

Abstract
DNA methylation is essential to mammalian development, and dysregulation can cause serious pathological conditions. Key enzymes responsible for deposition and removal of DNA methylation are known, but how they cooperate to tightly regulate the methylation landscape remains a central question. Utilizing a knockin DNA methylation reporter, we performed a genome-wide CRISPR/Cas screen in human embryonic stem cells to discover DNA methylation regulators. The top screen hit was an uncharacterized gene, QSER1, which proved to be a key guardian of bivalent promoters and poised enhancers of developmental genes, especially those residing in DNA methylation valleys (or canyons). We further demonstrate genetic and biochemical interactions of QSER1 and TET1, supporting their cooperation to safeguard transcriptional and developmental programs from DNMT3-mediated de novo methylation.
Print Page Summary
Introduction:
DNA methylation is essential to mammalian development, and dysregulation can cause serious pathological conditions, including immunodeficiency-centromeric instability-facial anomalies (ICF) syndrome and microcephalic dwarfism. The DNMT and TET enzymes are responsible for addition and removal of DNA methylation, but how they coordinate to tightly regulate the methylation landscape remains a central question. Utilizing a knockin DNA methylation reporter, we performed a genome-wide CRISPR/Cas screen in human embryonic stem cells (hESCs) to discover DNA methylation regulators.
Rationale:
We focused on bivalent promoters, defined by the presence of both active (H3K4me3) and repressive (H3K27me3) histone marks and typically occupied by Polycomb-repressive complexes 1 and 2 (PRC1 and PRC2). In stem or progenitor cells, bivalent promoters are believed to maintain developmental regulators in a “poised state” ready for activation upon differentiation, and they are sensitive to DNA hypermethylation in dysfunctional cellular contexts such as cancer or aging. Creation of a knockin DNA methylation reporter line provided a unique opportunity to visualize epigenetic alterations otherwise “invisible” in terms of gene expression changes in the stem cell state. Using the bivalent PAX6 P0 promoter as a representative locus, we aimed to discover mechanisms that regulate DNA methylation at regions with similar chromatin features, which would inform not only gene regulation during development but also epigenetic dysregulation in disease.
Results:
Our screen successfully identified known methylation regulators such as TET1, TDG, and KDM2B, but also functionally uncharacterized genes including QSER1. Like the TET proteins, QSER1 safeguards bivalent promoters and poised enhancers (marked by H3K4me1 but not H3K27ac) against hypermethylation. However, distinct from the more general protective effect of the TET proteins on regulatory regions, QSER1 preferentially protects broad PRC2-bound/H3K27me3-marked regions and DNA methylation valleys (DMVs). Also known as DNA methylation canyons, DMVs identify large (≥ 5kb) hypomethylated regions present in cells of many lineages and conserved across vertebrates. They are enriched with bivalent promoters, developmental genes, and transcription factors including PAX6 and the HOX genes. QSER1 and TET1 showed high correlation in genomic occupancy measured by ChIP-seq, and both were high at DMVs, whereas the binding of de novo methyltransferases DNMT3A/3B was excluded in DMVs and relatively enriched in the flanking regions. Further proteomic and genomic analyses revealed that QSER1 and TET1 share many common interacting proteins, depend on each other for efficient recruitment to DNA, and cooperate to limit the encroachment of DNMT3A/3B in DMVs. In addition, deleting DNMT3B reversed the hypermethylation in QSER1 knockout (KO) hESCs. Furthermore, combined KO of QSER1 and TET1 had a stronger impact on DNA methylation and gene expression than either single KO and resulted in a failure of hESC differentiation to PDX1+NKX6.1+ pancreatic progenitors.
Conclusion:
We show QSER1 cooperates with TET1 to safeguard transcriptional and developmental programs from DNMT3-mediated de novo methylation at important genomic loci, especially in DMVs and bivalent promoters where hypermethylation has been linked to developmental disorders and cancers. Our work highlights the utility of unbiased genome-wide screens and locus-specific epigenetic measurements, including, but not limited to, DNA methylation for probing the epigenetic regulatory mechanisms relevant to human health.
One Sentence Summary:
QSER1 cooperates with TET1 to safeguard DNA methylation valleys from DNMT3-mediated de novo methylation
Graphical Abstract
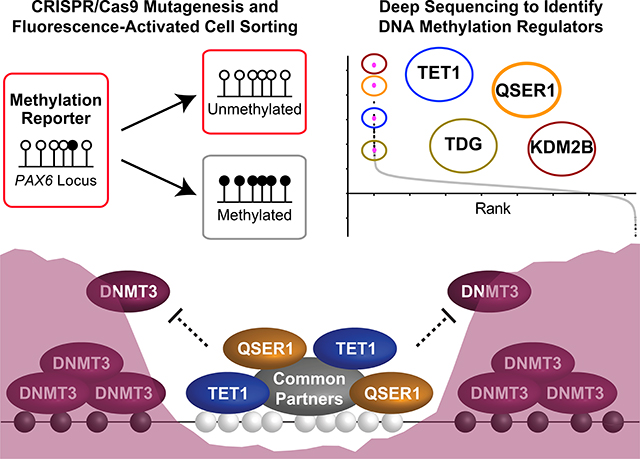
QSER1 safeguards DNA methylation valleys from de novo methylation. A fluorescence reporter hESC line (indicated by the red outline for fluorescence) was generated to track DNA methylation levels of the PAX6 promoter region in a genome-scale CRISPR/Cas9 screen for regulators of DNA methylation. A top screen hit, QSER1, cooperates with TET1 to antagonize DNMT3-mediated de novo methylation (depicted by the purple shade) at DMVs.
DNA methylation is essential to mammalian development, and aberrant DNA methylation is the cause of many genetic diseases, including immunodeficiency-centromeric instability-facial anomalies syndrome (ICF) and microcephalic dwarfism (1–4). There is also strong evidence to suggest that dysregulated DNA methylation plays a causative role in cancer, aging, and neurodegenerative disorders (4–6). The DNA methyltransferases DNMT3A and DNMT3B execute de novo methylation by adding a methyl group to cytosine of CpG dinucleotides, creating 5-methylcytosine (5mC), and DNMT1 is responsible for maintaining methylation following DNA replication (7). The Ten-eleven translocation (TET) family of enzymes (TET1, TET2, and TET3) initiate DNA demethylation through the progressive oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), all of which can be removed actively by DNA repair mechanisms or passively through DNA replication (8). In mice, knockout (KO) of Dnmtl, Dnmt3b, or triple knockout (TKO) of all three Tet enzymes causes embryonic lethality, and Dnmt3a null mice die shortly after birth (9–12). In human embryonic stem cells (hESCs), deletion of DNMT1 results in rapid depletion of global DNA methylation and cell death (13). DNMT3A/3B double KO and TET TKO hESCs are viable but show aberrant methylation at focal regions of the genome, including CpG islands (CGIs) and CGI shores of developmental genes (13, 14). Together, these studies demonstrate the essential role of DNA methylation in development. They further show that whether a particular CpG is targeted for or protected against methylation is not random, but rather a function of its genomic and cellular context.
Despite knowledge of the methylation and demethylation enzymes, the mechanisms responsible for their coordinated actions, especially at regulatory regions such as promoters and enhancers, are not well understood. Regional modulation of CpG methylation is regulated in part by the recruitment of TETs. TET1 and TET3 bind to CpG-rich DNA through the CXXC domain, and additional proteins have been proposed to assist in TET recruitment (8, 15, 16). Not mutually exclusive from the recruitment of TETs, crosstalk with histone modifications and histone modifying enzymes could also contribute to the regulation of CpG methylation. In particular, TET TKO hESCs showed prominent gain of DNA methylation at bivalent promoters (e.g., the PAX6 P0 promoter), domains with both active (H3K4me3) and repressive (H3K27me3) chromatin marks (17). A prominent feature of bivalent promoters is the occupancy of Polycomb group (PcG) proteins, which form two main repressive complexes, PRC1 and PRC2. The non-canonical PRC1.1 complex component Kdm2b has been shown to protect Polycomb-bound promoters from DNA hypermethylation in mouse embryonic stem cells (mESCs) (18), and a similar role has been implicated for another complex component Bcor in the hematopoietic system (19). However, opposing roles have been suggested for PRC2, which is responsible for deposition of H3K27me3: an early study showed that PRC2 core component EZH2 is required for CpG methylation of Polycomb-bound promoters through the recruitment of DNMTs (20); yet several recent studies suggest that PRC2 could also antagonize DNA methylation (21–23). Therefore, there are few unified models to explain the regional modulation of CpG methylation at bivalent promoters and other regulatory regions.
In stem or progenitor cells, bivalent promoters are believed to maintain developmental regulators in a “poised state” ready for activation upon differentiation (17), and they are susceptible to DNA hypermethylation in dysfunctional cellular contexts such as cancer or aging (24, 25). In an effort to discover regulators of DNA methylation at bivalent promoters in hESCs, we performed a genome-wide CRISPR/Cas screen that utilized a sensitive DNA methylation reporter system (26). Our screen identified known methylation regulators such as TET1, TDG, and KDM2B, but also functionally uncharacterized genes including QSER1 that safeguards DNA from hypermethylation. We show that QSER1 preferentially protects bivalent promoters and poised enhancers, many of which reside in DNA methylation valleys (DMVs). Also known as DNA methylation canyons, DMVs identify large, conserved regions of low DNA methylation in cells of diverse lineages (27–29). Further mechanistic enquiry revealed that QSER1 and TET1 depend on each other for efficient recruitment to DNA, and together they inhibit the DNA binding of DNMT3A/3B and safeguard developmental programs. These fmdings help explain the region-specific hypermethylation observed in TET loss-of-function studies and provide a model for how human cells maintain low methylation at important genomic loci, especially in DMVs where hypermethylation has been linked to developmental disorders and cancers (28–31).
Results
A genome-wide CRISPR/Cas screen for regulators of DNA methylation
We used the PAX6 P0 promoter as a representative locus to uncover regulators of DNA methylation at bivalent promoters and additional regulatory regions with similar genomic features (Fig. 1A, S1A). To visualize locus-specific methylation changes, we utilized a recently developed reporter system, which uses the methylation-sensitive minimal Snrpn promoter to translate the methylation levels of a neighboring region into expression of the fluorescent protein tdTomato (26). In the H1 iCas9 hESC line, which expresses Cas9 upon doxycycline treatment (32, 33), we targeted the reporter construct into the endogenous PAX6 locus just upstream of the promoter region that is hypermethylated in TET TKO hESCs (Fig. 1A, S1A). The resulting hESC line, named iCas9;PAX6-DmetdTom, displayed ~75% tdTomato-positive (tdTomato+) cells, consistent with the expected hypomethylated state of the locus (Fig. 1B), and the integrated reporter construct did not affect PAX6 expression upon neuroectoderm differentiation (Fig. S1B). In the days following TET1 targeting using lentiviruses expressing TET1-targeting gRNAs, the reporter hESCs showed increasing levels of DNA methylation at both the endogenous PAX6 region and the integrated Snrpn promoter (Fig. 1C), which were accompanied by a concordant decrease of tdTomato+ cells (Fig. 1D, S1C). Further supporting the fidelity of this reporter, clonal TET1 KO hESCs (around 20 days after targeting) had almost no tdTomato+ cells and showed nearly 100% methylation levels in the CpGs examined at the Snrpn promoter and the endogenous PAX6 region (Fig. 1C, S1C–D).
Fig. 1.

A genome-wide CRISPR/Cas screen for regulators of DNA Methylation. (A) Knockin strategy for generating iCas9;PAX6-DmetdTom reporter in reference to DNA methylation at PAX6 locus from Whole Genome Bisulfite Sequencing in WT (black) and TET TKO (blue) hESCs (14). LoxP-flanked puromycin was excised with transient expression of Cre recombinase. (B) Immunofluorescence staining (with anti-RFP antibody) and flow cytometry for tdTomato expression. (C) Bisulfite sequencing of PAX6 promoter and inserted Snrpn promoter. TET1KO-D4 and D16 denote KO using lentivirus expressing TET1_gRNA#1 where cells were collected on day 4 (D4) or day 16 (D16). Day 1 (D1) denotes 5 days after doxycycline treatment. (D) Summary of flow cytometry of lentivirus TET1 KO cells. (E) Screen schematic: doxycycline induction for Cas9 expression and puromycin selection for integration of lentiviruses. (F) Screen results: MAGeCK RRA score vs. average of two highest Z-scores for each gene. Venn diagram indicates overlap between top 100 genes in each ranking method. Genes selected for validation and selected top hits are highlighted. (G) Summary of flow cytometry of 54 lines expressing gRNAs targeting genes for validation compared to non-targeting control line (n=2). P values = one-way ANOVA followed by Dunnet multiple comparisons test.
Having established the feasibility of using the tdTomato reporter as a sensor for methylation levels at the PAX6 locus, we proceeded with a pooled screening strategy established in our laboratory (34) (Fig. 1E). We first infected the iCas9;PAX6-DmetdTom reporter hESCs with the pooled lentiviral human Brunello library (35). After five days of Cas9 expression, cells were maintained for 15 additional days to allow for DNA methylation levels to change, and then tdTomato+ and tdTomato− cells were isolated through fluorescence-activated cell sorting (FACS) (Fig. S1E). The abundance of individual gRNAs in each population was determined by high-throughput sequencing. To identify hits, we used the MAGeCK robust ranking aggregation (RRA) algorithm (36) and also calculated the Z-score for each gRNA based on the ratio of gRNA reads in the tdTomato− versus tdTomato+ population (Fig. S1F–G). We identified 66 overlapping genes out of the top 100 hits from each method, and they included genes known to affect DNA methylation such as TET1, TDG, KDM2B, and BCOR (8, 18, 19) (Fig. 1F, Table S1). Among the 54 overlapping hits selected for validation, 40 genes (74%) significantly affected tdTomato expression when targeted using gRNAs expressed from lentiviral vectors (Fig. 1G, S1H).
QSER1 protects bivalent promoters and poised enhancers against hypermethylation
QSER1, the highest ranked gene in the screen (Fig. 1F), was of particular interest given that it is functionally uncharacterized. In mESCs, Qser1 was previously identified by mass spectrometry as a putative Tet1 binding partner, supporting a possible link with TET1 (15). To explore the function of QSER1, we created a clonal QSER1 KO line in the H1 hESC background (without the methylation reporter) (Fig. 2A). QSER1 KO caused significant hypermethylation at the PAX6 P0 promoter, similar to the degree of increase observed in the TET1 KO generated in the same H1 background (Fig. 2B). Methylome analysis showed that most differentially methylated regions (DMRs) were hypermethylated (Fig. 2C). The vast majority of hyper-methylated DMRs (hyper-DMRs) were in regions indicative of gene regulation such as DNase I-hypersensitive sites (DHS), promoters, and enhancers (Fig. 2C), which were predominantly bivalent promoters (marked by H3K4me3 and H3K27me3) and poised enhancers (marked by H3K4me1 but not H3K27ac) (Fig. 2D). Indeed, among promoter and enhancer types, bivalent promoters and poised enhancers showed the greatest increase in DNA methylation in QSER1 KO (Fig. 2E, S2A).
Fig. 2.

QSER1 protects bivalent promoters and poised enhancers against hypermethylation. (A) Schematic illustrating QSER1 targeting, 20-nt CRISPR sequence (green), PAM sequence (red), and mutation introduced (red letter). (B) Heatmap of DNA methylation levels at PAX6 promoter (n=3; chr11:31,840,696–31,840,802) from amplicon bisulfite sequencing. (C) Left: relative quantities of hyper- and hypo-DMRs. Right: relative quantities of hyper-DMRs that overlap with indicated genomic features. (D) Bar plots showing relative quantities of hyper-DMRs that overlap with each promoter or enhancer type. (E) Hexagon plots showing methylation levels for all CpGs (n=2986165), CpGs within bivalent promoters (n=283258), and CpGs within poised enhancers (n=103153) in QSER1 KO vs. WT. (F) 1-kb tile plot showing methylation levels for QSER1 KO vs. WT. (G) Bar plot showing percent of all tiles (expected) or hyper-tiles (observed in QSER1 KO) overlapped with each genomic region. (H) Enrichment of QSER1 KO hyper- or hypo-tiles in transcription factor binding sites and histone modifications. Data from aggregate of ENCODE ChIP-seq (49) and ChIP-seq in hESCs are indicated by triangles and circles, respectively, and listed in Table S6. (I) DNA methylation difference (QSER1 KO vs. WT) around promoters, peaks of designated ChIP-seq, and 5hmC-Seal. Window extends 5 kb from region center (binned 100 bp).
To further characterize genomic regions affected in QSER1 KO hESCs, we binned genome-wide methylation signals into 1-kb tiles (Fig. 2F). The hypermethylated tiles (hyper-tiles) in QSER1 KO showed greater changes in DNA methylation compared to hypomethylated tiles (hypo-tiles) (Fig. S2B). The hyper-tiles also showed a tendency to cluster, while the hypo-tiles distributed more randomly (Fig. S2C). Consistent with the DMRs, the hyper-tiles, but not the hypo-tiles, were enriched in regions identified as DHS, CGIs, and CGI shores (Fig. S2D). In particular, the hyper-tiles showed strong enrichment of bivalent promoters and poised enhancers (Fig. 2G). Furthermore, the hyper-tiles, but not the hypo-tiles, were significantly enriched in bindings sites of PRC2 components EZH2 and SUZ12 as well as the corresponding histone mark H3K27me3 (Fisher’s exact test followed by Benjamini-Hochberg FDR procedure, odds ratio > 2 and BH-FDR < 2.2e-16) (Fig. 2H). Significant enrichment was also observed in regions with 5hmC, H3K4me1/2/3, and the non-canonical PRC1.1 complex components KDM2B and BCOR as well as the associated UbH2A mark (Fig. 2H). Consistent with these findings, we observed robust ChIP-seq signals for H3K4me3, H3K27me3, EZH2, and BCOR as well as 5hmC signal at QSER1 KO hyper-DMRs (Fig. S2E), and QSER1 KO showed a greater increase of methylation in regions occupied by TET1, EZH2, and H3K27me3 (especially when overlapped with H3K4me3 or H3K4me1), but not in regions occupied by DNMT3B, H3K79me2, H3K9me3, or H3K27ac (Fig. 2I). Collectively, these results indicate that QSER1 preferentially protects Polycomb-bound, H3K27me3-marked regulatory regions such as bivalent promoters and poised enhancers.
QSER1 and TET1 protect developmental genes and DNA methylation valleys from hypermethylation
Comparison of QSER1 KO with TET1 KO hESCs (Fig. S3A) showed that there were fewer hyper-DMRs in QSER1 KO, but the vast majority of hyper-DMRs in QSER1 KO overlapped with TET1 KO hyper-DMRs (Fig. 3A), and the hyper-DMRs from both KOs shared similar genomic features (Fig. S3B). Similar overlaps were observed for genes associated with hypermethylation at their promoters or enhancers (Fig. 3B). Intriguingly, hypermethylated genes in QSER1 KO were highly enriched in pathways of transcription regulation, homeobox proteins, DNA binding, and developmental processes (Fig. 3B, S3C). Many prominent developmental genes were affected, including those encoding members of the HOX, FOX, GATA, NKX, and PAX family of transcription factors, and signaling molecules such as SHH and BMPs (Fig. 3B, Data S1). Similar gene functional groups were reported in Gene Ontology analysis for DMVs (22, 27–29), and recent studies show that DMVs are among the regions regulated by Tet1/2 in mouse epiblast, mESCs, and mouse embryonic fibroblasts (37–39). We found that many QSER1 KO and TET1 KO hyper-DMRs overlapped with previously defined DMV loci (28) (Fig. 3A, S3D), and 36% of DMVs contained at least one QSER1 KO hyper-DMR. Indeed, QSER1 KO and TET1 KO showed high correlation of CpG methylation at DMVs (Fig. S3E), and DMVs in both KOs shifted substantially towards hypermethylation compared to wildtype (WT) (Fig. 3C–E, S3F–G). We examined hyper-tiles based on whether they were affected in QSER1 KO, TET1 KO, or both (Fig. 3F). QSER1 KO hyper-tiles showed dramatic preferential overlap with DMVs compared to control tiles, whereas such preference was not observed for hyper-tiles affected only in TET1 KO (affected in TET1 KO but not in QSER1 KO) (Fig. 3F, S3H). Therefore, while TET proteins protect regulatory regions broadly (including DMVs), QSER1 has a more preferential effect on DMVs. Indeed, TET1 KO caused a greater increase in DNA methylation for most regulatory regions examined, but at DMVs, QSER1 KO showed the same increase as TET1 KO (Fig. S3I–J).
Fig. 3.

QSER1 and TET1 protect DNA methylation valleys. (A) Total and overlapping hyper-DMRs between QSER1 KO and TET1 KO. (B) Venn diagrams representing genes with hyper-DMR associated Promoters (left) or enhancers (right). Bar plots represent pathway enrichment analysis. Red line represents FDR cutoff 0.01. (C) Hexagon plots showing CpG methylation inside DMVs (KO vs. WT). (D) Cumulative fraction of DMVs according to average methylation of each DMV. (E) Bar plot showing DMVs in designated range of average methylation. (F) DNA methylation change at QSER1 KO and TET1 KO hyper-tiles. Number of hyper-tiles in each category is indicated. DMVs and broad H3K27me3 peak overlap are indicated. (G) Heatmaps of ChIP-seq signals (log2 ratio vs. input) at DMVs ranked by methylation increase in QSER1 KO. (H) Meta-signal plots of DNA methylation at DMV Classes and 10kb flanking regions for WT and QSER1 KO. (I-J) Box plots showing average width of H3K27me3 peaks that overlap with designated hyper-tiles and methylation difference (KO-WT) for tiles that overlap with designated H3K27me3 peaks. In box plots here and later, edges refer to the upper/lower quartiles and whiskers represent 1.5 interquartile range beyond edges; Mann-Whitney U-test was conducted. Pie charts show percentage of tiles that overlap with DMVs.
To further define features of the genomic regions affected by QSER1, we ranked DMVs in order of DNA methylation increase in QSER1 KO. Class I DMVs (with the greatest increase in DNA methylation) were higher for 5hmC, TET1-V5, EZH2, H3K27me3 signals, whereas Class III DMVs (with the least increase) had higher H3K4me3 and H3K36me3 signals (Fig. 3G–H, S4A–C). However, none of the histone marks were affected in QSER1 KO (Fig. S4D). EZH2 showed slightly increased occupancy, possibly a compensatory response based on its role in maintaining hypomethylation at DMVs in mESCs (22). We also observed that Class I and II DMVs were on average larger than Class III DMVs (Fig. S4E), suggesting that QSER1 may preferentially protect broad, H3K27me3-marked regions that overlap with DMVs. Supporting this notion, the size of H3K27me3 peaks that overlapped with QSER1 KO hyper-tiles were substantially larger than peaks that overlapped with hyper-tiles in the TET1 KO but not QSER1 KO (Fig. 3I), and QSER1 KO hyper-tiles preferentially overlapped with broad H3K27me3 peaks (>10kb) (Fig. 3F, S3H). Furthermore, 1-kb tiles that overlapped with H3K27me3 broad peaks showed significantly more hypermethylation in QSER1 KO compared to narrower peaks (Fig. 3J). The same was true for EZH2 peaks but not H3K4me3 peaks (Fig. S4F–H). By contrast, TET1 KO showed more hypermethylation in narrower (<5kb) H3K27me3 or EZH2 peaks (Fig. 3J, S4G). Therefore, compared to TET1, QSER1 preferentially protects broad PRC2/H3K27me3-marked regions, including DMVs, against hypermethylation.
QSER1 and TET1 show correlative genomic occupancy, mutual dependence, and complementary binding with DNMT3A/3B
We used CRISPR/Cas-mediated knockin to fuse a FLAG epitope tag (3XFLAG) to the C-terminus of the endogenous QSER1 protein (Fig. S5A–B). After confirming the nuclear location of QSER1 by FLAG immunostaining (Fig. 4A), we conducted FLAG ChIP-seq and identified QSER1 binding broadly in regulatory regions (Fig. S5C). QSER1 binding was much greater at QSER1 KO hyper-tiles compared to random tiles or hypo-tiles (Fig. 4B), supporting a direct role of QSER1 against hypermethylation. Furthermore, 98% of bivalent promoters, 99% of DMVs, and 76% of broad H3K27me3 peaks overlapped with QSER1 peaks, and QSER1 binding was significantly higher at these regions compared to control (Fig. 4C, S5D). Therefore, QSER1 acts as a chromatin factor to protect regulatory regions such as bivalent promoters and DMVs from hypermethylation.
Fig. 4.

Genomic occupancy of QSER1, TET1 and DNMT3A/3B. (A) QSER1–3XFLAG and TET1-V5 Immunofluorescence staining. (B-C) Violin plots quantifying QSER1 ChIP-seq signal at 1-kb tiles that overlap with the regions specified and random tiles (n=5,000). In all box plots inside violin plots here and later, edges refer to the upper/lower quartiles and whiskers represent 1.5 interquartile range beyond edges; Mann-Whitney U-test was conducted. (D) Heatmaps representing ChIP-seq signals at corresponding peaks. Peaks overlapping with DMVs are indicated. (E) 1-kb plots showing ChIP-seq signals correlation. Tiles overlapping with DMVs are red. (F) Meta-signal plot of ChIP-seq signals at DMVs. Min and Max values are indicated. (G) Density plots showing TET1 binding difference (QSER1 KO – WT) at DMV (left) or QSER1 KO hyper-DMR (right) overlapping tiles compared to control. (H) Heatmaps of TET1 binding at DMVs in WT and QSER1 KO ranked by methylation increase in QSER1 KO. (I) Differential TET1 binding (QSER1 KO – WT) at the three classes of DMVs. (J, L) Meta-signal plots of ChIP-seq signals at DMVs. (K, M) 1-kb plots comparing ChIP-seq signal change vs. methylation change (KO – WT). Tiles that overlap with KO hyper-DMRs are highlighted in red.
We further interrogated potential cooperative or antagonistic relationships between QSER1, TET1, and DNMT3A/3B. A TET1 V5 endogenously tagged line was created for TET1-V5 ChIP-seq (Fig. 4A, S5A–B). We observed significant overlap between QSER1 and TET1 occupancy (Fig. 4D) with 86% of TET1 peaks overlapping with QSER1 peaks, and QSER1 and TET1 peaks share similar genomic features (Fig. S5C). When examining 1-kb tiles globally, TET1 and QSER1 signals showed a very high correlation (Pearson correlation coefficient r = 0.93, p < 2.2e-16), and both were high in DMVs (Fig. 4E). These findings strongly support a cooperative relationship between QSER1 and TET1. In contrast, QSER1 and TET1 both showed a negative correlation with DNMT3A and DNMT3B signals (Fig. 4E, S5E). DNMT3A/3B was depleted in DMVs, broad H3K27me3 peaks, QSER1/TET1-bound regions and relatively enriched in the flanking regions (Fig. 4D–F, S5E–F). The complementary binding patterns suggest a competitive relationship of QSER1/TET1 with DNMT3A/3B.
Given that QSER1 and TET1 have overlapping genomic binding, we speculated that loss of QSER1 could affect the genomic targeting of TET1 or vice versa. Indeed, QSER1 KO caused decreased TET1 binding at DMVs (Fig. 4G–J), with the strongest decrease observed at Class I and II DMVs (Fig. 4H–I). Globally, we also observed decreased TET1 binding at 1-kb tiles that overlapped with hyper-DMRs (Fig. 4G, K). No effects were observed on overall TET1 protein levels (Fig. 4A, S5G) or 5hmC levels based on mass spectrometry (Fig. S5H). Similarly, we observed decreased QSER1 binding in TET1 KO without changes in QSER1 protein levels (Fig. 4L–M, S5G, S5I–J). The relative specificity of the decrease in TET1 binding at DMVs and regions that gained methylation in QSER1 KO suggests that QSER1 regulates DNA methylation at least partially through recruiting TET1.
QSER1 and TET1 cooperate to inhibit DNMT3A/3B DNA binding
To investigate biochemical mechanisms, we performed QSER1-FLAG and TET1-V5 ChIP followed by Mass Spectrometry (ChIP-MS) to identify chromatin-associated binding partners for each factor (Fig. 5A). In addition to detecting a significant enrichment for QSER1 in TET1 ChIP-MS, there was a strong overlap between the significantly enriched proteins in both ChIPs (Fig. 5B–C). Many of the overlapping enriched proteins have known interactions amongst themselves (Fig. S6A), strengthening the notion that QSER1 and TET1 could cooperate with common partners to mediate dynamic changes of DNA methylation in hESCs.
Fig. 5.

QSER1 and TET1 cooperate to inhibit DNMT3A/3B binding. (A) Schematic of ChIP-MS experiments. (B) Volcano plots showing identified and overlapping proteins in QSER1–3XFLAG and TET1-V5 IPs for ChIP-MS. Dotted lines represent the fold change and p value cutoffs for significantly enriched proteins. (C) Venn diagram showing the overlap of significantly enriched proteins in both IPs for ChIP-MS. (D) Hexagon plots showing methylation levels of CpGs inside DMVs for each KO or DKO compared to WT. (E, J) Cumulative fraction of DMVs plotted according to average methylation of each DMV. (F) Methylation difference (KO-WT) of 1-kb tiles that overlap with designated H3K27me3 peaks for specified genotypes. (G) Heatmaps of DNMT3A/3B ChIP-seq signals at DMVs in WT and QSER1/TET1 DKO ranked by methylation increase in DKO. (H) Meta-signal plots of ChIP-seq signals for DNMT3A/3B and DNA methylation levels at Class I DMVs in WT and QSER1/TET1 DKO. (I) 1-kb plots comparing ChIP-seq signal change vs. methylation change (QSER1/TET1 DKO – WT). Tiles that overlap with QSER1/TET1 DKO hyper-DMRs are highlighted in red.
Considering the evidence for a biochemical interaction between QSER1 and TET1 and that complementary KO decreased but did not completely ablate the DNA binding of either factor, we examined potential genetic interactions between QSER1 and TET1. QSER1 and TET1 double KO (DKO) hESCs showed more hypermethylation than either single KO with ~30% more hyper-DMRs than TET1 KO (Data S1) and clear additive effects at the PAX6 promoter region, DMVs, broad H3K27me3 peaks, and overlapping hyper-tiles (Fig. 5D–F, S6B–D). The complementary genomic binding patterns between QSER1/TET1 and DNMT3A/3B was suggestive of a competitive relationship. Indeed, we observed an increase in DNMT3A/3B binding, most apparent in the QSER1/TET1 DKO at DMVs and in regions where we observed DNA hypermethylation (Fig. 5G–I, S6E). These observed shifts in DNA binding could not be attributed to changes in the expression of the corresponding genes (Fig. S6F). To further test the hypothesis that de novo methylation is responsible for the hypermethylation in QSER1 KO, we generated a QSER1 /DNMT3B DKO line. The hypermethylation in QSER1 KO was reversed upon loss of DNMT3B, as exemplified by almost WT levels of DNA methylation in QSER1/DNMT3B DKO at DMVs and broad H3K27me3 peaks (Fig. 5F, 5J, S6C–D). These results demonstrate that QSER1 and TET1 cooperate at these broad regulatory regions to antagonize DNMT3-mediated de novo methylation.
QSER1 and TET1 cooperate to safeguard transcriptional and developmental programs
As observed in the TET TKO, the QSER1 KO, TET1 KO, or QSER1/TET1 DKO did not affect stem cell morphology or expression of pluripotency markers such as OCT4 and SOX2 (Fig. S7A–B). Only the QSER1/TET1 DKO showed a small decrease in cell proliferation (Fig. S7C). We conducted RNA sequencing (RNA-seq) to investigate the transcriptional consequences (Fig. 6A, Data S2). In all KO and DKO hESCs, genes with hyper-DMR associated promoters showed a significant shift towards decreased expression (Fig. 6B). Mirroring the DNA methylation results, QSER1/TET1 DKO had more differentially expressed genes (DEGs) than either single KO (Fig. S7D), where DEGs with hyper-DMR promoters showed stronger changes in the DKO (Fig. S7D–E). To further explore the developmental consequences, we performed spontaneous differentiation to embryoid bodies (EB) and directed differentiation to pancreatic progenitors followed by RNA-seq (Fig. 6A, S7F, Data S2). Similar transcriptional changes were observed (Fig. S7D–E), and importantly, QSER1/TET1 DKO cells showed a dramatic decline in differentiation efficiency to pancreatic progenitors based on the expression of key progenitor markers PDX1 and NKX6.1: PDX1+ cells were greatly decreased in both the early (PP1) and late (PP2) stages and NKX6.1+ cells were essentially absent at the PP2 stage (Fig. 6C–D, S7G–H). This decreased differentiation efficiency was accompanied by downregulation of pancreatic genes, many of which reside in DMVs and showed hypermethylation in at least one KO line (Fig. 6E–F, Data S1–2). For instance, PAX6 was downregulated in all KO and DKO lines. These findings further support a genetic interaction between TET1 and QSER1 and their cooperation to safeguard transcriptional and developmental programs.
Fig. 6.

QSER1 and TET1 safeguard transcriptional and developmental programs. (A) Schematic showing strategy of differentiation of hESCs. (B) Violin plots showing log2 fold change of expression (KO vs. WT) in ES stage for genes with or without hyper-DMR associated promoters for specified genotypes. (C) Representative flow cytometry plots for PDX1 and NKX6.1 expression at PP1 and PP2 stages. Two clonal lines each from WT, QSER1 KO, TET1 KO, and three QSER1/TET1 DKO clonal lines were used in 2–3 independent differentiation experiments each. (D) Representative immunofluorescence staining of PDX1 and FOXA2 at PP1 for specified genotypes. (E) Quantitative real-time PCR results, shown in relative fluorescence units (RFU) relative to one WT replicate, for selected DEGs at PP1 (genes overlapping with DMVs are ONECUT1, PAX6, PDX1, SOX9, SOX17). Data are mean +/− SD (n=3). Star denotes p < .05 by one-way ANOVA followed by Dunnet multiple comparisons test vs. WT control. (F) DNA methylation (black), TET1 ChIP-seq (blue), QSER1 ChIP-seq (orange), and DNMT3A/3B ChIP-seq (magenta) for designated genotypes at selected genes. Hg19 coordinates are shown along with DMV regions (grey).
Discussion
Control of DNA methylation is essential for gene regulation and genomic stability in the mammalian cell. Although it is clear that loss of TET enzymes compromises the integrity of the methylome, it is unclear how robust, region-specific regulation is achieved. The creation of a DNA methylation reporter line provided a unique opportunity to visualize epigenetic alterations at the PAX6 locus, otherwise “invisible” in terms of gene expression changes in the stem cell state. This direct readout combined with the efficiency of gene editing in iCas9 hESCs led to the comprehensive identification of factors that protect the PAX6 bivalent promoter from hypermethylation, including the well-known genes TET1, TDG, and KDM2B, as well as unknown genes such as QSER1. Some identified genes could have affected tdTomato levels independent of DNA methylation and will require further investigation. Our analysis of QSER1 KO hESCs revealed that QSER1 protects many bivalent promoters and poised enhancers from hypermethylation, demonstrating a marked preference for these regulatory elements over other types of promoters or enhancers. Overlapping with these genomic regions, QSER1 KO had the most significant impact at DMVs. Fittingly, the PAX6 locus chosen for the integration of the methylation reporter is within a DMV region, so future studies may be able to employ other representative loci to identify regulators that act on regions with different genomic features. Furthermore, we discovered that QSER1 and TET1 have many common chromatin-associated interacting partners and rely on each other for efficient recruitment to DNA. QSER1/TET1 DKO causes increased DNMT3A/3B binding at hypermethylated regions, and DNMT3B KO in the QSER1 KO background reversed the hypermethylation phenotype. These findings support a model where QSER1 and TET1 bind to DNA in a common complex and cooperate to inhibit the binding of DNMT3A/3B and therefore de novo methylation.
Given the mechanistic link between QSER1 and TET1, we now have a better understanding of the region-specific hypermethylation observed in TET loss-of-function studies (12, 14, 40). However, there exists the new question of how QSER1 demonstrates preferential protection of broad, Polycomb-bound regions that overlap with DMVs. We did not observe a decrease in EZH2 binding at DMVs in the QSER1 KO, arguing against the possibility that QSER1 could be providing its protective effects through PRC2. Another possibility is that uninhibited DNMT3 activity generally leads to preferential hypermethylation of DMVs, feasibly a result of DMVs typically having exceptionally low occupancy of DNMT3. This idea would be consistent with DNMT3A PWWP domain gain-of-function mutations, found in microcephalic dwarfism patients, that cause hypermethylation at bivalent domains and DMVs in patient cells and in mice (31, 41). We speculate that broad, PRC2-bound regions generally exist in a poised, open-chromatin state where TET could be vulnerable to antagonizing DNMT3 activities. Therefore, the main function of QSER1 could be to stabilize TET-bound complexes within this context, fortifying the antagonistic relationship between TET and DNMT3 and ensuring the proper boundaries of DNA methylation at important developmental genes.
Defects in the epigenome can cause numerous diseases (42), and understanding the complex regulation of DNA methylation can help us uncover vulnerabilities to pathogenic alterations. The loss of DNA methylation regulatory factors such as TET2 has been unambiguously linked to the development of certain hematological cancers (43). However, many other aberrant epigenetic states, including the DMV hypermethylation frequently observed in cancers (28–30), lack a validated molecular mechanism. The identification of regulators such as QSER1 may assist in understanding the progression and significance of this epigenetic dysregulation in broad biological contexts. Supporting this idea, QSER1 was identified as a susceptibility locus to Parkinson’s disease and type 2 diabetes in patient studies (44–46), which could be significant considering the pancreatic differentiation defects observed in our KO studies. In addition, de novo mutations of PRR12, a functionally uncharacterized paralog of QSER1, lead to developmental disability (47, 48). Given the multitude of regions and epigenetic mechanisms affected in pathological conditions, our work highlights the utility of genome-scale screens and locus-specific epigenetic measurements (not limited to DNA methylation) for probing the regulatory mechanisms relevant to human health.
Supplementary Material
Data S1. (separate file). List of QSER1 KO, TET1 KO, and QSER1/TET1 DKO hyper-DMRs. Hyper-DMRs of different genotypes are separated into different sheets. Hg19 genomic coordinates, regulation classification, regulation subtype, whether or not the region overlaps with a DMV, the Entrez Gene ID of the closest gene, the distance in bp to the closest gene, and the corresponding gene symbol are indicated.
Data S2. (separate file). Differential expression analysis results for QSER1 KO, TET1 KO, and QSER1/TET1 DKO compared to WT in ES, EB, and PP1 cell states. Genes are sorted by log2 fold change of expression (KO versus WT). Column Descriptions: gene identifiers; mean normalized counts averaged over all samples from both conditions; the log2 fold change; standard error estimate for the log2 fold change estimate; Wald statistic; p value for the statistical significance of this change; p value adjusted for multiple testing with the Benjamini-Hochberg procedure, which controls false discovery rate.
Acknowledgments:
We thank Vivian Bardwell and Micah Gearhart for providing anti-BCOR antibody, and Alex Kentsis, Dinshaw Patel, and Lorenz Studer for helpful advice.
Funding: this study was funded in part by Starr Tri-I Stem Cell Initiative (grant no. 2016–032), NIH/NIDDK (grant no. R01DK096239), NIH/NCI MSKCC Cancer Center Support Grant (grant no. P30CA008748), American Diabetes Association (grant no. 1–19-IBS-125), NIH T32 Training Grants (grant no. T32HD060600 to G.D. and T32GM008539 to B.P.R.), a Frank Lappin Horsfall Jr Fellowship (to G.D.), a Howard Hughes Medical Institute Medical Research Fellowship (to N.V.), a postdoctoral fellowship from a NYSTEM grant (no. DOH01-TRAIN3–2015-2016–00006 to D.Y.), NIH/NHGRI (R01 HG006827 to C.H.). C.H. is a Howard Hughes Medical Institute Investigator.
Footnotes
Competing interests: C.H. is an inventor on patent/patent application (US-8741567-B2) held/submitted by The University of Chicago that covers 5hmC-Seal. 5hmC-Seal has been licensed to Active Motif and Epican by the University of Chicago. C.H. is a shareholder of Epican Genetech, a scientific founder and scientific advisory board member of Accent Therapeutics, Inc. O.E. advises and holds equity in Owkin, Freenome, One Three Biotech and Volastra Therapeutics.
Data and materials availability: the H1 hESC line is available from WiCell under a material transfer agreement. Sequencing data is available at GEO accession GSE150072. Mass spectrometry raw data are available at the free online repository Chorus (https://chorusproject.org/) under the Project number 1693.
References and Notes:
- 1.Goll MG, Bestor TH, Eukaryotic cytosine methyltransferases. Annu Rev Biochem 74, 481–514 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Smith ZD, Meissner A, DNA methylation: roles in mammalian development. Nat Rev Genet 14, 204–220 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Schmitz RJ, Lewis ZA, Goll MG, DNA Methylation: Shared and Divergent Features across Eukaryotes. Trends Genet 35, 818–827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenberg MVC, Bourc’his D, The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol 20, 590–607 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Luo C, Hajkova P, Ecker JR, Dynamic DNA methylation: In the right place at the right time. Science 361, 1336–1340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gangisetty O, Cabrera MA, Murugan S, Impact of epigenetics in aging and age related neurodegenerative diseases. Front Biosci (Landmark Ed) 23, 1445–1464 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Dan J, Chen T, Genetic Studies on Mammalian DNA Methyltransferases. Adv Exp Med Biol 945, 123–150 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Wu X, Zhang Y, TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18, 517–534 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Li E, Bestor TH, Jaenisch R, Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926 (1992). [DOI] [PubMed] [Google Scholar]
- 10.Okano M, Bell DW, Haber DA, Li E, DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Dai HQ et al. , TET-mediated DNA demethylation controls gastrulation by regulating Lefty-Nodal signalling. Nature 538, 528–532 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Dawlaty MM et al. , Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev Cell 29, 102–111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao J et al. , Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet 47, 469–478 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verma N et al. , TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat Genet 50, 83–95 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costa Y et al. , NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 495, 370–374 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Z et al. , EGR1 recruits TET1 to shape the brain methylome during development and upon neuronal activity. Nat Commun 10, 3892 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voigt P, Tee WW, Reinberg D, A double take on bivalent promoters. Genes Dev 27, 1318–1338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulard M, Edwards JR, Bestor TH, FBXL10 protects Polycomb-bound genes from hypermethylation. Nat Genet 47, 479–485 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Tara S et al. , Bcor insufficiency promotes initiation and progression of myelodysplastic syndrome. Blood 132, 2470–2483 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vire E et al. , The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Hagarman JA, Motley MP, Kristjansdottir K, Soloway PD, Coordínate regulation of DNA methylation and H3K27me3 in mouse embryonic stem cells. PLoS One 8, e53880 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y et al. , Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol 19, 18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das PP et al. , PRC2 Is Required to Maintain Expression of the Maternal Gtl2-Rian-Mirg Locus by Preventing De Novo DNA Methylation in Mouse Embryonic Stem Cells. Cell Rep 12, 1456–1470 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernhart SH et al. , Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci Rep 6, 37393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rakyan VK et al. , Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res 20, 434–439 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stelzer Y, Shivalila CS, Soldner F, Markoulaki S, Jaenisch R, Tracing dynamic changes of DNA methylation at single-cell resolution. Cell 163, 218–229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Long HK et al. , Epigenetic conservation at gene regulatory elements revealed by non- methylated DNA profiling in seven vertebrates. Elife 2, e00348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie W et al. , Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeong M et al. , Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet 46, 17–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su J et al. , Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol 19, 108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heyn P et al. , Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nat Genet 51, 96–105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez F et al. , An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell 15, 215–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi ZD et al. , Genome Editing in hPSCs Reveals GATA6 Haploinsufficiency and a Genetic Interaction with GATA4 in Human Pancreatic Development. Cell stem cell 20, 675–688 e676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li QV et al. , Genome-scale screens identify JNK-JUN signaling as a barrier for pluripotency exit and endoderm differentiation. Nat Genet 51, 999–1010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doench JG et al. , Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature biotechnology 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li W et al. , MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiehle L et al. , Tet1 and Tet2 Protect DNA Methylation Canyons against Hypermethylation. Mol Cell Biol 36, 452–461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu T et al. , DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol 19, 88 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y et al. , Dynamic epigenomic landscapes during early lineage specification in mouse embryos. Nat Genet 50, 96–105 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Lu F, Liu Y, Jiang L, Yamaguchi S, Zhang Y, Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev 28, 2103–2119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sendzikaite G, Hanna CW, Stewart-Morgan KR, Ivanova E, Kelsey G, A DNMT3A PWWP mutation leads to methylation of bivalent chromatin and growth retardation in mice. Nat Commun 10, 1884 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zoghbi HY, Beaudet AL, Epigenetics and Human Disease. Cold Spring Harb Perspect Biol 8, a019497 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rasmussen KD, Helin K, Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 30, 733–750 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Latourelle JC et al. , Genomewide association study for onset age in Parkinson disease. BMC Med Genet 10, 98 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barbitoff YA et al. , Identification of Novel Candidate Markers of Type 2 Diabetes and Obesity in Russia by Exome Sequencing with a Limited Sample Size. Genes (Basel) 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahajan A et al. , Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 50, 1505–1513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leduc MS et al. , De novo apparent loss-of-function mutations in PRR12 in three patients with intellectual disability and iris abnormalities. Hum Genet 137, 257–264 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Cordova-Fletes C et al. , A de novo t(10;19)(q22.3;q13.33) leads to ZMIZ1/PRR12 reciprocal fusion transcripts in a girl with intellectual disability and neuropsychiatric alterations. Neurogenetics 16, 287–298 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Gerstein MB et al. , Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. (separate file). List of QSER1 KO, TET1 KO, and QSER1/TET1 DKO hyper-DMRs. Hyper-DMRs of different genotypes are separated into different sheets. Hg19 genomic coordinates, regulation classification, regulation subtype, whether or not the region overlaps with a DMV, the Entrez Gene ID of the closest gene, the distance in bp to the closest gene, and the corresponding gene symbol are indicated.
Data S2. (separate file). Differential expression analysis results for QSER1 KO, TET1 KO, and QSER1/TET1 DKO compared to WT in ES, EB, and PP1 cell states. Genes are sorted by log2 fold change of expression (KO versus WT). Column Descriptions: gene identifiers; mean normalized counts averaged over all samples from both conditions; the log2 fold change; standard error estimate for the log2 fold change estimate; Wald statistic; p value for the statistical significance of this change; p value adjusted for multiple testing with the Benjamini-Hochberg procedure, which controls false discovery rate.