

Abstract
Adsorption kinetic equation has been derived assuming that the process follows the behavior of a heterogeneous chemical reaction at the solid–liquid interface. This equation is converted into the Langmuir isotherm at equilibrium and describes well the unsteady-state adsorption process. Based on that, a working equation has been developed, which gives adsorption-rate-constant independent of operating parameters including concentration. Also, a kinetic model expressed as a sum of first- and second-order systems available in the literature has been applied (modified with the interface reaction concept) to determine the adsorption rate constant. Both methods gave similar results. Three dimensionless numbers have been developed to determine and distinguish pseudo-first-order and pseudo-second-order kinetics justified from the viewpoint of chemical kinetics. It is shown that curve-fitting with a high correlation coefficient could validate an empirical kinetic model, but the fitted model parameters could not automatically be related to chemical kinetic parameters if the model itself is not grounded on well-defined chemical kinetics. Finally, it is concluded that the currently applied empirical approach could not provide reliable data for comparison among similar systems, while the Langmuir kinetic equation developed based on the concept of heterogeneous reaction would be a good basis for standardization of the method for adsorption system characterization.
1. Introduction
Rapid industrialization has led to the production of a devastating amount of wastewater. Adsorption process seems to be an effective method to manage this problem. In the last few decades, an enormous number of papers have been published on wastewater treatment by adsorption process using locally available, naturally abundant, and practically noncommercial products (with a few exceptions for commercial activated carbon) such as tea-waste,1,2 leaves,3,4 agricultural residues,5,6 biomasses,7−10 waste minerals,11,12 etc. The task of these works consists mainly of the search for suitable adsorbents characterized by equilibrium and kinetic parameters. Most of the adsorption systems dealing with wastewater treatment follow the well-known Langmuir adsorption equilibrium model, but for studying the adsorption kinetics, the authors use some pseudo-first-order (PFO) or pseudo-second-order (PSO) empirical models, and the fitted values (pseudo-rate constants) appear to be dependent on operating parameters (initial concentration, adsorbent amount, W, and solution volume V) and they are not correlated to the Langmuir parameters at all. We must recognize that for conducting an adsorption process in an optimal regime, the adsorption rate constants (like those of a chemical reaction) must be known to be independent of operating parameters, and the equilibrium equation must be a special case of the kinetic equation. This is the element to which the decade-long research on developing an adsorbent system for wastewater treatment, with a few exceptions,13−29 has not been paid the deserved attention.
Islam et al.13 developed an adsorption kinetic model based on the Langmuir adsorption concept, in which for a given adsorption dosage, wa = W/V, three parameters, namely, maximum adsorption density (adsorption capacity) (q∞) and concentration-independent adsorption and desorption rate constants (ka and kd), completely described both adsorption kinetics and equilibrium simultaneously.
The research group of Azizian14−17,19 is the most active one in the experimental work equipped with a theoretical interpretation of adsorption data. In ref (14), the author concluded that the system followed PFO kinetics for high initial adsorbate concentration and PSO kinetics for low initial adsorbate concentration and sufficiently small reaction times. In ref (15), they concluded that the correlation coefficient could not be the only criterion to distinguish between the PFO and the PSO kinetics. In ref (16), the author introduced a two-site PSO adsorption model for the heterogeneous surface. In ref (17), the authors developed further a geometric approach initially proposed by Kuan et al.18 to determine the adsorption rate constant and also proposed a mixed Langmuir–Freundlich model, for which the reaction became of n-th order with respect to the active site. In ref (19), the authors applied extended geometric methods and claimed to have obtained a concentration-independent adsorption constant. But still the adsorption and desorption rate constants remained adsorption dosage-dependent.
Extensive theoretical works on adsorption kinetics have been reported by a research team in refs (20−24). In ref (20), they propose a general kinetic equation based on the statistical rate theory (SRT), which is transformed into the PFO and PSO kinetic equations for surface-reaction-controlled adsorption processes. In ref (21), they conclude that the PFO and PSO equations are simple approximations of different theoretical approaches. In ref (22), they show that the PSO kinetic equation describes the process efficiently close to equilibrium. In ref (23), they proposed a two-resistance model for sorption kinetics controlled simultaneously by intraparticle diffusion and surface reaction. In ref (24), they express the PSO constant as a function of the initial sorbate concentration, the progress of the sorption process, and the solid/solution ratio.
Liu and Shen25 made excellent rearrangement of the adsorption kinetic equation as a sum of the first- and second-order kinetic components. The rate constants corresponding to the PFO and PSO components (kH1 and kH2) were dependent on operating parameters.
Marczewski26 proposed a new integrated kinetic Langmuir (IKL) equation to describe the general kinetic behavior of the adsorption process. In a further work, the author made vigorous and complicated mathematical treatment to determine pure PSO kinetics.27
Salvestrini28 developed a method for the determination of the concentration-independent adsorption rate constant, ka, but that method required highly complicated estimations. Also, the adsorption constant, ka, is again dependent on operating parameters, as the starting equation does not take into proper account the solid–liquid-phase mass balance (interface reaction). Guo and Wang29 chose a mixed-order model to describe Langmuir adsorption kinetics and solved the equation with a 4–5-order Runge–Kutta method using a MatLab program.
As mentioned above, the models describing adsorption kinetics in aqueous media are becoming more and more complicated. In refs (13, 19, 28), it has already been established that by applying the conventional Langmuir kinetic model, one could estimate concentration-independent adsorption constants. Search for some correlation between pseudo-rate constants and adsorption rate constants might merely satisfy scientific curiosity. Discussion on pseudo-order kinetics, however, is necessary to convince research teams active in the field to abandon the empirical approach in treating adsorption kinetic data. Moreover, the PFO and PSO kinetics are distinguished based on some data and methods of treatment, which have been disputed by Canzano et al.30 and Simonin.31 Since the last four decades, a huge number of adsorbent–adsorbate systems have been studied, but since the pseudo-rate constants are operating-parameter-dependent, there is no scope of comparing the adsorption kinetics. To design and operate a plant in an optimal regime, the rate constants should be available as independent of operating parameters. The formulations described in refs (13, 19, 28) give concentration-independent adsorption constants, but they are dependent on the adsorption amount, W, and solution volume, V. This would mean that different researchers would obtain the same concentration-independent value of rate constants for themselves, but these values would differ from one another, as they would design the batch experiments independently with different W and V. Thus, for a given adsorption system, different researchers would report different concentration-independent rate constants.
The method of determination of adsorption constants might be standardized by fixing the adsorbent dosage, but such fixation might not be optimum for different adsorbents as they might show largely varying adsorption capacities. To avoid such disadvantages, the Langmuir adsorption kinetics may be described as an interface reaction. With the interface reaction model, the adsorption rate constant will appear independent of operating parameters. The prime task of this study is to develop a simple methodology for determining the adsorption rate constant, ka, which, together with the Langmuir equilibrium parameters—namely, (i) maximum adsorption capacity conventionally denoted q∞ (kg adsorbate/kg adsorbent) and (ii) Langmuir equilibrium constant, K (m3/kg)—will characterize the equilibrium as well as the kinetic state of an adsorption system. A set of these three parameters would serve as a basis for comparing the adsorption efficiencies of different adsorbent–adsorbate systems.
The secondary task of the work is (i) to derive a working equation to determine the “rate constants of the first- and the second-order components” of the “hybrid-order” kinetic equation and to relate them to the Langmuir constants in terms of operating parameters, and (ii) finally, to find well-defined conditions under which Langmuir kinetics is reduced to “true” PFO and PSO kinetics. Dimensionless numbers will be defined to determine the conditions under which the adsorption processes could be described by the true pseudo-first-order and pseudo-second-order kinetics justified from the viewpoint of chemical kinetics. The secondary task is undertaken only to establish the findings of the prime task as the simplest and most reliable method for characterizing adsorbate–adsorbent systems. The theoretical ground and the procedure for determining the rate constants defined through the mentioned kinetic models have been discussed in detail and illustrated through the same data available in the literature.
2. Theoretical
2.1. Langmuir Model for Equilibrium Adsorption and Independent Empirical Model for Kinetics: Empirical Approach (EA)
This is the approach that has been applied indiscriminately in the literature, where the kinetic model is separately treated from the equilibrium one. To distinguish it from the others, this approach will be called the “Empirical Approach (EA)” henceforth.
2.1.1. Langmuir Adsorption Equilibrium Model
The Langmuir model adapted to the liquid system is represented by the following relation
| 1 |
where qe is the adsorption density at equilibrium (kg adsorbate A/kg adsorbent), q∞ is the adsorption capacity of the adsorbent or the maximum achievable adsorption density (kg adsorbate A/kg adsorbent), CA,e is the equilibrium adsorbate concentration in the liquid phase (kg/m3), and K is the Langmuir equilibrium constant (m3/kg). Equation 1 can be linearized as follows
| 2 |
A CA,e/qe vs CA,e plot will give the Langmuir parameters, q∞ and K. In fact, the determination of these two equilibrium parameters is a mandatory step in the analysis of Langmuir adsorption kinetics for all three approaches to be discussed in the following section.
2.1.2. Kinetic Model of EA
In EA, the kinetic models are expressed as PFO32 and PSO33 equations in terms of (qe – q), where q is the adsorption density at any time, t. The PFO equation is expressed as follows
| 3 |
where kE1 (s–1) is the apparent adsorption rate constant for the PFO adsorption process (as per EA).
Integrating eq 3 for t = 0, q = 0, one obtains
| 4 |
If a −ln(1 – q/qe) vs t plot gives a straight line, the value of kE1 is determined from the slope. In fact, with q vs t data available at hand, the derivative of q could be estimated graphically or numerically and eq 3 itself could be applied to determine kE1 on a dq/dt vs (qe – q) plot. This option, however, is not practiced. Since the correlation coefficient for linear fit is the only criterion for the assertion of the pseudo-rate order, the change in the reference frame for linear fit might also be used to diffuse any confusion distinguishing pseudo-rate orders. In this study, both options will be applied.
The PSO adsorption kinetics33 is represented by the following relation
| 5 |
where kE2 (s–1) is the apparent rate constant for a PSO adsorption process as per EA.
Integrating eq 5 for t = 0, q = 0 and rearranging, one derives
| 6 |
Equation 6 is the most popular form of the integrated adsorption kinetic equation for determining the rate constant of PSO adsorption kinetics. If a t/q vs t plot gives a straight line, the value of kE2 is determined from the intercept of the plot. Similar to the PFO kinetics, the value of kE2 could also be determined from a dq/dt vs (qe – q)2 plot described by eq 5.
The q vs t curves are of saturation type. The adsorption density q gradually increases with a continuously decreasing slope until it attains an ultimate value, qe, depending on the initial concentration, adsorbent mass, and solution volume. Such curves can be described empirically as follows
| 7 |
and/or
| 8 |
Equations 7 and 8 are standard empirical equations for describing saturation-type curves. The PFO kinetic equation (eq 3) and its linearized form (eq 4) are virtually the differential and the linearized form, respectively, of eq 7 with a = kE1. Moreover, the PSO kinetic equation (eq 5) and its linearized form (eq 6) are virtually the same for eq 8 with b = kE2qe (for details, see Appendix A in the Supporting Information SI). Both a and b (and consequently, kE1 and kE2) depend on the initial concentration (and also on operating parameters).
Although both eqs 7 and 8 generally describe the saturation curves well, in many adsorption kinetic experiments, saturation is reached within a short time, and there are very few data recorded in the steep-rising zone and the other data are available only in the near-equilibrium zone.34−36 It might be expected that eq 7 (which corresponds to eq 3) containing an exponential term would describe the adsorption curves reaching the equilibrium with a very steep slope comparatively better. The reverse picture, however, is observed in reality. All such curves are undisputedly fitted to eq 6, which is derived from eq 8, and the kinetics is recognized as PSO. Canzano et al.,30 however, have excellently demonstrated that the fit of the near-equilibrium data to eq 6 is a straight line irrespective of the sorption kinetics. Simonin31 also concludes that the inclusion of several data points from the near-equilibrium zone might lead to an erroneous decision that the adsorption kinetics follows the PSO model. For a good adsorbent, the equilibrium is reached very quickly and data points far from equilibrium are rare, and more data points are taken from near-equilibrium. Despite the conclusive finding of Canzano et al.30 and Simonin31 that near-equilibrium data have to be avoided in determining the order of kinetics, most of the authors still have been concluding about kinetics based on such data. Also, there are cases when both PFO and PSO models describe the adsorption kinetic data equally well, but again with curve-fittings with a higher coefficient of correlation, usually, it is concluded to be PSO kinetics. In this study, both the differential and integral forms of the pseudo-order kinetic equation will be tested for cross-checking. In chemical kinetics, there must be some well-defined demarcation line between the PFO and PSO reaction kinetics. Discussion on such a demarcation line will be discussed later in Section 2.4.
2.2. Adsorption–Desorption as a Physicochemical Interface Reaction: Interface Reaction Approach (IRA)
The subject matter of this subsection is related to the prime task of this work, i.e., to work out a simple method to characterize the adsorption–adsorbate system. For this purpose, a theoretical model will be developed considering the adsorption process as a physicochemical interaction at the solid–liquid interface following the laws of chemical kinetics, and this approach will be termed the “Interface Reaction Approach (IRA)” in this work.
In a batch adsorption process, a preweighed amount of adsorbent, W (kg), is added to an adsorbate solution of volume V and initial concentration CA,0. The system is kept under vigorous stirring to keep the adsorbate concentration, CA, uniform throughout the liquid phase. At some predetermined time intervals, samples are collected from the vessel/reactor to control the remaining adsorbate in the liquid phase.
Assuming that there is no diffusion layer, and the interface represents a sharp boundary between the solid and liquid phases (i.e., the process is adsorption kinetics-limited), the adsorption kinetics is represented by a reversible interface reaction of the type
| 9 |
where ka (m3/s) and kd (kg/s) are the adsorption and desorption rate constants, respectively, the suffixes (l) and (s) denote the liquid and solid phases, respectively, and S represents the active site or “a collection of sites” capable of hosting a single molecule of the adsorbate. It should be mentioned that the Langmuir model was initially proposed to describe adsorption in the gas–solid system with an assumption of a 1:1 ratio for the adsorbate molecules and the active sites. When the model is adapted to the solid–liquid system, due to the large size of the adsorbate, S might not represent a single site but rather a collection of sites performing the same function as a single site does in the gas–liquid system.
In the differential form, the mass-balance equation concerning the adsorbate mass in the whole solid–liquid system is as follows
| 10 |
Referring to the physicochemical interaction at the solid–liquid interface in eq 9, the sorption–desorption kinetics can be described as follows
| 11 |
or
| 12 |
where CA (kg/m3) and CAS (kg/kg) are the adsorbate concentrations at time t in the liquid and solid phases, respectively, and CS represents the unoccupied site concentration in the solid phase and is expressed as the amount of adsorbate (kg) that could still be accommodated in 1 kg of adsorbent. Although the adsorption takes place at the solid surface, the concentration in the solid phase is expressed in terms of the mass of the adsorbent (assuming that the surface area is proportional to the mass of the adsorbent). It is worth mentioning that the form of writing kinetics of a chemical reaction by multiplying the rate of change in concentration by weight or volume (as done in eqs 11 or 12) is not much common in the analysis of chemical kinetics. However, this is the key to finding operating-condition-independent adsorption rate constants, which have been so desired by all researchers in the field of intensive adsorption kinetics studies since the last four decades. Interface reaction is the rate of transfer of a species from one phase to another.37 The first term on the right-hand side of eqs 11 or 12 is the rate of transfer of species A (kg/s) from the liquid to the solid phase, and the second term is the rate for the reverse course. This exchange of mass is manifested in the change of concentration of the species in both phases simultaneously. On the left-hand side of eq 11, dCAS/dt represents the rate of change of concentration (kg/kg) of species A in the solid phase, while in eq 12, dCA/dt represents the rate of change of concentration (kg/m3) of species A in the liquid phase. Therefore, multiplying the rate of change of adsorbate concentration in the solid phase (i.e., the rate of adsorption) by W on the left-hand side of eq 11 or multiplying the rate of change of adsorbate concentration in the liquid phase by V on the left-hand side of eq 12 is mandatory to define the adsorption and desorption constants independent of operating parameters, and this makes eqs 11 and 12 different from all other formulations available in the literature studying adsorption kinetics in waste treatment. In an adsorption experiment, the rate of change in adsorbate concentration is not monitored on the adsorbent surface but rather in the liquid volume. Therefore, one can also apply eq 12 for analysis and will get the same values for the rate constants.
Using conventional symbols used in the adsorption process, CAS ≡ q and Cs ≡ q∞ – q, eq 11 takes the form
| 13 |
At equilibrium, CA = CA,e, q = qe, and dq/dt = 0, and after some algebraic manipulations, eq 13 is converted into the Langmuir isotherm equation (eq 1) with ka/kd = K, and eq 13 itself is the corresponding Langmuir adsorption kinetic equation.
2.2.1. Derivation of the Working Equation for Studying Adsorption Kinetics in IRA
Integrating the mass-balance equation (eq 10) for the adsorbate in the solid–liquid system, one has
| 14 |
where wa is the adsorption dosage (kg/m3) and CA,0 is the initial concentration of the adsorbate in the liquid phase.
Equation 14 is substituted in eq 13, and then the adsorption kinetic equation takes the following form
| 15 |
Equation 15 can now be integrated for q analytically by a conventional method, and fitting the experimental data to the solution equation and knowing that K = ka/kd, one can determine the values of ka and kd. For details, the readers are referred to ref (10). To make a better analogy with eqs 3 or 5, eq 15 is rearranged as follows
| 16 |
or
| 17 |
where
RQ is called the “reaction quotient.”
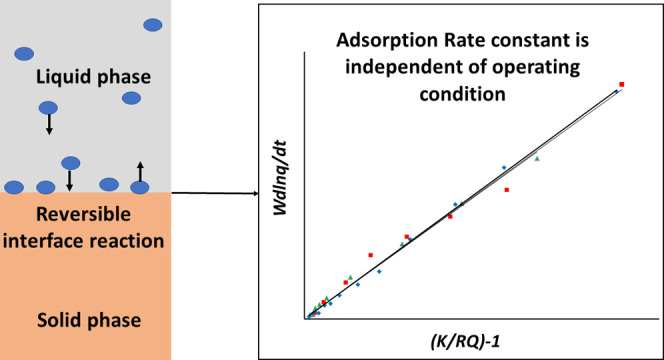
The linearity of the 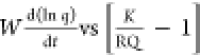 plot will
validate the Langmuir kinetic
model, and from the slope of the line, one will get kd (and subsequently, ka).
In fact, with the application of the equilibrium relation in eq 1 and the working equation
of adsorption kinetics (eq 17), the three basic Langmuir parameters, namely, q∞, K, and ka, will be known, and the adsorbent–adsorbate system
is well-characterized.
plot will
validate the Langmuir kinetic
model, and from the slope of the line, one will get kd (and subsequently, ka).
In fact, with the application of the equilibrium relation in eq 1 and the working equation
of adsorption kinetics (eq 17), the three basic Langmuir parameters, namely, q∞, K, and ka, will be known, and the adsorbent–adsorbate system
is well-characterized.
2.3. Adsorption Kinetic Equation in the Hybrid-Order Approach (HOA)
The subject matter of this subsection is related to the secondary task of this work. For this purpose, the adsorption kinetics described by eq 13 will be presented as a sum of the first-and second-order reaction components, and a methodology will be developed to determine the adsorption rate constant. This approach of data analysis will be termed “HOA” in this work, and then, the results from IRA will be compared with those obtained by EA and HOA.
2.3.1. Derivation of the Kinetic Equation as a Sum of the First- and Second-Order Reaction Components
Rewriting eq 15 for the equilibrium condition, one obtains
| 18 |
Equation 18 is a quadratic one, and solving it for qe, two probable expressions are obtained (eq 19 with a ± sign).
| 19 |
with
| 20 |
From the two expressions for qe in eq 19, that one is accepted for which it has got physical significance. Thus, for a given batch adsorption system, with the known Langmuir parameters, K, and adsorption capacity, q∞, the equilibrium adsorption density, qe, can be predicted by eq 19 for a given adsorbent mass, initial adsorbate concentration, and liquid volume. Both the signs (positive and negative) in eq 19 give a positive value for qe (which are justified as a mathematical solution), but with a positive sign, it will give an unacceptable value in the physical sense (e.g., higher than q∞ or some other irrational value like that corresponding to the adsorbed amount higher than the total adsorbate amount in the system). Only the negative sign will give a value that is acceptable in the physical sense. Thus, the acceptable option of eq 19 is eq 21.
| 21 |
Combining eqs 15, 18, and 21, we have
| 22 |
with
| 23–25 |
The derivation of eq 22 along with the expressions for kH1 and kH2 is available in refs (25, 29). As in this work, the adsorption process is viewed as a solid–liquid interface reaction (eq 9) and the adsorption kinetics is described in conformity with changes of the adsorbate concentration in the liquid and solid phases (eqs 11 and 12); for ease of the readers, the derivation of the equation and the modified expressions for kH1 and kH2 with the correction for material balance in the solid–liquid system are given in details in Appendix B (Supporting Information).
As seen in eq 23, the “so-called rate constants” kH1 and kH2, respectively, for the first- and second-order components of the reaction kinetics are related to the adsorption rate constants of the Langmuir kinetic model and also depend on the operating parameters. Thus, these rate constants could be considered as technological rate constants for the adsorbate–adsorbent system valid for given initial concentrations and operating parameters (adsorbent amount W and liquid volume V). Equation 22, however, could be used as an indirect method for estimating the adsorption rate constant, ka. It can be noted that the ratio kH1/kH2 in eq 23 does not depend on the adsorption rate constant but rather depends on Δ (i.e., on the equilibrium and the operating parameters of the process) (eq 20).
2.3.2. Determination of the Technological Rate Constant, kH1
Equation 22 can be rearranged as follows
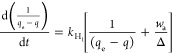 |
26 |
A d(1/(qe – q))/dt vs 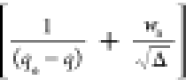 plot will
give the value of kH1. Finally,
the value of ka will be estimated following
the relation expressed by eq 23.
plot will
give the value of kH1. Finally,
the value of ka will be estimated following
the relation expressed by eq 23.
2.4. PFO or PSO Kinetics in IRA
In subsection 2.1, we have already introduced the PFO (eq 3) and PSO (eq 5) kinetic models as per the empirical approach (EA), in which the pseudo-rate constants are not correlated to the rate constants that define the equilibrium process. The chemical kinetics of a complex process could reduce to some pseudo-first-order or pseudo-second-order kinetics under some special operating conditions, but in such cases, the rate constants preserve their significance as those in the complex processes, i.e., the pseudo-rate constants remain closely related to the rate constant of the precursor complex processes. Tien and Ramarao38 and Zhang39 have discussed in detail the conditions under which the Langmuir kinetic equation is reduced to PFO and PSO order models and shown the errors made in this approximation. In the following subsection, we shall find out the conditions under which the adsorption kinetic equation (eq 15) will be converted into pseudo-first-order and pseudo-second-order equations from the viewpoint of interface reaction kinetics.
2.4.1. Condition 1 for PFO Kinetics in IRA
Combining eqs 15 and 18, and making some algebraic manipulation, one obtains
| 27 |
As q increases from 0 to qe, the term waq/CA,0 assumes the value from 0 to N1 = waqe/CA,0, and now if N1 is much less than unity, eq 27 is approximated to
| 28 |
with
| 29 |
where kI,a is the rate constant of true PFO adsorption kinetics in IRA under the condition N1 = waqe/CA,0 ≪ 1, and this PFO rate constant (unlike kE1 in the empirical approach, EA) is well-correlated with ka through eq 29. Practically, kE1 is averaged over the whole range of θ (=q/qe) from 0 to 1 to satisfy an empirical relation representing a saturation curve. Note that two different symbols (kI and kE with additional subscripts a, 1, etc.) have been used in this work to denote pseudo-rate constants. This has been done deliberately to differentiate the pseudo-order reactions from the viewpoints of IRA and EA. Under the condition of N1 ≪1, kE1 in EA coincides with kI,a in IRA.
2.4.1.1. Physical Significance of the Dimensionless Number, N1
As defined, N1 is equal to waqe/CA,0 = (Wqe)/(VCA,0). Thus, N1 is the ratio between the amount (kg) of adsorbate in the solid phase at equilibrium and that in the liquid phase dissolved initially (which is virtually the total amount of adsorbate in the whole system). In other words, at equilibrium, it is the part of the adsorbate in the solid phase in a solid–liquid system. Under a given operating condition (W, V, CA,0), N1 assumes a constant value independent of the degree of completion, θ (=q/qe), of the adsorption process.
2.4.2. Condition 2 for PFO Kinetics in IRA
Again combining eqs 15 and 18, we have
| 30 |
As q increases from 0 to qe, the term q/q∞ assumes the value from 0 to N2 = qe/q∞, and now if N2 is much less than unity, eq 30 is approximated to
| 31 |
with
| 32 |
where kI,b is also the rate constant of PFO adsorption kinetics in IRA under the condition N2 = qe/q∞ ≪ 1. Under this condition, kE1 in EA coincides with kI,b in IRA.
2.4.2.1. Physical Significance of the Dimensionless Number N2
As defined, N2 is equal to qe/q∞. Thus, it is the ratio between the amount of adsorption at equilibrium and the adsorption capacity of the adsorbent. In terms of active sites, it is the degree of occupation of active sites at equilibrium. Under a given operating condition (W, V, CA,0), N2 assumes a constant value independent of the degree of completion, θ, of the adsorption process.
2.4.3. Reaction-Order-Determining Common Dimensionless Number in IRA
Equation 15 can also be rewritten as follows
| 33 |
with
| 34, 35 |
Comparing eq 34 with eq 21, we have
| 36, 37 |
Substituting eq 36 into eq 33 and after some simple algebraic manipulation, we have
| 38 |
Let us define a reaction-order-determining dimensionless number, NOD, such that
| 39 |
The value of NOD will depend on NOD,0 and a factor fθ. For a change of θ from 0 to 0.9 (keeping a 10% margin to reach qe), the factor fθ varies from 1 to 10.
2.4.3.1. Physical Significance of the Dimensionless Number, NOD
The value of NOD depends on two factors: NOD,0 and fθ. The dimensionless number, NOD,0, is defined in eq 39, and combining eqs 39, 21, and 1, it can be expressed as follows
| 40 |
The derivation of eq 40 is given in details in Appendix C in the Supporting Information. As seen in eq 40, NOD,0 is a complex function of the dimensionless numbers, N1 and N2, and under a given operating condition (W, V, CA,0), NOD,0 will also assume a constant value independent of the degree of completion, θ, of the adsorption process. NOD, however, depends on the factor, fθ, as well, which varies with θ. In terms of the dimensionless number, NOD, one can rewrite eq 38 as follows
| 41 |
If NOD ≫ 1 in the θ-range of (0, 0.9), eq 41 is approximated to eq 42
| 42 |
where kIc is also a rate constant of PFO adsorption kinetics in IRA. For NOD ≫ 1, kI,c in IRA coincides with kH1 in HOA and also with kE1 in EA.
If NOD ≪ 1 in the θ-range of (0, 0.9), then eq 41 is approximated to eq 43
| 43 |
where kII is the rate constant of PSO adsorption kinetics in IRA under the condition NOD ≪ 1 for θ in the range of (0, 0.9). For NOD ≪ 1, kII in IRA coincides with kH2 in HOA and also with kE2 in EA.
Thus, the three dimensionless numbers will determine under what conditions the adsorption reaction will follow true PFO or PSO kinetics (justified from the viewpoint of chemical kinetics), and the adsorption rate constant could be estimated from the pseudo- rate constants in EA.
2.4.4. Pseudo-Rate Constants of EA in the Light of the Dimensionless Number NOD
As discussed in the preceding three subsections, the PFO and PSO adsorption processes are well-defined in the IRA and are characterized by the corresponding rate constants (kIa, kIb, or kIc for PFO, and kII for PSO), and these pseudo-rate constants have a well-defined correlation with ka. Then, the question arises as to what do the PFO and PSO adsorption kinetics in EA with the corresponding rate constants, kE1 and kE2, stand for? What information could one get from these rate constants?
Since the 1980s, enormous numbers of papers have been published in scientific journals on adsorption in aqueous media, in which adsorption kinetics have been treated as PFO or PSO (as per EA). In subsection 2.1, it has been mentioned that the saturation-type experimental curves could almost always be described with some approximation with eqs 7 or 8, and the PFO (eq 3) and PSO (eq 5) equations in EA are derived from these equations.
The three dimensionless numbers, N1, N2, and NOD, can determine under what conditions an adsorption kinetic equation can be transformed into the PFO or the PSO equation. If such conditions are not met, the adsorption rate constant has to be determined from the general equation (eq 17). In EA, there is no buffer zone, and the kinetic data must satisfy either the PFO or PSO model. Thus, the corresponding rate constants, kE1 and kE2, could be related to ka only when the PFO or PSO is justified by chemical kinetics (in terms of the dimensionless numbers). Beyond that, there is no theoretical ground to correlate kE1 and kE2 to ka.
The adsorption kinetic data could be fitted to PFO kinetics (eq 4) for N1 ≪ 1, N2 ≪ 1, or NOD ≫ 1, and the fitted value of kE1 could be equated to kI,a, kI,c, or kI,c (whatever applies), respectively, and ka can be evaluated. Compromising with the condition, N1 ≪ 1, N2 ≪ 1, or NOD ≫ 1, will result in a ka value with more and more uncertainty. For NOD ≪ 1, the adsorption kinetic data could be fitted to PSO kinetics (eq 6), and the fitted value of kE2 could be evaluated, and finally, by equating kE2 to kII, ka can be evaluated.
The choice of a maximum value as “much less than unity” or a minimum value as “much higher than unity” determines the preciseness of the analysis. The value of NOD depends on the factor fθ, which increases from 0 to 10 for an increase in θ from 0 to 0.9 (eq 39). That will mean, if NOD,0 = 8 (for example), the value of NOD will increase from 8 to 80 in the θ-range of 0–0.9. Also, if NOD,0 = 0.02, the value of NOD will increase from 0.02 to 0.2 for the same θ-range. Avoiding any discussion on experimental errors in kinetic data acquisition, for the present analysis, we accept that for N1 < 0.2, N2 < 0.2, or NOD,0 > 8, the process will follow PFO kinetics, and for NOD,0 < 0.02, the process will follow PSO kinetics.
3. Results and Discussion
3.1. Validation of the Model Generated from IRA and HOA
Before stepping up into the application of any form of adsorption kinetics model, it must be remembered that the acquisition of data for equilibrium analysis (eq 1) is much easier and more precise than that of the data for kinetics analysis. This is because the samples for equilibrium data could be collected and analyzed at the end of the experiment after an undetermined period of time to ensure that equilibrium has been reached. This is not the case with the collection of adsorption kinetics data. Distinguishable q vs t data is collected easily for slow adsorption processes when the sampling time interval is much smaller than the time for reaching the equilibrium. Each sample undergoes centrifuging for some time (2–3 min) to separate the adsorbent particles from the sample solution (during that time the adsorption continues, but not counted) before being analyzed with a UV spectrometer. Thus, from sampling to analysis, it always requires some time, and the sampling is necessary at some reasonable time interval to acquire well-distinguishable data points.
When the adsorption process is very fast, it may not be possible to collect an adequate number of well-distinguishable q vs t data points. In that case, the analysis has to be done with very few numbers of data points. The data points near equilibrium may be appealing for some theoretical analyses,40 but those experienced in the experimental work on adsorption must admit that near equilibrium, the data are sometimes a bit fluctuating, the increment rate is very low, and sometimes confusion may arise about whether or not the changes are within the experimental error. Such data are not much suitable for precise kinetic studies. Canzano et al.30 and Simonin31 have made a thorough analysis of the consequence of the inclusion of data near equilibrium and concluded that such inclusion may lead to a confusing conclusion about the order of adsorption kinetics. The data near equilibrium are acquired for a long period of time only to ascertain the value of equilibrium adsorption, and not to follow the kinetics. Consequently, the treatment of equilibrium data with eq 1 is more precise than that of the kinetics ones.
3.2. Illustration of IRA and HOA with Literature Data and the Determination of ka
Four absorbent–adsorbate systems (each designated by a number from 1 to 4, and each system is subdivided into groups “a–c” based on the variation in operating conditions) under investigation have been presented in Table 1 along with their equilibrium parameters. The data have been collected from the plots of the source materials using the software WebPlotDigitizer and retreated to verify the preciseness of the collection. As seen in Table 1, the equilibrium values reported in the source and those determined in this work are satisfactory in the same range.
Table 1. Absorbent–Adsorbate Systems under Investigation along with the Langmuir Equilibrium Parameters.
| designation | adsorption system | W × 104 (kg) | V × 104 (m3) | CA,0 (kg/m3) | T (K) | q∞ (q∞) | K (m3/kg) (K) | source |
|---|---|---|---|---|---|---|---|---|
| system 1a | aGAC-MV | 0.8 | 0.5 | 0.001 | 298.15 | 0.095 (0.093) | 380.76 (378.5) | (19) |
| system 1b | 0.002 | |||||||
| system 1c | 0.003 | |||||||
| system 2a | aCA-B_AC composite beads-MB | 2 | 2 | 0.20 | 303.15 | 0.756 (0.755) | 21.5 (21.0) | (41) |
| system 2b | 0.30 | |||||||
| system 2c | 0.50 | |||||||
| system 3a | Durian shell-MB | 6 | 1 | 0.20 | 303.15 | 0.289 (0.283) | 19.7 (21.2) | (42) |
| system 3b | 0.25 | |||||||
| system 3c | 0.30 | |||||||
| system 4a | Cadmium II-C. vulgaris | 0.75 | 1 | 0.10 | 303.15 | 0.091 (0.089) | 22 (21.4) | (43) |
| system 4b | 313.15 | 0.083 (0.085) | 15 (14.1) | |||||
| system 4c | 323.15 | 0.077 (0.077) | 9.5 (9.3) |
Abbreviations: GAC, granular activated carbon; MV, methyl violet; CA_B_AC, calcium alginate–bentonite–activated carbon; MB, methylene blue. In parentheses are the values re-estimated in the present work.
3.3. Determination of ka as per IRA and HOA
The adsorption kinetic data of all four
systems in Table 1 have
been fitted to eq 17 as per IRA and to eq 26 as per HOA to determine the adsorption/desorption rate constants, kd and ka (from the
relation K = ka/kd). Figures 1 and 2 present the W(d ln q/dt) vs (K/RQ – 1) and the d(1/(qe – q))/dt vs 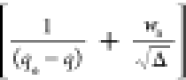 plots with kinetic data of the system 1c.
For the economy of spaces, similar plots for some representative systems
have been presented in Appendix D (Supporting Information). The results
from the curve-fittings for the validation of IRA and HOA are presented
in Table 2.
plots with kinetic data of the system 1c.
For the economy of spaces, similar plots for some representative systems
have been presented in Appendix D (Supporting Information). The results
from the curve-fittings for the validation of IRA and HOA are presented
in Table 2.
Figure 1.

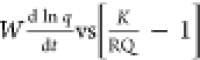
Figure 2.

HOA validation (eq 26): d(1/(qe – q))/dt vs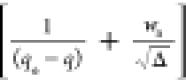 plot for the system 1c.
plot for the system 1c.
Table 2. Adsorption and Desorption Rate Constants, ka and kd, of the Adsorption Systems Determined by IRA and HOA.
| parameters
as per IRA |
parameters
as per HOA |
|||||
|---|---|---|---|---|---|---|
| adsorption system | characteristic change in operating condition | ka × 109 (m3/s) | kd × 109 (kg/s) | kH1 × 105 (s–1) | ka × 109 (m3/s) | kd × 109 (kg/s) |
| system 1a | CA,0 = 0.001 kg/m3 | 182 | 0.48 | 35.1 | 183 | 0.48 |
| system 1b | CA,0 = 0.002 kg/m3 | 181 | 0.48 | 35.6 | 187 | 0.49 |
| system 1c | CA,0 = 0.003 kg/m3 | 185 | 0.49 | 35.0 | 184 | 0.48 |
| system 2a | CA,0 = 0.20 kg/m3 | 18.4 | 0.86 | 6.37 | 20.1 | 0.94 |
| system 2b | CA,0 = 0.30 kg/m3 | 16.6 | 0.77 | 4.58 | 16.5 | 0.77 |
| system 2c | CA,0 = 0.50 kg/m3 | 15.4 | 0.71 | 3.63 | 16.9 | 0.79 |
| system 3a | CA,0 = 0.20 kg/m3 | 5.97 | 0.30 | 1.57 | 5.90 | 0.30 |
| system 3b | CA,0 = 0.25 kg/m3 | 5.93 | 0.30 | 1.58 | 6.11 | 0.31 |
| system 3c | CA,0 = 0.30 kg/m3 | 6.21 | 0.31 | 1.61 | 6.42 | 0.33 |
| system 4a | T = 303.15 K | 334 | 15.2 | 70.3 | 389 | 17.7 |
| system 4b | T = 313.15 K | 390 | 26.0 | 79.7 | 360 | 24.2 |
| system 4c | T = 323.15 K | 440 | 46.3 | 104 | 363 | 38.3 |
As seen in Table 2, the adsorption and desorption rate constants, ka and kd, of a given system determined by the two approaches, IRA and HOA (corrected with material balance), are satisfactorily close to each other. Obtaining of the close values for ka and kd is quite expected as both approaches have their origin in the interface reaction kinetic equation (eq 13). The working equation in IRA (eq 17) is formed directly from eq 13, while that in HOA (eq 26) is formed from eq 22, which was derived again from eq 13.
The adsorption rate constants of system 1 (determined at three different initial concentrations but at a constant temperature) are close to one another and could be averaged as (1.83 ± 0.06) × 10–07 m3/s and (4.8 ± 0.2) × 10–10 kg/s, respectively, in IRA and (1.85 ± 0.05) × 10–07 and (4.9 ± 0.2) × 10–10 kg/s, respectively, in HOA. Similar is the case with systems 2 and 3, the ka values of which could be averaged to consider them as concentration-independent parameters. “The rate constant of a chemical reaction does not depend on concentration” is described even in textbooks of physical chemistry.44 As the adsorption process is viewed as a physicochemical interaction and its kinetics is described by the law of chemical kinetics, the adsorption (also desorption) rate constant must be concentration-independent, and it has become such within data treatment (also acquisition) error. The rate constant determined in this method could be a basis for comparison with other similar systems
With an increase in temperature, both the adsorption and desorption rate constants increase simultaneously (see ka and kd values for system 4 in columns 3 and 4, Table 2), but the equilibrium constant K gradually decreases (see column 8, Table 1). Obviously, the increment rate of the desorption process is higher than that of the adsorption process, and the whole adsorption process is an exothermic one.
Figure 3a,b presents the temperature dependence, respectively, of the adsorption constant, ka (m3/s), and the equilibrium constant K (recalculated into the SI unit, m3/mol) of system 4. The data are fitted to eqs 44 and 45, respectively.44
| 44, 45 |
where ka,0 is a constant for the system, R (kJ/(mol K)) is the universal gas constant, EA (kJ/mol) is the energy of activation, ΔH (kJ/mol) is the enthalpy, and ΔS (kJ/(mol K)) is the entropy of the process.
Figure 3.
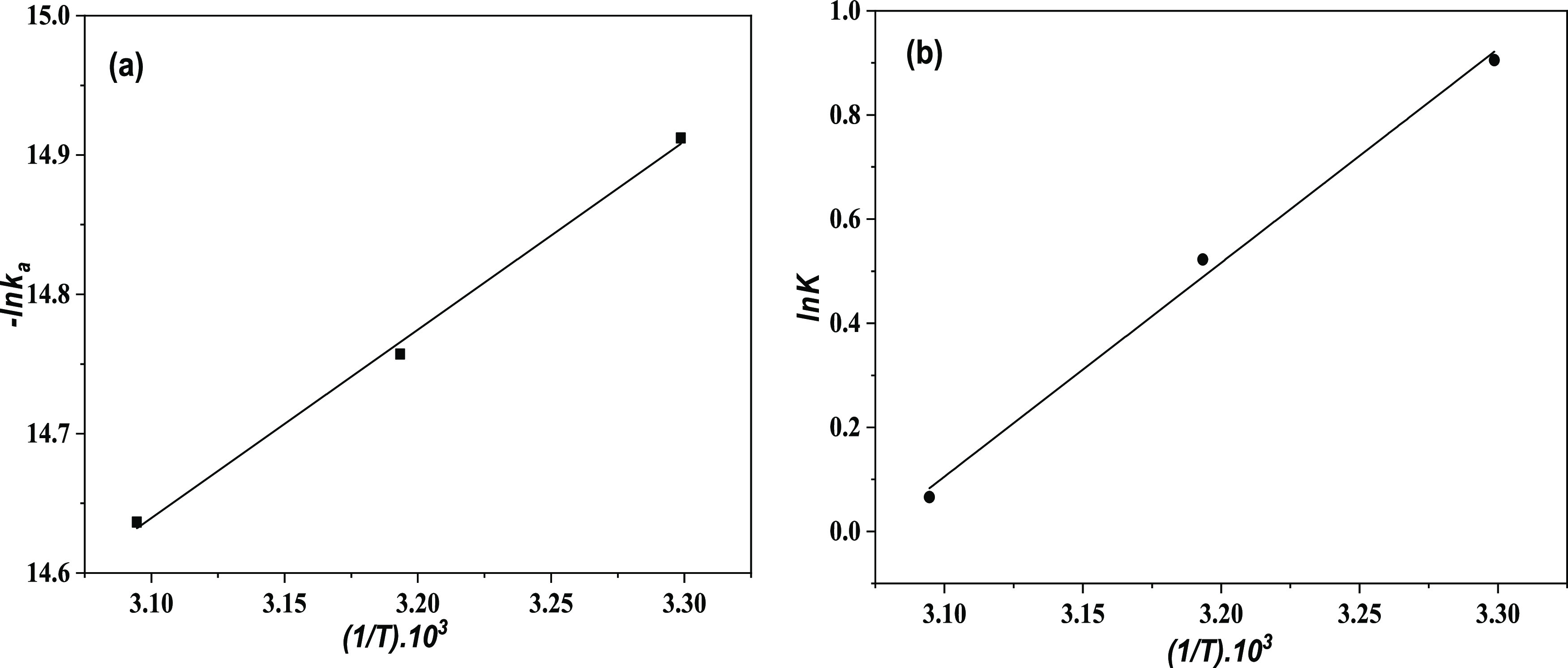
Temperature dependence of (a) ka (m3/s) and (b) K (in m3/mol).
From the slope and intercept of the lines in Figure 3a,b, the energy parameters are estimated as follows: EA = 11.2 kJ/mol, ΔH = −34.2 kJ/mol, and ΔS = −105.0 kJ/(mol K). The evaluated parameters confirm that the process is exothermic and takes place with a decrease in entropy. However, in the temperature range of 303–323 K, the change in Gibb’s energy, ΔG, is negative and the process is spontaneous.
3.4. Validation of PFO and PSO Kinetics in EA
For the validation of the PFO and PSO models in EA, four plots, namely, (i) dq/dt vs (qe – q) (eq 3) and (ii) −ln(1 – q/qe) vs t (eq 4) for the PFO, and (iii) dq/dt vs (qe – q)2 (eq 5) and (iv) t/q vs t (eq 6) for the PSO kinetics, have been drawn for each of the systems. The fitted values of kE1 and kE2 from these plots along with the correlation coefficients of the fittings are presented in Table S1 (Supporting Information). The same for −ln(1– q/qe) vs t (eq 4) and t/q vs t (eq 6) reported in the source materials have also been inserted in Table S1 for comparison.
As seen in Table S1 (Supporting Information), for some subsystems, the difference in the estimated kE1 and kE2 values between the source and the present work is a bit high, indicating that the collection of kinetic data from the plots of the source material might not always be precise enough (the collection of equilibrium data was more precise). However, as an illustrative material, it does not have much impact on the analysis as the analysis is performed based on the values retreated in this work. From the data treatment in this work, it is found that in PFO kinetics, the fitted value of kE1 and also the correlation coefficient for model validation do not differ much for a change in the form of the model equation from eq 4 to eq 3 (see the R2 values for all subsystems in columns 3 and 4 in Table S1). This, however, could not be said for PSO, in which the correlation coefficient for model validation shows substantial changes for a change in the form of the model equation from eq 6 to eq 5. With poor curve-fitting, the fitted value of kE2 is also not much reliable (see the R2 values for subsystems 2a–c, 3a–c, and 4b,c in columns 6 and 7 in Table S1). Thus, the application of cross-checking especially for PSO kinetics with eq 5 against eq 6 is quite justified. Cross-checking gives the opportunity to decide with more confidence whether a system is empirically more inclined to PFO or PSO kinetics. There are systems that satisfy both PFO and PSO kinetic models with good correlation coefficients (e.g., see correlation coefficients for systems 1c, 3a, and 4a). Such cases in IRA are quite absurd, as in a chemical reaction, the transformation of a complex kinetic equation into both the PFO and PSO forms simultaneously is mutually excluding. On the contrary, in EA, it is quite acceptable that a kinetic equation may be described in some cases by both the PFO and PSO models satisfactorily because a saturation-type curve could be described by both eqs 7 and 8 simultaneously. Satisfactory curve-fitting to an empirical equation, however, is not adequate to help determine the adsorption rate constants. For this purpose, the requirements to dimensionless numbers for defining PFO or PSO kinetics have to be satisfied.
3.5. Reliability of Application of Pseudo-Rate Constants, kE1 and kE2, in EA for the Estimation of Adsorption Rate Constants ka
As proposed in subsection 2.4.4, for practical purposes, we shall accept that for N1 < 0.2, N2 < 0.2, or NOD,0 > 8 (theoretically, N1 ≪1, N2 ≪ 1, or NOD ≫ 1) the process will follow PFO kinetics, and for NOD,0 < 0.02 (correspondingly, NOD,max < 0.2, and theoretically, NOD ≪ 1), the process will follow PSO kinetics. Such a compromise with the limiting values of the dimensionless numbers N1, N2, and NOD will bring some uncertainty in the estimated value of ka. The higher the compromise we make, the higher will be the uncertainty in the ka value.
Table 3 presents the dimensionless numbers N1 (column 3), N2 (column 5), and NOD,0 (column 7) for all systems (column 1) under investigation along with the ka value determined by IRA (column 2) for comparison. Whether the condition N1 < 0.2, N2 < 0.2, or NOD,0 > 8 is maintained or not, the corresponding ka values for PFO kinetics have been estimated by eq 29 (with kI,a = kE1), eq 32 (with kI,b = kE1), or eq 42 (with kI,c = kE1), whatever applies, and put in columns 4, 6, and 8, respectively. The values of kE1 and kE2 are determined from dq/dt vs (qe – q) (eq 3) and dq/dt vs (qe – q)2 (eq 5) plots, respectively, which were used in estimating the ka values. The systems under discussion were all of PFO type (although in source articles some of them were recognized as PSO type). Thus, whether the condition NOD,0 < 0.02 is maintained or not, the ka value has been estimated with eq 43 (with kII = kE2) and put in column 9. The ka values estimated by maintaining the conditions imposed on the dimensionless numbers are underlined.
Table 3. ka-Values Estimated from kE1 and kE2 in EA, with or without Fulfillment of the Requirements to Dimensionless Numbers as Indicators for PFO or PSO Kineticsa.
| PFO
kinetics: N1 < 0.2 |
PFO
kinetics: N2 < 0.2 |
PFO
kinetics: NOD,0 > 8 PSO kinetics: NOD,0 < 0.02 |
||||||
|---|---|---|---|---|---|---|---|---|
| system no. | ka × 109 from IRA | N1 | ka × 109 | N2 | ka × 109 | NOD,0 | *ka,1 × 109 | **ka,2 × 109 |
| 1a | 182 | 0.96 | 8460 | 0.01 | 203 | 146.5 | 205 | 37 450 |
| 1b | 181 | 0.98 | 6630 | 0.01 | 199 | 76.9 | 202 | 22 200 |
| 1c | 185 | 0.94 | 5407 | 0.01 | 195 | 54.8 | 198 | 14 900 |
| 2a | 18.4 | 0.93 | 58.3 | 0.25 | 18.0 | 3.4 | 22.8 | 43.2 |
| 2b | 16.6 | 0.95 | 31.9 | 0.38 | 13.8 | 1.9 | 20.0 | 68.8 |
| 2c | 15.4 | 0.87 | 20.5 | 0.58 | 14.0 | 0.98 | 26.2 | 43.2 |
| 3a | 5.97 | 0.97 | 42.35 | 0.11 | 5.95 | 8.25 | 6.65 | 71.8 |
| 3b | 5.93 | 0.97 | 34.7 | 0.14 | 5.85 | 6.4 | 6.73 | 56.7 |
| 3c | 6.21 | 0.97 | 31.3 | 0.17 | 6.15 | 5.2 | 7.29 | 48.9 |
| 4a | 334 | 0.37 | 375 | 0.55 | 481 | 3.6 | 403 | 2270 |
| 4b | 390 | 0.32 | 441 | 0.51 | 569 | 5.2 | 442 | 4690 |
| 4c | 440 | 0.23 | 515 | 0.41 | 649 | 8.9 | 493 | 9210 |
*ka,1 and **ka,2 values were calculated from kE1 and kE2, respectively.
As we see in Table 3, no system satisfies the condition N1 < 0.2, and consequently, ka could not be predicted for any system from kE1 under this criterion. The subsystems 1a–c and 3a–c satisfy the condition N2 < 0.2 and hence follows condition 2 for PFO kinetics in IRA. Consequently, the ka values estimated for the systems (column 5) from kE1 values agree satisfactorily with the corresponding ones in column 2 determined by IRA.
Again, subsystems 1a–c, 3a, and 4c satisfy the condition NOD,0 > 8 and hence follow condition 3 for PFO kinetics in IRA. Consequently, ka values estimated for the systems (column 8) from kE1 values agree satisfactorily with the corresponding ones in column 2 determined by IRA. No subsystem satisfies the condition for PSO kinetics in IRA, and the ka value estimated for the systems (column 9) from kE2 values is abnormally high and not acceptable.
It is interesting to observe that although most of the subsystems do not satisfy condition NOD,0 > 8, still the ka values estimated for the systems (column 8) from kE1 values are well within the acceptable range of the corresponding ka values in column 2 determined by IRA. It should be remembered that NOD depends on θ, and if NOD,0 = 3.5 (for example), NOD varies from 3.5 to 35 for a change in θ from 0 to 0.9 with NOD = 7 for θ = 0.5. Thus, if EA is applied for kinetic data in the θ-range of 0.5–0.9, the requirements to NOD for PFO kinetics will be fulfilled. However, it will be a very exhaustive job to sort out the kinetic data from a suitable θ-range, while a simple but chemical kinetics-justified method has been worked out in IRA for determining ka.
From the exhaustive analysis and discussion, it has become quite clear that the EA applied by researchers will give ka values if the pseudo-rate constants are correlated with ka within a defined range of the dimensionless number, NOD. The question arises, does it deserve to treat the adsorption kinetic data with EA at all, while through proper scrutiny, it might be found that most of the previously accepted PSO kinetics are wrongly recognized. With the inclusion of the interface reaction concept, the adsorption rate constant has become concentration- as well as operating-parameter-independent, and thus, although tested only for four adsorption systems, the IRA appears to be very perspective for the characterization of adsorbents operating in liquid media. In developing the present model, no diffusion layer is considered and the system is considered surface-reaction-controlled. It is curious to verify to what extent the model could describe systems with micro- and mesoporous adsorbents and how the IRA could supplement the intraparticle diffusion model. Now, it is high time to standardize the characterization method of the adsorbent–adsorbate system in an aqueous medium. The characterization should be such that one system could be comparable to a similar one in terms of equilibrium and kinetic parameters. The empirical approach as it has been implemented in practice could not serve the comparison purpose. Both IRA and HOA (with correction of material balance, eq 10) could serve the purpose for surface-reaction-controlled processes, but the IRA is a comparatively straightforward method for determining ka, while HOA is an indirect method passing through the determination of the technological rate constants. Further research on adsorbent materials with different porosities would definitely lead to the standardization of characterization methods for adsorbent–adsorbate systems.
4. Conclusions
-
(1)
Both the interface reaction kinetics and the hybrid-order approaches considering mass balance in the solid–liquid system give reliable operating-parameter-independent adsorption rate constants. The first approach provides a straightforward way to obtain that, while the second, although justified from the viewpoint of chemical kinetics, follows a somewhat tortuous path to achieve the goal.
-
(2)
The empirical approach, which was widely practiced in the last few decades, in the form of a pseudo-first-order or pseudo-second-order kinetic model, does not provide significant information and could not be correlated to the adsorption rate constant reliably.
-
(3)
Three dimensionless numbers have been proposed to identify whether the adsorption kinetics could be described by the pseudo-first-order and the pseudo-second-order models. Applying one of these numbers, namely, the “order-determining dimensionless number,” it is established that none of the four systems under investigation followed pseudo-second-order kinetics, while in the source literature, three of them were recognized as such.
-
(4)
A standardized method of characterizing the kinetic behavior of adsorption systems is the demand of the time when effective adsorbents are expected to be in operation for wastewater treatment. For this purpose, systematic intensive studies are recommended on various adsorbent–adsorbate systems and to find conditions to correlate IRA and the intraparticle diffusion model.
Acknowledgments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01449.
Conversion of standard equations for saturation type curves into PFO and PSO kinetic equations; derivation of the kinetic equation (eq 22) in HOA; derivation of the expression for the dimensionless number, NOD,0 (eq 40); validation of IRA and HOA for systems 2c, 3a, and 4a; validation of PFO and PSO kinetics in EA for systems 2c, 3a, and 4a; and table with data for pseudo-rate constants obtained from linear fit to eqs 3–6 (PDF)
Author Contributions
This manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Uddin M. T.; Islam M. A.; Mahmud S.; Rukanuzzaman M. Adsorptive Removal of Methylene Blue by Tea Waste. J. Hazard. Mater. 2009, 164, 53–60. 10.1016/j.jhazmat.2008.07.131. [DOI] [PubMed] [Google Scholar]
- Eroğlu H.; Yapici S.; Nuhoğlu Ç.; Varoğlu E. An Environmentally Friendly Process; Adsorption of Radionuclide Tl-201 on Fibrous Waste Tea. J. Hazard. Mater. 2009, 163, 607–617. 10.1016/j.jhazmat.2008.07.076. [DOI] [PubMed] [Google Scholar]
- Han R.; Zou W.; Yu W.; Cheng S.; Wang Y.; Shi J. Biosorption of Methylene Blue from Aqueous Solution by Fallen Phoenix Tree’s Leaves. J. Hazard. Mater. 2007, 141, 156–162. 10.1016/j.jhazmat.2006.06.107. [DOI] [PubMed] [Google Scholar]
- Uddin M. T.; Rukanuzzaman M.; Khan M. R.; Islam M. A. Adsorption of Methylene Blue from Aqueous Solution by Jackfruit (Artocarpus Heteropyllus) Leaf Powder: A Fixed-Bed Column Study. J. Environ. Manage. 2009, 90, 3443–3450. 10.1016/j.jenvman.2009.05.030. [DOI] [PubMed] [Google Scholar]
- Ajmal M.; Rao R. A. K.; Khan M. A. Adsorption of Copper from Aqueous Solution on Brassica Cumpestris (Mustard Oil Cake). J. Hazard. Mater. 2005, 122, 177–183. 10.1016/j.jhazmat.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Ahmad A. A.; Hameed B. H. Reduction of COD and Color of Dyeing Effluent from a Cotton Textile Mill by Adsorption onto Bamboo-Based Activated Carbon. J. Hazard. Mater. 2009, 172, 1538–1543. 10.1016/j.jhazmat.2009.08.025. [DOI] [PubMed] [Google Scholar]
- Russo M. E.; Di Natale F.; Prigione V.; Tigini V.; Marzocchella A.; Varese G. C. Adsorption of Acid Dyes on Fungal Biomass: Equilibrium and Kinetics Characterization. Chem. Eng. J. 2010, 162, 537–545. 10.1016/j.cej.2010.05.058. [DOI] [Google Scholar]
- Antony G. S.; Manna A.; Baskaran S.; Puhazhendi P.; Ramchary A.; Niraikulam A.; Ramudu K. N. Non-Enzymatic Reduction of Cr (VI) and It’s Effective Biosorption Using Heat-Inactivated Biomass: A Fermentation Waste Material. J. Hazard. Mater. 2020, 392, 122257 10.1016/j.jhazmat.2020.122257. [DOI] [PubMed] [Google Scholar]
- Hassan M. M.; Carr C. M. Biomass-Derived Porous Carbonaceous Materials and Their Composites as Adsorbents for Cationic and Anionic Dyes: A Review. Chemosphere 2021, 265, 129087 10.1016/j.chemosphere.2020.129087. [DOI] [PubMed] [Google Scholar]
- Gurav R.; Bhatia S. K.; Choi T. R.; Choi Y. K.; Kim H. J.; Song H. S.; Lee S. M.; Lee Park S.; Lee H. S.; Koh J.; Jeon J. M.; Yoon J. J.; Yang Y. H. Application of Macroalgal Biomass Derived Biochar and Bioelectrochemical System with Shewanella for the Adsorptive Removal and Biodegradation of Toxic Azo Dye. Chemosphere 2021, 264, 128539 10.1016/j.chemosphere.2020.128539. [DOI] [PubMed] [Google Scholar]
- Gupt C. B.; Bordoloi S.; Sekharan S.; Sarmah A. K. Adsorption Characteristics of Barmer Bentonite for Hazardous Waste Containment Application. J. Hazard. Mater. 2020, 396, 122594 10.1016/j.jhazmat.2020.122594. [DOI] [PubMed] [Google Scholar]
- Li Y.; Gao B.; Wu T.; Wang B.; Li X. Adsorption Properties of Aluminum Magnesium Mixed Hydroxide for the Model Anionic Dye Reactive Brilliant Red K-2BP. J. Hazard. Mater. 2009, 164, 1098–1104. 10.1016/j.jhazmat.2008.09.009. [DOI] [PubMed] [Google Scholar]
- Islam M. A.; Khan M. R.; Mozumder S. I. Adsorption Equilibrium and Adsorption Kinetics: A Unified Approach. Chem. Eng. Technol. 2004, 27, 1095–1098. 10.1002/ceat.200402084. [DOI] [Google Scholar]
- Azizian S. Kinetic Models of Sorption: A Theoretical Analysis. J. Colloid Interface Sci. 2004, 276, 47–52. 10.1016/j.jcis.2004.03.048. [DOI] [PubMed] [Google Scholar]
- Azizian S.; Yahyaei B. Adsorption of 18-Crown-6 from Aqueous Solution on Granular Activated Carbon: A Kinetic Modeling Study. J. Colloid Interface Sci. 2006, 299, 112–115. 10.1016/j.jcis.2006.01.058. [DOI] [PubMed] [Google Scholar]
- Azizian S. A Novel and Simple Method for Finding the Heterogeneity of Adsorbents on the Basis of Adsorption Kinetic Data. J. Colloid Interface Sci. 2006, 302, 76–81. 10.1016/j.jcis.2006.06.034. [DOI] [PubMed] [Google Scholar]
- Azizian S.; Haerifar M.; Basiri-Parsa J. Extended Geometric Method: A Simple Approach to Derive Adsorption Rate Constants of Langmuir-Freundlich Kinetics. Chemosphere 2007, 68, 2040–2046. 10.1016/j.chemosphere.2007.02.042. [DOI] [PubMed] [Google Scholar]
- Kuan W. H.; Lo S. L.; Chang C. M.; Wang M. K. A Geometric Approach to Determine Adsorption and Desorption Kinetic Constants. Chemosphere 2000, 41, 1741–1747. 10.1016/S0045-6535(00)00054-0. [DOI] [PubMed] [Google Scholar]
- Azizian S.; Haerifar M.; Bashiri H. Adsorption of Methyl Violet onto Granular Activated Carbon: Equilibrium, Kinetics and Modeling. Chem. Eng. J. 2009, 146, 36–41. 10.1016/j.cej.2008.05.024. [DOI] [Google Scholar]
- Rudzinski W.; Plazinski W. Kinetics of Solute Adsorption at Solid/Solution Interfaces: A Theoretical Development of the Empirical Pseudo-First and Pseudo-Second Order Kinetic Rate Equations, Based on Applying the Statistical Rate Theory of Interfacial Transport. J. Phys. Chem. B 2006, 110, 16514–16525. 10.1021/jp061779n. [DOI] [PubMed] [Google Scholar]
- Plazinski W.; Rudzinski W.; Plazinska A. Theoretical Models of Sorption Kinetics Including a Surface Reaction Mechanism: A Review. Adv. Colloid Interface Sci. 2009, 152, 2–13. 10.1016/j.cis.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Rudzinski W.; Plazinski W. On the Applicability of the Pseudo-Second Order Equation to Represent the Kinetics of Adsorption at Solid/Solution Interfaces: A Theoretical Analysis Based on the Statistical Rate Theory. Adsorption 2009, 15, 181–192. 10.1007/s10450-009-9167-8. [DOI] [Google Scholar]
- Plazinski W.; Rudzinski W. A Novel Two-Resistance Model for Description of the Adsorption Kinetics onto Porous Particles. Langmuir 2010, 26, 802–808. 10.1021/la902211c. [DOI] [PubMed] [Google Scholar]
- Plazinski W.; Dziuba J.; Rudzinski W. Modeling of Sorption Kinetics: The Pseudo-Second Order Equation and the Sorbate Intraparticle Diffusivity. Adsorption 2013, 19, 1055–1064. 10.1007/s10450-013-9529-0. [DOI] [Google Scholar]
- Liu Y.; Shen L. From Langmuir Kinetics to First- and Second-Order Rate Equations for Adsorption. Langmuir 2008, 24, 11625–11630. 10.1021/la801839b. [DOI] [PubMed] [Google Scholar]
- Marczewski A. W. Analysis of Kinetic Langmuir Model. Part I: Integrated Kinetic Langmuir Equation (IKL): A New Complete Analytical Solution of the Langmuir Rate Equation. Langmuir 2010, 26, 15229–15238. 10.1021/la1010049. [DOI] [PubMed] [Google Scholar]
- Marczewski A. W. Application of Mixed Order Rate Equations to Adsorption of Methylene Blue on Mesoporous Carbons. Appl. Surf. Sci. 2010, 256, 5145–5152. 10.1016/j.apsusc.2009.12.078. [DOI] [Google Scholar]
- Salvestrini S. Analysis of the Langmuir Rate Equation in Its Differential and Integrated Form for Adsorption Processes and a Comparison with the Pseudo First and Pseudo Second Order Models. React. Kinet. Mech. Catal. 2018, 123, 455–472. 10.1007/s11144-017-1295-7. [DOI] [Google Scholar]
- Guo X.; Wang J. A General Kinetic Model for Adsorption: Theoretical Analysis and Modeling. J. Mol. Liq. 2019, 288, 111100 10.1016/j.molliq.2019.111100. [DOI] [Google Scholar]
- Canzano S.; Iovino P.; Leone V.; Salvestrini S.; Capasso S. Use and Misuse of Sorption Kinetic Data: A Common Mistake That Should Be Avoided. Adsorpt. Sci. Technol. 2012, 30, 217–225. 10.1260/0263-6174.30.3.217. [DOI] [Google Scholar]
- Simonin J. P. On the Comparison of Pseudo-First Order and Pseudo-Second Order Rate Laws in the Modeling of Adsorption Kinetics. Chem. Eng. J. 2016, 300, 254–263. 10.1016/j.cej.2016.04.079. [DOI] [Google Scholar]
- Lagergren S. About the Theory of So-Called Adsorption of Soluble Substances, Kungliga Svenska Vetenskapsakademiens. Handingarl 1898, 24, 1–39. [Google Scholar]
- Ho Y. S.; McKay G. Pseudo-Second Order Model for Sorption Processes. Process Biochem. 1999, 34, 451–465. 10.1016/S0032-9592(98)00112-5. [DOI] [Google Scholar]
- Namasivayam C.; Kavitha D. Removal of Congo Red from Water by Adsorption onto Activated Carbon Prepared from Coir Pith, an Agricultural Solid Waste. Dyes Pigm. 2002, 54, 47–58. 10.1016/S0143-7208(02)00025-6. [DOI] [Google Scholar]
- Piccin J. S.; Gomes C. S.; Feris L. A.; Gutterres M. Kinetics and Isotherms of Leather Dye Adsorption by Tannery Solid Waste. Chem. Eng. J. 2012, 183, 30–38. 10.1016/j.cej.2011.12.013. [DOI] [Google Scholar]
- Ghasemi M.; Ghasemi N.; Zahedi G.; Alwi S. R. W.; Goodarzi M.; Javadian H. Kinetic and Equilibrium Study of Ni(II) Sorption from Aqueous Solutions onto Peganum Harmala-L. Int. J. Environ. Sci. Technol. 2014, 11, 1835–1844. 10.1007/s13762-014-0617-9. [DOI] [Google Scholar]
- Islam M. A.; Buschatz H.; Paul D. Non-Equilibrium Surface Reactions - A Factor in Determining Steady State Diffusion Flux. J. Membr. Sci. 2002, 204, 379–384. 10.1016/S0376-7388(02)00064-9. [DOI] [Google Scholar]
- Tien C.; Ramarao B. V. Further Examination of the Relationship between the Langmuir Kinetics and the Lagergren and the Second-Order Rate Models of Batch Adsorption. Sep. Purif. Technol. 2014, 136, 303–308. 10.1016/j.seppur.2014.08.013. [DOI] [Google Scholar]
- Zhang J. Physical Insights into Kinetic Models of Adsorption. Sep. Purif. Technol. 2019, 229, 115832 10.1016/j.seppur.2019.115832. [DOI] [Google Scholar]
- Ezzati R. Derivation of Pseudo-First-Order, Pseudo-Second-Order and Modified Pseudo-First-Order Rate Equations from Langmuir and Freundlich Isotherms for Adsorption. Chem. Eng. J. 2020, 392, 123705 10.1016/j.cej.2019.123705. [DOI] [Google Scholar]
- Benhouria A.; Islam M. A.; Zaghouane-Boudiaf H.; Boutahala M.; Hameed B. H. Calcium Alginate-Bentonite-Activated Carbon Composite Beads as Highly Effective Adsorbent for Methylene Blue. Chem. Eng. J. 2015, 270, 621–630. 10.1016/j.cej.2015.02.030. [DOI] [Google Scholar]
- Chandra T. C.; Mirna M. M.; Sudaryanto Y.; Ismadji S. Adsorption of Basic Dye onto Activated Carbon Prepared from Durian Shell: Studies of Adsorption Equilibrium and Kinetics. Chem. Eng. J. 2007, 127, 121–129. 10.1016/j.cej.2006.09.011. [DOI] [Google Scholar]
- Aksu Z. Equilibrium and Kinetic Modelling of Cadmium(II) Biosorption by C. Vulgaris in a Batch System: Effect of Temperature. Sep. Purif. Technol. 2001, 21, 285–294. 10.1016/S1383-5866(00)00212-4. [DOI] [Google Scholar]
- Atkins P.; De Paula J.. Elements of Physical Chemistry; Macmillan, 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.