

Abstract
SMAD4 encodes a member of the SMAD family of proteins involved in the TGF-β signaling pathway. Potentially heritable, autosomal dominant, gain-of-function heterozygous variants of SMAD4 cause a rare developmental disorder, the Myhre syndrome, which is associated with a wide range of developmental and post-developmental phenotypes that we now characterize as a novel segmental progeroid syndrome. Whole-exome sequencing of a patient referred to our International Registry of Werner Syndrome revealed a heterozygous p.Arg496Cys variant of the SMAD4 gene. To investigate the role of SMAD4 mutations in accelerated senescence, we generated cellular models overexpressing either wild-type SMAD4 or mutant SMAD4-R496C in normal skin fibroblasts. We found that cells expressing the SMAD4-R496C mutant exhibited decreased proliferation and elevated expression of cellular senescence and inflammatory markers, including IL-6, IFNγ, and a TGF-β target gene, PAI-1. Here we show that transient exposure to TGF-β, an inflammatory cytokine, followed by chronic IFNγ stimulation, accelerated rates of senescence that were associated with increased DNA damage foci and SMAD4 expression. TGF-β, IFNγ, or combinations of both were not sufficient to reduce proliferation rates of fibroblasts. In contrast, TGF-β alone was able to induce preadipocyte senescence via induction of the mTOR protein. The mTOR inhibitor rapamycin mitigated TGF-β-induced expression of p21, p16, and DNA damage foci and improved replicative potential of preadipocytes, supporting the cell-specific response to this cytokine. These findings collectively suggest that persistent DNA damage and cross-talk between TGF-β/IFNγ pathways contribute to a series of molecular events leading to cellular senescence and a segmental progeroid syndrome.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11357-020-00318-6.
Keywords: Myhre syndrome, Gain of function SMAD4 mutations, TGF-β, IFNγ, DNA damage, Cellular senescence, Segmental progeroid syndromes
Introduction
The term “progeroid” refers to phenotypes that resemble either a single major feature of biological aging, defined as “unimodal progeroid syndromes,” or multiple features of aging, defined as “segmental progeroid syndromes” (1). We propose that Myhre syndrome (MS) is an example of the latter. It is a rare genetic syndrome with a wide range of skeletal, cardiovascular, central nervous system, ocular, and inner ear abnormalities (2). These include several features that overlap with what is observed in the Werner syndrome (WS), although without the seminal diagnostic feature of early-onset bilateral cataracts (3). MS is caused by heterozygous missense mutations in the SMAD4 gene. At the molecular level, most of the SMAD4 mutations involve the Ile500 residue, namely, p.Ile500Thr, p.Ile500Val, and p.Ile500Met, located in the MAD homology 2 domain (4) or, less commonly, arginine substitution for cysteine (p.Arg496Cys) (5). The majority of these mutations are sporadic and de novo (4, 6), although there has been a recent report of a pedigree with familial recurrence of p.Arg496Cys (7). Subsequently, the SMAD4 variants involving Ile500 were also found in laryngotracheal stenosis, arthropathy, prognathism, and short stature syndrome (LAPS) (8), indicating the high degree of phenotypic variability often seen in autosomal dominant progeroid syndromes.
The SMAD4 gene encodes a protein involved in the signal transduction of TGF-β, a component of the senescence-associated secretory phenotype (SASP) (9). A defect of SMAD4 ubiquitination with impaired TGF-β signaling was thought to be the disease mechanism of Ile500 mutations based on cell biological studies (4). Structural analysis of the pArg496Cys variant suggested that a conformational change that affects the formation of the SMAD4 complex and/or impaired SMAD4 ubiquitination results in perturbation of TGF-β signaling as a result of enhanced levels of non-ubiquitinated SMAD4 (5). Neither MS nor LAPS syndrome was recognized as progeroid syndromes, however, and has not been studied in the context of aging.
WS is one of the most extensively studied segmental progeroid syndromes. Cells derived from WS patients display senescent cell morphology at early passages, slow proliferation rates, enhanced rates of DNA damage accumulation, dysfunctional telomeres, and increased markers of chronic inflammation (10, 11). Pro-inflammatory cytokines, including TNFα, IL-1α IL-6, IL-8, and IFNγ, have been shown to reinforce senescence via autocrine/paracrine mechanisms (12–14). Genomic instability due to defective DNA repair machinery is one of the major disease mechanisms of segmental progeroid syndromes, including WS. However, involvement of the pathways and molecular events of chronic inflammation in the pathogenesis of these disorders remains largely unknown.
TGF-β/SMAD signaling is one of the dynamic pathways known to regulate programmed cell death and cellular senescence and has been cited as a contributor to cardiac aging (15, 16). TGF-β signaling is regulated by the SMAD complex. Upon activation, SMAD proteins are translocated to the nucleus, where they control and regulate TGF-β signal transduction. In particular, SMAD4 is known to play a central role in regulating downstream targets of TGF-β (17, 18). Signaling by TGF-β, a pleotropic cytokine, contributes to various cellular processes, including senescence, cell proliferation, differentiation, and apoptosis, and does so in diverse cell types (19, 20). A number of studies have emphasized the importance of TGF-β signaling in the regulation of senescence both in vivo and in vitro (15, 19, 20). IFNγ also reportedly induces cellular senescence through regulation of TGF-β/SMAD signaling (21). In addition, elevated levels of TGF-β expression are often observed during aging and age-related pathologies, including diabetes, atherosclerosis, and obesity (22–24). TGF-β/SMAD signaling, however, has not been investigated in the context of the pathogenesis of segmental progeroid syndromes.
Here we report on a patient referred to the International Registry of Werner Syndrome (IRWS) as a reputed “mild case” of the Werner syndrome. Exome sequencing, while failing to confirm mutations at the RecQ locus responsible for that syndrome, did reveal an allele with a SMAD4 pathogenic mutation. Subsequent cell biological studies supported TGF-β/SMAD-mediated accelerated senescence as an important disease mechanism.
Methods
Patient samples
The patient was referred to the International Registry of Werner Syndrome (IRWS) (http://www.wernersyndrome.org) for molecular diagnosis of their progeroid syndrome. Prior to the initiation of the study, written informed consent was given by the patient.
Upon arrival of the blood samples to the registry, genomic DNA was isolated from the patients’ blood samples and Epstein-Barr virus-mediated lymphoblastoid cell lines (LCLs) were established from whole blood, as previously described (25). This study was approved by the University of Washington Institutional Review Board.
Exome sequencing and analysis
A library of DNA fragments was constructed and enriched for protein and RNA coding portions of the human genome using the Exome v1.0 (Integrated DNA Technologies) capture system. Paired-end sequencing of the enriched library was performed using rapid run v2.0 (Illumina) chemistry on a HiSeq 2500 (Illumina) sequencer according to the manufacturer’s recommended protocol. The resulting sequences were aligned to the human genome reference (hg19) using the Burrows-Wheeler Aligner (BWA) and variants identified with the Genome Analysis Tool Kit (GATK). Genbank accession number NG_013013.2 was used for analyses for the SMAD4 gene. Variants were annotated using an in-house software tool based on SnpEff (26).
Cell culture
82-6 is a primary human foreskin fibroblast line derived from a newborn (27, 28). 78-85 is a primary skin fibroblast line derived from a Japanese WS patient carrying a homozygous WRN mutation c.3139-1G>C (29, 30). Human adipocyte progenitors, also termed adipose-derived stem cells or preadipocytes (31), were isolated from abdominal subcutaneous adipose tissue biopsies from nine healthy subjects undergoing surgery to donate a kidney (age 37 ± 6 years, male, BMI 26 ± 2). Studies of preadipocytes were approved by the Mayo Clinic Institutional Review Board. All cells were maintained in Dulbecco’s modified Eagle medium, 10% fetal calf serum, and 1x penicillin/streptomycin in an environment 5% CO2, 5% O2, and 37 °C. For serial passage, cells were cultured until they reached 90% confluence, whereupon they were sub-cultured at 1–3 × 103/cm2. Cell numbers were determined by hemocytometer to calculate the number of doublings (log 2 of the fold increase of cell numbers) and cumulative population doublings (27, 28).
For cytokine stimulation, cells were treated with TGF-β (50 ng/ml) or IFNγ (20 ng/ml, 400 Units/ml) (PeproTech, Rocky Hill, NJ) or a combination of TGF-β and IFNγ or with TGF-β for 30 min and then washed twice with culture media, and then stimulated with IFNγ for 6 days. For mTOR inhibition experiments, preadipocytes were pre-treated with rapamycin (10 μM) (R8781, Sigma, St Louis, MO) for 30 min before stimulation with TGF-β (20 ng/ml) for 7 days.
Establishing and culturing SMAD4-overexpressing fibroblasts
The lentiviral construct of sequence-verified human wild type SMAD4, pLX304-SMAD4, was obtained commercially (# OH6085-213573632, Dharmacon, Lafayette, CO) (32). In pLX304-SMAD4, full-length SMAD4 cDNA with a V5 tag at it C-terminal end was driven by the CMV promoter. The p.Arg496Cys variant found in the patient was introduced to pLX304-SMAD4 by PCR-based site-directed mutagenesis using iProof HF polymerase (#172-5310, Bio-Rad, Hercules, CA) to generate pLX304-SMAD4-R496C. Primers used for the site-directed mutagenesis are indicated below, where bold underlined nucleotides are the sites of mutagenesis.
SMAD4 Arg496Cys F 5’-TTGATGACCTTTGTCGCTTATGC-3’
SMAD4 Arg496Cys R 5’-GCATAAGCGACAAAGGTCATCAA-3’
The presence of R497C mutation in pLX304-SMAD4-R496C was confirmed by Sanger sequencing. Lentiviruses of pLX304-SMAD4 and pLX304-SMAD4-R406C were produced in the 293T cell line (define or give reference) using a Trans-Lentiviral ORF Packaging System (# TLP5916, Dharmacon) according to the manufacturer’s instruction and were stored at − 80 °C. Viral supernatants with 1 μg/μl polybrene were applied to the exponentially growing 82-6 cultures for 4 h each for 3 successive infections. Cultures were then selected with 1 μg/μl Blasticidin-S for 7 days to establish 82-6-SMAD4 and 82-6-SMAD4-R496C. Presence of the wild-type transgene and the R496C mutation was verified by Sanger sequencing in genomic DNAs of established cell lines using primers spanning from exon 11 to exon 12, as shown below.
SMAD4_A5’-GATTTGCGTCAGTGTCATCG-3’
SMAD4_B5’-AAGGTTGTGGGTCTGCAATC-3’
Western blotting analysis
Western blotting analyses were performed using a commercial anti-SMAD antibody (1:1000 dilution, #MA5-15682, Thermo Fisher Scientific, Waltham, MA), anti-V5 antibody (1:10000 dilution, #R960-25, Thermo Fisher), anti-phospho-mTOR (Ser2448) (1:1000, #2971, Cell Signaling Technology, Danvers, MA), anti-phospho-p70S6kinase (Thr389) (1:1000, #9206, Cell Signaling), anti-p21 (1:1000, #2947, Cell Signaling), anti-β-actin (1:4000, #A1978; Sigma), and biotinylated anti-mouse IgG antibody (#BA-9200, Vector Laboratories, Burlingame, CA), as described previously (26).
Immunocytochemistry and image processing
Immunofluorescence staining and image analysis of double strand break foci were performed as described previously (28). The antibodies used were anti-53BP1 (1:100 dilution, #NB100-304, Novus Biologicals), phospho-H2AX (1:500, #9718, Cell Signaling), and secondary antibody, goat anti-rabbit Alexa Fluor 594 (1:200, #A11080, Invitrogen Molecular Probes, Eugene, OR). Stained images were captured using an EVOS XL fluorescence microscope. The mean numbers of foci per nucleus were manually determined within a minimum of 200 cells.
BrdU incorporation assay
To determine the fraction of proliferating cells, a 5-bromo-2deoxyuridine (BrdU) incorporation assay was carried out using a BrdU labeling and detection kit (11 296 736 001, Roche Applied Science, Indianapolis, IN, USA). To do so, cells were labeled for 48–72 h and processed according to the manufacturer’s instructions.
Real-time PCR
Quantitative PCR was performed using a TaqMan gene expression assay system (Hs00194619_m1, Applied Biosystem) and normalized to GAPDH mRNA (Hs02758991_g1, Applied Biosystem), as previously described (33). Briefly, total RNA was isolated from the cultured cell pellet using Trizol (Thermo Fisher). Reverse transcription was carried out with 2 μg total RNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative PCR was performed with Applied Biosystem 7500. All primers were obtained from Thermo Scientific. TATA-binding protein (TBP) or Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as the internal control.
Senescence associated β-galactosidase staining (SA-β-gal)
Cell SA-β-gal staining was performed with Senescence β-Galactosidase staining kit (Cell Signaling, #9860) following manufacturer’s instruction. Briefly, the cells were washed twice with PBS, fixed with 1 mL of 1× fixing solution per well (to each 35 mm well) and incubated at room temperature for 10 min. After removing fixing solution, the cells were washed twice again with PBS, and incubated overnight with 1 mL of freshly prepared β-Galactosidase staining solution per well at 37 °C overnight, without CO2 and protected from light. The percentage of cells positive for SA-β-gal was determined in 200 cells per sample in triplicates.
Results
Case report
The patient (IRWS #LA1010) is a 55-year-old male with tight skin of extremities, high-pitched voice, short stature, premature thinning of hair, hearing loss, diabetes mellitus, osteoporosis, and possible hypogonadism. He did not have ocular cataracts, a cardinal sign of the WS. Parental consanguinity was denied. The patient was referred to the IRWS with a suspected “mild form” of WS. Sanger sequencing of WRN exons did not show any pathological variants. Western blotting analysis using an antibody against the C-terminal region of the WRN protein showed normal amounts of WRN protein of the correct size. Exome sequencing revealed a heterozygous c.1486C>T, p.Arg496Cys substitution, which was confirmed by Sanger sequencing (Fig. 1a) (“c” refers to cDNA position, and “p” refers to protein number). The patient has two sons. His 18-year-old son exhibits some tightness of his extremities and a high-pitched voice, while his 20-year-old son does not have these findings. We, however, were unable to obtain biological samples from the sons.
Fig. 1.
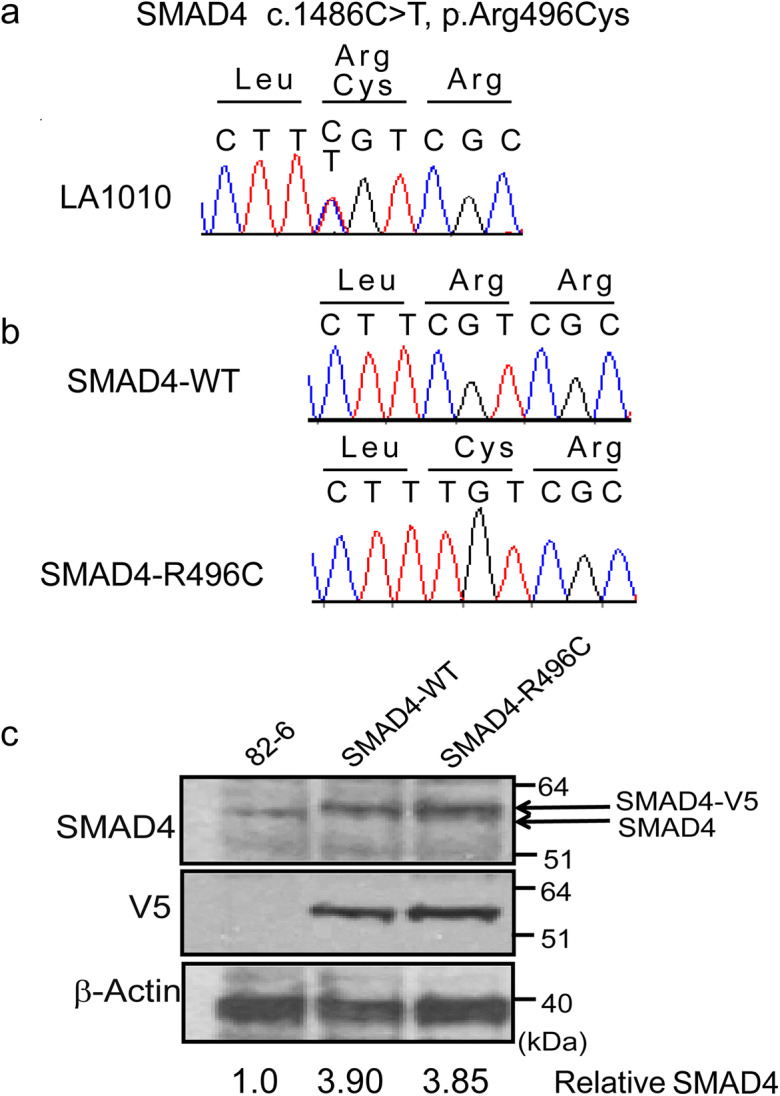
SMAD4 mutation in the progeroid patient. a Sanger sequencing confirmation of the SMAD4 mutation in exon 12 of the Registry# LA1010 patient. b Sanger confirmation of introduced SMAD4 cDNA constructs. c Western blotting analysis of SMAD4, V5 tag, and actin as loading control
Due to the unavailability of a skin biopsy from our patient, we generated a cellular model of SMAD4 R496C by overexpressing the mutant protein in normal human foreskin fibroblasts, 82-6 (82-6-SMAD4-R496C). As a control, 82-6-SMAD4-WT, which overexpressed the wild-type SMAD4, was also generated. Sanger sequencing of the p.Arg496Cys region spanning multiple exons of SMAD4 cDNA in established blasticidin-S resistant cultures showed the expected c.1486C, p.Arg496 in SMAD4-WT, and c.1486T, p.Cys496 in SMAD4-R496C (Fig. 1b). Western blotting analysis of SMAD4 protein in established lines showed the doublet of SMAD4 in transfected lines express, one corresponding to endogenous SMAD seen in the control line, and the slightly larger one corresponding to SMAD4-V5 from the transgene (Fig. 1c). Western blotting analysis using an antibody against V5 tag (14 amino acid long peptide) to detect overexpressed SMAD4 only showed the V5 band in transfected lines. After normalizations, the total amounts of SMAD4 proteins in transfected lines were approximately 3.8–3.9-fold higher compared to the control line. These data confirmed the successful establishment of SMAD4-overexpressing fibroblasts.
Reduced replicative lifespan of primary human fibroblasts expressing SMAD4 p.R496C
In order to determine the potential cell biological consequences of this SMAD4 mutation, we first assessed growth characteristics of mutant cultures, starting at passage 10, the time of cell line establishment. As shown in Fig. 2a, the cumulative population doubling times (CPD) of SMAD4-R496C mutant decreased with time compared to control cells. At passage 15, doubling times of 82-6 and its genetically engineered SMAD4-WT and SMAD4-R496C counterparts were 0.58, 0.48, and 0.34 doublings/day, respectively, and cell numbers in confluent cultures in 100-mm dishes were 2.32 × 106, 2.17 × 106, and 1.08 × 106 (Fig. 2a). After passage 19, all three cell lines ceased to proliferate, at which point, the CPD of SMAD4-R496C mutant cells was 12.4, which was markedly less than the 17.5 and 16.9 CPD observed in 82-6 and SMAD4-WT, respectively (Fig. 2b). Pairwise t-test confirmed the statistically significant difference in the comparison between 82-6 and 82-6_SMAD4-R496C (p < 0.001), but not between 82-6 and 82-6_SMAD4-WT (p = 0.51). The reduced replicative lifespan of SMAD4-R496C, but not of SMAD4-WT, was consistent with this being a molecular/cellular mechanism responsible for this progeroid syndrome. Next, we assessed expression of cellular senescence and proliferative markers, including p16, p21, and Ki67. The mRNA levels of p16 and p21 were increased > 4-fold and > 5-fold, respectively, in mutant compared to control or SMAD4-WT cells (Fig. 2c). Consistent with this, expression of the proliferative marker Ki67 was substantially reduced in SMAD4-R496C mutant compared to control cells (Fig. 2c). Quantitative analysis of the cell cycle regulators, CDK2 and CDC25A, indicated reduced expression in SMAD4-R496C compared to controls, providing further evidence for repressed or halted cell cycle progression and enforcement of cellular senescence (Fig. S1).
Fig. 2.
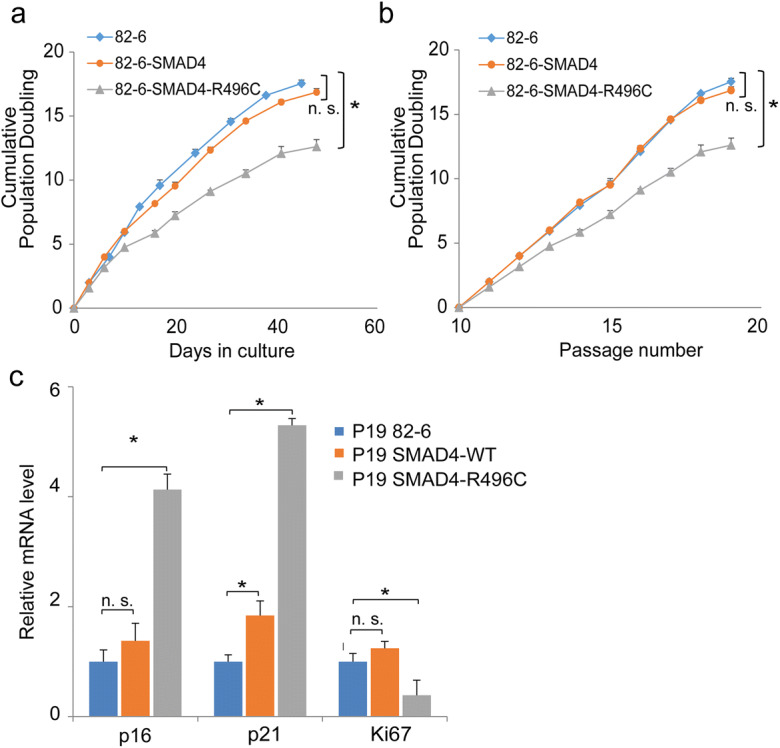
Growth characteristics of SMAD4 fibroblast lines. a–b Cumulative population doublings (CPD) of control or the parental 82-6 (rhombus), SMAD4-WT (circle), and SMAD4-R496C lines (triangle) plotted against days in culture (a) and passage (b). * donates statistical significance (p < 0.01) (c). Real-time PCR analysis of 82-6, SMAD4-WT, and SMAD4-R496C at passage 19 for p16, p21, and Ki67. *denotes statistical significance (p < 0.05), n.s. denotes not significant. Error bars represent SEM
Increased expression of IL-6/IFNγ and senescence makers in SMAD4-R496C
Genomic instability is one of the common mechanisms of human progeroid syndromes, including those caused by mutations in WRN, LMNA, POLD1, SPRTN, ERCC4, and CTC1, as documented in our Registry (26, 28, 34–37). We therefore examined the accumulation of double strand break foci in early passages of SMAD4-overexpressing fibroblast using 53BP1 foci as markers of DNA damage. At passage 16, the average number of 53BP1 foci per nucleus in SMAD4-WT was 2.8, similar to the 2.7 result found in the parental 82-6 cultures (p = 0.45). There was a significant increase to 4.2 in SMAD4-R496C compared to 82-6; however, (p = 3.42x10-13) (Fig. 3a). CPDs at the time of 53BP1 staining (passage 16) were 12.1 for 82-6, 12.3 for SMAD4-WT, and 9.2 for SMAD4-R496C (Fig. 2b). This suggests that, as seen in other progeroid syndromes associated with genomic instability, accumulation of DNA damage may be involved in the cell growth suppression in SMAD4 mutant fibroblasts.
Fig. 3.

Increased DNA damage foci and cytokine expression in SMAD4 mutant fibroblasts. a DNA damage and increased expression of senescence markers in SMAD4-R496C. DNA damage in the fraction of cells with indicated numbers of nuclear 53BP1 foci are shown for three fibroblast lines by immunostaining. b Quantitative real-time PCR for PAI-1 and IL-6 in 82-6, SMAD4-WT, and SMAD4-R496C at passage 10. c Relative mRNA expression of IFNγ in 82-6, SMAD4-WT, and SMAD4-R496C at passages 19 by real-time PCR. *denotes statistical significance (*p < 0.05), n. s. denotes not significant. Error bars represent SEM
DNA damage is also an established inducer of chronic inflammation (38). We therefore assessed the expression of a set of inflammatory genes. Expression of PAI-I (plasminogen activator inhibitor 1, also known as SERPINE1), a marker of cellular senescence, was increased by ~ 10-fold in 82-6-SMAD-R496C and ~ 4-fold 82-6-SMAD4-WT compared to the control (Fig. 3b). Similarly, IL-6 was increased by ~ 10-fold in SMAD4-R496C, while only by ~ 4-fold in SMAD4-WT relative to controls (Fig. 3b). In late passages, we found significantly increased IFNγ mRNA in 82-6-SMAD4-R496C (Fig. 3c). We observed the trend of the increase in IFNγ mRNA in 82-6-SMAD4-WT, but it did not rise to the statistical significance. These findings are in agreement with the larger increase of p16 and p21 and reciprocal decrease of Ki67 in SMAD4-R496C compared to SMAD4-WT (Fig. 2c). These data collectively support the hypothesis that the presence of R496C mutation in SMAD4 contributes to the accelerated senescence in fibroblasts.
Evidence of TGF-β/SMAD4 signals during the pathogenesis of Werner syndrome: a typical segmental progeroid syndrome
We previously reported that genes associated TGF-β and IFNγ pathways were up-regulated in fibroblasts of Werner syndrome patients (11). We therefore sought to test the involvement of the TGF-β/SMAD pathway in fibroblast cultures derived from a patient with Werner syndrome (29, 30). Real-time PCR analysis of serially passaged primary Werner fibroblasts (78-85) displayed increased and sustained expression of SMAD4 at levels substantially higher compared to normal fibroblast (Fig. 4a). Consistent with this, the growth curve of Werner cells displayed an increased doubling time with passage compared to control normal fibroblasts (Fig. 4b). At passage 14, the population doubling time of Werner cells was markedly increased compared to corresponding controls, suggesting proliferative arrest and accelerated senescence of Werner cells, as described previously (30, 39). Increased senescence of Werner cells was further supported by increased numbers of SA-beta positive cells (Fig. 4c) and expression of IL-6, STAT1, and IFNγ (Fig. 4d). These results demonstrate the association of TGF-β/SMAD-IFNγ signaling pathway and the accelerated fibroblast senescence in this classic adult-onset segmental progeroid syndrome
Fig. 4.
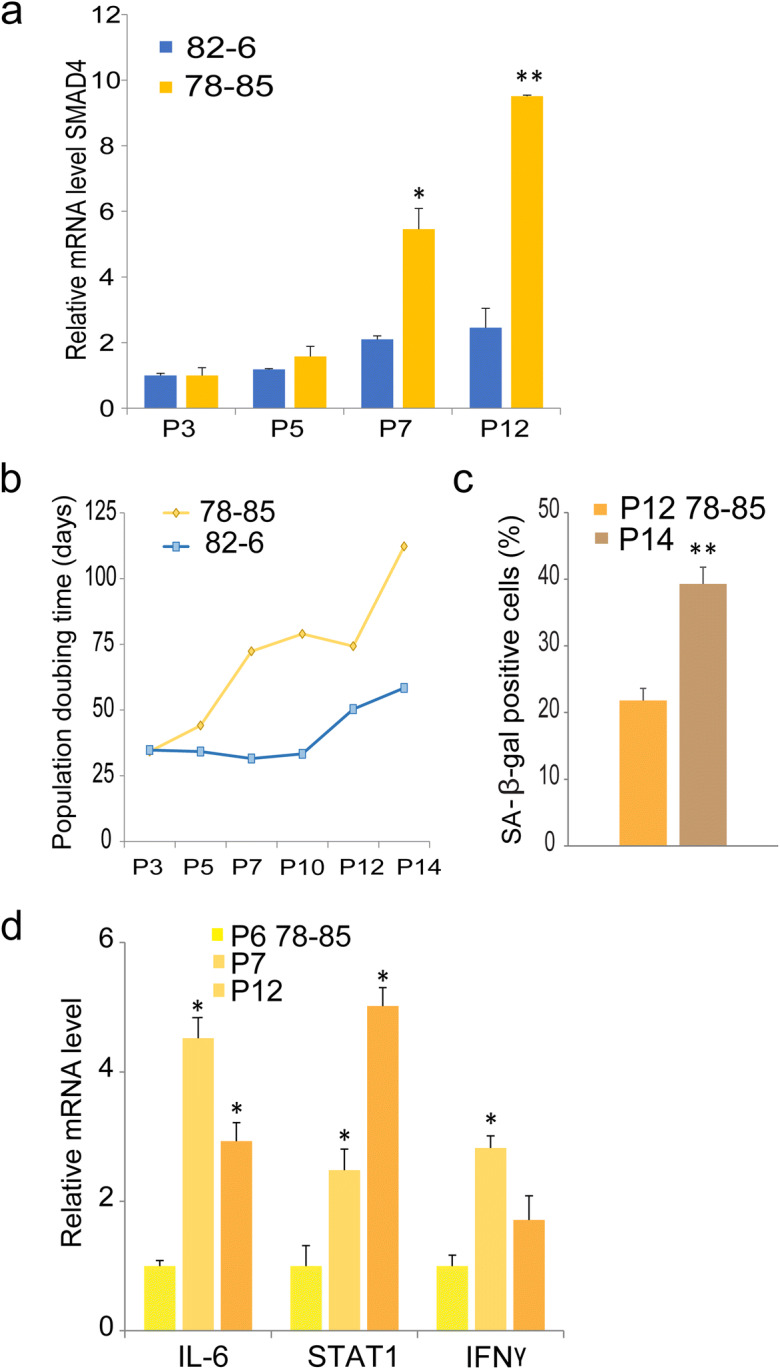
Accelerated senescence and increased expression of SMAD4 in Werner syndrome fibroblasts. a SMAD4 levels were quantified in serially passaged normal skin fibroblasts, 82-6, and Werner skin fibroblasts, 78-85. p indicates passage number. Fold induction of SMAD4 mRNA was quantified by real-time PCR. b Growth curves represent population doubling times of 78-85 Werner cells or 82-6 normal fibroblasts cells at the indicated passage numbers. c Percentage of SA-β-gal positive cells in 78-85 Werner cells. d Relative mRNA expression of IL-6, STAT1, and IFNγ by real-time-PCR in Werner cells at the passage numbers indicated. *denotes statistical significance (*p < 0.05, ** < 0.01). Error bars represent SEM
Transient TGF-β coupled with chronic IFNγ exposure accelerates rates of senescence in fibroblasts
Given the above evidence for the role of this patient’s SMAD mutation in accelerating fibroblast senescence, we initiated studies to extend our exploration of the role of the TGF-β/SMAD pathway in the regulation of senescence. The elevated expression of IL-6 and IFNγ in SMAD4-R496C prompted us to consider a role for interactions between IFNγ and TGF-β pathways in the regulation of cell proliferation. We therefore explored chronic effects of the pro-inflammatory cytokines, TGF-β, and IFNγ, alone or in combination. Exposure of human fibroblasts to TGF-β and IFNγ alone or in combination did not elicit differences in proliferation rates compared to controls (Fig. S2), suggesting that continuous presence of TGF-β with IFNγ could potentially inhibit the anti-proliferative effects of these cytokines. However, given that TGF-β and IFNγ are also known for their opposing and suppressing cellular activities (40, 41), we pre-treated the cells with TGF-β for a short time period followed by chronic exposure to IFNγ (Fig. 5a). Transient exposure to TGF-β along with prolonged exposure to IFNγ resulted in decreased proliferation of the fibroblasts after 6 days (Fig. 5b). This result was supported by observations of decreased numbers of BrdU positive cells (Fig. 5c). We also found increased expression of senescence markers, including p21, p16, and SA-β-gal positive cells, after 6 days of this treatment paradigm (Fig. 5d and e). Taken together, our results demonstrate that transient exposure to TGF-β along with prolonged exposure to IFNγ results in acceleration of cell senescence and decreased proliferative capacity of skin fibroblast cells.
Fig. 5.

Transient TGF-β and chronic IFNγ exposure accelerates senescence in human fibroblasts. a Schematic model of experiment. Cells were pulsed with TGF-β (20 ng/ml) for 30 min and then washed 2× with warm media followed by IFNγ (20 ng/ml) for 6 days. b Growth curves of fibroblasts treated with TGF-β for 30 min washed 2× with medium followed by IFNγ stimulation for 6 days. Untreated cells were used as control. c 72-h BrdU incorporation in cultures treated with TGF-β for 30 min followed by IFNγ for 6 days. d Real-time PCR quantification of p21, and p16 in cells stimulated with TGF-β or IFNγ, the combination of both, or treated with TGF-β for 30 min and IFNγ for 6 days. Data represent means of three independent experiments. e Percentages of SA-β-gal positive control cells or cells treated with TGF-β for 30 min and IFNγ for 6 days. Data represent the mean of two experiments. *denotes statistical significance (*p < 0.05, ** < 0.01). Error bars represent SEM
Synergy between TGF-β and IFNγ pathways
The DNA damage response is implicated as a central regulator of cellular senescence and its associated inflammation (42, 43). Transient TGF-β exposure followed by chronic IFNγ resulted in formation of multiple DNA damage foci and increased percentage of γH2AX, as revealed by immunostaining (Fig. 6a and b). As the SMAD4-R496C mutant displayed potential cross-talk between TGF-β and IFNγ signaling, we next sought to explore the expression of genes associated with these pathways. Using quantitative RT-PCR, we quantified the expression of SMAD4 and IL-6 in cells treated with TGF-β or IFNγ alone or after transient treatment with TGF-β, followed by prolonged treatment with IFNγ. IL-6 expression was found to be increased in cells treated with IFNγ alone or transiently with TGF-β followed by prolonged IFNγ. The combination both cytokines, however, failed to induce IL-6, suggesting antagonistic roles for these cytokines (Fig. 6c). Expression of SMAD4 and the anti-apoptotic marker BCL2 were drastically increased only in cells treated transiently with TGF-β and chronic IFNγ, compared to TGF-β or IFNγ alone (Fig. 6c and d). These results indicate transient TGF-β expression in synergy with IFNγ is necessary for expression of SMAD4 and BCL2. We also observed increased expression of the downstream TGF-β targets, PAI-1(Fig. 6e) and COL1A1 (Fig. S3), indicating that acute TGF-β expression initiates a cascade of events, including feedback loops, to sustain expression of its downstream target genes. These results collectively indicate that synergistic actions of transient TGF-β and IFNγ pathways are required for the establishment of senescence and associated inflammation.
Fig. 6.
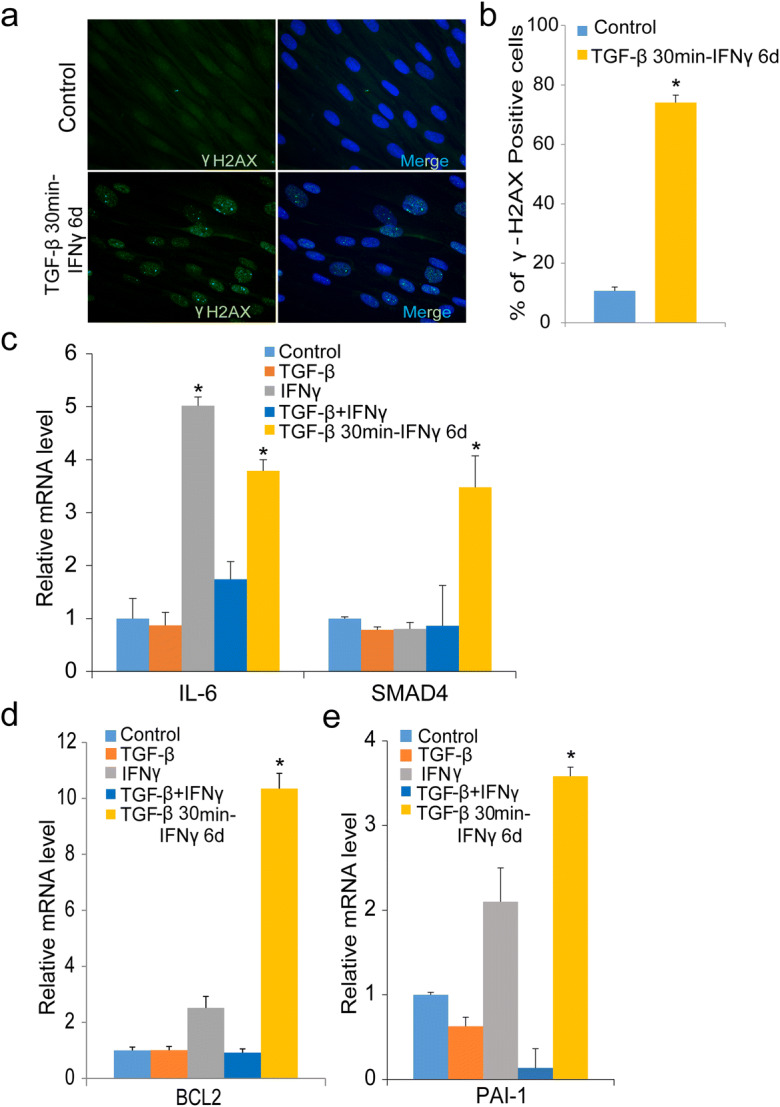
DNA damage and increased expression of TGF-β/IFNγ targets in cytokine-treated human fibroblasts. a Immunofluorescence detection of γH2AX foci in untreated or cells treated with TGF-β for 30 min and IFNγ for 6 days; scale bar 10 μm. The γH2AX foci are also shown in merged images of nuclear DAPI staining. b Percentage of γH2AX foci in untreated or cells treated with TGF-β for 30 min and IFNγ for 6 days. c IL-6 and SMAD4 expression determined by real-time PCR in untreated cells or cells treated with TGF-β or IFNγ alone or in combination or with transient TGF-β for 30 min and IFNγ for 6 days. d mRNA expression of the pro-apoptotic gene BCL2 with TGF-β, IFNγ alone, the combination of both, or with transient TGF-β for 30 min and IFNγ for 6 days as measured by real-time PCR. e Real-time PCR analysis of PAI-1 expression in conditions as described *denotes statistical significance (*p < 0.05). Error bars represent SEM
Cell-specific effects of TGF-β on proliferation: in preadipocytes, TGF-β alone was sufficient to accelerate senescence via activation of the mTOR pathway
The TGF-β pathway is known to play a major regulatory role in adipogenesis and insulin resistance. For example, several publications demonstrated associations of hyperglycemia with increased activity of TGF-β (24, 44). TGF-β has been noted to be increased in the adipose tissues of obese subjects exhibiting insulin resistance (45). We also found elevated expression of TGF-β in adipocytes and preadipocytes following treatment with high levels of glucose and insulin (Fig. S4). To understand cell-specific effects of TGF-β, we chose to investigate preadipocytes initially. To ascertain effects of TGF-β on cell proliferation, we treated preadipocytes with varying concentrations of TGF-β for 7 days. Human preadipocytes stimulated with recombinant TGF-β displayed flattened morphology and reduced proliferation (Fig. 7a). These results correlated with increased senescence markers, including p21, p16, and GLB1, a gene responsible for SA-β-gal activity (senescence-associated beta-galactosidase) (Fig. 7b). Although the Ki67 expression showed a trend of decrease in TGF-β treated culture, the difference was not significant.
Fig. 7.

TGF-β accelerates senescence in cultures of preadipocytes via regulation of the mTOR pathway. a Representative images of untreated or control cells or cells stimulated with TGF-β 10 ng/ml or 50 ng/ml for 7 days. b Quantification of mRNA of p21, p16, Ki67, and GLB1 in control cells or cells stimulated with TGF-β (50 ng/ml) for 7 days by real-time PCR. Data represent mean of three independent experiments. c Western blotting analysis of phosphorylated mTOR in cells treated with TGF-β (20 ng/ml) for indicated times. d Cells pre-treated with the mTOR inhibitor rapamycin (10 μm) 30 min prior to stimulation with TGF-β (50 ng/ml) for 7 days. Protein extracts were analyzed for phosphorylated mTOR, phosphorylated p70S6Kinase, and p21. β-actin was used as loading control. e Real-time PCR analysis of control cells or cells treated with rapamycin (10 μm) and TGF-β (50 ng/ml) or TGF-β (50 ng/ml) alone for 7 days. p16, Ki67, and CDK1 were normalized to GADPH, which was used as an internal control. Data represent mean of three independent experiments. f Immunofluorescence staining of γH2AX foci with nuclear DAPI staining in control or treated with TGF-β (50 ng/ml), or cells treated with rapamycin (10 μm) and TGF-β (50 ng/ml) for 7 days. Scale bar 10 μm. g Percentage of γH2AX foci in control or cells treated with TGF-β (50 ng/ml), or cells treated with combination of rapamycin (10 μm) and TGF-β (50 ng/ml) for 7 days. *denotes statistical significance (*p < 0.05), n.s. denotes not significant. Error bars represent SEM
The mTOR pathway is known to play important regulatory roles in adipogenesis, adipose tissue function, and adipose tissue aging (46). We therefore initiated studies of mTOR regulation in TGF-β-mediated preadipocyte senescence. Western blotting analysis showed that TGF-β stimulation led to rapid and early activation of the phosphorylated-mTORC1 pathway (Fig. 7c), while phosphorylated mTOR remained constant for 7 days (Fig. 7d). We then demonstrated that rapamycin, an mTOR inhibitor, mitigated TGF-β-induced phosphorylation of mTOR together with p70s6kinase and p21 (Fig. 7d). Rapamycin reduced TGF-β-induced senescence markers, such as p16, and improved the cell cycle and proliferative markers (Fig. 7e). Moreover, pre-treatment with rapamycin reduced TGF-β-mediated DNA damage, as revealed by decreased γH2AX foci compared to cells stimulated with TGF-β alone (Fig. 7f and g). In parallel experiments, we also found that rapamycin improved expression of the cell cycle regulatory and repair genes that are required for cellular proliferation and cell cycle progression (Fig. S5). In summary, our results demonstrate that, in contrast to human fibroblasts, TGF-β alone was capable of accelerating preadipocyte senescence. Moreover, the senescent phenotype involved activation of the mTOR pathway.
Discussion
In this study, we present a patient with features reported in MS, including tight skin of extremities, short stature, hoarse voice, hearing loss, and premature thinning of hair. There is overlap of these features with Werner syndrome, although this patient lacked the cardinal WS features of bilateral cataracts before age 30 years, premature graying, or skin ulcerations. By exome sequencing, we identified a heterozygous mutation in SMAD4 at c.1486C>T, p.Arg496Cys. While heterozygous loss of function mutations in SMAD4 have been reported in the syndrome of juvenile polyposis syndrome and hereditary hemorrhagic telangiectasia (HHT) (47), there has been a growing literature documenting dominant, gain-of-function mutations in SMAD4 as the basis for MS. In a family with laryngotracheal stenosis, cardiovascular, and skeletal features, the same p.Arg496Cys missense substitution was reported (7). Given that MS is primarily a developmental disorder, this emphasizes the important role of development in our understanding of the biology and pathobiology of aging, a neglected area of research and one that was recently emphasized in the case of Werner syndrome (48). While most cases of MS have manifested as de novo mutations, there have been instances of autosomal dominant inheritance (7). Our patient did, in fact, have a son said to have a MS phenotype. Unfortunately, however, we could not obtain a biological sample from the son to confirm that diagnosis.
SMAD4 has an essential role in the canonical signaling through both the TGF-β and BMP pathways (49). In order to elucidate the molecular mechanisms underlying the pathogenesis of this disorder, we generated an in vitro model of the SMAD4-R496C mutation and its wild-type counterpart in normal diploid human skin fibroblasts. Overexpression of the SMAD4 mutant led to persistent DNA damage foci and reduced proliferative capacity of these cells. This is consistent with the general recognition that SMAD4 is a potent tumor suppressor (50). Moreover, a recent publication has documented neoplasia in MS (51). In addition, sustained DNA damage and increased senescence-associated inflammatory genes, such as IL-6, IFNγ, and PAI-1, correlated with the diminished proliferative capacity of SMAD4 mutant cells.
IFNγ and TGF-β are two major cytokines implicated in the regulation of aging and cellular senescence (20, 52). TGF-β is known to exert both growth inhibitory and proliferative effects in several cell types (53). Similarly, exposure to IFNγ is known to accelerate senescence and increase DNA damage and ROS via the TGF-β/SMAD signaling pathway (21). Conversely, we found that stimulation with TGF-β, IFNγ, or combinations of both, failed to induce proliferative arrest in skin fibroblasts. Considering that IFNγ and TGF-β have opposing roles and are known for their antagonistic and suppressive mechanisms (40, 41, 54), we speculate that continuous presence of both cytokines could potentially alleviate their anti-proliferative effects. On the other hand, our results demonstrate a model in which transient or acute TGF-β exposure with prolonged exposure to IFN-γ initiates persistent DNA damage signaling and SMAD4 expression to promote senescence. Reciprocal regulation and cross-talk between the TGF-β and IFNγ pathways have been previously observed (55, 56). Our results suggest that (1) acute TGF-β signals could be internalized and recycled at low levels and (2) transient TGF-β together with IFNγ initiates a positive feedback/autocrine loop via SMAD4 that regulates and sustains senescence-associated features. Additional research will be required to explain how acute TGF-β signals are involved in mediating downstream signaling events leading to permanent cell cycle arrest.
The limitation of our study concerns with the significance of TGF-β/SMAD signaling in wide ranges of progeroid syndromes. At this point, we were unable to address the mechanistic link involved in regulating SMAD4 and TGF-β in various other progeroid syndromes. In addition we have not assessed how the modulation of TGF-β/SMAD4 pathway may impact regulations of the genes associated to progeroid phenotypes. Nonetheless, our results demonstrate the association of TGF-β/SMAD4 and IFNγ pathways in segmental progeroid syndrome and its central role for driving transcription of genes that are necessary for establishing features of senescence.
As noted in “BrdU incorporation assay” section, we turned our attention to preadipocyte cultures as an initial study to investigate the extent to which the molecular pathways that regulate replication differ among distinct types of somatic cells. We were aware that TGF-β is a potent inhibitor of adipocyte differentiation and adipogenesis (57) and that TGF-β/ SMAD signaling leads to adipocyte dysfunction, which is often associated with obesity, insulin resistance, and diabetes (24). We found that, in cultures of preadipocytes, TGF-β alone was sufficient to induce senescence via activation the mTOR pathway, suggesting that the cellular responses to cytokines and their modes of regulation can vary depending on the cell type. This is consistent with previous reports documenting a critical role for TGF-β in the regulation of senescence (15, 20, 21). Disruption of the mTOR pathway with rapamycin rescued cells from DNA damage and improved replicative potential.
Persistent DNA damage and inflammation play critical roles during cellular senescence and the biology of aging, including roles in acceleration of rates of genomic instability (42). We also noted decreased expression of genes involved in DNA repair during TGF-β-mediated senescence, further supporting the role of genomic instability as one of the hallmarks of aging (58). Our report has highlighted the importance of the TGF-β/SMAD4-IFNγ network and DNA damage in the genesis of cellular senescence, an interest sparked by the referral to our Registry of a patient thought to have a “mild form” of the Werner syndrome, but who turned out to have a gain of function single allele mutation at the SMAD4 locus. In summary, to the best of our knowledge, this study is the first report showing that a SMAD4-R496C mutation links DNA damage and features of senescence. This is in agreement with the report describing the emergence of neoplasia in some carriers of such mutations (51). Our study also revealed synergy between the TGF-β and IFNγ pathways, which could act as a potential driver for the establishment of cellular senescence and underlie a progeroid syndrome. Further understanding of downstream events along the TGF-β/IFNγ pathway in regulating senescence will likely shed light on potential therapeutic targets for progeroid syndromes and age-related pathologies.
Supplementary Information
(PDF 580 kb)
Acknowledgments
The authors are grateful to Ms. Lin Lee for administrative and laboratory assistance, Ms. Nino Giorgadze for isolation of primary human preadipocytes, and Dr. Tamar Pirtskhalava for insightful discussions.
Author contributions
RKP designed and performed experiments, analyzed data, and wrote the manuscript. DH, JZ, and YY performed experiments and analyzed the data. GP and FH contributed clinical description of the patients and interpretation of clinical data. TM conducted exome analysis. TT and JLK contributed preadipocyte studies, supervised the project, and prepared and revised the manuscript. GMM and JO supervised the project and designed the study and prepared and revised the manuscript.
Funding
This work was supported in part by JSPS KAKENHI 17H04037 (JO) and NIH grants R01CA210916 (GMM/JO), R37AG013925 (JLK/TT), and P01AG062413 (JLK/TT), and Robert and Arlene Kogod (JLK/TT), the Connor Group (JLK/TT), Robert J. and Theresa W. Ryan (JLK/TT), and the Noaber Foundation (JLK/TT).
Data availability
Data that support the findings of this study are available from the corresponding author upon request.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflicts of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hisama FM, Oshima J, Martin GM. How research on human progeroid and antigeroid syndromes can contribute to the Longevity Dividend Initiative. Cold Spring Harb Perspect Med. 2016;6(4):a025882. doi: 10.1101/cshperspect.a025882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gürsoy S, Hazan F, Öztürk T, Ateş H. Novel ocular and inner ear anomalies in a patient with Myhre syndrome. Mol Syndromol. 2020;10(6):339–343. doi: 10.1159/000504829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oshima J, Sidorova JM, Monnat RJ., Jr Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev. 2017;33:105–114. doi: 10.1016/j.arr.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Goff C, Mahaut C, Abhyankar A, Le Goff W, Serre V, Afenjar A, et al. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat Genet. 2011;44(1):85–88. doi: 10.1038/ng.1016. [DOI] [PubMed] [Google Scholar]
- 5.Caputo V, Bocchinfuso G, Castori M, Traversa A, Pizzuti A, Stella L, Grammatico P, Tartaglia M. Novel SMAD4 mutation causing Myhre syndrome. Am J Med Genet A. 2014;164A(7):1835–1840. doi: 10.1002/ajmg.a.36544. [DOI] [PubMed] [Google Scholar]
- 6.Burglen L, Heron D, Moerman A, Dieux-Coeslier A, Bourguignon JP, Bachy A, et al. Myhre syndrome: new reports, review, and differential diagnosis. J Med Genet. 2003;40(7):546–551. doi: 10.1136/jmg.40.7.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meerschaut I, Beyens A, Steyaert W, De Rycke R, Bonte K, De Backer T, et al. Myhre syndrome: a first familial recurrence and broadening of the phenotypic spectrum. Am J Med Genet A. 2019;179(12):2494–2499. doi: 10.1002/ajmg.a.61377. [DOI] [PubMed] [Google Scholar]
- 8.Michot C, Le Goff C, Mahaut C, Afenjar A, Brooks AS, Campeau PM, et al. Myhre and LAPS syndromes: clinical and molecular review of 32 patients. Eur J Hum Genet. 2014;22(11):1272–1277. doi: 10.1038/ejhg.2013.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyazawa K, Miyazono K (2017) Regulation of TGF-beta family signaling by inhibitory Smads. Cold Spring Harb Perspect Biol. 9(3) [DOI] [PMC free article] [PubMed]
- 10.Sugimoto M. A cascade leading to premature aging phenotypes including abnormal tumor profiles in Werner syndrome (review) Int J Mol Med. 2014;33(2):247–253. doi: 10.3892/ijmm.2013.1592. [DOI] [PubMed] [Google Scholar]
- 11.Tang W, Robles AI, Beyer RP, Gray LT, Nguyen GH, Oshima J, Maizels N, Harris CC, Monnat RJ., Jr The Werner syndrome RECQ helicase targets G4 DNA in human cells to modulate transcription. Hum Mol Genet. 2016;25(10):2060–2069. doi: 10.1093/hmg/ddw079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freund A, Orjalo AV, Desprez PY, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16(5):238–246. doi: 10.1016/j.molmed.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kandhaya-Pillai R, Miro-Mur F, Alijotas-Reig J, Tchkonia T, Kirkland JL, Schwartz S. TNFalpha-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging (Albany NY). 2017;9(11):2411–2435. doi: 10.18632/aging.101328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lyu G, Guan Y, Zhang C, Zong L, Sun L, Huang X, Huang L, Zhang L, Tian XL, Zhou Z, Tao W. Addendum: TGF-beta signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging. Nat Commun. 2018;9(1):4134. doi: 10.1038/s41467-018-06710-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schuster N, Krieglstein K. Mechanisms of TGF-beta-mediated apoptosis. Cell Tissue Res. 2002;307(1):1–14. doi: 10.1007/s00441-001-0479-6. [DOI] [PubMed] [Google Scholar]
- 17.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19(8):1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 19.Morikawa M, Derynck R, Miyazono K (2016) TGF-beta and the TGF-beta family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 8(5) [DOI] [PMC free article] [PubMed]
- 20.Tominaga K, Suzuki HI (2019) TGF-beta Signaling in Cellular Senescence and Aging-Related Pathology. Int J Mol Sci. 20(20) [DOI] [PMC free article] [PubMed]
- 21.Hubackova S, Kucerova A, Michlits G, Kyjacova L, Reinis M, Korolov O, Bartek J, Hodny Z. IFNgamma induces oxidative stress, DNA damage and tumor cell senescence via TGFbeta/SMAD signaling-dependent induction of Nox4 and suppression of ANT2. Oncogene. 2016;35(10):1236–1249. doi: 10.1038/onc.2015.162. [DOI] [PubMed] [Google Scholar]
- 22.Krieglstein K, Miyazono K, ten Dijke P, Unsicker K. TGF-beta in aging and disease. Cell Tissue Res. 2012;347(1):5–9. doi: 10.1007/s00441-011-1278-3. [DOI] [PubMed] [Google Scholar]
- 23.Toma I, McCaffrey TA. Transforming growth factor-beta and atherosclerosis: interwoven atherogenic and atheroprotective aspects. Cell Tissue Res. 2012;347(1):155–175. doi: 10.1007/s00441-011-1189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, Zerfas P, Zhigang D, Wright EC, Stuelten C, Sun P, Lonning S, Skarulis M, Sumner AE, Finkel T, Rane SG. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab. 2011;14(1):67–79. doi: 10.1016/j.cmet.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang S, Lee L, Hanson NB, Lenaerts C, Hoehn H, Poot M, Rubin CD, Chen DF, Yang CC, Juch H, Dorn T, Spiegel R, Oral EA, Abid M, Battisti C, Lucci-Cordisco E, Neri G, Steed EH, Kidd A, Isley W, Showalter D, Vittone JL, Konstantinow A, Ring J, Meyer P, Wenger SL, Herbay A, Wollina U, Schuelke M, Huizenga CR, Leistritz DF, Martin GM, Mian IS, Oshima J. The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat. 2006;27(6):558–567. doi: 10.1002/humu.20337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mori T, Yousefzadeh MJ, Faridounnia M, Chong JX, Hisama FM, Hudgins L, Mercado G, Wade EA, Barghouthy AS, Lee L, Martin GM, Nickerson DA, Bamshad MJ, University of Washington Center for Mendelian Genomics. Niedernhofer LJ, Oshima J. ERCC4 variants identified in a cohort of patients with segmental progeroid syndromes. Hum Mutat. 2018;39(2):255–265. doi: 10.1002/humu.23367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubio MA, Kim SH, Campisi J. Reversible manipulation of telomerase expression and telomere length. Implications for the ionizing radiation response and replicative senescence of human cells. J Biol Chem. 2002;277(32):28609–28617. doi: 10.1074/jbc.M203747200. [DOI] [PubMed] [Google Scholar]
- 28.Saha B, Cypro A, Martin GM, Oshima J. Rapamycin decreases DNA damage accumulation and enhances cell growth of WRN-deficient human fibroblasts. Aging Cell. 2014;13(3):573–575. doi: 10.1111/acel.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore). 1966;45(3):177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Oshima J, Campisi J, Tannock TC, Martin GM. Regulation of c-fos expression in senescing Werner syndrome fibroblasts differs from that observed in senescing fibroblasts from normal donors. J Cell Physiol. 1995;162(2):277–283. doi: 10.1002/jcp.1041620213. [DOI] [PubMed] [Google Scholar]
- 31.Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD, Kirkland JL. Mechanisms and metabolic implications of regional differences among fat depots. Cell metabolism. 2013;17(5):644–656. doi: 10.1016/j.cmet.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X, Boehm JS, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8(8):659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saha B, Zitnik G, Johnson S, Nguyen Q, Risques RA, Martin GM, et al. DNA damage accumulation and TRF2 degradation in atypical Werner syndrome fibroblasts with LMNA mutations. Front Genet. 2013;4:129. doi: 10.3389/fgene.2013.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiorillo C, D'Apice MR, Trucco F, Murdocca M, Spitalieri P, Assereto S, Baratto S, Morcaldi G, Minetti C, Sangiuolo F, Novelli G. Characterization of MDPL Fibroblasts carrying the recurrent p.Ser605del mutation in POLD1 gene. DNA Cell Biol. 2018;37(12):1061–1067. doi: 10.1089/dna.2018.4335. [DOI] [PubMed] [Google Scholar]
- 35.Lessel D, Hisama FM, Szakszon K, Saha B, Sanjuanelo AB, Salbert BA, Steele PD, Baldwin J, Brown WT, Piussan C, Plauchu H, Szilvássy J, Horkay E, Högel J, Martin GM, Herr AJ, Oshima J, Kubisch C. POLD1 Germline mutations in patients initially diagnosed with Werner syndrome. Hum Mutat. 2015;36(11):1070–1079. doi: 10.1002/humu.22833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic-Terzic I, Lopez-Mosqueda J, Philipp M, Sim JCH, Smith KR, Oehler J, Cabrera E, Freire R, Pope K, Nahid A, Norris F, Leventer RJ, Delatycki MB, Barbi G, von Ameln S, Högel J, Degoricija M, Fertig R, Burkhalter MD, Hofmann K, Thiele H, Altmüller J, Nürnberg G, Nürnberg P, Bahlo M, Martin GM, Aalfs CM, Oshima J, Terzic J, Amor DJ, Dikic I, Ramadan K, Kubisch C. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet. 2014;46(11):1239–1244. doi: 10.1038/ng.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sargolzaeiaval F, Zhang J, Schleit J, Lessel D, Kubisch C, Precioso DR, Sillence D, Hisama FM, Dorschner M, Martin GM, Oshima J. CTC1 mutations in a Brazilian family with progeroid features and recurrent bone fractures. Mol Genet Genomic Med. 2018;6(6):1148–1156. doi: 10.1002/mgg3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ioannidou A, Goulielmaki E, Garinis GA. DNA Damage: from chronic inflammation to age-related deterioration. Front Genet. 2016;7:187. doi: 10.3389/fgene.2016.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin GM, Sprague CA, Epstein CJ. Replicative life-span of cultivated human cells. Effects of donor's age, tissue, and genotype. Lab Invest. 1970;23(1):86–92. [PubMed] [Google Scholar]
- 40.Ghosh AK, Yuan W, Mori Y, Chen S, Varga J. Antagonistic regulation of type I collagen gene expression by interferon-gamma and transforming growth factor-beta. Integration at the level of p300/CBP transcriptional coactivators. J Biol Chem. 2001;276(14):11041–11048. doi: 10.1074/jbc.M004709200. [DOI] [PubMed] [Google Scholar]
- 41.Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397(6721):710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- 42.Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Zglinicki T, Saretzki G, Ladhoff J. d'Adda di Fagagna F, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev. 2005;126(1):111–117. doi: 10.1016/j.mad.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 44.Wu L, Derynck R. Essential role of TGF-beta signaling in glucose-induced cell hypertrophy. Dev Cell. 2009;17(1):35–48. doi: 10.1016/j.devcel.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samad F, Yamamoto K, Pandey M, Loskutoff DJ. Elevated expression of transforming growth factor-beta in adipose tissue from obese mice. Mol Med. 1997;3(1):37–48. [PMC free article] [PubMed] [Google Scholar]
- 46.Magdalon J, Festuccia WT. Regulation of adiposity by mTORC1. Einstein (Sao Paulo). 2017;15(4):507–511. doi: 10.1590/S1679-45082017RB4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin É, Westermann CJJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363(9412):852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 48.Martin GM, Poot M, Haaf T. Lessons for aging from Werner syndrome epigenetics. Aging (Albany NY). 2020;12(3):2022–2023. doi: 10.18632/aging.102829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389(6651):631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 50.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29(2):117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 51.Lin AE, Alali A, Starr LJ, Shah N, Beavis A, Pereira EM, Lindsay ME, Klugman S. Gain-of-function pathogenic variants in SMAD4 are associated with neoplasia in Myhre syndrome. Am J Med Genet A. 2020;182(2):328–337. doi: 10.1002/ajmg.a.61430. [DOI] [PubMed] [Google Scholar]
- 52.Kim KS, Kang KW, Seu YB, Baek SH, Kim JR. Interferon-gamma induces cellular senescence through p53-dependent DNA damage signaling in human endothelial cells. Mech Ageing Dev. 2009;130(3):179–188. doi: 10.1016/j.mad.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Alexander PB, Wang XF (2017) TGF-beta family signaling in the control of cell proliferation and survival. Cold Spring Harb Perspect Biol. 9(4) [DOI] [PMC free article] [PubMed]
- 54.Eickelberg O, Pansky A, Koehler E, Bihl M, Tamm M, Hildebrand P, et al. Molecular mechanisms of TGF-(beta) antagonism by interferon (gamma) and cyclosporine A in lung fibroblasts. FASEB J. 2001;15(3):797–806. doi: 10.1096/fj.00-0233com. [DOI] [PubMed] [Google Scholar]
- 55.Ishida Y, Kondo T, Takayasu T, Iwakura Y, Mukaida N. The essential involvement of cross-talk between IFN-gamma and TGF-beta in the skin wound-healing process. J Immunol. 2004;172(3):1848–1855. doi: 10.4049/jimmunol.172.3.1848. [DOI] [PubMed] [Google Scholar]
- 56.Strober W, Kelsall B, Fuss I, Marth T, Ludviksson B, Ehrhardt R, Neurath M. Reciprocal IFN-gamma and TGF-beta responses regulate the occurrence of mucosal inflammation. Immunol Today. 1997;18(2):61–64. doi: 10.1016/s0167-5699(97)01000-1. [DOI] [PubMed] [Google Scholar]
- 57.Choy L, Skillington J, Derynck R. Roles of autocrine TGF-beta receptor and Smad signaling in adipocyte differentiation. J Cell Biol. 2000;149(3):667–682. doi: 10.1083/jcb.149.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 580 kb)
Data Availability Statement
Data that support the findings of this study are available from the corresponding author upon request.