

Abstract
Objective:
Chronic hemolysis is a hallmark of sickle cell disease (SCD) and a driver of vasculopathy; however, the mechanisms contributing to hemolysis remain incompletely understood. While xanthine oxidase (XO) activity has been shown to be elevated in SCD, its role remains unknown. XO binds endothelium and generates oxidants as a byproduct of hypoxanthine and xanthine catabolism. We hypothesized that XO inhibition decreases oxidant production leading to less hemolysis.
Approach and Results:
Wild-type mice were bone marrow (BM) transplanted with control (AA) or sickle (SS) Townes BM. After 12 weeks, mice were treated with 10 mg/kg/day of febuxostat (Uloric®), FDA-approved XO inhibitor, for 10 weeks. Hematological analysis demonstrated increased hematocrit, cellular hemoglobin, and red blood cells, with no change in reticulocyte percentage. Significant decreases in cell-free hemoglobin and increases in haptoglobin suggest XO inhibition decreased hemolysis. Myographic studies demonstrated improved pulmonary vascular dilation and blunted constriction, indicating improved pulmonary vasoreactivity, whereas pulmonary pressure and cardiac function were unaffected. The role of hepatic XO in SCD was evaluated by BM transplanting hepatocyte-specific XO knockout mice with SS Townes BM. However, hepatocyte-specific XO knockout, which results in >50% diminution in circulating XO, did not affect hemolysis levels or vascular function, suggesting hepatocyte-derived elevation of circulating XO is not the driver of hemolysis in SCD.
Conclusions:
Ten weeks of febuxostat treatment significantly decreased hemolysis and improved pulmonary vasoreactivity in a mouse model of SCD. Although hepatic XO accounts for >50% of circulating XO, it is not the source of XO driving hemolysis in SCD.
Keywords: xanthine oxidase endothelium, sickle cell disease, hemolysis, endothelium, oxidant stress, Vascular Biology, Oxidant Stress
INTRODUCTION
Sickle cell disease (SCD) is a major global hemoglobinopathy that affects approximately 300,000 newborns worldwide each year.1 It is estimated that there are approximately 100,000 affected individuals in the United States;1 however, in other parts of the world, such as Sub-Sahara Africa, SCD is endemic with ~230,000 new incidences in 2010,2–4 leading the World Health Organization and United Nations to classify SCD as a global public health problem.1, 3
SCD presents as multi-organ dysfunction that is the consequence of a single genetic mutation in the ß-subunit of hemoglobin (Hb).2, 5 Under low pH or deoxygenation conditions, the point mutation in the Hb gene triggers Hb polymerization due to the hydrophobicity change of the amino acid substitution and subsequent sickling of red blood cells (RBCs).2, 6, 7 Sickled RBCs cause vasooclusive crises and RBC hemolysis, releasing cell free Hb and heme into circulation.2, 8 Free Hb and heme generate a pro-inflammatory environment that results in endothelial dysfunction, hyperviscosity, and RBC-leukocyte-platelet interaction on the endothelial surface.6, 8–10 Despite having a direct impact on Hb structure, function, and RBC shape, the mutation also causes complications such as pulmonary and systemic vasculopathy,7 pulmonary and relative systemic hypertension,6 acute chest syndrome,6 heart failure,10 and renal failure,6 which result in accelerated mortality.11 Treatment options for SCD remain limited, with only four FDA-approved drugs: hydroxyurea,6, 9, 12, 13 L-glutamine,9, 14 crizanlizumab,15 and voxelotor.16, 17 This is due in part to the poor understanding of the molecular drivers of hemolysis in SCD and therefore lack of small molecule drugs that can inhibit hemolysis in SCD patients. Chronic blood transfusions are also used to treat SCD; however, benefits for patients with pulmonary hypertension remain inconclusive.18, 19
Xanthine oxidase (XO) has been shown to be increased in several studies involving human SCD patients and mouse models of SCD;20–22 however, the role of XO in SCD is yet to be established. XO and xanthine dehydrogenase (XDH) are two forms of the enzyme collectively known as xanthine oxidoreductase (XOR).23, 24 XOR is transcribed and translated as XDH which catalyzes the oxidation of hypoxanthine to xanthine and xanthine to uric acid at the Mo-cofactor while reducing NAD+ to NADH at the FAD-cofactor. During inflammation, oxidation of critical cysteine residues and/or limited proteolysis converts XDH to XO which catalyzes the same reactions at the Mo-cofactor, yet reduces oxygen to superoxide (O2•-) and hydrogen peroxide (H2O2).20
XDH is transcribed and translated primarily in the liver; however, following hepatic stressors such as ischemia, hypoxia, or inflammation, XDH is released from the liver and enters circulation where it is rapidly converted to XO by plasma proteases.20, 25 XO binds with considerable affinity (Kd = 6 nM) to distal endothelial surfaces via electrostatic interactions with glycosaminoglycans (GAGs), thus increasing XO concentration in organs with low basal levels of the enzyme and amplifying O2•- or H2O2 production at the endothelial surface.26, 27 While XO directly produces O2•- or H2O2, other reactive species such as peroxynitrite and hydroxyl radical can also be generated via reactions with nitric oxide (NO) and iron respectively.20 This oxidative milieu at the endothelial surface can lead to endothelial and tissue damage, altered signaling, and diminished NO bioavailability.23 ROS produced on the endothelial surface or in circulation can cause RBC membrane fragility, resulting in hemolysis.28 Therefore, we hypothesized that XO inhibition would decrease hemolysis in a mouse model of SCD.
MATERIAL AND METHODS
Please see the Major Resources Table and extended methods in the Supplemental Materials.
Animals
C57BL/6J (58 wild type; stock #000664;) mice were ordered from The Jackson Laboratory (Bar Harbor, ME). Colonies of Xdhfloxed/floxedAlb-1Cre/Wt (7 hepatocyte specific Xdh knockout)29, Xdhfloxed/floxedAlb-1Wt/Wt (8 littermate FLX controls)29, and Townes knock‐in sickle (SS) and their control (AA) mice, were bred and maintained at the University of Pittsburgh. All Xdhfloxed/floxed mice were bred on a C57BL/6J background. All Townes mice were bred on a C57BL/6J and 129S background.30 Only 6–13-week-old male mice were used for experiments because there is currently no indication that male and female mice have major differences in hemolysis in SCD. In the AA control Townes mice, both murine alpha globin genes are knocked out and replaced by human alpha and beta globin (Hb) transgenes in the mouse locus. The SS sickle mice contain two copies of the human beta sickle globin transgene.31 All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. All mice were supplied standard laboratory diet (#5234) and drinking water according to the “Guide for the Care and Use of Laboratory Animals” of the Department of Laboratory Animal Research at the University of Pittsburgh. All mice were housed in pathogen-free conditions.
Bone Marrow (BM) Transplanted Chimeric Mice
The genotype of the Xdhfloxed/floxed mice was confirmed by PCR analysis of DNA obtained from tail tissue using a Cre-specific probe. Genotypes of the Townes mice were confirmed by peripheral blood Hb electrophoresis. Hb electrophoresis was performed using the reagents provided in the Hemoglobin (E) and Hemoglobin (E) Acid Kits (Sebia Capillary System; Lisse, France), in accordance with the manufacturer’s guidelines. Briefly, 5 μL of the red blood cell (RBC) pellet was lysed in 65 μL of Sebia hemolysis solution. Samples were run on an agarose gel and stained for identification of hemoglobin fraction by band location. Phoresis software was used to quantify the density of the bands. Four different groups of BM chimeras were generated: C57BL/6J/AA, C57BL/6J/SS, Xdhfloxed/floxedAlb-1Cre/Wt/SS, and Xdhfloxed/floxedAlb-1Wt/Wt/SS. C57BL/6J/AA mice received BM cells from Townes AA donor mice and C57BL/6J/SS, Xdhfloxed/floxedAlb-1Cre/Wt/SS, and Xdhfloxed/floxedAlb-1Wt/Wt/SS mice received BM cells from Townes SS donor mice. BM transplants were performed as previously described.32 Briefly, BM cells were isolated from the femurs and tibias of Townes AA or SS donor mice. C57BL/6J (8 weeks old), Xdhfloxed/floxedAlb-1Cre/Wt (6–13 weeks old), or Xdhfloxed/floxedAlb-1Wt/Wt (6–13 weeks old) recipient mice were lethally irradiated (two 500–550 rad doses, 3 hours apart) and AA or SS BM (1.5–2.5×106 cells) was injected retro-orbitally immediately following the second irradiation. Following BM transplant, chimeras were housed in autoclaved cages and provided 0.2% neomycin drinking water for 2 weeks, followed by normal autoclaved drinking water for the remainder of the experiment. At 12 weeks post-BM transplant, 50–100 μL of blood was collected via retro-orbital eye bleed to assess BM engraftment of the chimeric mice by Hb electrophoresis using the Sebia Hb Electrophoresis kit described above. Only chimeras with ≥80% of the donor Hb phenotype were used for subsequent experiments. At 12 weeks post-BM transplant C57/AA and C57/SS mice were given the clinically relevant dose of febuxostat in drinking water (0.05 mg/mL or ~10mg/kg/day) for 10 weeks. BM engraftment was confirmed 0, 3- and 10-weeks post-initiation of febuxostat treatment. Ten weeks post-BM engraftment, mice were weighed and euthanized via heart puncture following right heart catheterization. Blood was collected via heart puncture (200–500 μL) in 5% EDTA. Mice were perfused with 5 mL of PBS and organs were collected and flash frozen in liquid nitrogen. Mice were then perfused with 5 mL of 4% paraformaldehyde (PFA) in PBS and organs were weighed and fixed in PFA overnight, then transferred to 70% ethanol for paraffin embedding.
Blood Collection and Hematological Phenotyping
Blood samples were obtained at 0, 3, 6, and 10 weeks post-engraftment via retro-orbital eye bleed. Blood samples were obtained immediately following euthanasia via heart puncture. For retro-orbital eye bleeds, mice were anesthetized with 1% isoflurane and heparin-coated microcapillary tubes were used to collect 50–100 μL of blood. Blood was collected in sterile tubes containing 5% EDTA and maintained at room temperature until analysis of complete blood count (within 2–4 hours). Complete blood counts were measured using a Heska (HemaTrue Inc.; Miami Lakes, FL, U.S.A.) according to manufacturer instructions. Reticulocytes were measured from whole blood using flow cytometry. Thiazole Orange (80 pM in 1xPBS) was used to stain reticulocytes. Samples were incubated overnight at 4°C and protected from light. The samples were analyzed using flow cytometry, and FACSDIVA software was used for data collection. Whole blood was spun down at 2500xg for 10 minutes at 4°C. Plasma was separated from the RBC pellet, aliquoted for additional experiments, and stored at −80°C. Plasma haptoglobin (Hp) levels were measured by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (Crystal Chem; Elk Grove Village, IL). Plasma hemopexin (Hx) levels were measured by ELISA according to the manufacturer’s instructions (AssayPro; St. Charles, MO). Heme-containing species and their metabolites including methemoglobin, oxyhemoglobin, and hemin were measured in plasma using UV-visible spectral deconvolution as previously described.33
Xanthine Oxidase (XO) Activity
Human blood samples from healthy controls (non-SCD pediatric patients undergoing surgery) or SCD patients were obtained from Children’s of Alabama Hospital according to University of Alabama at Birmingham Institutional Review Board approved protocols.33 Blood from AA, AS, and SS chimeric mice was collected via heart puncture. Blood from AA and SS chimeric mice treated with febuxostat was collected via retro-orbital eye bleed at 0 and 3 weeks of treatment and via heart puncture at 10 weeks of treatment. Whole mouse blood was spun down at 2500xg for 10 minutes to collect plasma. Liver, lung, and kidney tissue was collected after perfusion with 5 mL of PBS. Plasma and tissue XO activity was measured as previously described.29 Briefly, XO activity was evaluated by electrochemical detection (ESA Coul-Array System) of uric acid via reverse-phase high-performance liquid chromatography.
Purine Metabolite Analysis by LC/MS-MS
Purine metabolite analysis was done as previously described.34 Briefly, 20 μL of plasma was combined with 80 μL of methanol on ice for 15 minutes to precipitate proteins. The plasma/methanol mixture was centrifuged at 13,300 rpm at 4°C for 15 minutes, followed by lyophilization of the supernatant. To reconstitute the metabolites, 200 μL of 5 mM ammonium acetate in 95% water, 5% acetonitrile, and 0.5% acetic acid was passed through a 0.45-μm PDVF filter. The extracts were analyzed by a Shimadzu LC Nexera X2 UHPLC coupled with a QTRAP 5500 LC MS/MS (AB Sciex). An ACQUITY UPLC BEH Amide analytic column (2.1 X 50 mm, 1.7 μm, Waters) was used for chromatographic separation. The extracted MRM peaks were integrated using MultiQuant 3.0.2 software (AB Sciex).34
Statistics
Outliers were identified using a ROUT analysis with a Q=1% and excluded from the data presented. When two groups were included, an unpaired Student’s t test was used unless otherwise noted. An unpaired Student’s t test with a Welch’s correction was used if the standard deviation of the two groups was significantly different. Normality was assessed using the Shapiro-Wilk normality test. If the two groups were not normally distributed, a Mann-Whitney test was performed. When three or more groups were included, a one-way ANOVA with Dunnett’s multiple comparison test was used when comparing multiple groups to a control group or Sidak’s multiple comparison test was used when making multiple comparisons. When comparing two groups over time or across several doses a two-way ANOVA with Sidak’s multiple comparison test was used. Significance was defined as a P-value less than 0.05.
RESULTS
Febuxostat Treatment Significantly Decreases Plasma and Tissue XO Activity
Previous reports have shown a significant increase in XO activity in human patients with SCD compared to healthy controls, as well as in the Townes mouse model of SCD.20 To confirm these findings, we obtained plasma samples from pediatric patients with or without SCD and observed a significant increase (8-fold) in the plasma XO activity of patients with SCD compared to the healthy controls (Figure 1A). We also measured plasma XO activity in WT mice transplanted with bone marrow from AA control, AS sickle trait, and SS sickle Townes mice and observed a 1.3-fold increase in the AS sickle trait and a 1.5-fold increase in the SS sickle mice (Figure 1B). In order to evaluate the role of XO in SCD, 8-week-old wild-type (WT) mice were bone marrow transplanted with AA control or SS sickle bone marrow from Townes mice (Figure 1C, Supplemental Figure IA). After 12 weeks, mice were fully engrafted with the Townes bone marrow (Supplemental Figure IB). The mice were then split into four groups: AA, AA + febuxostat, SS, and SS + febuxostat. They were treated with a clinically relevant dose of febuxostat (10 mg/kg/day) in their drinking water for 10 weeks. Blood was collected at 0, 3, 6, and 10 weeks post initiation of febuxostat treatment, with 10 weeks being the endpoint of the study (Figure 1C, Supplemental Figure IA). Febuxostat treatment resulted in significant decreases in plasma, liver, lung, and kidney XO activity in the AA control (Supplemental Table I) and in the SS sickle mice (Figure 1D, Supplemental Table II). XO inhibition was confirmed by measuring hypoxanthine, xanthine, and urate levels. No change in hypoxanthine or xanthine, and a significant decrease in urate was observed in the AA control (Supplemental Figure IC) and a significant increase in hypoxanthine, no change in xanthine, and a significant decrease in urate was observed in the SS sickle mice (Figure 1E–G) treated with febuxostat. Oxidant load was also assessed via a coumarin boronate assay (CBA). There was a significant decrease in oxidant load in the febuxostat treated SS sickle mice compared to the SS sickle mice (Figure 1H). No difference in body weight between the SS and SS + febuxostat groups was observed (Figure 1I); however, there was a significant 2.4 g weight loss in the AA + febuxostat group compared to the AA control mice (Supplemental Figure ID). There was approximately a 7-fold difference in spleen weight between the AA and SS mice; however, febuxostat treatment did not alter the AA or SS spleen weight (Supplemental Figure IE).
Figure 1. Febuxostat treatment inhibits XO activity in chimeric SCD mice.
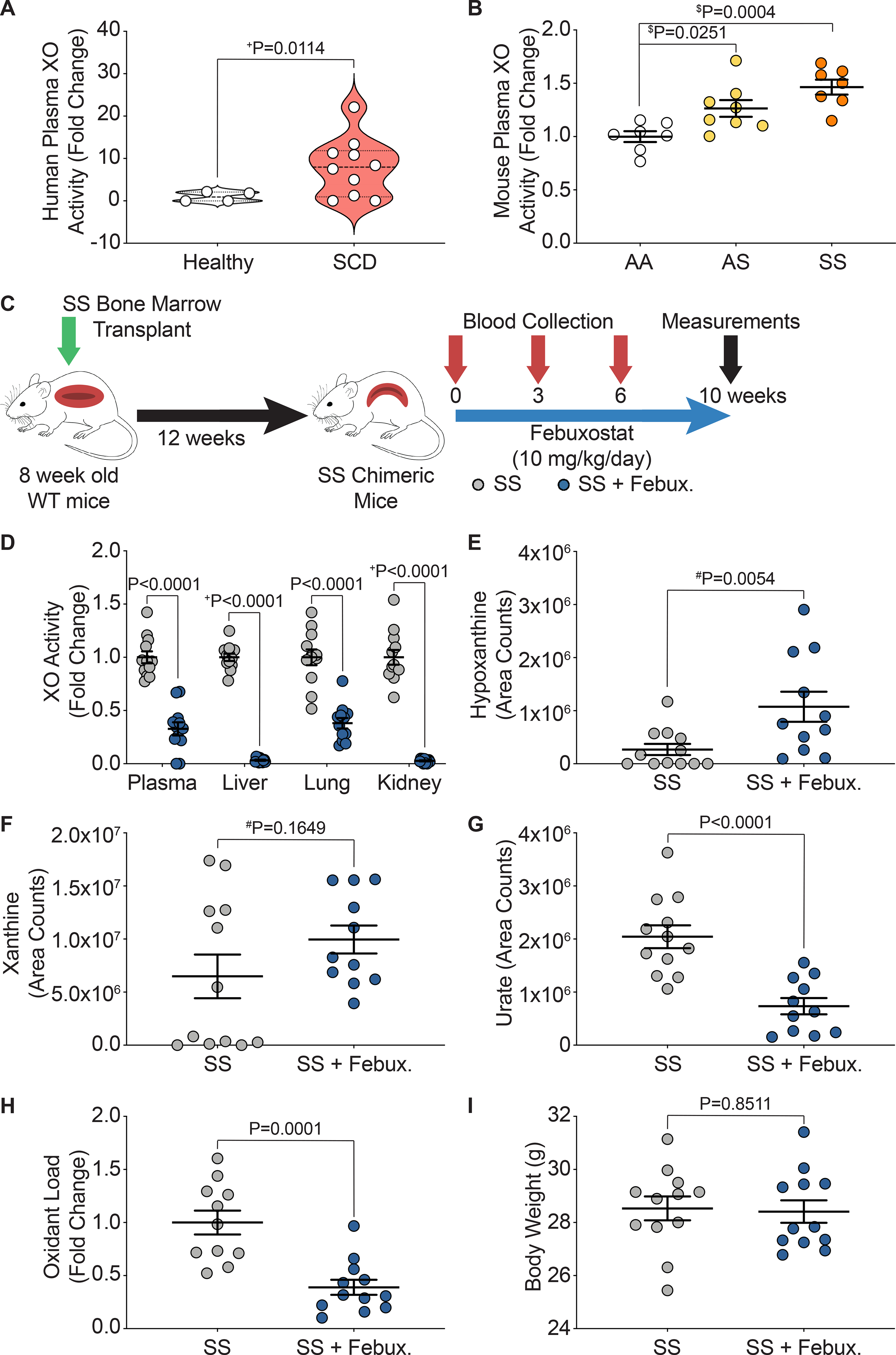
XO activity was assessed in A) human sickle cell disease patients compared to healthy controls and B) chimeric AA control, AS sickle trait, and SS sickle mice. C) Experimental design. D) XO activity of plasma, liver, lung, and kidney after 10 weeks of febuxostat treatment. LC/MS-MS was used for purine metabolite analysis of E) hypoxanthine, F) xanthine, and G) urate. H) A CBA assay was used to measure plasma oxidant load. I) Febuxostat treatment did not alter body weight. Values are mean ± SEM using an unpaired Student’s t test unless otherwise noted. +Values are mean ± SEM using an unpaired Student’s t test with Welch’s correction. $Values are mean ± SEM using a one-way ANOVA with Dunnett’s multiple comparisons test. #Values are mean ± SEM using a Mann-Whitney test. XO, xanthine oxidase; SCD, sickle cell disease; CBA, coumarin boronate acid; WT, wild type; febux, febuxostat.
XO Inhibition Decreased Hemolysis in Chimeric Sickle Mice
Key hematological parameters altered in SCD were measured as a delta change from 0 to 10 weeks of febuxostat treatment (Figure 2A). In the SS sickle mice, significant increases in hematocrit, cellular hemoglobin, RBCs, and platelets were observed over the ten weeks of treatment (Figure 2B–E). A complete analysis of blood parameters is included for the AA control and SS sickle mice in Supplemental Table III and Supplemental Table IV, respectively, at 0, 3, 6, and 10 weeks of febuxostat treatment. Additionally, the febuxostat-treated AA mice demonstrated significant diminution in white blood cells at three weeks and a significant increase in red blood cell distribution width (RDW) at 6 and 10 weeks of treatment compared to the AA mice (Supplemental Table III). No additional differences were observed in the hematology between SS and febuxostat treated SS mice (Supplemental Table IV). Despite these hematological improvements, there was no difference in reticulocytes, the RBC precursors (Figure 2F). To further assess hemolysis, cell free hemoglobin and cell free hemin were measured along with acute phase proteins that scavenge these species, haptoglobin and hemopexin, respectively, after 10 weeks of febuxostat treatment (Figure 3A, Supplemental Figure IIA). In the SS sickle mice, febuxostat treatment resulted in a significant decrease in cell free hemoglobin concentration (Figure 3B) which was primarily due to a loss in methemoglobin levels rather than a loss in oxyhemoglobin levels (Supplemental Figure IIB–C). There was no difference in plasma cell free hemin concentration or hemopexin concentration in the SS sickle mice (Figure 3D–E). In the AA control mice, no difference in methemoglobin, oxyhemoglobin, cell free hemoglobin, cell free hemin, haptoglobin, or hemopexin were observed (Supplemental Figure IIB–G).
Figure 2. XO inhibition improves hematological parameters.
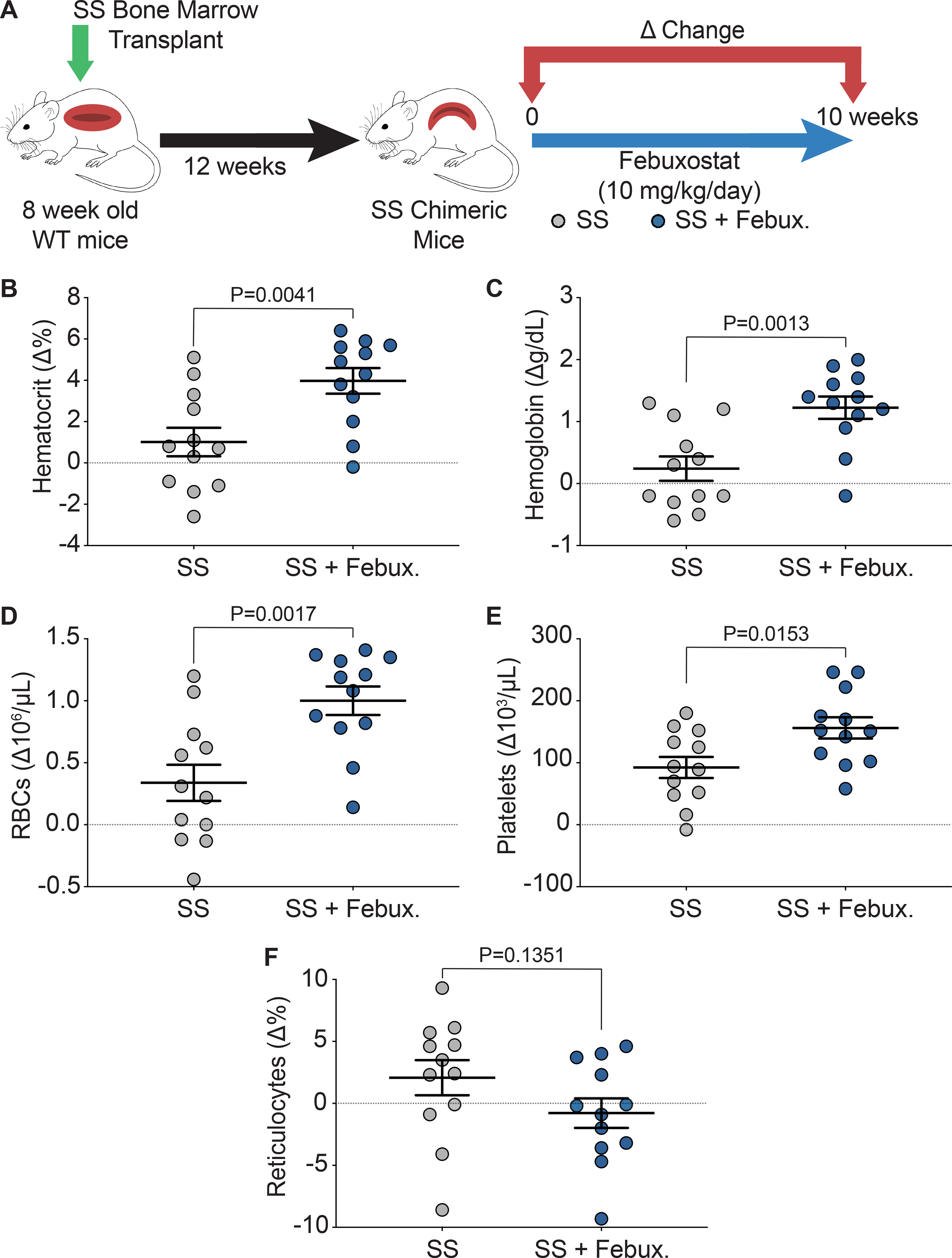
A) Experimental design. B-E) Complete blood counts shown as a delta change from 0 to 10 weeks of treatment. F) Flow cytometry was used to measure the delta change of reticulocyte percentage from 0 to 10 weeks of treatment. Values are mean ± SEM using an unpaired Student’s t test. WT, wild type; febux, febuxostat; RBCs, red blood cells.
Figure 3. XO inhibition decreases hemolysis.
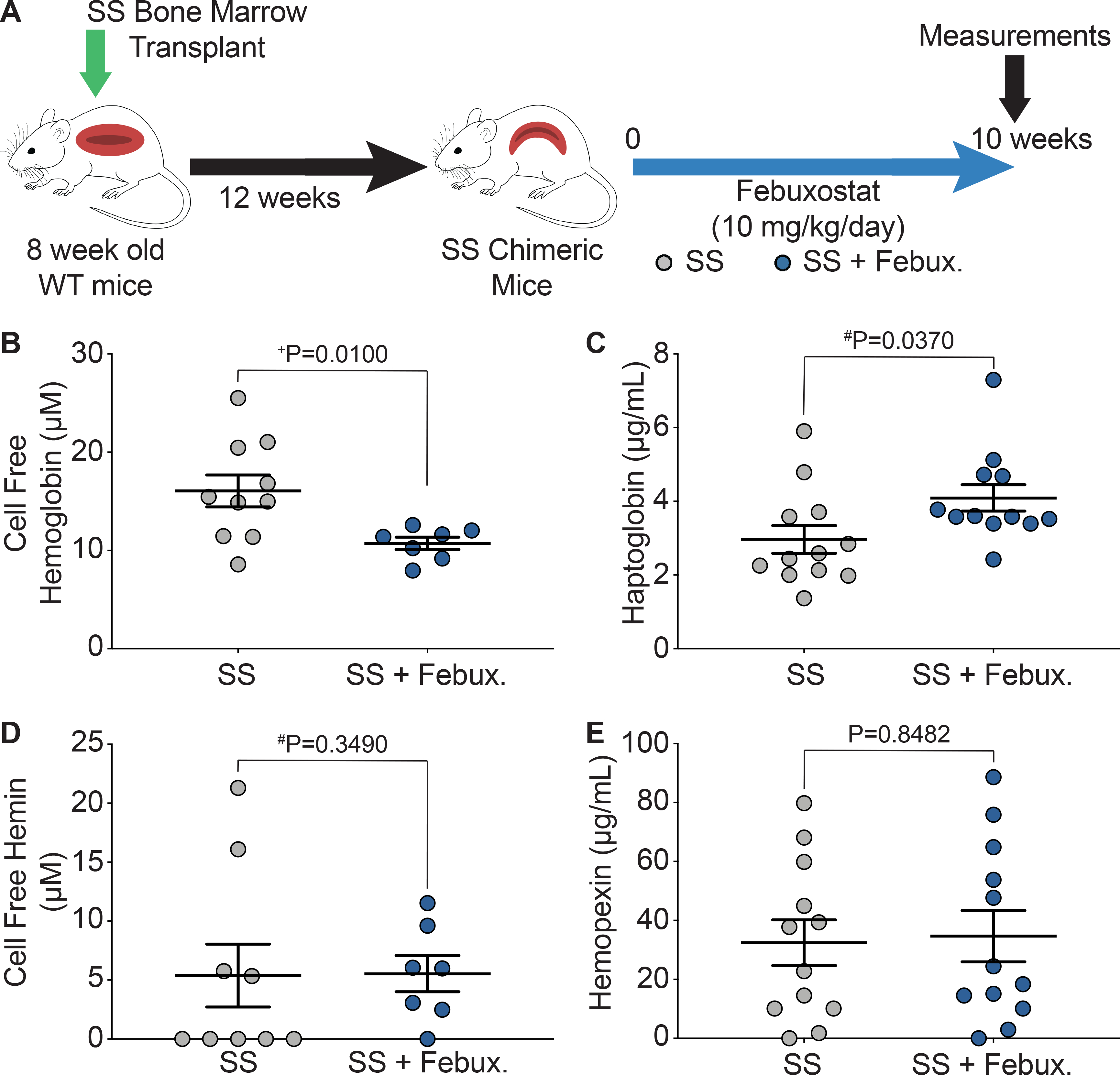
A) Experimental design. B) UV-visible spectral deconvolution was used to measure plasma cell free hemoglobin, a combination of oxyhemoglobin and methemoglobin, after 10 weeks of treatment. C) An ELISA was used to measure plasma haptoglobin concentration after 10 weeks of treatment. D) UV-visible spectral deconvolution was used to measure plasma cell free hemin concentration after 10 weeks of treatment. E) An ELISA was used to measure plasma hemopexin concentration after 10 weeks of treatment. Values are mean ± SEM using an unpaired Student’s t test unless otherwise noted. +Values are mean ± SEM using an unpaired Student’s t test with Welch’s correction. #Values are mean ± SEM using a Mann-Whitney test. WT, wild type; febux, febuxostat; ELISA, enzyme-linked immunosorbent assay.
XO Inhibition Improves Pulmonary Vasoreactivity in Chimeric Sickle Mice
Wire myography was used to assess isolated pulmonary, mesenteric, and thoracodorsal (TDA) vasoreactivity in SS sickle (Figure 4A) and AA control mice (Supplemental Figure IIIA). Febuxostat treatment significantly decreased constriction in pulmonary, mesenteric, and TDA arteries of SS sickle mice (Figure 4B–D). In the AA control mice, febuxostat treatment significantly reduced constriction in pulmonary vessels, but no differences in mesenteric or TDA constriction were observed (Supplemental Figure IIIB–D). A cumulative acetylcholine dose response was used to assess endothelium dependent dilation of the three vascular beds. In the SS sickle mice, febuxostat treatment significantly improved the dilation of the pulmonary vessels and decreased the EC50 compared to the untreated vessels (Figure 4E); however, this effect was not observed with vessels from the systemic circulation as no difference was observed in the mesenteric or TDA vessel dilation (Figure 4F–G). No differences in dilation were observed in the AA control mice (Supplemental Figure IIIE–G).
Figure 4. XO inhibition decreases constriction across multiple vascular beds, but only improves dilation in pulmonary arteries.
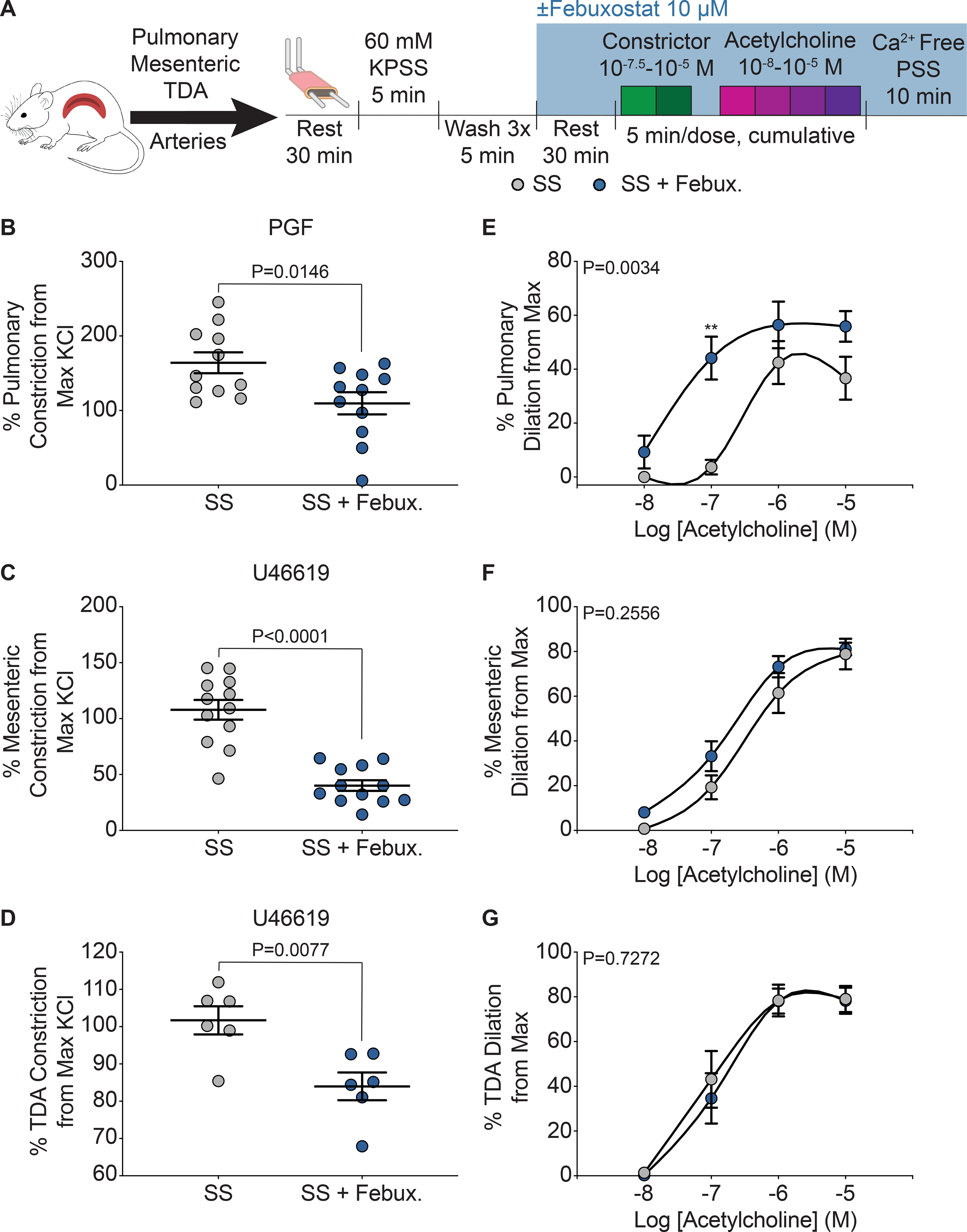
A) Ex vivo wire myography was used to asses vasoreactivity of pulmonary, mesenteric, and TDA arteries. B) Pulmonary (n=8), C) mesenteric (n=12), and D) TDA (n=6) constriction was measured by normalizing to maximum KCl response. Values are mean ± SEM using an unpaired Student’s t test. An acetylcholine dose response was used to measure dilation of E) pulmonary, F) mesenteric, and G) TDA arteries. Values are mean ± SEM using a two-way ANOVA with Sidak’s multiple comparisons test. **P<0.01. TDA, thoracodorsal; KPSS; potassium physiological salt solution; PSS, physiological salt solution; febux, febuxostat; max, maximum; PGF, prostaglandin F2α; XO, xanthine oxidase.
Improvements in Pulmonary Vasoreactivity Did Not Manifest into Cardiac Changes
We hypothesized that improved pulmonary vasoreactivity would also result in decreased pulmonary artery pressure and improved cardiac function. To assess cardiac function, closed chest right heart catheterization, right ventricle (RV) weight, and left ventricle + septum (LV + S) weight were measured after ten weeks of treatment. Additionally, echocardiograms were performed at 0 and 10 weeks of treatment and changes in parameters are shown as a delta change from baseline to ten weeks of treatment (Figure 5A). Febuxostat treatment did not alter the RV maximum pressure or heart rate of the SS sickle mice (Figure 5B–C). A complete list of parameters from the RV catheterization is included in Supplemental Table V. No differences were observed in the AA control or SS sickle mice when treated with febuxostat; however, significant increases in RV maximum dP/dt and RV contractile index were observed in the SS mice in comparison to the AA mice (Supplemental Table V). The RV was separated from the LV + S and weighed and normalized to tibia length in order to assess hypertrophy of the ventricles. No significant differences in RV or LV + S weight were observed in the SS sickle (Figure 5D–E) or the AA control (Supplemental Figure IVA–B) mice. Trichrome staining was also performed to measure fibrosis of the right and left ventricles. Fibrosis was observed in the SS sickle and SS sickle mice treated with febuxostat; however, when quantified no difference in fibrosis was observed in the RV or LV + S (Supplemental Figure IVC–E). Echocardiograms were performed before and after 10 weeks of febuxostat treatment in order to assess the effects of febuxostat treatment on cardiac function. No differences were observed in end systolic volume, end diastolic volume, stroke volume, or cardiac output in the SS sickle mice (Figure 5F–I). A complete list of right ventricle echocardiogram parameters is included in Supplemental Table VI. Febuxostat treatment significantly increased the tricuspid valve peak velocity of early diastolic transmitral flow to peak velocity of early diastolic mitral annular motion ratio (TV E/e’) in the SS sickle mice but did not affect any other measurement (Supplemental Table VI). In comparing the SS sickle to the AA control mice, the SS sickle mice had significantly increased tricuspid valve peak velocity of early diastolic transmitral flow (TV E) and the ratio of tricuspid valve peak velocity of early diastolic transmitral flow to peak velocity of late transmitral flow (TV E/A) (Supplemental Table VI). A complete list of left ventricle echocardiogram parameters is included in Supplemental Table VII. Febuxostat treatment did not significantly alter any left ventricle parameters assessed in the AA control or SS sickle mice (Supplemental Table VII). SS febuxostat treated mice had significantly decreased isovolumic contraction time (IVCT) and significantly increased cardiac output, LV mass, LV mass corrected, and left ventricle anterior wall thickness during systole and diastole (LVAW;s, and LVAW;d) compared to AA febuxostat treated mice (Supplemental Table VII).
Figure 5. XO inhibition did not affect pulmonary pressure or cardiac function.
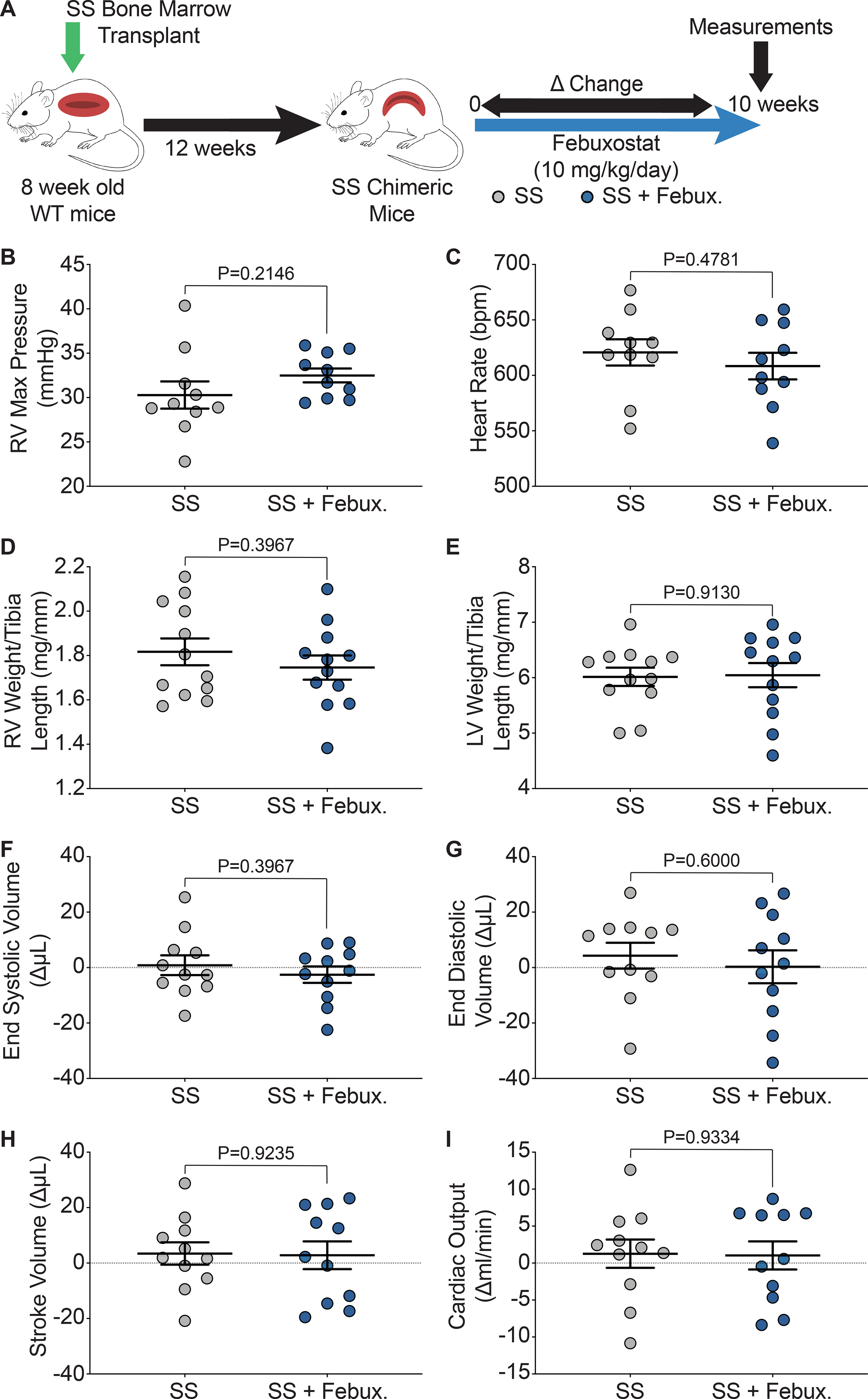
A) Experimental design. Closed chest right heart catheterization was used to measure B) RV maximum pressure and, C) heart rate after 10 weeks of treatment. D) RV weight and E) LV + septum weight was normalized to tibia length. Echocardiogram was used to asses cardiac function as a delta change from 0 to 10 weeks of febuxostat treatment: F) systolic volume, G) diastolic volume, H) stroke volume, and I) cardiac output. Values are mean ± SEM using an unpaired Student’s t test. WT, wild type, febux, febuxostat; RV, right ventricle; max, maximum; LV, left ventricle; XO, xanthine oxidase.
Hepatic XO is not the Driver of Hemolysis in Chimeric Sickle Mice
In order to evaluate the role of hepatic derived XO during hemolysis in SCD mice, 6–13-week old, Xdhfloxed/floxedAlb-1Cre/Wt (HXdh−/−, hepatocyte-specific XO KO) and Xdhfloxed/floxedAlb-1Wt/Wt (Xdhfl/fl, littermate control) mice were bone marrow transplanted with SS sickle bone marrow from Townes mice (Figure 6A, Supplemental Figure VA). After 12 weeks, the mice were fully engrafted with the SS sickle Townes bone marrow (Supplemental Figure VB). The mice were aged an additional 10 weeks to match the experimental timeline of the febuxostat treated groups. Blood was collected at 0, 3, 6, and 10 weeks post-engraftment with 10 weeks being the final endpoint of the study (Figure 6A). Hepatocyte-specific XO KO (HXdh−/−) significantly decreased plasma and liver XO activity but did not impact lung or kidney XO activity levels (Figure 6B, Supplemental Table VIII). XO inhibition was further evaluated in plasma via measurement of hypoxanthine, xanthine, and urate concentrations. Consistent with pharmacological XO inhibition the HXdh−/− mice had no change in hypoxanthine, and a significant decrease in xanthine and urate concentration compared to the Xdhfl/fl mice (Supplemental Figure VC). Hepatocyte-specific XO KO did not alter the body weight of the mice compared to the Xdhfl/fl mice (Supplemental Figure VD). In contrast to the febuxostat treated SS sickle mice, there was no significant difference in hematocrit, hemoglobin, RBCs, platelets, or any hematological parameter measured between the HXdh−/− and Xdhfl/fl mice (Figure 6C–F, Supplemental Table IX). Additional measurements of hemolysis confirmed hepatocyte-specific XO KO has no effect on hemolysis. No differences were observed in haptoglobin, cell free hemoglobin (methemoglobin and oxyhemoglobin), cell free hemin, or hemopexin (Figure 6G–H, Supplemental Figure VE–G). Wire myography was again used to asses pulmonary, mesenteric, and TDA vasoreactivity in the Xdhfl/fl and HXdh−/− mice. Despite the improvement in pulmonary artery dilation observed in the febuxostat treated mice, the Xdhfl/fl and HXdh−/− pulmonary arteries had comparable dilation (Figure 6I). There was no difference in pulmonary, mesenteric, or TDA constriction or mesenteric or TDA dilation (Supplemental Figure VIA–F). Finally, we evaluated changes in cardiac function and pulmonary pressure between the Xdhfl/fl and HXdh−/− mice. No difference was observed in RV or LV + S weight normalized to tibia length (Supplemental Figure VIH–I) and closed chest RV catheterization parameters (Supplemental Table V). Echocardiograms of Xdhfl/fl and HXdh−/− mice showed a significant increase in fractional area change (FAC) in the HXdh−/− mice (Supplemental Table VI), but no other differences in RV or LV parameters (Supplemental Tables VI–VII). FAC is a measure of the percent RV area change between systole and diastole, and an increase in the HXdh−/− mice could suggest improved RV function.
Figure 6. Hepatocyte-specific XO knockout did not decrease hemolysis or alter pulmonary vasoreactivity.
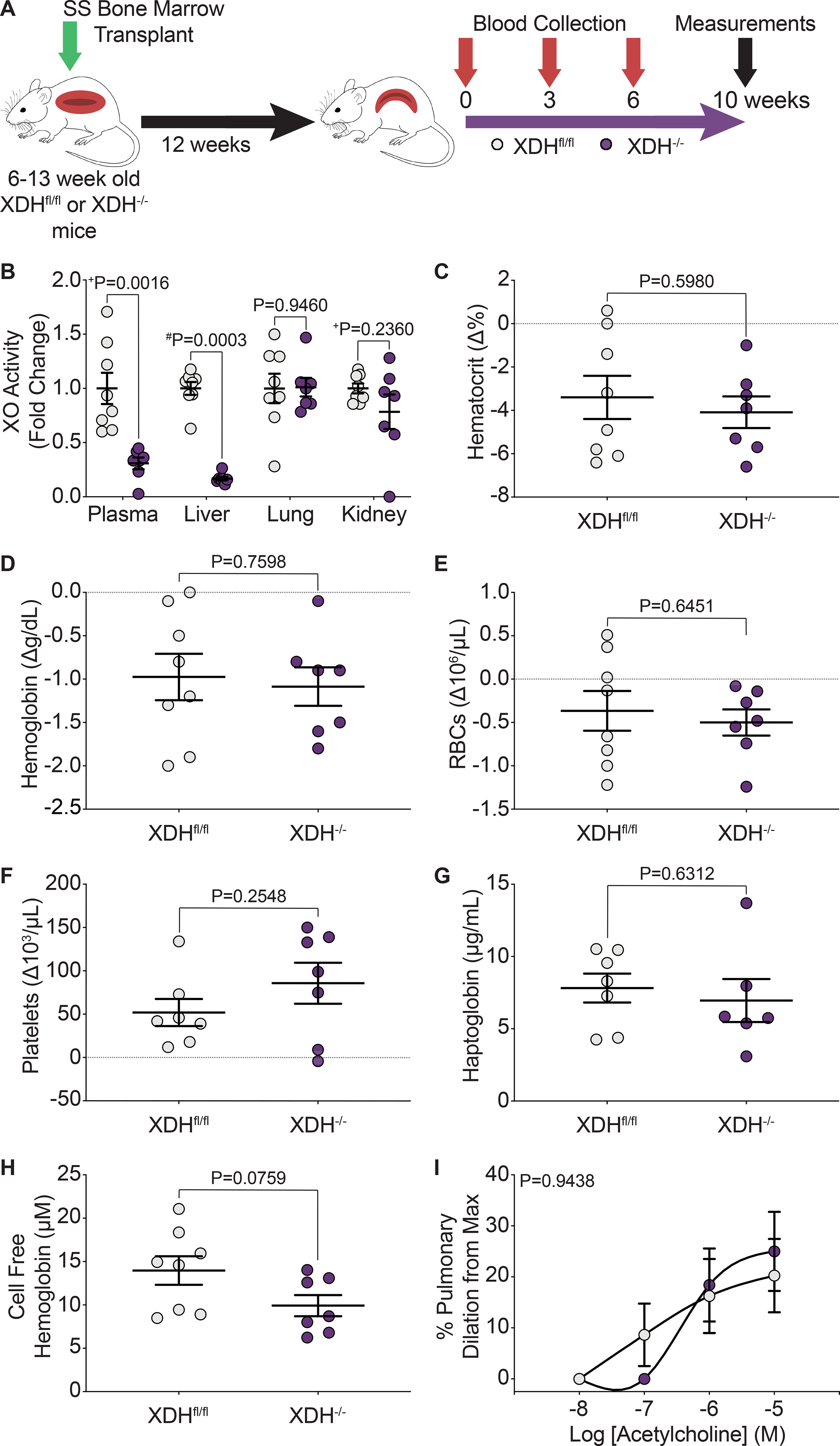
A) Experimental design. B) HPLC was used to measure plasma, liver, lung, and kidney XO activity. Complete blood counts show the hematological parameters C) hematocrit, D) hemoglobin, E) RBCs, and F) platelets, as a delta change from 0 to 10 weeks post-engraftment. G) An ELISA was used to measure plasma haptoglobin concentration 10 weeks post-engraftment. H) UV-visible spectral deconvolution was used to measure plasma cell free hemoglobin, a combination of oxyhemoglobin and methemoglobin, 10 weeks post-engraftment. Values are mean ± SEM using an unpaired Student’s t test unless otherwise noted. +Values are mean ± SEM using an unpaired Student’s t test with Welch’s correction. #Values are mean ± SEM using a Mann-Whitney test. I) An acetylcholine dose response was used to measure dilation of pulmonary arteries 10 weeks post-engraftment (Xdhfl/fl n=8, Xdh−/− n=7). Values are mean ± SEM using a two-way ANOVA with Sidak’s multiple comparisons test. XDH, xanthine dehydrogenase; XO, xanthine oxidase; RBCs, red blood cells; max, maximum; HPLC, high performance liquid chromatography ELISA, enzyme linked immunosorbent assay.
DISCUSSION
SCD affects approximately 300,000 new patients worldwide each year;1 however, treatment options are limited, with no FDA-approved drugs specifically inhibiting hemolysis. The molecular drivers of hemolysis are not well understood, leaving a gap in the overall understanding of the disease and a target for therapeutics that could improve sickle pathologies. We have identified XO as a key driver of hemolysis in a chimeric mouse model of sickle cell disease. Bone marrow transplanted SS sickle mice treated with febuxostat, an FDA-approved XO inhibitor, have increased hematocrit, cellular hemoglobin, and RBC number (Figure 2B–D), with no impact on reticulocyte percentage (Figure 2F). Since there is no difference in reticulocyte percentage, the improvements in hematocrit, cellular hemoglobin, and RBCs is likely due to less hemolysis, rather than increased RBC production; however, it is also possible that RBC storage sites such as the liver, are releasing red cells in response to the hepatocellular damage mediated by SCD. Decreased hemolysis is further supported by an increase in haptoglobin concentration and a decrease in cell free hemoglobin (Figure 3B–C). Less cell free hemoglobin and an increase in haptoglobin, a hemoglobin scavenging protein, suggest less hemoglobin is being released from RBCs, thus less haptoglobin is required for clearance. No difference was observed in hemopexin or cell free hemin concentration (Figure 3D–E). We hypothesize that only changes in haptoglobin/cell free hemoglobin occurred as inhibition of circulating XO decreases reactive oxygen species (ROS) production, thus limiting the oxidation of heme molecules and preventing their release from hemoglobin. This is crucial as H2O2 and O2•-, both products of XO, are noted to be intimately involved in degrading hemoglobin and oxidizing histidine and cysteine heme binding residues, resulting in heme dissociation.35, 36 It has been shown that ferric heme interacts with H2O2 to produce biliverdin IXα and carbon monoxide, similar to the canonical heme degrading enzyme, heme oxygenase-1.37 ROS play a critical role in hemoglobin modulation as O2•- can react with hemoglobin to form methemoglobin or oxyhemoglobin and under certain conditions may lead to release of heme molecules; however, it has not yet been determined if XO-derived ROS directly act on hemoglobin.37 Thus, we have identified XO as a specific target capable of mediating hemolysis in chimeric SCD mice. The exact mechanism in which XO inhibition decreases hemolysis was not identified in this study; however, we hypothesize a reduction in oxidant production from XO activity protects RBCs from ROS-induced damage and hemolysis. Further studies will explore this hypothesis.
Vascular injury and pulmonary hypertension are major complications experienced by SCD patients.6, 10 By inhibiting XO activity, we were able to significantly reduce constriction in three vascular beds (pulmonary, mesenteric, and TDA) of chimeric SCD mice (Figure 4B–D). Less constriction of the vessels will allow for larger vessel diameter and therefore, improved blood flow in the febuxostat treated mice. Additionally, XO inhibition was able to significantly improve endothelium-dependent pulmonary artery dilation, while having no effect on mesenteric or TDA arteries (Figure 4E–G). This suggests that XO inhibition was able to specifically protect the endothelium in the pulmonary vasculature. It is also important to note the two key physiological differences in the pulmonary vasculature compared to other vascular beds that may impact our results: 1) pulmonary circulation has a lower pressure than systemic circulation and 2) pulmonary artery dilation is primarily NO dependent, while systemic circulation has multiple mechanisms of dilation.38
XO is released into circulation and binds to the surface of endothelial cells via electrostatic interactions with GAGs, specifically heparin sulphate (HS) and chondroitin sulfate (CS).26, 39, 40 Therefore, endothelial bound XO concentration is dependent on the composition of the glycocalyx of each vascular bed. GAG composition varies depending on the vascular bed and can also be altered under disease conditions. For example, HS and CS composition percentage is affected in human patients with lung cancer;41 however, to our knowledge GAG composition in SCD remains unexplored. We posit that differences in GAG composition between vascular beds is responsible for the pulmonary-specific improvement in endothelial-dependent vasodilation. Alternatively, XOR produced in lung endothelial cells could be released and adhere to the endothelium resulting in endothelial damage. Despite improved pulmonary vasoreactivity, there was no indication of improved pulmonary pressure or cardiac function (Figure 5). Based on our results, 10 weeks of XO inhibition has no effect on pulmonary pressure or cardiac function in sickle mice. It is possible that a longer treatment time may be required in order to impact pulmonary pressure and cardiac output or that the vessels become tolerant to the febuxostat treatment requiring more acute studies. Hematologic improvements did not occur prior to 10 weeks of treatment (Supplemental Table IV), also affirming the potential value of a long-term model.
A hepatocyte-specific XO knockout mouse model demonstrated that ~50% of circulating XO is released from the liver.29 Therefore, we bone marrow transplanted Xdhfloxed/floxedAlb-1Wt/Wt (Xdhfl/fl, littermate control) and Xdhfloxed/floxedAlb-1Cre/Wt (HXdh−/−, hepatocyte-specific XO KO) mice with SS sickle Townes bone marrow in order to evaluate the effects of hepatic-derived XO in SCD. Global pharmacologic inhibition of XO resulted in marked improvements in hematology and ex vivo pulmonary vasoreactivity; however, HXdh−/− mice showed no indications of reduced hemolysis or alteration in pulmonary vasoreactivity compared to Xdhfl/fl mice (Figure 6). This suggests hepatic-derived XO is not the driver of hemolysis in SCD or there are compensatory effects due to the loss of hepatic XO from birth. An alternative source of XO, such as endothelial-derived XO, may be responsible for causing hemolysis in SCD. The presence and activity of XOR in endothelial cells has been confirmed by numerous studies.42–44 Endothelial-derived XOR could be an important driver of hemolysis and vascular dysfunction in SCD because endothelial XO could produce oxidants that damage endothelial cells and when released into circulation generate oxidants that induce hemolysis. Exploring this hypothesis is warranted. One limitation to our febuxostat studies is the possibility that febuxostat could have off-target effects. Although febuxostat has a high affinity for XO (Ki = 0.12 nM), it has also been shown to bind and inhibit ATP binding cassette transporter (ABCG2), which has significant expression in the small intestine and kidney.45, 46 ABCG2 inhibition could be important in our model as it can control protoporphyrin IX (PPIX) efflux from cells.45 PPIX is an important intermediate in the heme biosynthesis pathway;47 therefore, interference with PPIX efflux via off-target effects of febuxostat could potentially play a role in our model.
In conclusion, we identified XO as a key driver of hemolysis in SCD. Febuxostat, an FDA-approved drug, is able to improve hematological parameters and pulmonary vascular function by decreasing hemolysis in chimeric SCD mice. We propose that modulation of XO activity could be used alone or in combination with current SCD therapies to improve the hemolysis-driven pathologies of SCD patients. While we determined hepatic XO is not the driver of hemolysis in SCD, future studies will define the role of endothelial-derived XO.
Supplementary Material
HIGHLIGHTS.
XO is a key driver of hemolysis in SCD
XO inhibition improved ex-vivo pulmonary vasoreactivity
Hepatic-derived XO is not the driver of hemolysis in SCD
ACKNOWLEDGEMENTS
The Center for Biologic Imaging at the University of Pittsburgh was utilized for imaging and image analysis of the trichrome staining.
SOURCES OF FUNDING
This work was supported, in whole or in part, by National Institutes of Health Grants: F31 HL149241 (H.M.S); R25 HL128640–03 (K.C.W); 1S10OD023684–01A1 (B.M) K01 HL133331 (D.A.V); R01 HL106192 and U01HL117721 (S.O.A); NIH R01 DK124510–01 and American Heart Association 19TPA34850089 (E.E.K); NIH R01 EY026030 and Retina Research Foundation (J.D); and R01 HL133864, R01 HL128304 and American Heart Association Established Investigator Award (A.C.S).
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- SCD
sickle cell disease
- Hb
hemoglobin
- RBCs
red blood cells
- XO
xanthine oxidase
- XDH
xanthine dehydrogenase
- XOR
xanthine oxidoreductase
- GAGs
glycosaminoglycans
- NO
nitric oxide
- BM
bone marrow
- PFA
paraformaldehyde
- CBA
coumarin boronate acid
- Hp
haptoglobin
- ELISA
enzyme linked immunosorbent assay
- Hx
hemopexin
- TDA
thoracodorsal
- PSS
physiological salt solution
- PGF
prostaglandin F2α
- RV
right ventricle
- PV
pressure volume
- WT
wild type
- RDW
red cell distribution width
- LV
left ventricle
- ROS
reactive oxygen species
- HS
heparin sulphate
- CS
chondroitin sulphate
- ABCG2
ATP binding cassette transporter
- PPIX
protoporphyrin IX
- HPLC
high performance liquid chromatography
- Febux
febuxostat
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Pinto VM, Balocco M, Quintino S and Forni GL. Sickle cell disease: a review for the internist. Intern Emerg Med. 2019;14:1051–1064. [DOI] [PubMed] [Google Scholar]
- 2.Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, Panepinto JA, Weatherall DJ, Costa FF and Vichinsky EP. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. [DOI] [PubMed] [Google Scholar]
- 3.Williams TN. Sickle Cell Disease in Sub-Saharan Africa. Hematol Oncol Clin North Am. 2016;30:343–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, Temperley WH, Williams TN, Weatherall DJ and Hay SI. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381:142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sundd P, Gladwin MT and Novelli EM. Pathophysiology of Sickle Cell Disease. Annu Rev Pathol. 2019;14:263–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mburu J and Odame I. Sickle cell disease: Reducing the global disease burden. Int J Lab Hematol. 2019;41 Suppl 1:82–88. [DOI] [PubMed] [Google Scholar]
- 7.Kato GJ, Steinberg MH and Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127:750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novelli EM and Gladwin MT. Crises in Sickle Cell Disease. Chest. 2016;149:1082–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meier ER. Treatment Options for Sickle Cell Disease. Pediatr Clin North Am. 2018;65:427–443. [DOI] [PubMed] [Google Scholar]
- 10.Nader E, Romana M and Connes P. The Red Blood Cell-Inflammation Vicious Circle in Sickle Cell Disease. Front Immunol. 2020;11:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lubeck D, Agodoa I, Bhakta N, Danese M, Pappu K, Howard R, Gleeson M, Halperin M and Lanzkron S. Estimated Life Expectancy and Income of Patients With Sickle Cell Disease Compared With Those Without Sickle Cell Disease. JAMA Netw Open. 2019;2:e1915374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP and Bonds DR. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317–22. [DOI] [PubMed] [Google Scholar]
- 13.Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377:1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niihara Y, Miller ST, Kanter J, et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med. 2018;379:226–235. [DOI] [PubMed] [Google Scholar]
- 15.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med. 2017;376:429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howard J, Hemmaway CJ, Telfer P, et al. A phase 1/2 ascending dose study and open-label extension study of voxelotor in patients with sickle cell disease. Blood. 2019;133:1865–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vichinsky E, Hoppe CC, Ataga KI, et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med. 2019;381:509–519. [DOI] [PubMed] [Google Scholar]
- 18.Turpin M, Chantalat-Auger C, Parent F, et al. Chronic blood exchange transfusions in the management of pre-capillary pulmonary hypertension complicating sickle cell disease. Eur Respir J. 2018;52. [DOI] [PubMed] [Google Scholar]
- 19.Estcourt LJ, Hopewell S, Trivella M, Hambleton IR and Cho G. Regular long-term red blood cell transfusions for managing chronic chest complications in sickle cell disease. Cochrane Database Syst Rev. 2019;2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, Tousson A, Gladwin MT, Patel RP, Tarpey MM, Batinic-Haberle I, White CR and Freeman BA. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc Natl Acad Sci U S A. 2001;98:15215–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osarogiagbon UR, Choong S, Belcher JD, Vercellotti GM, Paller MS and Hebbel RP. Reperfusion injury pathophysiology in sickle transgenic mice. Blood. 2000;96:314–20. [PubMed] [Google Scholar]
- 22.Pritchard KA Jr., Ou J, Ou Z, Shi Y, Franciosi JP, Signorino P, Kaul S, Ackland-Berglund C, Witte K, Holzhauer S, Mohandas N, Guice KS, Oldham KT and Hillery CA. Hypoxia-induced acute lung injury in murine models of sickle cell disease. Am J Physiol Lung Cell Mol Physiol. 2004;286:L705–14. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt HM, Kelley EE and Straub AC. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol. 2019;21:101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pacher P, Nivorozhkin A and Szabo C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58:87–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parks DA and Granger DN. Xanthine oxidase: biochemistry, distribution and physiology. Acta Physiol Scand Suppl. 1986;548:87–99. [PubMed] [Google Scholar]
- 26.Adachi T, Fukushima T, Usami Y and Hirano K. Binding of human xanthine oxidase to sulphated glycosaminoglycans on the endothelial-cell surface. Biochem J. 1993;289 ( Pt 2):523–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houston M, Estevez A, Chumley P, Aslan M, Marklund S, Parks DA and Freeman BA. Binding of xanthine oxidase to vascular endothelium. Kinetic characterization and oxidative impairment of nitric oxide-dependent signaling. J Biol Chem. 1999;274:4985–94. [DOI] [PubMed] [Google Scholar]
- 28.Diederich L, Suvorava T, Sansone R, Keller TCS, Barbarino F, Sutton TR, Kramer CM, Lückstädt W, Isakson BE, Gohlke H, Feelisch M, Kelm M and Cortese-Krott MM. On the Effects of Reactive Oxygen Species and Nitric Oxide on Red Blood Cell Deformability. Frontiers in Physiology. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harmon DB, Mandler WK, Sipula IJ, et al. Hepatocyte-Specific Ablation or Whole-Body Inhibition of Xanthine Oxidoreductase in Mice Corrects Obesity-Induced Systemic Hyperuricemia Without Improving Metabolic Abnormalities. Diabetes. 2019;68:1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Donnell BJ, Guo L, Ghosh S, Shah FA, Strollo PJ Jr., McVerry BJ, Gladwin MT, Ofori-Acquah SF, Kato GJ and O’Donnell CP. Sleep phenotype in the Townes mouse model of sickle cell disease. Sleep Breath. 2019;23:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan TM, Ciavatta DJ and Townes TM. Knockout-transgenic mouse model of sickle cell disease. Science. 1997;278:873–6. [DOI] [PubMed] [Google Scholar]
- 32.Wood KC, Durgin BG, Schmidt HM, Hahn SA, Baust JJ, Bachman T, Vitturi DA, Ghosh S, Ofori-Acquah SF, Mora AL, Gladwin MT and Straub AC. Smooth muscle cytochrome b5 reductase 3 deficiency accelerates pulmonary hypertension development in sickle cell mice. Blood Adv. 2019;3:4104–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oh JY, Hamm J, Xu X, Genschmer K, Zhong M, Lebensburger J, Marques MB, Kerby JD, Pittet JF, Gaggar A and Patel RP. Absorbance and redox based approaches for measuring free heme and free hemoglobin in biological matrices. Redox Biol. 2016;9:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chao JR, Knight K, Engel AL, Jankowski C, Wang Y, Manson MA, Gu H, Djukovic D, Raftery D, Hurley JB and Du J. Human retinal pigment epithelial cells prefer proline as a nutrient and transport metabolic intermediates to the retinal side. J Biol Chem. 2017;292:12895–12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dawson RB, Rafal S and Weintraub LR. Absorption of hemoglobin iron: the role of xanthine oxidase in the intestinal heme-splitting reaction. Blood. 1970;35:94–103. [PubMed] [Google Scholar]
- 36.Donegan RK, Moore CM, Hanna DA and Reddi AR. Handling heme: The mechanisms underlying the movement of heme within and between cells. Free radical biology & medicine. 2019;133:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagababu E and Rifkind JM. Heme degradation by reactive oxygen species. Antioxid Redox Signal. 2004;6:967–78. [DOI] [PubMed] [Google Scholar]
- 38.Suresh K and Shimoda LA. Lung Circulation. Compr Physiol. 2016;6:897–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pritsos CA. Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chem Biol Interact. 2000;129:195–208. [DOI] [PubMed] [Google Scholar]
- 40.Radi R, Rubbo H, Bush K and Freeman BA. Xanthine oxidase binding to glycosaminoglycans: kinetics and superoxide dismutase interactions of immobilized xanthine oxidase-heparin complexes. Arch Biochem Biophys. 1997;339:125–35. [DOI] [PubMed] [Google Scholar]
- 41.Li G, Li L, Joo EJ, Son JW, Kim YJ, Kang JK, Lee KB, Zhang F and Linhardt RJ. Glycosaminoglycans and glycolipids as potential biomarkers in lung cancer. Glycoconj J. 2017;34:661–669. [DOI] [PubMed] [Google Scholar]
- 42.Phan SH, Gannon DE, Varani J, Ryan US and Ward PA. Xanthine oxidase activity in rat pulmonary artery endothelial cells and its alteration by activated neutrophils. Am J Pathol. 1989;134:1201–11. [PMC free article] [PubMed] [Google Scholar]
- 43.Sohn HY, Krotz F, Gloe T, Keller M, Theisen K, Klauss V and Pohl U. Differential regulation of xanthine and NAD(P)H oxidase by hypoxia in human umbilical vein endothelial cells. Role of nitric oxide and adenosine. Cardiovasc Res. 2003;58:638–46. [DOI] [PubMed] [Google Scholar]
- 44.Rieger JM, Shah AR and Gidday JM. Ischemia-reperfusion injury of retinal endothelium by cyclooxygenase- and xanthine oxidase-derived superoxide. Exp Eye Res. 2002;74:493–501. [DOI] [PubMed] [Google Scholar]
- 45.Kast RE, Skuli N, Sardi I, Capanni F, Hessling M, Frosina G, Kast AP, Karpel-Massler G and Halatsch ME. Augmentation of 5-Aminolevulinic Acid Treatment of Glioblastoma by Adding Ciprofloxacin, Deferiprone, 5-Fluorouracil and Febuxostat: The CAALA Regimen. Brain Sci. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyata H, Takada T, Toyoda Y, Matsuo H, Ichida K and Suzuki H. Identification of Febuxostat as a New Strong ABCG2 Inhibitor: Potential Applications and Risks in Clinical Situations. Front Pharmacol. 2016;7:518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sachar M, Anderson KE and Ma X. Protoporphyrin IX: the Good, the Bad, and the Ugly. J Pharmacol Exp Ther. 2016;356:267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.