

Abstract
Sleep is conserved across all species and impaired sleep is a common trait of the diseased brain. The quality of sleep decreases as we age and disruption of the regular sleep architecture is a frequent antecedent to the onset of dementia in neurodegenerative diseases. The glymphatic system, which clears the brain of protein waste products, is mostly active during sleep. Yet the glymphatic system degrades with age, suggesting a causal relationship between sleep disturbance and symptomatic progression in the neurodegenerative dementias. The ties that bind sleep, aging, glymphatic clearance and protein aggregation have shed new light on the pathogenesis of a broad range of neurodegenerative diseases, for which glymphatic failure may comprise a therapeutically targetable final common pathway.
Little can replace the rejuvenating feeling of a good night’s sleep. Our mood and affect, ability to attend, focus and problem solve, are all directly linked to how well we sleep. Indeed, the benefits of sleep are cumulative; they are not restricted to the morning hours, or even to a given day. Good sleepers live longer, weigh less, have a reduced incidence of psychiatric disorders, and remain cognitively intact longer (1–4).
Why do we sleep?
The idea that our brain rests during sleep so as to preserve energy was both posited and rejected in the 1950s, when electroencephalgraphic (EEG) recordings of brain activity made it clear that rapid eye movement (REM) sleep, which comprises about 20% of normal sleep, is linked to cortex-wide neuronal activation (5, 6). In fact, energy consumption declines by only 15% in the remaining non-REM (NREM) periods of sleep. Borbely proposed 40 years ago that the sleep-wake cycle is determined by the interaction of two processes: a circadian oscillator, which cycles with the solar day, and a homeostatic drive for sleep (7). A key element in that model is that a sleep deficit - i.e., sleep deprivation – causes a quantifiable “pressure to go to sleep.” The subject’s subsequent NREM sleep is both longer and deeper than normal, and the antecedent sleep loss can be identified post hoc by an increase in EEG slow wave activity during recovery sleep (8). Slow wave activity is a wave of synchronous local neural firing, that typically begins in the frontal cortex and propagates posteriorly, roughly every second during NREM sleep (9). One of the predictions of the Borbely model is that daytime sleep is lighter, since it is not aligned with the circadian clock, and hence fails to fulfill the homeostatic function of sleep. This prediction has been supported by numerous studies of night shift workers, who as a group are predisposed to stress, obesity, cognitive deficits and an elevated risk of neurodegenerative diseases (10–13). One of the most prominent current models of sleep - posits that the purpose of sleep is to restore synaptic homeostasis (14). The synaptic homeostasis hypothesis of sleep is based on the observations that wakefulness is associated with the sustained potentiation of excitatory transmission, as well as with the structural expansion of postsynaptic dendritic spines (15, 16). The larger size of spines during wakefulness increases their postsynaptic currents, and thereby strengthens excitatory transmission. This model is supported by the observation that sleep deprivation is linked to an increased risk of seizures in predisposed individuals (17). It is only during subsequent recovery sleep that excitatory transmission tone and spine volume fall, each returning to its sleep-associated baseline (18).
Recent studies have offered molecular insights into the synaptic homeostasis hypothesis, by mapping the impact of the sleep-wake cycle on synaptic gene expression (19, 20). These studies showed that genes involved in synaptic signaling were predominantly transcribed prior to mice waking up, while transcripts of genes involved in metabolism rose a few hours prior to the expected bedtime. Thus, the circadian clock dictates the transcription of genes in anticipation of the tasks appropriate for the time of day. Similarly, translation of mRNAs into proteins largely followed transcription, so that proteins involved in synaptic signaling were produced during wakefulness, while those involved in metabolism were translated during sleep. Surprisingly though, when the mice were kept awake longer than normal, the translation of proteins involved in synaptic signaling continued during sleep deprivation, concurrently with a suppression of production of proteins involved in metabolism (19, 20). Thus, the behavioral state, rather than the circadian clock, controls synaptic protein production. Under continued wakefulness, those proteins involved in synaptic signaling are continuously made, while proteins needed for restorative metabolic processes are not translated. Thus, extended wakefulness is associated with a dysregulation of translation that enables the sustained potentiation of excitatory transmission; this supports a critical homeostatic role of sleep that cannot occur in the awake state. It is intriguing to speculate that the depth of recovery sleep, detected as slow wave activity, controls the translation of proteins needed to restore metabolic homeostasis.
The glymphatic and lymphatic systems
A fundamental tenet of brain homeostasis is that protein clearance must approximate protein synthesis. Is removal of protein waste also controlled by the sleep wake cycle? Until 2012 it was believed that the brain, unique among organs, was recycling all of its own protein waste (21). Only a small number of proteins were known to be transported across the blood-brain barrier, and these did not include most of the major proteins made or shed by brain cells (22). Lacking lymphatic vessels or any overt pathways for fluid export, it was unclear how protein waste might exit the mature brain parenchyma. The default conclusion was that the classical cellular protein degradation pathways, autophagy and ubiquitination, must be responsible for all CNS protein recycling (23).
This supposition, that the brain must recycle its own wastes, was questioned by the discovery of the glymphatic system (24). The glymphatic system is a highly organized cerebrospinal fluid transport system that shares several key functions with the lymphatic vessels of peripheral tissues, including the export of excess interstitial fluid and proteins (Fig. 1a). Indeed, both the brain’s cerebrospinal fluid (CSF) and peripheral lymph are drained together into the venous system, from which protein waste is removed and recycled by the liver (25). Yet brain tissue itself lacks histologically-distinct lymphatic vessels. Rather, fluid clearance from the brain proceeds via the glymphatics, a structurally distinct system of fluid transport that utilizes the perivascular spaces created by the vascular endfeet of astrocytes (26). The endfeet surround arteries, capillaries and veins serving as a second wall plastering the entire cerebral vascular bed. The perivascular spaces are open, fluid-filled tunnels that offer little resistance to flow. This is in sharp contrast to the disorientingly crowded and compact architecture of adult brain tissue, called the neuropil, through which interstitial fluid flow is necessarily slow and restricted; a marsh flowing to the glymphatics’ creeks and then rivers (27). The glymphatics’ perivascular tunnels are directly connected to the subarachnoid spaces surrounding the brain, from which CSF is rapidly driven into deep regions of the brain by the cardiac rhythm-linked pulsations of the arterial wall (28). The vascular endfeet of astrocytes, a major subtype of glial cells, surround the perivascular spaces and can be regarded as open gates for fluid influx into the neuropil. The astrocytic endfeet are connected by gap junctions, and almost 50% of their plasma membrane facing the vessel wall is occupied by square arrays composed of the water channel AQP4 (29). Deletion of AQP4 channels reduces both the influx of CSF tracers and the efflux of solutes from the neuropil (24, 30, 31). Given its functional similarities to the peripheral lymphatic system, we coined this astrocyte-regulated mechanism of brain fluid transport the glymphatic (glial-lymphatic) system.
Fig. 1. The brain glymphatic system is a highly organized fluid transport system.

(A) The vascular endfeet of astrocytes create the perivascular spaces through which CSF enters the brain and pervades its interstitium. CSF enters these perivascular spaces from the subarachnoid space, and is propelled by arterial pulsatility deep into the brain, from where CSF enters the neuropil, facilitated by the dense astrocytic expression of the water channel AQP4, which is arrayed in nanoclusters within the endfeet. CSF mixes with fluid in the extracellular space and leaves the brain via the perivenous spaces, as well as along cranial and spinal nerves. Interstitial solutes, including protein waste, are then carried through the glymphatic system and exported from the central nervous system via meningeal and cervical lymphatic vessels. (B) Amyloid-ß plaque formation is associated with an inflammatory response, including reactive micro- and astrogliosis with dispersal of AQP4 nanoclusters. An age-related decline in CSF production, the decrease in perivascular AQP4 polarization, gliosis and plaque formation all impede directional glymphatic flow, and thereby impair waste clearance. Of note, vascular amyloidosis might be initiated by several mechanisms. Amyloid-ß might be taken up from the CSF by vascular smooth muscle cells expressing the low-density lipoprotein receptor-related protein 1 (LRP1) (111). Alternatively, amyloid deposition might be initiated by the backflow of extracellular fluid containing amyloid-ß into the periarterial space from the neuropil – rather than proceeding on to the perivenous spaces - due to an increase in hydrostatic pressure on the venous side, or because of an inflammation-associated loss of AQP4 localization to astrocytic endfeet.
It is important to note that fluid transport through the glymphatic system is directionally polarized, with influx along penetrating arteries, fluid entry into the neuropil supported by AQP4, and efflux along the peri-venous spaces, as well as along the cranial and spinal nerves (24, 32–34). Besides its vectorial nature, glymphatic clearance is temporally regulated and cyclically so, in that fluid transport is enabled by sleep and suppressed during wakefulness. Brain fluid transport initiates and proceeds during NREM sleep, and CSF tracer influx correlates with the prevalence of EEG slow wave activity (35, 36). Fluid flow through the glymphatic system is thus inextricably linked with sleep, to the extent that flow appears to stop with the onset of wakefulness. In this regard, slow wave activity predominates in early hours of asleep, and is a direct measure of the sleep pressure, increasing with antecedent sleep deprivation (8). As such, waste removal is likely most efficient in the early hours of sleep, and especially during recovery sleep after prolonged wakefulness (37). Yet it is easy to imagine why the awake state might be incompatible with active parenchymal fluid flow. Wakefulness relies on the precision of synaptic transmission in both time and space. Active flow might be expected to increase glutamate spillover during synaptic activity, resulting in the bystander activation of local synapses, and hence in a loss of both the temporal and spatial fidelity of synaptic transmission. A recent analysis showed that glymphatic flow is also regulated by circadian rhythmicity, such that fluid transport peaks during the sleep phase of diurnal activity, and falls during the active phase, independent of the light cycle. This rhythm is supported by the temporally-regulated localization of AQP4 via the dystrophin associated complex, providing a dynamic link to the molecular circadian clock (38),
The glymphatic-lymphatic system is a functionally integrated unit
Upon discovery and characterization of the glymphatic system, it became quickly apparent that glymphatic efflux pathways needed to be more comprehensively defined. At about that time, two other groups reported that classical lymphatic vessels draining brain interstitial CSF might also be identified in the dura – the fibrous external layer of the meningeal membranes (39, 40). The meningeal lymphatic vessels are separated from CSF by the arachnoid membrane – an internal meningeal layer whose cells comprise a tight fluid barrier by virtue of their dense expression of tight junctions, identified by their expression of claudin-11 (41). Yet the glymphatic and meningeal lymphatic systems are clearly connected: CSF tracers can exit the CNS via the meningeal lymphatic vessels, particularly by way of the lymph vessels draining the ventral aspect of the brain draining to the cervical lymph nodes (39, 40, 42). Indeed, CSF exit from the CNS by way of the meningeal lymph vessels, as well as via both cranial and spinal nerve roots, is rapid; contrast agents can be detected in the deep cervical lymph nodes within minutes after CSF delivery (42–45). Nonetheless, proteins and tracers can circulate back into the brain along the peri-arterial spaces, suggesting that our understanding of flow vectors in the CNS is incomplete; more work is clearly needed to comprehensively account for all of the paths by which extracellular fluid and its solutes are cleared from the adult brain (46). Regardless of its precise efflux pathways, CSF ultimately drains into the cervical lymphatic vasculature, by which it returns to the venous system. Accordingly, in a murine model of Alzheimer disease, amyloid-b was present in high concentrations in the cervical and axillary lymph nodes, at levels analogous to those in the brain, and yet was either undetectable or barely so in the spleen and other peripheral tissues (47). Accordingly, a large proportion of brain waste proteins and metabolites might then be expected to pass through, and be cleared, by the cervical lymphatics. It is in this regard important to note that lymphatic vessels undergo atrophy in aging (48, 49), so that one may speculate that lymphatic drainage of CSF may pose a checkpoint – and with aging, a bottleneck - for brain protein clearance. In this regard, VEGFC-overexpression induced spouting of the meningeal lymphatic vessels and slowed the cognitive decline in a mouse model of Alzheimer disease (50). Conversely, both UV photoablation of meningeal lymphatic vessels and mechanical ligation of cervical lymphatics aggravated amyloid plaque formation in the same mouse models of Alzheimer disease (50, 51). Thus, the glymphatic and lymphatic systems are intimately connected, both structurally and functionally, such that interference with fluid transport at any segment or node within their integrated network risks upstream fluid stasis, and hence the aggregation of proteins otherwise destined for clearance.
Why do proteins aggregate in neurodegenerative diseases?
Aging is also associated with a steep fall in glymphatic flow in the brains of both rodents and humans. CSF inflow of larger tracers are reduced by up to 85% in aged wild-type mice, while contrast clearance in human brain tissue was inversely correlated to age in all subjects studied (50, 52–54). The decrease in glymphatic flow in old mice is in part mediated by mislocation of AQP4 water channels away from the vascular wall (52) and possible by atrophy of meningeal lymphatic vessel (42). On top of aged-related decreases in brain fluid transport, glymphatic CSF influx and CSF clearance are each reduced in early stages of amyloid deposition in the APP/PS1 model of Alzheimer disease, compared with littermate controls; CSF clearance continues to decline as the amyloid burden increases (Fig. 1b). Infusion of amyloid-β into CSF acutely reduced glymphatic activity in wild-type mice suggesting a direct toxic effect (50, 55). The suppressive effects of both age and amyloid-ß overexpression on glymphatic flow can be extended to other experimental rodent models of neurodegeneration; both traumatic brain injury and Parkinson’s disease are similarly linked to a sustained reduction of glymphatic fluid transport (56–58). Importantly, most of these age-related primary neurodegenerative diseases involve disorders of protein processing and aggregation. The hallmark feature of these proteinopathies are the fibrillary aggregates of misfolded or hyperphosphorylated proteins (59). The protein aggregates can range in size from oligomers to large fibrillary structures. These aggregation-prone proteins include amyloid-ß in Alzheimer Disease; phosphorylated tau in frontotemporal dementia (FTD) and chronic traumatic encephalopathy, as well as in Alzheimer’s; α-synuclein in Parkinson’s, Lewy body disease, and the multisystem atrophies; mutant huntingtin in Huntington’s disease, and TDP-43 in ALS and FTD (60). Although the specific protein species differ in the different neurodegenerative disorders, in most cases their protein aggregates are formed in part by the interactions of intermolecular β-sheet-rich strands. Once a seed is formed, the aggregates attract monomers of the same protein, as well as other proteins which may be preferentially bound and entrapped (60).
To understand why aging predisposes to these proteinopathies, we need to consider those conditions that favor nucleation, the growth of protein aggregates, and their subsequent seeding to neighboring cells. Proteins self-assemble and aggregate depending upon a number of factors, among which are structure, concentration, ionic strength and local pH, as well as their interactions with nucleating interfaces, such as phospholipid membranes (61, 62). Ex vivo aggregation can be induced by simply mixing hydrophobic nanoparticles into an aqueous solution containing proteins (63). A lack of fluid flow, i.e. stagnation, or its opposite, shear stress, can also promote aggregation (64, 65), which can occur at a distance from the protein source - for example, along the cerebral vasculature (Fig. 1) (66). Depending upon the protein, each of these factors, alone or in combination, can lead to self-aggregation with the formation of stable β-sheet-rich strands. Reduced glymphatic clearance might then be predicted to increase the risk of protein aggregation, given the combination of locally stagnant fluid flow together with the elevated extracellular concentration of the protein of interest.
Spread of protein aggregates: A possible role of the glymphatic system?
The recent discovery that specific misfolded and aggregated proteins can propagate and spread in a prion-like fashion has sparked considerable interest (67). It has been generally posited that seeding occurs across regions that are synaptically connected (68). However, the evidence for synaptic spread is largely based upon post hoc analysis of anatomic networks; it remains unclear how synaptic relationships per se can mediate seeding. The arguments for synaptic spread are somewhat weakened by the fact that aggregate spread happens in both antero- and retrograde directions across regions that are anatomical neighbors (68). An alternative hypothesis is that aggregates simply spread via the extracellular spaces, and that the age-dependent reduction in glymphatic flow, with its attendant fluid stagnation, raises the local protein concentration to a level that favors aggregation. In support of this hypothesis, the suppression of glymphatic flow by deletion of AQP4 water channels sharply increased both amyloid-ß plaque formation and cognitive deficits in a mouse model of Alzheimer disease (69). Similarly, in human subjects, efflux of CSF containing amyloid-ß and phosphor-tau is reduced in patients with Alzheimer disease, compared to age-matched controls. The suppression of CSF in Alzheimer disease is so substantial that it can possible serve as a biomarker (70).
What then do we know about the spread of protein aggregates on a macroscopic scale? In Alzheimer disease, amyloid-ß deposition typically first deposit in the basal portions of the frontal, temporal, and occipital lobes. Later the plaques spread to include the hippocampus and posterior parietal cortex, initially sparing both the motor and sensory cortex. These latter regions are first recruited in the final stages of the disease, along with subcortical gray matter regions. Yet the cognitive decline of Alzheimer disease correlates more closely with the later-occurring tauopathy and microglial activation, than with the earlier amyloid-ß plaque formation (71, 72). In the initial stages of AD, phosphorylated tau deposits in the entorhinal cortex, followed by the hippocampus and dorsal thalamus, while the neocortex becomes involved later. In Parkinson’s disease and Lewy body disease, α-synuclein aggregates initially spread through the brainstem and olfactory bulb followed by limbic structures, and only thence to the neocortex. (Fig. 3A). In each of these cases, the aggregates initially deposit at the ventral base of the forebrain and midbrain, and then extend rostrally and dorsally to the cortex.
Fig. 3. Prion-like spread of protein aggregates and proposed role of glymphatic transport.
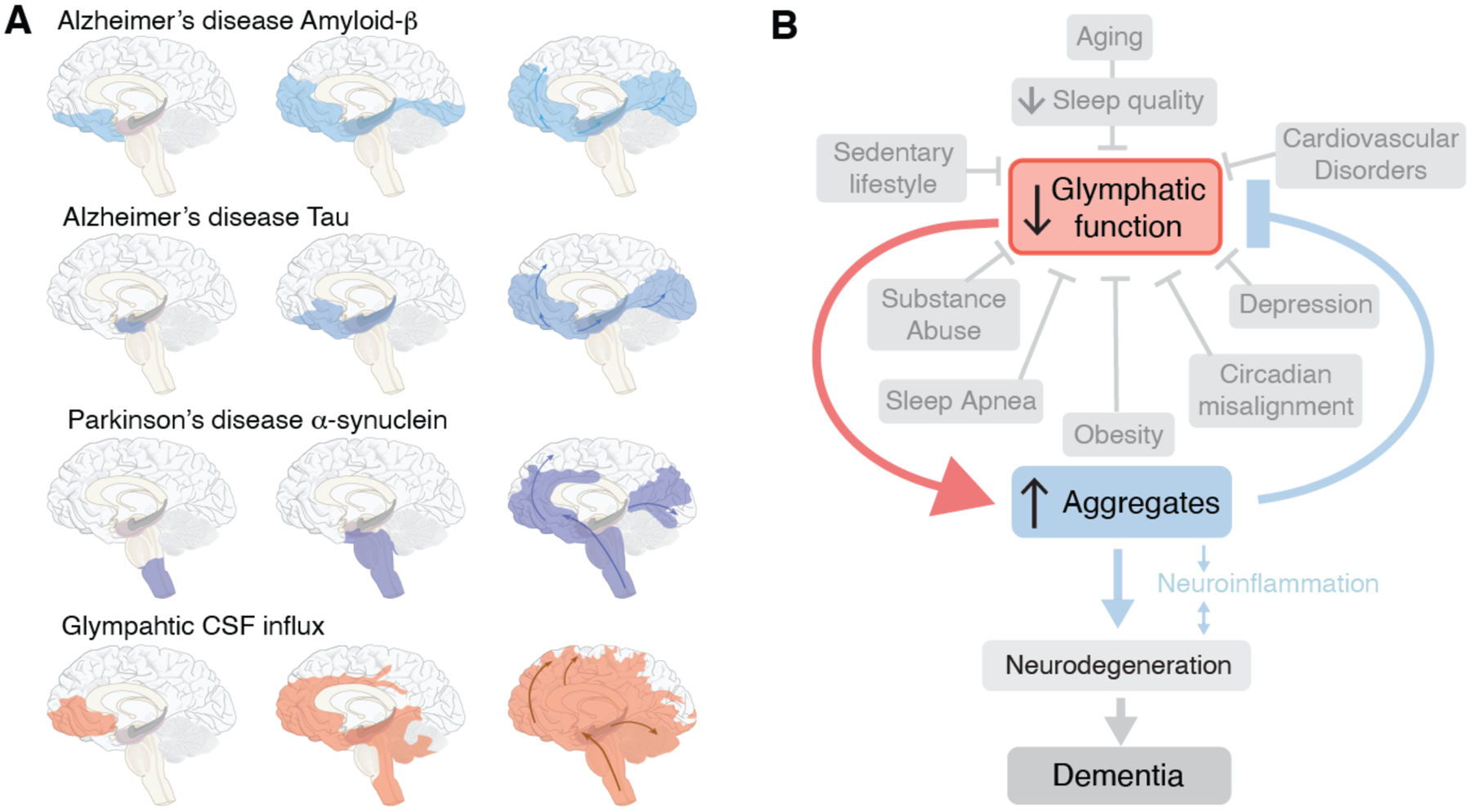
(A) Seeding and prion-like spread of protein aggregates (amyloid-ß and tau) in Alzheimer disease, and α-synuclein in Parkinson disease, relative to the distribution of glymphatic influx of a CSF tracer after intrathecal delivery (67). Prion-like spread of protein aggregates includes an extracellular component and thereby the possibility that the seeds are transported by the glymphatic system. (B) In this model, the glymphatic system resides at the intersection of a broad scope of disorders, which share an association with diminished brain fluid clearance. In addition, normal aging is also linked to a sharp decrease in the quality of sleep and in glymphatic flow. In turn, the stagnation of glymphatic flow, and hence that of extracellular proteins, contribute to protein aggregation, with misfolding and seeding, leading in turn to local inflammation, neuronal loss, and ultimately dementia.
How does this pattern of spread compare to glymphatic CSF inflow? (Fig. 3A)(67, 73). Neuroimaging studies from the Eide group have shown that intrathecally-delivered contrast agents first are propelled into the brain along the large cerebral arteries entering the mediobasal frontal lobe and cingulate cortex along the anterior cerebral artery, insula via the middle cerebral artery and the limbic structures, including the hippocampus and entorhinal cortex via the posterior circulation. The contrast agent remains trapped in the same regions for prolonged periods of time, especially if an underlying pathology is present (74, 75). The accumulation of low molecular weight CSF contrast agents (< 1 kDa) supports the notion that much larger proteins also get trapped in the tortuous extracellular spaces of deep brain regions.
While the conditions by which pathogenic proteins may become entrapped and aggregate in glymphatic channels remain unclear, the geographic spread of aggregates in Alzheimer and Parkinson diseases clearly mirrors the pattern of glymphatic inflow in the human brain, as mapped by MRI. In fact, the geographic pattern of macroscopic aggregate formation closely resembles that of entrapped CSF contrast agents during challenge of glymphatic flow in those brains (Fig. 3B). On that basis, we propose that trapping of aggregation-prone proteins in the extracellular space, rather that synaptic connectivity, is responsible for the patterns of protein spread in at least some proteinopathies. As such, the regional variations in the path of seeding noted across the different types of neurodegenerative diseases may reflect region- and patient-specific variability in the rates of neuronal production of amyloid, tau, and α-synuclein. Of note, while proteins involved in neurodegenerative diseases may normally be either intracellular or extracellular in nature, all are present in the extracellular space. Sampling of CSF and extracellular fluid have documented that amyloid-ß, tau and a-synuclein are all present outside the cytosol. These proteins all lack N-terminal signal sequences, so that unconventional mechanisms must be responsible for their release (76). In each of these cases, it is unclear whether oligomers or the larger protein aggregates comprise the principal neurotoxic species (60). While no consensus has been reached, several studies have highlighted the critical role of oligomers as both directly toxic, and as niduses for macromolecular aggregation. Immune therapies have attempted to ‘clear” the extracellular space and CSF of amyloid-ß in Alzheimer patients. The failure of these clinical trials may reflect the relatively late initiation of treatment or that the antibody load was not enough to clear sufficient amyloid-ß to yield clinical benefit. Alternatively though, it is possible that the underlying model of direct, aggregation-associated neurotoxicity is fundamentally incorrect, in AD as well as more broadly (77).
Sleep, aging, neurodegeneration and the glymphatic system
The most significant risk factor for developing protein aggregation – just as it is for developing dementia - is age (78). With the glymphatic in mind, it is of interest to note that sleep quality decreases as a function of normal aging. Insomnia is more frequent with age, and total sleep duration becomes shorter and more interrupted. Perhaps more critically, older individuals rarely enter deep NREM, stage 3 sleep. Most NREM sleep in people over 60 years is light, consisting of the more superficial stages 1–2 (79) (Fig. 2). Thus, the aged brain spends less time in NREM sleep, potentially causing a catastrophic decline in clearance of brain waste, as the efficacy of glymphatic fluid transport correlates directly with the prevalence of slow wave activity (36). The age-related impairment in sleep quality may thus be causally involved in the increased incidence and accelerated course of neurodegenerative disease in the elderly, whose disrupted sleep architecture and depth may sharply diminish the clearance of brain fluid and its attendant export of protein waste, leading in turn to the stagnant interstitial flow that favors aggregate formation.
Fig. 2. Sleep architecture in young and old subjects.
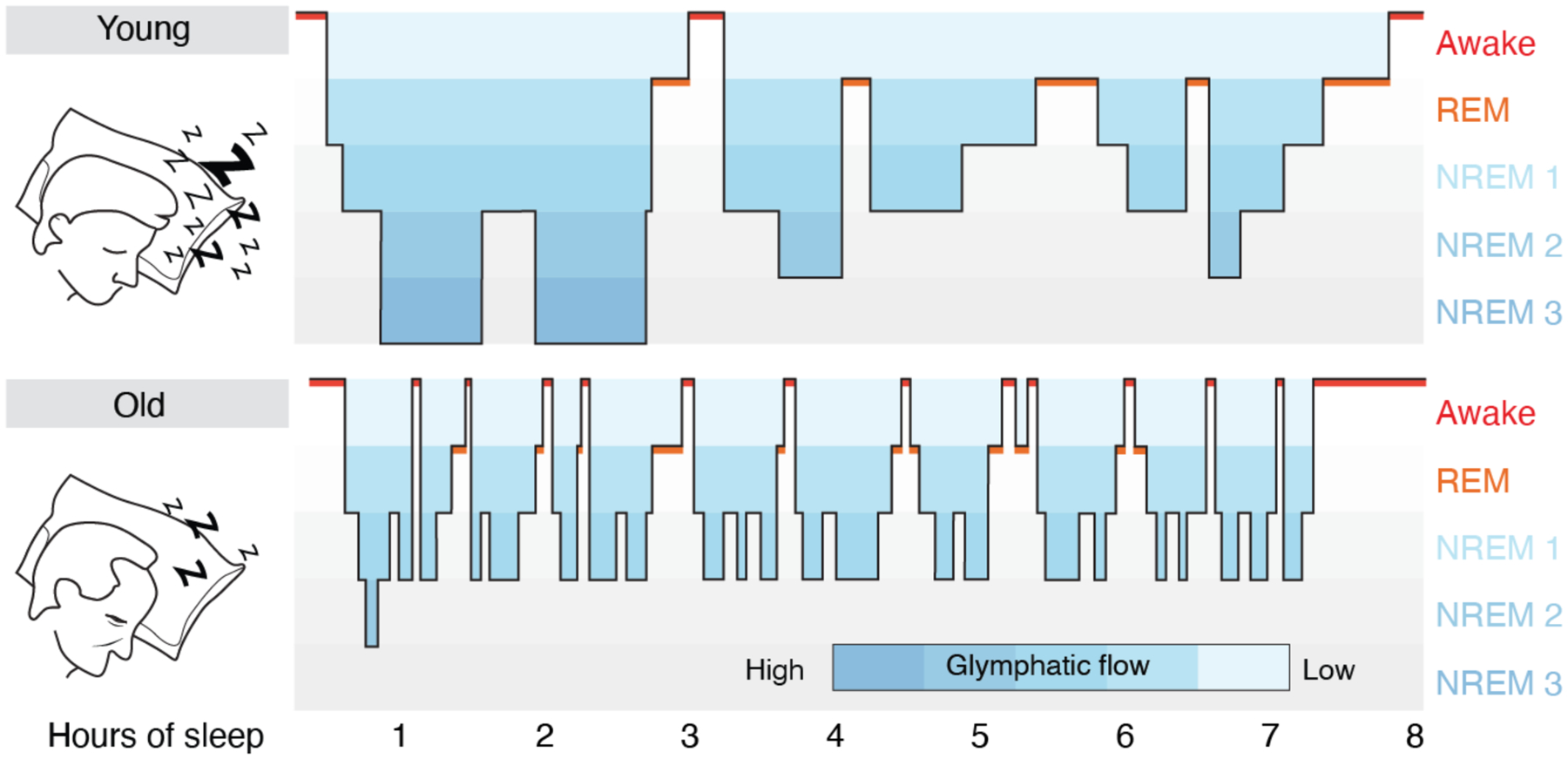
Hypnograms are constructed from EEG recordings and display the cyclic transitions between sleep stages. The two schematic hypnograms illustrate the sleep architecture of a young and an old subject that transitions spontaneously between awake, REM and NREM (stage 1–3) sleep. Stages 1 NREM sleep is light sleep whereas stage 3 NREM sleep is the deepest sleep stage and characterized by slow wave EEG activity. Deep stage 3 NREM sleep dominates in the early phases of sleep, whereas REM sleep is more frequent in the later phases of sleep in young subjects. Sleep spindles are most frequent in stage 2 NREM sleep. In subjects older than 60 years, sleep is often interrupted by short awake episodes, and older subjects do not typically enter stage 3 NREM sleep; total sleep time decreases by 10 min for each decade of life (79). Blue coloring indicates the proposed efficacy of glymphatic clearance based on data collected in rodents (35, 36). The lack of stage 3 NREM sleep, the frequent interruptions of stage 1–2 NREM sleep, and the shorter total sleep time, all serve to decrease glymphatic activity in aging. Critically, a number of disorders and conditions can suppress glymphatic function during NREM sleep (Fig. 3B), further exacerbating the effects of glymphatic dysfunction in neurodegenerative disease.
On top of the deterioration of sleep architecture in aging, the neurodegenerative diseases, including Alzheimer disease, Parkinson disease, Huntington disease, the multisystem atrophies, and the frontotemporal dementias, are all associated with sleep disturbances (80). The best characterized among these are the sleep pathologies associated with Parkinson disease, in which REM sleep disturbances often precede the onset of motor symptoms by several years, even decades (80, 81). Future work should define whether sleep disturbances that preceded the clinical diagnosis contribute to aggregate seeding and whether the sleep disturbances during disease progression accelerate the aggregate spread. It would seem axiomatic that a stronger focus on age-related impairment of sleep quality should benefit the growing elderly population.
AQP4 polymorphisms in sleep and neurodegeneration
The polarized expression of the water channel, AQP4, in the vascular endfeet of astrocytes facilitates glymphatic fluid transport and amyloid-ß export in rodents (24, 30)(Fig. 1). In man, genetic variation in AQP4 has been shown to impact both sleep and amyloid-β burden (82). A recent study established a link between AQP4, sleep and the effects of prolonged wakefulness on cognitive function. The study demonstrating that a common single nucleotide polymorphism (SNP) of AQP4 was linked to changes in slow wave activity during NREM sleep, that were mirrored by changes in daytime sleepiness as well as in altered reaction times during extended wakefulness (83). Yet AQP4 SNPs have also been associated with the rate of cognitive decline in longitudinally-followed cohorts of AD patients (84). Patients with two specific AQP4 SNPs exhibited slower cognitive decline after AD diagnosis, while conversely, cognitive decline progressed more rapidly in subjects with two other AQP4 SNPs (85). Structurally, the integrity of perivascular AQP4 localization was found to degrade with AD, while it was preserved by subjects older than 85 years, who remained cognitively intact (84). Similarly, the expression of a cluster of transcripts encoding proteins associated with astrocytic end-feet predicted lower levels of cortical phospho-tau in human subjects (86). Indeed, a recent study reported that deletion of AQP4 accelerated amyloid plaque formation in a murine model of Alzheimer disease (69). Thus, although AQP4 is expressed only in astrocytes, and not in amyloid-producing neurons, considerable evidence indicates that AQP4 modulates both sleep architecture, the tolerance to sleep deprivation, amyloid-ß accumulation, and the progression of Alzheimer disease. Targeting the brain’s waste removal system may thus an attractive approach for alleviating the waste burden of the proteinopathies, because aggregation-prone proteins are removed by bulk flow, without the requirement for specific transporters.
Glymphatic failure links cardiovascular with neurodegenerative disease
Neurodegenerative diseases are hardly the only cause of dementia. It has been known for decades that poor cardiovascular health negatively affects cognitive abilities (87, 88), while cardiovascular fitness positively correlates with cognition in young adults (89) and preserves cognitive performance with aging (90). Why then is a healthy heart so important for higher brain function? It has been shown that glymphatic function is suppressed in hypertensive rats (91, 92). It is also well-establish that the quality of sleep is compromised in cardiovascular diseases (93) perhaps providing a link to impaired glymphatic clearance and subsequent protein aggregation and dementia (94).
We also propose that a healthy cardiovascular system, besides its role in delivering energy metabolites to the brain, plays a hitherto-unappreciated role in the clearance of neurotoxic wastes from the brain. In particular, we have found that the brain’s fluid transport system is designed to take advantage of cardiac pulsatility to drive CSF transport in the neuropil (28). To wit, the ejection pressure of blood from the left ventricle is partly absorbed by the elastic arterial wall of the aorta. As the ejected blood transits the arteries, it enlarges the arterial diameter as its pulse wave propagates downstream (28). About 20–25% of the total ejected blood volume enters CNS via the paired internal carotid and posterior cerebral arteries. Pulsatility in these large caliber arteries constantly transmits pressure waves along the axis of the major vessels, as well as through the soft brain tissue (Fig. 4). The motion of the brain is locally supplemented by the pulsatility of the penetrating arteries, as they enter the brain from the CSF-filled subarachnoid space, thereby driving CSF into the neuropil along the periarteral spaces (24). It should not be surprising that heart disease associated with reduced cardiac output, including such common entities as congestive heart failure and atrial dysrhythmias (95), are associated with diminished glymphatic flow, since the pulsatility of the cerebral arteries and hence the driving forces within the glymphatic system arereduced. Indeed, the cognitive decline frequently noted in patients with low cardiac ejection fraction, so often attributed to low cerebral perfusion, may also reflect poor glymphatic flow and incomplete waste clearance, as well as a consequent predisposition to aggregate formation and still-slower glymphatic flow as a result (95).
Fig. 4. Arterial pulsatility propels fluid flow in the brain.
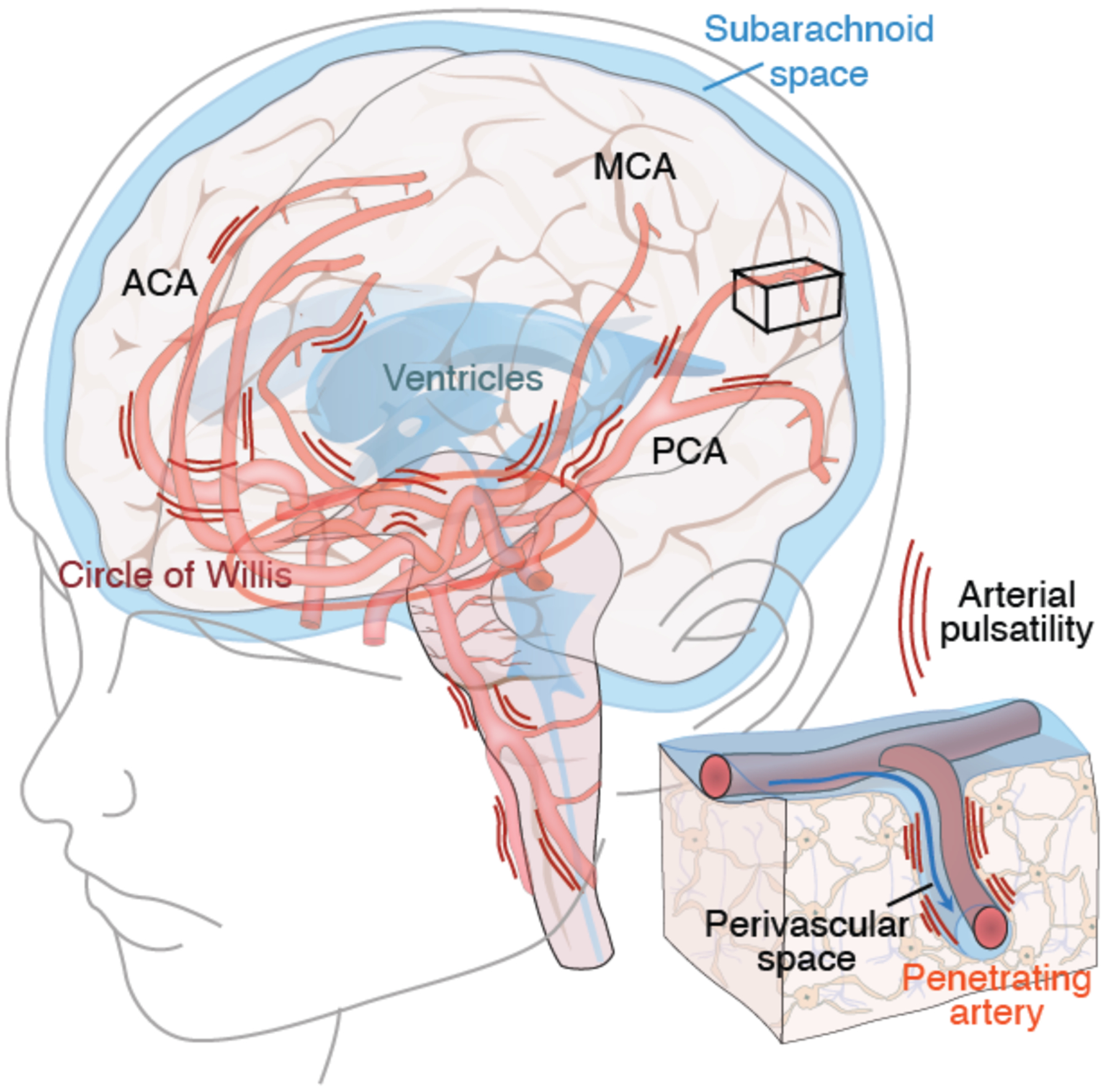
The brain receives 20–25% of cardiac output, yet comprises ~2% of total body weight. The large caliber arteries of the circle of Willis are positioned in the CSF-containing basal cisterns below the ventral surface of the brain. Arterial pulsatility provides the motive force for CSF transit into the perivascular spaces surrounding the major arteries, while respiration and slow vasomotion contribute to sustaining its flow (112). The anterior (ACA), middle (MCA), and posterior (PCA) arteries transport CSF to the penetrating arteries (insert), from which CSF is then driven into the neuropil via the still-contiguous perivascular spaces. Cardiovascular diseases associated with reduced cardiac output, such as left heart failure and atrial arrhythmias, reduce arterial wall pulsatility, resulting in less CSF flow. In addition, thickening of the arterial wall in small vessel disease, hypertension and diabetes all reduce arterial wall compliance and hence pulsatility. Each of these fundamentally cardiovascular disorders serves to attenuate glymphatic flow, providing a potential causal link between these vascular etiologies and Alzheimer disease (113).
Small vessel disease (SVD) is a vascular disorder that targets the small cerebral vessels, in which penetrating arterioles undergo progressive thickening of their walls (96). Deterioration of the vascular bed may occur alone or in combination with other pathologies (97), leading to progressive demyelination and loss of white matter (98). SVD is common in hypertensive patients, many of whom are concurrently diabetic or smokers, and it progresses silently for years before dementia is clinically evident (99). Hypertension in particular induces hypertrophy of vascular smooth muscle cells, with a stiffening of the arterial wall that dampens arterial wall pulsatility and compliance, thus reducing convective perivascular flow (94, 100). The stiffening of perivascular glycocalyx of diabetic patients accomplishes much the same (101), and the two disorders are in frequent combination as the incidence of obesity, a predisposing factor and comorbidity to both, increases worldwide. SVD is linked glymphatic dysfunction in experimental models (91), and may potentiate the progression of neurodegenerative dementias in precisely the same patients at risk for SVD-associated vascular dementia. It is no wonder then that the clinical distinctions between Alzheimer’s and the vascular dementias often is blurred by their frequent co-association (102).
Conclusion
Fundamentally, these studies have served to highlight the benefits of a good night’s sleep. To be sure, sleep is an evolutionary conserved mechanism that serves multiple purposes, with benefits to the homeostatic support of the cardiovascular system, immune system and memory (103–105). Yet the most fundamental incentive for the brain to sleep lies in its own self-preservation: Only the sleeping brain is capable of efficiently cleaning up the waste products generated during active wakefulness. amyloid-ß, tau and α-synuclein are all present at higher levels in the brain extracellular fluid and CSF during wakefulness than during sleep, and sleep deprivation increases their levels further (106–108). Indeed, PET imaging showed that a single night of sleep deprivation resulted in a significant increase in Amyloid-β burden in the hippocampus and thalamus (109). We need sleep to clear proteins from the brain extracellular space, less they aggregate, impede flow, and in a vicious positive feedback cycle, potentiate further fibril polymerization, which together with inflammation then suppresses glymphatic flow in the most affected regions.
Together, these observations suggest a causal linkage between the sleep-wake cycle and its regulation of fluid flow via the glymphatics, and the modulation thereby of the balance between protein clearance and aggregation. As such, they suggest a basis for the increased incidence of aggregation-related disorders seen with aging, the appearance of which tracks age-related declines in both vascular health and glymphatic patency. The neurodegenerative dementias may thus be viewed as the products of a final common pathway that integrates the dysfunction of any and all of these closely interdependent upstream mechanisms (Fig. 3B). These various processes are linked in their regulation by the brain’s glymphatic system, whose directed regulation in turn may present bold new therapeutic opportunities for the disease-modifying treatment of patients with these disorders (75). In particular, the development of small molecule agonists of glymphatic efflux might present opportunities to slow disease progression in the aggregation disorders, just as the optimization of cardiovascular health might be expected to delay disease onset. These systems are intimately connected, so that modulation of glymphatic flow and hence protein clearance from the brain will ultimately require a deeper understanding of the dependence of both glymphatic and lymphatic flow on intracardiac pressures. Indeed, the high incidence of interstitial lung disease that is emerging in patients with resolved COVID19 may predict a higher risk of aggregation disorders for these individuals going forward, as their greater risk of long-term heart failure may predict their impaired lymphatic and hence glymphatic flow. In that regard, recent advances in neuroimaging have provided multiple approaches to map the human glymphatic system, and hence to assess its functional competence in the context of disease, as well as of the effects thereof on sleep-dependent glymphatic cyclicity (72, 73, 108). The diagnostic neuroimaging of glymphatic function via such “glymphograms” may indeed provide both a means to predict the risk of developing proteinopathies, and an approach by which to evaluate the efficacy of glymphatic flow-directed treatments as they are developed.
Until then though, the most assured means of preserving effective glymphatic clearance is to just get a good night’s sleep.
Acknowledgments:
We would like to thank Dan Xue for assistance with illustrations and Chiara Cirelli and Natalie Beschorner for discussions. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 742112) and was funded by the Lundbeck and Novo Nordisk Foundations.
Footnotes
Conflict of interest statement: The authors declare no conflict of interest
References
- 1.Mander BA et al. , Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal-dependent memory in aging. Nature neuroscience 16, 357–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spiegelhalder K, Regen W, Nanovska S, Baglioni C, Riemann D, Comorbid sleep disorders in neuropsychiatric disorders across the life cycle. Current psychiatry reports 15, 364 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Spira AP, Chen-Edinboro LP, Wu MN, Yaffe K, Impact of sleep on the risk of cognitive decline and dementia. Curr Opin Psychiatry 27, 478–483 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leigh L, Hudson IL, Byles JE, Sleeping difficulty, disease and mortality in older women: a latent class analysis and distal survival analysis. Journal of sleep research 24, 648–657 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Aserinsky E, Kleitman N, Regularly occurring periods of eye motility, and concomitant phenomena, during sleep. Science 118, 273–274 (1953). [DOI] [PubMed] [Google Scholar]
- 6.Hobson JA, Sleep is of the brain, by the brain and for the brain. Nature 437, 1254–1256 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Borbely AA, A two process model of sleep regulation. Human neurobiology 1, 195–204 (1982). [PubMed] [Google Scholar]
- 8.Rodriguez AV et al. , Why Does Sleep Slow-Wave Activity Increase After Extended Wake? Assessing the Effects of Increased Cortical Firing During Wake and Sleep. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 12436–12447 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massimini M, Huber R, Ferrarelli F, Hill S, Tononi G, The sleep slow oscillation as a traveling wave. The Journal of neuroscience : the official journal of the Society for Neuroscience 24, 6862–6870 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajaratnam SM, Howard ME, Grunstein RR, Sleep loss and circadian disruption in shift work: health burden and management. The Medical journal of Australia 199, S11–15 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Kecklund G, Axelsson J, Health consequences of shift work and insufficient sleep. Bmj 355, i5210 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Itani O et al. , Short sleep duration, shift work, and actual days taken off work are predictive life-style risk factors for new-onset metabolic syndrome: a seven-year cohort study of 40,000 male workers. Sleep medicine 39, 87–94 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Bokenberger K et al. , Shift work and risk of incident dementia: a study of two population-based cohorts. European journal of epidemiology 33, 977–987 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tononi G, Cirelli C, Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G, Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nature neuroscience 11, 200–208 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Huber R et al. , Human cortical excitability increases with time awake. Cerebral cortex 23, 332–338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Logothetis J, Milonas I, Bostantzopoulou S, Sleep Deprivation as a Method of EEG Activation. European neurology 25(suppl 2), 134–140 (1986). [DOI] [PubMed] [Google Scholar]
- 18.de Vivo L et al. , Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science 355, 507–510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noya SB et al. , The forebrain synaptic transcriptome is organized by clocks but its proteome is driven by sleep. Science 366, (2019). [DOI] [PubMed] [Google Scholar]
- 20.Bruning F et al. , Sleep-wake cycles drive daily dynamics of synaptic phosphorylation. Science 366, (2019). [DOI] [PubMed] [Google Scholar]
- 21.Rubinsztein DC, The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Ballabh P, Braun A, Nedergaard M, The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiology of disease 16, 1–13 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Thibaudeau TA, Anderson RT, Smith DM, A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nature communications 9, 1097 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iliff JJ et al. , A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Science translational medicine 4, 147ra111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nedergaard M, Neuroscience. Garbage truck of the brain. Science 340, 1529–1530 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M, Signaling at the gliovascular interface. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 9254–9262 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tithof J, Kelley DH, Mestre H, Nedergaard M, Thomas JH, Hydraulic resistance of periarterial spaces in the brain. Fluids and barriers of the CNS 16, 19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mestre H et al. , Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nature communications 9, 4878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagelhus EA, Ottersen OP, Physiological roles of aquaporin-4 in brain. Physiological reviews 93, 1543–1562 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mestre H et al. , Aquaporin-4-dependent glymphatic solute transport in the rodent brain. eLife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mestre H et al. , Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 367, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braun M, Iliff JJ, The impact of neurovascular, blood-brain barrier, and glymphatic dysfunction in neurodegenerative and metabolic diseases. International review of neurobiology 154, 413–436 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Wang X et al. , An ocular glymphatic clearance system removes beta-amyloid from the rodent eye. Science translational medicine 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mestre H, Mori Y, Nedergaard M, The Brain’s Glymphatic System: Current Controversies. Trends in neurosciences, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie L et al. , Sleep drives metabolite clearance from the adult brain. Science 342, 373–377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hablitz LM et al. , Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci Adv 5, eaav5447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hauglund L, Pavan C, Nedergaard M, Cleaning the sleeping brain – the potential restorative function of the glymphatic system. Current Opinion in Physiology 15 1–6 (2020). [Google Scholar]
- 38.Hablitz L et al. , Circadian control of brain glymphatic/lymphatic fluid flow. Nature communications (in press), (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Louveau A et al. , Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aspelund A et al. , A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. The Journal of experimental medicine 212, 991–999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brochner CB, Holst CB, Mollgard K, Outer brain barriers in rat and human development. Frontiers in neuroscience 9, 75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahn JH et al. , Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 572, 62–66 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Ma Q et al. , Rapid lymphatic efflux limits cerebrospinal fluid flow to the brain. Acta neuropathologica 137, 151–165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma Q, Decker Y, Muller A, Ineichen BV, Proulx ST, Clearance of cerebrospinal fluid from the sacral spine through lymphatic vessels. The Journal of experimental medicine 216, 2492–2502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacob L et al. , Anatomy and function of the vertebral column lymphatic network in mice. Nature communications 10, 4594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Louveau A et al. , Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. The Journal of clinical investigation 127, 3210–3219 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pappolla M et al. , Evidence for lymphatic Abeta clearance in Alzheimer’s transgenic mice. Neurobiology of disease 71, 215–219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scallan JP, Zawieja SD, Castorena-Gonzalez JA, Davis MJ, Lymphatic pumping: mechanics, mechanisms and malfunction. The Journal of physiology 594, 5749–5768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jakic B, Kerjaschki D, Wick G, Lymphatic Capillaries in Aging. Gerontology, 1–8 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Da Mesquita S et al. , Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 560, 185–191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L et al. , Deep cervical lymph node ligation aggravates AD-like pathology of APP/PS1 mice. Brain pathology 29, 176–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kress BT et al. , Impairment of paravascular clearance pathways in the aging brain. Annals of neurology 76, 845–861 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benveniste H et al. , The Glymphatic System and Waste Clearance with Brain Aging: A Review. Gerontology 65, 106–119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Y et al. , Impairment of the Glymphatic Pathway and Putative Meningeal Lymphatic Vessels in the Aging Human. Annals of neurology 87, 357–369 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Peng W et al. , Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiology of disease 93, 215–225 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iliff JJ et al. , Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 34, 16180–16193 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou W et al. , Blocking meningeal lymphatic drainage aggravates Parkinson’s disease-like pathology in mice overexpressing mutated alpha-synuclein. Translational neurodegeneration 8, 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sundaram S et al. , Establishing a framework for neuropathological correlates and glymphatic system functioning in Parkinson’s disease. Neuroscience and biobehavioral reviews 103, 305–315(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soto C, Unfolding the role of protein misfolding in neurodegenerative diseases. Nature reviews. Neuroscience 4, 49–60 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Soto C, Pritzkow S, Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nature neuroscience 21, 1332–1340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burke KA, Yates EA, Legleiter J, Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Frontiers in neurology 4, 17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yates EA et al. , Specific domains of Abeta facilitate aggregation on and association with lipid bilayers. Journal of molecular biology 425, 1915–1933 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Kopp MRG, Capasso Palmiero U, Arosio P, A Nanoparticle-Based Assay To Evaluate Surface-Induced Antibody Instability. Molecular pharmaceutics 17, 909–918 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Trumbore CN, Shear-Induced Amyloid Formation in the Brain: I. Potential Vascular and Parenchymal Processes. Journal of Alzheimer’s disease : JAD 54, 457–470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roeters SJ et al. , Evidence for Intramolecular Antiparallel Beta-Sheet Structure in Alpha-Synuclein Fibrils from a Combination of Two-Dimensional Infrared Spectroscopy and Atomic Force Microscopy. Scientific reports 7, 41051 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weller RO, Pathology of cerebrospinal fluid and interstitial fluid of the CNS: significance for Alzheimer disease, prion disorders and multiple sclerosis. Journal of neuropathology and experimental neurology 57, 885–894 (1998). [DOI] [PubMed] [Google Scholar]
- 67.Jucker M, Walker LC, Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Henderson MX et al. , Spread of alpha-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nature neuroscience 22, 1248–1257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu Z et al. , Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Molecular neurodegeneration 10, 58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Leon MJ et al. , Cerebrospinal Fluid Clearance in Alzheimer Disease Measured with Dynamic PET. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 58, 1471–1476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hong S et al. , Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Terada T et al. , In vivo direct relation of tau pathology with neuroinflammation in early Alzheimer’s disease. Journal of neurology 266, 2186–2196 (2019). [DOI] [PubMed] [Google Scholar]
- 73.Brundin P, Melki R, Kopito R, Prion-like transmission of protein aggregates in neurodegenerative diseases. Nature reviews. Molecular cell biology 11, 301–307 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ringstad G et al. , Brain-wide glymphatic enhancement and clearance in humans assessed with MRI. JCI insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ringstad G, Vatnehol SAS, Eide PK, Glymphatic MRI in idiopathic normal pressure hydrocephalus. Brain : a journal of neurology 140, 2691–2705 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Davis AA, Leyns CEG, Holtzman DM, Intercellular Spread of Protein Aggregates in Neurodegenerative Disease. Annual review of cell and developmental biology 34, 545–568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knopman DS, Sifting through a failed Alzheimer trial: What biomarkers tell us about what happened. Neurology 90, 447–448 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Hoyer S, Age as risk factor for sporadic dementia of the Alzheimer type? Annals of the New York Academy of Sciences 719, 248–256 (1994). [DOI] [PubMed] [Google Scholar]
- 79.Landolt HP, Borbely AA, Age-dependent changes in sleep EEG topography. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology 112, 369–377 (2001). [DOI] [PubMed] [Google Scholar]
- 80.Malhotra RK, Neurodegenerative Disorders and Sleep. Sleep medicine clinics 13, 63–70 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Malkani R, Attarian H, Sleep in Neurodegenerative Disorders. Current sleep medicine reports 1, 81–90 (2015). [Google Scholar]
- 82.Rainey-Smith SR et al. , Genetic variation in Aquaporin-4 moderates the relationship between sleep and brain Aβ-amyloid burden. Translational psychiatry 8, 47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ulv Larsen SM et al. , Haplotype of the astrocytic water channel AQP4 is associated with slow wave energy regulation in human NREM sleep. PLoS biology 18, e3000623 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zeppenfeld DM et al. , Association of Perivascular Localization of Aquaporin-4 With Cognition and Alzheimer Disease in Aging Brains. JAMA neurology 74, 91–99 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Burfeind KG et al. , The effects of noncoding aquaporin-4 single-nucleotide polymorphisms on cognition and functional progression of Alzheimer’s disease. Alzheimer’s & dementia 3, 348–359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Simon MJ et al. , Transcriptional network analysis of human astrocytic endfoot genes reveals region-specific associations with dementia status and tau pathology. Scientific reports 8, 12389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Knopman D et al. , Cardiovascular risk factors and cognitive decline in middle-aged adults. Neurology 56, 42–48 (2001). [DOI] [PubMed] [Google Scholar]
- 88.Leritz EC, McGlinchey RE, Kellison I, Rudolph JL, Milberg WP, Cardiovascular Disease Risk Factors and Cognition in the Elderly. Curr Cardiovasc Risk Rep 5, 407–412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aberg MAI et al. , Cardiovascular fitness is associated with cognition in young adulthood. Proceedings of the National Academy of Sciences of the United States of America 106, 20906–20911 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wendell CR et al. , Cardiorespiratory fitness and accelerated cognitive decline with aging. J Gerontol A Biol Sci Med Sci 69, 455–462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mortensen KN et al. , Impaired Glymphatic Transport in Spontaneously Hypertensive Rats. The Journal of neuroscience : the official journal of the Society for Neuroscience 39, 6365–6377 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xue Y et al. , Concomitant enlargement of perivascular spaces and decrease in glymphatic transport in an animal model of cerebral small vessel disease. Brain research bulletin 161, 78–83 (2020). [DOI] [PubMed] [Google Scholar]
- 93.Gonzaga C, Bertolami A, Bertolami M, Amodeo C, Calhoun D, Obstructive sleep apnea, hypertension and cardiovascular diseases. Journal of human hypertension 29, 705–712 (2015). [DOI] [PubMed] [Google Scholar]
- 94.Mestre H, Kostrikov S, Mehta RI, Nedergaard M, Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clinical science 131, 2257–2274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vogels RL et al. , Profile of cognitive impairment in chronic heart failure. Journal of the American Geriatrics Society 55, 1764–1770 (2007). [DOI] [PubMed] [Google Scholar]
- 96.Wardlaw JM, Smith C, Dichgans M, Small vessel disease: mechanisms and clinical implications. The Lancet. Neurology 18, 684–696 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Iadecola C, The pathobiology of vascular dementia. Neuron 80, 844–866 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zlokovic BV, Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nature reviews. Neuroscience 12, 723–738 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wardlaw JM, Smith C, Dichgans M, Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. The Lancet. Neurology 12, 483–497 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wardlaw JM et al. , Perivascular spaces in the brain: anatomy, physiology and pathology. Nature reviews. Neurology 16, 137–153 (2020). [DOI] [PubMed] [Google Scholar]
- 101.Jiang Q et al. , Impairment of the glymphatic system after diabetes. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 37, 1326–1337 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sweeney MD et al. , Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 15, 158–167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Toda H, Williams JA, Gulledge M, Sehgal A, A sleep-inducing gene, nemuri, links sleep and immune function in Drosophila. Science 363, 509–515 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McAlpine CS et al. , Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 566, 383–387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Niethard N, Burgalossi A, Born J, Plasticity during Sleep Is Linked to Specific Regulation of Cortical Circuit Activity. Frontiers in neural circuits 11, 65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bateman RJ et al. , Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nature medicine 12, 856–861 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kang JE et al. , Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Holth JK et al. , The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363, 880–884 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shokri-Kojori E et al. , beta-Amyloid accumulation in the human brain after one night of sleep deprivation. Proceedings of the National Academy of Sciences of the United States of America 115, 4483–4488 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fultz NE et al. , Coupled electrophysiological, hemodynamic, and cerebrospinal fluid oscillations in human sleep. Science 366, 628–631 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kanekiyo T, Liu CC, Shinohara M, Li J, Bu G, LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 16458–16465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kiviniemi V et al. , Ultra-fast magnetic resonance encephalography of physiological brain activity - Glymphatic pulsation mechanisms? Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 36, 1033–1045 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rius-Perez S, Tormos AM, Perez S, Talens-Visconti R, Vascular pathology: Cause or effect in Alzheimer disease? Neurologia 33, 112–120 (2018). [DOI] [PubMed] [Google Scholar]