


Abstract
Proper activation of DNA repair pathways in response to DNA replication stress is critical for maintaining genomic integrity. Due to the complex nature of the replication fork (RF), problems at the RF require multiple proteins, some of which remain unidentified, for resolution. In this study, we identified the N-methyl-D-aspartate receptor synaptonuclear signaling and neuronal migration factor (NSMF) as a key replication stress response factor that is important for ataxia telangiectasia and Rad3-related protein (ATR) activation. NSMF localizes rapidly to stalled RFs and acts as a scaffold to modulate replication protein A (RPA) complex formation with cell division cycle 5-like (CDC5L) and ATR/ATR-interacting protein (ATRIP). Depletion of NSMF compromised phosphorylation and ubiquitination of RPA2 and the ATR signaling cascade, resulting in genomic instability at RFs under DNA replication stress. Consistently, NSMF knockout mice exhibited increased genomic instability and hypersensitivity to genotoxic stress. NSMF deficiency in human and mouse cells also caused increased chromosomal instability. Collectively, these findings demonstrate that NSMF regulates the ATR pathway and the replication stress response network for genome maintenance and cell survival.
INTRODUCTION
Precise and complete duplication of the genome during each cell division cycle is critical to maintain genomic stability and cell survival. Failures in this process lead to accumulation of mutations and chromosomal instability and eventually lead to cancer and other genetic diseases (1,2). DNA replication errors, spontaneous chemical reactions, exogenous environmental agents, some anticancer therapeutics and oncogene overexpression induce DNA replication stress (DRS), which causes DNA replication forks (RFs) to slow or stall (3). In response to these genotoxic stresses, eukaryotic cells induce replication stress response pathways to cope with DNA damage for proper cell-cycle progression.
During the replication stress response, numerous changes take place at RFs. First, minichromosome maintenance (MCM) helicase uncouples from replicative DNA polymerase producing persistent long single-stranded DNA (ssDNA) in the genome. ssDNA is rapidly detected and bound by replication protein A (RPA), a heterotrimeric ssDNA-binding complex, to form RPA-coated ssDNA (RPA-ssDNA), a crucial platform of the DNA damage response (DDR) (4). In addition to stabilizing ssDNA, the RPA-ssDNA complex serves as a protein–protein binding platform for the coordination of multiple DNA repair events, including recruitment of ATR-interacting protein (ATRIP) and the pre-mRNA processing factor 19 (PRP19)-cell division cycle 5-like (CDC5L) complex, which then recruits ataxia telangiectasia and Rad3-related protein (ATR) to stressed RFs. The RPA-ssDNA complex also acts as a platform for recruitment of the Rad17-replication factor C (RFC) complex to DNA damage sites, after which RFC loads the 9-1-1 checkpoint complex and DNA topoisomerase 2-binding protein 1 (TopBP1). In the RPA-ssDNA complex, ATR is activated by direct interaction with TopBP1 (5,6) or Ewing tumor-associated antigen 1 (ETAA1), which is independent of TopBP1 (7). Activated ATR phosphorylates and activates checkpoint kinase 1 (Chk1), which stabilizes and restarts stalled RFs by regulating various downstream effectors (8).
N-methyl-D-aspartate (NMDA) receptor synaptonuclear signaling and neuronal migration factor (NSMF), also called Jacob, was originally found to be involved in neuronal plasticity and development. Mutations in the NSMF gene were found in patients with Kallmann syndrome (KS), which is characterized by idiopathic hypogonadotropic hypogonadism (IHH) associated with anosmia or hyposmia (9–12). It has been shown that knockdown of NSMF results in reduced migration of gonadotropin-releasing hormone (GnRH)-positive neurons from the olfactory bulb to the hypothalamus during early neuronal development (13). NSMF contains a nuclear localization signal domain and is shuttled to the nucleus via extracellular signal-regulated kinase (ERK)-mediated phosphorylation. Transmission of synaptic NMDA receptor signaling to the nucleus by NSMF is critical for hippocampal dendrite development and synapse formation (14). In addition, it has been suggested that NSMF nuclear localization is involved in neuronal dysfunction resulting from amyloid-β signaling (15,16). Although NSMF is highly expressed in the brain and although most NSMF functional studies have focused on neuronal development, NSMF is also present in multiple adult tissues, including the kidney, liver, lung, brain and heart, but little is known about the function of NSMF in these tissues (9,17).
In this study, we uncovered a novel function of NSMF as a regulator of the DNA replication stress response. We found that NSMF expressed exclusively in the nucleus interacts with CDC5L in non-neuronal cells. NSMF rapidly localizes to stalled RFs and acts as a scaffold to stabilize the RPA-CDC5L-ATR/ATRIP complex for full activation of the ATR signaling pathway. We also demonstrated that NSMF knockout (KO) mice show increased chromosomal instability and greater sensitivity to genotoxic treatment. Taken together, we propose that NSMF directly promotes RF recovery to ensure genome stability.
MATERIALS AND METHODS
Generation of NSMF KO mice
NSMF KO mice were generated from embryonic stem (ES) cells harboring the Nsmf mutation Nsmftm1a(KOMP)Wtsi that were purchased from the Knock-Out Mouse Project (KOMP; University of California, Davis, CA). The NSMF KO was confirmed by genotyping, qRT-PCR and western blot. Primers for genotyping were Nsmf-A: 5′-GGC CCT GAG GTT ATT GAT GC-3′, Nsmf-B: 5′-GCT TGG CTT GAG GTG GTC TC-3′ and Nsmf-C: 5′-TCG TGG TAT CGT TAT GCG CC-3′. Primers for qRT-PCR were Nsmf-F: 5′-ACT CGG CTT TTT CTA GCC TTG-3′ and Nsmf-R: 5′-ACA ATG CTG GTG AGG TTC TTG-3′. Mice used in this study were bred and maintained at the In Vivo Research Center at Ulsan National Institute of Science and Technology (UNIST), and all procedures were approved by the Institutional Animal Care and Use Committee (UNISTIACUC-18-08).
Cell culture and stable cell lines
HeLa and human embryonic kidney (HEK) 293T cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). The cell lines were maintained in Dulbecco modified Eagle’s medium (DMEM; Invitrogen, CA) supplemented with 10% fetal bovine serum (FBS; Gibco, Franklin Lakes, NJ) and 1% penicillin/streptomycin (Gibco) at 37°C under 5% v/v CO2. To generate the NSMF KO cell line, Hela cells were cotransfected with CRISPR/Cas9, a single guide RNA targeting NSMF, and donor plasmids by nucleofection (Lonza, Basel, Switzerland). After 48 h, high GFP-expressing cells were sorted into 96-well plates with a FACSAria Fusion (BD Biosciences, San Jose, CA). The presence of insertions/deletions that cause frameshifts in both alleles was confirmed by sequencing. Hela cells with stable NSMF expression were obtained upon antibiotic selection with 3 μg/ml puromycin (cat# ant-pr; InvivoGen, San Diego, CA) for 2 weeks. Clones were pooled into a single population to avoid clonal heterogeneity. All cell lines were routinely tested for mycoplasma contamination (MP0025; Sigma-Aldrich, St. Louis, MO).
Plasmids
The GFP-NSMF and SFB-NSMF, NSMF D-1, D-2, D-3, D-4 and D-5 deletion mutant expression plasmids were created using GFP-, mCherry- or SFB-tagged mammalian expression vectors. CDC5L and ATRIP genes were purchased from the Korea Human Gene Bank. Myc-CDC5L, CDC5L D-1, D1-1, D1-2, D-2, D-3 and D-4 deletion mutant expression plasmids were created using a Myc-tagged mammalian expression vector. GFP-CDC5L and GFP–ATRIP expression plasmids were created using a GFP-tagged mammalian expression vector. The Myc-RPA2 expression plasmid was created using a Myc-tagged mammalian expression vector and GST-RPA2. Guide RNA plasmids for human NSMF were generated by cloning guide sequences into pX330 (plasmid number 42230; Addgene). The target sequences for gene editing were selected using CRISPOR (www.crispor.tefor.net). The target sequences for each guide RNA were as follows: Human NSMF guide #1: 5′- CGTAGCAGCCGTTGGAGACG -3′ and Human NSMF guide #2: 5′- CAGCTTCTTGCGGCGACCGG -3′. All constructs and primers are summarized in Supplementary Table S1.
Transfection and small interfering RNAs (siRNAs)
Transient transfections with plasmid DNA and siRNAs were performed using XtremeGENE™ HP (Roche, Basel, Switzerland) and Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA), respectively, according to the manufacturer’s instructions. Control siRNAs were described previously (18). The following custom siRNA sequences NSMF #1: 5′ UGAGCAAAGUGAACCCAGAtt 3′, NSMF #2: 5′ UCUGGGUUCACUUUGCUCAtc 3′, NSMF #3: 5′ CAGAUGAUCGAGACGUACUtt 3′, PRP19 #1: 5′ GCCAAGUUCCCAACCAAGUtt 3′, PRP19 #2: 5′ CAGAAGAGCUCAGCAAAUAtt 3′, PRP19 #3: 5′ CCUUCUCUGAGAAUGGUUAtt 3′, PRP19 #4: 5′ CAAGUUCAUCGCUUCAACAtt 3′, PRP19 #5: 5′ GACAGAAGCCUCAAGUUCUtt 3′, RFWD3 #1: 5′ GCAGUCAUGUGCAGGAGUUtt 3′, RFWD3 #2: 5′ GGAAACAGGCCGAGUUAGAtt 3′, RFWD3 #3: 5′ GGACCUACUUGCAAACUAUtt 3′ and CDC5L : 5′ GAUGGAAUGGCAGACUAUAtt 3′ were synthesized by Bioneer Inc. (Korea).
Preparation of MEFs
NSMF WT and KO MEFs were isolated from a pregnant mouse on day E14.5 as described previously (19). Briefly, on day E14.5, head and internal organs were removed from the embryos. The remainder of the embryo bodies were minced with razor blades into small fragments and digested using Trypsin-EDTA (cat# 25200-072; Gibco) to obtain cells for culture. MEFs were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin.
Antibodies and dilution factors
Anti-γH2AX and anti-H2B antibodies were described previously (20,21) The dilutions of the various antibodies used for western blot analysis were as follows: 1:1000, anti-NSMF (OAAN03468; Aviva Systems Biology, San Diego, CA); 1:1000, anti-CDC5L (612362; BD biosciences); 1:1000, anti-FLAG (F3165; Sigma-Aldrich); 1:2000, anti-Myc (11814150001; Roche); 1:5000, anti-GFP (632380; Clontech, Mountain View, CA); 1:2000, anti-β-actin (A5316; Sigma-Aldrich); 1:2000, anti-α-tubulin (05-829; Millipore, Burlington, MA); 1:1000, anti-RAD51 (ab63801; Abcam, Cambridge, United Kingdom); 1:1000, anti-RPA2 (A300-244A; Bethyl Laboratories, Inc., Montgomery, TX); 1:1000, anti-RPA2 (S4/8) (A300-244A; Bethyl Laboratories, Inc.); 1:1000, anti-BRCA1 (SC-6954; Santa Cruz, Dallas, TX); 1:1000, anti-53BP1 (#4937; Cell Signaling Technology, Danvers, MA); 1:1000, anti-ATRIP (A300-670A; Bethyl Laboratories, Inc.); 1:1000, anti-ATR (A300-137A; Bethyl Laboratories, Inc.); 1:1000, anti-PRP19 (ab27692; Abcam, Cambridge, United Kingdom); 1:250, anti-RFWD3 (A301-397A; Bethyl laboratories, Inc.); 1:1000, anti-CHK1(SC-8408; Santa Cruz, Dallas, TX); 1:1000, anti-phosphoCHK1 (S317, #2344; Cell Signaling Technology, Danvers, MA); Horseradish peroxidase-conjugated secondary antibodies specific to rabbit (A0545; Sigma-Aldrich) or mouse (A9917; Sigma-Aldrich) IgG were used at a dilution of 1:2000. The dilutions of the various antibodies used for indirect immunofluorescence analysis were as follows: 1:1000, anti-RPA2 (MA1-26418; Thermo Fisher Scientific); 1:100, anti-RPA2 (S4/8, A300-244A; Bethyl Laboratories, Inc.); 1:100, anti-RPA2 (S33, A300-246A; Bethyl Laboratories, Inc.).
Immunoprecipitation
For immunoprecipitation, cells were washed with ice-cold phosphate buffered saline (PBS) and then lysed in NETN buffer [0.5% Nonidet P-40, 20 mM Tris pH 8.0, 50 mM NaCl, 50 mM NaF, 100 μM Na3VO4, 1 mM dithiothreitol (DTT), and 50 μg/ml phenylmethylsulfonyl fluoride (PMSF)] at 4°C for 10 min. Crude lysates were cleared by centrifugation at 14 000 rpm at 4°C for 5 min, and supernatants were incubated with protein A-agarose-conjugated primary antibodies or FLAG-M2 affinity gel (A2220; Sigma-Aldrich). The immunocomplexes were washed three times with NETN buffer and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Western blotting was performed using the antibodies indicated in the figures. Proteins were visualized using secondary horseradish peroxidase-conjugated antibodies (Enzo Life Sciences, New York, NY) and enhanced chemiluminescence reagent (Thermo Fisher Scientific). Signals were detected using an automated imaging system (ChemiDoc™; Bio-Rad Laboratories, Hercules, CA).
Chromatin fraction analysis
Cells were collected and washed in PBS. The collected cells were lysed in NETN buffer at 4°C for 30 min. Crude lysates were cleared by centrifugation at 13 000 rpm at 4°C for 10 min and the pellet was resuspended in 0.2 N HCl for 1 h. The resuspended mixture was centrifuged at 13 000 rpm at 4°C for 10 min, and the supernatant chromatin fraction was neutralized with 1 M Tris-HCl (pH 8.0) for western blotting. We used histone 2B (H2B) as a control for the chromatin fraction.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) based NSMF interactome analysis
To identify NSMF-associated proteins, HEK293T cells were transfected with S-FLAG-streptavidin binding peptide (SFB)-tagged NSMF plasmid. SFB-NSMF and associated proteins were immunoprecipitated from cell lysates using FLAG-M2 affinity gel (Sigma-Aldrich), and the immunoprecipitated proteins were separated by SDS-PAGE. After staining with colloidal Coomassie blue, bands were sliced from the protein gel and in-gel tryptic digestion was performed as described by Shevchenko et al. (22). The tryptic digests were separated by online reversed-phase chromatography using a Thermo Scientific Eazy nano LC 1200 UHPLC equipped with an autosampler using a reversed-phase peptide trap Acclaim PepMapTM 100 (75 μm inner diameter, 2 cm length) and a reversed-phase analytical column PepMapTM RSLC C18 (75 μm inner diameter, 15 cm length, 3 μm particle size), both from Thermo Scientific, followed by electrospray ionization at a flow rate of 300 nl min-1. Samples were eluted using a split gradient of 3–50% solution B (80% ACN with 0.1% FA) in 60 min and 50–80% solution B in 10 min followed column wash at 100% solution B for 10 min. The chromatography system was coupled in line with an Orbitrap Fusion Lumos mass spectrometer. The mass spectrometer was operated in a data-dependent mode with the 120 000 resolution MS1 scan (375–1500 m/z), AGC target of 5e5 and max injection time of 50 ms. Peptides above threshold 5e3 and charges 2–7 were selected for fragmentation with dynamic exclusion after 1 time for 15 s and 10 ppm tolerance. Spectra were searched against the uniprot-human DB using the Proteome Dsciverer Sorcerer 2.1 with SEQUEST-based search algorithm, and comparative analysis of proteins identified in this study was performed using Scaffold 4 Q+S. All MS/MS samples were analyzed using the Sequest Sorcerer platform (Sagen-N Research, San Jose, CA). Sequest was set up to search the Homo sapiens protein sequence database (20612 entries; UniProt [http://www.uniprot.org/]), which includes frequently observed contaminants from trypsin digestion. Sequest was searched with a fragment ion mass tolerance of 1.00 Da and a parent ion tolerance of 10.0 ppm. Carbamidomethylation of cysteine was specified as a fixed modification in Sequest. Oxidation of methionine, acetylation of the N-terminus and phosphorylation of serine, threonine and tyrosine were specified as variable modifications. Scaffold (Version 4.10.0; Proteome Software Inc., Portland, OR) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they were established with >97.0% probability and a false discovery rate (FDR) < 1.0% by the Scaffold Local FDR algorithm. Protein identifications were accepted if they were established with >95.0% probability, an FDR < 1.0%, and contained at least two identified peptides. Protein probabilities were assigned by the ProteinProphet algorithm (23). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins were annotated with GO terms from NCBI (downloaded 14 April 2019).
Proteins that interacted directly or indirectly with NSMF were identified and were functionally annotated using DAVID (24). A Bonferroni stepdown test was applied, and only GO terms and pathways with P values < 0.05 were considered for analysis. IPA software version 01–08 (Qiagen Bioinformatics) was used for annotations and pathway analyses. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (25) partner repository with the dataset identifier PXD (PXD021942)
Cell cycle analysis
NSMF WT and KO HeLa cells were treated with 2 mM HU or 20 nM CPT. After 24 h, cells were washed and fresh medium was added. Cells were harvested every 2 h after addition of fresh medium for 18 h. For each sample, cells were detached with trypsin-EDTA and fixed in cold 70% ethanol overnight at -20°C. Fixed cells were washed, treated with RNase A and stained with propidium iodide for 4 h at 4°C. Data analysis and acquisition were evaluated using a NovoCyte Flow Cytometer (ACEA Biosciences, Inc., San Diego, CA) with NovoExpress software.
Laser microirradiation and imaging of cells
For laser microirradiation, cells were grown on 35 mm glass bottom dishes (MatTek Corporation, Ashland, MA). Laser microirradiation was carried out using a Nikon A1 laser microdissection system equipped with a 37°C chamber and CO2 module (Nikon, Tokyo, Japan). Selected regions were microirradiated for 20 iterations with a fixed wavelength ultraviolet A laser (405 nm) with 100% power to induce localized DNA breaks. For GFP or mCherry-tagged proteins, time-lapse images were acquired at 10 s time intervals after laser microirradiation. The fluorescent intensities on the laser strips were recorded with NIS elements C software (Nikon) and analyzed with ImageJ software. The fluorescence values for >10 cells from three independent experiments were normalized to the original signal and plotted as fluorescence versus time using GraphPad Prism software.
For immunostaining, cells were fixed with 3% paraformaldehyde for 10 min and permeabilized with 0.5% Triton X-100 in PBS for 5 min at room temperature. Samples were blocked with 5% goat serum and then incubated with primary antibody for 1 h. Fixed cells were incubated with the indicated antibodies diluted in PBS supplemented with 10% fetal bovine serum at 4°C overnight. After three washes with 0.05% Triton X-100 in PBS, fluorescent-conjugated secondary antibodies (Thermo Fisher Scientific) were added and incubated for 30 min. Cells were mounted using ProLong® Gold antifade reagent (Vector Laboratories, Burlingame, CA). Confocal images were acquired with an LSM880 confocal microscope (Carl Zeiss, Oberkochen, Germany). Image acquisition and analysis were performed with ZEN2.1 software.
Cell survival assay
One-hundred or two-hundred NSMF WT or KO HeLa cells in a 30 mm diameter plate were treated with the indicated doses of DNA damaging agents. After 14 d, cells were washed with PBS, fixed and stained with 2% (w/v) methylene blue. Images of colonies with defined scales were analyzed in ImageJ to determine the area of each colony.
Homologous recombination assay
The homologous recombination assay was performed as described previously with some adaptations (18). U2OS DR-GFP cells were treated with the indicated siRNAs in 24-well plates, and after 24 h, they were transfected with 0.4 μg of I-SceI per well. Seventy-two hours after I-SceI transfection, cells were trypsinized, and the percentages of GFP positive cells were determined by flow cytometry. The paired t-test was performed for statistical analysis.
DNA end resection assay
Quantification of DNA end resection assay was performed as described previously (26). Genomic DNA was extracted from AsiSI-ER-U2OS cells transfected with the indicated siRNAs. The cells were trypsinized, centrifuged and resuspended with 37°C 0.6% low-gelling point agarose in PBS at a concentration of 1.2 × 107 cells/ml. A 50 μl cell suspension was dropped on a piece of parafilm to generate a solidified agar ball, which was then transferred to a 2 ml tube. The agar ball was treated with 1 ml of ESP buffer (0.5 M EDTA, 2% N-lauroylsarcosine, 1 mg/ml proteinase-K, 1 mM CaCl2, pH 8.0) for 20 h at 16°C with rotation, followed by treatment with 1 ml of HS buffer (1.85 M NaCl, 0.15 M KCl, 5 mM MgCl2, 2 mM EDTA, 4 mM Tris, 0.5% Triton X-100, pH 7.5) for 20 h at 16°C with rotation. After washing with 1 ml of PBS (Sigma) for 6 h at 4°C with rotation, the agar ball was melted by placing the tube in a 68°C heat block for 10 min. The melted sample was diluted 15-fold with 68°C ddH2O. The DNA was then digested with restriction enzymes (HindIII for a negative control and BsrGI (for measuring resection). The level of ssDNAs generated by DNA end resection at the specific AsiSI sites (DSBs) was evaluated by qPCR using the primers and probes shown in Supplementary Table S1. The percentage of ssDNA (ssDNA%) generated by resection at the DSBs was determined as previously described (27). Briefly, for each sample, a △Ct value was calculated by subtracting the Ct value of the mock-digested sample from the Ct value of the digested sample. The ssDNA% was calculated with the following equation: ssDNA% = 1/(2(△Ct − 1) + 0.5) × 100 (28).
Analysis of metaphase chromosomes
For chromosomal analysis, cells were incubated with 1 μg/ml colcemid for 4 h and then metaphase cells were harvested by trypsinization. The cells were incubated in 75 mM KCl for 15 min at 37°C and then fixed twice with methanol:acetic acid (3:1). Cells were dropped onto glass microscope slides, aged and then stained with 5% Giemsa. Images were acquired using a fluorescence microscope (BX53; Olympus, Tokyo, Japan). At least 35 metaphase cells were chosen at random for each condition analyzed. The unpaired t-test was performed for statistical analysis.
DNA fiber combing analysis
DNA combing analysis was performed as described previously (29). To analyze HU-induced deceleration of RF progression, cells were pre-labeled with 100 μM CldU for 20 min and then washed with media three times. After washing, cells were treated with 2 mM HU for 2 h. Cells were washed again and then incubated in fresh medium containing 250 μM IdU for 30 min. To analyze CPT-induced deceleration of RF progression, cells were pre-labeled with 100 μM CldU for 30 min, washed and labeled with 250 μM IdU for 30 min with or without 1 μM CPT. Cells were harvested by trypsinization and embedded in a low-melting agarose plug at a density of 4 × 105 cells/plug. The plugs were lysed in lysis buffer (100 mM EDTA, 1% N-lauroyl-sarcosine, 10 mM Tris-Cl pH 8.0, 1 mg/ml proteinase K), melted at 68°C for 20 min in the presence of 0.5 M 2-(N-morpholino)-ethanesulfonic acid (pH 5.5) and β-agarase, and cooled to 42°C. After β-agarase digestion, DNA was combed on PBS containing 0.1% Tween® 20, Alexa Fluor®-conjugated secondary antibodies (Thermo Fisher Scientific) were added, and the mixture was incubated for 60 min at room temperature (RT). Slides were mounted using ProLong® Gold antifade reagent (Vector Laboratories). Confocal images were acquired with an Axio Observer 7 (Carl Zeiss). Image acquisition and analysis were performed with ImageJ software.
γ-Irradiation and mouse survival analysis
To determine the sensitivity to irradiation, Nsmf +/+ and Nsmf-/- mice (12 weeks old, male) were exposed to a single 7 Gy dose of whole body γ-irradiation using a Gammacell 3000 Elan, a cesium-137 gamma animal irradiator (Best Theratronics, Ottawa, ON, Canada). After irradiation, survival was monitored by daily inspection. Kaplan–Meier survival curves and the log-rank test were used to analyze survival rates.
Erythrocyte micronucleus assay
We performed a micronucleus assay as described previously (30). Briefly, Nsmf+/+ and Nsmf-/- mice (6 weeks old, male) were treated with 100 mg/kg HU or were exposed to 1 Gy of γ-irradiation using a cesium-137 gamma animal irradiator (Best Theratronics). Fifty milliliters of peripheral blood was collected from the retro-orbital sinus before irradiation and every 24 h after irradiation for 3 d. The blood was transferred into a tube containing 300 ml heparin solution on ice, fixed with pre-cooled (-80°C) methanol and stored at -80°C for at least 12 h. Samples were then stained with anti-CD71-FITC (cat# 11-0711-85; Invitrogen) and propidium iodide and analyzed using a FACSVerse flow cytometer and BD FACSuite software (BD Biosciences). Data analysis was performed using FlowJo software.
Quantification and statistical analysis
Statistical analysis was performed using Prism 8 (GraphPad Software, San Diego, CA). The Student’s t-test was used to compare two data groups with normal distributions and similar variances. Multiple group comparisons were performed using one-way ANOVA. Technical and biological triplicates of each experiment were performed. P values < 0.05 were considered statistically significant.
RESULTS
NSMF is a novel binding partner of CDC5L in the DNA damage response (DDR) network
To investigate the biological functions of NSMF in other tissues, we affinity-purified proteins that interact with NSMF in a HEK293T cell line overexpressing S-FLAG-streptavidin binding peptide (SFB)-tagged NSMF and analyzed them by mass spectrometry. After subtracting nonspecific binding proteins, we identified 355 proteins that specifically interacted with NSMF (Figure 1A). Functional-enrichment analysis of the NSMF interactome using ingenuity pathway analysis (IPA) revealed several distinct clusters of functionally related proteins and multi-protein complexes involved in 11 pathways, including the G2/M DNA damage checkpoint, nucleotide excision repair (NER), and ataxia telangiectasia mutated (ATM) signaling pathways, which are all involved in DNA damage repair (Figure 1B,C). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using Database for Annotation, Visualization and Integrated Discovery (DAVID) also revealed enrichment of a number of pathways involved in RNA and DNA metabolism, including the spliceosome, ribosome biogenesis, RNA transport, RNA polymerase and DNA replication (Supplementary Figure S1).
Figure 1.

NSMF is identified as a novel CDC5L-binding protein. (A) Heat map representing the spectral count of NSMF-associated proteins identified by a 1D proteomics screen in HEK293T cells. The normalized enrichment of the unique peptide counts with control beads is shown for proteins with >9 unique peptide counts out of a total of 335 identified proteins. CDC5L and its binding partners PRPF19, SPF27 and PLRG1, which form the PRP19/CDC5L complex, are shown in red. (B) IPA canonical pathway enrichment analysis of the 335 proteins identified as NSMF-binding proteins. The DDR pathways are presented in red. The ratio indicates the number of genes in the NSMF-binding protein list divided by the total number of genes in the same pathway. (C) Enrichment map of the IPA canonical pathway for NSMF-binding proteins showing the interaction network. The three main categories of NSMF-associated proteins by interaction analysis, G2/M DNA damage check point regulation, ATM signaling and NER signaling, are shown in red. (D) The interaction between exogenous NSMF and exogenous CDC5L. Cell extracts from HEK293T cells expressing SFB-NSMF and Myc-CDC5L were immunoprecipitated with anti-Flag beads and analyzed by immunoblotting with the indicated antibodies. (E) The interaction between exogenous NSMF and endogenous CDC5L. Cell extracts from HEK293T cells expressing SFB-NSMF were immunoprecipitated with anti-Flag beads and analyzed by immunoblotting with the indicated antibodies. (F) The interaction between endogenous NSMF and endogenous CDC5L. Cell extracts from HeLa cells were immunoprecipitated using control IgG or anti-NSMF antibodies and analyzed by immunoblotting with the indicated antibodies. (G and H) Schema of NSMF WT and NSMF deletion mutants used to identify the minimal region of NSMF required for interaction with CDC5L. (G) Schematic diagram of the NSMF deletion mutants. Boxes indicate the nuclear localization signal (NLS). (H) Cell extracts from HEK293T cells expressing either Flag-NSMF WT or a Flag-NSMF deletion mutant and Myc-CDC5L were immunoprecipitated with an anti-Myc antibody and analyzed by immunoblotting with the indicated antibodies. (I and J) Schema of Myc-CDC5L WT and Myc-CDC5L deletion mutants used to identify the minimal region of CDC5L required for interaction with NSMF. (I) Schematic diagram of the CDC5L deletion mutants. The boxes represent the Myb domain, the CDC5p/cef1 domain and the NLS. (J) HEK293T cells were transfected with Myc-CDC5L WT or a Myc-CDC5L deletion mutant and Flag-NSMF. Cell lysates were immunoprecipitated with anti-Flag antibody and analyzed by immunoblotting with the indicated antibodies.
The interactome analysis indicated that CDC5L binds strongly to NSMF. Other PRP19/CDC5L complex components that were identified as NSMF-binding proteins (Figure 1A) included PRPF19, SPF27 and pleiotropic regulator 1 (PLRG1), which functions as a ubiquitin ligase during the DDR (31,32). Although multiple additional DDR factors were identified as NSMF-binding proteins (Figure 1A), we focused on the interaction between NSMF and CDC5L to investigate the functional role of NSMF in the DDR. We used immunoprecipitation to confirm the interaction between NSMF and CDC5L in cells overexpressing both SFB-NSMF and Myc-CDC5L (Figure 1D) or SFB-NSMF alone (Figure 1E). We also confirmed that endogenous NSMF binds CDC5L (Figure 1F). To identify the regions of NSMF required for CDC5L binding, we generated a series of Flag-tagged NSMF-deletion mutants (NSMF-D1 to NSMF-D5; Figure 1G), cotransfected cells with each mutant and Myc-CDC5L, and performed co-immunoprecipitation (Figure 1H). Full-length NSMF (NSMF WT), NSMF-D1 and NSMF-D2 interacted with CDC5L, whereas NSMF-D3, NSMF-D4 and NSMF-D5 did not, indicating that amino acids 250–523 of NSMF are required for CDC5L binding (Figure 1H). We also constructed a series of CDC5L-deletion mutants (CDC5L-D1 to CDC5L-D4) and used them in co-immunoprecipitation assays to determine the regions of CDC5L required for NSMF binding (Figure 1I). The co-immunoprecipitation results suggested that the C-terminal region of CDC5L (residues 655–795) interacts with NSMF (Figure 1J). Together with the interactome results, these data demonstrate that NSMF is a novel binding partner of CDC5L and may be involved in the DDR.
The central region of NSMF is required for translocation to DNA lesions
The PRP19/CDC5L complex localizes to DNA lesions (32). NSMF binding to CDC5L suggests that NSMF might also translocate to sites of DNA damage. To test this, we evaluated NSMF localization in HeLa cells before and after inducing DNA damage by laser microirradiation. Enrichment of GFP-NSMF at DNA lesions occurred rapidly, peaked within seconds, and diminished after ∼15 min (Supplementary Figure S2A). Furthermore, GFP-NSMF colocalized with γH2AX, which is also enriched at DNA lesions (Supplementary Figure S2B) (33). To test whether NSMF and CDC5L colocalize at DNA lesions, we monitored the localization of GFP-NSMF and mCherry-CDC5L in cotransfected HeLa cells. We found that GFP-NSMF and mCherry-CDC5L rapidly translocated with similar kinetics to sites of DNA damage (Figure 2A). NSMF also accumulated on chromatin treated with the DNA-damaging agents hydroxyurea (HU) and camptothecin (CPT; Figure 2B). To investigate the recruitment of NSMF to DNA replication sites, we examined NSMF cellular localization following replication stress. NSMF localization was apparent at RPA foci after 2 h of HU treatment and reached a maximum after 10 h (Figure 2C and Supplementary Figure S2C).
Figure 2.

NSMF localizes to DNA lesions via the central residues of NSMF. (A) The kinetics of GFP-NSMF and mCherry-CDC5L recruitment to DNA lesions were examined by live cell imaging. The relative signal intensities are quantified and summarized in the bottom panel. The highest intensity on any lesion was set at 100% for each cell, and the recruitment kinetics were plotted. The average intensity for ≥10 cells for each condition is presented graphically. Data represent the mean ± SD. (B) HeLa cells were treated with 1 μM CPT for 2 h or with 2 mM HU for 16 h and then harvested as whole-cell extracts (WCEs) or fractionated into soluble or chromatin-bound protein fractions. Each fraction was analyzed by western blot with the indicated antibodies. (C) HeLa cells transfected with GFP-NSMF expression plasmids were treated with 2 mM HU for 16 h and then incubated in fresh media for 3 h. Colocalization of GFP-NSMF and RPA2 was determined by immunofluorescence with an RPA2 antibody. Magnified insets show colocalization of NSMF and RPA2 foci. (D) HeLa cells were transfected with expression plasmids for GFP-NSMF WT or GFP-NMSF deletion mutants. After 24 h, the cells were laser microirradiated, and recruitment of GFP-NSMF proteins to DNA lesions was examined by live cell imaging. Relative intensities of the lesions were quantified, and the recruitment kinetics are plotted in the right panel. The average intensity for ≥10 cells for each condition is presented graphically. Data represent the mean ±SEM of two independent experiments. (E) A schematic diagram of NSMF WT and the NSMF A3A4 mutant. Black boxes indicate the NLS. (F) HeLa cells were transfected with GFP-NSMF WT or the GFP-NSMF A3A4 mutant. The percentages of GFP-NSMF cells recruited to DNA lesions are presented graphically. More than 10 cells were analyzed for each condition. Data represent the mean ± SEM of two independent experiments. (G) Hela cells were transfected with control or CDC5L siRNA. After 24 h, the cells were transfected with GFP-NSMF and incubated for another 24 h. The cells were then laser microirradiated, and recruitment of the GFP-NSMF proteins to DNA lesions was examined by live cell imaging. The relative intensities of the lesions were quantified. The recruitment kinetics are plotted on the bottom panel. The average intensity of ≥ 20 cells for each condition is presented graphically. Data represent the mean ± SEM of two independent experiments.
To determine the specific regions of NSMF that are responsible for translocation to sites of DNA damage, we analyzed the accumulation of GFP-tagged NSMF-deletion mutants at DNA lesions over time (Figure 2D). Although the accumulation of NSMF-D1, NSMF-D2 and NSMF-D5 was reduced by ∼60% compared with that of NSMF WT, those mutants retained some ability to localize at DNA lesions. By contrast, NSMF-D4 completely lost its ability to be recruited to DNA lesions, and NSMF-D3 was recruited slowly and weakly in comparison with the other mutants. These results indicate that the central residues (250–402) of NSMF are responsible for CDC5L interaction and for NSMF recruitment to sites of DNA damage (Figure 2D).
We next asked whether CDC5L affects NSMF recruitment to sites of DNA damage. We found that CDC5L-knockdown cells showed a clear reduction in NSMF translocation to DNA lesions (Figure 2G). To determine whether NSMF-D3 and NSMF-D4 mutants were unable to localize to DNA lesions because of altered protein conformation, we tested the ability of the central region of NSMF alone (NSMF-A3A4, residues 250–402, Figure 2E) to localize to DNA lesions. We found that this region was able to localize to DNA lesions, indicating that the protein conformation was sufficiently maintained for localization to occur after deletion of the other parts of NSMF (Figure 2F).
Next, to determine whether NSMF recruitment to DNA lesions depends on other known DDR pathways, we tested NSMF recruitment in the presence inhibitors of ATM kinase, ATR kinase, DNA-dependent protein kinase (DNA-PK) and poly (ADP-ribose) polymerase (PARP). The PARP1 inhibitor olaparib strongly inhibited NSMF recruitment to DNA lesions, whereas ATM, ATR and DNA-PK inhibitors did not (Supplementary Figure S2D). Consistently, NSMF recruitment to DNA lesions was impaired in PARP1 KO HEK293T cells (Supplementary Figure S2E). PARP inhibition also reduced CDC5L translocation to DNA lesions (Supplementary Figure S2F). Collectively, these data demonstrate that NSMF–CDC5L complexes translocate to sites of DNA damage in a PARP1-dependent manner.
NSMF is involved in the replication stress response
To explore the role of NSMF during the DDR, we generated an NSMF KO cell line using CRISPR/Cas9. We confirmed the KO by direct sequencing and the resulting loss of NSMF expression by western blot (Figure 3A and Supplementary Figure S3A,B). RPA2 is phosphorylated at multiple sites, including Ser33, Ser4/Ser8 and Thr21, in response to DNA damage (34,35). We therefore examined whether NSMF affects RPA2 phosphorylation upon replication stress. In response to HU-induced replication stress, RPA2 foci were slightly reduced, and phospho-RPA2 foci were dramatically reduced, in NSMF KO cells compared with those in NSMF WT cells (Figure 3B–F). Consistently, in response to HU or CPT treatment, phosphorylation of RPA2 Ser33 and Ser4/Ser8 was reduced by ∼50% in the chromatin fractions of NSMF KO cells compared with that in WT cells (Figure 3G,H). In addition, CHK1 phosphorylation induced by HU or CPT was reduced in NSMF KO cells compared with that in WT cells (Supplementary Figure S3C,D). To confirm that the reduced RPA2 phosphorylation was due solely to the loss of NSMF, we created a complemented NSMF KO strain that expresses Flag-NSMF WT (NSMF KO#2). We found that Flag-NSMF expression restored HU-induced RPA2 phosphorylation in the chromatin fraction of the NSMF KO#2 cells (Supplementary Figure S3E,F).
Figure 3.

NSMF is involved in the replication stress response. (A) The NMSF gene was deleted from HeLa cells using CRISPR/Cas9. Expression of NSMF in WT and NSMF KO HeLa-cell extracts was analyzed by western blot. (B–F) Localization of RPA2 and phospho-RPA2 (S4/S8 or S32) was determined by immunofluorescence. NSMF WT and KO HeLa cells were treated with 2 mM HU for 16 h, fixed and stained with the indicated antibodies. RPA2 and phospho-RPA2 intensities per nucleus were quantified for each sample using ZEN Blue software (Carl Zeiss). The numbers above each sample indicate the number of nuclei analyzed. Data are presented as the mean ± SD; ***P < 0.001, ****P < 0.0001. (G and H) NMSF WT and KO HeLa cells were treated with either 2 mM HU for 16 h or 1 μM CPT for 2 h. The cells were fractionated, and phosphorylation of RPA2 in the chromatin-bound protein fraction was determined by western blot with the indicated antibodies. The levels of phospho-RPA2 (S4/S8 or S32) and total RPA were quantified using ImageJ software. Quantification of the western blots represents an average of three independent experiments. Data represent the mean ± SEM of three independent experiments; *P < 0.05, **P < 0.01, ****P < 0.0001. (I and J) Sensitivity of NSMF KO HeLa cells to DNA replication-blocking agents. NSMF WT and KO HeLa cells were plated and treated with the indicated concentrations of HU or CPT. The numbers of surviving colonies were counted 14 days after treatment. Data represent the mean ± SEM of three independent experiments; **P < 0.01, ***P < 0.001.
In addition to replication stress, phosphorylation of RPA2 and CHK1 in response to ATR activation is triggered by a broad spectrum of DNA damage, including double-strand break (DSB). To examine whether NSMF is involved in DSB repair, we performed a homologous recombination (HR) reporter assay using a direct repeat (DR)-GFP reporter system in which expression of I-SceI endonuclease generates a DSB that knocks out GFP expression, which is restored when the break is repaired by HR (36). NSMF knockdown with three different siRNAs decreased the fraction of GFP-positive DR-GFP cells, suggesting that NSMF participates in HR for DSB repair (Supplementary Figure S3G). Next, we performed a quantitative resection assay using an AsiSI–ER–U2OS reporter system to measure the effect of NSMF on ssDNA production (26). Knockdown of NSMF reduced DNA end resection at AsiSI-induced DSBs to a level similar to that achieved by depletion of CtIP, the key HR cofactor for the essential catalytic enzyme MRE11 (Supplementary Figure S3H, I). Because defects in the DDR to replication stress leads to increased genomic instability and susceptibility to genotoxic agents, we next tested whether NSMF deficiency sensitizes cells to HU and CPT, which block DNA replication. We found that NSMF KO cells were hypersensitive to CPT and HU compared with WT cells (Figure 3I,J and Supplementary Figure S3J,K). These results suggest that NSMF regulates RPA2 phosphorylation in response to replication stress and is therefore important for cell survival.
NSMF is important for RF progression under replication stress
Regulation of RPA function is critical to activate ATR signaling in order to stabilize and protect stalled RFs during replication stress. Because NSMF regulates RPA2 phosphorylation during replication stress, we investigated the effects of NSMF function in the replication stress response. In the absence of replication stress, the cell-cycle profiles of NSMF KO cells and WT cells did not differ (Figure 4A). However, when the cells were exposed to replication stress induced by HU or CPT, the progression of S phase was delayed in NSMF KO cells compared with that in WT cells (Figure 4B and Supplementary Figure S4A). To test whether restart of stalled RFs during replication stress is altered in NSMF KO cells, we performed a DNA combing assay. We found that NSMF KO cells exhibited defective RF restart in the absence of HU and exposure to HU further increased the number stalled forks in NSMF KO cells compared with that in WT cells (Figure 4C–E). In addition, adding back Flag-NSMF to NSMF KO cells restored RF restart rates to WT levels (Figure 4C–E).
Figure 4.

NSMF is important for recovery from replication stress. (A) Basal cell-cycle profiles of NSMF WT and KO cells were measured by flow cytometry. (B) Effect of NSMF on cell-cycle progression after HU treatment. NMSF WT and KO cells were treated with 2 mM HU for up to 24 h, transferred to fresh media and harvested at the indicted times. The cells were stained with propidium iodide (PI) and subjected to fluorescence-activated cell sorting (FACS) analysis for DNA content analysis. Cell-cycle profiles and relative distributions (percent of total) of G1, S and G2-M phases are shown. (C–E) WT, NSMF KO or NSMF KO cells complemented with Flag-NSMF were labeled with CldU for 20 min, incubated with or without 2 mM HU for 2 h, and labeled with IdU for 30 min. DNA fiber images were captured using Axio Observer (Carl Zeiss). (C) Representative images of replication tracts. (D and E) Percentages of stalled (D) and restarted (E) replication forks were calculated and graphed. Data represent the mean ± SEM from two independent experiments; *P < 0.05, **P < 0.01. (F) WT cells, NSMF KO cells or NSMF KO cells complemented with Flag-NSMF were labeled with CldU for 20 min, incubated with 2 mM HU for 2 h and labeled with IdU for the indicated times. DNA fiber images were captured using Axio Observer (Carl Zeiss). (G) Percentages of restarted and stopped forks were calculated and graphed. Data represent the mean ± SEM from two independent experiments. **P < 0.01, ****P < 0.0001.
Next, to evaluate whether NSMF participates in RF progression, we measured the newly synthesized track length of RFs under replication stresses. The replication track length after HU or CPT treatment was significantly shorter in NSMF KO cells than in WT cells (Supplementary Figure S4B–G). We also observed that adding back NSMF to NSMF KO cells restored the rate of RF progression to a level comparable to that of WT cells (Supplementary Figure S4B,C and F,G). To further dissect role of NSMF in RF progression, we investigated whether depletion of NSMF affects the speed of RF restart by DNA fiber analysis, measuring fork restart at 10 min intervals after release from 2 h of HU treatment (Figure 4F). NSMF KO cells displayed reduced rates of RF restart and increased numbers of stopped folks, both of which were restored upon addback of WT NSMF (Figure 4G). These results demonstrate that NSMF KO caused the restart of stalled RFs at DNA lesions to fail or be delayed, which would be expected to impede the recovery of RFs from replication stress.
NSMF regulates RPA2/ATRIP-ATR complex formation in response to replication stress
To determine the mechanism of NSMF function during replication stress, we asked whether NSMF recruits DDR factors to RPA-ssDNA. First, we evaluated recruitment of the ATRIP-ATR complex to RPA-ssDNA in response to replication stress. We found that compared with WT cells, NSMF KO cells displayed slower recruitment of GFP-ATRIP to laser-induced DNA lesions and reduced ATRIP-ATR accumulation on chromatin in response to HU treatment (Figure 5A,B). Complementation by mCherry-NSMF WT, but not by the NSMF-D4 mutant defective in localization to DNA lesions, restored the recruitment of GFP-ATRIP in NSMF KO cells (Figure 5C). Consistently, expression of NSMF WT, but not the NSMF D4 mutant, rescued HU-induced RPA phosphorylation in NSMF-depleted cells (Supplementary Figure S5A). These results suggest that NSMF plays an important role in the recruitment of ATRIP-ATR complexes to DNA lesions for ATR activation.
Figure 5.

NSMF directs recruitment of ATRIP-ATR to DNA lesions by mediating the interaction between RPA2 and ATRIP-ATR. (A) NSMF WT and KO HeLa cells were transfected with GFP-ATRIP expression plasmids. After 24 h, the cells were microirradiated, and recruitment of GFP-ATRIP to DNA lesions was examined by live cell imaging. (B) NSMF WT and KO HeLa cells were treated with or without 2 mM HU for 16 h. The cells were fractionated, and the chromatin-bound protein fraction was subjected to western blot with the indicated antibodies. (C) Cotransfection of GFP-ATRIP with the mCherry-vec, mCherry-NSMF-WT or mCherry-NSMF-D4 mutant plasmid in NSMF KO#1 and KO#2 HeLa cells. After 24 h, the transfected cells were microirradiated, and recruitment of ATRIP and NSMF to DNA lesions was examined by live cell imaging. (D and E) HeLa-cell lysates were immunoprecipitated with anti-IgG, anti-ATRIP or anti-RPA2 antibodies. Endogenous interactions between NSMF and ATRIP or RPA2 were determined by western blot with the indicated antibodies. (F and G) HEK293T cells were cotransfected with a Flag-NSMF–deletion mutant and either GFP-ATRIP or Myc-RPA2. Immunoprecipitation using an anti-FLAG antibody and western blot with the indicated antibodies were performed to identify the NSMF regions that interact with ATRIP (F) or RPA2 (G). (H) HEK293T cells were transfected with GFP-ATRIP or Myc-RPA2 and either FLAG-NSMF WT or FLAG-NSMF D2. Immunoprecipitation with an anti-FLAG antibody and western blot with the indicated antibodies were performed. (I) Endogenous RPA2 in NSMF WT and KO HeLa-cell lysates was immunoprecipitated with anti-RPA2 antibody, and western blot with the indicated antibodies was performed.
ATRIP recognizes DNA lesions through interactions with RPA-ssDNA complexes (37). Therefore, we hypothesized that NSMF would interact with ATRIP and/or RPA2. Consistent with our hypothesis, we observed endogenous interactions between NSMF and ATRIP or RPA2 in HeLa-cell lysates by co-immunoprecipitation (Figure 5D,E). To identify the specific region of NSMF that binds with ATRIP or RPA2, we overexpressed WT or mutant FLAG-NSMF along with GFP-ATRIP or Myc-RPA2 and then performed immunoprecipitation. We found that the NSMF-D2 mutant lacking amino acids 75–238 of WT NSMF, which are well conserved across different species (Supplementary Figure S5B), failed to interact with ATRIP-ATR complex or with RPA2 (Figure 5F–H). To further narrow down the binding region, we generated three additional deletion mutants: NSMF D2-1, NSMF D2-2 and NSMF D2-3 (Supplementary Figure S5C). Co-immunoprecipitation results with these mutants suggested that amino acids 188–238 of NSMF are crucial for binding to ATRIP-ATR complex and RPA2 (Supplementary Figure S5D). Furthermore, we found that the interaction between RPA2 and ATRIP was dramatically reduced in NSMF KO cells (Figure 5I). Collectively, these results suggest that NSMF modulates RPA2 and ATRIP-ATR complex formation in response to replication stress.
NSMF mediates RPA2 ubiquitination under replication stress
The multiple interactions of NSMF with PRP19/CDC5L, ATR/ATRIP and RPA2 suggest that NSMF may mediate formation of RPA complexes with other proteins. We found that NSMF depletion attenuated the CDC5L/ATR interaction (Figure 6A). PRP19/CDC5L ubiquitinates RPA2 for ATR activation (32). Therefore, we checked the ubiquitination status of RPA2 in response to replication stress after silencing NSMF expression. In agreement with previous results, ubiquitination of RPA2 increased in response to HU treatment; however, NSMF deficiency led to a significant reduction in HU-induced RPA2 ubiquitination (Figure 6B,C). In addition, upon HU treatment, NSMF expression increased lysine-63-mediated RPA2 ubiquitination (Figure 6D,E). These results suggest that NSMF mediates RPA complex formation with PRP19/CDC5L and ATR to facilitate RPA2 ubiquitination in the replication stress pathway.
Figure 6.

NSMF mediates RPA2 ubiquitination. (A) Endogenous ATR in NSMF WT or KO HeLa cell lysates was immunoprecipitated with anti-ATR antibody and western blotting with the indicated antibodies was performed. (B) NSMF WT and KO HeLa cells were transfected with Myc-RAP2 and HA-ubiquitin and were treated with or without 2 mM HU for 3 h. Cells were lysed in denaturing conditions, diluted and ubiquitinated proteins were immunoprecipitated with anti-HA antibody and subjected to western blotting with the indicated antibodies. (C) NSMF WT and KO HeLa cells transfected with HA-ubiquitin were treated with or without 2 mM HU for 3 h. Cells were lysed in denaturing conditions, diluted, and ubiquitinated proteins were immunoprecipitated with anti-HA antibody. Endogenous RPA2 ubiquitination was detected by immunoblotting with the indicated antibodies. (D) HeLa cells transfected with HA-ubiquitin and Flag-NSMF were treated with or without 2 mM HU for 3 h. Cells were lysed in denaturing conditions, diluted, ubiquitinated proteins were immunoprecipitated with anti-HA antibody, and RPA2 ubiquitination was detected by immunoblotting with the indicated antibodies. (E) HeLa cells transfected with Myc-RPA2 and Flag-NSMF, plus the WT, lysine mutant K48R, or K63R form of HA-ubiquitin, were treated with or without 2 mM HU for 3 h. Cells were lysed in denaturing conditions, diluted and ubiquitinated proteins and immunoprecipitated with anti-HA antibody. RPA2 ubiquitination was detected by immunoblotting with the indicated antibodies.
Two E3 ligases for RPA2, RFWD3 and PRP19, have been reported (32,38). We investigated the impacts of RFWD3 and PRP19 on RPA2 ubiquitination by combinatorial depletion. Knockdown of PRP19 or RFWD3 alone reduced RPA ubiquitination (Supplementary Figure S6A, B), and combinational depletion of both ligases further reduced RPA ubiquitination in an additive manner (Supplementary Figure S6C). Although RFWD3 depletion inhibited RPA2 ubiquitination more than PRP19 depletion did, both E3 ligases contributed to RPA2 ubiquitination. Together, these results suggest that NSMF is important for RPA ubiquitination in response to replication stress.
NSMF is important for genome maintenance and survival after DNA damage in vivo
To investigate the role of NSMF in the DDR pathway in vivo, we generated an NSMF KO mouse (Figure 7A–D). We found that NSMF is expressed in many mouse tissues, including the brain (Supplementary Figure S7A). Next, we confirmed that NSMF KO results in defective interaction between RPA2 and ATRIP in mouse embryonic fibroblasts (MEFs; Figure 7E). Compared with WT cells, NSMF-deficient MEF cells and NSMF KO HeLa cells exhibited increased chromosomal instability that increased further upon HU treatment (Figure 7F,G; Supplementary Figure S7B,C). We also counted the number of erythrocytes with micronuclei after HU treatment because micronuclei represent abnormal mitotic cell division in hematopoietic cells due to unrepaired chromosomal breaks. We found that the percentage of micronucleated reticulocytes increased after intraperitoneal injection of 100 mg/kg HU and peaked at 48 h after HU injection (Figure 7I). The NSMF KO mice exhibited a higher number of micronuclei compared with WT mice, suggesting increased genomic instability in the absence of NSMF (Figure 7H,I). Similarly, the percentage of micronucleated reticulocytes increased in NSMF KO mice after 1 Gy of ionizing radiation (IR) treatment (Supplementary Figure S7D,E). Furthermore, after exposure to 7 Gy of IR, NSMF KO mice had significantly shorter lifespans than WT mice, indicating that NSMF is important for the repair of IR-induced DNA damage and survival (Supplementary Figure S7F).
Figure 7.
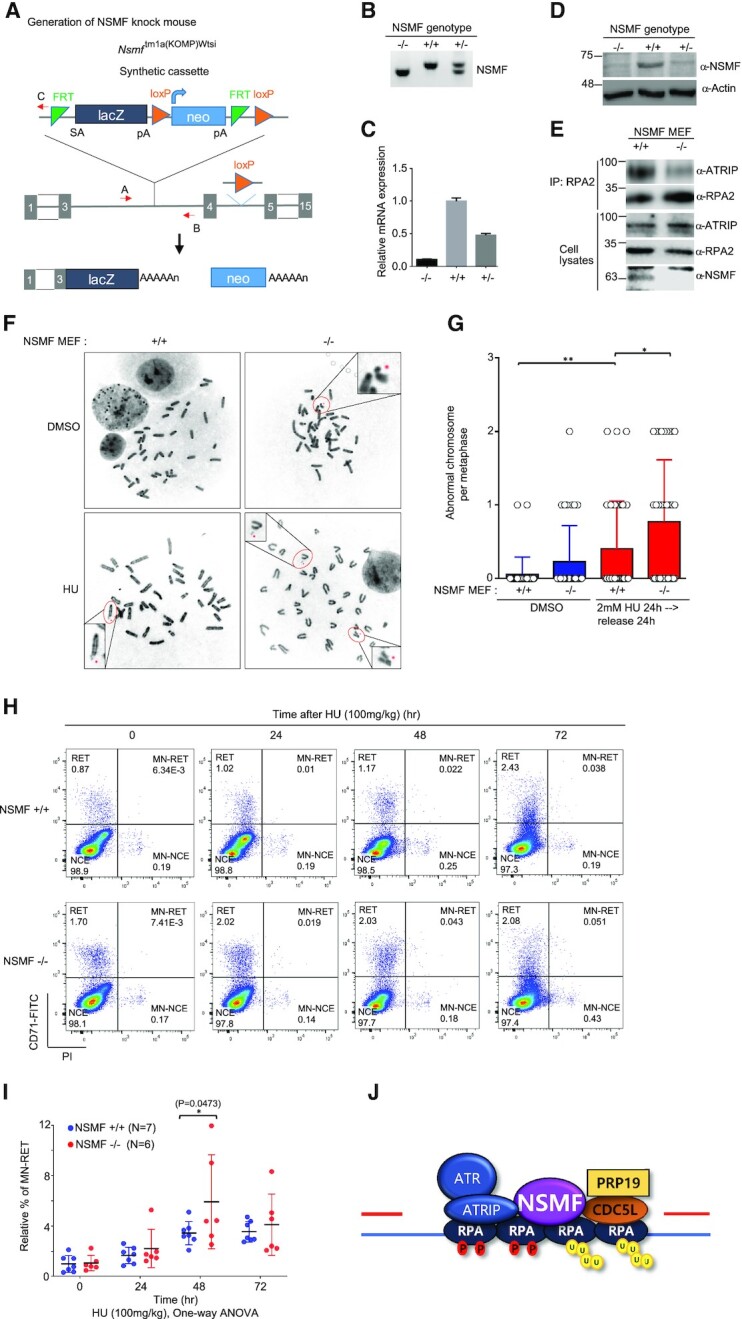
NSMF participates in genome maintenance and mouse survival in response to in vivo DNA damage. (A) Diagram describing generation of the Nsmf KO allele using a gene targeting vector. The vector consists of a Flp-recombinase target (FRT)-flanked selection cassette inserted between the third and fourth exons of Nsmf. The Nsmf transcript is trapped through the splice acceptor (SA) element and truncated through the SV40 polyadenylation signal (pA). lacZ, β-galactosidase; Neo, Neomycin resistance gene. The red arrow is the primer for genotyping. (B) Genotyping of Nsmf+/+, Nsmf+/- and Nsmf-/- mice using tail DNA. The sizes of the PCR products for the WT and mutant alleles are 343 and 292 bp, respectively. (C and D) Confirmation of Nsmf removal by qRT-PCR (C) and western blot analysis (D) using MEF mRNA and protein lysates, respectively. (E) Impaired interaction between RPA2 and ATRIP in NSMF KO MEF cells. Endogenous RPA2 in NSMF WT and KO MEF cell lysates was immunoprecipitated with anti-RPA2 antibody, and western blotting with the indicated antibodies was performed. (F and G) NSMF WT or KO cells were treated with 2 mM HU for 24 h, transferred to fresh media and subjected to metaphase spreading. (F) Representative images of spontaneous and HU-induced chromosome instability in NSMF WT and KO MEFs. The red dots indicate chromosomal abnormalities, such as chromosome breakage, fragments and radial figures. (G) The numbers of abnormal chromosomes per cell were plotted. Thirty-five metaphase cells were counted for each condition. Data are presented as mean ± SD; *P < 0.05, **P < 0.01. (H) Representative flow cytometry plots of micronucleus (MN) assays performed 0, 24, 48 or 72 h after the intraperitoneal injection of Nsmf +/+ or Nsmf-/- mice with 100 mg/kg HU; NCE, erythrocyte; RET, reticulocyte. (I) Quantification of induced MN formation in reticulocytes (MN-RET). Percentages of MN-RET were calculated as percentages of total RET. Data are presented as mean ± SD, with n > 6 mice per group. Statistical analysis: two-way analysis of variance, *P = 0.047. (J) Diagram showing that NSMF mediates recruitment of ATRIP/ATR and PRP19/CDC5L to repair stalled RFs.
DISCUSSION
In response to DNA replication stress, multiple DDR proteins are recruited to RFs to coordinate DNA replication and repair precisely, spatially and temporally. RPA-coated ssDNA recruits and activates DDR signaling through ATR/ATRIP, RAD17, RAD9 and TopBP1. Thus, proper assembly and modification of RPA-coated ssDNA is important for tight regulation of the ATR kinase-signaling pathway. In response to DNA damage, the RPA complex at the RF functions as a signaling hub to drive multiple post-translational modifications, such as phosphorylation, SUMOylation and ubiquitination, all of which differentially coordinate and regulate checkpoint and DDR pathways spatially and temporally (39). Despite their importance, the proteins that interact with RPA-ssDNA, as well as the regulation of the post-translational modifications that coordinate the DDR to replication stress, are poorly understood. We identified NSMF as a key regulator of RPA-dependent ATR signaling that promotes the restart and progression of stalled RFs during replication stress. NSMF rapidly translocates to DNA lesions and acts as a scaffold to bridge the interactions among RPA, CDC5L and ATR/ATRIP in order to modulate phosphorylation and ubiquitination of RPA2 for ATR signaling in response to DNA replication stress.
We found that NSMF regulates formation of the RPA complex for activation of ATR and the replication stress response. This conclusion is strongly supported by data showing that NSMF translocates to sites of DNA damage and forms complexes with CDC5L, ATRIP and RPA2. NSMF promotes the interaction between RPA2 and ATR/ATRIP and recruits ATR/ATRIP to DNA lesions resulting from replication stress (Figure 5). Recruitment of the ATR/ATRIP-kinase and PRP19/CDC5L-ligase complexes induced phosphorylation and ubiquitination of RPA, modifications that have been shown to be capable of functional crosstalk (40). ATR/ATRIP-induced phosphorylation of RPA promotes PRP19 recruitment onto RPA-ssDNA, and PRP19 exerts its ubiquitin ligase activity on the RPA-ssDNA platform to further facilitate recruitment of ATR-ATRIP and autophosphorylation of ATR. This positive feedback phosphorylation–ubiquitination cascade facilitates full ATR activation for RF protection, genome maintenance and cell survival (32,38). NSMF deficiency reduced RPA2 phosphorylation and ubiquitination, impaired ATR activation, and caused defects in stalled RF restart and progression. NSMF stabilizes ATR/ATRIP and PRP19/CDC5L complexes on RPA-ssDNA to modulate the RPA phosphorylation–ubiquitination cascade that activates ATR signaling (Figure 7J).
Our results show that rapid recruitment of NSMF and CDC5L to sites of DNA damage is dependent on PARP1 (Figure 2). PARP1-mediated PARylation is a critical event early in the response to DNA damage. The formation of PAR chain remodeling complexes causes condensed chromatin to switch to an open conformation, providing a docking platform for the ordered recruitment of DNA repair factors (41). Upon detection of DNA damage, PARP1 is activated rapidly and can synthesize long PAR chains within 10 s (42,43). Similarly to PARP1, NSMF and CDC5L were rapidly recruited to sites of DNA damage within 10 s (Figure 2). PARP1 inhibition prevents the recruitment of DNA repair factors and interferes with DNA replication, fork stalling and lead replication-fork collapse (44). PARP1-dependent rapid and transient recruitment of NSMF and CDC5L to DNA lesions supports our notion that NSMF recruits signaling molecules to regulate ATR signaling at an early stage of the replication stress response.
In this study, we showed the two ligases PRP19 and RFWD3 contributed to ubiquitination of RPA2 in response to replication stress. Interestingly, we observed that NSMF depletion reduced accumulation of both PRP19 and RFWD3 on chromatin in response to HU treatment. Furthermore, interactions of RPA2 with RFWD3 and PRP19 were decreased in NSMF-depleted cells (data not shown). It is possible that depletion of NSMF may cause significant changes in replisome architecture and reduce the association of the PRP19 and RFWD3 complex with RPA.
NSMF depletion dramatically reduced ATRIP association with RPA (Figure 5I) but reduced ATR-specific phosphorylation of RPA S33 by only 60%. Phosphorylation of multiple sites of RPA by various kinases such as ATR, ATM, DNA-PK and CDK is interconnected through processes such as priming. In fact, these kinases phosphorylate a large number of partially overlapping substrates and collaboratively regulate downstream targets (45). It is therefore possible that other kinases can compensate for the absence of ATR to achieve RPA phosphorylation. TopBP1-mediated and ETAA1-mediated pathways function in parallel to activate ATR signaling in response to replication stress (6,7,46). Both the TopBP1 pathway and the ETAA1 pathway are dependent on RPA-coated ssDNA for ATR activation in mammalian cells. Because NSMF forms complexes with ATRIP/ATR and PRP-19/CDC5L on RPA-ssDNA to modulate ATR activation, NSMF may also interact with upstream regulators of the ATR activation pathway. It is important to determine which pathway NSMF is involved in for ATR activation.
ATR kinase functions as a master regulator that directs DNA damage-specific signaling cascades via direct phosphorylation of downstream effector proteins in the DDR pathway. ATR is critical for ensuring the fidelity of DNA replication in response to replication-associated damage by preventing RF collapse, promoting the restart of stalled forks, and suppressing new origin firing. Consistent with the role of ATR, NSMF depletion inhibits RF progression and cell-cycle progression and increases the sensitivity of cells to HU and CPT. Although ATR is primarily a replication stress-response kinase, it is activated by DSBs and other types of DNA damage (47). We demonstrated that NSMF depletion causes defects in HR repair and DNA end resection (Supplementary Figure S3G–I). In addition to HU treatment, we found that NSMF KO in mice caused increased genomic sensitivity to γ-irradiation and shorter lifespans compared with those in WT mice, suggesting that NSMF regulates ATR activation at any DSB or ssDNA at stalled forks, regardless of how these arise (Supplementary Figure S7D–F).
Most NSMF studies have focused on neuronal functions, although NSMF is expressed in other tissues the including kidney, liver, lung and heart (Supplementary Figure S7A). The cellular and molecular functions of NSMF in those tissues are unclear, however (9,17). Using quantitative mass spectrometry analysis of NSMF interactomes, we found that NSMF is an important regulatory factor in the replication stress response. Under replication stress, NSMF deficiency caused defects in stalled RF restart and RF progression (Figure 4) and hypersensitivity to RF-blocking and DNA-damaging agents (Figure 3I,J), indicating the importance of NSMF in repairing RF-associated DNA damage. Deletion of proteins in the ATR signaling pathway, including ATR, ATRIP, RAD17, TopBP1 or ETAA1, caused complete or partial embryonic lethality (46,48,49), although NSMF KO mice were viable, fertile and appeared normal. Recently, zinc finger protein 161 (ZFP161) was found to act as a scaffold to promote the association between RPA2 and the ATR/ATRIP complex for ATR activation (50). Because NSMF and ZFP161 appear to act in a similar manner, it is possible that ZFP161 and NSMF work redundantly to regulate ATR activation. It is possible that these two proteins function in parallel, distinct pathways to regulate ATR activation in response to various replication stresses. Abnormal regulation of replication-stress signaling is associated with cancer and other human diseases. Inhibition of ATR kinase has shown promise as a therapeutic approach in cancer treatment, exacerbating oncogene-induced replication stress to specifically sensitize cancer cells to DNA damage (51,52). NSMF is less important under normal growth conditions, but it is an important DDR factor for survival and genome maintenance under stressful conditions with increased DNA damage. Further studies are needed to elucidate the role of NSMF in cancer and other diseases linked to the replication stress response.
In summary, we demonstrated that NSMF is a regulator of the DNA replication stress response. In response to replication stress, NSMF promotes cell survival and genome maintenance by functioning as a scaffold protein to bridge the RPA complex with ATP/ATRIP and PRP19/CDC5L, resulting in ATR activation through the RPA phosphorylation-ubiquitination cascade.
DATA AVAILABILITY
The mass spectrometry proteomics data which used in our manuscript have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021942. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Young Chan Chae (ychae@unist.ac.kr).
Supplementary Material
Contributor Information
Min Kyung Ju, Department of Life Sciences, Ulsan National University of Science and Technology (UNIST), Ulsan 44919, Republic of Korea.
Kyeong Jin Shin, Department of Life Sciences, Ulsan National University of Science and Technology (UNIST), Ulsan 44919, Republic of Korea.
Joo Rak Lee, Department of Life Sciences, Ulsan National University of Science and Technology (UNIST), Ulsan 44919, Republic of Korea.
Keon Woo Khim, Department of Life Sciences, Ulsan National University of Science and Technology (UNIST), Ulsan 44919, Republic of Korea.
Eun A. Lee, Center for Genomic Integrity Institute for Basic Science (IBS), UNIST, Ulsan 44919, Republic of Korea.
Jae Sun Ra, Center for Genomic Integrity Institute for Basic Science (IBS), UNIST, Ulsan 44919, Republic of Korea.
Byung-Gyu Kim, Center for Genomic Integrity Institute for Basic Science (IBS), UNIST, Ulsan 44919, Republic of Korea.
Han-seul Jo, Neurodegenerative Diseases Research Group, Korea Brain Research Institute, Daegu 41062, Republic of Korea.
Jong Hyuk Yoon, Neurodegenerative Diseases Research Group, Korea Brain Research Institute, Daegu 41062, Republic of Korea.
Tae Moon Kim, Center for Genomic Integrity Institute for Basic Science (IBS), UNIST, Ulsan 44919, Republic of Korea.
Kyungjae Myung, Center for Genomic Integrity Institute for Basic Science (IBS), UNIST, Ulsan 44919, Republic of Korea; Department of Biomedical Engineering, Ulsan National University of Science and Technology (UNIST), Ulsan 44919, Republic of Korea.
Jang Hyun Choi, Department of Life Sciences, Ulsan National University of Science and Technology (UNIST), Ulsan 44919, Republic of Korea.
Hongtae Kim, Department of Life Sciences, Ulsan National University of Science and Technology (UNIST), Ulsan 44919, Republic of Korea; Center for Genomic Integrity Institute for Basic Science (IBS), UNIST, Ulsan 44919, Republic of Korea.
Young Chan Chae, Department of Life Sciences, Ulsan National University of Science and Technology (UNIST), Ulsan 44919, Republic of Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
UNIST [1.200094.01]; National Research Foundation of Korea funded by the Ministry of Science and ICT [2018R1A2B2003129, 2020R1A2C1011284, 2016M3A9D5A01952411]; Ministry of Education [2018R1A6A1A03025810]; Institute for Basic Science [IBS-R022-D1]. Funding for open access charge: Institute for Basic Science [IBS-R022-D1].
Conflict of interest statement. None declared.
REFERENCES
- 1. Zeman M.K., Cimprich K.A.. Causes and consequences of replication stress. Nat. Cell Biol. 2014; 16:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Halazonetis T.D., Gorgoulis V.G., Bartek J.. An oncogene-induced DNA damage model for cancer development. Science. 2008; 319:1352–1355. [DOI] [PubMed] [Google Scholar]
- 3. Negrini S., Gorgoulis V.G., Halazonetis T.D.. Genomic instability–an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010; 11:220–228. [DOI] [PubMed] [Google Scholar]
- 4. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Delacroix S., Wagner J.M., Kobayashi M., Yamamoto K., Karnitz L.M.. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007; 21:1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumagai A., Lee J., Yoo H.Y., Dunphy W.G.. TopBP1 activates the ATR-ATRIP complex. Cell. 2006; 124:943–955. [DOI] [PubMed] [Google Scholar]
- 7. Haahr P., Hoffmann S., Tollenaere M.A., Ho T., Toledo L.I., Mann M., Bekker-Jensen S., Raschle M., Mailand N.. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016; 18:1196–1207. [DOI] [PubMed] [Google Scholar]
- 8. Saldivar J.C., Cortez D., Cimprich K.A.. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017; 18:622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miura K., Acierno J.S., Seminara S.B. Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (IHH). J. Hum. Genet. 2004; 49:265–268. [DOI] [PubMed] [Google Scholar]
- 10. Trarbach E.B., Baptista M.T., Garmes H.M., Hackel C.. Molecular analysis of KAL-1, GnRH-R, NELF and EBF2 genes in a series of Kallmann syndrome and normosmic hypogonadotropic hypogonadism patients. J. Endocrinol. 2005; 187:361–368. [DOI] [PubMed] [Google Scholar]
- 11. Pitteloud N., Quinton R., Pearce S., Raivio T., Acierno J., Dwyer A., Plummer L., Hughes V., Seminara S., Cheng Y.Z.et al.. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J. Clin. Invest. 2007; 117:457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sykiotis G.P., Plummer L., Hughes V.A., Au M., Durrani S., Nayak-Young S., Dwyer A.A., Quinton R., Hall J.E., Gusella J.F.et al.. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc. Natl. Acad. Sci. USA. 2010; 107:15140–15144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kramer P.R., Wray S.. Novel gene expressed in nasal region influences outgrowth of olfactory axons and migration of luteinizing hormone-releasing hormone (LHRH) neurons. Genes Dev. 2000; 14:1824–1834. [PMC free article] [PubMed] [Google Scholar]
- 14. Karpova A., Mikhaylova M., Bera S., Bar J., Reddy P.P., Behnisch T., Rankovic V., Spilker C., Bethge P., Sahin J.et al.. Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell. 2013; 152:1119–1133. [DOI] [PubMed] [Google Scholar]
- 15. Ronicke R., Mikhaylova M., Ronicke S., Meinhardt J., Schroder U.H., Fandrich M., Reiser G., Kreutz M.R., Reymann K.G.. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging. 2011; 32:2219–2228. [DOI] [PubMed] [Google Scholar]
- 16. Gomes G.M., Dalmolin G.D., Bar J., Karpova A., Mello C.F., Kreutz M.R., Rubin M.A.. Inhibition of the polyamine system counteracts beta-amyloid peptide-induced memory impairment in mice: involvement of extrasynaptic NMDA receptors. PLoS One. 2014; 9:e99184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kramer P.R., Wray S.. Nasal embryonic LHRH factor (NELF) expression within the CNS and PNS of the rodent. Brain Res. Gene Expr. Patterns. 2001; 1:23–26. [DOI] [PubMed] [Google Scholar]
- 18. Lee N.S., Chung H.J., Kim H.J., Lee S.Y., Ji J.H., Seo Y., Han S.H., Choi M., Yun M., Lee S.G.et al.. TRAIP/RNF206 is required for recruitment of RAP80 to sites of DNA damage. Nat. Commun. 2016; 7:10463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qiu L.Q., Lai W.S., Stumpo D.J., Blackshear P.J.. Mouse embryonic fibroblast cell culture and stimulation. Bio Protoc. 2016; 6:e1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim H., Chen J.J., Yu X.H.. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007; 316:1202–1205. [DOI] [PubMed] [Google Scholar]
- 21. Kim H., Huang J., Chen J.J.. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat. Struct. Mol. Biol. 2007; 14:710–715. [DOI] [PubMed] [Google Scholar]
- 22. Shevchenko A., Tomas H., Havlis J., Olsen J.V., Mann M.. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006; 1:2856–2860. [DOI] [PubMed] [Google Scholar]
- 23. Nesvizhskii A.I., Keller A., Kolker E., Aebersold R.. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003; 75:4646–4658. [DOI] [PubMed] [Google Scholar]
- 24. Huang D.W., Sherman B.T., Tan Q., Kir J., Liu D., Bryant D., Guo Y., Stephens R., Baseler M.W., Lane H.C.et al.. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007; 35:W169–W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M.et al.. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019; 47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hwang S.Y., Kang M.A., Baik C.J., Lee Y., Hang N.T., Kim B.G., Han J.S., Jeong J.H., Park D., Myung K.et al.. CTCF cooperates with CtIP to drive homologous recombination repair of double-strand breaks. Nucleic Acids Res. 2019; 47:9160–9179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou Y., Caron P., Legube G., Paull T.T.. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res. 2014; 42:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zierhut C., Diffley J.F.. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008; 27:1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park S.H., Kang N., Song E., Wie M., Lee E.A., Hwang S., Lee D., Ra J.S., Park I.B., Park J.et al.. ATAD5 promotes replication restart by regulating RAD51 and PCNA in response to replication stress. Nat. Commun. 2019; 10:5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Balmus G., Karp N.A., Ng B.L., Jackson S.P., Adams D.J., McIntyre R.E.. A high-throughput in vivo micronucleus assay for genome instability screening in mice. Nat. Protoc. 2015; 10:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ajuh P., Kuster B., Panov K., Zomerdijk J.C., Mann M., Lamond A.I.. Functional analysis of the human CDC5L complex and identification of its components by mass spectrometry. EMBO J. 2000; 19:6569–6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marechal A., Li J.M., Ji X.Y., Wu C.S., Yazinski S.A., Nguyen H.D., Liu S., Jimenez A.E., Jin J., Zou L.. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol. Cell. 2014; 53:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bonner W.M., Redon C.E., Dickey J.S., Nakamura A.J., Sedelnikova O.A., Solier S., Pommier Y.. GammaH2AX and cancer. Nat. Rev. Cancer. 2008; 8:957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brush G.S., Morrow D.M., Hieter P., Kelly T.J.. The ATM homologue MEC1 is required for phosphorylation of replication protein A in yeast. Proc. Natl. Acad. Sci. USA. 1996; 93:15075–15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brush G.S., Kelly T.J.. Phosphorylation of the replication protein A large subunit in the Saccharomyces cerevisiae checkpoint response. Nucleic Acids Res. 2000; 28:3725–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Soo Lee N., Jin Chung H., Kim H.J., Yun Lee S., Ji J.H., Seo Y., Hun Han S., Choi M., Yun M., Lee S.G.et al.. TRAIP/RNF206 is required for recruitment of RAP80 to sites of DNA damage. Nat. Commun. 2016; 7:10463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zou L., Elledge S.J.. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003; 300:1542–1548. [DOI] [PubMed] [Google Scholar]
- 38. Elia A.E., Wang D.C., Willis N.A., Boardman A.P., Hajdu I., Adeyemi R.O., Lowry E., Gygi S.P., Scully R., Elledge S.J.. RFWD3-dependent ubiquitination of RPA regulates repair at stalled replication forks. Mol. Cell. 2015; 60:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marechal A., Zou L.. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015; 25:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dubois J.C., Yates M., Gaudreau-Lapierre A., Clement G., Cappadocia L., Gaudreau L., Zou L., Marechal A.. A phosphorylation-and-ubiquitylation circuitry driving ATR activation and homologous recombination. Nucleic Acids Res. 2017; 45:8859–8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gibson B.A., Kraus W.L.. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012; 13:411–424. [DOI] [PubMed] [Google Scholar]
- 42. Mortusewicz O., Ame J.C., Schreiber V., Leonhardt H.. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007; 35:7665–7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haince J.F., McDonald D., Rodrigue A., Dery U., Masson J.Y., Hendzel M.J., Poirier G.G.. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008; 283:1197–1208. [DOI] [PubMed] [Google Scholar]
- 44. Liao H., Ji F., Helleday T., Ying S.. Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018; 19:e46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Serrano M.A., Li Z., Dangeti M., Musich P.R., Patrick S., Roginskaya M., Cartwright B., Zou Y.. DNA-PK, ATM and ATR collaboratively regulate p53-RPA interaction to facilitate homologous recombination DNA repair. Oncogene. 2013; 32:2452–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bass T.E., Luzwick J.W., Kavanaugh G., Carroll C., Dungrawala H., Glick G.G., Feldkamp M.D., Putney R., Chazin W.J., Cortez D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 2016; 18:1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marechal A., Zou L.. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013; 5:a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ruzankina Y., Pinzon-Guzman C., Asare A., Ong T., Pontano L., Cotsarelis G., Zediak V.P., Velez M., Bhandoola A., Brown E.J.. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007; 1:113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jeon Y., Ko E., Lee K.Y., Ko M.J., Park S.Y., Kang J., Jeon C.H., Lee H., Hwang D.S.. TopBP1 deficiency causes an early embryonic lethality and induces cellular senescence in primary cells. J. Biol. Chem. 2011; 286:5414–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim W., Zhao F., Wu R., Qin S., Nowsheen S., Huang J., Zhou Q., Chen Y., Deng M., Guo G.et al.. ZFP161 regulates replication fork stability and maintenance of genomic stability by recruiting the ATR/ATRIP complex. Nat. Commun. 2019; 10:5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Josse R., Martin S.E., Guha R., Ormanoglu P., Pfister T.D., Reaper P.M., Barnes C.S., Jones J., Charlton P., Pollard J.R.et al.. ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 2014; 74:6968–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Komura K., Yoshikawa Y., Shimamura T., Chakraborty G., Gerke T.A., Hinohara K., Chadalavada K., Jeong S.H., Armenia J., Du S.Y.et al.. ATR inhibition controls aggressive prostate tumors deficient in Y-linked histone demethylase KDM5D. J. Clin. Invest. 2018; 128:2979–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data which used in our manuscript have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021942. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Young Chan Chae (ychae@unist.ac.kr).