

Abstract
Selective autophagy requires the autophagy receptor specifically localizing to the target for degradation. In the budding yeast, Atg39 and Atg40 function as an autophagy receptor for the endoplasmic reticulum (ER)-selective autophagy, referred to as ER-phagy. The expression level of the ATG39 gene is increased in response to ER stress and nitrogen starvation. Under unstressed conditions, ATG39 transcription is repressed by Mig1/2 repressors. ER stress activates Snf1 AMP-activated protein kinase (AMPK), which negatively regulates Mig1/2 and consequently derepresses ATG39 transcription. However, ATG39 expression is still induced by ER stress and nitrogen starvation in the absence of Snf1, suggesting that additional molecules are involved in regulation of ATG39 expression. Here, we identify Msn2/4 transcription factors as an activator of ATG39 transcription. Not only ATG39 promoter activity but also ER-phagy are downregulated by loss of Msn2/4 and disruption of Msn2/4-binding consensus sequences located in the ATG39 promoter. We also find that the cAMP-dependent protein kinase pathway is involved in Msn2/4-mediated transcriptional regulation of ATG39. Our results suggest that yeast ER-phagy is appropriately controlled through modulation of the expression level of the ER-phagy receptor involving multiple signaling pathways and transcription factors.
Subject terms: Autophagy, Cell signalling, Organelles
Introduction
Autophagy is an evolutionarily conserved process which degrades intracellular components1,2. Autophagy is categorized into two types, macroautophagy and microautophagy. In macroautophagy, double-membrane vesicles termed autophagosomes are generated to transport target constituents into degradative organelles (the vacuole in the budding yeast). On the other hand, microautophagy transports the targets without forming autophagosomes. Macroautophagy is further categorized into two types, non-selective macroautophagy and selective macroautophagy, which degrade intracellular components non-specifically and specifically, respectively3–5. Selective macroautophagy includes mitophagy, pexophagy, and ER-phagy, which specifically degrade mitochondria, peroxisomes, and the endoplasmic reticulum (ER), respectively.
In selective macroautophagy, the autophagy receptor localizing to the target organelle is required for recruiting the core autophagy-related proteins that function in the autophagosome formation3–5. In the budding yeast, Atg39 and Atg40 have been identified as an autophagy receptor specific for ER-phagy6. The budding yeast ER consists of two distinct regions, the perinuclear ER and the cortical ER, which are connected by cytoplasmic ER7. Atg39 and Atg40 show the distinct localization pattern in cells treated with rapamycin, a compound that mimics nitrogen starvation6: Atg39 specifically localizes to the perinuclear ER; Atg40 predominantly localizes to the cortical and cytoplasmic ER. Reflecting their localization pattern, the perinuclear ER is mainly degraded by Atg39 after rapamycin treatment, whereas the cortical and cytoplasmic ER is mainly degraded by Atg406. ER-phagy is also initiated by ER stress, the condition where aberrant proteins accumulate in the ER lumen and membrane8,9. However, differently from rapamycin treatment, Atg39 plays a major role in degradation of both the perinuclear ER and the cortical and cytoplasmic ER during ER stress response9.
Regulation of the expression levels of the core autophagy-related proteins and the autophagy receptors is crucial for modulating the activity of autophagy8–14. The core autophagy-related proteins are induced when cells are cultivated under nitrogen-starved conditions. The mRNA levels of the core autophagy-related genes are controlled at both the transcriptional and post-transcriptional levels11–14. Like the core autophagy-related proteins, the expression levels of autophagy receptors are elevated under conditions that induce selective autophagy6,15,16. The expression levels of ATG39 and ATG40 are upregulated by nitrogen starvation and ER stress6,8,9. ATG40 expression is regulated by the Pho23-Rpd3L histone deacetylase complex8. We have previously identified Snf1 AMP-activated protein kinase (AMPK) and two closely related transcriptional repressors, Mig1 and Mig2, as a regulator of ATG39 expression9. Under unstressed conditions, Mig1 and Mig2 redundantly repress ATG39 promoter activity. ER stress leads to activation of Snf1, which consequently promotes nuclear export of Mig1 and Mig2 to derepress the ATG39 promoter. However, the existence of additional molecules regulating ATG39 promoter activity has been predicted from the following observations: ER stress upregulated ATG39 expression in the mig1 mig2 double mutant cells; ATG39 induction in response to nitrogen starvation was normally occurred in the snf1 mutant cells.
In this study, we identified two closely related transcription factors, Msn2 and Msn4, as a regulator of ATG39 expression by a genetic approach using the reporter cells. Loss of Msn2 and Msn4 diminished ATG39 induction caused by ER stress and nitrogen starvation. Activation of the ATG39 promoter induced by ER stress and nitrogen starvation was significantly inhibited by disruption of Msn2/4-binding consensus sequences located in the ATG39 promoter. Mutations in the MSN2 and MSN4 genes reduced the activity of ER-phagy under ER-stressed and nitrogen-starved conditions. We also found that loss of Pde1/2 cyclic AMP (cAMP) phosphodiesterases downregulates ATG39 promoter activity and ER-phagy. These results suggest that ER-phagy is positively regulated by Msn2/4 through activation of the ATG39 promoter and that Msn2/4 themselves are negatively regulated by protein kinase A (PKA).
Results
Development of the reporter cell to monitor ATG39 promoter activity
We previously showed that the ATG39 promoter is activated by ER stress and nitrogen starvation9. Activation of the ATG39 promoter caused by ER stress is only partially reduced by snf1 mutation. Furthermore, the ATG39 promoter is normally activated by nitrogen starvation in the snf1 mutant cells. Thus, additional molecules should regulate ATG39 promoter activity. To measure ATG39 promoter activity indirectly by monitoring the cell growth and further identify the regulator of ATG39 transcription genetically, we constructed the PATG39-HIS3 reporter which expresses the HIS3 gene under the control of the ATG39 promoter. Previously, we found that ATG39 promoter activity under unstressed conditions is enhanced by reg1 mutation which causes Snf1 hyperactivation9,17–19. Therefore, we first compared growth between wild-type and reg1 mutant cells when PATG39-HIS3 was provided as a sole functional HIS3 gene. Compared with wild-type cells, reg1 mutant cells grew slowly on synthetic defined media containing histidine (SD + His) (Fig. 1A). Nevertheless, reg1 mutant cells could grow on synthetic defined media lacking histidine (SD-His) at a level comparable to wild-type cells. Furthermore, reg1 mutant cells grew better than wild-type cells in the presence of 3-amino-1H-1,2,4-triazole (AT), a competitor of the HIS3 gene product. Increased growth observed in reg1 mutant cells was suppressed by snf1 mutation, indicating that this reg1 phenotype is caused by hyperactivation of Snf1. We performed similar experiments using the PMCM2-HIS3 construct which expresses the HIS3 gene under the control of the MCM2 promoter. The promoter activity of MCM2 under unstressed conditions was relatively higher than that of ATG39 and was unaffected by reg1 mutation (Supplementary Fig. 1). Consistent with this, histidine depletion had little effects on the growth of wild-type and reg1 mutant cells harboring PMCM2-HIS3 (Fig. 1A). These results indicate that the PATG39-HIS3 reporter can be utilized in monitoring ATG39 promoter activity.
Figure 1.

ATG39 expression is increased by Msn2/4 overexpression. (A) Effects of Snf1 activity on growth of cells harboring the PATG39-HIS3 reporter. Wild-type (WT) and indicated mutant strains harboring the PATG39-HIS3 or PMCM2-HIS3 reporters were spotted onto synthetic defined media lacking or containing histidine (His) and 3-amino-1H-1,2,4-triazole (AT), and incubated at 25 °C. (B) Effects of MSN2/4 overexpression on growth of cells harboring the PATG39-HIS3 reporter. Wild-type (WT) cells harboring the PATG39-HIS3 reporter and the plasmids with or without the MSN2/4 genes were spotted onto synthetic defined media lacking or containing histidine (H) and 3-amino-1H-1,2,4-triazole (AT), and incubated at 25 °C. (C) Effects of MSN2/4 overexpression on expression of the PATG39-GFP reporter. Wild-type strains harboring the PATG39-GFP reporter and the plasmids with or without the MSN2/4 genes were grown at 25 °C until exponential phase and then harvested. The GFP mRNA levels were quantified by qRT-PCR analysis, and relative mRNA levels were calculated using ACT1 mRNA. The values are plotted as the fold change from cells harboring the empty plasmids. The data show mean ± SEM (n = 3). **P < 0.01 as determined by Student’s t-test. (D,E) Effects of MSN2/4 overexpression on the mRNA levels of ATG39 (D) and ATG40 (E). Wild-type strains harboring the plasmids with or without the MSN2/4 genes were grown at 25 °C until exponential phase and then harvested. The mRNA levels were quantified by qRT-PCR analysis, and relative mRNA levels were calculated using ACT1 mRNA. The values are plotted as the fold change from cells harboring the empty plasmids. The data show mean ± SEM (n = 3). **P < 0.01 as determined by Student’s t-test. NS, not statistically significant (P > 0.05).
Msn2/4 overexpression facilitates ATG39 promoter activity
We screened a multicopy genomic library to identify genes whose overexpression facilitates growth of cells harboring PATG39-HIS3 on synthetic defined media lacking histidine and containing AT (SD-His + AT). We isolated 16 plasmids which facilitate cell growth on SD-His + AT and were categorized into 4 groups (Supplementary Table 1). The group I was comprised of 8 plasmids that carry genomic regions including the ATR1 (AminoTriazole Resistance 1) gene20, suggesting that these plasmids confer AT resistance without upregulating HIS3 expression. The group II included 4 plasmids, which contain the MSN2 gene encoding a transcription factor18,21. The groups III and IV were comprised of 3 and 1 plasmids, respectively, and they carried a gene known to encode a negative regulator of the RAS/protein kinase A (PKA) pathway, PDE2 or GPB118,22,23. PKA negatively regulates Msn2 through phosphorylation18,24,25. Therefore, we hypothesized that Msn2 overexpression upregulates ATG39 transcription and that overexpression of Pde2 and Gpb1 activates Msn2 by downregulating the PKA activity. To examine whether Msn2 overexpression really increases growth of cells harboring PATG39-HIS3 on SD-His + AT, we constructed a multicopy plasmid expressing Msn2 alone. Overexpression of Msn2 did not change the growth rate of the reporter cell on SD + His but increased it on SD-His + AT (Fig. 1B). The budding yeast has a gene closely related to MSN2, MSN418,21. Therefore, we tested whether Msn4 is also involved in ATG39 expression. Msn4 overexpression facilitated growth of the reporter cell on SD-His + AT, but not on SD + His (Fig. 1B).
We next investigated whether the mRNA expressed from the ATG39 promoter is increased by Msn2/4 overexpression using the PATG39-GFP reporter which expresses the GFP gene under the control of the ATG39 promoter9. The quantitative real-time RT-PCR (qRT-PCR) analysis showed that GFP expression from the PATG39-GFP reporter was enhanced by Msn2/4 overexpression (Fig. 1C). We next examined the effect of Msn2/4 overexpression on the expression levels of endogenous genes. ATG39 expression was upregulated by Msn2/4 overexpression (Fig. 1D). However, Msn2/4 overexpression had no effect on the expression level of ATG40, another gene encoding an ER-phagy receptor (Fig. 1E). These results suggest that Msn2/4 overexpression specifically increases ATG39 transcription.
Msn2/4 activate ATG39 promoter activity during ER stress response and nitrogen starvation
ATG39 expression is induced by ER stress and nitrogen starvation6,9. We therefore asked if Msn2/4 are involved in ATG39 induction caused by ER stress. In wild-type cells, ATG39 mRNA was markedly increased by treatment with tunicamycin, which causes ER stress by inhibiting N-linked glycosylation (Fig. 2A). ATG39 induction was diminished in the msn2 msn4 double mutant cells, although it was normally occurred in the msn2 and msn4 single mutant cells (Fig. 2A and Supplementary Fig. 2A). We also found that msn2 msn4 double mutations reduced ATG39 induction caused by dithiothreitol (DTT), which causes ER stress by inhibiting the disulfide bond formation (Fig. 2B). We next examined whether Msn2/4 are involved in ATG39 induction caused by nitrogen starvation. ATG39 induction was diminished in the msn2 and msn4 single mutant cells (Supplementary Fig. 2B). The msn2 and msn4 mutations synergistically reduced ATG39 induction (Fig. 2C). We also measured ATG40 mRNA levels and found that ATG40 induction in response to ER stress and nitrogen starvation was unaffected by msn2 msn4 double mutations (Supplementary Fig. 3). These results suggest that Msn2/4 are specifically involved in ATG39 induction.
Figure 2.

ATG39 expression is reduced by msn2 msn4 mutations. (A–C) The ATG39 mRNA levels in msn2 msn4 mutant. Wild-type (WT) and msn2 msn4 mutant strains were grown at 25 °C until exponential phase and treated with 3 μg/ml tunicamycin (TM) (A) or 6 mM dithiothreitol (DTT) (B) or incubated under nitrogen-starved conditions (C) for the indicated time. The ATG39 mRNA levels were quantified by qRT-PCR analysis, and relative mRNA levels were calculated using ACT1 mRNA. The values are plotted as the fold change from wild-type cells at the time of ER stressors addition or nitrogen removal. The data show mean ± SEM (n > 4). *P < 0.05 and **P < 0.01 as determined by Student’s t-test. (D,E) Expression of PATG39-GFP reporter in msn2 msn4 mutant. Wild-type (WT) and indicated mutant strains were grown at 25 °C until exponential phase and treated with 3 μg/ml tunicamycin (TM) (D) or incubated under nitrogen-starved conditions (E) for the indicated time. The ATG39 mRNA levels were quantified by qRT-PCR analysis, and relative mRNA levels were calculated using ACT1 mRNA. The values are plotted as the fold change from wild-type cells at the time of TM addition or nitrogen removal. The data show mean ± SEM (n > 3). **P < 0.01 as determined by Student’s t-test.
ER stress and nitrogen starvation enhance ATG39 promoter activity9. Therefore, we examined whether Msn2/4 are involved in activation of the ATG39 promoter in response to ER stress and nitrogen starvation. GFP expression from the PATG39-GFP reporter was increased by ER stress and nitrogen starvation; however, their induction was significantly diminished by msn2 msn4 double mutations (Fig. 2D,E). These results suggest that during ER stress response and nitrogen starvation, Msn2/4 positively regulate ATG39 expression via its promoter.
Msn2/4 regulate ATG39 promoter activity via the STRE elements
Msn2/4 activate transcription of stress responsive genes by binding to the STRE element, 5′-CCCCT-3′, located in the promoter of their target genes26,27. Our analysis using JASPAR, a database of transcription factor binding profiles (http://jaspar.genereg.net/), revealed that two putative STRE elements exist in the ATG39 promoter (Fig. 3A). To examine whether the STRE elements are required for Msn2/4 to upregulate ATG39 transcription, we mutated them in the PATG39-GFP reporter. Msn4 overexpression failed to increase GFP expression from the PATG39-GFP reporter mutated in the STRE elements (Fig. 3B). We next asked whether the STRE elements are important for activation of the ATG39 promoter in response to ER stress. Activation of the ATG39 promoter was diminished by either of the mutations in two STRE elements (Supplementary Fig. 4A). They synergistically reduced ATG39 promoter activity during ER stress response (Fig. 3C and Supplementary Fig. 4A). Similar results were obtained when the ATG39 promoter was activated by nitrogen starvation (Fig. 3D and Supplementary Fig. 4B). These results suggest that Msn2/4 positively regulate ATG39 expression through the STRE elements located in its promoter.
Figure 3.

Msn2/4 positively regulate ATG39 promoter activity through the STRE elements. (A) Two putative STRE elements in ATG39 promoter region. (B) Effects of MSN4 overexpression on expression of the PATG39-GFP reporter mutated in putative STRE elements. Wild-type (WT) strains harboring the PATG39-GFP reporter mutated in putative STRE elements and the plasmids with or without the MSN4 gene were grown at 25 °C until exponential phase, and then harvested. The GFP mRNA levels were quantified by qRT-PCR analysis, and relative mRNA levels were calculated using ACT1 mRNA. The values are plotted as the fold change from cells harboring the plasmids without the MSN4 gene. The data show mean ± SEM (n = 4). NS, not statistically significant (P > 0.05), as determined by Student’s t-test. (C,D) Effects of mutations in putative STRE elements on ATG39 upregulation induced by ER stress and nitrogen starvation. Wild-type (WT) cells harboring wild-type or mutated PATG39-GFP reporters were grown at 25 °C until exponential phase and treated with 3 μg/ml tunicamycin (TM) (C) or incubated under nitrogen-starved conditions (D) for the indicated time. The GFP mRNA levels were quantified by qRT-PCR analysis, and relative mRNA levels were calculated using ACT1 mRNA. The values are plotted as the fold change from wild-type cells harboring the integration which expresses GFP under the control of wild-type ATG39 promoter at the time of TM addition or nitrogen removal. The data show mean ± SEM (n = 3). **P < 0.01 as determined by Student’s t-test.
Msn2/4 positively regulate ER-phagy
Next, we investigated whether ATG39 induction mediated by Msn2/4 is important for activation of ER-phagy. We previously revealed that Atg39 acts as a major ER-phagy receptor in ER stress response9: diminished ATG39 induction in response to ER stress reduces degradation of Sec63 ER transmembrane protein. Therefore, we investigated the role of Msn2/4 in ER stress-induced ER-phagy using the strain which expresses the carboxyl-terminally GFP-tagged Sec63 (Sec63-GFP)6,9. GFP is resistant to the vacuole-resident proteases, and thus autophagic degradation of GFP-tagged protein yields free GFP28. When wild-type cells expressing Sec63-GFP were treated with tunicamycin, free GFP production from Sec63-GFP was observed9 (Fig. 4A). Compared with wild-type cells, free GFP was decreased in the msn2 msn4 double mutant cells (Fig. 4A). This result suggests that Msn2/4 are involved in autophagic degradation of Sec63-GFP caused by ER stress. We next examined whether msn2 msn4 double mutations generally reduce autophagic activities during ER stress response. We monitored non-selective autophagy using strains that express the cytoplasmic Pgk1 tagged by GFP (Pgk1-GFP)29. Autophagic degradation of Pgk1-GFP in the msn2 msn4 double mutant cells was comparable to that in wild-type cells (Fig. 4B). This result suggests that Msn2/4 are dispensable for activation of non-selective autophagy during ER stress response.
Figure 4.

Msn2/4 positively regulate ER-phagy. (A) Sec63-GFP degradation in ER-stressed msn2 msn4 mutant. Wild-type (WT) and msn2 msn4 mutant strains harboring GFP-tagged SEC63 were grown at 25 °C until exponential phase and treated with 3 μg/ml tunicamycin (TM) for 18 h. Extracts prepared from each cell were immunoblotted with anti-GFP antibodies. The intensities of free GFP were measured and normalized to the Sec63-GFP level. The values are plotted as the fold change from wild-type cells. The data show mean ± SEM (n = 3). *P < 0.01 as determined by Student’s t-test. (B) Pgk1-GFP degradation in ER-stressed msn2 msn4 mutant. Wild-type (WT) and msn2 msn4 mutant strains harboring GFP-tagged PGK1 were grown at 25 °C until exponential phase and treated with 3 μg/ml tunicamycin (TM) for 18 h. Extracts prepared from each cell were immunoblotted with anti-GFP antibodies. The intensities of free GFP were measured and normalized to the Pgk1-GFP level. The values are plotted as the fold change from wild-type cells. The data show mean ± SEM (n = 3). NS, not statistically significant (P > 0.05), as determined by Student’s t-test. (C) Sec63-GFP degradation in nitrogen-starved msn2 msn4 mutant. Wild-type (WT) and msn2 msn4 mutant strains harboring GFP-tagged SEC63 were grown at 25 °C until exponential phase and incubated under nitrogen-starved conditions for 18 h. Extracts prepared from each cell were immunoblotted with anti-GFP antibodies. The intensities of free GFP were measured and normalized to the Sec63-GFP level. The values are plotted as the fold change from wild-type cells. The data show mean ± SEM (n = 3). NS, not statistically significant (P > 0.05), as determined by Student’s t-test. (D) Effects of msn2 msn4 mutations on Sec63-GFP degradation in the atg40 mutant cells. Indicated mutant strains harboring GFP-tagged SEC63 were grown at 25 °C until exponential phase and incubated under nitrogen-starved conditions for 18 h. Extracts prepared from each cell were immunoblotted with anti-GFP antibodies. The intensities of free GFP were measured and normalized to the Sec63-GFP level. The values are plotted as the fold change from wild-type cells. The data show mean ± SEM (n = 4). **P < 0.01 as determined by Student’s t-test. (E) Sec63-GFP degradation in cells expressing ATG39 under the control of mutated ATG39 promoter. The atg39 mutant strains harboring GFP-tagged SEC63 and the integration which expresses ATG39 under the control of wild-type or mutated ATG39 promoter were grown at 25 °C until exponential phase and treated with 3 μg/ml tunicamycin (TM) for 18 h. Extracts prepared from each cell were immunoblotted with anti-GFP antibodies. The intensities of free GFP were measured and normalized to the Sec63-GFP level. The values are plotted as the fold change from cells harboring the wild-type ATG39 integration. The data show mean ± SEM (n = 4). **P < 0.01 as determined by Student’s t-test.
We next examined whether Msn2/4 are involved in nitrogen starvation-induced ER-phagy. Nitrogen starvation caused Sec63-GFP degradation in the msn2 msn4 double mutant cells at a level comparable to that in wild-type cells (Fig. 4C). This result seems to be consistent with our previous observation that degradation of Sec63-GFP in response to nitrogen starvation is unaffected by the atg39 single mutation9. We previously revealed that Sec63-GFP degradation in response to nitrogen starvation is partly blocked by atg40 single mutation, and this atg40 phenotype is enhanced by atg39 mutation9. Accordingly, the inhibitory effect of msn2 msn4 double mutations on nitrogen starvation-induced ER-phagy might be detected only in an atg40 mutant background. Alternatively, reduced ATG39 induction in the msn2 msn4 double mutant cells might be enough to activate Atg39-dependent ER-phagy normally, since ATG39 induction caused by nitrogen starvation is significantly greater than that caused by ER stress (Fig. 2A–C). Therefore, we compared degradation of Sec63-GFP in response to nitrogen starvation between the atg40 single mutant and the atg40 msn2 msn4 triple mutant cells. The msn2 msn4 double mutations decreased Sec63-GFP degradation in nitrogen-starved atg40 mutant cells (Fig. 4D). These results suggest that Msn2/4 are involved in ER-phagy induced by ER stress and nitrogen starvation.
We next investigated whether transcriptional activation of the ATG39 gene mediated by Msn2/4 is important for regulation of ER-phagy. To test this, we generated strains expressing Atg39 under the control of the ATG39 promoter mutated in the STRE elements. Sec63-GFP degradation in response to ER stress was decreased when Atg39 was expressed from the ATG39 promoter mutated in the STRE elements (Fig. 4E). This result suggests that the control of ATG39 promoter activity via the STRE elements is critical for regulation of ER-phagy.
The PKA pathway is involved in regulation of ATG39 expression and ER-phagy
We previously revealed that Snf1 is involved in transcriptional activation of ATG39 during ER stress response9. To examine the relationship between Msn2/4 and Snf1, we constructed the msn2 msn4 snf1 triple mutant cells. ATG39 induction after ER stress treatment was decreased by the msn2 msn4 snf1 triple mutations compared with the msn2 msn4 double mutations and the snf1 single mutation (Supplementary Fig. 5). This result suggests that Msn2/4 and Snf1 independently regulate ATG39 transcription.
Here, we isolated the multicopy plasmid carrying the PDE2 gene by the genetic screen. Pde2 is a cyclic AMP (cAMP) phosphodiesterase and downregulates the PKA activity by decreasing the level of cAMP18,22. It is well-known that PKA negatively regulates Msn2/418,24,25. These previous findings raised the possibility that Pde2 positively regulates ATG39 expression and ER-phagy upstream of Msn2/4. We first examined the effect of Pde2 overexpression on growth of cells harboring PATG39-HIS3. Introduction of a multicopy plasmid expressing Pde2 facilitated growth of the reporter cell on SD-His + AT (Supplementary Fig. 6A). We also found that mRNAs from endogenous ATG39 gene and the PATG39-GFP reporter were increased by introduction of a multicopy plasmid expressing Pde2 (Fig. 5A and Supplementary Fig. 6B). These results suggest that ATG39 promoter activity is upregulated by Pde2 overexpression. Next, we examined whether ATG39 upregulation caused by Pde2 overexpression depends on the function of Msn2/4. In the msn2 msn4 double mutant cells, Pde2 overexpression failed to increase GFP expression from the PATG39-GFP reporter (Fig. 5A). Furthermore, mutations of the STRE elements in the PATG39-GFP reporter significantly inhibited GFP upregulation caused by Pde2 overexpression (Fig. 5B). These results suggest that Msn2/4 are required for Pde2 to upregulate ATG39 promoter activity. We next investigated whether Pde2 is involved in ATG39 induction in response to ER stress and nitrogen starvation. The budding yeast possesses another cAMP phosphodiesterase which is encoded by the PDE1 gene30, and Pde1 functions redundantly with Pde2 to regulate the cAMP level18,30,31. Therefore, we constructed the pde1 pde2 double mutant cells. Similar to msn2 msn4 double mutations, pde1 pde2 double mutations partly but significantly diminished ATG39 induction caused by ER stress and nitrogen starvation (Fig. 5C,D). These results suggest that Pde1/2 positively regulate ATG39 expression under ER-stressed and nitrogen-starved conditions.
Figure 5.

Pde1/2 positively regulate ATG39 expression via Msn2/4. (A) Effects of PDE2 overexpression on expression of the PATG39-GFP reporter in the msn2 msn4 mutant cells. Wild-type (WT) and msn2 msn4 mutant strains harboring the PATG39-GFP reporter and the plasmids with or without the PDE2 genes were grown at 25 °C until exponential phase and then harvested. The GFP mRNA levels were quantified by qRT-PCR analysis, and relative mRNA levels were calculated using ACT1 mRNA. The values are plotted as the fold change from wild-type cells harboring the empty plasmids. The data show mean ± SEM (n = 4). **P < 0.01 as determined by Student’s t-test. (B) Effects of PDE2 overexpression on expression of mutated PATG39-GFP reporter. Wild-type (WT) cells harboring wild-type or mutated PATG39-GFP reporters and the plasmids with or without the PDE2 genes were grown at 25 °C until exponential phase and then harvested. The GFP mRNA levels were quantified by qRT-PCR analysis, and relative mRNA levels were calculated using ACT1 mRNA. The values are plotted as the fold change from cells harboring wild-type PATG39-GFP reporter and the empty plasmids. The data show mean ± SEM (n = 4). **P < 0.01 as determined by Student’s t-test. (C,D) The ATG39 mRNA levels in pde1 pde2 mutant. Wild-type (WT) and pde1 pde2 mutant strains were grown at 25 °C until exponential phase and treated with 3 μg/ml tunicamycin (TM) (C) or incubated under nitrogen-starved conditions (D) for the indicated time. The ATG39 mRNA levels were quantified by qRT-PCR analysis, and relative mRNA levels were calculated using ACT1 mRNA. The values are plotted as the fold change from wild-type cells at the time of TM addition or nitrogen removal. The data show mean ± SEM (n > 3). *P < 0.05 and **P < 0.01 as determined by Student’s t-test. (E) Cellular localization of Msn2. Wild-type (WT) and pde1 pde2 mutant strains harboring GFP-tagged MSN2 were grown at 25 °C until exponential phase and treated with 3 μg/ml tunicamycin (TM) for 3 h, and subjected to microscopy. The fluorescence intensities were measured, and then the ratios (N/C) of the fluorescence intensity per unit area in the nucleus/that in the cytoplasm were calculated. The graphs show mean ± SEM (n = 30). **P < 0.01 as determined by Student’s t-test. NS, not statistically significant (P > 0.05). Scale bar, 10 μm. (F) Sec63-GFP degradation in ER-stressed pde1 pde2 mutant. Wild-type (WT) and pde1 pde2 mutant strains harboring GFP-tagged SEC63 were grown at 25 °C until exponential phase and treated with 3 μg/ml tunicamycin (TM) for 18 h. Extracts prepared from each cell were immunoblotted with anti-GFP antibodies. The intensities of free GFP were measured and normalized to the Sec63-GFP level. The values are plotted as the fold change from wild-type cells. The data show mean ± SEM (n = 3). *P < 0.01 as determined by Student’s t-test.
PKA negatively regulates Msn2/4 by promoting their nuclear export18,25. Previous studies have already revealed that Msn2 accumulates in the nucleus in response to nitrogen starvation and its nuclear accumulation is clearly inhibited by hyperactivation of PKA32,33. Additionally, it has been reported that ER stress leads to inactivation of PKA34. Therefore, we examined whether ER stress causes nuclear accumulation of Msn2 using the strains which express the carboxyl-terminally GFP-tagged Msn2 (Msn2-GFP). Under normal conditions, Msn2-GFP distributed throughout the cytoplasm and nucleus (Fig. 5E). Msn2-GFP accumulated in the nucleus after ER stress treatment. However, nuclear accumulation of Msn2-GFP induced by ER stress was clearly inhibited in the pde1 pde2 double mutant cells. We also found that nuclear accumulation of Msn2 is caused by nitrogen starvation and inhibited by pde1 pde2 double mutations (Supplementary Fig. 6C). These results suggest that the PKA pathway negatively regulates ATG39 transcription by promoting the nuclear export of Msn2/4.
Finally, we investigated whether Pde1/2 are involved in regulation of ER-phagy. Sec63-GFP degradation in response to ER stress was modestly diminished in the pde1 pde2 double mutant cells (Fig. 5F), suggesting that Pde1/2 positively regulate ER stress-induced ER-phagy. We also monitored non-selective autophagy in the pde1 pde2 double mutant cells. Pgk1-GFP degradation in response to ER stress was significantly reduced by pde1 pde2 double mutations (Supplementary Fig. 6D). This result is consistent with the previous findings that non-selective autophagy is inhibited by activation of PKA33,35. These results suggest that the PKA pathway not only regulates ATG39 transcription via Msn2/4 but also influences general autophagic activities.
Discussion
In this study, we identified two closely related transcriptional activators, Msn2 and Msn4, as a positive regulator of ATG39 transcription. We also found that the ATG39 promoter contains two Msn2/4-binding consensus sequences termed STRE, whose disruption downregulates ATG39 promoter activity to a similar extent as msn2 msn4 double mutations. These results suggest that ATG39 expression is transcriptionally activated via the STRE elements by Msn2 and Msn4 (Fig. 6). We also isolated Pde2 cAMP phosphodiesterase as a positive regulator of ATG39 transcription. Pde2 functions redundantly with Pde1 to downregulate the PKA activity18. PKA negatively regulates Msn2 and Msn4 by promoting their nuclear export18,25. Thus, Pde1 and Pde2 positively regulate Msn2 and Msn4. Consistently, the pde1 pde2 double mutants exhibited the phenotypes similar to that observed in the msn2 msn4 double mutants: ATG39 induction in response to ER stress and nitrogen starvation was diminished in the pde1 pde2 double mutant cells. We also found that ATG39 promoter activity could not be activated by Pde2 overexpression when the MSN2 and MSN4 genes were deleted or the STRE elements were mutated. Furthermore, nuclear accumulation of Msn2 in response to ER stress and nitrogen starvation was clearly inhibited by pde1 pde2 double mutations. These results suggest that Pde1/2 positively regulate Msn2/4-mediated ATG39 transcription probably through PKA. Additionally, it is suggested that PKA regulates autophagy through both Msn2/4-dependent and -independent mechanisms from the following observations: (1) pde1 pde2 double mutations inhibited autophagic degradation of the Sec63 ER transmembrane protein more severely than msn2 msn4 double mutations (Figs. 4A, 5F); (2) pde1 pde2 double mutations inhibited autophagic degradation of the Pgk1 cytoplasmic protein, but msn2 msn4 double mutations did not (Fig. 4B and Supplementary Fig. 6D). It has been reported that PKA directly phosphorylates and negatively regulates Atg1, a kinase essential for activation of autophagy1,2,36. Thus, the PKA pathway not only regulates ER-phagy by controlling Msn2/4-mediated ATG39 transcription but also modulates the activity of the core autophagy-related machinery (Fig. 6).
Figure 6.
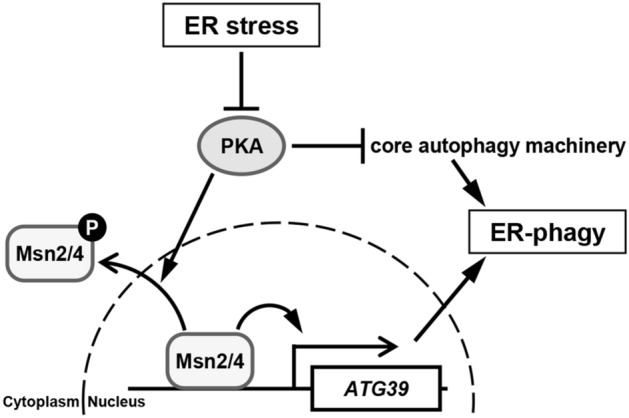
Proposed model for Msn2/4 in ER stress-induced ER-phagy.
A previous study showed that ER stress downregulates the PKA activity34. Here, we observed nuclear accumulation of Msn2 in ER-stressed cells. These findings suggest that ER stress inactivates PKA and consequently promotes Msn2 nuclear accumulation to induce ATG39 transcription. Nuclear accumulation of Msn2 under nitrogen-starved conditions has been observed32,33. However, there is little compelling evidence that the PKA activity is controlled by nitrogen availability. It is well-known that nitrogen starvation leads to inactivation of target of rapamycin complex 1 (TORC1)37,38. A previous study revealed that TORC1 promotes nuclear export of Msn239. Therefore, it is possible that during nitrogen starvation, Msn2 nuclear accumulation and consequent ATG39 transcriptional induction are mediated by inactivation of TORC1, rather than inactivation of PKA.
ATG39 transcription was partly activated by ER stress and nitrogen starvation even when the MSN2 and MSN4 genes were deleted or the STRE elements were mutated. We previously revealed that ATG39 transcription is repressed by Mig1 and Mig2 transcription factors and this repression itself is negatively regulated by Snf1 AMPK9. We found here that unlike snf1 mutation, msn2 msn4 double mutations diminished ATG39 induction in response to nitrogen starvation. Furthermore, snf1 mutation downregulated ATG39 induction caused by ER stress in the msn2 msn4 double mutant cells. These results suggest that Msn2/4 regulate ATG39 transcription in a manner independent of Snf1. Additionally, our findings that ATG39 induction in response to ER stress was still observed in the snf1 msn2 msn4 triple mutant cells predicted the existence of unidentified activators for ATG39 expression. Here, we developed a genetic screen to identify the positive regulator of ATG39 transcription. However, we could not isolate any candidates except Msn2/4 transcription factors and negative regulators of the PKA pathway such as Pde2 and Gpb1. All factors we identified here upregulate ATG39 transcription through the STRE elements located in its promoter. Therefore, it is expected that unidentified positive regulators may be further isolated using a reporter in which the STRE elements are mutated. Such investigation would enhance our understandings of transcriptional regulation of autophagy receptors and its importance in regulation of the activity of selective autophagy.
Methods
Plasmids
The YCplac33-PATG39-GFP, YCplac33-PMCM2-GFP and YCplac33-PATG39-ATG39 plasmids were described previously9. The PATG39-HIS3 construct was generated as follows. A 990-bp genomic fragment containing the ATG39 promoter was amplified from the YCplac33-PATG39-GFP plasmid by PCR with the following primers: 5′-CTCTAGAGGATCCCCGGGAAAAACTGTGCTCCTAGCAG-3′ and 5′-GGCTGGTTCTGCCATTTTAGGTCCGACAACTCG-3′. A DNA fragment encoding HIS3MX6 followed by the ADH1 terminator (TADH1) was amplified from the pFA6a-GFP-HIS3MX6 vector by PCR with the following primers: 5′-ATGGCAGAACCAGCCCAAAAAAAGC-3′ and 5′-TCGAGCTCGGTACCCGGGCAGTATAGCGACCAGCATTCACATACG-3′. The amplified 5′ upstream sequences of the ATG39 gene, together with the HIS3MX6-TADH1 DNA fragment, were fused to the YCplac33 vector by In-Fusion cloning kits (Takara), yielding the YCplac33-PATG39-HIS3 plasmid. Similarly, the YCplac33-PMCM2-HIS3 plasmid was constructed. The primers used to amplify PMCM2 are 5′-CTCTAGAGGATCCCCGGGAAAGGCGTCTATCCACTTGC-3′ and 5′-GGCTGGTTCTGCCATCACTTATATTGTAGTTGTG-3′. The MSN2, MSN4 and PDE2 genes were amplified from the Saccharomyces cerevisiae W303 derivative by PCR with the following primers: 5′-CTCTAGAGGATCCCCGGGGTTTCCAGCGAAAGAGACAG-3′ and 5′-TCGAGCTCGGTACCCGGGTAAAAGTAGCAAACTGGTAG-3′ for MSN2; 5′-CTCTAGAGGATCCCCGGGAAACCCGAGCTAGAACTAGG-3′ and 5′-TCGAGCTCGGTACCCGGGAGCAAACGTCGTACCAATCC-3′ for MSN4; 5′-CTCTAGAGGATCCCCGGGATTATCTTTGTAATCTGCAG-3′ and 5′-TCGAGCTCGGTACCCGGGCTTTGCATATACCAACACAG-3′ for PDE2. The amplified MSN2, MSN4 and PDE2 genes were fused to the YEplac181 vector by In-Fusion cloning kits (Takara), yielding the YEplac181-MSN2, YEplac181-MSN4 and YEplac181-PDE2 plasmids, respectively. Mutations of STREs in the ATG39 promoter were generated by oligonucleotide-directed PCR using the following primers: 5′-CCTCCGGACCATCTACCCCGGTGTG-3′ and 5′-TAGATGGTCCGGAGGGTTAACTGTC-3′ for STRE1 mutation; 5′-GCGCAAAGATGTTACCGCAAAATGG-3′ and 5′-GTAACATCTTTGCGCGACAGCTGC-3′ for STRE2 mutation. To generate the PATG39(STREmut)-GFP and PATG39(STREmut)-ATG39 integrations, the inserts in the YCplac33 plasmids were subcloned into the pRS306 vector. Plasmids used in this study are described in Supplementary Table 2.
Strains
Standard procedures were followed for yeast manipulations40. SD(–N) medium (0.17% (w/v) yeast nitrogen base without amino acids and ammonium sulfate and 2% (w/v) glucose) was used to induce nitrogen starvation. Yeast strains harboring the complete gene deletions (MSN2, MSN4, PDE1 and PDE2) and carboxyl-terminally GFP-tagged MSN2 were generated by a PCR-based method as described previously41. Primer sets were designed such that 46 bases at the 5′ end of primers were complementary to those at the corresponding region of the target gene, and 20 bases at their 3′ end were complementary to the pFA6a sequence, 5′-TGCAGTACTCTGCGGGTGTATACAG-3′ or 5′-ATTTGACTGTATTACCAATGTCAGC-3′. All strains produced by a PCR-based method were verified by colony PCR amplification to confirm that replacement had occurred at the expected locus. Strains carrying the PATG39(STREmut)-GFP and PATG39(STREmut)-ATG39 integrations were constructed by integrating the linearized pRS306-PATG39(STREmut)-GFP and pRS306-PATG39(STREmut)-ATG39 plasmids, respectively. Strains used in this study are listed in Supplementary Table 3.
Identification of genes activating the PATG39-HIS3 reporter
Wild-type cells harboring the PATG39-HIS3 reporter poorly grew on synthetic defined media that lack histidine and contain 3-amino-1H-1,2,4-triazole (AT) due to low expression of HIS3MX6. To identify genes whose overexpression allows cells harboring the PATG39-HIS3 reporter to form colonies on synthetic defined media lacking histidine, the multicopy genomic libraries were screened as follows. A Saccharomyces cerevisiae YEp13 genomic library was transformed into cells harboring the PATG39-HIS3 reporter. Cells were plated onto synthetic defined media lacking leucine and allowed to grow by incubation at 30 °C for 2 days. Transformants grown on synthetic defined media lacking leucine were transferred on synthetic defined media lacking histidine and containing AT and allowed to grow by incubation at 30 °C. Plasmids were collected from colonies that grew on selective media, and the ends of genomic inserts were sequenced.
RNA isolation and RT-PCR
Preparation of total RNA and generation of cDNA were performed as described previously19. The cDNAs were quantitated by a quantitative real-time RT-PCR (qRT-PCR) method using QuantStudio 5 real-time PCR systems (Applied Biosystems) with TB Green Premix Ex Taq (Takara), and levels of gene expression were normalized to ACT1 expression. Primers used to analyze the mRNA level are described in Supplementary Table 4.
Protein extraction, western blot analysis and antibodies
Preparation of protein extracts and Western blot analysis were performed as described previously19. Anti-GFP antibody from mouse IgG1κ (clones 7.1 and 13.1) (Roche) was used. Detection and quantification were carried out by using the Odyssey Imaging Systems (LI-COR Biosciences). Statistical analysis was performed with Excel (Microsoft).
Microscopy
To visualize GFP-tagged Msn2 in living cells, cells were grown at 25 °C until exponential phase and treated with 3 μg/ml tunicamycin (TM) for 3 h. Cells were then harvested, suspended in SD medium, and observed immediately using a Keyence BZ-X700 microscope (Keyence) with a PlanAproλ 100 × NA 1.45 oil objective lens (Nikon). Fluorescence intensities were quantified using Hybrid Cell Count BZ-H2C software (Keyence).
Supplementary Information
Acknowledgements
We thank the members of our laboratory for their helpful suggestions and feedback. This research was supported by JSPS KAKENHI Grant Number 19K06632 (to T.M.) and the Takeda Science Foundation (to T.M.).
Author contributions
T.M. designed research strategies, performed experiments, analyzed data, and wrote manuscript. K.I. provided solutions.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-91480-0.
References
- 1.Reggiori F, Klionsky DJ. Autophagic processes in yeast: Mechanism, machinery and regulation. Genetics. 2013;194:341–361. doi: 10.1534/genetics.112.149013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohsumi Y. Historical landmarks of autophagy research. Cell. Res. 2014;24:9–23. doi: 10.1038/cr.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 4.Farré JC, Subramani S. Mechanistic insights into selective autophagy pathways: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2016;17:537–552. doi: 10.1038/nrm.2016.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018;20:233–242. doi: 10.1038/s41556-018-0037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mochida K, et al. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522:359–362. doi: 10.1038/nature14506. [DOI] [PubMed] [Google Scholar]
- 7.Friedman JR, Voeltz GK. The ER in 3D: A multifunctional dynamic membrane network. Trends Cell Biol. 2011;21:709–717. doi: 10.1016/j.tcb.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui Y, et al. COPII subunit acts with an autophagy receptor to target endoplasmic reticulum for degradation. Science. 2019;365:53–60. doi: 10.1126/science.aau9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizuno T, Muroi K, Irie K. Snf1 AMPK positively regulates ER-phagy via expression control of Atg39 autophagy receptor in yeast ER stress response. PLoS Genet. 2020;10:e1009053. doi: 10.1371/journal.pgen.1009053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aihara M, et al. Tor and the Sin3-Rpd3 complex regulate expression of the mitophagy receptor protein Atg32 in yeast. J. Cell. Sci. 2014;127:3184–3196. doi: 10.1242/jcs.153254. [DOI] [PubMed] [Google Scholar]
- 11.Bartholomew CR, et al. Ume6 transcription factor is part of a signaling cascade that regulates autophagy. Proc. Natl. Acad. Sci. USA. 2012;109:11206–11210. doi: 10.1073/pnas.1200313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin M, et al. Transcriptional regulation by Pho23 modulates the frequency of autophagosome formation. Curr. Biol. 2014;24:1314–1322. doi: 10.1016/j.cub.2014.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernard A, et al. Rph1/KDM4 mediates nutrient-limitation signaling that leads to the transcriptional induction of autophagy. Curr. Biol. 2015;25:546–555. doi: 10.1016/j.cub.2014.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu G, et al. A conserved mechanism of TOR-dependent RCK-mediated mRNA degradation regulates autophagy. Nat. Cell. Biol. 2015;17:930–942. doi: 10.1038/ncb3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Motley AM, Nuttall JM, Hettema EH. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 2012;31:2852–2868. doi: 10.1038/emboj.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hedbacker K, Carlson M. SNF1/AMPK pathways in yeast. Front. Biosci. 2008;13:2408–2420. doi: 10.2741/2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broach JR. Nutritional control of growth and development in yeast. Genetics. 2012;192:73–105. doi: 10.1534/genetics.111.135731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizuno T, Masuda Y, Irie K. The Saccharomyces cerevisiae AMPK, Snf1, negatively regulates the Hog1 MAPK pathway in ER stress response. PLoS Genet. 2015;11:e1005491. doi: 10.1371/journal.pgen.1005491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanazawa S, Driscoll M, Struhl K. ATR1, a Saccharomyces cerevisiae gene encoding a transmembrane protein required for aminotriazole resistance. Mol. Cell Biol. 1988;8:664–673. doi: 10.1128/mcb.8.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Estruch F, Carlson M. Two homologous zinc finger genes identified by multicopy suppression in a SNF1 protein kinase mutant of Saccharomyces cerevisiae. Mol. Cell Biol. 1993;13:3872–3881. doi: 10.1128/MCB.13.7.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sass P, Field J, Nikawa J, Toda T, Wigler M. Cloning and characterization of the high-affinity cAMP phosphodiesterase of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 1986;83:9303–9307. doi: 10.1073/pnas.83.24.9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harashima T, Heitman J. The Galpha protein Gpa2 controls yeast differentiation by interacting with kelch repeat proteins that mimic Gbeta subunits. Mol. Cell. 2002;10:163–173. doi: 10.1016/S1097-2765(02)00569-5. [DOI] [PubMed] [Google Scholar]
- 24.Smith A, Ward MP, Garrett S. Yeast PKA represses Msn2p/Msn4p-dependent gene expression to regulate growth, stress response and glycogen accumulation. EMBO J. 1998;17:3556–3564. doi: 10.1093/emboj/17.13.3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Görner W, et al. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 1998;12:586–597. doi: 10.1101/gad.12.4.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmitt AP, McEntee K. Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 1996;93:5777–5782. doi: 10.1073/pnas.93.12.5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martínez-Pastor MT, et al. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE) EMBO J. 1996;15:2227–2235. doi: 10.1002/j.1460-2075.1996.tb00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meiling-Wesse K, Barth H, Thumm M. Ccz1p/Aut11p/Cvt16p is essential for autophagy and the cvt pathway. FEBS Lett. 2002;526:71–76. doi: 10.1016/S0014-5793(02)03119-8. [DOI] [PubMed] [Google Scholar]
- 29.Welter E, Thumm M, Krick R. Quantification of nonselective bulk autophagy in S. cerevisiae using Pgk1-GFP. Autophagy. 2010;6:794–797. doi: 10.4161/auto.6.6.12348. [DOI] [PubMed] [Google Scholar]
- 30.Nikawa J, Sass P, Wigler M. Cloning and characterization of the low-affinity cyclic AMP phosphodiesterase gene of Saccharomyces cerevisiae. Mol. Cell Biol. 1987;7:3629–3636. doi: 10.1128/MCB.7.10.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma P, Wera S, Van Dijck P, Thevelein JM. The PDE1-encoded low-affinity phosphodiesterase in the yeast Saccharomyces cerevisiae has a specific function in controlling agonist-induced cAMP signaling. Mol. Biol. Cell. 1999;10:91–104. doi: 10.1091/mbc.10.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999;402:689–692. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- 33.Schmelzle T, Beck T, Martin DE, Hall MN. Activation of the RAS/cyclic AMP pathway suppresses a TOR deficiency in yeast. Mol. Cell Biol. 2004;24:338–351. doi: 10.1128/MCB.24.1.338-351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pincus D, Aranda-Díaz A, Zuleta IA, Walter P, El-Samad H. Delayed Ras/PKA signaling augments the unfolded protein response. Proc. Natl. Acad. Sci. USA. 2014;111:14800–14805. doi: 10.1073/pnas.1409588111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Budovskaya YV, Stephan JS, Reggiori F, Klionsky DJ, Herman PK. The Ras/cAMP-dependent protein kinase signaling pathway regulates an early step of the autophagy process in Saccharomyces cerevisiae. J. Biol. Chem. 2004;279:20663–20071. doi: 10.1074/jbc.M400272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Budovskaya YV, Stephan JS, Deminoff SJ, Herman PK. An evolutionary proteomics approach identifies substrates of the cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA. 2005;102:13933–13938. doi: 10.1073/pnas.0501046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/S0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 38.González A, Hall MN. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017;36:397–408. doi: 10.15252/embj.201696010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santhanam A, Hartley A, Düvel K, Broach JR, Garrett S. PP2A phosphatase activity is required for stress and Tor kinase regulation of yeast stress response factor Msn2p. Eukaryot. Cell. 2004;3:1261–1271. doi: 10.1128/EC.3.5.1261-1271.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaiser CA, Adams A, Gottschling DE. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- 41.Longtine MS, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.