

Abstract
Mitochondrial Ca2+ transport is essential for regulating cell bioenergetics, Ca2+ signaling and cell death. Mitochondria accumulate Ca2+ via the mitochondrial Ca2+ uniporter (MCU), whereas Ca2+ is extruded by the mitochondrial Na+/Ca2+ (mtNCX) and H+/Ca2+ exchangers. The balance between these processes is essential for preventing toxic mitochondrial Ca2+ overload. Recent work demonstrated that MCU activity varies significantly among tissues, likely reflecting tissue-specific Ca2+ signaling and energy needs. It is less clear whether this diversity in MCU activity is matched by tissue-specific diversity in mitochondrial Ca2+ extrusion. Here we compared properties of mitochondrial Ca2+ extrusion in three tissues with prominent mitochondria function: brain, heart and liver. At the transcript level, expression of the Na+/Ca2+/Li+ exchanger (NCLX), which has been proposed to mediate mtNCX transport, was significantly greater in liver than in brain or heart. At the functional level, Na+ robustly activated Ca2+ efflux from brain and heart mitochondria, but not from liver mitochondria. The mtNCX inhibitor CGP37157 blocked Ca2+ efflux from brain and heart mitochondria but had no effect in liver mitochondria. Replacement of Na+ with Li+ to test the involvement of NCLX, resulted in a slowing of mitochondrial Ca2+ efflux by ~70%. Collectively, our findings suggest that mtNCX is responsible for Ca2+ extrusion from the mitochondria of the brain and heart, but plays only a small, if any, role in mitochondria of the liver. They also reveal that Li+ is significantly less effective than Na+ in driving mitochondrial Ca2+ efflux.
Keywords: Mitochondria, Ca2+ transport, NCLX, NCX, hippocampal neurons
Graphical Abstract
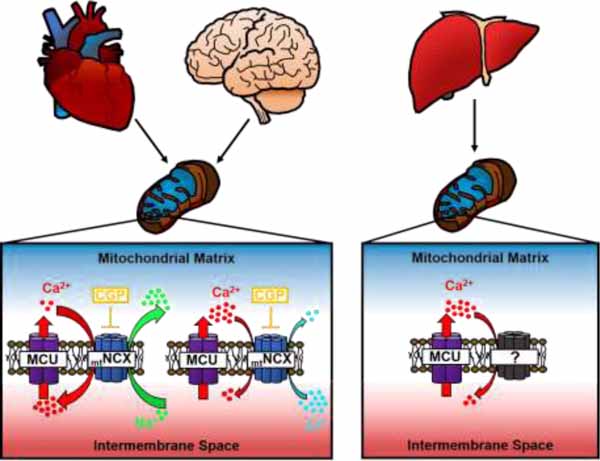
1. Introduction
Mitochondrial Ca2+ transport is essential for many cellular functions including the regulation of cell bioenergetics, Ca2+ signaling and cell death and survival [1–5]. Ca2+ uptake by mitochondria is mediated by the mitochondrial Ca2+ uniporter (MCU) complex, which functions as a Ca2+-selective and Ca2+-gated ion channel, whereas Ca2+ extrusion from mitochondria is controlled by Na+/Ca2+ and H+/Ca2+ exchangers (mtNCX and mtHCX, respectively) [3, 4, 6, 7]. Recent analysis in isolated mitochondria has demonstrated that MCU activity varies significantly among tissues, with some of the lowest activity reported for heart mitochondria, and higher activities found in mitochondria from liver and skeletal muscle [8, 9]. In addition, mitochondria from different tissues displayed a difference in the Ca2+ activation threshold and cooperativity [9]. This variability in mitochondrial Ca2+ uptake likely reflects attuning of the mitochondrial Ca2+ transport machinery to tissue-specific Ca2+ signaling and energy needs, and is fundamental for optimal performance of mitochondria in a given tissue [3, 8–10].
Ca2+ that accumulates in mitochondria during cellular activity is subsequently released back into the cytosol via mtNCX and mtHCX [3, 4, 6]. Earlier work in isolated mitochondria suggested that mtNCX plays a prominent role in removing Ca2+ from mitochondria in excitable tissues such as the brain, heart and skeletal muscle, whereas mtHCX is likely responsible for mitochondrial Ca2+ extrusion in non-excitable tissues such as liver, kidney and lung [6, 11, 12]. However recent identification of the protein NCLX (a product of gene Slc8b1) as a molecule likely responsible for the mtNCX transport has provided a new molecular perspective on the role of mtNCX in various tissues [13–19]. Notably, NCLX was found to be expressed in the liver, kidney and lung at levels similar or higher than those found in brain and heart [10], suggesting that the mtNCX mechanism could contribute to mitochondrial Ca2+ extrusion in both excitable and non-excitable tissues [20, 21]. The role of mtNCX is especially prominent in neurons, where it can significantly prolong elevation of the cytosolic Ca2+ concentration ([Ca2+]cyt) and regulate Ca2+-dependent processes such as synaptic transmission and gene expression [22–24]. Equally important, Ca2+ extrusion mediated by mtNCX is critical for preventing toxic Ca2+ build-up in mitochondria and protecting neurons and cardiomyocytes from cell death in the context of either excessive stimulation or ischemia [18, 25, 26]. Similarly, in non-excitable tissues, mtNCX was found to provide crucial support for the function and survival of cells, specifically for brown fat adipocytes in the context of adrenergic stimulation [27]. Thus, a precisely controlled balance between the activities of the mitochondrial Ca2+ uptake and efflux machinery is essential for preventing toxic mitochondrial Ca2+ overload and cell death. This concept also raises another key question of whether the tissue-specific variability in the activity of mitochondrial Ca2+ uptake is paralleled by a similarly diverse and tissue-specific activity of mitochondrial Ca2+ extrusion.
Thus, we sought to address this important question by carrying out a quantitative comparison of Ca2+ extrusion from isolated mitochondria obtained from the three tissues with prominent mitochondria function: brain, heart and liver. A second objective of this study was to re-assess the role of mtNCX in excitable and non-excitable tissues. To this end, we developed an assay based on the use of a low-affinity fluorescent Ca2+ indicator Ca2+-Green 5N (Kd~15 µM), for monitoring Ca2+ extrusion from isolated mitochondria under standardized and highly controlled recording conditions. A property unique to NCLX among the Na+/Ca2+ exchangers is that Li+ is nearly as effective as Na+ in driving its transmembrane transport of Ca2+ [28–31]. This unique feature of NCLX can be utilized to gain further insight into the molecular nature of the mitochondrial Ca2+ extrusion machinery. Thus, as a part of our assessment, we examined the sensitivities of mitochondrial Ca2+ extrusion to Li+ as well as to the mtNCX inhibitor CGP37157, which is a widely accepted pharmacological tool for studying mtNCX activity [32–34]. Our analysis revealed that Na+ strongly activated Ca2+ efflux from isolated brain and heart mitochondria, which was inhibited by CGP37157. In contrast, Na+ had no effect on Ca2+ efflux from isolated liver mitochondria, and the efflux was also insensitive to CGP37157. These findings were corroborated in intact primary neurons and hepatocytes by simultaneous measurement of the cytosolic and mitochondrial Ca2+ concentrations ([Ca2+]cyt and [Ca2+]mt, respectively). Although Li+ was able to promote Ca2+ efflux from both brain and heart mitochondria, the rate of Li+-driven Ca2+ efflux was only ~30% of that induced by Na+. Collectively, these findings suggest that mtNCX is the principal mechanism involved in extruding Ca2+ from mitochondria of the brain and heart, but that it does not play a significant role in mediating Ca2+ efflux from liver mitochondria. Our data also reveal that Li+ is significantly less effective than Na+ in its ability to drive mitochondrial Ca2+ efflux.
2. Materials and methods
2.1. Mitochondria isolation and measurement of Ca2+ efflux
All experiments involving mice were approved by the University of Iowa Institutional Animal Care and Use Committee and were carried out in strict accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Every effort was made to minimize the number of mice used and their suffering. Mitochondria were isolated from adult (6–10 weeks) C57BL/6J mice, using a protocol similar to one that was previously described, with some modifications [35]. Briefly, mice were anesthetized, swiftly decapitated and the brain was quickly removed. Brains were chopped and then homogenized in ice-chilled mitochondrial isolation buffer (MIB; 225 mM mannitol, 75 mM sucrose, 2 mM K2HPO4, 5 mM HEPES, 1 mM EGTA, 0.1% FAF BSA, pH 7.2 w/ KOH) using a tissue grinder (E2355, Eberbach; Ann Arbor MI). Homogenate was centrifuged at 1500 g (5 min) to pellet unlysed cells, and then the supernatant was additionally centrifuged at 21,000 g (10 min) to pellet crude mitochondria. Pelleted mitochondria were resuspended in 3.5 mL of 15% Percoll solution, which was layered on top of 3.7 mL 24% and 1 mL 40% Percoll solutions in MIB. The solutions were then centrifuged at 30,700 g for 8 min. Non-synaptosomal mitochondria were collected from a band between the 24% and 40% Percoll layers. Mitochondria were then resuspended in a 1:5 Percoll and MIB solution, and centrifuged at 6900 g for 10 min. The mitochondrial pellet was then resuspended in EGTA- and BSA-free MIB and then each sample was subjected to bicinchoninic acid assay (BCA) analysis.
For the isolation of heart and liver mitochondria, essentially the same protocol was used as for the brain mitochondria, but with slight modifications. Briefly, mice were anesthetized and swiftly decapitated, after which the tissue was quickly harvested. Tissue was chopped and homogenized (see above) in ice-chilled isolation buffer 1 (IB1; 225 mM mannitol, 75 mM sucrose, 10 mM HEPES, 1 mM EGTA, 0.1% FAF BSA, pH 7.4 with KOH). The homogenate was centrifuged at 700 g (10 min), and then the supernatant was centrifuged at 18,900 g (10 min). The pellet was resuspended in isolation buffer 2 (IB2; 225 mM mannitol, 75 mM sucrose, 10 mM HEPES, 0.1 mM EGTA, pH 7.4 w/ KOH) and then centrifuged at 18,900 g (10 min). Then, the pellet was resuspended in EGTA-free IB2 and BCA analysis was performed.
Mitochondrial Ca2+ flux was monitored using the low-affinity fluorescent Ca2+ indicator Calcium Green 5N (Kd~15 µM) and measuring changes in the extramitochondrial Ca2+ concentration. Specifically, mitochondria were suspended in a respiration buffer (Res; 150 mM KCl, 2 mM K2HPO4, 0.5 mM MgCl2, 5 mM malate, 5 mM glutamate, 0.1 mM ADP, 10 mM HEPES, 10 μM EGTA, and 1 μM Calcium Green 5N) in a 96 well, black/clear, tissue culture treated plate (Falcon). Calcium Green 5N was excited at 506 nm and fluorescence collected at 532 nm using a SpectraMax M2 Microplate Reader (Molecular Devices). For a standard experiment, mitochondria were loaded with a bolus of Ca2+ (100 µM), followed by addition of the MCU inhibitor Ru360 (10 µM) to prevent mitochondrial Ca2+ uptake, thereby enabling to study mitochondrial Ca2+ efflux in isolation. To probe for the role of mtNCX, 20 mM of NaCl was added to mitochondria in the continuous presence of Ru360. This concentration of Na+ was chosen based on the reported mtNCX Km~8–10 mM for Na+ and the findings that 20 mM Na+ produces maximal activation of mtNCX in various tissues [6]. The rate of Ca2+ efflux was quantified as nmol Ca2+ per minute per mg of mitochondrial protein during the first min of any given conditions (maximal slope). For calculating changes in extramitochondrial Ca2+ concentration, Calcium Green 5N fluorescence was calibrated by adding standard amounts of Ca2+ to mitochondria-free recording (respiration) buffer.
2.2. cDNA Plasmid Constructs and FIV Lentivirus
The plasmids encoding Flag-tagged mouse MCU and NCLX constructs were obtained from OriGene (Cat.#MR218926 for MCU-Flag, and Cat.#MR223948 for NCLX-Flag). The plasmid encoding the mitochondrial Ca2+ indicator mito-RGECO1 was obtained from Addgene (Cat.#46021). The Mito-V5-DsRed construct was modified from pDsRed2-Mito (Takara) by insertion of a V5 epitope tag. The GFP-KDEL plasmid construct was previously described [36] and kindly provided by Dr. Tom Rutkowski (University of Iowa).
For preparation of the mito-RGECO1 FIV (feline immunodeficiency virus) lentivirus, the corresponding cDNA sequence was PCR amplified (forward primer: 5’-GAGGTCTATATAAGCAGAGC-3’; reverse primer: 5’-GACGTCGACGAATTCGAGGCTGATCAGCGGTTTAAAC-3’), cut using EcoRI and NheI, and ligated into the EcoRI and SpeI sites of the lentiviral shuttle vector pFIV3.2-CAG-mcs. The lentivirus (1×107 - 5×108 transforming units/ml) was produced by the University of Iowa Viral Vector Core as previously described [23, 24].
2.3. Culture and transfection of mouse hippocampal neurons
Primary cultures of hippocampal neurons were prepared from C57BL/6J mouse neonates (P0–1) and transfected as previously described [37]. Briefly, the mice were swiftly decapitated, the brain was removed, the hippocampi were dissected in Neurobasal Plus medium supplemented with 20 mM HEPES (pH 7.3), and the tissue was digested in a trypsin solution (1 mg/mL) for 10 min at 25°C. Cells were washed in fresh medium and mechanically dissociated by sequential trituration with fire-polished Pasteur pipettes of increasingly smaller bore size. Cells were then plated onto 25 mm glass coverslips, pre-coated with poly-L-ornithine (0.2 mg/mL) and laminin (50 μg/mL) in a 6-well plate. The cultures were grown in Neurobasal Plus Medium with B27 Plus Supplement and penicillin-streptomycin (50 U/mL and 50 μg/mL, respectively) in a 5% CO2 incubator at 37°C. Every 3–4 days, 50% of the media was replaced. After 6–7 DIV, hippocampal neurons were transfected with the mitochondrial Ca2+ indicator plasmid mito-RGECO1 using an FIV lentivirus [38]. Cells were used for imaging on DIV 10–14.
2.4. Culture and transfection of primary mouse hepatocytes
Hepatocytes were isolated from 6–8 weeks old C57BL/6J mice. Hepatocytes were isolated using a two-step collagenase perfusion as previously described [39] with slight modifications. Briefly, the abdomen of an anesthetized mouse was opened exposing the inferior vena cava (IVC). The IVC was cannulated with a 24-guage IV-catheter and perfusion was started with 37ºC pre-warmed perfusion buffer (HBSS -Ca2+, -Mg2+, - phenol red (Thermo Fisher 14175095); 25 mM HEPES (Thermo Fisher #15630080); 0.5 M EGTA; 1X penicillin/streptomycin (Thermo Fisher #10378016)) at 6 mL/min. The portal vein was quickly cut and the liver was observed to blanch immediately. After 5–8 minutes perfusion buffer was changed to 37ºC pre-warmed digestion buffer (HBSS +Ca2+, +Mg2+, - phenol red (Thermo Fisher #14025092); 25 mM HEPES; 0.2% BSA (Fraction V); 1X penicillin/streptomycin; 100 U/mL Collagenase IV (Worthington Biochemical #LS004188)) at 6 mL/min. Approximately 3–5 times during perfusion and digestions, the portal vein was gently clamped for 5 seconds using forceps and the liver was allowed to slightly swell. After 8 minutes, perfusion was halted, and the liver was carefully excised and placed in a 10 cm culture dish containing 10 mL of digestion buffer. The liver capsule was disrupted, and the liver was gently shaken liberating the hepatocytes into the excess digestion media. This hepatocyte suspension was filtered through a 100 µM cell strainer into 30 mL of washing media (DMEM low glucose (Thermo Fisher #11885076), 5% FBS, 10 mM HEPES, 1X penicillin/streptomycin). The hepatocyte suspension was centrifuged at 50 x g for 5 minutes, and the pellet was gently resuspended in 25 mL of washing media. This process was repeated 3 more times. The final hepatocyte pellet was resuspended in plating media (William’s E media (Thermo Fisher #12551032), 1X B27 supplement (Thermo Fischer #17504044), 10 nM dexamethasone, 1X Glutamax (Thermo Fischer #35050061), 1X penicillin/streptomycin), and cells were counted, and viability was assessed using trypan blue. Approximately 8.0×105 cells/cm2 hepatocytes were plated onto collagen coated coverslips. Hepatocytes were allowed to attach for 3.5 hours before plating media was changed to long-term maintenance media (William’s E media, 1X B27 supplement, 1X Glutamax, 1X penicillin/streptomycin, 20 µM forskolin (Tocris #1099), 10 µM SB431542 (Tocris #1614), 0.5 µM IWP2 (Tocris #3533), 1 µM DAPT (Tocris #2634), 0.1 µM LDN193189 (Tocris #6053) [40]. After 24 hrs in culture, hepatocytes were transfected with the mito-RGECO1 plasmid using the PromoFectin-Hepatocyte transfection kit (PromoCell; Cat.# PK-CT-2000-HEP) according to the manufacturer protocol, and used for imaging in 2–3 days after the transfection.
2.5. Ca2+ imaging in hippocampal neurons and primary hepatocytes
Simultaneous monitoring of the Ca2+ concentrations in the cytosol ([Ca2+]cyt) and mitochondria ([Ca2+]mt) of cultured hippocampal neurons and hepatocytes was performed as previously described [37]. On the day of imaging, transfected cell cultures were loaded with the cytosolic Ca2+ indicator Fura-2/AM (2 μM, 30 min). Cells were then placed in a flow-through perfusion chamber and mounted on an inverted IX-71 Olympus microscope (Olympus, Japan). Cells were perfused with a HEPES-buffered Hank’s salt solution of the following composition: 140 mM NaCl, 5 mM KCl, 1.3 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, pH 7.4 with KOH (310 mOsm/kg with sucrose). [Ca2+]cyt and [Ca2+]mt elevations in neurons were elicited by brief (15 s) depolarization using 30 mM KCl, and in hepatocytes, by application of 100 μM ATP (1 min).
Fura-2 and mito-RGECO1 were sequentially excited at 340 nm (12 nm bandpass), 380 nm (12 nm bandpass), and 550 nm (12 nm bandpass) using a Polychrome V monochromator (TILL Photonics, Germany) and a 40x oil-immersion objective (NA=1.35, Olympus). Excitation/emission fluorescence was separated using a dual fluorophore beamsplitter (FF493/574-Di01, Semrock; Rochester NY) while signal was collected using a dual emission filter (FF01–512/630, Semrock) and an IMAGO CCD camera (640×480 pixels; Till Photonics, Germany). Fluorescent images were acquired at a rate of 0.5 Hz. Fura-2 fluorescence ratio (R=340/380 nm) was converted to [Ca2+]cyt using the equation: Kdβ((R-Rmin)/(R-Rmax)) as previously described [41, 42]. Rmin, Rmax and β were calculated by applying 10 μM ionomycin in either Ca2+-free buffer (1 mM EGTA) or Ca2+-containing (1.3 mM) HEPES-buffered Hank’s salt solution as previously described [38]. Using this approach, the calibration values were determined as follows: Rmin=0.19, Rmax=2.98, β=6.77. A Kd value of 275 nM was used for Fura-2 [41, 42]. Changes in [Ca2+]mt were quantified as ΔF/F0 = (F-F0)/F0, where F is current fluorescence intensity (λex = 550 nm) and F0 is the fluorescence intensity at baseline. At each wavelength, fluorescence was corrected for background as measured in an area free of cells.
2.6. Confocal microscopy and image analysis of NCLX and MCU localization in AML12 cells
Mouse liver AML12 cells [43] were purchased from ATCC (Cat.# CRL-2254), and after thawing and 3–6 passages, plated on collagen coated glass coverslip in Dulbecco’s modified Eagle’s medium/Ham’s F12 with 10 mM HPEPES (Gibco) supplemented with 10% fetal bovine serum (GIBCO), a 1x mixture of insulin, transferrin and selenium (ITS, GIBCO), 40 ng/ml dexamethasone (sigma) , and penicillin/streptomycin (1%) (GIBCO). The cells were co-transfected with plasmids encoding either MCU-Flag or NCLX-Flag together with the mitochondrial and endoplasmic reticulum (ER) reporters, Mito-V5-DsRed and GFP-KDEL, respectively, using Lipofectamine 2000 (Thermofisher) according to the manufacturer’s protocol. Two days after the transfection, cells were fixed with 4% paraformaldehyde in PBS, and subjected to immunofluorescence staining as previously described [23, 44, 45] using the following primary antibodies: rabbit polyclonal anti-GFP (Abcam Cat.# ab290; 1:500), mouse monoclonal anti-V5 (Invitrogen, Cat.# 14–6796-82; 1:1000), mouse monoclonal anti-Flag(DDK) (OriGene, Cat.#TA50011–100; 1:1000). Secondary antibodies conjugated to Alexa Fluor 488, 555, and 647 were targeted to each of the primary antibodies respectively (Thermofisher, 1:1000). After staining, the coverslips were mounted onto microscope slides using ProLong fluoromount with DAPI (Invitrogen Cat.# P36962). AML12 cells were imaged on an inverted Olympus FV-3000 confocal microscope using a 100x objective (UPLXAPO100X, Olympus, NA=1.45). Following image collection, all images were processed through a constrained iterative deconvolution program in the cellSens software (Olympus) designed for the FV-3000 microscope. The colocalization of the NCLX or MCU with the mitochondria and ER was quantified as the Pearson’s correlation coefficient using the JaCoP plug-in for ImageJ software [46] as previously described [45]. Analysis was performed on a single z-plane of each cell imaged, which was selected by finding the approximate center of the cells using Dapi.
2.7. qRT-PCR
RNA was isolated from adult (6–10 weeks) C57BL/6J mice, as previously described [24]. Briefly, mice were anesthetized and swiftly decapitated, after which the hippocampi, cortex, heart and liver were isolated. Tissue was placed in Trizol and homogenized, and then an additional 20% of total volume of chloroform was added. Homogenates were centrifuged at 13,200 rpm for 5 min, and then supernatant was removed and diluted 1:1 in 70% EtOH. RNA was extracted using the Qiagen RNeasy kit (Qiagen) and converted to cDNA using an Iscript-cDNA synthesis kit (Bio-Rad). mRNA expression was determined using the Step One Plus Real-time PCR System (Applied Biosystems) and the fast SYBR green master mix kit (ThermoFisher). The primer sequences were as follows. Mouse NCLX: 5’-ATACTGGAGACGGCGTCTGGGA-3’ (forward) and 5’-CTGCGGCAGTCCGGATTCCTC-3’ (reverse); mouse GAPDH: 5’-AGGTCGGTGTGAACGGATTTG-3’ (forward) and 5’-TGTAGACCATGTAGTTGAGGTCA-3’ (reverse). Data were normalized to GAPDH and analyzed using the 2ΔΔCt method.
2.8. Reagents
Ru360 was purchased from Calbiochem (Sigma-Millipore; 557440–1 SET). CGP37157, forskolin, SB431542, IWP2, DAPT and LDN193189 were purchased from Tocris (Biotechne; 1114–10 mg). Fura-2A/M (F1201) and Calcium Green 5-N (C3737) were purchased from Thermofisher. Percoll was purchased from GE Healthcare Bio-Sciences (17–0891-01). All other reagents were purchased from Sigma-Aldrich/RPI .
2.9. Statistical analysis.
Data were analyzed by Student’s t test (for comparing two groups of data; two-tailed with Welch’s correction) or one-way ANOVA with Bonferroni’s post hoc test (for comparing >2 groups of data), and for non-parametric testing (qRT-PCR data), Kruskal Wallis test was used followed by Dunn’s multiple-comparisons test (GraphPad Prism v7 or v8 for Windows). Means ± SEM plotted throughout, and significance levels are abbreviated as follows: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
3. Results
3.1. NCLX is expressed in the mouse brain, heart and liver
As the first step, we used quantitative RT-PCR to compare the expression of NCLX mRNA in the brain, heart and liver of the mouse. As shown in Figure 1, NCLX mRNA expression was similar in brain and heart, but significantly higher in the liver. A similar NCLX expression profile was recently reported for human tissues [10].
Figure 1. NCLX expression in excitable and non-excitable tissues.
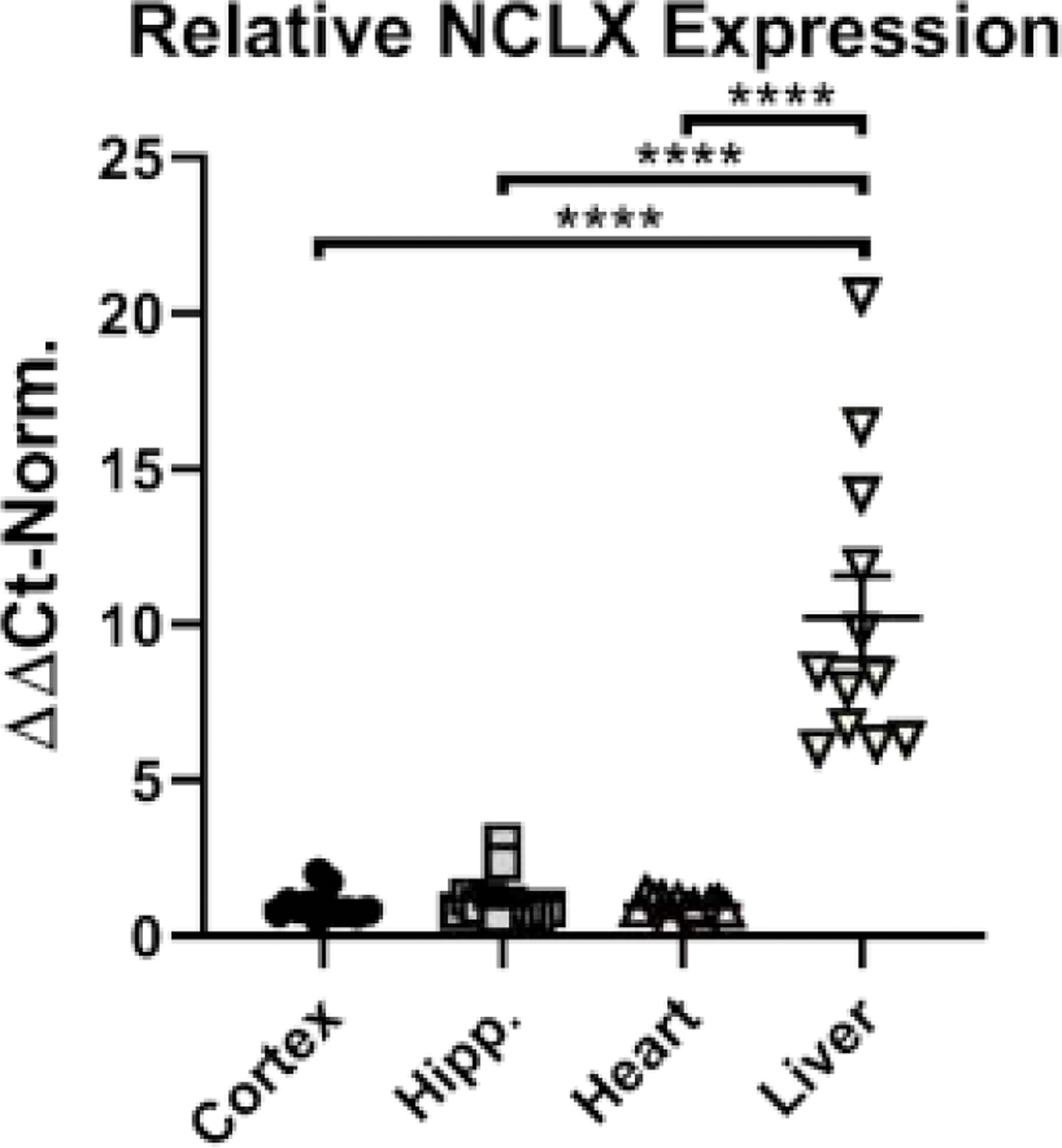
Quantification of NCLX expression in adult mouse cortex, hippocampus (Hipp.), heart and liver using real-time PCR. Data are presented as mean ± SEM, n=12 mice, analyzed using Kruskal-Wallis with Dunn’s multiple comparison post-hoc test. **** p<0.001
3.2. Na+-dependent Ca2+ extrusion from isolated brain mitochondria is inhibited by CGP37157 and slowed by Na+ replacement with Li+
Next, we used isolated brain mitochondria to determine the properties of mitochondrial Ca2+ efflux (Fig. 2). In these experiments, isolated brain mitochondria were loaded with Ca2+ in Na+-free solution and then treated with the MCU inhibitor Ru360 (10 µM) to block mitochondrial Ca2+ uptake, enabling measurements of mitochondrial Ca2+ efflux in isolation. Addition of 20 mM NaCl to the extramitochondrial solution rapidly induced strong Ca2+ efflux from mitochondria. This Na+-induced Ca2+ efflux was blocked by application of the mtNCX inhibitor CGP37157 (Fig.2B-D). As shown in Fig. 2, the addition of 20 mM LiCl also stimulated mitochondrial Ca2+ efflux that was sensitive to CGP37157. However, mitochondrial Ca2+ efflux triggered by Li+ was significantly slower than that induced by Na+ (Fig. 2C and D).
Figure 2. Unlike Na+, Li+ fails to effectively stimulate Ca2+ efflux from mouse brain mitochondria.
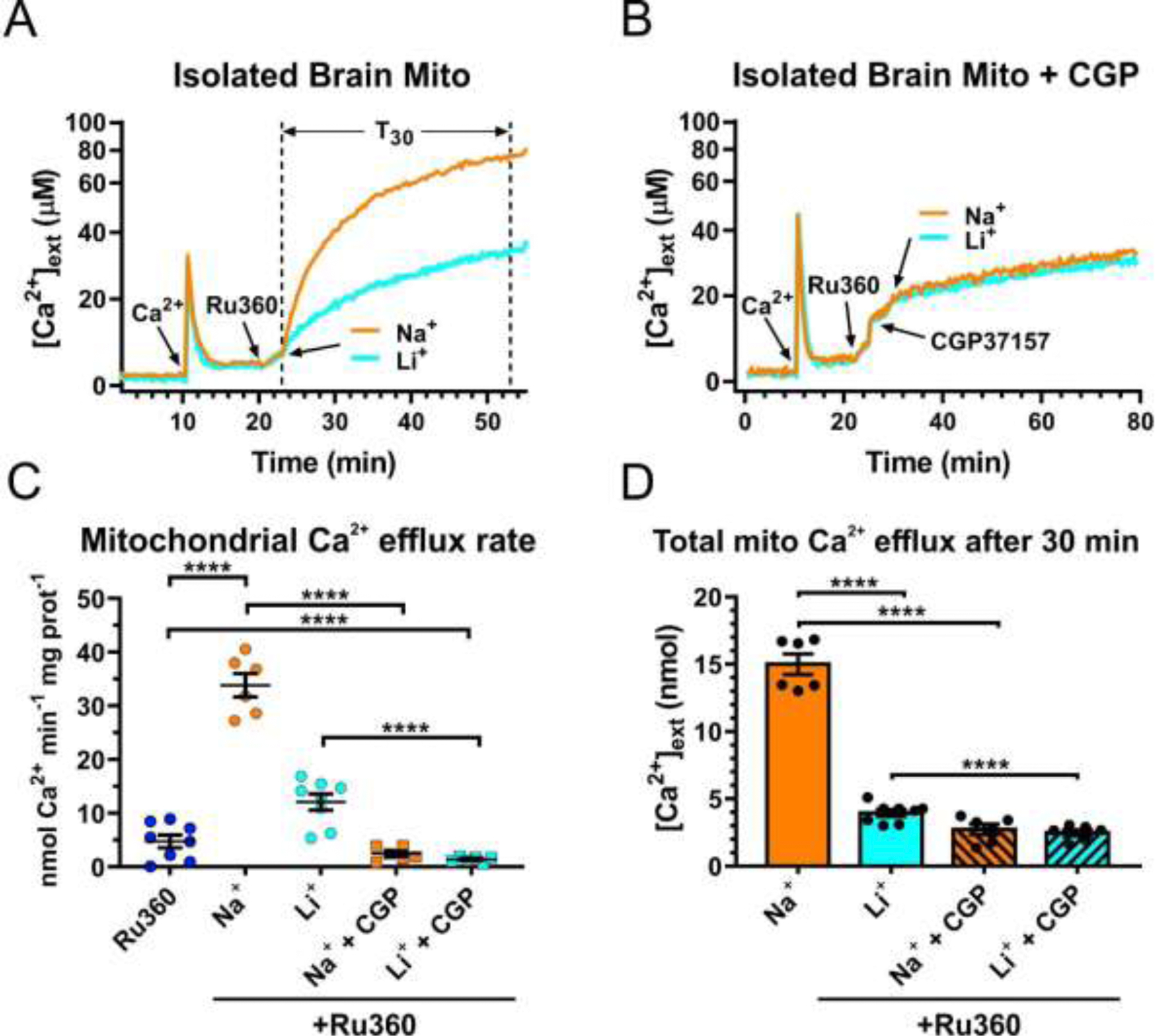
(A) and (B), Representative traces of extracellular Ca2+ ([Ca2+]ext) in a suspension of isolated brain mitochondria illustrating the protocol for measuring Na+ (Li+)-induced Ca2+ efflux from mitochondria in the absence (A) or presence (B) of the mtNCX inhibitor CGP37157 (30 µM). First, a pulse of Ca2+ (100 µM; arrow) was added to a suspension of mitochondria, followed by addition of the inhibitor of mitochondrial Ca2+ uniporter, Ru360 (10 µM; arrow) to block mitochondrial Ca2+ uptake; then either 20 mM Na+ (orange line) or Li+ (blue line) was added to induce Ca2+ efflux from mitochondria. This concentration of Na+ was chosen based on the reported mtNCX Km~8–10 mM for Na+ and the findings that 20 mM Na+ produces maximal activation of mtNCX in various tissues [6]. (C) Summary of the maximal rates of mitochondrial Ca2+ efflux induced by Ru360 alone (dark blue circles), 20 mM Na+ without (orange circles) or with 30 µM CGP37157 (orange squares), 20 mM Li+ without (light blue circles) or with 30 µM CGP37157 (light blue squares), in the continuous presence of 10 µM Ru360. (D) The amount of Ca2+ extruded from mitochondria during a 30 min period (T30 in (A), dotted vertical lines) after the addition of 20 mM Na+ (orange bar), Li+ (blue bar), without or with 30 µM CGP37157 (striped bars). Data are presented as mean ± SEM, n=6–8 independent experiments; ****p<0.0001, one-way ANOVA with Bonferroni’s post hoc test.
3.3. Mitochondrial Ca2+ efflux in hippocampal neurons is inhibited by Na+ replacement with Li+ and by CGP37157
Our findings that replacement of Na+ with Li+ significantly slowed mitochondrial Ca2+ efflux from isolated brain mitochondria (Fig. 2) was somewhat surprising given reports that NCLX-driven Ca2+ transport shows similar sensitivity to Na+ and Li+ [28–31]. Therefore, we decided to further test the effect of Li+ on mitochondrial Ca2+ extrusion in intact cells, using cultured primary mouse hippocampal neurons as a model. In these experiments, we simultaneously monitored [Ca2+]mt and [Ca2+]cyt using fluorescent Ca2+ indicators, mito-RGECO1 and Fura-2, respectively, as previously described [38, 47]. [Ca2+]cyt and [Ca2+]mt responses were induced by depolarization (30 mM KCl, 15 s), either under control conditions (140 mM NaCl in extracellular solution) or after extracellular Na+ had been completely replaced with Li+ (Fig. 3). Given that voltage-gated Na+ channels are highly permeable to Li+, this ion was expected to enter the cell upon depolarization [48, 49]. However unlike Na+, Li+ is negligibly transported out of the cell by the Na+/K+-ATPase [50, 51], which is expected to promote substitution of intracellular Na+ with Li+.
Figure 3. Substitution of Li+ for Na+ markedly inhibits mitochondrial Ca2+ efflux in hippocampal neurons.
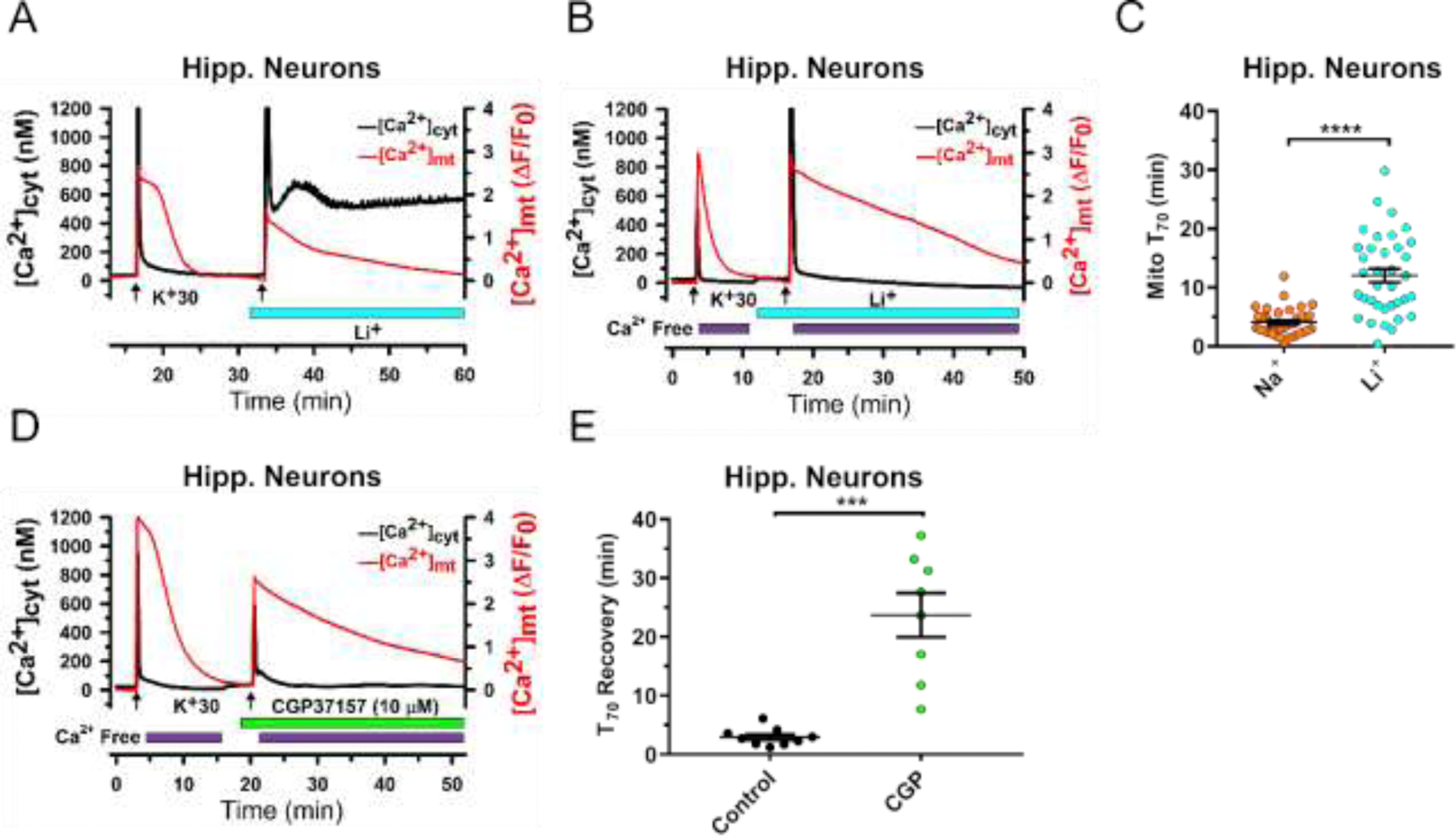
(A) and (B), Representative traces from experiments with simultaneous monitoring of [Ca2+]cyt (Fura-2; black) and [Ca2+]mt (mito-RGECO1; red) in cultured hippocampal neurons. [Ca2+]cyt and [Ca2+]mt transients were induced by brief depolarization using 30 mM KCl (K+30, 15 s, arrows). Substitution of Li+ for Na+ in the extracellular solution (0 mM Na+, 140 mM Li+) is indicated by blue bar under the traces. In (B), extracellular Ca2+ was removed (0 mM Ca2+, 100 µM EGTA) immediately after K+30 stimulation (purple bars). (C), Mitochondrial Ca2+ efflux kinetics was quantified by measuring the duration of 70% [Ca2+]mt recovery relative to its peak elevation (T70), and compared between the control conditions (140 mM NaCl in extracellular solution) and following Na+ replacement with Li+ in the extracellular solution. Data are presented as mean ± SEM, n=36; ****p<0.0001, two-tailed paired Student’s t-test. (D) Representative traces of [Ca2+]cyt (black) and [Ca2+]mt (red) show the effect of the mtNCX inhibitor CGP37157 (10 μM; green bar) on mitochondrial Ca2+ efflux in hippocampal neurons. (E), Mitochondrial Ca2+ efflux kinetics was quantified by measuring the duration of 70% [Ca2+]mt recovery relative to its peak elevation (T70), and compared before and after treatment with CGP37157. Data are presented as mean ± SEM, n=9; ***p<0.001, two-tailed paired Student’s t-test.
Under the control conditions, depolarization induced rapid Ca2+ transients in both the cytosolic and mitochondrial compartments, followed by recovery to baseline levels. Recovery of the [Ca2+]mt elevation was slower than the recovery of [Ca2+]cyt signal, consistent with the previous reports [38, 47]. When extracellular Na+ was replaced with Li+, the inhibition of Ca2+ clearance was prominent in both mitochondria and the cytosol (Fig. 3A). Given that changes in [Ca2+]cyt can affect mitochondrial Ca2+ transport, Li+-induced slowing of mitochondrial Ca2+ efflux could potentially have been a consequence of the prolonged [Ca2+]cyt elevation, with the latter being caused by reversal of the plasma membrane Na+/Ca2+ exchanger in the absence of extracellular Na+. To rule out this potential effect of elevated [Ca2+]cyt on mitochondrial Ca2+ efflux, we repeated the Na+ replacement experiments with extracellular Ca2+ removed immediately at the end of the depolarization. This approach indeed normalized recovery of [Ca2+]cyt in Li+-containing extracellular solution (Fig. 3B). Notably, although recovery of [Ca2+]cyt following depolarization was rapid under these conditions, the replacement of Na+ with Li+ resulted in marked slowing of recovery of [Ca2+]mt (Fig. 3B and C). These findings in intact hippocampal neurons were similar to those in isolated brain mitochondria (Fig. 2). Specifically, T70 of [Ca2+]mt recovery increased from 4.1 ± 0.4 min in controls (Na+-containing solution) to 12.1 ± 1.1 min (p<0.0001; Student’s t-test) in the cells treated with Li+-containing extracellular solution (Fig. 3C). As positive controls, we tested the effect of the mtNCX inhibitor CGP37157, and found that in cells treated with the inhibitor [Ca2+]mt recovery was significantly slowed (Fig. 3D and E). Overall, these data are consistent with our observations for isolated brain mitochondria (Fig.2), and they suggest that first, mtNCX is a major mechanism responsible for mitochondrial Ca2+ efflux in neurons, and second, that Li+ is significantly less effective than Na+ in driving this Ca2+ efflux.
3.4. Na+-dependent Ca2+ efflux from isolated heart mitochondria is inhibited by CGP37157 and slowed by Na+ replacement with Li+
We also examined the properties of mitochondrial Ca2+ extrusion in another excitable tissue, the heart (Fig. 4). As described for the experiments using mitochondria isolated from brain (Fig.2), isolated heart mitochondria were loaded with Ca2+ in a Na+-free solution and then treated with the MCU inhibitor Ru360 (10 µM) to block Ca2+ uptake. As with the brain mitochondria, 20 mM NaCl stimulated a strong Ca2+ efflux from heart mitochondria (Fig. 4A). However, the rate of Ca2+ efflux from heart mitochondria was significantly slower (12.3 ± 0.4 nmol Ca2+ min−1 x mg protein−1; n=6) than the rate of Ca2+ efflux from brain mitochondria (Fig.2C; 33.8 ± 2.2 nmol Ca2+ min−1 x mg protein−1; n=6; p<0.0001; ANOVA with Bonferroni’s post hoc test). Na+-induced Ca2+ efflux from heart mitochondria was blocked by the mtNCX inhibitor CGP37157 (30 µM; Fig. 4). Addition of 20 mM LiCl was also able to stimulate Ca2+ extrusion from heart mitochondria, although at a markedly slower rate than 20 mM NaCl (Fig. 4). Thus, similar with brain mitochondria, Ca2+ efflux from heart mitochondria is driven by Na+, blocked by CGP37157, and significantly less sensitive to Li+ than Na+ as an exchange partner.
Figure 4. Na+-induced Ca2+ extrusion from isolated heart mitochondria is inhibited by Li+ and CGP37157.
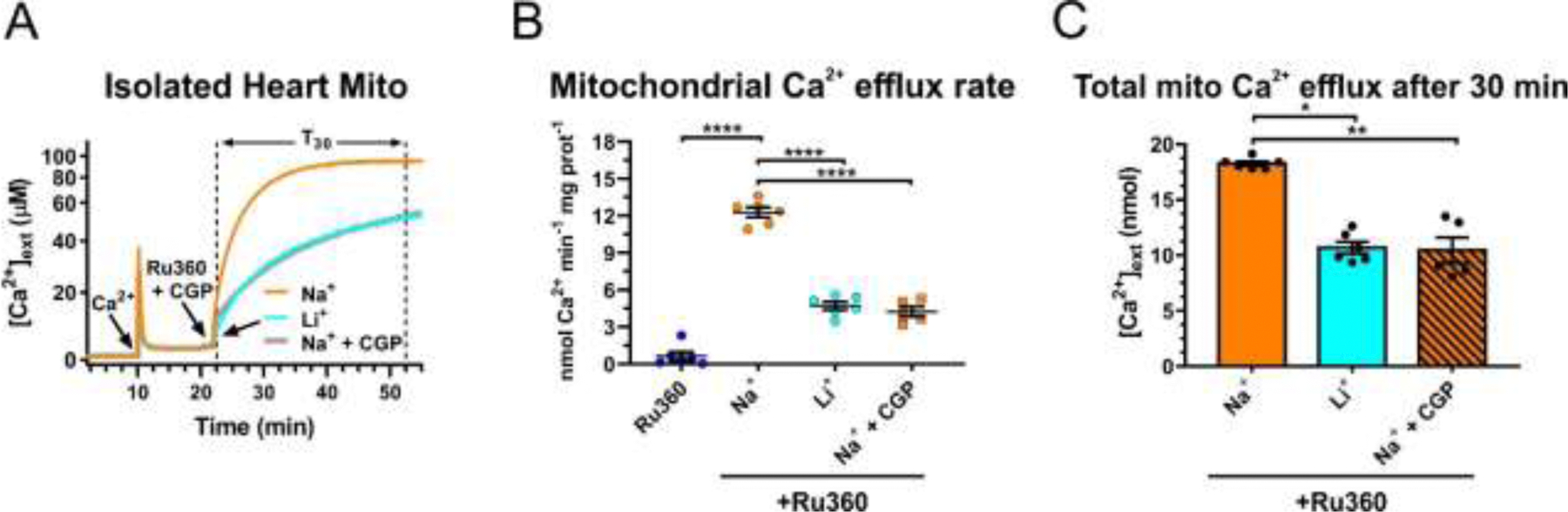
(A) Representative traces of extracellular Ca2+ ([Ca2+]ext) in a suspension of mitochondria isolated from the heart, illustrating the protocol for measuring Na+ (Li+)-induced Ca2+ efflux from mitochondria. A pulse of Ca2+ (100 µM; arrow) was added to the suspension of mitochondria, followed by addition of the inhibitor of mitochondrial Ca2+ uniporter, Ru360 (10 µM; arrow) to block mitochondrial Ca2+ uptake, and in some instances mtNCX inhibitor CGP37157 (30 µM, gray line). Then either 20 mM Na+ (orange line) or Li+ (blue line) was added to induce Ca2+ efflux from mitochondria in the continuous presence of Ru360. (B) Quantification of the maximal rates of mitochondrial Ca2+ efflux induced by Ru360 alone (dark blue circles), 20 mM Na+ without (orange circles) or with 30 µM CGP37157 (orange squares), and 20 mM Li+ (light blue circles), in the continuous presence of 10 µM Ru360. (C) The amount of Ca2+ extruded from mitochondria during a 30 min period (T30 in (A), dotted vertical lines) after the addition of 20 mM Na+ (orange bar), Li+ (blue bar), or Na+ plus 30 µM CGP37157 (striped bar). Data are presented as mean ± SEM, n=5–8 independent experiments; *p<0.05, **p<0.01, ****p<0.0001, one-way ANOVA with Bonferroni’s post hoc test.
3.5. Ca2+ efflux from isolated liver mitochondria is insensitive to Na+ or CGP37157
Whether or not mitochondria from non-excitable tissue, such as liver, could utilize an electrogenic mtNCX mechanism has been a matter of debate [6, 11, 12, 20]. Thus, we applied the same protocol that we established for brain and heart mitochondria (Figs. 2 and 4) to isolated liver mitochondria (Fig. 5A) and monitored Ca2+ efflux from this organelle. Similar to the results for brain and heart mitochondria, application of Ru360 caused a small but detectable Ca2+ efflux from liver mitochondria (1.8 ± 0.3 nmol Ca2+ min−1 x mg protein−1; n=7; Fig. 5B). However, in contrast to the findings for brain and heart mitochondria, the addition of 20 mM Na+ failed to significantly stimulate Ca2+ extrusion (1.7±0.1 nmol Ca2+ min−1 x mg protein−1; n=12; Fig. 5B) compared to Ru360 alone (p>0.05; one-way ANOVA with Bonferroni’s post hoc test), and the mtNCX inhibitor CGP37157 had no effect. Li+ also failed to stimulate Ca2+ extrusion from liver mitochondria (Fig. 5).
Figure 5. Effects of Na+, Li+ and CGP37157 on Ca2+ efflux from liver mitochondria.
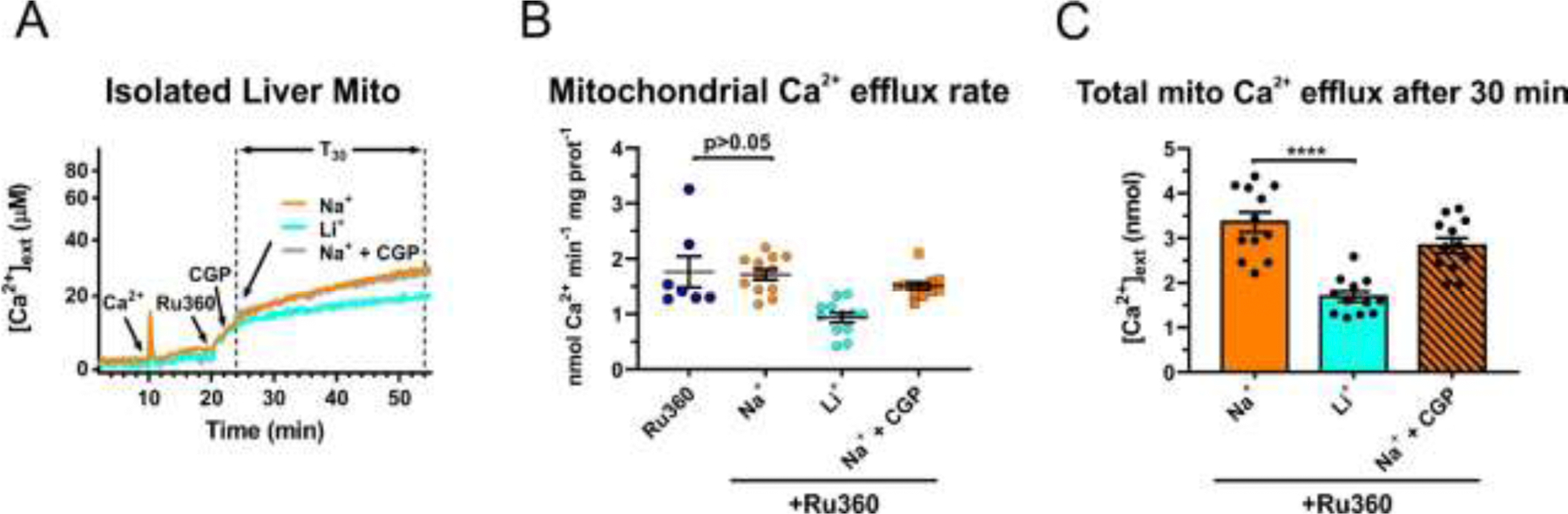
(A) Representative traces of extracellular Ca2+ ([Ca2+]ext) in a suspension of isolated liver mitochondria illustrating the protocol for measuring Na+ (Li+)-induced Ca2+ efflux from mitochondria. First a pulse of Ca2+ (100 µM; arrow) was added to a suspension of mitochondria, followed by addition of the MCU inhibitor, Ru360 (10 µM; arrow) to block mitochondrial Ca2+ uptake, and in some instances mtNCX inhibitor CGP37157 (30 µM, gray line); then either 20 mM Na+ (orange line) or Li+ (blue line) was added to induce Ca2+ efflux from mitochondria, all in continuous presence of Ru360. (B) Quantification of the maximal rates of mitochondrial Ca2+ efflux induced by Ru360 alone (dark blue circles), 20 mM Na+ without (orange circles) or with 30 µM CGP37157 (orange squares), and 20 mM Li+ (light blue circles), in the continuous presence of 10 µM Ru360. (C) The amount of Ca2+ extruded from mitochondria during a 30 min period (T30 in (A), dotted vertical lines) after the addition of 20 mM Na+ (orange bar), Li+ (blue bar), or Na+ plus 30 µM CGP37157 (striped bar). Data are presented as mean ± SEM, n=7–12 independent experiments; one-way ANOVA with Bonferroni’s post hoc test.
These results suggest that in contrast to brain and heart mitochondria, mtNCX does not significantly contribute to mitochondrial Ca2+ efflux in the liver.
3.6. Mitochondrial Ca2+ efflux in primary mouse hepatocytes is slow and insensitive to CGP37157
To further examine the implications of distinctly slow Ca2+ efflux from liver mitochondria at a single cell level, we simultaneously monitored [Ca2+]cyt and [Ca2+]mt in primary mouse hepatocytes (Fig. 6), using the same approach as described for hippocampal neurons (Fig. 3). [Ca2+]cyt and [Ca2+]mt responses were induced by ATP (100 µM), which is a well-known activator of Ca2+ signaling in hepatocytes via P2Y and also likely P2X receptors [52–55]. As shown in Fig. 6A-B, ATP produced a large [Ca2+]cyt elevation that rapidly recovered toward the baseline. In contrast, the recovery of [Ca2+]mt was very slow, taking on average more than 20 min to return to the baseline (Fig. 6A). Notably, the process of [Ca2+]mt recovery in hepatocytes was significantly slower (T70=19.7 ± 2.7 min; n=9) than that in hippocampal neurons (T70=4.1 ± 0.4 min; n=36; p<0.0001; Student’s t-test). The rate of mitochondrial Ca2+ efflux in hepatocytes was not significantly affected by the treatment with the mtNCX inhibitor CGP37157 (Fig. 6C).
Figure 6. Mitochondrial Ca2+ efflux in primary mouse hepatocytes is slow and insensitive to CGP37157.
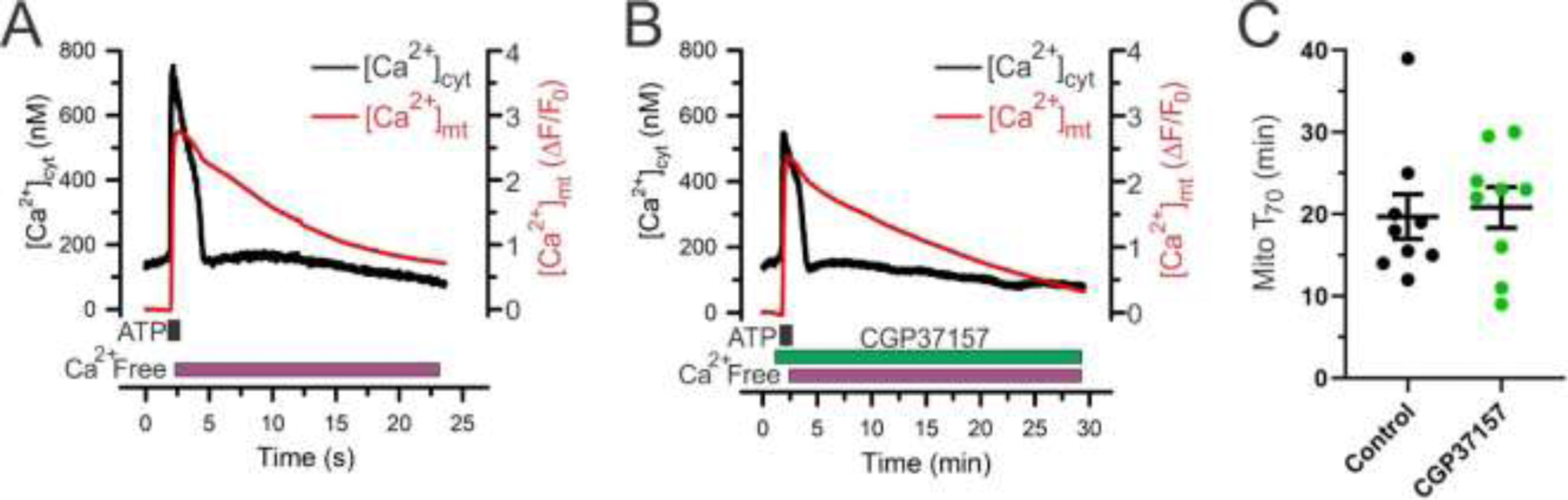
(A, B), Examples of simultaneous measurements of [Ca2+]cyt (Fura-2; black) and [Ca2+]mt (mito-RGECO1; red) in primary mouse hepatocytes in response to 100 µM ATP (1 min) in the absence (A) or presence (B) of the mtNCX inhibitor CGP37157 (10 µM). (C) Comparison of mitochondrial Ca2+ efflux kinetics studied under control conditions (n=9; black) or in the presence of 10 µM CGP37157 (n=9; green), which was quantified by measuring the duration of 70% [Ca2+]mt recovery relative to its peak elevation (T70). The T70 values were obtained from the experiments as those shown in (A) and (B). CGP37157 treatment did not significantly change the [Ca2+]mt recovery kinetics compared to the control (p=0.77; unpaired Student’s t-test). Data are presented as mean ± SEM.
Overall, these results are consistent with the observations in isolated liver mitochondria and suggest that mtNCX does not play a significant role in mediating mitochondrial Ca2+ efflux in the liver.
3.7. Extramitochondrial localization of NCLX overexpressed in liver cells
It is thought that NCLX is responsible for the mtNCX transport in various tissues [20, 21]. Therefore, our observations that the mtNCX mechanism does not significantly contribute to mitochondrial Ca2+ efflux in the liver was surprising, given a relatively high expression of NCLX mRNA in the liver (Fig. 1). However, several groups have reported extramitochondrial localization and function of NCLX [28, 56–58], which could potentially help to explain our results. To test the possibility of extramitochondrial localization of NCLX in the liver, we co-expressed a Flag-tagged NCLX in the mouse liver cell line AML12 [43] together with mitochondria-targeted DsRed and ER-targeted GFP reporter plasmids. Co-localization analysis using confocal microscopy and Pearson’s correlation coefficient [46, 59] showed that a large fraction of NCLX was localized outside of mitochondria (Fig. 7). In contrast, Flag-tagged MCU demonstrated mitochondria-restricted subcellular localization in AML12 cells.
Figure 7. Extramitochondrial localization of NCLX overexpressed in liver AML12 cells.
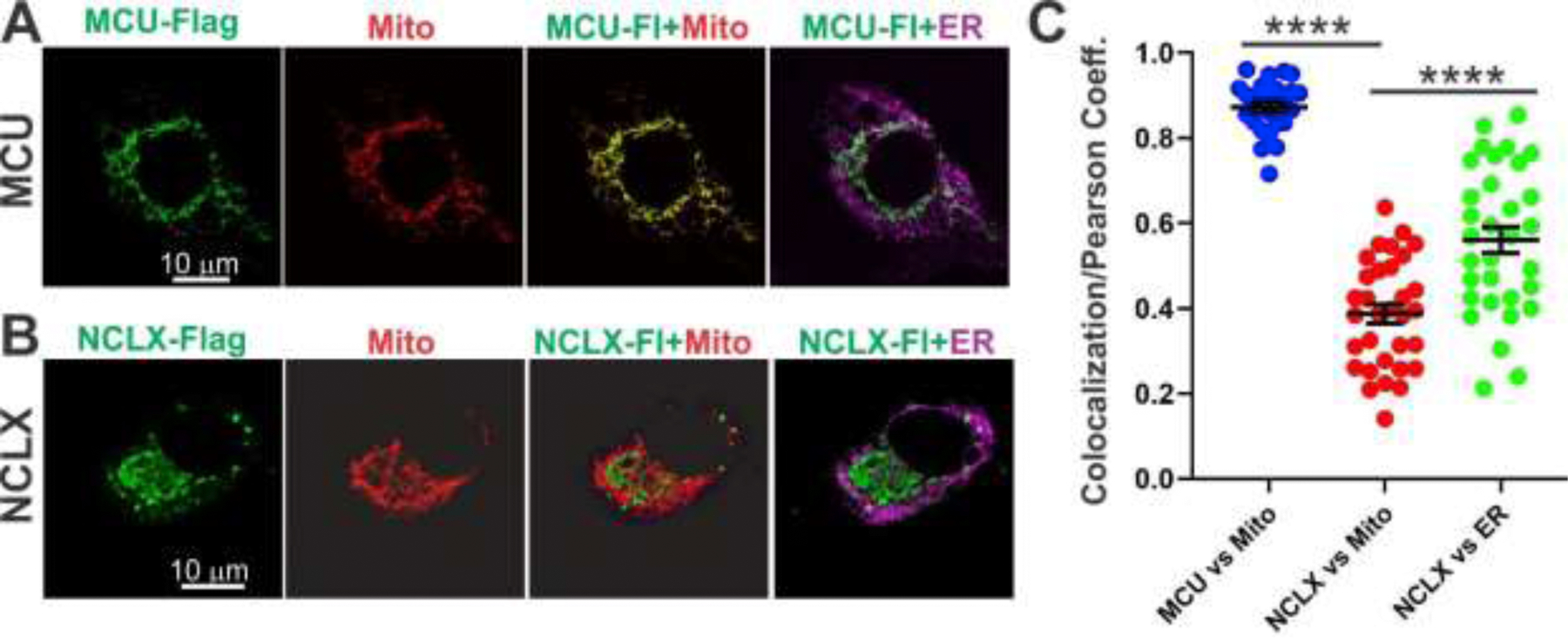
(A, B) Subcellular localization of MCU-Flag (A, green) and NCLX-Flag (B, green) in mouse liver AML12 cells relative to mitochondria (mito-V5-DsRed; red) and ER (GFP-KEDL; magenta). (C) Summary of colocalization of MCU-Flag with mitochondria (blue; n=24 cells), NCLX-Flag with mitochondria (red; n=32 cells) and NCLX-Flag ER (green; n=32 cells) quantified by using the Pearson’s correlation coefficient [46, 59]. Data are presented as mean ± SEM; ****p<0.0001, one-way ANOVA with Bonferroni’s post hoc test.
4. Discussion
This study demonstrates that the properties of mitochondrial Ca2+ extrusion differ significantly between the brain, heart and liver. More specifically, our findings suggest that mtNCX is the principal mechanism responsible for Ca2+ extrusion from heart and brain mitochondria, but not in liver, where contribution of this mechanism was undetectable. This conclusion is supported by robust effects of Na+ and mtNCX inhibitor CGP37157 on mitochondrial Ca2+ efflux in both brain and heart mitochondria, and the absence of any effects of these treatments in liver mitochondria. We also found that Li+ was significantly less effective than Na+ in driving mitochondrial Ca2+ efflux, as apparent from a 70% difference in the rate of Ca2+ efflux triggered by these ions. Another key finding was that NCLX was expressed in all the tissues tested, including brain, heart and liver, with the highest level detected in the liver.
Our finding that mtNCX plays an important role in the brain and heart, but not in the liver, is consistent with earlier work by Carafoli and colleagues [11, 60]. Our conclusions are further strengthened by the distinctive effects of the mtNCX inhibitor CGP37157 [32–34] both in isolated mitochondria and intact cells (Fig.2–6). Quantitative comparison under similar conditions showed that the rate of Ca2+ efflux from brain mitochondria was nearly 3-fold greater than that found for heart mitochondria (33.8 ± 2.2 and 12.3 ± 0.4 nmol Ca2+ min−1 x mg protein−1, respectively). These values are consistent with rates previously reported for mitochondria of the rat heart [60], but exceed those for the mitochondria of rat and guinea pig brain [11, 61]. The latter discrepancy could potentially be explained by differences in mitochondrial Ca2+ load, ADP concentration and species used [62]. Importantly, our findings that Ca2+ efflux from liver mitochondria with or without Na+ was approximately 10-fold slower than Na+-dependent mitochondrial Ca2+ efflux from heart and brain mitochondria, are in good agreement with findings from previous studies [11, 21, 60].
Although consistent with previous work on isolated mitochondria [11, 21, 60], the lack of a detectable contribution of mtNCX in the liver was surprising in the context of high NCLX expression in this organ (Fig. 1). Our findings in mouse tissues are in good agreement with the NCLX expression profiling in human tissues, which also showed that NCLX expression in the liver is higher than that in the brain and heart [10] (Table 1 in this publication). In addition to the proposed role of NCLX in mitochondria, previous work suggested that this protein (also known as NCKX6) functions in the plasma membrane and possibly other cell membranes and organelles, including the endoplasmic reticulum [28, 56–58]. Thus, the apparent discrepancy between NCLX expression and a lack of its contribution to mitochondrial Ca2+ efflux in the liver might be potentially explained by the extra-mitochondrial localization of NCLX in the liver. Our finding that NCLX localization was mostly extramitochondrial in liver AML12 cells (Fig. 7) is consistent with this idea. The mechanisms that regulate NCLX trafficking within the cell remain largely unknown, and future work is needed to clarify some of the conflicting evidence about its cellular localization [28, 56–58].
In this study, we took advantage of the reported ability of NCLX to utilize either Li+ or Na+ to transport Ca2+ through an exchange mechanism [28–31], testing the consequences of Na+ substitution with Li+ on the rate of mitochondrial Ca2+ efflux. Our conclusion that Li+ is much less effective than Na+ in driving mitochondrial Ca2+ extrusion is supported by experiments using both isolated mitochondria and intact cells (Figs. 2–4). For the experiments in cultured hippocampal neurons, we simultaneously measured [Ca2+]mt and [Ca2+]cyt. Given that cytosolic Ca2+ changes can significantly impact mitochondrial Ca2+ transport, simultaneous monitoring of [Ca2+]mt and [Ca2+]cyt in the same cell provided an important control for potential effects of ion substitution on cytosolic Ca2+ signaling. This approach also enabled us to optimize experimental conditions for selective manipulation of mitochondrial Ca2+ transport while preserving the amplitude and kinetics of cytosolic Ca2+ responses. We found that under these controlled conditions, replacement of Na+ with Li+ led to marked slowing of Ca2+ efflux from mitochondria (Fig. 3). Importantly, these findings in intact cells were consistent with those from isolated brain and heart mitochondria (Figs. 2 and 4), with the latter allowing direct monitoring of Ca2+ fluxes in and out of mitochondria. As expected, the mtNCX inhibitor CGP37157 blocked mitochondrial Ca2+ efflux in both intact cells (Fig. 3) and isolated mitochondria (Figs. 2 and 4).
Our finding that Li+ was significantly less effective than Na+ in driving Ca2+ efflux from brain and heart mitochondria was somewhat unexpected given the proposed role of NCLX in mediating Na+-dependent Ca2+ extrusion from mitochondria [13–18]. Previous work using intact or permeabilized cells had suggested that NCLX is able to exchange Ca2+ for Li+ nearly as effectively as for Na+, as well as identified the amino acids within NCLX that are critical for this Li+ sensitivity [28–31]. However, our results regarding the effects of Li+ in isolated mitochondria are consistent with those from studies by others using isolated mitochondria obtained from the heart and vascular smooth muscles [60, 63]. Although the original, and commonly cited, article by the Carafoli group reported that both Na+ and Li+ can stimulate Ca2+ efflux from isolated heart mitochondria [64], a later publication by the same group provided quantitative analysis, which showed that the rate of Li+-driven mitochondrial Ca2+ efflux was only ~25–30% of that driven by Na+ [60] (Fig.4 in that publication). Similarly, measurements in mitochondria isolated from vascular smooth muscles demonstrated that the rate of Li+-driven mitochondrial Ca2+ efflux was only ~10% of that driven by Na+ [63]. Thus, Li+ is markedly less effective than Na+ in driving mtNCX-mediated Ca2+ efflux from mitochondria.
Utilization of the mtNCX mechanism in excitable cells such as neurons and cardiomyocytes is well suited to their patterns of physiological activity and Ca2+ signaling [9, 22, 65–69]. Indeed, high frequency [Ca2+]cyt spikes in cardiomyocytes associated with every heart beat (up to 400–600 beats/min in rodents), and similarly frequent [Ca2+]cyt transients produced by bursts of action potentials in neurons, necessitate rapid Ca2+ unloading from mitochondrial to prevent toxic accumulation of Ca2+ in the matrix. The mtNCX transport is also aided by Na+ entering through voltage-gated Na+ channels, which could potentially elevate the intracellular Na+ concentration to above 10 mM [70, 71], a value comparable to the mtNCX Km for Na+ (8–10 mM) [6]. In the case of liver, a much slower rate of mitochondrial Ca2+ extrusion in the liver might be optimal for the relatively infrequent [Ca2+]cyt spikes that characterize hepatocytes [9, 72]. This is expected to prolong Ca2+ retention within the matrix after a single [Ca2+]cyt transient (Fig. 6), allowing for more efficient use of Ca2+ signals to support stimulation of Ca2+-dependent dehydrogenases and ATP synthesis. Future studies will be needed to clarify the specific contributions of mtNCX and mtHCX mechanisms to Ca2+ signaling in excitable and non-excitable cells, and more generally, to identify molecular mechanisms that enable the coordination of mitochondrial Ca2+ uptake and release, processes that are tailored to unique characteristics of Ca2+ signaling and the metabolic needs of a particular cell type or tissue.
HIGHLIGHTS.
Mitochondrial Na+/Ca2+ exchanger is active in brain and heart, but not in liver
NCLX is expressed in brain, heart and liver
Li+ is significantly less effective than Na+ in driving mitochondrial Ca2+ efflux
Acknowledgements:
This work was supported by National Institutes of Health grants NS087068 and NS096246 (Y.M.U.) and DK116624 (S.S. and Y.M.U.). J.E.R. was supported by AHA predoctoral fellowship 15PRE25310013 and a postdoctoral fellowship through NIH T32 Grant HL007638, G.C.W. was supported by a predoctoral fellowship through NIH T32 Grant GM067795, and M.N. was supported by NIH F31 predoctoral fellowship NS106773. We would like to thank Drs. Guo and Merrill for their help and advices.
Abbreviations:
- MCU
mitochondrial Ca2+ uniporter
- mtNCX
mitochondrial Na+/Ca2+ exchanger
- mtHCX
mitochondrial H+/Ca2+ exchanger
- NCLX
Na+/Ca2+/Li+ exchanger
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing financial interests.
References
- [1].Nicholls DG, Mitochondria and calcium signaling, Cell calcium, 38 (2005) 311–317. [DOI] [PubMed] [Google Scholar]
- [2].Glancy B, Balaban RS, Role of mitochondrial Ca2+ in the regulation of cellular energetics, Biochemistry, 51 (2012) 2959–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kamer KJ, Mootha VK, The molecular era of the mitochondrial calcium uniporter, Nat Rev Mol Cell Biol, 16 (2015) 545–553. [DOI] [PubMed] [Google Scholar]
- [4].De Stefani D, Rizzuto R, Pozzan T, Enjoy the Trip: Calcium in Mitochondria Back and Forth, Annu Rev Biochem, 85 (2016) 161–192. [DOI] [PubMed] [Google Scholar]
- [5].Usachev YM, Mitochondrial Ca2+ Transport in the Control of Neuronal Functions: Molecular and Cellular Mechanisms in: Gribkoff VK, Jonas EA, Hardwick JM (Eds.) The Functions, Disease-Related Dysfunctions, and Therapeutic Targeting of Neuronal Mitochondria, Wiley, Hoboken, NJ, 2016, pp. 103–129. [Google Scholar]
- [6].Bernardi P, Mitochondrial transport of cations: channels, exchangers, and permeability transition, Physiological reviews, 79 (1999) 1127–1155. [DOI] [PubMed] [Google Scholar]
- [7].Drago I, Pizzo P, Pozzan T, After half a century mitochondrial calcium in‐ and efflux machineries reveal themselves, The EMBO Journal, 30 (2011) 4119–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fieni F, Bae Lee S, Jan YN, Kirichok Y, Activity of the mitochondrial calcium uniporter varies greatly between tissues, Nature Communications, 3 (2012) 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Paillard M, Csordas G, Szanda G, Golenar T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spat A, Hajnoczky G, Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca(2+) Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU, Cell Rep, 18 (2017) 2291–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D, MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake, Cell Death & Differentiation, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Crompton M, Moser R, LÜDi H, Carafoli E, The Interrelations between the Transport of Sodium and Calcium in Mitochondria of Various Mammalian Tissues, European Journal of Biochemistry, 82 (1978) 25–31. [DOI] [PubMed] [Google Scholar]
- [12].Boyman L, Williams GS, Khananshvili D, Sekler I, Lederer WJ, NCLX: the mitochondrial sodium calcium exchanger, J Mol Cell Cardiol, 59 (2013) 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I, NCLX is an essential component of mitochondrial Na+/Ca2+ exchange, Proceedings of the National Academy of Sciences, 107 (2010) 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Parnis J, Montana V, Delgado-Martinez I, Matyash V, Parpura V, Kettenmann H, Sekler I, Nolte C, Mitochondrial exchanger NCLX plays a major role in the intracellular Ca2+ signaling, gliotransmission, and proliferation of astrocytes, J Neurosci, 33 (2013) 7206–7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kostic M, Ludtmann MHR, Bading H, Hershfinkel M, Steer E, Chu CT, Abramov AY, Sekler I, PKA Phosphorylation of NCLX Reverses Mitochondrial Calcium Overload and Depolarization, Promoting Survival of PINK1-Deficient Dopaminergic Neurons, Cell reports, 13 (2015) 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nita II, Caspi Y, Gudes S, Fishman D, Lev S, Hersfinkel M, Sekler I, Binshtok AM, Privileged crosstalk between TRPV1 channels and mitochondrial calcium shuttling machinery controls nociception, Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1863 (2016) 2868–2880. [DOI] [PubMed] [Google Scholar]
- [17].Nita II, Hershfinkel M, Kantor C, Rutter GA, Lewis EC, Sekler I, Pancreatic beta-cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria, FASEB J, 28 (2014) 3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, Tsai EJ, Molkentin JD, Chen X, Madesh M, Houser SR, Elrod JW, The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability, Nature, 545 (2017) 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pathak T, Gueguinou M, Walter V, Delierneux C, Johnson MT, Zhang X, Xin P, Yoast RE, Emrich SM, Yochum GS, Sekler I, Koltun WA, Gill DL, Hempel N, Trebak M, Dichotomous role of the human mitochondrial Na(+)/Ca2(+)/Li(+) exchanger NCLX in colorectal cancer growth and metastasis, Elife (Cambridge), 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Haworth RA, Hunter DR, Berkoff HA, Na+ releases Ca2+ from liver, kidney and lung mitochondria, FEBS Letters, 110 (1980) 216–218. [DOI] [PubMed] [Google Scholar]
- [21].Wingrove DE, Gunter TE, Kinetics of mitochondrial calcium transport. I. Characteristics of the sodium-independent calcium efflux mechanism of liver mitochondria, J Biol Chem, 261 (1986) 15159–15165. [PubMed] [Google Scholar]
- [22].García-Chacón LE, Nguyen KT, David G, Barrett EF, Extrusion of Ca(2+) from mouse motor terminal mitochondria via a Na(+)–Ca(2+) exchanger increases post-tetanic evoked release, The Journal of Physiology, 574 (2006) 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Medvedeva YV, Kim M-S, Usachev YM, Mechanisms of Prolonged Presynaptic Ca2+ Signaling and Glutamate Release Induced by TRPV1 Activation in Rat Sensory Neurons, The Journal of Neuroscience, 28 (2008) 5295–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kim M-S, Usachev YM, Mitochondrial Ca2+ Cycling Facilitates Activation of the Transcription Factor NFAT in Sensory Neurons, The Journal of Neuroscience, 29 (2009) 12101–12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, Abramov AY, PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death, Mol Cell, 33 (2009) 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stanika RI, Villanueva I, Kazanina G, Andrews SB, Pivovarova NB, Comparative impact of voltage-gated calcium channels and NMDA receptors on mitochondria-mediated neuronal injury, The Journal of Neuroscience, 32 (2012) 6642–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Assali EA, Jones AE, Veliova M, Acin-Perez R, Taha M, Miller N, Shum M, Oliveira MF, Las G, Liesa M, Sekler I, Shirihai OS, NCLX prevents cell death during adrenergic activation of the brown adipose tissue, Nat Commun, 11 (2020) 3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, Silverman WF, Argaman M, Sekler I, Lithium-Calcium Exchange Is Mediated by a Distinct Potassium-independent Sodium-Calcium Exchanger, Journal of Biological Chemistry, 279 (2004) 25234–25240. [DOI] [PubMed] [Google Scholar]
- [29].Lytton J, Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport, Biochem J, 406 (2007) 365–382. [DOI] [PubMed] [Google Scholar]
- [30].Khananshvili D, The SLC8 gene family of sodium-calcium exchangers (NCX) - structure, function, and regulation in health and disease, Mol Aspects Med, 34 (2013) 220–235. [DOI] [PubMed] [Google Scholar]
- [31].Roy S, Dey K, Hershfinkel M, Ohana E, Sekler I, Identification of residues that control Li+ versus Na+ dependent Ca2+ exchange at the transport site of the mitochondrial NCLX, Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1864 (2017) 997–1008. [DOI] [PubMed] [Google Scholar]
- [32].Thayer SA, Usachev YM, Pottorf WJ, Modulating Ca2+ clearance from neurons, Front Biosci, 7 (2002) d1255–1279. [DOI] [PubMed] [Google Scholar]
- [33].Cox DA, Matlib MA, A role for the mitochondrial Na(+)-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria, The Journal of biological chemistry, 268 (1993) 938–947. [PubMed] [Google Scholar]
- [34].Cox DA, Conforti L, Sperelakis N, Matlib MA, Selectivity of Inhibition of Na+-Ca2+ Exchange of Heart Mitochondria by Benzothiazepine CGP-37157, Journal of Cardiovascular Pharmacology, 21 (1993) 595–599. [DOI] [PubMed] [Google Scholar]
- [35].Hamilton J, Brustovetsky T, Rysted JE, Lin Z, Usachev YM, Brustovetsky N, Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria, J Biol Chem, 293 (2018) 15652–15663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kneen M, Farinas J, Li Y, Verkman AS, Green fluorescent protein as a noninvasive intracellular pH indicator, Biophys J, 74 (1998) 1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wu J, Prole David L., Shen Y, Lin Z, Gnanasekaran A, Liu Y, Chen L, Zhou H, Chen SRW, Usachev Yuriy M., Taylor Colin W., Campbell Robert E., Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum, Biochemical Journal, 464 (2014) 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rysted JE, Lin Z, Usachev YM, Techniques for Simultaneous Mitochondrial and Cytosolic Ca2+ Imaging in Neurons, in: Strack S, Usachev YM (Eds.) Techniques to Investigate Mitochondrial Function in Neurons, Humana Press, New York, 2017, pp. 151–178. [Google Scholar]
- [39].Gray LR, Sultana MR, Rauckhorst AJ, Oonthonpan L, Tompkins SC, Sharma A, Fu X, Miao R, Pewa AD, Brown KS, Lane EE, Dohlman A, Zepeda-Orozco D, Xie J, Rutter J, Norris AW, Cox JE, Burgess SC, Potthoff MJ, Taylor EB, Hepatic Mitochondrial Pyruvate Carrier 1 Is Required for Efficient Regulation of Gluconeogenesis and Whole-Body Glucose Homeostasis, Cell Metab, 22 (2015) 669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Xiang C, Du Y, Meng G, Soon Yi L, Sun S, Song N, Zhang X, Xiao Y, Wang J, Yi Z, Liu Y, Xie B, Wu M, Shu J, Sun D, Jia J, Liang Z, Sun D, Huang Y, Shi Y, Xu J, Lu F, Li C, Xiang K, Yuan Z, Lu S, Deng H, Long-term functional maintenance of primary human hepatocytes in vitro, Science, 364 (2019) 399–402. [DOI] [PubMed] [Google Scholar]
- [41].Grynkiewicz G, Poenie M, Tsien RY, A new generation of Ca2+ indicators with greatly improved fluorescence properties, Journal of Biological Chemistry, 260 (1985) 3440–3450. [PubMed] [Google Scholar]
- [42].Shuttleworth TJ, Thompson JL, Effect of temperature on receptor-activated changes in [Ca2+]i and their determination using fluorescent probes, Journal of Biological Chemistry, 266 (1991) 1410–1414. [PubMed] [Google Scholar]
- [43].Wu JC, Merlino G, Fausto N, Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha, Proc Natl Acad Sci U S A, 91 (1994) 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Merrill RA, Dagda RK, Dickey AS, Cribbs JT, Green SH, Usachev YM, Strack S, Mechanism of Neuroprotective Mitochondrial Remodeling by PKA/AKAP1, PLoS Biol, 9 (2011) e1000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Strack S, Cribbs JT, Allosteric modulation of Drp1 mechanoenzyme assembly and mitochondrial fission by the variable domain, J. Biol. Chem, 287 (2012) 10990–11001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bolte S, Cordelieres FP, A guided tour into subcellular colocalization analysis in light microscopy, J Microsc, 224 (2006) 213–232. [DOI] [PubMed] [Google Scholar]
- [47].Wu J, Prole DL, Shen Y, Lin Z, Gnanasekaran A, Liu Y, Chen L, Zhou H, Chen SR, Usachev YM, Taylor CW, Campbell RE, Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum, Biochem J, 464 (2014) 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hille B, The permeability of the sodium channel to metal cations in myelinated nerve, The Journal of general physiology, 59 (1972) 637–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Reiser G, Scholz F, Hamprecht B, Pharmacological and electrophysiological characterization of lithium ion flux through the action potential sodium channel in neuroblastoma X glioma hybrid cells, J Neurochem, 39 (1982) 228–234. [DOI] [PubMed] [Google Scholar]
- [50].Gordon TR, Kocsis JD, Waxman SG, Electrogenic pump (Na+/K(+)-ATPase) activity in rat optic nerve, Neuroscience, 37 (1990) 829–837. [DOI] [PubMed] [Google Scholar]
- [51].Glitsch HG, Electrophysiology of the sodium-potassium-ATPase in cardiac cells, Physiol Rev, 81 (2001) 1791–1826. [DOI] [PubMed] [Google Scholar]
- [52].Schlosser SF, Burgstahler AD, Nathanson MH, Isolated rat hepatocytes can signal to other hepatocytes and bile duct cells by release of nucleotides, Proc Natl Acad Sci U S A, 93 (1996) 9948–9953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dixon CJ, White PJ, Hall JF, Kingston S, Boarder MR, Regulation of human hepatocytes by P2Y receptors: control of glycogen phosphorylase, Ca2+, and mitogen-activated protein kinases, J Pharmacol Exp Ther, 313 (2005) 1305–1313. [DOI] [PubMed] [Google Scholar]
- [54].Emmett DS, Feranchak A, Kilic G, Puljak L, Miller B, Dolovcak S, McWilliams R, Doctor RB, Fitz JG, Characterization of ionotrophic purinergic receptors in hepatocytes, Hepatology, 47 (2008) 698–705. [DOI] [PubMed] [Google Scholar]
- [55].Bartlett PJ, Gaspers LD, Pierobon N, Thomas AP, Calcium-dependent regulation of glucose homeostasis in the liver, Cell Calcium, 55 (2014) 306–316. [DOI] [PubMed] [Google Scholar]
- [56].Cai X, Lytton J, Molecular Cloning of a Sixth Member of the K+-dependent Na+/Ca2+ Exchanger Gene Family, NCKX6, Journal of Biological Chemistry, 279 (2004) 5867–5876. [DOI] [PubMed] [Google Scholar]
- [57].Palty R, Hershfinkel M, Yagev O, Saar D, Barkalifa R, Khananshvili D, Peretz A, Grossman Y, Sekler I, Single α-Domain Constructs of the Na+/Ca2+ Exchanger, NCLX, Oligomerize To Form a Functional Exchanger, Biochemistry, 45 (2006) 11856–11866. [DOI] [PubMed] [Google Scholar]
- [58].Han YE, Ryu SY, Park SH, Lee KH, Lee SH, Ho WK, Ca(2+) clearance by plasmalemmal NCLX, Li(+)-permeable Na(+)/Ca(2+) exchanger, is required for the sustained exocytosis in rat insulinoma INS-1 cells, Pflugers Arch, 467 (2015) 2461–2472. [DOI] [PubMed] [Google Scholar]
- [59].Cordelieres FP, Bolte S, Experimenters’ guide to colocalization studies: finding a way through indicators and quantifiers, in practice, Methods Cell Biol, 123 (2014) 395–408. [DOI] [PubMed] [Google Scholar]
- [60].Crompton M, KÜNzi M, Carafoli E, The Calcium-Induced and Sodium-Induced Effluxes of Calcium from Heart Mitochondria, European Journal of Biochemistry, 79 (1977) 549–558. [DOI] [PubMed] [Google Scholar]
- [61].Nicholls DG, Calcium transport and proton electrochemical potential gradient in mitochondria from guinea-pig cerebral cortex and rat heart, Biochemical Journal, 170 (1978) 511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Vitorica J, Satrústegui J, The role of ADP in the modulation of the calcium-efflux pathway in rat brain mitochondria, Biochemical Journal, 225 (1985) 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tokunaga H, Hollenberg NK, Graves SW, Sodium-Dependent Calcium Release from Vascular Smooth Muscle Mitochondria, Hypertension Research, 23 (2000) 39–45. [DOI] [PubMed] [Google Scholar]
- [64].Carafoli E, Tiozzo R, Lugli G, Crovetti F, Kratzing C, The release of calcium from heart mitochondria by sodium, Journal of Molecular and Cellular Cardiology, 6 (1974) 361–371. [DOI] [PubMed] [Google Scholar]
- [65].Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song LS, Zingman LV, Anderson ME, Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart, Proc Natl Acad Sci U S A, 112 (2015) 9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Werth JL, Thayer SA, Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons, J Neurosci, 14 (1994) 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shutov LP, Kim MS, Houlihan PR, Medvedeva YV, Usachev YM, Mitochondria and plasma membrane Ca2+-ATPase control presynaptic Ca2+ clearance in capsaicin-sensitive rat sensory neurons, J Physiol, 591 (2013) 2443–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Hogan QH, Sprick C, Guo Y, Mueller S, Bienengraeber M, Pan B, Wu HE, Divergent effects of painful nerve injury on mitochondrial Ca(2+) buffering in axotomized and adjacent sensory neurons, Brain Res, 1589 (2014) 112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lu S-G, Zhang X, Gold MS, Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons, The Journal of Physiology, 577 (2006) 169–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rose CR, Ransom BR, Regulation of intracellular sodium in cultured rat hippocampal neurones, J Physiol, 499 ( Pt 3) (1997) 573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Rose CR, Kovalchuk Y, Eilers J, Konnerth A, Two-photon Na+ imaging in spines and fine dendrites of central neurons, Pflugers Arch, 439 (1999) 201–207. [DOI] [PubMed] [Google Scholar]
- [72].Hajnoczky G, Robb GL, Seitz MB, Thomas AP, Decoding of cytosolic calcium oscillations in the mitochondria, Cell, 82 (1995) 415–424. [DOI] [PubMed] [Google Scholar]