage of single-copy marker genes of each taxon to that of all taxa, which naturally avoids genome-size and copy-number corrections5.
In summary, we suggest that the whole microbiome research community should pay more attention to potentially misleading biological conclusions arising from the issue of ignoring the distinction between sequence and taxonomic abundances. In particular, we suggest that end users should be more careful in interpretating sequence abundance data. If we identified a low abundance of viruses/bacterial phages using Kraken or other similar profilers, then we should be aware of the potential abundance under-estimation. Similarly, if a fungus showed a high abundance in the data, it could be over-estimated especially when calculating bacteria-fungi ratio. Going forward, in future development or evaluation of metagenomic profilers, the type of the relative abundance should be strictly distinguished and labeled, especially when the sequence abundance is the default output. This would substantially improve the comparison of abundance estimations of metagenomic profilers and enhance the reproducibility and biological interpretation of microbiome studies. In most microbiome studies, we first need to address questions related to who are there and how abundant they are in a microbial community. In other words, taxonomic abundance is what we need first. Considering the challenges in genome-length and ploidy corrections for DNA-to-DNA methods, and the biological relevance of taxonomic abundance, the development of DNA-to-Marker methods should be more encouraged and appreciated by the whole microbiome research community.
Methods
Simulation of microbiome profiles.
In the simulation of microbiome profiles based on different species number (from 10 to 500 metagenomes representing different species), the abundance was created randomly from a log-normal distribution using “rlnorm” function in R language with parameters: meanlog = 0 and sdlog = 1, and 10 repeats were simulated for each species count. In the simulation of microbiome profiles for alpha diversity calculation, 100 profiles were simulated for each habitat, and species number in different habitats were set up as: 10–50 (vaginal), 50–100 (skin), 100–150 (gut), 150–200 (oral), 200–300 (building). The representative species in each specific habitat were selected based on the set of microbial species identified in the HMP38 and by Hsu et al.39.
Simulation of sequencing reads.
Firstly, the 25 microbiome profiles (five for each habitat) were simulated using the above method. Then the simulation of sequencing data is illustrated as the process in Fig.1a: Given a specified species composition (taxonomic abundance), their sequence abundance can be inferred accordingly (taxonomic abundance equals to sequence abundance divide by their genome length) and “Wgsim” (https://github.com/lh3/wgsim) was then used (with default parameters) to simulate the sequences. The selection of genomes for simulated data was based on the intersection between MetaPhlAn2 and mOTUs2 reference database and Bracken’s database to avoid database biases.
Currently, there are many more DNA-to-DNA profilers (e.g., Bracken and Kraken2) than DNA-to-Marker profilers (e.g., MetaPhlAn2 and mOTU2). In this paper we focused on two DNA-to-DNA profilers for the following reasons. First, as representative DNA-to-DNA methods, Bracken and Kraken/Kraken2 demonstrated the best performance in previous benchmarking studies6, 8, 20, and have been cited in more than one thousand microbiome studies. Second, mOTU2 and MetaPhlAn2 do not support custom reference databases, and the reference database is a critical factor affecting profiler performance. As such we decided to use the intersection of organisms in mOTU2, MetaPhlaAn2, and Kraken2 reference databases as the source for our simulation data. Introducing more DNA-to-DNA profilers could further reduce the reference database size of the simulated data and affect the diversity of genome sizes (Fig.S6).
Alpha and beta diversity calculation.
Alpha diversity calculation e.g. Shannon and Simpson indices were performed in R language by the “Vegan 2.5-6” package. As for the beta diversity, we employed “Vegan 2.5-6” for distance/dissimilarity calculation e.g. L1 (“Manhattan” in vegdist function), L2 (“Euclidean”) and BC (“Bray”), while rJSD and rAD were calculated by self-programmed script (see code availability). In the ordination analyses, R packages “ade4 1.7-15”, “Rtsne 0.15”, “ape 5.4-1” and “umap 0.2.6.0” were used to conduct the NMDS, t-SNE, PCoA and UMAP analyses separately. Since the iterative algorithm of NMDS, t-SNE and UMAP find different solutions depending on the starting point of the calculation (which is a randomly chosen configuration) we performed 101 repeats of NMDS, t-SNE, UMAP and their Procrustes test, the median result (sorting by the Mote-Caro test) was selected for presentation of similarity and p-value in Fig.5, Fig.S4 and Fig.S5. The ordination analyses based on the ground truth of the sequence abundance and taxonomic abundance for the 500 profiles (from five habitats) were conducted separately before Procrustes analysis.
Robust Aitchison distance calculation.
We applied DEICODE (https://github.com/biocore/DEICODE) to calculate the robust Aitchison distance (rAD) to benchmark the performance of metagenomics profilers. DEICODE represents a form of Aitchison Distance that is robust to high levels of sparsity. It preprocesses the compositional data using the centered log-ratio (CLR) transform only on the non-zero values of the data (hence no pseudo counts are used). Then it performs dimensionality reduction through robust PCA based on the non-zero values of the data. The Euclidean distance of the robust CLR-transformed abundance profiles (i.e., rAD) was finally employed to evaluate the performance of metagenomic profilers. To avoid the impact of false positives on the benchmarking results, we further filtered out false positives in all output taxonomic profiles and compared the performance of different profilers using rAD calculated from the true positives only. This is termed as the modified rAD in Fig.3. For other evaluation measures, the same procedure was performed and presented in Fig.S1.
Mantel Test.
Mantel test was used as a correlation test to determine the correlation between two beta diversity (BC, rJSD, L1, L2 and rAD) matrices based on sequence abundance and taxonomic abundance. In order to calculate the correlation, the matrix values of both matrices are ‘unfolded’ into long column vectors, which are then used to determine correlation. Permutations (n=9999) of one matrix are used to determine significance. Whether distances between samples in one matrix are correlated with the distances between samples in the other matrix is revealed by the p-value.
Procrustes analysis.
Procrustes analysis (by R package “ade4 1.7-15”) typically takes as input two coordinate matrices with matched sample points, and transforms the second coordinate set by rotating, scaling, and translating it to maximize the similarity between corresponding sample points in the two shapes. It allows us to determine whether we would come to same conclusions on the beta diversity, regardless of which distance/dissimilarity measure was used to compare the samples. To assess the significance level of observed similarity between two matrices, empirical p-values are calculated using a Monte Carlo simulation. Basically, sample labels are shuffled in one of the coordinate matrices, and then the similarity between them is re-computed for 9999 times. Here, similarity is calculated as the sum of the squared residual deviations between sample points for each measurement. The proportion of similarity values that are equal to or lower than the observed similarity value is then the Monte Carlo or empirical p-value.
Data availability.
All the simulated datasets can be downloaded here: https://figshare.com/projects/Challenges_in_Benchmarking_Metagenomic_Profilers/79916
Code availability.
R scripts used in this paper is available at https://github.com/shihuang047/re-benchmarking
Supplementary Material
Acknowledgements.
Research reported in this publication was supported by grants R01AI141529, R01HD093761, R01AG067744, UH3OD023268, U19AI095219, and U01HL089856 from National Institutes of Health. This work was also supported by IBM Research through the AI Horizons Network, UC San Diego AI for Healthy Living program in partnership with the UC San Diego Center for Microbiome Innovation.
Footnotes
Competing interests. The authors declare no competing interests.
Reference:
- 1.Knight R et al. Best practices for analysing microbiomes. Nature Reviews Microbiology 16, 410–422 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Ye SH, Siddle KJ, Park DJ & Sabeti PC Benchmarking metagenomics tools for taxonomic classification. Cell 178, 779–794 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu B, Gibbons T, Ghodsi M, Treangen T & Pop M Accurate and fast estimation of taxonomic profiles from metagenomic shotgun sequences. BMC genomics 12 Suppl 2, S4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arumugam M et al. Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milanese A et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nature communications 10, 1014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wood DE, Lu J & Langmead B Improved metagenomic analysis with Kraken 2. Genome biology 20, 257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wood DE & Salzberg SL Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome biology 15, R46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu J, Breitwieser FP, Thielen P & Salzberg SL Bracken: estimating species abundance in metagenomics data. Peerj Computer Science 3, e104 (2017). [Google Scholar]
- 9.Kostic AD et al. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nature biotechnology 29, 393–396 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menzel P, Ng KL & Krogh A Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nature communications 7, 11257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchfink B, Xie C & Huson DH Fast and sensitive protein alignment using DIAMOND. Nature methods 12, 59–60 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Truong DT et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nature methods 12, 902–903 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Segata N et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nature methods 9, 811–814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sunagawa S et al. Metagenomic species profiling using universal phylogenetic marker genes. Nature Methods 10, 1196–1199 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Nurk S, Meleshko D, Korobeynikov A & Pevzner PA metaSPAdes: a new versatile metagenomic assembler. Genome research 27, 824–834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li D et al. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Mavromatis K et al. Use of simulated data sets to evaluate the fidelity of metagenomic processing methods. Nature methods 4, 495–500 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Meyer F et al. Assessing taxonomic metagenome profilers with OPAL. Genome biology 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sczyrba A et al. Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software. Nature methods 14, 1063–1071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McIntyre ABR et al. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome biology 18, 182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindgreen S, Adair KL & Gardner PP An evaluation of the accuracy and speed of metagenome analysis tools. Scientific reports 6, 19233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen F, Mackey AJ, Vermunt JK & Roos DS Assessing performance of orthology detection strategies applied to eukaryotic genomes. PloS one 2, e383–e383 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soppa J Polyploidy in archaea and bacteria: about desiccation resistance, giant cell size, long-term survival, enforcement by a eukaryotic host and additional aspects. Journal of molecular microbiology and biotechnology 24, 409–419 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Mendell JE, Clements KD, Choat JH & Angert ER Extreme polyploidy in a large bacterium. Proceedings of the National Academy of Sciences of the United States of America 105, 6730–6734 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gloor GB, Macklaim JM, Pawlowsky-Glahn V & Egozcue JJ Microbiome Datasets Are Compositional: And This Is Not Optional. Frontiers in microbiology 8, 2224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aitchison J On criteria for measures of compositional distance. Mathematical Geology 24, 365–379 (1992). [Google Scholar]
- 27.Martino C et al. A Novel Sparse Compositional Technique Reveals Microbial Perturbations. mSystems 4, e00016–00019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aitchison J, Barceló-Vidal C, Martín-Fernández JA & Pawlowsky-Glahn V Logratio Analysis and Compositional Distance. Mathematical Geology 32, 271–275 (2000). [Google Scholar]
- 29.Breitwieser FP, Lu J & Salzberg SL A review of methods and databases for metagenomic classification and assembly. Briefings in bioinformatics 20, 1125–1136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Legendre P, Borcard D & Peres-Neto PR Analyzing beta diversity: partitioning the spatial variation of community composition data. Ecological Monographs 75, 435–450 (2005). [Google Scholar]
- 31.Mantel N The detection of disease clustering and a generalized regression approach. Cancer research 27, 209–220 (1967). [PubMed] [Google Scholar]
- 32.Faith DP, Minchin PR & Belbin L Compositional dissimilarity as a robust measure of ecological distance. Vegetatio 69, 57–68 (1987). [Google Scholar]
- 33.Legendre P & Gallagher ED Ecologically meaningful transformations for ordination of species data. Oecologia 129, 271–280 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Hinton G Visualizing High-Dimensional Data Using t-SNE. Vigiliae Christianae 9, 2579–2605 (2008). [Google Scholar]
- 35.McInnes L & Healy J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. The Journal of Open Source Software 3, 861 (2018). [Google Scholar]
- 36.Dray S, Chessel D & Thioulouse J Procrustean co-inertia analysis for the linking of multivariate datasets. Écoscience 10, 110–119 (2003). [Google Scholar]
- 37.Digby P & Kempton R Multivariate Analysis of Ecological Communities. Population and Community Biology (1987). [Google Scholar]
- 38.Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu T et al. Urban Transit System Microbial Communities Differ by Surface Type and Interaction with Humans and the Environment. mSystems 1, e00018–00016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the simulated datasets can be downloaded here: https://figshare.com/projects/Challenges_in_Benchmarking_Metagenomic_Profilers/79916