

SUMMARY
Molecular mechanisms underlying adaptive targeted therapy resistance in pancreatic ductal adenocarcinoma (PDAC) are poorly understood. Here, we identify SETD5 as a major driver of PDAC resistance to MEK1/2 inhibition (MEKi). SETD5 is induced by MEKi resistance and its deletion restores refractory PDAC vulnerability to MEKi therapy in mouse models and patient-derived xenografts. SETD5 lacks histone methyltransferase activity but scaffolds a co-repressor complex, including HDAC3 and G9a. Gene silencing by the SETD5 complex regulates known drug resistance pathways to reprogram cellular responses to MEKi. Pharmacological co-targeting of MEK1/2, HDAC3, and G9a sustains PDAC tumor growth inhibition in vivo. Our work uncovers SETD5 as a key mediator of acquired MEKi therapy resistance in PDAC and suggests a context for advancing MEKi use in the clinic.
Graphical Abstract
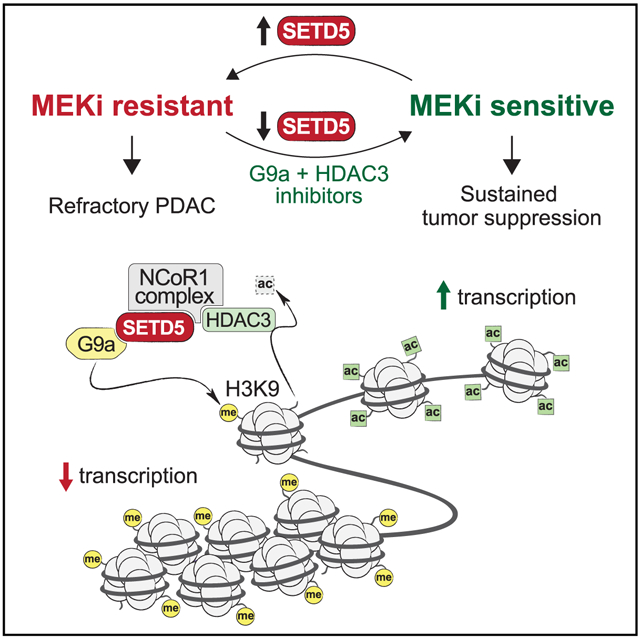
In Brief
In pancreatic ductal adenocarcinoma (PDAC), a major roadblock in therapies targeting the KRAS-MAPK pathway, such as MEK1/2 inhibition (MEKi), is the rapid emergence of resistance. Wang et al. identify a clinically actionable epigenetic pathway mediated by SETD5 to drive PDAC resistance to MEKi.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest major cancer types, killing on a yearly basis more than 430,000 patients worldwide (Rawla et al., 2019). The 5-year survival rate of PDAC is <8%, with progress in improving outcomes lagging behind most other neoplastic diseases (Siegel et al., 2019). The difficult prognosis of PDAC is due to the disease typically being diagnosed at an advanced stage, at which point surgical resection is not beneficial, and the absence of effective medical options (Ryan et al., 2014). The vast majority of PDAC cases are driven by oncogenic activating mutations in KRAS (Almoguera et al., 1988). However, besides recent promising data with KRAS(G12C)-specific inhibitors (a mutation found in <1% of PDAC) (Canon et al., 2019), drugging the KRAS mutations typically associated with PDAC has been unsuccessful (Stephen et al., 2014). As an alternative approach, drug discovery efforts have focused on targeting downstream KRAS effector pathways. In particular, drugs targeting the mitogen-activated protein kinase (MAPK) signaling cascade, a canonical pathway downstream of KRAS, have been clinically explored (Collisson et al., 2012; Manchado et al., 2016; Sun et al., 2017; Zhao and Adjei, 2014). MEK inhibibion (MEKi)-based therapies have achieved FDA approval for some cancer types; however, clinical trials for PDAC have been less encouraging (Bodoky et al., 2012; Infante et al., 2014). The failure of MEKi in PDAC is likely due to adaptive signaling and the development of therapy resistance (Ponz-Sarvise et al., 2019). Thus, understanding the mechanisms underlying resistance acquisition in PDAC to targeted therapies is likely to lead to improved treatment modalities (Sun et al., 2017). In this context, pathways implicated in promoting MEKi resistance in PDAC include cellular programs that regulate oxidative phosphorylation and mitochondrial function, autophagy, lysosome activity, and compensatory induction of other pathways, such as phosphoinositide 3-kinase (PI3K) signaling, receptor tyrosine kinase signaling pathways (e.g., ERBB and FGFR1), and YAP1-regulated pathways (Bryant et al., 2019; Kapoor et al., 2014; Kinsey et al., 2019b; Manchado et al., 2016; Perera et al., 2015; Ponz-Sarvise et al., 2019; Shao et al., 2014; Viale et al., 2014). While treatment regimens using dual inhibition of MEK and certain resistance pathways (e.g., PI3K and EGFR) have to date been ineffective (Chung et al., 2017; Ko et al., 2016), ongoing trials co-targeting autophagy and oxidative phosphorylation are pending (Kinsey et al., 2019a; Molina et al., 2018). However, whether there are underlying clinically actionable epigenetic-based mechanisms regulating general resistance programs is not known.
The chromatin-associated protein SETD5 contains a catalytic methyltransferase SET domain and is thus annotated as a candidate protein lysine methyltransferase (KMT) (Husmann and Gozani, 2019). However, whether SETD5 is an active enzyme is unclear. The SETD5 gene is commonly mutated in patients with intellectual disabilities and autism spectrum disorders (Deliu et al., 2018; Grozeva et al., 2014). SETD5 knockout mice die early in development due to cardiovascular defects and other abnormalities and SETD5 deletion in embryonic stem cells impaired proliferation and differentiation with altered gene expression (Deliu et al., 2018; Osipovich et al., 2016; Sessa et al., 2019). Setd5 haploinsufficiency also leads to aberrant gene expression in neuronal tissue and is associated with cognitive and behavioral defects in mice (Deliu et al., 2018; Sessa et al., 2019). Finally, independent Sleeping Beauty transposon mutagenesis-based in vivo screens identified SETD5 as a common insertion site that cooperates with KRAS to accelerate pancreatic carcinogenesis (Mann et al., 2012; Perez-Mancera et al., 2012). However, functions for SETD5 in cancer are largely unexplored.
RESULTS
Identification of SETD5 as a Candidate Regulator of PDAC Cell Resistance to MEKi
To explore possible connections between chromatin regulation, protein methylation, and the development of targeted MEKi therapy resistance in Ras-driven pancreatic cancer, we performed a high-content small hairpin RNA (shRNA) screen (Kampmann et al., 2014). The pancreatic cancer cell line MiaPaCa2, which harbors mutant KRAS (Sulahian et al., 2019), was transduced with a pooled high-coverage library containing 25 independent shRNAs directed against each of 95 known and putative human methyltransferase genes, including the vast majority of known KMTs present in the human genome (see schematic Figure 1A). After transduction, cells were treated with the MEKi trametinib or vehicle control and differences in shRNA abundance after 12 days were used to identify candidate genes influencing the drug response (Figures 1A and 1B) (Sulahian et al., 2019). Notably, out of the 2,375 shRNAs in the library, the ones that rendered cells most sensitive to trametinib targeted the candidate histone KMT SETD5 (Figure 1B; Table S1). The direct depletion of SETD5 in MiaPaCa2 cells (Figure 1C) attenuated cellular proliferation, although to a lesser degree than trametinib treatment (Figure 1D). Combining SETD5 depletion with trametinib treatment effectively inhibited cell proliferation (Figure 1D), with SETD5 depletion decreasing the half-maximum inhibitory concentration of trametinib in MiaPaCa2 and five additional human PDAC cell lines by roughly 2.5-fold (Figures 1E and S1A-S1E). Consistent with these results, computational analysis of publicly available gene expression data suggests that SETD5 is overexpressed in pancreatic cancer (Figure S1F). Furthermore, SETD5 immunohistological signal is high in human PDAC samples relative to healthy tissue and this signal negatively correlates with patient survival (Figures S1G and S1H). Based on these data and previous studies (Mann et al., 2012; Perez-Mancera et al., 2012) we postulated a role for SETD5 in PDAC pathology.
Figure 1. Identification of SETD5 as a Candidate Regulator of PDAC Cell Resistance to MEKi.

(A) Schematic of the screen to identify methyltransferases conferring sensitivity to the MEKi trametinib. MiaPaCa2 pancreatic cancer cells infected with a pooled high-coverage shRNA library were split into two subpopulations and treated as indicated. The frequency of shRNA-encoding constructs in each subpopulation was determined by deep sequencing. See also Table S1.
(B) shRNAs targeting SETD5 sensitize cells to MEKi. A quantitative resistance phenotype ρ was calculated for each shRNA based on the sequencing frequency in the two subpopulations. The graph compares the distribution of ρ for shRNAs targeting a gene of interest (shown here SETD5) to the ρ distribution for negative control shRNAs using the Mann-Whitney U test that yielded a p value for the gene.
(C) Western analysis with the indicated antibodies of whole-cell extracts (WCEs) from MiaPaCa2 cells ± MEKi and depleted for SETD5 by Cas9/sgRNA (sgSETD5) or control (sgControl). Actin shown as a loading control.
(D) SETD5 knockdown synergizes with MEKi to attenuate cell proliferation. Confluency of MiaPaCa2 cells as in (C) treated for 120 h with MEKi (trametinib 15 nM) or vehicle control. Data are represented as mean ± SEM of three technical replicates in two independent experiments. *p < 0.033, **p < 0.002, ***p < 0.0001 by two-way ANOVA with Tukey’s testing for multiple comparisons.
(E) SETD5 depletion increases MiaPaCa2 cells sensitivity to MEKi. Cellular viability in response to trametinib at the indicated doses in MiaPaCa2 cells ± SETD5. The calculated geometric mean half-maximum inhibitory concentration (IC50) values for MEKi are shown. Data represented as mean ± SEM of three technical replicates in two independent experiments.
(F) Schematic of acquisition of pancreatic tissue biopsies from Kras;p53 PDAC mouse model through abdominal laparotomy. The tumor biopsy cores were removed from mice before treatment (first biopsy, naive tumor, red) after initial treatment with MEKi (second biopsy, MEKi-responsive tumor, green) and upon tumor relapse with increased volume (third biopsy, MEKi-resistant tumor, purple). Representative magnetic resonance imaging (MRI) scans to analyze tumor volume in Kras;p53 mutant mice area shown. Scale bars, 10 mm.
(G) SETD5 expression increases in PDAC tumors upon the development of MEKi resistance. Western analysis with the indicated antibodies of PDAC tissue biopsies from Kras;p53 mouse model as described in (F). Three independent and representative samples are shown for each biopsy stage.
See also Figure S1.
We generated conditional Setd5LoxP/LoxP knockout mice to test the role of SETD5 in cancer in vivo. Setd5LoxP/LoxP mice develop normally, are viable, and fertile (data not shown). Deletion of Setd5 in the pancreas using the pancreas-specific Cre-recombinase-expressing strain Ptf1aCre/+ (Kawaguchi et al., 2002) (Figure S2A) resulted in no apparent developmental consequences (data not shown). To investigate the role of SETD5 in KRAS-driven PDAC development, we used the Ptf1a+/Cre; Kras+/LSL-G12D;p53LoxP/LoxP (Kras;p53) mutant model in which morbid PDAC develops with 100% penetrance 6–8 weeks after birth (Bardeesy et al., 2006; Hingorani et al., 2005). In this aggressive malignancy model, Setd5 deletion resulted in a modest extension in median survival relative to control (Figures S2B and S2C; data not shown). These data suggest a more specialized role for SETD5 in tumor response to therapy rather than broadly regulating tumor development.
To directly explore a role for SETD5 in vivo in tumor responses to RAS-pathway modulation, we obtained serial biopsies of PDAC tissue from Kras;p53 mice: one sample before trametinib treatment, and two additional samples taken early and late during the course of the treatment protocol (Figure 1F). Tumor size was monitored by magnetic resonance imaging (MRI) (Liu et al., 2019) to optimize consistent biopsy acquisition of (1) naive (untreated), (2) trametinib-responsive, or (3) trametinib-resistant cancer tissues (Figure 1F). Serial biopsies were obtained by laparotomy (Sastra and Olive, 2014) to mitigate potential tumor tissue heterogeneity. Inhibition of ERK1/2 phosphorylation was observed at time points 2 and 3, indicating that trametinib remained effective despite progression of resistant tumors (Figure 1G); consistent with PDAC resistance mechanisms not acting via MAPK signaling reactivation (Kapoor et al., 2014; Shao et al., 2014). Notably, SETD5 expression is higher in the trametinib-refractory tumor samples compared with the trametinib-responsive and naive PDAC samples (Figure 1G). A similar increase in SETD5 expression is observed in PDAC biopsies upon prolonged exposure to selumetinib, an independent MEKi (Banerji et al., 2010) (Figures S2D and S2E). Moreover, the expression of SETD5 mRNA and protein increases in cells derived from naive murine PDAC tumors (here to referred to as KPCN cells) cultured to develop resistance to different MEK inhibitors relative to control-treated cells (Figures S2F and S2G). Thus, increased SETD5 expression correlates with the development of resistance to a variety of MEK inhibitors both in cells and in vivo in a widely used PDAC mouse model.
SETD5 Depletion Re-sensitizes Resistant PDAC to Trametinib in PDX and Mouse Models In Vivo
Cell lines were established from trametinib-resistant PDAC tumors (hereto referred to as KPCR cells) to test in vivo responsiveness of trametinib-resistant allografts to MEKi ± SETD5 (Figures 2A and 2B). The control allograft tumors are resistant to trametinib and expand rapidly in the presence of drug, whereas SETD5-depletion restores sensitivity to trametinib as allograft tumor growth is impaired (Figure 2C). A patient-derived xenograft (PDX)-based model was next developed to explore the development of MEKi resistance in human PDAC (schematic, Figure 2D). Growing PDAC PDX tumors of approximately 200 mm3 were exposed to trametinib until tumor expansion recommenced (~5 weeks), indicating drug resistance. Cells isolated from the trametinib-resistant PDX tumor ± SETD5 (Figure 2E) were then re-tested in xenograft tumor studies in the presence of trametinib. As expected, xenografts from relapsed trametinib-resistant tumors grow robustly despite trametinib; however, MEKi treatment impaired growth of SET5-knockdown tumors (Figure 2F).
Figure 2. SETD5 Depletion Re-sensitizes Resistant PDAC to Trametinib in PDX and Mouse Models In Vivo.

(A) Schematic of generation of PDAC allografts in syngeneic mice established from MEKi-resistant tumor biopsies (as in Figure 1F) ± SETD5. Trametinib treatment schedule (MEKi, 0.3 mg/kg by intraperitoneal injection once daily) is shown.
(B) Western blots with the indicated antibodies of a representative sample for each condition described in (A) are shown. Actin is shown as a loading control.
(C) SETD5 depletion re-sensitizes PDAC allografts to MEKi. Quantification of mouse allograft tumor volume growth in syngeneic mice (n = 8 mice, for each treatment group). *p < 0.033, **p < 0.002, ***p < 0.0001 by two-way ANOVA with Tukey’s testing for multiple comparisons. Data are represented as mean ± SEM.
(D) Schematic to generate MEKi-resistant primary human PDAC for PDX studies. Patient tumor samples were grafted subcutaneously to immunocompromised NSG mice. Once tumor volume reached 200 mm3, mice were treated with trametinib 0.3 mg/kg by intraperitoneal injection once daily until tumor growth relapsed (~5 weeks), indicating drug resistance. Resistant cells were modified to express Cas9/sgRNA targeting SETD5 (sgSETD5) or control (sgControl) and tested for xenograft growth with treatment as shown.
(E) Western blots with the indicated antibodies of PDX samples in different stages described in (D). A representative sample for each condition is shown. Actin is shown as a loading control.
(F) SETD5 depletion restores refractory PDAC PDX tumor sensitivity to MEKi. Tumor volume quantification of MEKi-resistant patient-derived PDAC xenografts described in (D) in immunocompromised mice (n = 8 mice, for each treatment group). *p < 0.033, **p < 0.002, ***p < 0.0001 by two-way ANOVA with Tukey’s testing for multiple comparisons. Data are represented as mean ± SEM.
(G) Schematic of dual-recombinase (Flp/Frt, Cre/LoxP) system to acutely delete SETD5 in vivo in aggressive PDAC. Activation of KrasFSF-G12D and deletion of p53Frt/Frt alleles in mouse pancreata (Pdx1Flp) results in development of malignant PDAC. Time-specific tamoxifen-mediated Rosa26FSF-CreERT2 activation allows for recombination of the conditional Setd5LoxP/LoxP allele with loss of SETD5 expression (SETD5KO) in established PDAC. Control animals that received vehicle express wild-type SETD5WT. Subsequently mice were treated with placebo (vehicle) or trametinib (MEKi, 0.3 mg/kg, intraperitoneal injection once daily).
(H) Treatment schedule for administration of tamoxifen, MEKi or placebo (vehicle) in the system described in (G).
(I) Deletion of SETD5 in established PDAC cooperates with MEKi to suppress tumor growth. Quantification of PDAC volume change based on MRI scans (detailed procedure in the STAR Methods) in mice described in (G and H) (n = 9 mice for each experimental group). Boxes, 25th to 75th percentiles; whiskers, minimum to maximum; center line, median; *p < 0.033, **p < 0.002, ***p < 0.0001 by two-way ANOVA with Tukey’s testing for multiple comparisons. Scale bars, 100 μm.
(J and K) (J) Quantification of proliferation (Ki67+ cells), and (K) cleaved caspase-3 (cl.Caspase3+ cells) a marker of apoptosis in samples as in (I). Boxes, 25th to 75th percentile; whiskers, minimum to maximum; center line, median; arrowheads, positive cleaved caspase-3 cells; *p < 0.033, **p < 0.002, ***p < 0.0001 by two-way ANOVA with Tukey’s testing for multiple comparisons. Data are represented as mean ± SEM. Scale bars, 100 μm.
(L) Western blots with the indicated antibodies of the indicated pancreatic tissue lysates. Two independent and representative samples are shown for each genotype. Actin is shown as a loading control.
See also Figure S2.
To address if maintenance of trametinib resistance in PDAC tumors in vivo actively requires SETD5 expression, a dual-recombinase approach was used to delete Setd5 in established tumors (Schonhuber et al., 2014). PDAC tumors were formed using Pdx1Flp;KrasFSF-G12D;p53Frt/Frt;R26FSF-CreER;Setd5LoxP/LoxP mice, in which Pdx1-driven expression of Flp-recombinase causes pancreas-specific expression of oncogenic KRAS, deletion of p53, and expression of tamoxifen-inducible Cre (CreER) (schematic, Figure 2G). Setd5 was deleted in tumors that reached ~150 mm3 by treating mice with tamoxifen to activate CreER, which is expressed exclusively in the Flp-recombined epithelial pancreatic cells (Rosa26FSF-CreER) (Figures 2H and S2H). Tumors were then treated with trametinib or vehicle (control) and tumor volume change evaluated by MRI (Figure 2H). The single intervention of trametinib treatment or SETD5 deletion in established PDAC each modestly slowed but did not eliminate tumor growth (Figures 2I-2K and S2I). Notably, combining administration of tamoxifen (to knockout Setd5) with trametinib halted tumor growth and caused some tumors to regress in size (Figures 2I-K and S2I). Analyses of tumor biopsy lysates showed that SETD5 protein was not expressed in tamoxifen-treated mice and that trametinib inhibited ERK1/2 phosphorylation (Figure 2L).
SETD5 Lacks Intrinsic Histone Lysine Methyltransferase Activity
While SET domain proteins are frequently active KMTs, the SET domain of SETD5 lacks key conserved residues that bind to the methyl donor S-adenosyl methionine and are present on all known active SET enzymes (Figure S3A) (Mas et al., 2016). Surprisingly, SETD5 was recently reported to be a robust H3K36 trimethylase (Sessa et al., 2019). However, the recombinant SET domain of SETD5 did not methylate recombinant histone H3, recombinant nucleosome, or purified histones (Figure 3A). In contrast, the positive control catalytic domain of the H3K9 KMT G9a methylated all three histone substrates (Figure 3A). We did not detect methylation on any histone residue, including H3K36, using the same SETD5 derivative (murine SETD5 amino acids 1–423) and reaction conditions reported in (Sessa et al., 2019) (Figures 3B and S3B). In contrast, H3K36 was methylated by SETD2, the principle human enzyme that generates physiologic H3K36me3 (Husmann and Gozani, 2019) (Figures 3B and S3B). Finally, in contrast to several positive controls (Figures S3C-S3F), full-length recombinant SETD5 did not methylate histones (Figure 3C).
Figure 3. SETD5 Has No Intrinsic Histone Lysine Methyltransferase Activity but Is Present in a Complex that Methylates H3K9.

(A) SETD5 does not methylate histones or nucleosomes. In vitro methylation assays with recombinant SETD5SET (GST-SETD5 residues 1–520) or positive control G9aSET (GST-G9a SET domain) on recombinant histone H3 (rH3), purified calf thymus histones (CTH), or recombinant nucleosomes (rNuc) substrates as indicated. Top panel, [3H]S-adenosyl methionine is the methyl donor and methylation visualized by autoradiography and indicated as H3me. Bottom panel, Coomassie stain of proteins in the reaction.
(B) SETD5 does not methylate poly-nucleosomes, whereas SETD2 does. In vitro methylation assays as in (A) with hSETD5 (amino acids [aa]: 1–415) (human SETD5 residues 1–415), mSETD5 (aa: 1–423) (murine SETD5 residues 1–423 as in Sessa et al., 2019), and positive control SETD2SET (GST-SETD2 SET domain) on rNucpoly (H3.3-containing recombinant poly-nucleosomes as in Sessa et al., 2019). Top panel, autoradiogram of methylation assay. Bottom panel, Coomassie stain of proteins in the reaction. Asterisk indicates cleaved H3 breakdown product.
(C) Full-length SETD5 does not methylate nucleosomes. In vitro methylation assays as in (A) with full-length SETD5 on rNuc as indicated. Top panel, autoradiogram of methylation assay. Bottom panel, Coomassie stain of proteins in the reaction.
(D) Methylation of H3 by the SETD5 complex. In vitro methylation assay as in (A) on recombinant nucleosomes with tandem-affinity-purified SETD5 complex (SETD5com) from 293T cells. MLL4 complex (MLL4com) used as a positive control. Top panel, autoradiogram of methylation assay. Bottom panel, Coomassie stain of proteins in the reaction as indicated.
(E) SETD5com primarily di-methylates H3K9. Selected ion chromatograms for non-, mono-, di-, and tri-methyl H3K9 peptides from trypsin digestion of SETD5com methylation reactions on recombinant nucleosomes. High-pressure liquid chromatogrphy elution profiles show a 10-ppm mass window around expected peptide masses, peptide sequence KSTGGKAPR, K9 is underlined; m/z are 535.3037 (H3K9me0), 542.3115 (H3K9me1), 521.3062 (H3K9me2), and 528.3140 (H3K9me3). Arrows indicate elution peaks of non-, mono-, di-, and tri-methylated H3K9 peptides in the profiles and percent methyl state is indicated. See also Figure S3G.
(F) SETD5com methylates H3K9 but not any other H3 lysine residue. Summary of H3 lysine methylation states detected in (E) by tandem mass spectrometry. See also Figure S3H.
(G) The SETD5 SET domain is dispensable for SETD5com H3K9 methylation activity. Methylation assays as in (D) with the indicated V5-SETD5-Flag derivatives. Top panel, schematic of SETD5 constructs with the position of the SET domains shown: SETD5, full-length SETD5; SETD5ΔSET, N-terminal truncation; SETD5ΔC, C-terminal truncation. Left panel, Coomassie stain of proteins in the reaction, Right panel, western analysis with H3K9 methylation detected using the indicated antibodies. SETD5 constructs were detected with anti-Flag.
(H) SETD5 co-purifies with the NCoR1-HDAC3 complex and G9a. Silver stain of SETD5com after first and second purification steps. The identity of associated proteins as indicated on the right was determined by mass spectrometry. See also Table S3.
(I) SETD5 interacts with the NCoR1-HDAC3 complex, G9a, and GLP in 293T cells. Co-immunoprecipitation (coIP) experiments in 293T cells expressing Flag-SETD5 and using the indicated antibodies for the IPs (anti-Flag for SETD5) and western analyses. Input, nuclear extract.
(J) CoIP experiments as in (I) with Flag-tagged wild-type SETD5 or the indicated derivatives as in (G). Input, 293T nuclear extract.
Some KMTs are active only in the context of a macromolecular complex (i.e., MLL1-4) (Husmann and Gozani, 2019); indeed, like the positive control MLL4 complex, tandem-tag affinity purified SETD5 (hereto referred to as SETD5com) from 293T cells methylated nucleosomes on H3 (Figure 3D). Tandem mass spectrometry analysis of the SETD5com-catalyzed reaction identified primarily H3K9 dimethylation (Figures 3E and S3G). Methylation was not detected on any other lysine residue of H3, including H3K36 (Figures 3F and S3H). Notably, the SET domain of SETD5 is dispensable for catalyzing H3K9 methylation, as a complex purified with a C-terminal SETD5 derivative lacking the SET domain (SETD5ΔSET) methylates H3K9, and a complex purified with an N-terminal SETD5 derivative containing the SET domain but missing the C terminus (SETD5ΔC) lacks methylation activity (Figure 3G). We note that SETD5 did not methylate any of over 9,000 proteins present on a protein array (Mazur et al., 2014) and it had no activity on fractionated pancreatic cancer cell lysates, suggesting that SETD5 does not methylate a non-histone protein (Figures S3I and S3J; data not shown). Thus, SETD5 does not methylate H3K36 and indeed lacks intrinsic histone methylation activity (see Discussion). These data also suggest that an H3K9-specific KMT associates with SETD5.
SETD5 Forms a Distinct Co-repressor Complex with NCoR1/HDAC3 and G9a/GLP
Analysis of SETD5com by mass spectrometry identified components of the NCoR1-HDAC3 complex, which is known to interact with SETD5 (Osipovich et al., 2016), and the H3K9 KMTs G9a and GLP (Tachibana et al., 2001) but no other KMTs (Figure 3H; Table S2; data not shown). The interactions between SETD5 and the NCoR1-HDAC3 complex and with G9a/GLP were also observed in immunoprecipitation (IP)-western analyses (Figure 3I). In reverse IPs, HDAC3 and G9a each separately immunoprecipitated SETD5; however, G9a and GLP were not in the HDAC3 IP, and components of the NCoR1-HDAC3 complex were not detected in the G9a IP (Figure 3I). These data suggest that SETD5 abundance is low compared with that of the NCoR1-HDAC3 complex and G9a/GLP as all three components are all only present in the SETD5 IP. Furthermore, the observed interactions of SETD5 with two repressive histone-modifying activities (histone deacetylation and H3K9 methylation) suggests that SETD5 scaffolds a distinct co-repressor complex. Finally, SETD5 and SETD5ΔSET, but not SETD5ΔC, interact with HDAC3 and G9a/GLP (Figure 3J). Thus, the C terminus region of SETD5 is necessary for H3K9 methylation and mediates the interaction with HDAC3, G9a/GLP.
We postulated that G9a and GLP, two KMTs that generate H3K9me1/2 and interact with SETD5, are responsible for the methylation activity associated with SETD5com. To test this idea, SETD5com was purified from control 293T cells or 293T cells in which G9a and GLP were co-depleted (293DKD; Figure 4A). As expected, SETD5com purified from control cells methylated H3K9; however, SETD5com purified from 293TDKD cells does not methylate H3K9 (Figure 4B). Furthermore, in vitro methylation assays in the presence of the selective G9a/GLP inhibitory compound UNC0638 (Vedadi et al., 2011) abrogated the activity of the positive control full-length G9a and of SETD5-com (Figure 4C). Together, these data suggest that H3K9 methylation by SETD5com is mediated by the KMTs G9a/GLP.
Figure 4. G9a/GLP Mediate SETD5com Methylation Activity.
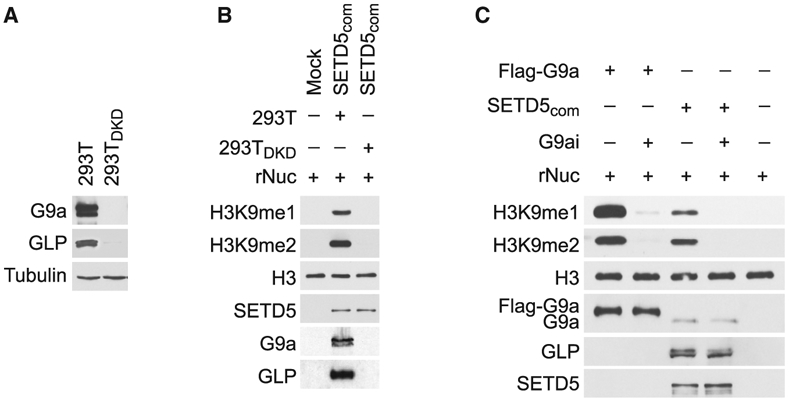
(A) Generation of G9a/GLP-depleted 293T cells. Western analysis with the indicated antibodies of control (293T) or G9a/GLP co-depleted 293T cell lysates (293TDKD). Tubulin is shown as a loading control.
(B) Methylation of H3K9 by SETD5com requires G9a and GLP. Western analysis with the indicated antibodies of in vitro methylation assay as in Figure 3D using SETD5com purified from 293T or 293TDKD cells.
(C) Methylation of H3K9 by SETDcom is inhibited by the G9a/GLP inhibitor (UNC0638, G9ai). Western analysis with the indicated antibodies of in vitro methylation assay on recombinant nucleosomes with G9a and SETD5com ± UNC0638 (2 μM).
The association of SETD5 with the NCoR1-HDAC3 co-repressor complex suggests a model in which SETD5 coordinates placement of repressive methylation at H3K9me by G9a/GLP with removal of activating histone acetylation marks by HDAC3. To test this hypothesis, deacetylase assays using SETD5com from control or HDAC3-depleted cells (Figure 5A) was performed on HeLa-purified nucleosomes, which contain a large array of existing histone modifications. As shown in Figure 5B, H3K9 acetylation (H3K9ac) was removed by SETD5com in an HDAC3-dependent manner. Moreover, the selective HDAC3 inhibitor RGFP966 (Xu et al., 2009) blocks H3K9ac deacetylation by SETD5com, whereas the HDAC8 inhibitor PCI-34051 does not (Balasubramanian et al., 2008) (Figure 5C). In contrast to H3K9ac, SETD5com does not deacetylate the reported HDAC3 substrates H4K5ac and H4K8ac (Figures 5B and 5C) (Vermeulen et al., 2004). To the best of our knowledge, the selectivity of HDAC3 on histones has not been comprehensively characterized. Therefore, we assembled an active recombinant HDAC3 complex (rHDAC3 and the DAD domain of NCoR1 [Guenther et al., 2001] [Figure 5D]) and performed in vitro deacetylation assays on HeLa-purified nucleosomes. The HDAC3 complex deacetylated a broad range of lysine-acetylated substrates on nucleosomes, including the published substrates H4K5ac and H4K8ac (Figure 5E). The difference in deacetylation activity on HeLa nucleosomes between SETD5com and rHDAC suggest that SETD5com imposes substrate selectivity upon HDAC3. To test this, a side-by-side comparison of SETD5com and rHDAC3 complex deacetylation activity was performed on a library of recombinant nucleosomes designed to harbor a single acetylation modification on 1 of 11 different lysine residues known to be modified on H3 and H4. In this system, the rHDAC3 complex deacetylated all of the lysine-acetylated nucleosomes besides H3K36ac, whereas SETD5com only deacetylated H3K9ac and H3K27ac (Figures 5F-5H and S4A-S4I). Thus, the in vitro catalytic activity of HDAC3, in the context of the SETD5 complex, is restricted to acetylated H3K9 and H3K27, two residues that when methylated are markers of silenced chromatin (Husmann and Gozani, 2019).
Figure 5. HDAC3-Selective Deacetylation of H3K9Ac by SETD5com.

(A) Generation of HDAC3-depleted 293T cells. Western analysis with the indicated antibodies of control (293T) or HDAC3-depleted 293T cell lysates (HDAC3KD). Tubulin is shown as a loading control.
(B) SETD5com possess HDAC3-dependent lysine deacetylation activity. Western analysis with the indicated antibodies of in vitro histone deacetylation assay on HeLa-purified nucleosomes using SETD5com purified from 293T or HDAC3KD cells.
(C) HDAC3-dependent SETD5com lysine deacetylation activity is inhibited by a selective HDAC3 inhibitor. In vitro deacetylation assays as in (B) ± the selective HDAC3 inhibitor (RGF9966, 1 μM) or ± the selective HDAC8 inhibitor (PCI-34051, 1.5 μM). SETD5ΔC does not interact with HDAC3 (see Figure 3J) and serves as a negative control.
(D) Coomassie stain of active recombinant HDAC3 complex (contains HDAC3 and the DAD domain of NCoR1, labeled as rHDAC3) purified from E. coli.
(E) HDAC3 has broad deacetylation activity on histones. In vitro histone deacetylation assay on HeLa-purified nucleosomes with rHDAC3 complex analyzed by western blots with the indicated antibodies.
(F–H) HDAC3 selectively deacetylates H3K9Ac and H3K27Ac in the context of SETD5com. (F) Summary of deacetylation assays using SETDcom or rHDAC3 on a library of recombinant nucleosomes designed to harbor a single lysine acetylation as indicated. (G and H) Western analysis with the indicated antibodies of deacetylation assays on H3K9Ac rNuc (G) and H3K18Ac rNuc (H). Figure S4 shows the other nine modified nucleosomes summarized in (F).
The SETD5 Complex Regulates H3K9 Modification and MEKi Resistance in PDAC Cells
SETD5 expression increases in cells and tumors upon the development of resistance to MEKi (see Figures 1G and S2D-S2H). The levels of SETD5 protein are also higher in KPCR cells (derived from trametinib-resistant murine PDAC tumors) compared with KPCN cells (derived from naive murine PDAC tumors) (Figure 6A). In contrast, the levels of SETD5-associated proteins, such as HDAC3 and G9a are equivalent in KPCN and KPCR lysates (Figure 6A). Like in 293T cells (see Figure 3), SETD5 interacts with the NCoR1-HDAC3 complex and G9a in KPCR cells (Figure 6B; we were unable to identify an antibody that reliably detected murine GLP). In addition, analysis of KPCR cell lysates by size-exclusion chromatography identified co-enrichment of SETD5, the NCoR1-HDAC3 complex, and G9a within the same high-molecular-weight fractions (Figure S5A). These data suggest that the rate-limiting component in the assembly of SETD5com is SETD5, whose expression increases in MEKi-resistant cells and tumors.
Figure 6. SETD5 Coordinates a Targeted Therapy Resistance Program in PDAC Cells.

(A) Increased levels of SETD5 but not associated proteins in MEKi-resistant PDAC cells. Western analysis with the indicated antibodies of WCEs from KPCN cells (derived from naive murine PDAC) and KPCR cells (derived from trametinib-resistant murine PDAC).
(B) SET5com interactions occur in KPCR cells. IPs of endogenous SETD5 complex (or IgG control) from nuclear extracts of KPCR followed by western analysis with the indicated antibodies. Input, nuclear extract.
(C and D) SETD5 depletion inhibits proliferation of KPCR cells but has no impact on KPCN cells. Western analysis (top panel) and proliferation assays (bottom panel) in KPCN (C) and KPCR (D) cell lines depleted for SETD5 with four independent sgRNAs (sgSETD51-4) or control (sgControl). KPCR cells are cultured in the presence of 0.2 μM trametinib. Error bars represent mean ± SD from three independent experiments. ***p < 0.001; n.s., not significant, two-tailed unpaired Student’s t test.
(E) SETD5 depletion does not cause bulk chromatin modification changes. Western analysis of cells in (D) with the indicated antibodies.
(F) SETD5 is a transcriptional repressor. Volcano plot of RNA-seq comparison between KPCR cells grown in the presence of trametinib ± SETD5 (three biological replicates for each condition). SETD5 depletion caused increasing expression of 329 genes shown in red (fold change log2 ≤ −0.5 and p < 0.05 by Wald test) and decreasing expression of 93 genes shown in green (fold change log2 ≥ 0.5 and p < 0.05 by Wald test). False discovery rate (FDR) values are provided (detailed description in the STAR Methods).
(G) KEGG analysis of SETD5-repressed genes. The most significantly enriched KEGG terms associated with the 329 genes upregulated by SETD5 ablation in KPCR cancer cells are shown.
(H) Decreased total cellular glutathione (GSH) levels in SETD5 knockdown cells. Analysis of total cellular GSH levels in KPCR cells ± SETD5. Left panel, western analysis of WCE with indicated antibodies. Right panel, total GSH levels (see the STAR Methods). Error bars represent mean ± SD from three independent experiments, **p < 0.01 by two-tailed unpaired Student’s t test.
(I) Reconstitution of SETD5 knockdown cells with CRISPR-resistant SETD5. Western analysis of KPCR WCEs ± SETD5 and complemented with CRISPR-resistant SETD5.
(J) Reconstitution of SETD5-mediated target gene repression. Real-time qPCR analysis of the indicated mRNAs from cells in (I). Real-time qPCR data were normalized to Actb and are presented as fold change relative to the control. Error bars represent mean ± SD from three independent experiments. **p < 0.01, ***p < 0.001 by two-tailed unpaired Student’s t test.
(K) SETD5 directly regulates promoter chromatin of target genes. Top panel, schematic of general gene structure and site of primers used to study the GSTA1 (left panel) and PDK4 (right panel) gene loci. Real-time qPCR of chromatin immunoprecipitation (ChIP-qPCR) analysis of SETD5, H3K9ac, and H3K9me2 occupancy at the promoter (p1) and gene body (p2) of the Gsta1 and Pdk4 loci in control (sgControl), SETD5-deficient (sgSETD5) or reconstituted (sgSETD5 + SETD5) KPCR cells. The data are plotted as percent enrichment relative to input. Error bars represent mean ± SEM from three independent experiments, **p < 0.01, ***p < 0.001; n.s., not significant, by two-tailed unpaired Student’s t test.
See also Figure S6B.
While SETD5 knockdown has no impact on proliferation of KPCN cells in culture (Figure 6C), depletion of SETD5 in KPCR cells inhibits proliferation (Figure 6D). In these cells, bulk levels of H3K9me2, H3K9ac, and H3K27ac are not affected by SETD5 knockdown (Figure 6E), suggesting that potential SETD5-dependent regulation of chromatin modifications is localized rather than general. Notably, no change in H3K36me3 levels was observed upon SETD5 depletion in KPCR cells (Figure 6E) or in several other cell types, including the ones used in (Sessa et al., 2019) (Figures S5B-S5E); whereas knockdown of SETD2, the only validated mammalian H3K36me3 KMT, does deplete H3K36me3 (Figures S5C-S5E; see Discussion). Finally, the RAS-pathway factor SHOC2 is a major regulator of PDAC cell sensitivity to MEKi (Sulahian et al., 2019) and depletion of SHOC2 sensitized KPCN cells to even low dose MEKi, but unlike SETD5, SHOC2 loss had no effect on KPCR viability (Figures S5F-S5H).
Comparison of the transcriptomes of KPCR cells grown in the presence of trametinib ± SETD5 show that SETD5 loss is associated with gene repression, with SETD5 knockdown increasing expression of 329 genes and decreasing expression of 93 genes (Figure 6F). These data are consistent with SETD5 being associated with a co-repressor complex and two canonical repressive histone-modifying activities. KEGG pathway analysis of the de-repressed genes identified key functional pathways (e.g., cytochrome P450 pathway and glutathione [GSH] metabolism) that confer drug resistance in cancer (Figure 6G; see below) (Bansal and Simon, 2018; Noll et al., 2016). For example, several genes in multiple top functional pathways are enzymes that metabolize GSH (Figures 6G and S6A). While GSH initially plays a role in preventing cancer development, elevation of GSH levels is a mechanism used by many malignancies to promote chemotherapy resistance (Bansal and Simon, 2018). In this context, depletion of SETD5 in KPCR cells decreases total cellular GSH levels (Figure 6H). Notably, treatment of the KPCR-SETD5 knockdown cells with N-acetylcysteine, which counteracts GSH depletion, partially rescues the inhibition of proliferation caused by SETD5 loss (Figures S6B and S6C). Also present in the de-repressed gene group is Pdk4 (Figure S6A and Table S3); pyruvate dehydrogenase kinase 4 (PDK4) inhibits entry into the TCA cycle to oppose resistance to RAS-pathway signaling ablation in PDAC and targeted therapy in EGFR mutant lung cancer (Sun et al., 2014; Viale et al., 2014). The regulation of many key SETD5 target genes (e.g., GSTT1 [GSH-metabolizing enzyme], PDK4, and others) was observed in independent SETD5 knockdown cell lines (Figure S6A). Moreover, complementation of KPCR-SETD5 knockdown cells with sgRNA-resistant SETD5 restores repression of several genes (Figures 6I and 6J). These data suggest that SETD5 directly regulates an MEKi-resistance gene expression program in PDAC cells.
Next, chromatin immunoprecipitation assays were performed to test whether the SETD5-regulated genes are direct targets of SETD5com. SETD5 occupancy at the promoter of the target genes (GSTA1, PKD4, and GM3776) is eliminated in SETD5 knockdown cells, whereas SETD5 signaling within the coding sequence of these genes showed low signal irrespective of SETD5 expression (Figures 6K and S6B). Consistent with these results, at target gene promoters, H3K9ac levels increase and H3K9me2 levels decrease upon SETD5 knockdown, and this SETD5-dependent acetyl-methyl switch at H3K9 is not observed within the coding region (Figures 6K and S6B). Finally, adding back SETD5 reconstituted the repressive chromatin environment at target gene promoters (Figures 6K and S6B). Together, these data suggest a model in which SETD5—via coordinated deacetylation and methylation of H3K9 at chromatin targets—orchestrates a transcriptional repression program to promote PDAC resistance to MEKi.
Small-Molecule Inhibitors of G9a and HDAC3 Re-sensitize Resistant PDAC to Trametinib Therapy
Our model predicts that blocking SETD5’s associated histone-modifying activities could functionally mirror SETD5 knockdown and render refractory PDAC re-sensitized to MEKi. To test this idea, KPCR cells were treated with different combinations of trametinib with the selective inhibitors of G9a/GLP (UNC0638) and HDAC3 (RGFP966) (Vedadi et al., 2011; Xu et al., 2009). The combination of all three drugs (hereto referred to as TripleTx) significantly inhibited KPCR cell proliferation, whereas trametinib/UNC0638 ortrametinib/RGFP966 had only a modest impact compared with trametinib alone (Figures 7A and S7A). Transcriptome profiling comparing KPCR cells treated with TripleTx versus trametinib alone showed that TripleTx increased expression of 452 genes and decreased expression of 320 genes (Figure S6C). Functional analysis of the upregulated genes included pathways similar to those seen with SETD5 depletion, including cytochrome P450 and glutathione metabolism pathways and individual genes, such as Pdk4 (Figures S6D and S6E). Indeed, there was highly significant overlap in the upregulated gene sets elicited by TripleTx and sgSETD5/MEKi conditions (Figure 7B; overlap of negatively regulated genes was limited and less significant, Figure S6F). KEGG analysis of the shared upregulated gene set showed enrichment in the same functional pathways as observed with the individual upregulated gene sets (Figure 7C, compare with Figures 6G and S6D). Gene set enrichment analysis of the de-repressed genes under both conditions (sgSETD5/MEKi and TripleTx) showed strong overlap with several categories implicated in drug resistance (e.g., OXPHOS-related processes and glutathione metabolism), cancer cell phenotypes (e.g., apoptosis), and chromatin-silencing in pancreatic cancer (Figures 7D and S6G). These data suggest that the chromatin landscape at SETD5-target genes is regulated in a similar fashion by G9a/HDAC3 inhibition as with SETD5 depletion. Indeed, while TripleTx had no impact on SETD5 chromatin occupancy at the promoters of GSTA1 and PDK4 genes, the treatment increased H3K9ac signal and decreased H3K9me2 signal (Figure 7E). Thus, the combined action of selective G9a and HDAC3 inhibition may functionally phenocopy SETD5 depletion with respect to mitigating reprogramming of PDAC into a MEKi-resistant state.
Figure 7. Pharmacological Blockade of G9a and HDAC3 Sustains Trametinib Therapy Inhibition of PDAC Tumor Growth.

(A) Combination pharmacologic blockade of MEK, G9a, and HDAC3 inhibits proliferation of MEKi-resistant PDAC cells. Proliferation assay in KPCR cells treated with MEKi (0.2 μM), MEKi (0.2 μM) + G9ai (0.6 μM), MEKi (0.2 μM) + HDAC3i (0.6 μM), or MEKi + G9ai (0.6 μM) + HDAC3i (0.6 μM) (TripleTx). Error bars represent mean ± SD from three independent experiments. ***p < 0.001; n.s., not significant, by two-tailed unpaired Student’s t test.
(B) Significant overlap in the transcriptional changes triggered by SETD5 depletion and inhibition of G9a and HDAC3. Venn diagram showing the overlap of de-repressed gene sets from SETD5 depletion (see Figure 6F) and TripleTx-treated KPCR cells. p value by hypergeometric test.
(C) KEGG analysis of overlapping gene set in (B). The most significantly enriched KEGG terms associated with the genes de-repressed by SETD5 depletion and TripleTx treatment in MEKi-resistant KPCR cancer cells are shown.
(D) Gene set enrichment analysis analysis of RNA-seq data of MEKi-treated SETD5 knockdown (sgSETD5) versus control (sgControl) and TripleTx-treated versus control (MEKi-treated) KPCR cells. FDR values are provided (detailed description in the STAR Methods).
(E) ChIP-qPCR analysis as in Figure 6K of SETD5, H3K9ac, and H3K9me2 at the promoter or gene body of Gsta1 and Pdk4 (p1 and p2) in TripleTx- or MEKi-treated KPCR cells. The data are plotted as percent enrichment relative to input. Error bars represent mean ± SEM from three independent experiments, *p < 0.05, ***p < 0.001; n.s., not significant, by two-tailed unpaired Student’s t test.
(F) Treatment schedule for administration of MEKi, G9ai + HDACi, MEKi + G9ai + HDACi combination (TripleTx), or vehicle (control) via intraperitoneal injection once per day to Kras;p53 mutant mice. Animals undergoing monotherapy also received placebo (vehicle) so that all arms of the trial received equal volume injections.
(G) Waterfall plot of individual pancreatic tumor volume dynamics after 14 and 42 days of treatment. Note that a fraction of animals in the MEKi and in the G9ai + HDAC3i treatment arms presented with morbidity, necessitating euthanasia.
(H) Representative MRI scan to analyze tumor volume in Kras;p53 mutant mice treated with G9A, HDAC3, and MEK inhibitors. Scale bars, 1 cm.
(I) Kaplan-Meier survival curves of Kras;p53 mutant mice from enrollment time in control (vehicle) (n = 8, median survival = 22 days), G9ai + HDAC3i (n = 9, median survival = 26 days), MEKi (n = 8 median survival = 51 days), and TripleTx (n = 8, median survival = 73 days) treatment groups. *p < 0.05, ***p < 0.001; n.s., not significant, by log rank test for significance.
(J) Tumor volume quantification of patient-derived PDAC xenografts in mice (n = 8 mice, for each treatment group). Mice undergoing monotherapy also received vehicle. *p < 0.033, **p < 0.002, ***p < 0.0001 by two-way ANOVA with Tukey’s testing for multiple comparisons. Data are represented as mean ± SEM.
See also Figure S7.
The therapeutic efficacy on PDAC in vivo of TripleTx was tested in the Kras;p53 mouse model (Bardeesy et al., 2006). Treatment was initiated in tumors of ~150 mm3 in size as ascertained by MRI and then biweekly tumor growth and survival was monitored (Figures 7F and S7B-S7E). Four treatment arms were used: (1) vehicle control, (2) G9a/HDAC3 inhibition, (3) trametinib, and (4) TripleTx. At 2 weeks, G9a/HDAC3 inhibition modestly attenuated tumor growth compared with the vehicle control, whereas trametinib alone and triple therapy halted tumor growth or caused tumor regression (Figures 7G and 7H). By 6 weeks, all of the mice in the control and G9a/HDAC3 inhibition treatment arms were deceased (Figures 7G-7I). Tumors in the trametinib alone treatment arm showed significant growth by 6 weeks, indicating drug resistance emergence (Figures 7G-7I). In contrast, tumors in the TripleTx regimen were smaller than when treatment was initiated (Figures 7G and 7H). Consistent with this, TripleTx nearly tripled lifespan relative to the control group, with reduced tumor burden even at death (Figure 7I). Thus, combining drugs that inhibit G9a/GLP, HDAC3, and MEK1/2 results in a sustained potent antitumor response in an aggressive model of PDAC in mice.
To investigate the efficacy of the triple therapy in human pancreatic cancer, PDX studies using two independent primary PDAC patient samples were performed (Figures 7J and S7F). Once the xenograft tumors reached a volume of 200 mm3, treatment with the same four arms as above commenced and growth monitored until humane euthanasia was required. G9a/HDAC3 inhibition was not therapeutically effective with either PDX sample (Figures 7J and S7F). With trametinib treatment, tumors are initially sensitive but over time became resistant (Figures 7J and S7F). In contrast, TripleTx significantly slowed tumor progression for the full duration of the treatment protocol, well after the other treatment conditions had failed (Figures 7J and S7F). Taken together, these data suggest that inhibition of G9a and HDAC3 with small-molecule drugs renders PDAC tumors vulnerable to MEK inhibition, potentially due to blockade of an SETD5-orchestrated epigenetic resistance program.
DISCUSSION
Here, we identify SETD5 as a chromatin-based master regulator of adaptive targeted therapy resistance in pancreatic cancer (Figure S7H). SETD5 belongs to the SET domain family of proteins, a family that contains many bona fide histone lysine methyltransferases (Husmann and Gozani, 2019). However, there are also many SET proteins with divergent catalytic activities (e.g., histidine methylation [Wilkinson et al., 2019]) or several that are not active enzymes (Husmann and Gozani, 2019). Here, we investigated SETD5 methylation activity in vitro on histone and nucleosomal substrates and failed to detect any intrinsic activity (Figures 3A-3C and S3B-S3F). These findings are consistent with previous studies in which SETD5 deletion in cells or in mice did not cause bulk histone lysine methylation changes (Deliu et al., 2018; Mas et al., 2016; Osipovich et al., 2016). In contrast, Sessa et al. (2019) recently classified SETD5 as a robust H3K36 trimethylase in multiple contexts, and concluded that SETD5, not SETD2, is the main physiologic H3K36me3-generating enzyme in neuronal stem cells (NSCs) (see Figure 8E in Sessa et al., 2019). In our study, we failed to reproduce any of the reported key results on H3K36me3 from (Sessa et al., 2019), whereas our positive controls (i.e., SETD2) behaved as expected (Figures 3B and S3B-S3F). For example, SETD5 loss in NSCs did not alter H3K36 methylation (Figure S5E); in contrast, SETD2 knockdown did (Figure S5E). Thus, the preponderance of evidence does not support a role for SETD5 in the direct regulation of H3K36 methylation—in cancer or in an intellectual disability/neuronal development context.
We also found that while HDAC3 normally deacetylates a broad spectrum of histone acetyl-lysine substrates, when partnered with SETD5, HDAC3 is converted from a relatively promiscuous enzyme into a selective one (Figure 5F). This suggests a model in which the SETD5-HDAC3-G9a co-repressor complex couples selective deacetylation of H3K9ac with methylation of this residue at target genes, such as Pdk4, the repression of which promotes tumor drug resistance. The mechanism proposed here of an H3K9 acetyl-methyl switch for gene expression regulation is established for other silencing activities, such as the CtBP co-repressor complex (Shi et al., 2003). HDAC3 also has activity on H3K27ac-nucleosomes, suggesting that the SETD5 complex may also regulate drug resistance programming by targeting chromatin modifications at enhancer regions.
PDAC is virtually always driven by oncogenic mutant KRAS. Thus, clinically actionable strategies that can restore the therapeutic efficacy of downstream KRAS pathway inhibitors have the potential to have considerable impact upon the treatment of PDAC (Sun et al., 2017). In our study we used selective inhibitors of G9a and HDAC3 to mirror SETD5 depletion in sustaining the therapeutic benefit of MEKi in different PDAC models. While less selective HDAC3 inhibitors are in various stages of clinical trials for a variety of indications, at present, an HDAC3-specific inhibitor, such as RGFP966 has not been evaluated for safety in patients. Similarly, tolerance for G9a/GLP inhibitors are yet to be tested in patients. In our pre-clinical experiments, the combination of RGFP966 and UNC0642 did not have any adverse effects, with no toxicity in mice, and indeed in the PDX model the treatment largely alleviated the onset of cachexia, an important co-morbidity that contributes to PDAC mortality (Figure S7G). These findings suggest that pharmacologic blockage of SETD5-associated activities has the potential of being translatable into a clinical setting. In summary, our study identifies a chromatin-based mechanism mediated by SETD5 in the establishment of PDAC resistance to FDA-approved medicines that target the MAPK pathway.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pawel K. Mazur (pkmazur@mdanderson.org).
Materials Availability
Plasmids and antibodies generated in this study will be available upon request.
Data and Code Availability
The accession number for the RNA-seq datasets reported in this paper is paper is NCBI GEO: GSE142046. This study did not generate any unpublished code, software, or algorithm.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Ptf1a+/Cre, Pdx1Flp, Kras+/LSL-G12D, Kras+FSF-G12D, p53LoxP/LoxP, p53Frt/Frt, Rosa26FSF-CreER mice have been described before (Hingorani et al., 2003; Jonkers et al., 2001; Kawaguchi et al., 2002; Lee et al., 2012) (Schonhuber et al., 2014). Conditional Setd5LoxP/LoxP gene (NCBI Reference Sequence: NM_144877.1) knockout mice were generated in this study using a targeting vector obtained from KOMP repository (PGS00019_A_B10) (Skarnes et al., 2011). The targeting vector includes the Neo cassette flanked by Frt sites and exons 3 to 6 sequence were flanked by LoxP sites. The linearized vector was subsequently delivered to ES cells (C57BL/6) via electroporation, followed by drug selection, PCR screening, and Southern blot confirmation. Correctly targeted ES clones were selected for blastocyst microinjection, followed by founder mice production. Founders were confirmed as germline-transmitted via crossbreeding with wild-type animals. In conjunction with germ line transmission of the mutant allele the self-excising Neo cassette was deleted. Mice were in a mixed C57BL/6;129/Sv background, and we systematically used littermates as controls in all the experiments. Immunocompromised NSG mice (NOD.SCID-IL2Rg−/−) were utilized for transplantation studies. All experiments were performed on balanced cohorts of male and female mice as our initial data did not indicate significant differences in disease progression or response to treatment between females or males. All animals were numbered and experiments were conducted in a blinded fashion. After data collection, genotypes were revealed and animals assigned to groups for analysis. For treatment experiments mice were randomized. None of the mice with the appropriate genotype were excluded from this study or used in any other experiments. Mice had not undergone prior treatment or procedures. All mice were fed a standard chow diet ad libitum and housed in pathogen-free facility with standard controlled temperature, humidity, and light-dark cycle (12 h) conditions with no more than 5 mice per cage under the supervision of veterinarians, in an AALAC-accredited animal facility at the University of Texas M.D. Anderson Cancer Center. All animal procedures were reviewed and approved by the MDACC Institutional Animal Care and Use Committee (IACUC 00001636, PI: Mazur).
Cell Lines and Primary Cell Cultures
293T (female, embryonic kidney) cells were grown in DMEM medium supplemented with 10% fetal calf serum. MiaPaCa2 (male, 65 years old, pancreatic cancer), PSN1 (male, age not reported, pancreatic cancer), Panc1 (male, 56 years old, pancreatic cancer) and CaPan1 (male, 40 years old, pancreatic cancer), YAPC (male, 43 years old, pancreatic cancer), KP4 (male, 5-0 years old, pancreatic cancer), DANG (female, 68 years old, pancreatic cancer) cells were cultured in DMEM supplemented with 10% fetal bovine serum, glutamine, and 100 U/mL penicillin/streptomycin. Neural Stem Cells (NSCs) were derived from telencephalic cortex of embryos at E14.5. Embryonic cortices were dissociated, fragmented in Hank’s Balanced Salt Solution with 1% Penicillin/Streptomycin and digested with papain (10 U/ml) and cysteine (1 mM) in HBSS with 0.5 mM EDTA. Primary cultures of mouse embryonic hippocampal neurons were prepared from E17.5 C57BL/6 wildtype mice according to the methods as described in (Sessa et al., 2019). All cells were cultured at 37°C in a humidified incubator with 5% CO2. Cell lines were authenticated by short tandem repeat profiling and tested negative for mycoplasma (PromoKine).
Patient-Derived Cancer Xenografts and Mouse Allografts
Surgically resected tumor specimens were obtained from patients with histologically confirmed pancreatic cancer blinded for age and gender. All surgically resected tumors were collected after written patient consent and in accordance with the institutional review board-approved protocols of the University of Texas M.D. Anderson Cancer Center (PA19-0435, PI: Mazur). Patient-derived xenograft tumors were generated by transplanting small tumor fragments isolated directly from surgical specimens subcutaneously into mice as we established previously (Kim et al., 2009). In each case we first propagated the sample in NSG mice. For reconstitution assays, collected PDX tumors were minced using a razor blade and digested in collagenase digestion buffer at 37°C for 1 hour. Cells were passed through 100 μm and 40 μm cell strainers and centrifuged for 1200 rpm for 8 min. Cells were incubated in RBC lysis buffer for 2 min and then resuspended in 6 mL of media and spun through 0.5 mL of serum layered on the bottom of the tube to remove cellular debris. Contaminating human or mouse hematopoietic and endothelial cells (CD45, Ter119, CD31) are depleted using biotin conjugated anti-mouse CD45, CD31 and Ter119 antibodies and separated on a MACS LS column using anti biotin microbeads. Next, the cells were collected, mixed with matrigel (1:1) and transplanted to the flanks of NSG mice. When tumors became palpable, they were calipered every 3 days to monitor growth kinetics. Tumor volume was calculated using the formula: Volume = (width)2 x length / 2 where length represents the largest tumor diameter and width represents the perpendicular tumor diameter.
METHOD DETAILS
Pancreatic Cancer Mouse Models
For pancreatic cancer development studies we used Ptf1a+/Cre; Kras+/LSL-G12D; p53LoxP/LoxP (Kras;p53) and Ptf1a+/Cre; Kras+/LSL-G12D; p53LoxP/LoxP; Setd5loxP/loxP (Kras;p53;Setd5) mice, which develop aggressive disease. Mice were followed for signs of disease progression. At the end of the experiment, tumors were processed for biochemical, histological and immunohistochemical analysis. Histopathological analysis was conducted on de-identified slides based on the classification consensus (Bailey et al., 2016).
For the two-stage tumorigenesis studies sequential genetic manipulation of the murine pancreas was accomplished through a combined Flp/Frt and Cre/LoxP system as previously reported (Schonhuber et al., 2014). Briefly, PdxFlp; KrasFSF-G12D; p53Frt/Frt; Rosa26FSF-CreER; Setd5LoxP/LoxP mutant mice develop pancreatic cancer with high penetrance. Mice were monitored by Magnetic resonance imaging (MRI), as described below for tumor development. At the age of 8 weeks mice received Tamoxifen (1 mg in 100 μl corn oil) per intraperitoneal injections on 3 consecutive days when tumor volumes had reached approximately 150 mm3. Control animals underwent the same procedure but received vehicle only treatment. Successful recombination of Setd5LoxP/LoxP was confirmed by PCR on DNA isolated from tumor biopsies and loss of SETD5 expression was verified by immunoblotting of whole cell lysate of tumor biopsies. For therapy studies mice were treated as indicated with Trametinib (0.3 mg/kg daily, IP), Selumetinib (50 mg/kg daily, IP), SCH772984 (25 mg/kg daily, IP), UNC0642 (G9a/GLP inhibitor, 5 mg/kg daily, IP), RGFP966 (HDAC3 inhibitor, 10 mg/kg daily, IP) or vehicle 10% (2-hydroxypropyl)-β-cyclodextrin. Animals undergoing monotherapy also received placebo (vehicle).
Magnetic Resonance imaging
Magnetic Resonance Imaging (MRI) experiments were performed on PdxFlp; KrasFSF-G12D; p53Frt/Frt; Rosa26FSF-CreER; Setd5LoxP/LoxP and Kras;p53 mutant mice at indicated age. Before imaging, mice were anesthetized by continuous gaseous infusion of 2% isoflurane for at least 10 min using a veterinary anesthesia system. During imaging, the dose was kept at 2% isoflurane, animal temperature was maintained and continuously monitored, respiratory and ECG monitoring were performed using an MRI-compatible physiological monitoring system and eyes were protected with an eye ointment. MRI was performed using the Biospec USR70/30, a small animal experimental MR imaging system based on an actively-shielded 7 T magnet with a 30 cm bore and cryo-refrigeration. The system is equipped with 6 cm inner-diameter gradients that deliver a maximum gradient field of 950 mT m−1. A 3.5 cm inner-diameter linear birdcage coil transmits and receives the MR signal. For image acquisition, T2-weighted, respiratory gated, multi-slice imaging will be performed with respiration held to under 25 breaths per minute to minimize motion artefacts in the abdomen. The rapid acquisition with relaxation enhancement (RARE) T2-weighted pulse sequence was modified to include an effective Te (time of echo) of 38 ms, echo train length 9.5 ms, and number of averages equal to 4 in both the coronal and axial planes with a total TR (time repetition) of 2000 ms. A three-orientation (axial, sagittal, and coronal) scout image using a fast, low-angle single shot sequence was obtained to localize the mouse pancreas. Between 18 and 20 coronal and axial slices were acquired per mouse with a slice thickness of 0.7 mm and slice spacing of 1 mm to cover the entire pancreas. In plane, pixel sizes of 0.156 mm × 0.156 mm with a matrix size of 256 × 192 and field of view (FOV) of 40 mm × 30 mm was chosen to minimize in plane partial volume effects, maintain a FOV sufficient to cover the abdomen, while also providing sufficient throughput for the experiment. MR images were analyzed using an open source Horos processing software. Pancreas tumor burden was measured by tracing the outer border of the region of suspected lesions on each slice after image intensities were normalized. Analysis was conducted on de-identified images. Tumor volume (V) was assessed, using three-dimensional volumetric measurements according to the modified Simpson rule. In all contiguous transverse images, the area of tumor (A) in each slice was multiplied by the slice profile (0.7 mm slice thickness plus 1 mm intersection gap), and total tumor volume was automatically calculated by summation of the adjacent volume according to the formula:
where Ts is the thickness of each slice, i is the individual slice number and n is the total number of slices.
Histology and Immunohistochemistry
Tissue specimens were fixed in 4% buffered formalin for 24 hours and stored in 70% ethanol until paraffin embedding. 3 μm sections were stained with hematoxylin and eosin (HE) or used for immunohistochemical studies. Human tissue sections were collected in accordance with the institutional review board-approved protocols of the University of Texas M.D. Anderson Cancer Center (PA19-0435, PI: Mazur). Immunohistochemistry (IHC) was performed on formalin-fixed, paraffin embedded mouse and human tissue sections using a biotin-avidin method as described before (Mazur et al., 2014). The following antibodies were used (at the indicated dilutions): cleaved caspase 3 (1:100), Ki67 (1:1,000) and SETD5 (1:100). Sections were developed with DAB and counterstained with hematoxylin. Pictures were taken using a PreciPoint M8 microscope equipped with the PointView software. Analysis of the tumor area and IHC analysis was done using ImageJ software. Quantification of SETD5 IHC chromogen intensity was performed by measuring the reciprocal intensity of the chromogen stain as previously described (Nguyen, 2013). Briefly, standard RGB color images acquired from bright field microscopy have a maximum intensity of value 250 (represented by white, unstained areas) as measured by the standard intensity function in the open source ImageJ Fiji software. We subtracted the intensity of a stained tissue sample from 250, thereby deriving a reciprocal intensity that is directly proportional to the amount of chromogen present.
Meta-Analysis of Gene Expression
Meta-analysis of public PDAC data sets. We downloaded raw data for gene expression studies (7 pancreatic cancer) from the NCBI GEO and EBI ArrayExpress. After re-annotating the probes, each data set was normalized separately using gcRMA. We applied two meta-analysis approaches to the normalized data. The meta-analysis approach was previously described (Khatri et al., 2013). Briefly, the first approach combines effect sizes from each data set into a meta-effect size to estimate the amount of change in expression across all data sets. For each gene in each data set, an effect size was computed using Hedges’ adjusted g. If multiple probes mapped to a gene, the effect size for each gene was summarized using the fixed effect inverse-variance model. We combined study-specific effect sizes to obtain the pooled effect size and its standard error using the random effects inverse-variance technique. We computed z-statistics as a ratio of the pooled effect size to its standard error for each gene and compared the result to a standard normal distribution to obtain nominal p values that were corrected for multiple hypotheses testing using false discovery rate (FDR). We used a second non-parametric meta-analysis that combines p values from individual experiments to identify genes with a large effect size in all data sets. Briefly, we calculated a t-statistic for each gene in each study. After computing one-tail p values for each gene, they were corrected for multiple hypotheses using FDR. Next, we used Fisher’s sum of logs method, which sums the logarithm of corrected p values across all data sets for each gene and compares the sum against a chi-square distribution with 2k degrees of freedom, where k is the number of data sets used in the analysis.
Transfection and Viral Transduction
Transient expression was performed using TransIT-293 following the manufacturer’s protocol. For CRISPR/Cas9 knockouts, virus particles were produced by co-transfection of 293T cells with the pLentiCRISPR v2 (with puromycin selection) construct expressing indicated sgRNAs, pMD2.G and psPAX2 in a ratio of 5:2:3 by mass. 48 hours after transfection, target cells were transduced with 0.45 μm filtered viral supernatant and 8 μg/mL polybrene. Cells were selected 24 hours after media replacement with 2 μg/mL puromycin or 250 μg/mL hygromycin B. For SETD5 reconstitution, virus particles were produced by co-transfection of 293T cells with the pLenti CMV Hygro DEST (w117-1) expressing human SETD5, pMD2.G and psPAX2 in a ratio of 5:2:3 by mass. 48 hours after transfection, target cells were transduced with 0.45 μm filtered viral supernatant and 8 μg/mL polybrene. Cells were selected 24 hours after media replacement with 250 μg/mL hygromycin B, after one week selection, the cells expressing human SETD5 (with sgRNA tolerance synonymous mutation) were transduced with sgControl and sgSETD5 virus (sgSETD5-2), Cells were selected 24 hours after media replacement with 2 μg/mL puromycin. After 5 days selection, cells were harvested for western blot, RNA purification or ChIP.
Plasmids
Full length human SETD5 (NP_001073986.1), SETD5ΔSET (aa 511-1442), SETD5ΔC (aa 1-520) were cloned into pQCXIH vector with V5 tag at N-terminal with PreScission Protease cutting site and FLAG tag at C-terminal for sequential immunoprecipitation. Stable cells were generated using pLenti CMV Hygro DEST(w117-1); pLentiCRISPRv2 was used for CRISPR/Cas9 knockouts in cell lines. For bacterial expression, human SETD5ΔC, human SETD5 (aa 1-415), murine SETD5 (aa 1-423) (NP_766593), SETD2-SET (aa 1418-1714) (NP_054878.5), G9a (aa 913-1193) (NP_006700.3), NSD2 (aa 942-1240) (NP_579877.1) and MLL4 (aa 2551-2715) (NP_055542.1) were cloned into pGEX-6P-1, ASH2L (NP_001098684), RBBP5 (NP_005048), WDR5 (NP_060058.1), DPY30 (NP_115963) were cloned into pet28a; for Sf9 insect cell expression, human Flag-SETD5, PRC2 complex (Flag-EZH2 (NP_001190176.1), EED (NP_003788.2), SUZ12 (NP_056170), RbAp46 (NP_002884.1) and RbAp48 (NP_005601.2)), Flag-HDAC3 (NP_003874.2) and GST-NCoR1-DAD domain (aa 397-503) (NP_006302) were cloned into pFastbac1.
Pooled shRNA Screen
We have generated lentiviral shRNA sub-library (Bassik et al., 2009, 2013) containing 25 independent shRNAs directed against one of 95 known and putative human methyltransferase genes, including the vast majority of known protein lysine methyltransferase (KMTs) present in the human genome (shRNA targeting sequences are listed in Table S1). In addition, the library contains 1,000 negative control shRNAs that have the same overall base composition as the other shRNAs, but do not match the sequence of any human transcript. MiaPaCa2 cells were transduced with the lentivirus pool containing shRNAs as described previously (Bassik et al., 2009, 2013). Infected cells were expanded and split into two flasks. In one flask, cells were grown in the presence of 10 nM Trametinib (MEKi) for 12 days, while in the other flask, cells were grown in vehicle (DSMO). Untreated cells were diluted to a density of 500,000 cells/ml each day. MEKi-treated cells were diluted to a density of 500,000 cells/ml as needed. After the cell culture period, untreated and MEKi-treated cells were collected. Genomic DNA was isolated, and shRNA encoding-constructs were counted by deep sequencing as described previously (Bassik et al., 2009, 2013) and frequencies of shRNA-encoding cassettes were determined by next-generation sequencing (Illumina HiSeq). The MEKi resistance screen was carried out in two independent replicates. The MEKi resistance ρ conferred by an individual shRNA was calculated as described previously (Bassik et al., 2013). The set of ρ values of all shRNAs for a given gene were compared to the set of ρ values for the negative control shRNAs, and the significance by Mann-Whitney U test for enrichment (resistance) or depletion (sensitivity) was calculated (see also Table S1).
Immunoblot Analysis and Immunoprecipitation
For western blot analysis, cells were lysed in RIPA buffer with 1 mM PMSF and protease inhibitor cocktail. Protein concentration was determined using the Pierce Coomassie Plus Assay. Protein samples were resolved by SDS-PAGE and transferred to a PVDF membrane (0.45 μm). The following antibodies were used (at the indicated dilutions): ERK1/2 (1:2,000), phospho-ERK1/2 (1:5,000), SETD5 (1:1,000, this paper), SETD2 (1:1,000), SHOC2 (1:1,000), G9a (1:1,000), GLP (1:1,000), HDAC3 (1:1,000), NCoR1 (1:1,000), TBL1 (1:1,000), TBL1XR1 (1:1,000), GPS2 (1:1,000), GST (1:5,000), Flag (1:1000), Actin (1:10,000), Tubulin (1:2,000), H3 (1:5,000), H3.3 (1:1,000), H4 (1:3000), H3K9ac (1:2,000), H3K36me1 (1:5,000), H4K5ac (1:2,000), H4K8ac (1:2,000), all the other histone modification antibodies (1:1,000). Secondary antibodies were used at 1:5,000 or 1:10,000 dilution. Protein bands were visualized using ECL detection reagent.
For immunoprecipitation, nuclear extracts were incubated with specific antibody overnight at 4°C, antibodies for immunoprecipitation were used at the indicated amount: 5 μg SETD5 (lab generated), 3 μg HDAC3 (Abcam), 5 μg G9a (Bethyl). Extracts were then incubated with protein A Sepharose beads for 3 hours at 4°C; For tandem-affinity purification (TAP), nuclear extracts were incubated with 50 μl Anti-V5 Agarose affinity gel for 5 hours, after washing, proteins were cleaved from beads using 5 μg PreScission Protease for 3 hours, cleaved proteins were incubated with 20 μl Anti-FLAG M2 affinity gel for another 5 hours and eluted using 0.25 mM 3XFLAG peptides for 1 hour at 4°C. Proteins were resolved by SDS–PAGE, transferred to PVDF membrane and analyzed by western blot.
Generation of SETD5 Antibody
Human SETD5 (aa 1-131, NP_001073986.1) was cloned into pGEX-6P-1 and expressed in BL21 E.coli. SETD5 protein fragment was purified using GST Glutathione Sepharose 4B resin and cleaved from the beads using PreScission Protease. The purified protein fragment was used to immunize rabbits using standard methods (performed by Genemed Synthesis, Inc.)
Expression and Purification of Recombinant Proteins
GST fusion proteins were expressed in BL21 E.coli by overnight culture at 20°C in LB medium (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl) supplemented with 0.1 mM IPTG, purified using Glutathione Sepharose 4B and eluted in 10 mM reduced glutathione or cut from beads using PreScission Protease. Insect expression was done according to manufacturer’s protocol Bac-to-Bac Baculovirus Expression System. Next, the cell lysates were incubated with Anti-FLAG M2 affinity gel for 5 hours, after washing, proteins were cleaved from beads using 5 μg PreScission Protease for 3 hours. for HDAC3/NCoR1-DAD purification, the complex was cleaved from beads using PreScission Protease for 3 hours, cleaved proteins were incubated with Glutathione Sepharose 4B for 2 hours and eluted in 10 mM reduced glutathione. The proteins purified were snap frozen and stored in −80°C or used in in vitro reaction assays directly. Protein concentrations were measured using Coomassie assay.
ProtoArray, Methylation and Deacetylation Assays
In vitro methylation assays were performed similar to as described in (Mazur et al., 2014) by combining 0.5-2 μg recombinant enzymes or 2 μg SETD5 complex and 1-2 μg substrates (bulk histones, recombinant H3, recombinant nucleosomes) in a methyltransferase buffer (50 mM Tris pH 8.0, 20 mM KCl, 5 mM MgCl2, and 10% glycerol) supplemented with 0.1 mM S-adenosyl-methionine (SAM) or 2 μCi of tritiated AdoMet. The reaction mixtures were incubated overnight at 30°C. Reactions were analyzed by SDS-PAGE, followed by autoradiography, Coomassie stain, western blot or mass spectrometry. In vitro methylation assays with SETD2SET, human SETD5 (aa 1-415) and murine SETD5 (aa 1-423) were performed using the reaction buffer (50 mM Tris pH 8.8, 5 mM MgCl2, 4 mM DTT) supplemented with 50 μM S-adenosyl-methionine (SAM) or 2 μCi of tritiated AdoMet. The reactions using enzymes purified from insect cells and 293T cells were supplied with AMI-1 (40 μg/mL) to inhibit activity of protein arginine N-methyltransferases potentially interfering with the experiment. For in vitro deacetylation assay 0.1 μg recombinant HDAC3/NCoR1-DAD complex or 1 μg SETD5 complex and 1 μg of HeLa nucleosomes or 0.2 μg of synthesized nucleosomes were incubated in a histone deacetylase reaction buffer (50 mM Tris pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2) at 27°C for 5 hours. Reactions were analyzed by SDS-PAGE, followed by western blot.
Gel Filtration
Gel filtration chromatography was performed using Superose 6 Increase 10/300GL prepacked column. Nuclear extracts (1 mL) were loaded on the equilibrated column and eluted with buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA) at flow rate of 0.25 ml/min, collecting fractions, 0.4 mL each tube. Fractions were analyzed by SDS-PAGE, followed by western blot.
Mass Spectrometry
Recombinant nucleosomes were separated by SDS-PAGE (15%) and stained using InstantBlue Protein Stain without methanol. Histone H3 gel bands were cut into small pieces and subjected to chemical derivatization with propionic anhydride using the same protocol as previously described (Sidoli et al., 2016; Sidoli and Garcia, 2017). Peptides were separated by high pressure liquid chromatography (HPLC) using Dionex Ultimate 3000 LC-system and analyzed with an Orbitrap Elite mass spectrometer. Full MS spectra were acquired in the Orbitrap mass analyzer and MS/MS spectra were obtained by selection of top 20 ions followed by collision induced dissociation (CID) analysis of fragment ions in the ion trap. Methylation states of histones were manually inspected. Selected ion chromatograms for peptides spanning H3K9 were extracted using Xcalibur Qual Browser. The settings were as follows: Peptide H3 9-17(KSTGGKAPR), m/z 535.3037(me0), 542.3115(me1), 521.3062 (me2) and 528.3140(me3), 10p.p.m.
For SETD5 complex protein identification V5–SETD5-Flag tandem-affinity purified (TAP) material was separated by 10% SDS-PAGE and silver stained. Protein bands were excised from the gel and analyzed by mass spectrometry. Liquid chromatography-tandem mass spectrometry (LC–MS/MS) was performed using an Orbitrap Elite and data were analyzed using MaxQuant software.
Quantitative RT-PCR
For quantitative RT-PCR, RNA was extracted using TRIzol Reagent according to the manufacturer’s instructions, cDNA synthesis was obtained using the Superscript First-strand Synthesis kit. RT-qPCR was performed in triplicate with custom designed oligos using standard methods.
RNA-Sequencing and Data Analysis
Total RNA was extracted from KPCR cells (n = 3) (sgControl + Trametinib 0.2 μM, sgSETD5 + Trametinib 0.2 μM, or DMSO + Trametinib 0.2 μM, UNC0638 0.6 μM + RGFP966 0.6 μM + Trametinib 0.2 μM) using Trizol reagent. Total RNA was subjected to polyA selection using Dynabeads mRNA purification kit. 20 ng polyA RNA was utilized for library generation using SMARTer Stranded RNA-seq kit. The RNA-seq libraries were sequenced using Illumina HiSeq 3000 platform (pair end 150 bp analysis). RNA-seq data processing, low-quality and adapter-containing reads were trimmed using Trim-galore software package under paired-end mode, any reads shorter than 50 bp were removed. The remaining trimmed sequences were mapped to the reference genome (mm10) with hisat2 under default settings. We used htseq-count to count the mapped reads number on every mm10 Refseq transcript. Differential gene expression analysis was performed with DESeq2 software. Genes with p value ≤ 0.05, log2 fold change ≥ 0.5 were defined as up-regulated genes, and genes with p value ≤ 0.05 log2 fold change ≤ −0.5 were defined as down-regulated genes. Enrichment analysis was performed for KEGG pathways. Gene set enrichment analysis (GSEA) was performed using the Broad Institute of the Massachusetts Institute of Technology and Harvard University software. For both gene sets, we used the default parameters of the GSEA software package; the genes set permutation was used. In brief, the normalized enrichment score provides the degree to which a gene set is overrepresented at the top or bottom of a ranked list of genes. The false discovery rate q value (FDR) is the estimated probability that a gene set with a given NES represents a false positive finding, an FDR cutoff of 0.05 was used for all analysis.
Chromatin Immunoprecipitation (ChIP)
Cells were fixed with 1% formaldehyde for 10 min at room temperature before termination with 0.125 M glycine. Cells were then lysed in ChIP buffer (0.3% SDS, 300 mM NaCl, 1 mM EDTA, and 50 mM Tris-HCl pH 8.0), and cross-linked chromatin was sonicated to obtain DNA fragments around 250 bp. After sonication, dilute the 0.3% SDS to 0.1% SDS with ChIP dilution buffer (300 mM NaCl, 1 mM EDTA, and 50 mM Tris-HCl pH 8.0). Immunoprecipitations were performed using the diluted samples that were incubated at 4°C for 3 hours with following antibodies: SETD5 (3 μg, this paper), H3K9ac (2 μg), H3K9me2 (3 μg). Next, 10-15 μl protein A/G beads were added and incubated at 4°C for 5 hours. Then beads were treated with binding buffer (0.1% SDS, 300 mM NaCl, 10 mM EDTA, and 50 mM Tris-HCl pH 8.0) 4 times, wash buffer (1 mM EDTA, 50 mM Tris-HCl pH 8.0) twice, LiCl buffer (20 mM Tris, pH 7.4, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% Na-deoxycholate) once and TE buffer (10 mM Tris pH 8.0, 1 mM EDTA) once. DNA was eluted from beads using elution buffer (50 mM Tris pH 8.0, 10 mM EDTA, 1% SDS) and de-crosslinked at 65°C for 4 hours. DNA was recovered by PCR Purification Kit. RT-qPCR analyses were performed on immunoprecipitated DNA using specific primers. The results were presented as relative fold enrichment over the input.
GSH Assay
Reduced cellular glutathione (GSH) was determined enzymatically using a Glutathione assay kit according to manufacturer’s protocol. Briefly, 1 × 107 KPCR cells were washed with PBS, incubated with 500 μL MES reagent and sonicated for 1 min and then centrifuged at 13,500 rpm for 15 min at room temperature. Next, the samples were deproteinated by adding 500 μL of 10% metaphosphoric acid reagent and centrifuged at 13,500 rpm for 3 min at room temperature. Next, the supernatant was treated with 50 μL TEAM reagent. Total glutathione concentration was determined kinetically by measuring the formation of 5-thio-2-nitrobenzoic acid from 5, 5′-dithionitrobenzoic acid in the presence of NADPH and glutathione reductase fluorometrically at 405 nm.
Cell Assays
To analyze cell proliferation cancer cells were seeded at 2 × 105 cells/mL in triplicate in 6-well plates. Cell counts were acquired by Countess II FL automated cell counter at indicated days. After each counting, the cells were maintained at a density between 2-4 × 105 cells/mL. Trypan blue was used to stain non-viable cells. Cell numbers were expressed relative to 1 × 105 cells/mL. For analysis of cell response to MEKi we used the IncuCyte live cell imaging system. Cells were seeded at a concentration of 4,000 cells per well in 96-well plate and allowed to attach overnight. Next, MEKi at specified concentration was added and cells were analyzed every 4 hours for confluency.
To analyze SETD5 expression upon MEKi treatment mouse KPCN cells were treated with MEK1/2 inhibitors at low concentration of Binimetinib (10 nM), Pimasertib (10 nM), Selumetinib (10 nM) and Trametinib (5 nM) for the first week, then concentration was gradually increased to Binimetinib (0.6 μM), Pimasertib (1 μM), Selumetinib (0.5 μM) and Trametinib (0.2 μM) over the 4 weeks. After inhibitor treatment, total protein and RNA were isolated from the cells and analyzed by western blot and quantitative RT-qPCR.
siRNA Transfection
To perform SHOC2 depletion 5×105 KPCR and KPCN cells were transfected with 30 pmol siRNA oligo mix utilizing RNAimax transfection reagent for 48 hours according to manufacturer’s protocol. Following 48 hours post-transfection, cells were utilized to perform proliferation assay and western blot analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Please refer to the Figure Legends or the Experimental Details for description of sample size (n) and statistical details. All values for n are for individual mice or individual sample. Sample sizes were chosen based on previous experience with given experiments. Cell culture assays have been performed in triplicates and in two independent experiments, unless stated otherwise. Differences were analyzed by log-rank, two-tailed unpaired Student’s t test, Mann-Whitney U test, two-way ANOVA with Tukey’s testing for multiple comparisons using Prism 7 (GraphPad), p values ≤ 0.05 were considered significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Actin | Cell Signaling Technologies | Cat# 8457; RRID: AB_10950489 |
| Cleaved Caspase 3 | Cell Signaling Technologies | Cat# 9664; RRID: AB_2070042 |
| ERK1/2 | Cell Signaling Technologies | Cat# 4695: RRID: AB_390779 |
| Phospho-ERK1/2 | Cell Signaling Technologies | Cat# 4376; RRID: AB_331772 |
| SHOC2 | Cell Signaling Technologies | Cat# 53600; RRID: AB_2799440 |
| Tubulin | Millipore | Cat# 05-661; RRID: AB_309885 |
| Ki67 | BD Bioscience | Cat# 550609; RRID: AB_393778 |
| anti-Mouse HRP | Cell Signaling Technologies | Cat# 7076; RRID: AB_330924 |
| anti-Rabbit HRP | Cell Signaling Technologies | Cat# 7074; RRID: AB_2099233 |
| Peroxidase-conjugated Mouse Anti-Rabbit IgG, light chain specific | Jackson ImmunoResearch | Cat# 211-032-171; RRID: AB_2339149 |
| AffiniPure Goat Anti-Mouse IgG, light chain specific | Jackson ImmunoResearch | Cat# 115-005-174; RRID: AB_2338460 |
| Peroxidase-conjugated streptavidin | Jackson ImmunoResearch | Cat# 016-030-084; RRID: AB_2337238 |
| CD45-Biotin | eBiosciences | Cat# 13-0451-81; RRID: AB_466445 |
| CD31-Biotin | eBiosciences | Cat# 13-0319-80; RRID: AB_466422 |
| Ter119-Biotin | eBiosciences | Cat# 13-5921-81; RRID: AB_466796 |
| Biotin Micro Beads | Miltenyi Biotec | Cat# 130-042-401 |
| SETD5 | This paper | N/A |
| Histone H3 | EpiCypher | Cat# 13-0001 |
| Histone H3.3 | RevMab | Cat# 31-1058-00; RRID: AB_2716425 |
| Histone H4 | EMD Millipore Corporation | Cat# 05-858; RRID: AB_390138 |
| H3K4me1 | Cell Signaling Technologies | Cat# 5326; RRID: AB_10695148 |
| H3K4me2 | Cell Signaling Technologies | Cat# 9725; RRID: AB_10205451 |
| H3K4me3 | Epicypher | Cat# 13-0028 |
| H3K9me1 | Cell Signaling Technologies | Cat# 14186; RRID: AB_2798416 |
| H3K9me2 | Thermo Fisher Scientific | Cat# 710815; RRID: AB_2608303 |
| H3K27me1 | Epicypher | Cat# 13-0015 |
| H3K27me2 | Cell Signaling Technologies | Cat# 9728; RRID: AB_1281338 |
| H3K27me3 | Cell Signaling Technologies | Cat# 9733; RRID: AB_2616029 |
| H3K36me1 | Abclonal | Cat# A2364; RRID: AB_2764324 |
| H3K36me2 | Thermo Fisher Scientific | Cat# 701767; RRID: AB_2633024 |
| H3K36me3 | Thermo Fisher Scientific | Cat# MA5-24687; RRID: AB_2661912 |
| H3K4ac | Abclonal | Cat# A16078; RRID: AB_2763519 |
| H3K9ac | Epicypher | Cat# 13-0033 |
| H3K14ac | Abclonal | Cat# A7254; RRID: AB_2737401 |
| H3K18ac | Abclonal | Cat# A7257; RRID: AB_2767801 |
| H3K27ac | Cell Signaling Technologies | Cat# 8173; RRID: AB_10949503 |
| H3K36ac | Active motif | Cat# 39379; RRID: AB_2614977 |
| H4K5ac | Abcam | Cat# ab51997; RRID: AB_2264109 |
| H4K8ac | Abcam | Cat# ab45166; RRID: AB_732937 |
| H4K12ac | Abcam | Cat# ab46983; RRID: AB_873859 |
| H4K16ac | Abclonal | Cat# A5280; RRID: AB_2766099 |
| H3K9me2 | Thermo Fisher Scientific | Cat# 710815; RRID: AB_2608303 |
| TBL1XR1 | Abclonal | Cat# A7834; RRID: AB_2772539 |
| TBL1XR1 | Bethyl | Cat# A300-408A; RRID: AB_420967 |
| G9a | Santa Cruz Biotechnology | Cat# sc-515726 |
| G9a | Bethyl | Cat# A300-933A; RRID: AB_2097663 |
| GLP | Bethyl | Cat# A301-642A; RRID: AB_1210961 |
| HDAC3 | Millipore | Cat# 17-10238; RRID: AB_11205568 |
| HDAC3 | Abcam | Cat# ab32369; RRID: AB_732780 |
| NCoR1 | Bethyl | Cat# A301-145A; RRID: AB_873085 |
| TBL1 | Abcam | Cat# ab24548; RRID: AB_2199904 |
| TBL1 | Santa Cruz Biotechnology | Cat# sc-137006; RRID: AB_2199796 |
| GPS2 | Abcam | Cat# ab153986 |
| GST | (Shi et al., 2006) | N/A |
| FLAG | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Anti-FLAG M2 Affinity Gel | Sigma-Aldrich | Cat# A2220; RRID: AB_10063035 |
| Anti-V5 Agarose Affinity Gel | Sigma-Aldrich | Cat# A7345; RRID: AB_10062721 |
| Bacterial and Virus Strains | ||
| DH5 | Thermo Fisher Scientific | Cat# K4520-1 |
| BL21 | Thermo Fisher Scientific | Cat# C6070-03 |
| Sf9 | Thermo Fisher Scientific | Cat# 12659017 |
| Biological Sample | ||
| Human PDAC Tissue Array | MD Anderson Pathology | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RPMI 1640 Medium | Corning | Cat# MT10017CV |
| DMEM Medium | Corning | Cat# MT10040CV |
| Fetal bovine serum | Thermo Fisher Scientific | Cat# 10500056 |
| PBS | Corning | Cat# MT21031CV |
| HBSS | Thermo Fisher Scientific | Cat# 14025076 |
| Cysteine | Sigma-Aldrich | Cat# 30089 |
| Trypsin-EDTA 0.25% | Corning | Cat# MT25053CI |
| Geneticin | Thermo Fisher Scientific | Cat# 10131027 |
| Blasticidin S | Thermo Fisher Scientific | Cat# R21001 |
| Puromycin | Thermo Fisher Scientific | Cat# A1113802 |
| Hygromycin B | Corning | Cat# 30240CR |
| G418 Sulfate | Corning | Cat# MT30234CI |
| MACS separation columns | Miltenyi Biotech | Cat# 130-042-401 |
| Complete Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 4693159001 |
| Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | Cat# 78420 |
| Hydrogen Peroxide | Thermo Fisher Scientific | Cat# H325-500 |
| Sequencing Grade Modified Trypsin | Promega | Cat# V5113 |
| Glu-C | Promega | Cat# V1651 |
| TRIzol Reagent | Invitrogen | Cat# 15596018 |
| Forane (Isoflurane) | AbbVie | Cat# B506 |
| Papain | Worthington | Cat# LS003119 |
| Bovine Serum Albumin (BSA) | Thermo Fisher Scientific | Cat# BP9703100 |
| L-Reduced glutathione | Sigma-Aldrich | Cat# G4251-25G |
| S-adenosyl-methionine | New England Biolabs | Cat# B9003S |
| S-Adenosyl-l-[methyl-3H] methionine | American Radiolabeled Chemicals | Cat# ART0288 |
| AMI-1 | Sigma-Aldrich | Cat# 539209 |
| TransIT-293 | Mirus Bio | Cat# MIR-2706 |
| NP-40 | Sigma-Aldrich | Cat# I8896 |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat# P7626 |
| cOmplete, EDTA-free protease inhibitor | Sigma-Aldrich | Cat# 5056489001 |
| Glucose | Sigma-Aldrich | Cat# G8270 |
| Poly-L-lysine(PLL) | Sigma-Aldrich | Cat# P2636 |
| Neurobasal™ Medium | Thermo Fisher Scientific | Cat# 21103049 |
| B-27™ Supplement (50X) | Thermo Fisher Scientific | Cat# 17504044 |
| SUPERase In RNase Inhibitor | Thermo Fisher Scientific | Cat# AM2694 |
| RNasin Ribonuclease Inhibitors | Promega | Cat# N2511 |
| UNC0638 | Sigma-Aldrich | Cat# U4885 |
| UNC0642 | Selleckchem | Cat# S7230 |
| RGFP966 | Selleckchem | Cat# S7229 |
| Pimasertib (AS-703026) | Selleckchem | Cat# S1475 |
| Binimetinib | Selleckchem | Cat# S7007 |
| Selumetinib | Selleckchem | Cat# S1008 |
| SCH772984 | Selleckchem | Cat# S7101 |
| Trametinib (GSK1120212) | Selleckchem | Cat# S2673 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| HeLa Mononucleosomes | Epicypher | Cat# 16-0002 |
| Recombinant nucleosome | Epicypher | Cat# 16-0006 |
| Recombinant Polynucleosomes (H3.3) | Active motif | Cat# 31468 |
| 3X FLAG Peptide | Sigma-Aldrich | Cat# F4799 |
| CTH (Histones from calf thymus) | Sigma-Aldrich | Cat# 9064-47-5 |
| H3K4ac dNuc | Epicypher | Cat# 16-0342 |
| H3K9ac dNuc | Epicypher | Cat# 16-0314 |
| H3K14ac dNuc | Epicypher | Cat# 16-0343 |
| H3K18ac dNuc | Epicypher | Cat# 16-0372 |
| H3K23ac dNuc | Epicypher | Cat# 16-0364 |
| H3K27ac dNuc | Epicypher | Cat# 16-0365 |
| H3K36ac dNuc | Epicypher | Cat# 16-0378 |
| H4K5ac dNuc | Epicypher | Cat# 16-0352 |
| H4K8ac dNuc | Epicypher | Cat# 16-0353 |
| H4K12ac dNuc | Epicypher | Cat# 16-0312 |
| H4K16ac dNuc | Epicypher | Cat# 16-0354 |
| DMSO | Sigma-Aldrich | Cat# D5879 |
| (2-Hydroxypropyl)-β-cyclodextrin | Sigma-Aldrich | Cat# C0926 |
| Corn oil | Sigma-Aldrich | Cat# C8267 |
| PreScission Protease | This paper | N/A |
| Metaphosphoric acid | Sigma-Aldrich | Cat# 239275 |
| 16% Formaldehyde (w/v) | Sigma-Aldrich | Cat# F8775 |
| N-acetyl-L-Cysteine (NAC) | Sigma-Aldrich | Cat#A7250 |
| PVDF membrane (0.2 μm) | BioRad | Cat# 1620177 |
| PVDF membrane (0.45 μm) | Millipore | Cat# IPVH00010 |
| Glutathione Sepharose 4B | Sigma-Aldrich | Cat# GE17-0756-01 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| ZymoPURE Plasmid Miniprep Kit | Zymo | Cat# D4211 |
| ZymoPURE II Plasmid Maxiprep Kit | Zymo | Cat# D4203 |
| DNA PCR Purification Kit | Qiagen | Cat# 28106 |
| DAB Substrate Kit | Abcam | Cat# ab64238 |
| Vectastain ABC kit | Vector Laboratories | Cat# PK-6100 |
| BCA Protein Assay Kit | Pierce | Cat# 23227 |
| ECL Substrate | Amersham | Cat# RPN2106 |
| PCR Mycoplasma Test Kit I/C | PromoKine | Cat# PK-CA91-1096 |
| InstantBlue Protein Stain | Expedeon | Cat# ISB1L |
| SilverQuest Silver Staining Kit | Thermo Fisher Scientific | Cat# LC6070 |
| Site-directed mutagenesis kit | Agilent | Cat# 200523 |
| MACS LS column | Miltenyi Biotec | Cat# 130-042-401 |
| Glutathione Assay Kit | Cayman | Cat# 703002 |
| Superscript First-strand Synthesis kit | Invitrogen | Cat# 18091050 |
| SMARTer Stranded RNA-seq kit | Takara | Cat# 634839 |
| Dynabeads mRNA purification kit | Thermo | Cat# 61006 |
| PowerUP™SYBR™Green | Thermo Fisher Scientific | Cat# A25742 |
| CellfectinR II Reagent | Invitrogen | Cat# 10362 |
| InstantBlue Protein Stain ISB1L | Fisher / Expedeon | Cat# 07-300-150 |
| RNAimax Transfection Reagent | Thermo Fisher Scientific | Cat#13778030 |
| ON-TARGETplus Mouse Shoc2 siRNA | Dharmacon | Cat# LQ-059319-01-0002 |
| ON-TARGETplus Non-targeting Control | Dharmacon | Cat# D-001810-10-05 |
| Deposited Data | ||
| RNA-seq | This paper | NCBI GEO: GSE142046 |
| Experimental Models: Cell Lines | ||
| Human: 293T/17 | ATCC | Cat# CRL-11268 |
| Human: MiaPaCa2 | ATCC | Cat# CRL-1420 |
| Human: PSN1 | ATCC | Cat# CRL-3211 |
| Human: CaPan1 | ATCC | Cat# CRL-HTB-79 |
| Human: Panc1 — | ATCC | Cat# CRL-1469 |
| Human: YAPC | DSMZ | Cat# ACC-382 |
| Human: DANG | DSMZ | Cat# ACC-249 |
| Human: KP4 | RIKEN | Cat# RCB-1005 |
| Mouse: KPC (Kras;p53;Ptf1aCre) | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: KrasLSL-G12D | (Hingorani et al., 2003) | Strain# JAX 008179 |
| Mouse: p53LoxP/LoxP | (Jonkers et al., 2001) | Strain# JAX 008462 |
| Mouse: Ptf1aCre | (Kawaguchi et al., 2002) | MGI# 2387812 |
| Mouse: Setd5LoxP/LoxP | (Skarnes et al., 2011) | Cat# VG17502 |
| Mouse: KrasFSF-G12D | (Schonhuber et al., 2014) | MGI:5616879 |
| Mouse: p53Frt/Frt | (Lee et al., 2012) | Strain# JAX 017767 |
| Mouse: Pdx1Flp | (Schonhuber et al., 2014) | MGI# 5616872 |
| Mouse: ROSA26FSF-CreER | (Schonhuber et al., 2014) | MGI# 5616874 |
| Mouse: NOD.SCID-IL2Rg−/− (NSG) | The Jackson Laboratories | Strain# 005557 |
| Oligonucleotides | ||
| sgRNA non-targeting (control) 5’-CTTCGAAATGTCCGTTCGGT-3’ |
This paper | N/A |
| sgRNA SETD5-1 human and mouse 5’-TTTGTGCAGCCCTGAATCTG-3’ |
This paper | N/A |
| sgRNA SETD5-2 human and mouse 5’-GCAGTGCAACAGAAAGCT-3’ |
This paper | N/A |
| sgRNA SETD5-3 human and mouse 5’-CGGAAGCAGGACAACATATC-3’ |
This paper | N/A |
| sgRNA SETD5-4 human and mouse 5’-ACGCTCTTCTCATTAACTGC-3’ |
This paper | N/A |
| sgRNA SETD2-1 mouse 5’-AATGAACTGGGATTCCGACG-3’ |
This paper | N/A |
| sgRNA SETD2-2 mouse 5’-GGAAGAAGAACAAATCCCAC-3’ |
This paper | N/A |
| sgRNA HDAC3-1 human 5’-CAGACCACCAGCCCAGTTAA-3’ |
This paper | N/A |
| sgRNA HDAC3-2 human 5’-GTTGAAGGCATTAAGACTCT-3’ |
This paper | N/A |
| sgRNA G9a human 5’-GCGCCCCCATCTCAGCGG-3’ |
This paper | N/A |
| sgRNA GLP human 5’-GCGCAAGGGTCAACCCCC-3’ |
This paper | N/A |
| RT-qPCR SETD5 forward 5’-GAGAAAGAAACGGCGGGATC-3’ |
This paper | N/A |
| RT-qPCR SETD5 reverse 5’-TTTCTGCAGCTACATCCCCA-3’ |
This paper | N/A |
| RT-qPCR GSTA1 forward 5’-AAGAGAAGCCAAGACTGCCT-3’ |
This paper | N/A |
| RT-qPCR GSTA1 reverse 5’-TTCTTCACATTGGGGAGGCT-3’ |
This paper | N/A |
| RT-qPCR GSTA2 forward 5’-GAGCTTGATGCCAGCCTTCTGA-3’ |
This paper | N/A |
| RT-qPCR GSTA2 reverse 5’-TTCTCTGGCTGCCAGGATGTAG-3’ |
This paper | N/A |
| RT-qPCR GM3776 forward 5’-AGGTGTTGAAGAGCCATGGA-3’ |
This paper | N/A |
| RT-qPCR GM3776 reverse 5’-GGCTGCTGATTCTGCTCTTG-3’ |
This paper | N/A |
| RT-qPCR PDK4 forward 5’-TGGCTGGTTTTGGTTATGGC-3’ |
This paper | N/A |
| RT-qPCR PDK4 reverse 5’-GTTCTTCGGTTCCCTGCTTG-3’ |
This paper | N/A |
| RT-qPCR WNT7a forward 5’-TTCGGGAAGGAGCTCAAAGT-3’ |
This paper | N/A |
| RT-qPCR WNT7a reverse 5’-ATTCTGCTTGATCTCCCGGG-3’ |
This paper | N/A |
| RT-qPCR HTR3 forward 5’-AGTCCGCGGTACAAGTTCAA-3’ |
This paper | N/A |
| RT-qPCR HTR3 reverse 5’-ACCGGCTTCTGACATGATGA-3’ |
This paper | N/A |
| RT-qPCR Osgin1 forward 5’-ACAGACTTTGGAGGCAGCAT-3’ |
This paper | N/A |
| RT-qPCR Osgin1 reverse 5’-TTTCTTCCGCATCCAGTCTT-3’ |
This paper | N/A |
| RT-qPCR CD93 forward 5’-ATCAGTACAGCCCAACACCA-3’ |
This paper | N/A |
| RT-qPCR CD93 reverse 5’-ATACCTGCCTATCCCAAGCC-3’ |
This paper | N/A |
| RT-qPCR Serpinb1a forward 5’-TGTAAGTGGAGCCAGACCTG-3’ |
This paper | N/A |
| RT-qPCR Serpinbla reverse: 5’-GGAAGCGTGAATGGATGTCC-3’ |
This paper | N/A |
| RT-qPCR GSTT1 forward 5’-CCTGTGTGAGAGTGTGGCTA-3’ |
This paper | N/A |
| RT-qPCR GSTT1 reverse 5’-GCTCACCAAG GAAAACAGGG-3’ |
This paper | N/A |
| RT-qPCR GSTT2 forward 5’-GTGCCCAAGTCCACGAATAC-3’ |
This paper | N/A |
| RT-qPCR GSTT2 reverse 5’-TCCAGAGACATGAGATCCGC-3’ |
This paper | N/A |
| RT-qPCR CDH6 forward 5’-CTGAGCCGTTCGAAAAGGAG-3’ |
This paper | N/A |
| RT-qPCR CDH6 reverse 5’-TAATGAAGAGATCGCCCGCT-3’ |
This paper | N/A |
| RT-qPCR MIA2 forward 5’-CCGAGTCTTAGCCCTGAGAG-3’ |
This paper | N/A |
| RT-qPCR MIA2 reverse 5’-ATCTCGACTGCATCTCTGGG-3’ |
This paper | N/A |
| RT-qPCR CDH17 forward 5’-TAAGACCAACCCTCCAGCTG-3’ |
This paper | N/A |
| RT-qPCR CDH17 reverse 5’-CCATGAGAATCCAAGGCTGC-3’ |
This paper | N/A |
| RT-qPCR NDN forward 5’-CTAACTTTGCAGCCGAGGTC-3’ |
This paper | N/A |
| RT-qPCR NDN reverse 5’-GCTGCAGGATTTTAGGGTCA-3’ |
This paper | N/A |
| ChIP-qPCR GSTA1-p1 forward 5’-ACCCACAGAGAACTTGCAGA-3’ |
This paper | N/A |
| ChIP-qPCR GSTA1-p1 reverse 5’-CTCTCAAATTCGCCTGCCTC-3’ |
This paper | N/A |
| ChIP-qPCR GSTA1-p2 forward 5’-AAGAGAAGCCAAGACTGCCT-3’ |
This paper | N/A |
| ChIP-qPCR GSTA1-p2 reverse 5’-TCCCAGAAACTCAGTGTCCC-3’ |
This paper | N/A |
| ChIP-qPCR PDK4-p1 forward 5’-CTCCTCCCTCTCACCCTTTG-3’ |
This paper | N/A |
| ChIP-qPCR PDK4-p1 reverse 5’-GGCTCTGGGACTCTGAACTT-3’ |
This paper | N/A |
| ChIP-qPCR PDK4-p2 forward 5’-AGTGGTTCGGTGTCTGAGAG-3’ |
This paper | N/A |
| ChIP-qPCR PDK4-p2 reverse 5’-GATAGAAGCTGCTGACCCCT-3’ |
This paper | N/A |
| ChIP-qPCR GM3776-p1 forward 5’-GCTGAATCTGGTTTGGTGCA-3’ |
This paper | N/A |
| ChIP-qPCR GM3776-p1 reverse: 5’-ACGGTCTAGGGGTGAAAAGG-3’ |
This paper | N/A |
| ChIP-qPCR GM3776-p2 forward 5’-CAGCCGCTCCTTACAATTCC-3’ |
This paper | N/A |
| ChIP-qPCR GM3776-p2 reverse 5’-CATGGGCACTTGGTCAAACA-3’ |
This paper | N/A |
| Recombinant DNA | ||
| Plasmid: Setd5tm1a | EuMMCR | Cat# PGS00019_A_B10 |
| Plasmid: pLentiCRISPRv2 | Feng Zhang Lab | Cat# Addgene #52961 |
| Plasmid: pLentiCRISPRv2 hygro | Brett Stringer Lab | Cat# Addgene #98291 |
| Plasmid: psPAX2 | Trono Lab | Cat# Addgene #12260 |
| Plasmid: pMD2.G | Trono Lab | Cat# Addgene #12259 |
| Plasmid: pCMV-dR8.2 dvpr | Bob Weinberg Lab | Cat# Addgene #8455 |
| Plasmid: pCMV-VSV-G | Bob Weinberg Lab | Cat# Addgene #8454 |
| Plasmid: pUMVC | Bob Weinberg Lab | Cat# Addgene #8449 |
| Plasmid: pBABE-neo | Bob Weinberg Lab | Cat# Addgene #1767 |
| Plasmid: pWZL Blast GFP | Bob Weinberg Lab | Cat# Addgene #12269 |
| Plasmid: pGEX-6P-1 | GE Healthcare | Cat# 28-9546-48 |
| Plasmid: pcDNA3.1(+) | Thermo Fisher Scientific | Cat# V7020 |
| Plasmid: pENTR3C | Thermo Fisher Scientific | Cat# A10465 |
| Plasmid: pLenti6.2 V5 DEST | Thermo Fisher Scientific | Cat# V36820 |
| Plasmid: pLenti CMV Hygro DEST(w117-1) | Campeau and Kaufman lab | Cat# Addgene 17454 |
| Plasmid: pQCXIH | Clontech | Cat# 631516 |
| Plasmid: pet28a | Novagen | Cat# 69864-3 |
| Plasmid: pFastbac1 | Invitrogen | Cat# 10359-016 |
| Software and Algorithms | ||
| Prism 7 | GraphPad | https://www.graphpad.com/; RRID:SCR_002798 |
| Excel for Mac 2016 | Microsoft | https://www.microsoft.com/en-us/; RRID:SCR_016137 |
| PreciPoint M8 ViewPoint | PreciPoint | http://www.precipoint.com/microscopy-software/viewpoint/ |
| ImageJ – Fiji package | Freeware | http://fiji.sc; RRID:SCR_002285 |
| Origin Pro 8 | Microcal | https://www.originlab.com/RRID:SCR_002815 |
| Horos | GNU Lesser General Public License, Version 3.0 | https://www.horosproject.org/ |
| Gene Set Enrichment Analysis (GSEA) | (Subramanian et al., 2007) | http://software.broadinstitute.org/gsea/index.jsp RRID: SCR_003199 |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/ |
| Samtools | (Xu et al., 2009) | http://samtools.sourceforge.net/ |
| HISAT2 | (Kim et al., 2015) | https://ccb.jhu.edu/software/hisat2/index.shtml |
| DESeq2 | (Love et al., 2014) | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| BEDTools | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| DeepTools | (Ramirez et al., 2016) | https://deeptools.readthedocs.io/en/develop/ |
| Trim_galore | Babraham Bioinformatics | RRID:SCR_011847 |
| MaxQuant (v. 1.5.8.4) | (Cox and Mann, 2008) | http://www.coxdocs.org/doku.php?id=maxquant:start |
| Other | ||
| Superose 6 Increase 10/300GL column | Fisher / GE Healthcare | Cat# 29091596 |
| ProtoArray® Human Protein Microarray | Thermo Fisher Scientific | Cat# PAH0525101 |
| Orbitrap Elite | Thermo Fisher Scientific | N/A |
| Dionex Ultimate 3000 system | Thermo Fisher Scientific | N/A |
| Xcalibur Qual Browser | Thermo Fisher Scientific | N/A |
| Biospec USR70/30 | Bruker | N/A |
Highlights.
SETD5 is an epigenetic driver of pancreatic cancer resistance to MEK1/2 inhibition
SETD5 has no intrinsic methylation activity on histones, including at H3 lysine 36
A SETD5 co-repressor complex regulates a network of drug resistance pathways
Co-targeting of MEK1/2 and the SETD5 complex results in sustained tumor inhibition
Significance.
Oncogenic KRAS signaling is a hallmark of PDAC, a lethal malignancy with few treatment options. A major roadblock in deploying therapies targeting the KRAS-MAPK pathway is the rapid emergence of resistant cancer cells. Here, we show that SETD5 is a clinically actionable epigenetic driver of PDAC resistance to MEKi. We find that acute deletion of SETD5 in aggressive PDAC refractory to MEKi restores the vulnerability of tumors to targeted MEKi therapy. Pharmacologic blockade with inhibitors of histone-modifying activities associated with SETD5 disrupts a SETD5-driven resistance program and sustains inhibition of tumor growth by MEKi in pre-clinical murine and human models of PDAC. Together, our study reveals a potential clinical path for rationale testing of FDA-approved MEK inhibitors to treat PDAC.
ACKNOWLEDGMENTS
We thank members of the Gozani and Mazur laboratories for critical reading of the manuscript. This work was supported in part by grants from the NIH to P.K.M. and O.G. (CA236949), M.C.B. (1DP2HD084069), M.J.M., M.A.C., S.A.H. and M.C.K. (2R44GM116584). J.S. and O.G. from and the Lustgarten Foundation. A.M. by the MD Anderson Moonshot Program. P.K.M. by the Neuroendocrine Tumor Research Foundation, AACR, Lung Cancer Research Foundation, American Gastroenterological Association Research Foundation, DOD PRCRP Career Development Award (W81XWH1910761), University Cancer Foundation at MD Anderson Cancer Center, a CPRIT Scholar in Cancer Research (RR160078), and the Andrew Sabin Family Foundation Scholar. S.H. by a Deutsche Forschungsgemeinschaft Fellowship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.ccell.2020.04.014.
DECLARATIONS OF INTERESTS
O.G. is a co-founder of EpiCyphe and Athelas Therapeutics. M.C.K., M.J.M., M.A.C., and S.A.H. are employees and shareholders of EpiCypher.
REFERENCES
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, and Perucho M (1988). Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53, 549–554. [DOI] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, and Buggy JJ (2008). A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 22, 1026–1034. [DOI] [PubMed] [Google Scholar]
- Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB, Desar IM, Timmer-Bonte JN, Eckhardt SG, Lewis KD, et al. (2010). The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin. Cancer Res 16, 1613–1623. [DOI] [PubMed] [Google Scholar]
- Bansal A, and Simon MC (2018). Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol 217, 2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. (2006). Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. U S A 103, 5947–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassik MC, Kampmann M, Lebbink RJ, Wang S, Hein MY, Poser I, Weibezahn J, Horlbeck MA, Chen S, Mann M, et al. (2013). A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 152, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassik MC, Lebbink RJ, Churchman LS, Ingolia NT, Patena W, LeProust EM, Schuldiner M, Weissman JS, and McManus MT (2009). Rapid creation and quantitative monitoring of high coverage shRNA libraries. Nat. Methods 6, 443–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, and Tebbutt NC (2012). A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest. New Drugs 30, 1216–1223. [DOI] [PubMed] [Google Scholar]
- Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, et al. (2019). Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med 25, 628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223. [DOI] [PubMed] [Google Scholar]
- Chung V, McDonough S, Philip PA, Cardin D, Wang-Gillam A, Hui L, Tejani MA, Seery TE, Dy IA, Al Baghdadi T, et al. (2017). Effect of selumetinib and MK-2206 vs oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG S1115 study randomized clinical trial. JAMA Oncol. 3, 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort D, et al. (2012). A central role for RAF→MEK→ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Deliu E, Arecco N, Morandell J, Dotter CP, Contreras X, Girardot C, Kasper EL, Kozlova A, Kishi K, Chiaradia I, et al. (2018). Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat. Neurosci 21, 1717–1727. [DOI] [PubMed] [Google Scholar]
- Grozeva D, Carss K, Spasic-Boskovic O, Parker MJ, Archer H, Firth HV, Park SM, Canham N, Holder SE, Wilson M, et al. (2014). De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am. J. Hum. Genet 94, 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Barak O, and Lazar MA (2001). The SMRT and N-CoR co-repressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol 21, 6091–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. (2003). Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Husmann D, and Gozani O (2019). Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol 26, 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, Oh DY, Liu Y, Redhu S, Steplewski K, et al. (2014). A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 50, 2072–2081. [DOI] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, and Berns A (2001). Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet 29, 418–425. [DOI] [PubMed] [Google Scholar]
- Kampmann M, Bassik MC, and Weissman JS (2014). Functional genomics platform for pooled screening and generation of mammalian genetic interaction maps. Nat. Protoc 9, 1825–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al. (2014). Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158, 185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, and Wright CV (2002). The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet 32, 128–134. [DOI] [PubMed] [Google Scholar]
- Khatri P, Roedder S, Kimura N, De Vusser K, Morgan AA, Gong Y, Fischbein MP, Robbins RC, Naesens M, Butte AJ, et al. (2013). A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J. Exp. Med 210, 2205–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B,and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, and Gallick GE (2009). Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat. Protoc 4, 1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, et al. (2019a). Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med 25, 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, et al. (2019b). Publisher correction: protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med 25, 861. [DOI] [PubMed] [Google Scholar]
- Ko AH, Bekaii-Saab T, Van Ziffle J, Mirzoeva OM, Joseph NM, Talasaz A, Kuhn P, Tempero MA, Collisson EA, Kelley RK, et al. (2016). A multicenter, open-label phase II clinical trial of combined MEK plus EGFR inhibition for chemotherapy-refractory advanced pancreatic adenocarcinoma. Clin. Cancer Res 22, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CL, Moding EJ, Huang X, Li Y, Woodlief LZ, Rodrigues RC, Ma Y, and Kirsch DG (2012). Generation of primary tumors with Flp recombinase in FRT-flanked p53 mice. Dis. Model. Mech 5, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Hausmann S, Carlson SM, Fuentes ME, Francis JW, Pillai R, Lofgren SM, Hulea L, Tandoc K, Lu J, et al. (2019). METTL13 methylation of eEF1A increases translational output to promote tumorigenesis. Cell 176, 491–504.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchado E, Weissmueller S, Morris J.P.t., Chen CC, Wullenkord R, Lujambio A, de Stanchina E, Poirier JT, Gainor JF, Corcoran RB, et al. (2016). A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 534, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann KM, Ward JM, Yew CC, Kovochich A, Dawson DW, Black MA, Brett BT, Sheetz TE, Dupuy AJ, Australian Pancreatic Cancer Genome, I., et al. (2012). Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. U S A 109, 5934–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas YMS, Barbon M, Teyssier C, Demene H, Carvalho JE, Bird LE, Lebedev A, Fattori J, Schubert M, Dumas C, et al. (2016). The human mixed lineage leukemia 5 (MLL5), a sequentially and structurally divergent SET domain-containing protein with no intrinsic catalytic activity. PLoS One 11, e0165139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur PK, Reynoird N, Khatri P, Jansen PW, Wilkinson AW, Liu S, Barbash O, Van Aller GS, Huddleston M, Dhanak D, et al. (2014). SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 510, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, et al. (2018). An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med 24, 1036–1046. [DOI] [PubMed] [Google Scholar]
- Nguyen D (2013). Quantifying chromogen intensity in immunohistochemistry via reciprocal intensity. Nature Protocol Exchange. 10.1038/protex.2013.097. [DOI] [Google Scholar]
- Noll EM, Eisen C, Stenzinger A, Espinet E, Muckenhuber A, Klein C, Vogel V, Klaus B, Nadler W, Rosli C, et al. (2016). CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma. Nat. Med 22, 278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipovich AB, Gangula R, Vianna PG, and Magnuson MA (2016). Setd5 is essential for mammalian development and the co-transcriptional regulation of histone acetylation. Development 143, 4595–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. (2015). Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KA, Grutzmann R, Aust D, Rummele P, et al. (2012). The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponz-Sarvise M, Corbo V, Tiriac H, Engle DD, Frese KK, Oni TE, Hwang CI, Ohlund D, Chio IIC, Baker LA, et al. (2019). Identification of resistance pathways specific to malignancy using organoid models of pancreatic cancer. Clin. Cancer Res 25, 6742–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawla P, Sunkara T, and Gaduputi V (2019). Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J. Oncol 10, 10–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DP, Hong TS, and Bardeesy N (2014). Pancreatic adenocarcinoma. N. Engl. J. Med 371, 1039–1049. [DOI] [PubMed] [Google Scholar]
- Sastra SA, and Olive KP (2014). Acquisition of mouse tumor biopsies through abdominal laparotomy. Cold Spring Harb. Protoc 2014, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonhuber N, Seidler B, Schuck K, Veltkamp C, Schachtler C, Zukowska M, Eser S, Feyerabend TB, Paul MC, Eser P, et al. (2014). A next-generation dual-recombinase system for time- and host-specific targeting of pancreatic cancer. Nat. Med 20, 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa A, Fagnocchi L, Mastrototaro G, Massimino L, Zaghi M, Indrigo M, Cattaneo S, Martini D, Gabellini C, Pucci C, et al. (2019). SETD5 regulates chromatin methylation state and preserves global transcriptional fidelity during brain development and neuronal wiring. Neuron 104, 271–289.e3. [DOI] [PubMed] [Google Scholar]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, et al. (2014). KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, et al. (2006). ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, and Shi Y (2003). Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738. [DOI] [PubMed] [Google Scholar]
- Sidoli S, Bhanu NV, Karch KR, Wang X, and Garcia BA (2016). Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: from histone extraction to data analysis. J. Vis. Exp 10.3791/54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, and Garcia BA (2017). Characterization of individual histone post-translational modifications and their combinatorial patterns by mass spectrometry-based proteomics strategies. Methods Mol. Biol 1528, 121–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2019). Cancer statistics, 2019. CA Cancer J. Clin 69, 7–34. [DOI] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, and McCormick F (2014). Dragging ras back in the ring. Cancer Cell 25, 272–281. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Kuehn H, Gould J, Tamayo P, and Mesirov JP (2007). GSEA-P: a desktop application for gene set enrichment analysis. Bioinformatics 23, 3251–3253. [DOI] [PubMed] [Google Scholar]
- Sulahian R, Kwon JJ, Walsh KH, Pailler E, Bosse TL, Thaker M, Almanza D, Dempster JM, Pan J, Piccioni F, et al. (2019). Synthetic lethal interaction of SHOC2 depletion with MEK inhibition in RAS-driven cancers. Cell Rep. 29, 118–134.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Fang Y, Yin J, Chen J, Ju Z, Zhang D, Chen X, Vellano CP, Jeong KJ, Ng PK, et al. (2017). Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med 9, 10.1126/scitranslmed.aal5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Daemen A, Hatzivassiliou G, Arnott D, Wilson C, Zhuang G, Gao M, Liu P, Boudreau A, Johnson L, et al. (2014). Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab. 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Fukushima T, and Shinkai Y (2001). Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem 276, 25309–25317. [DOI] [PubMed] [Google Scholar]
- Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, et al. (2011). A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol 7, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Carrozza MJ, Lasonder E, Workman JL, Logie C, and Stunnenberg HG (2004). In vitro targeting reveals intrinsic histone tail specificity of the Sin3/histone deacetylase and N-CoR/SMRT corepressor complexes. Mol. Cell. Biol 24, 2364–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. (2014). Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson AW, Diep J, Dai S, Liu S, Ooi YS, Song D, Li TM, Horton JR, Zhang X, Liu C, et al. (2019). SETD3 is an actin histidine methyltransferase that prevents primary dystocia. Nature 565, 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Soragni E, Chou CJ, Herman D, Plasterer HL, Rusche JR, and Gottesfeld JM (2009). Chemical probes identify a role for histone deacetylase 3 in Friedreich’s ataxia gene silencing. Chem. Biol 16, 980–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, and Adjei AA (2014). The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol 11, 385–400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq datasets reported in this paper is paper is NCBI GEO: GSE142046. This study did not generate any unpublished code, software, or algorithm.