

Abstract
Asthma is a highly prevalent disorder characterized by chronic lung inflammation and reversible airways obstruction. Pathophysiological features of asthma include episodic and reversible airway narrowing due to increased bronchial smooth muscle shortening in response to external and host-derived mediators, excessive mucus secretion into the airway lumen, and airway remodeling. The aberrant airway smooth muscle (ASM) phenotype observed in asthma manifests as increased sensitivity to contractile mediators (EC50) and an increase in the magnitude of contraction (Emax); collectively these attributes have been termed “airways hyperresponsiveness” (AHR). This defining feature of asthma can be promoted by environmental factors including airborne allergens, viruses, and air pollution and other irritants. AHR reduces airway caliber and obstructs airflow, evoking clinical symptoms such as cough, wheezing and shortness of breath. G-protein-coupled receptors (GPCRs) have a central function in asthma through their impact on ASM and airway inflammation. Many but not all treatments for asthma target GPCRs mediating ASM contraction or relaxation. Here we discuss the roles of specific GPCRs, G proteins, and their associated signaling pathways, in asthma, with an emphasis on endogenous mechanisms of GPCR regulation of ASM tone and lung inflammation including regulators of G-protein signaling (RGS) proteins, G-protein coupled receptor kinases (GRKs), and β-arrestin.
Keywords: airway smooth muscle, GPCR, signal transduction, asthma, remodeling
1. Introduction
In asthma patients, AHR results from a complex interplay of inflammatory mediators, airway remodeling, and exaggerated ASM shortening in the medium-sized to small bronchioles. In about 50% of patients with asthma, type 2 immune responses to allergens evoke airway inflammation and AHR (Boonpiyathad, et al., 2019; Lambrecht & Hammad, 2015). Allergens activate innate immunity mediated by respiratory epithelial cells that secrete cytokines (e.g. IL-25, IL-33, and thymic stromal lymphopoietin [TSLP], among others) involved in migration and activation of leukocytes. Epithelial-derived mediators also activate airway resident immune cells including mast cells and type 2 innate lymphoid cells (ILC2s) (Lambrecht, et al., 2019) and induce specific adaptive immune responses eliciting secretion of immunoglobulin E (IgE). IgE crosslinks specific receptors (FcεR1) on MCs, promoting release of preformed substances (e.g. histamine, TNFα) from granules and newly synthesized mediators (leukotrienes, prostaglandins) that modulate bronchomotor tone (Komi, et al., 2020). IgE also increases the numbers of T helper type 2 (Th2) lymphocytes by stimulating activation and trafficking to the airways; ILC2 and Th2 lymphocytes then increase levels of type 2 cytokines, including IL-4, -5, and -13 (Lambrecht, et al., 2019). These and other mediators promote recruitment of eosinophils to the airways, mucous hypersecretion, and airway remodeling manifested by epithelial damage, collagen deposition, and ASM hypertrophy and hyperplasia (Fang, et al., 2020; Guida & Riccio, 2019). Collectively, these components facilitate development of AHR.
ASM is the pivotal structural cell of the airway that together with connective tissue determine bronchomotor tone (Anthracopoulos & Everard, 2020). The ASM layer is located just below the surface respiratory epithelium, within the lung parenchyma. Although the precise physiological functions of ASM remain unclear, investigators suggest that ASM: (1) spares distension and distortion of alveoli to prevent stretch-induced injury; (2) decreases anatomic dead space volume (the volume of ventilated air that does not participate in gas exchange); (3) promotes mucous clearance through airway compression; and (4) stabilizes the large airways during cough (Lam, et al., 2019). AHR is assessed clinically by spirometry, a technique that measures airflow at baseline and in response to inhaled bronchoconstrictors. The spirometric characteristics of asthma include: 1) an exaggerated response to bronchoconstrictors (e.g. methacholine (MCh), a muscarinic receptor agonist), manifested as both an increase in Emax and decreased EC50 as noted above; and 2) reversibility—improvement in airflow following inhalation of bronchodilators (e.g. beta2-adrenergic receptor [β2AR] agonists) (D’Urzo, et al., 2020; Davis, et al., 2018).
GPCRs expressed on ASM cells have a central role in the maintenance of airway tone by regulating ASM contraction and relaxation (Nayak & Penn, 2020). Other GPCRs expressed on lung structural cells and leukocytes recruited to the airways contribute to the development of allergic airway inflammation (Wendell, et al., 2020). In this review, we discuss airway cell GPCRs that regulate AHR and inflammation in asthma, with an emphasis on their associated intracellular signaling pathways. Specifically, we highlight host control mechanisms of GPCR signaling pathways including RGS proteins, GRKs, and β-arrestins.
2. Mediators and signaling mechanisms of ASM contraction and relaxation
2.1. Mediators affecting airway tone
Host-derived mediators within the airways modulate ASM shortening by activating specific GPCRs (Table 1). At homeostasis, acetylcholine (ACh) and neurokinins (substance P, neurokinin A) released from afferent sensory nerves of the autonomic nervous system (ANS) modulate basal airway tone by promoting ASM contractility (Bosse, et al., 2013; L. Y. Lee & Yu, 2014). Neural inputs released from postganglionic parasympathetic nerves can either augment airway constriction or reverse bronchoconstriction (L. Y. Lee & Yu, 2014). In allergic asthma, allergen exposure injures the respiratory epithelium, exposing sensory C-fibres that then release contractile agonists via an axon reflex. Some allergens, including proteases from fungi and house dust mites (HDM), directly activate epithelial and ASM cells through protease-activated receptors (PARs), eliciting autocrine and paracrine release of mediators such as endothelin-1 or prostaglandin (PGE2) that affect ASM tone by inducing bronchoconstriction or bronchorelaxation, respectively (Gauvreau, et al., 1999; Koziol-White, et al., 2020; Wong, et al., 2020; Yamada & Matsumoto, 2017). Airway-resident mast cells and recruited leukocytes (e.g. eosinophils, neutrophils, lymphocytes) release additional pro-contractile mediators acting on GPCRs expressed on ASM including histamine, bradykinin, cysteinyl leukotrienes, and thromboxanes (K. Lee, et al., 2020; Sasaki & Yokomizo, 2019; Yamauchi & Ogasawara, 2019) Several of these factors (e.g. bradykinin) can also induce bronchoconstriction indirectly by eliciting release of mediators (ACh, tachykinins) from airway sensory nerves (Barnes, 1992).
Table 1.
Regulators of airway contraction and relaxation
| Contraction | Relaxation | ||
|---|---|---|---|
| Endogenous regulators | Receptor | Bronchodilators | Receptor |
| Acetylcholine | M3 muscarinic | Acetylcholine | M2 |
| Adenosine | A1 | Adenosine | A2b |
| Bradykinin | BK1 | Adrenaline (Epinephrine) | β2-Adrenergic |
| Endothelin-1 | ETA/ETB | Endothelin-1 | ETB (epithelium) via PGE2/NO |
| Neurokinins | NK1/NK2 | Thrombin, trypsin | PAR2/4 via PGE2 |
| Thrombin, trypsin | PAR1/2/3 | Histamine | H2 |
| Serotonin | 5HT2 | Prostanoids | DP, EP, IP |
| Histamine | H1 | GABA | GABAA |
| Thromboxanes | TP | Vasoactive intestinal peptide | PVR1 |
| LTB4, Cysteinyl leukotrienes | BLT1/BLT2, CysLT1R | Nitric oxide | GC via cGMP |
| Prostanoids | TP, EP, FP | ||
| GABA | GABAB | ||
| Ca2+ | CaSR | ||
| H+ | OGR1 | ||
2.2. Mechanisms of GPCR-mediated ASM contraction
Transmembrane signaling by GPCRs sequentially activates receptors, G-proteins and effector molecules (Hepler & Gilman, 1992). Heterotrimeric G-proteins consist of three subunits: alpha (Gα), beta (Gβ), and gamma (Gγ), the latter two existing as a heterodimer. Agonist binding elicits a conformational change in the receptor that then activates heterotrimeric G-proteins through interactions with the carboxy-terminus of Gα subunit. Guanosine diphosphate (GDP), which binds Gα, is exchanged for guanosine triphosphate (GTP), promoting dissociation of Gα from the βγ dimer (Billington & Penn, 2003). Four distinct classes of Gα subunits bind Gβγ (Gi/o, Gs, Gq/11, G12/13), and the identity of Gα partially determines which downstream effectors become activated. Most GPCRs eliciting ASM contraction couple to heterotrimeric G-proteins containing Gαq and downstream calcium (Ca2+) signaling (Figure 1) (Lam, et al., 2019). Gαq-GTP and Gβγ can activate phospholipase C beta (PLCβ), an enzyme that catalyzes hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 releases Ca2+ from intracellular stores by binding its cognate receptor (IP3R) on the sarcoplasmic reticulum. Within the cytosol, Ca2+ binds to calmodulin, and in turn the Ca2+/calmodulin complex activates myosin light chain kinase (MLCK), culminating in myosin light chain phosphorylation on specific residues (Thr18 and Ser19 of myosin light chain 2). Phosphorylation of myosin light chain induces a conformational change in the protein that evokes cross-bridge cycling with actin filaments, promoting ASM shortening and force generation (Hong, et al., 2011). The sarcoplasmic/endoplasmic Ca2+-ATPase (SERCA), which actively pumps Ca2+ from the cytosol to the lumen of the SR, maintains homeostasis of intracellular Ca2+ (Mahn, et al., 2010). GPCR-independent membrane proteins also maintain intracellular Ca2+ concentrations including receptor-operated Ca2+ channels (ROCC), store-operated Ca2+channels (SOCC) and voltage-operated Ca2+ channels (VOCC) (Reyes-Garcia, et al. 2018). Additional pathways modify the sensitivity of ASM cells to steady-state intracellular Ca2+ concentrations. Ca2+ sensitivity occurs in part by myosin light chain phosphatase (MYPT-1) dephosphorylation of MLC. Gαq/11 and Gβγ activate guanine nucleotide exchange factors (GEFs) for the small GTPase RhoA to form RhoA-GTP (Cervantes-Villagrana, et al., 2019; Momotani & Somlyo, 2012; Strassheim, et al., 2019). The RhoA effector Rho associated coiled-coil forming kinase (ROCK) in turn phosphorylates MYPT-1, a modification that inhibits its activity (Guan, et al., 2013). A parallel pathway downstream of Gαq is mediated by DAG that promotes activation of protein kinase C (PKC) and phosphorylation of protein phosphatase 1 (CPI-17). CPI-17 can also inhibit MYPT-1 activity (Eto, 2009). When MYPT-1 activity is disabled, steady-state myosin light chain phosphorylation is increased, which sustains ASM shortening. Some pro-inflammatory GPCRs, such as those for thromboxanes, activate Gα12 and/or Gα13, G-proteins that can trigger the RhoA-MYPT-1 pathway through a distinct set of RhoGEFs including p115RhoGEF, LARG, and PDZ-RhoGEF (Yu & Brown, 2015).
Figure 1. Mechanisms of contraction in airway smooth muscle (ASM).
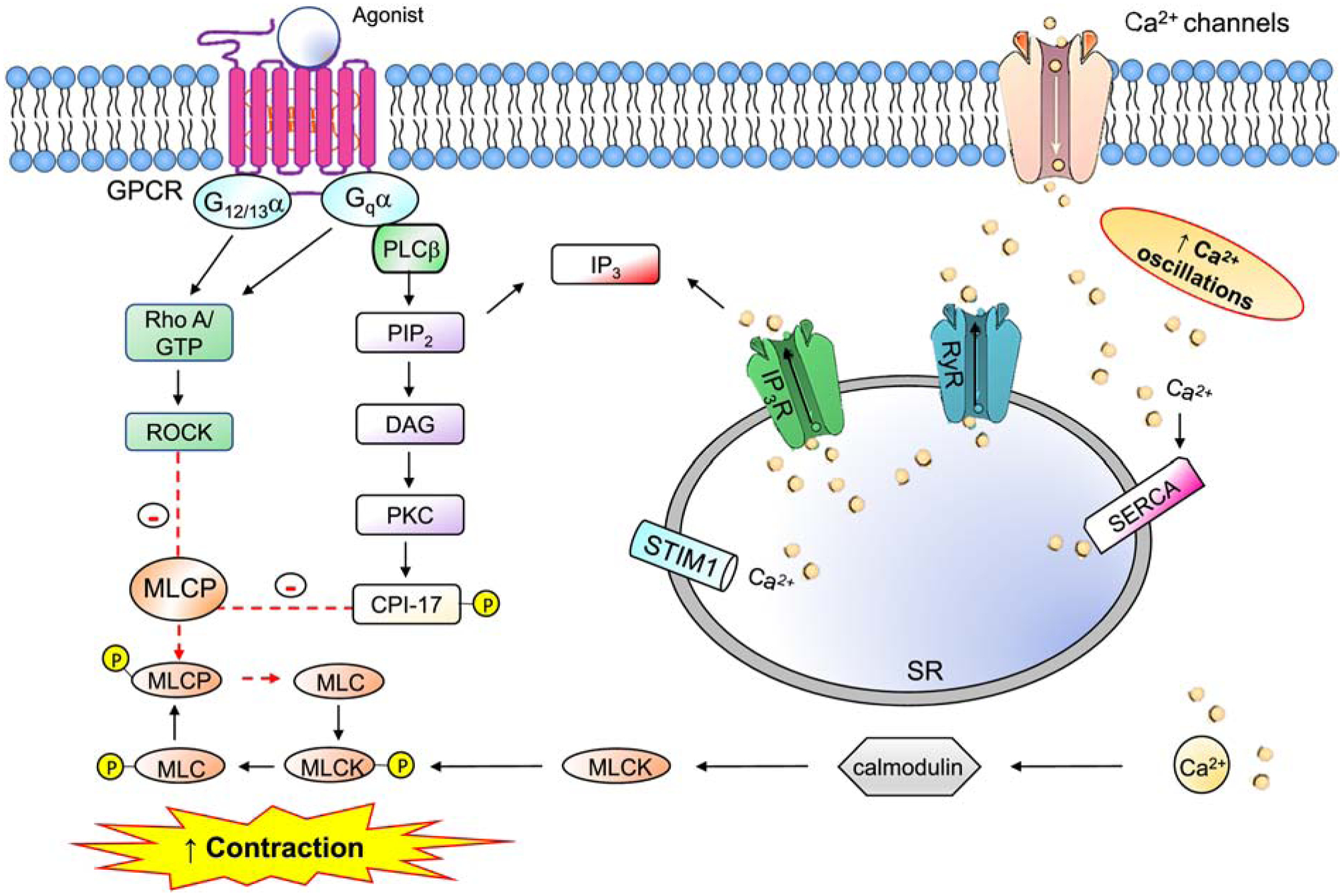
G-protein-coupled receptors (GPCRs) associated with Gαq activate phospholipase C (PLCβ), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2), and generate two lipid effectors: diacylglycerol (DAG) and inositol trisphosphate (IP3). IP3 liberates Ca2+ from the sarcoplasmic reticulum (SR) into the cytoplasm, stimulating Ca2+-calmodulin (Ca-CaM)-dependent myosin light chain kinase (MLCK). MLCK-mediated myosin light chain (MLC) phosphorylation promotes actin-myosin cross-bridging and ASM shortening. MLC phosphatase (MYPT-1) inhibits this pathway by dephosphorylating MLC. MYPT-1 is itself negatively regulated in a Ca2+-independent manner by: 1) RhoA/Rho kinase (ROCK)-mediated phosphorylation; 2) DAG-PKC-dependent activation of protein phosphatase 1 (CPI-17).
2.3. Mechanisms of GPCR-mediated ASM relaxation
Endogenous bronchodilators include catecholamines (epinephrine, norepinephrine), vasoactive intestinal peptide [VIP]) and prostaglandin E2 (PGE2) (Table 1) (Billington & Penn, 2003; Lam, et al., 2019). Host-derived mediators and beta 2 adrenergic receptor (β2AR) agonists, which are used for asthma treatment (e.g. short- or long-acting β2-adrenoceptor agonists [SABA, LABA], induce ASM relaxation by genomic actions (discussed further below) and by Gαs activation (Figure 2). Gs and Gβγ subunits stimulate membrane-bound adenylyl cyclase (AC), an enzyme that hydrolyzes ATP to cyclic adenosine monophosphate (cAMP). Increased cAMP levels may reverse excitation-contraction coupling in ASM through poorly defined mechanisms thought to involve at least two downstream effectors, protein kinase A (PKA) and the exchange protein directly activated by cAMP (Epac), a GEF for the small GTPase Rap1 (Roscioni, et al., 2011). PKA-mediated receptor phosphorylation of pro-contractile GPCRs evokes receptor internalization and degradation (Billington, et al., 2013). Epac decreases ASM shortening through several mechanisms including decreased RhoA activation (Roscioni, et al., 2011) and inhibition of store-operated Ca2+ entry (SOCE) (Cuinas, et al., 2016). cAMP is degraded by the family of phosphodiesterases (PDEs); non-selective inhibitors of phosphodiesterases such as theophylline increase steady-state cytosolic cAMP levels and have been exploited as add-on therapy for severe asthma (Barnes, 2013; Upham & Chung, 2018).
Figure 2. Mechanisms of relaxation in airway smooth muscle (ASM).
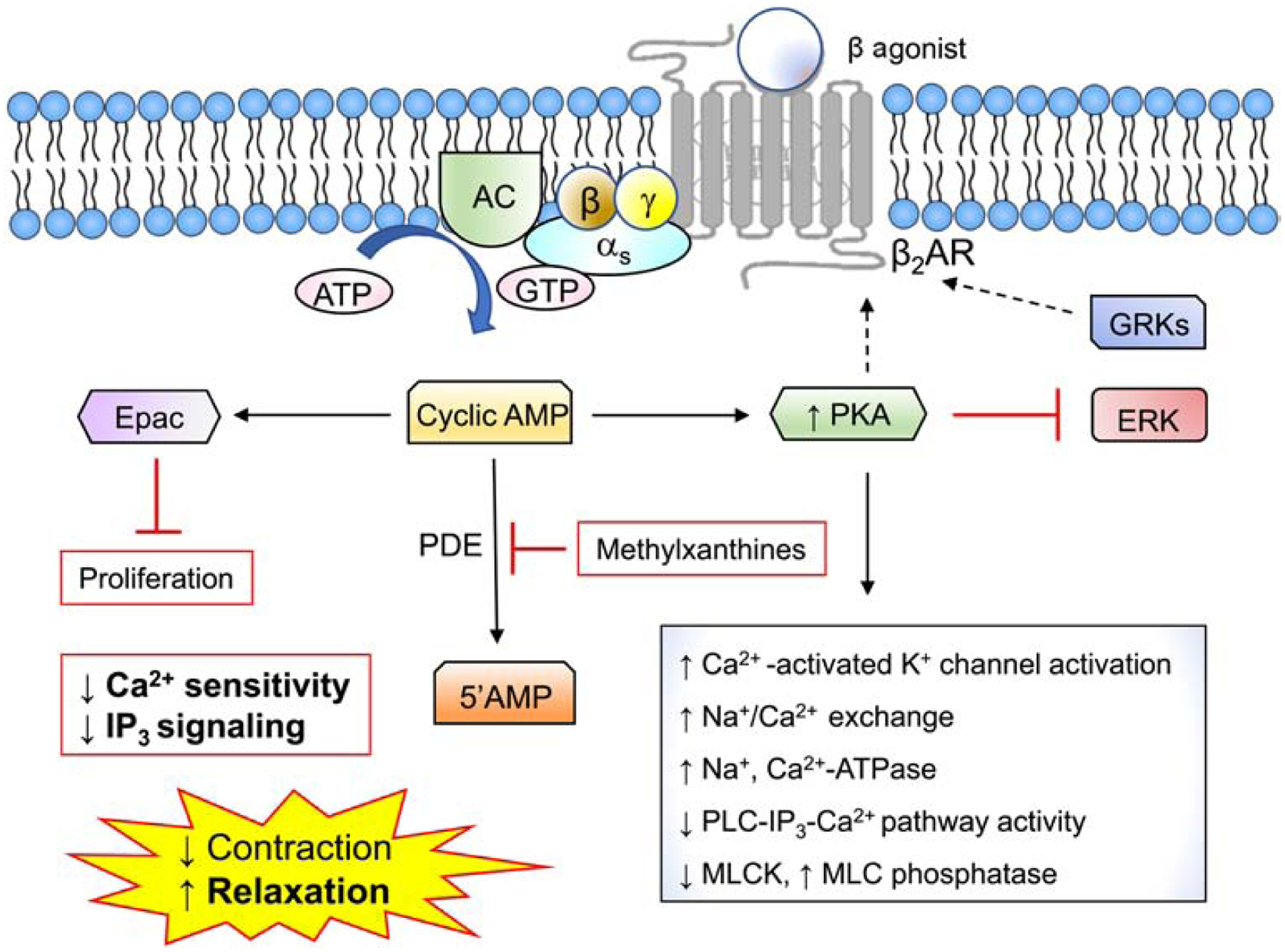
Stimulation of β2-adrenergic and other receptors coupled to Gαs promotes ASM relaxation by activating adenylyl cyclase to generate cyclic adenosine monophosphate (cAMP). cAMP-dependent activation of protein kinase A (PKA) antagonizes Ca2+ signaling through several potential mechanisms. The exchange protein directly activated by cAMP (Epac) inhibits ASM proliferation. Methylxanthines such as theophylline increase cytosolic cAMP levels by inhibiting cAMP-degrading cyclic nucleotide phosphodiesterase (PDE) enzymes.
3. Dysregulation of ASM GPCR signaling in asthma
3.1. Extrinsic and intrinsic regulation of ASM mechanics in asthma
Asthma-associated cytokines such as TNF-α and IL-13 can enhance ASM shortening by increasing aggregation of the sarcoplasmic reticulum protein stromal interaction molecule-1 (STIM1) that facilitates store-operated calcium entry by interacting with a Ca2+-activated Ca2+ membrane channel protein (Orai1) and thereby raising intracellular Ca2+ concentrations (Jia, et al., 2013). IL-13 or TNFα can also increase RhoA expression in ASM in a STAT6-dependent manner that promotes Ca2+ sensitivity (Goto, et al., 2010). Evidence points to intrinsic defects in contraction and/or relaxation signaling in asthma. For example, reduced expression of SERCA in ASM from patients with moderately severe asthma was associated with impaired recovery of bradykinin-induced intracellular Ca2+ flux (Mahn, et al., 2009). Increased expression of CPI-17 and RhoA has been detected in ASM from allergen-challenged mice (Sakai, et al., 2017). Others demonstrated increased expression and activity of the short transient receptor potential channel 3 (TRPC3) in ASM cells from allergen-challenged mice. TRPC3, a DAG-sensitive cation channel, can raise intracellular Ca2+ via activation of the PLC pathway and/or by sensing Ca2+ store depletion (Wang, et al., 2020).
The extent to which these and/or other abnormalities in ASM contribute to the development of AHR in asthma remains unclear. Ca2+ signaling in ASM cells from patients with asthma at homeostasis or in response to GPCR agonists was found to be essentially equivalent (Sweeney, et al., 2015) or reduced (Yang, et al., 2012) compared to that seen in cells from healthy controls. Others, however, found increased force generation in individual ASM cells from subjects with asthma relative to those from healthy donors at baseline and following histamine or MCh treatment (An, et al., 2016). Thus, the intrinsic mechanophenotype of ASM in asthma requires further study.
3.2. Therapeutic targeting of GPCR signaling in asthma
Although overproduction of inflammatory mediators in the airways is essential to the development of AHR, therapeutic inhibition of specific mediators (e.g. histamine, acetylcholine, leukotrienes) by selective GPCR antagonists has had a limited impact on symptoms in most patients (Wendell, et al., 2020). More recently, therapeutic targeting of G-proteins or other contraction signaling molecules in the contraction pathway (e.g. PKC, CPI, ROCK) has been explored in preclinical models. The compound FR900359 (FR) is a cyclic depsipeptide isolated from the roots of Ardisia crenatasims that selectively inhibits Gαq by blocking GDP-GTP exchange in the nucleotide binding pocket (Takasaki, et al., 2004). FR prevented bronchoconstriction in PCLS from humans and several other species, and topical administration of FR to the airways protected mice from development of allergen-associated AHR in experimental models of asthma with minimal effects on allergic lung inflammation and tissue remodeling (Matthey, et al., 2017). ROCK inhibitors such as Y-27632 and fasudil reduce allergen-induced AHR and airway inflammation in rodent models and inhibit agonist-induced bronchoconstriction in human precision cut lung slices (Zhang, et al., 2020).
4. Intracellular control of GPCR signaling: the regulators of G-protein signaling (RGS) superfamily
4.1. RGS protein discovery and mechanism of action
Intrinsic catalytic activity of the Gα subunit promotes hydrolysis of GTP to GDP that terminates GPCR signaling by re-associating Gα with Gβγ subunits, abrogating downstream effector activation. The index RGS protein, Sst2, was first discovered in the yeast Saccharomyces cerevisiae. Sst2 negatively regulates pheromone-induced responses of the GPCR Ste2 through direct interactions with the yeast Gα subunit, Gpa1 (Dohlman, et al., 1996). A large family of proteins with homology to Sst2 was discovered in Caenorhabditis elegans and mammals and termed the “Regulators of G-protein signaling” (RGS) (De Vries, et al., 1995; Druey, et al., 1996; Koelle & Horvitz, 1996; Siderovski, et al., 1996). Many RGS proteins act as GTPase activating proteins (GAPs) to markedly accelerate GTP hydrolysis by Gα and thereby limit the magnitude and duration of GPCR signaling (Figure 3) (Berman, et al., 1996; Watson, et al., 1996). While Gα has relatively weak enzymatic activity when assayed in vitro, the rate of deactivation of GPCR signaling observed in cells is much faster; the discovery of RGS proteins provided one explanation for this discordance (Ross & Wilkie, 2000). Over the past two decades, biochemical and structural studies have revealed that the GTPase accelerating (GAP) activity of RGS proteins is due to direct binding of conserved amino acids within the RGS domain, the motif common to all members of the family, to specific conserved residues in the so-called “switch” regions within Gα that are known to undergo a conformational change in the protein upon GTP binding and hydrolysis (Masuho, et al., 2020; Soundararajan, et al., 2008; Tesmer, et al., 1997). Although the RGS protein contributes no intrinsic enzymatic activity, it acts allosterically to stabilize a transition state conformation in Gα that enables a 100–1000-fold increase in the rate of GTP hydrolysis (Berman, et al., 1996). While RGS proteins with activity on Gα12/13 or Gαs proteins have been identified (Chen, et al., 2012; Zheng, et al., 2001), here we focus on those RGSs that function as GAPs for Gαi and Gαq as the functions(s) of the “non-canonical” RGS proteins in asthma have not been extensively characterized.
Figure 3. Biochemical actions of RGS proteins.
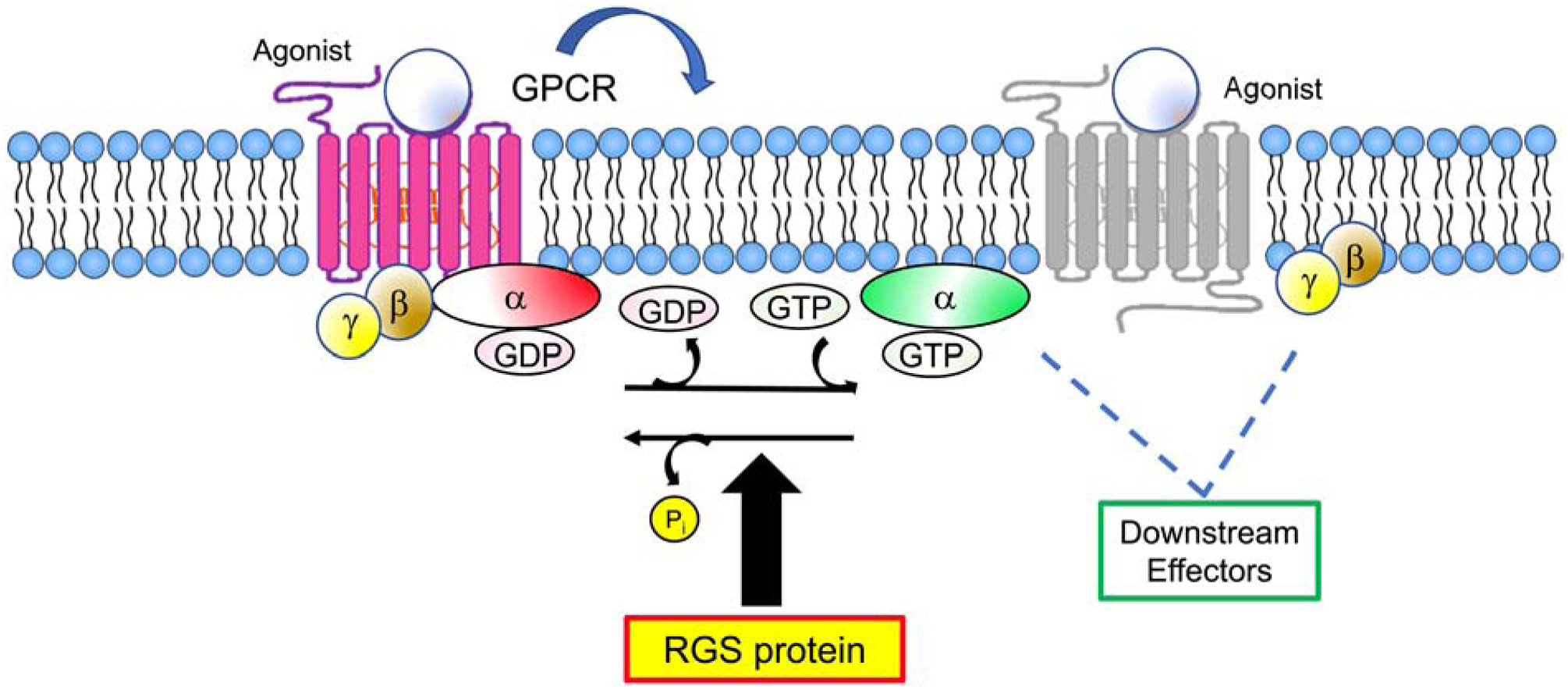
Activated GPCRs promote exchange of guanosine-5’-diphosphate (GDP) for guanosine-5’-triphosphate (GTP) on the Gα subunit of heterotrimeric G-proteins, resulting in dissociation of Gα from Gβγ dimers and effector activation. Gα hydrolyzes GTP to GDP, which promotes re-association of the G-protein heterotrimer and termination of signaling. RGS proteins interact with Gα in its transition state between GTP- and GDP-bound forms, an interaction which catalyzes hydrolysis and limits the duration and magnitude of GPCR signaling.
4.2. RGS protein structure
As a group, RGS proteins share homology primarily within the RGS domain, which is also referred to as the RGS box (Figure 4). The R4 subfamily of RGS proteins (RGS1–5, 8, 13, 16, 21) is mostly comprised of small-sized proteins containing little more than an RGS box. Many of the R4 RGS proteins have an amphipathic α-helix at the amino-terminus, which has been hypothesized to be involved in phospholipid binding and membrane localization (Bastin, et al., 2012). There is considerable biochemical and structural evidence that RGS2, amongst the R4 family, is uniquely attuned to serve as a GAP for Gαq (Kimple, et al., 2009; Nance, et al., 2013); however, recent comprehensive mapping studies have called into question this selectivity in a cellular environment (Masuho, et al., 2020). The RZ family (RGS17, RGS19, and RGS20) contains an amino-terminal cysteine-string, a motif that may be regulate subcellular trafficking and protein-protein interactions (Nunn, et al., 2006). This family of RGS proteins has higher affinity for Gαz relative to Gi/Go proteins. R7 RGS proteins exist in an obligate heterodimer with Gβ5 subunits, an interaction that is dependent upon the Gγ-like (GGL) domain (Snow, et al., 1998). Members of this subfamily also contain a disheveled-EGL10-Pleckstrin homology (DEP) domain, a motif that mediates interactions with protein partners that specify membrane localization and enhance GAP activity of the RGS protein, such as RGS9 Anchoring Protein (R9AP) and R7 Binding Protein (R7BP) (Jayaraman, et al., 2009). The R12 family is comprised of RGS10, RGS12, and RGS14. While RGS10 has no well-defined motifs other than the RGS domain, RGS12 has a PDZ region at the amino-terminus, a domain that facilitates direct interactions with the carboxy-terminus of GPCRs (Snow, et al., 2002). RGS12 also contains a phosphotyrosine binding domain (PTB domain), which interacts with specific phosphotyrosine residues in the pore-forming unit of Ca2+ channels (Richman, et al., 2005). Finally, RGS12 and RGS14 harbor: 1) a Ras binding domain (RBD), which modulates GAP activity through intramolecular interactions (Zhao, et al., 2013); 2) the Gαi/o-Loco (GoLoco) domain, a motif that inhibits GDP dissociation from Gα Siderovski & Willard, 2005). GoLoco motifs may have specific functions in cell polarity and the mitotic spindle orientation during asymmetric cell division (Liu, et al., 2018).
Figure 4. Domain structures of canonical regulator of G-protein signaling (RGS) proteins.
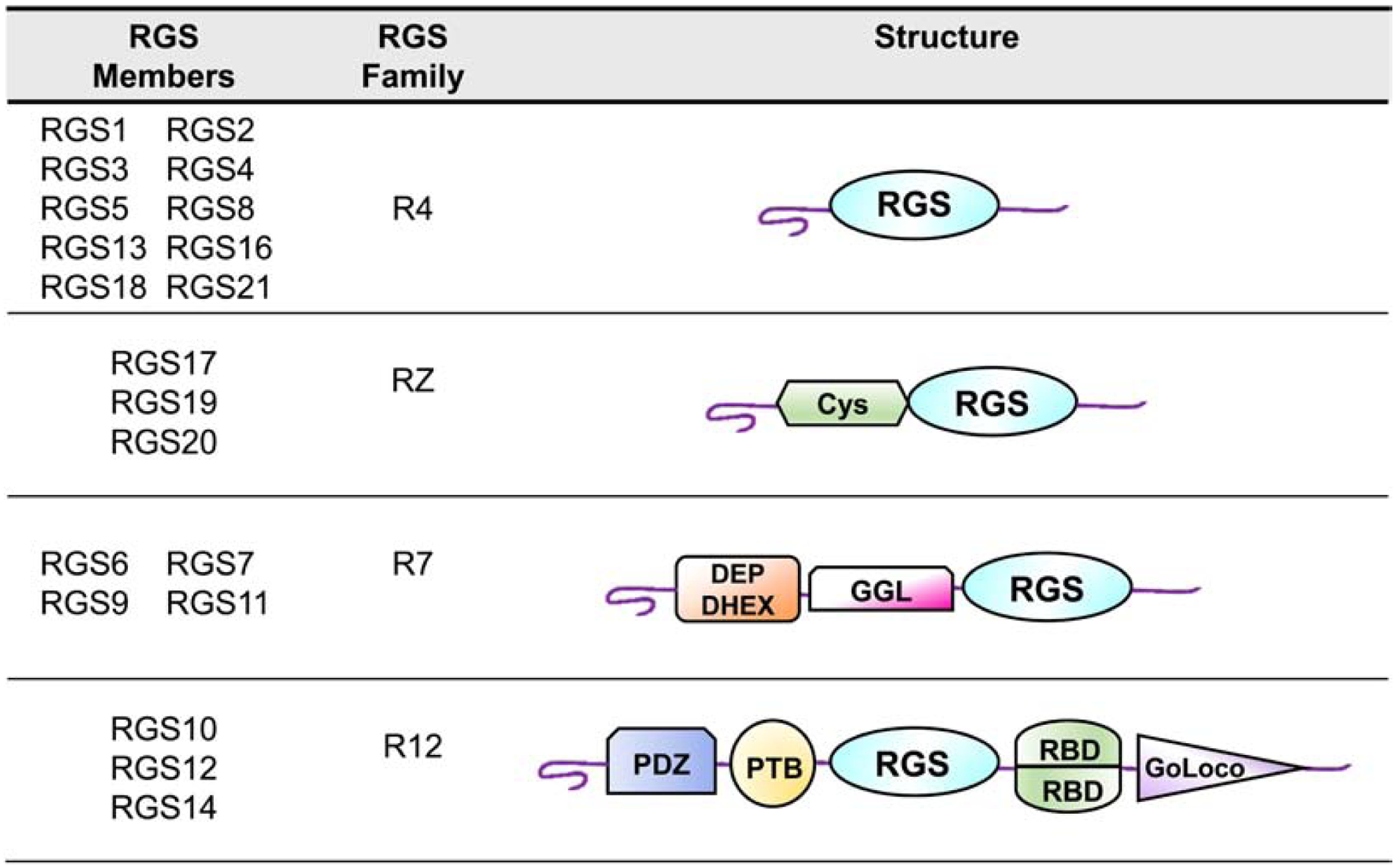
The classical RGS proteins are categorized into four subfamilies (R4, R7, RZ, and R12) based on sequence similarities. All subfamilies have the conserved RGS domain. The R4 group is the largest RGS subfamily and contains the RGS domain and an amino-terminal α-helix. The RZ family consists of RGS17, RGS19 and RGS20 and features an amino-terminal cysteine (Cys) string domain. In addition to the RGS domain, the R7 group contains a Dishevelled, Egl-10 and Pleckstrin (DEP) domain with a helical extension (DHEX) and the G-protein γ-like (GGL) motif. The structure of the R12 family, which includes RGS10, RGS12 and RGS14, is composed of two Ras-binding domains (RBDs), a phosphotyrosine-binding (PTB) domain, a PDZ (PSD95/ Dlg1/ ZO-1) domain and a GoLoco motif.
4.3. Physiological functions of RGS proteins
While the role(s) of many of the RGS family members in human health and disease, and more specifically in respiratory ailments, are unclear, defects in RGS protein function have been linked to human pathology. Mutations in RGS9–1 or its membrane anchoring protein (R9AP), which are highly expressed in the retina, were shown to cause bradyopsia, a rare condition that results in difficulty in adapting to changes in light and reduced ability to see moving objects. This results in prolonged activation of transducin, the Gα protein linked to rhodopsin photoreceptors (Nishiguchi, et al., 2004). Rare variants in RGS2, resulting in a loss of function (Q2L, D40Y, R44H, R188H) have been linked to hypertension in humans in genome-wide association studies (GWAS) (Zhang, et al., 2013). In functional studies, reconstitution of arteries from Rgs2−/− mice with wild type human RGS2 blunted angiotensin II-induced vasoconstriction ex vivo whereas introduction of each of the aforementioned mutants individually failed to suppress this response (Phan, et al., 2019).
Cellular and animal models have provided further insights into physiological functions of many RGS proteins (Table 2). A knock-in mouse carrying Gnai2 alleles encoding a point mutation in Gαi2 that renders it resistant to the GAP activity of all RGS proteins (G184S) has been utilized to interrogate functions of RGS proteins in diverse physiological systems (Neubig, 2015). These mice exhibit systemic abnormalities including resistance to diet-induced obesity due to increased insulin sensitivity (Huang, et al., 2008) and predisposition to bradycardia (Fu, et al., 2007). Although Gαi2 (G184S) mice have not yet been studied in the context of asthma, investigation of these mice has revealed functions of RGS proteins in immunity and inflammation. B and T lymphocytes from Gαi2 (G184S) mice displayed surprisingly poor chemotaxis to chemokines in vitro and altered trafficking patterns in vivo, findings which were associated with abnormal architecture of thymus and peripheral lymph nodes and defective antibody generation (Hwang, et al., 2015; Hwang, et al., 2017). Others found that macrophages from Gαi2 (G184S) mice have altered cytokine production and are biased towards an anti-inflammatory (M2) phenotype (Vural, et al., 2019). Together these findings suggest that RGS proteins may contribute to asthma and allergic airway inflammation by controlling Gαi-dependent leukocyte responsiveness to chemoattractants and cytokine secretion.
Table 2.
RGS proteins in the lung
| Protein | Expression in airway cells | Human tissue Expression | Main findings |
|---|---|---|---|
| RGS2 | ASM Bronchial epithelium Mouse T lymphocytes |
Bone marrow Adrenal gland Appendix Esophagus Endometrium Gall bladder |
RGS2 expression in ASM and epithelial cells induced by LABAs and glucocorticoids Decreased RGS2 expression in allergen-challenged mice and human asthma patients compared to controls RGS2 negatively regulates intracellular Ca2+ flux in ASM and airway epithelial cells RGS2 negatively regulates ciliary beat frequency in airway epithelial cells and inhibits acid-induced MUC5AC secretion Rgs2−/− mice have spontaneous and allergen- or cytokine- induced AHR associated with variable airway cytokine levels and airway remodeling depending on model used Rgs2−/− mice have impaired antiviral responses associated with impaired T lymphocyte cytokine production SNPs associated with decreased RGS2 promoter activity in asthma patients |
| RGS3 | Mouse T lymphocytes |
Heart Fat Lung Gall bladder Kidney Spleen |
T cells depleted of RGS3 have increased chemotaxis Rgs3 (ΔRGS) mice challenged with HDM have increased numbers of airway leukocytes, increased airway epithelial remodeling and peribronchial smooth muscle mass Increased numbers of CD4+ and CD8+ T cells in BALF of Rgs3 (ΔRGS) mice associated with reduced cell numbers in mediastinal lymph nodes |
| RGS4 | ASM Airway epithelium |
Brain Adrenal gland Gall bladder Esophagus Endometrium Prostate |
Increased RGS4 expression in airway epithelium and ASM correlates with asthma severity RGS4 is required for mitogen-induced human ASM cell cycle progression and proliferation RGS4 suppresses MUC5AC expression in airway epithelial cells in vitro and in mice tracheas ex vivo ASM cells from RGS4 transgenic mice have blunted GPCR-evoked intracellular Ca2+ flux and MLC phosphorylation than controls, associated with reduced allergen-evoked AHR and airway cytokine levels in vivo Rgs4−/− mice exhibit reduced allergen-associated AHR associated with increased airway PGE2 levels RGS4 antagonist reduces allergen- and aspirin-induced AHR in association with increased airway PGE2 levels |
| RGS5 | ASM | Adrenal gland Thyroid Fat Heart Gall bladder Urinary bladder |
RGS5 higher in ASM from asthma patients and in allergen-challenged mice compared to controls RGS5 expression reduced in ASM cells treated with LABAs compared to untreated cells ASM cells from Rgs5−/− mice have higher intracellular Ca2+ flux than those from wild-type cells, associated with spontaneous AHR RGS5 overexpression in PCLS inhibits carbachol-induced contraction |
| RGS13 | Peripheral blood mononuclear cells | Lymph node Duodenum Appendix Lung Small intestine Gall bladder |
Higher RGS13 expression in PBMCs from children with nonallergic asthma than cells from healthy controls |
| RGS16 | Mouse T lymphocytes | Thyroid Lung Gall bladder Lymph node Placenta Fat |
Increased RGS16 expression in activated and Th2 polarized T cells than in naïve cells In vitro-differentiated Th1 and Th2 cells from Rgs16−/− mice exhibit increased chemotaxis to chemokines compared to those from wild-type controls Rgs16−/− mice have increased Th2-mediated lung inflammation following Schistosoma egg antigen (SEA) challenge |
Expression patterns reflect top six tissues listed in descending order (Fagerberg, et al., 2014).
4.4. RGS proteins and allergic airway inflammation
Several RGS proteins are expressed in lung resident leukocytes and in inflammatory cells recruited to the airways in response to allergens. RGS2, RGS3, RGS13, and RGS16 have been detected in T lymphocytes. T cells from Rgs2−/− mice had impaired proliferation and IL-2 production in response to T cell-receptor or phorbol 12-myristate 13-acetate (PMA) stimulation compared to controls and reduced inflammatory responses to cutaneous infection with lymphocytic choriomeningitis (LCMV) (Oliveira-Dos-Santos, et al., 2000). Although the detailed mechanisms underlying this phenotype were not examined, these results suggest that RGS2 has a function in T-cell dependent antiviral immunity.
RGS13 is expressed in mast cells and inhibits IgE-mediated degranulation through mechanisms independent of its GAP activity (Bansal, et al., 2008a). Rgs13−/− mice had exaggerated IgE-dependent systemic and cutaneous anaphylaxis in vivo. However, the function of RGS13 in experimental asthma models remain unknown. In limited human studies, RGS2 and RGS13 expression was lower in peripheral blood mononuclear cells from subjects with asthma than in those from healthy controls (Raedler, et al., 2015; Xie, et al., 2012). Two variants in the promoter region of RGS2 that reduce promoter activity were over-represented in a cohort of patients with asthma relative to the healthy control group (Jiang, et al., 2015).
Several RGS proteins modulate leukocyte chemotaxis and immune cell trafficking to the lung in allergic responses. In addition to its role in mast cell activation, RGS13 inhibits mast cell chemotaxis to chemokines (Bansal, et al., 2008b). RGS3 is expressed in activated Th2 cells, and RGS3 knockdown by RNAi in T lymphocyte cell lines enhanced chemotaxis to the chemokines CXCL12 and CCL21 (Williams, et al., 2013). Lungs of HDM-challenged mice lacking the RGS domain of RGS3 [RGS3(ΔRGS)] had increased numbers of Th2 cells and eosinophils in the airways compared to controls and contained perivascular and peribronchial lymphoid aggregates (Williams, et al., 2013). Airway levels of type 2 cytokines were equivalent in WT and RGS3(ΔRGS) mice, but airway resistance was not examined in this study.
RGS16 has been detected in naïve mouse and human T lymphocytes and in higher amounts in activated (Beadling, et al., 1999) or Th2-polarized T cells (Shankar, et al., 2012). Th2 cells from Rgs16−/− mice exhibit increased chemotaxis in vitro towards CCL17 gradients than those from WT mice (Shankar, et al., 2012). These mice displayed increased lung fibrosis and granulomatous inflammation following challenge with Schistosoma mansoni egg antigen (SEA), which elicits a strong Th2-dependent immune response, compared to WT counterparts. Conversely, in a separate study of transgenic mice overexpressing RGS16 in T lymphocytes, numbers of adoptively transferred transgenic T cells were decreased in lungs of recipients following ovalbumin (OVA) challenge compared to wild-type cells, a result associated with diminished AHR (Lippert, et al., 2003). Together these findings imply that RGS16 controls Th2-dependent allergic airway inflammation and AHR.
5. Roles of RGS proteins in lung structural cells and AHR
Transcripts for RGS1–5, RGS8, and RGS10 have been detected in ASM cells isolated from healthy donors (Yang, et al., 2011). RGS2, RGS4, and RGS5 expression has been reported in mouse ASM (Balenga, et al., 2014; Xie, et al., 2012). RGS2 and RGS4 were detected within the bronchial epithelium of human lung biopsies by immunohistochemistry (Wong, et al., 2020; Xie, et al., 2012) and in cultured human airway epithelial cells (Holden, et al., 2014; Song, et al., 2009; Wong, et al., 2020). RGS protein expression in other lung cell types including endothelium, vascular smooth muscle, and alveolar pneumocytes has not been explored. In this section, we discuss potential functions of individual RGS proteins in asthma based on the results of mouse experimental models and human studies; we focus on the R4 subfamily since other RGS subfamilies remain unexplored in the lung.
5.1. RGS2
RGS2 expression was decreased in lungs of OVA-challenged mice compared to naïve mice; likewise, RGS2 expression in lung biopsies from asthma patients was lower than in those from healthy controls (Xie, et al., 2012). In both ASM and bronchial epithelial cells, RGS2 expression is synergistically induced by treatment with LABAs and glucocorticoids in vitro, and LABA/glucocorticoid pretreatment of either cell type reduced intracellular Ca2+ flux in response to GPCR agonists in an RGS2-dependent manner (Holden, et al., 2011; Holden, et al., 2014). Tracheal ASM cells isolated from Rgs2−/− mice displayed increased intracellular Ca2+ responses to ACh, and precision cut lung slices (PCLS) from these mice exhibited more MCh-induced bronchoconstriction ex vivo compared to controls (Xie, et al., 2012). Accordingly, several independent studies have shown that Rgs2−/− mice have increased airway resistance and lower compliance at homeostasis and following allergen (HDM, OVA), cytokine (IL-13), or lipopolysaccharide (LPS) challenge (George, et al., 2017; George, et al., 2018; Jiang, et al., 2015; Xie, et al., 2012) relative to wild-type controls. Taken together, these findings suggest that decreased RGS2 expression in the context of allergic airway inflammation and chronic anti-asthma therapy predisposes to AHR as a result of enhanced bronchoconstrictor signaling in ASM.
Studies have revealed other functions of RGS2 in airway cells. RGS2 knockdown in human airway epithelial cells enhanced ATP-evoked increases in intracellular Ca2+ and ciliary beat frequency in vitro (Nlend, et al., 2002). Others found that RGS2 knockdown increased secretion of the critical mucous protein MUC5AC mediated by the acid sensor ovarian cancer G-protein-coupled receptor 1 (OGR1) (Liu, et al., 2013). In mouse models of acute OVA challenge (Xie, et al., 2012) or those featuring chronic, repetitive allergen (HDM) challenges to the lower respiratory tract, eosinophil numbers and peribronchial smooth muscle thickness were increased in the airways of HDM-challenged Rgs2−/− mice compared to controls although overall histological appearance and mucous content of lungs were unchanged (George, et al., 2017). Thus, RGS2 may regulate airway remodeling and allergen clearance, both processes critical to the development of AHR.
5.2. RGS4
In contrast to RGS2, RGS4 expression is significantly increased in subjects with asthma compared to healthy controls, with expression that correlates with clinical disease severity (Wong, et al., 2020). The extent of RGS4 coverage increases proportionally with the degree of functional lung impairment; specifically, decreased FEV1, and with medication (corticosteroid) requirements for symptom control. Early mouse studies suggested that increased RGS4 expression in asthma might reduce susceptibility to AHR. Indeed, transgenic (Tg) mice featuring a bacterial artificial chromosome (BAC) that incorporates the native RGS4 promoter, which overexpressed RGS4 in both bronchial epithelium and ASM, had lower airway resistance at homeostasis and following Aspergillus fumigatus (Af) challenge in the acute model of experimental asthma compared to WT controls (Madigan, et al., 2018). While RGS4 overexpression had modest effects on allergic airway inflammation (reduced levels of type 2 cytokines IL-5 and IL-13), increased RGS4 expression was associated blunted PCLS contraction ex vivo and bronchoconstrictor signaling (Ca2+ flux, MLC phosphorylation) in ASM cells in vitro, suggesting that these mechanisms contributed to reduced AHR.
Unexpectedly, however, Rgs4−/− mice also had reduced AHR following Af challenge despite exhibiting: 1) increased airway contraction in PCLS ex vivo; 2) augmented ASM Ca2+ responses to bronchoconstrictors in vitro; 3) increased type 2 cytokines (IL-33, IL-13) in the airways (Wong, et al., 2020). The reduced susceptibility to AHR in the absence of RGS4 was associated with increased airway levels of PGE2; pretreatment of mice with indomethacin, which prevents PGE2 biosynthesis, restored AHR. RGS4 inhibited PAR2-mediated PGE2 secretion from human respiratory epithelial cells treated with protease-containing allergens in vitro (Wong, et al., 2020). In addition to its role as a bronchodilator, PGE2 has anti-inflammatory actions through its inhibition of MC, Th2, and ILC2 functions (Peebles, 2019). In this case, an RGS4 antagonist enhanced PAR2-dependent bronchorelaxation of airways in PCLS from naïve mice, a result suggesting that increased bronchodilation contributed to reduced lung resistance in Rgs4−/− mice in the model of experimental asthma (Wong, et al., 2020).
RGS4 may also affect other airway functions contributing to asthma pathogenesis. RGS4 negatively regulated ATP- or PGE2-induced MUC5AC secretion by airway epithelial cells in vitro (Song, et al., 2009a; Song, et al., 2009b). As discussed above, ASM hypertrophy and hyperplasia leads to increased peribronchial smooth muscle mass in chronic asthma (James, et al., 2012). RGS4 has an impact on ASM proliferation through several mechanisms including regulation of phosphoinositide 3-kinases (PI3Ks) independent of its GAP activity (Damera, et al., 2012). Some RGS proteins inhibit PI3K activity in several cell types through direct interactions with its regulatory p85α subunit (Bansal, et al., 2008a; Liang, et al., 2009). RGS4 coimmunoprecipitated with p85α in ASM cells, and RGS4 knockdown by siRNA reduced PI3K-dependent ASM proliferation induced by platelet-derived growth factor (PDGF) in association with decreased PI3 kinase activity (Damera, et al., 2012). Thus, the mechanism(s) by which RGS4 regulates ASM growth require further studies.
5.3. RGS5
Within the lung, RGS5 is found primarily in bronchial smooth muscle, with expression that is increased in ASM of lung biopsies from patients who died from acute asthma exacerbations compared to those from healthy donors (Yang, et al., 2012). Likewise, RGS5 expression was higher in allergen-challenged mice compared to naïve control mice (Yang, et al., 2012). In contrast to RGS2, RGS5 transcripts were lower in β2-agonist-treated human ASM than in untreated cells, and corticosteroids had no effect on RGS5 expression (Yang, et al., 2011). Human ASM cells treated with RGS5 siRNA increased peak Ca2+ flux in response to bradykinin and thrombin, but not endothelin (Yang, et al., 2011). Mouse tracheal ASM cells isolated from Rgs5−/− mice displayed increased ACh- or bradykinin-evoked intracellular Ca2+ flux whereas responses to some agonists (serotonin, thrombin) were equivalent to those seen in WT cells (Balenga, et al., 2014). Although further studies are needed to clarify these findings, these results suggest that RGS5 selectively regulates specific bronchoconstrictor-inducing GPCRs in ASM. While RGS proteins target G-proteins downstream of GPCRs, direct interactions with GPCRs may be one mechanism that contributes to RGS selectivity (Ghil, et al., 2014; Kim & Ghil, 2020). Similar to the phenotype of RGS2-deficient mice, Rgs5−/− mice develop spontaneous AHR at homeostasis, a finding that is exacerbated further by acute allergen (Af) challenge (Balenga, et al., 2014). However, in contrast to Rgs2−/− mice, these mice exhibited Af-induced airway inflammation (e.g. airway eosinophil numbers, mucin expression, type 2 cytokines) that was nearly indistinguishable from Af-challenged WT mice. Airway contraction in PCLS from naïve Rgs5−/− mice was significantly greater than that in slices from WT mice (Balenga, et al., 2014). In a separate study, lentiviral-mediated overexpression of RGS5 in PCLS from naïve Balb/c mice inhibited carbachol (a muscarinic receptor agonist similar to MCh)-induced contraction (Yang, et al., 2012). Together, these results suggest that RGS5 regulates AHR primarily by inhibiting ASM contraction rather than by reducing allergic inflammation.
6. G-protein-coupled receptor kinases (GRKs) and arrestins in asthma
6.1. GRK mechanisms of action
G-protein-coupled receptor kinases (GRKs) are another important group of intracellular regulators of ASM signaling and contraction. GRKs phosphorylate GPCRs to facilitate the binding of arrestin proteins. Arrestin binding in turn prompts receptor internalization and cessation of signaling (Gurevich & Gurevich, 2019). Conversely, arrestins may also activate Erk in a G-protein-independent manner (Gurevich & Gurevich, 2020). Ca2+-independent MLC phosphorylation by Erk has been reported, suggesting a mechanism whereby arrestins regulate contraction (Mansfield, et al., 2000). GRKs may also negatively regulate GPCR signaling through non-canonical functions. Although RGS homology domains have been identified in GRK2/3 (Carman, et al., 1999; Sterne-Marr, et al., 2004), both proteins lack appreciable GAP activity and are currently considered to regulate Gαq activity primarily by blocking its interactions with effectors. For example, GRK2 overexpression inhibited Gq-mediated PLCβ activation in both in vitro and in cell-based assays (Carman, et al., 1999).
6.2. GRKs in the lung
A limited number of studies have addressed the functions of GRKs in the lung. Variable expression of GRK2 and GRK5 has been reported in human and mouse bronchial smooth muscle (Kong, et al., 2008; W. C. Wang, et al., 2008). GRK2 overexpression in human ASM cells reduced β2 adrenergic receptor (AR)-induced cAMP accumulation (McGraw & Liggett, 1997; Penn, et al., 1998). Others found that GRK2 silencing by siRNA enhanced cAMP induced by β2-AR stimulation but had no effect on responses to PGE2 or adenosine A2b receptor-selective agonists (Kong, et al., 2008). GRK2 is also expressed in human and mouse mast cells (Guo, et al., 2011) GRK2 silencing either by gene deletion in mice or by shRNA in human cultured mast cells strongly inhibited IgE-mediated degranulation and cytokine production (Subramanian, et al., 2014). In contrast, overexpression of full-length WT or kinase-dead GRK2 or its RGS homology domain increased IgE-mediated degranulation of mouse mast cells by 50% compared to controls. This phenotype was associated with increased phosphorylation of Syk kinase, a critical effector downstream of the IgE receptor (FcεR1).
These studies suggest that GRK2 expressed in airway cells may contribute to the pathogenesis of AHR through multiple mechanisms; however, further advances have been limited by the lethality of genome-wide Grk2 deletion in mice. Tissue-specific GRK2 knockout mice have recently been generated that should facilitate future examination of its functions in allergen-induced AHR models (Steury, et al., 2018).
Human pharmacogenomic studies have also indicated that GRK variants may affect responses to asthma therapy. One variant of GRK5 (Q41L) was more prevalent in ASM cells from patients with asthma and African ancestry relative to other ethnic populations (W. C. Wang, et al., 2008). Overexpression of GRK5 (Q41L) in ASM cells enhanced β2AR phosphorylation and internalization compared to controls, results suggesting that a polymorphism in GRK5 may contribute to the development of resistance to β2-agonist treatment.
6.3. β-arrestin functions in asthma
The specific roles of β-arrestins in allergic asthma have been studied in mouse models. β2arr−−/−mice sensitized and challenged with OVA had lower AHR than WT controls, a decrease associated with reduced numbers of eosinophil and Th2 cells and type 2 cytokines in the airways (Nichols, et al., 2012; Walker, et al., 2003). CD4+ T cells isolated from bronchoalveolar lavage fluid (BALF) of β2arr−−/− mice exhibited decreased chemotaxis to CCL22 compared to controls, a result suggesting that β-arrestin-2 promotes inflammation in allergic asthma by increasing lymphocyte recruitment to the lung. A subsequent study of bone marrow chimeric mice demonstrated that engraftment of bone marrow from wild type mice onto irradiated β2arr−−/− mice was sufficient to elicit allergic airway inflammation and remodeling following OVA challenge but not to elicit AHR. Development of AHR in response to OVA challenge or intratracheal installation of IL-13 required expression of β-arrestin-2 on both hematopoietic and lung structural cells (Hollingsworth, et al., 2010). As β-arrestin-2 recruitment to β2-adrenergic receptors leads to receptor downregulation due to internalization (Lohse, et al., 1990), its absence in ASM cells may prevent β2-adrenergic receptor desensitization and thereby reduce AHR by promoting bronchorelaxation. Separate studies demonstrated that β-arrestin-2 knockdown by siRNA in ASM increased β2-adrenergic receptor-mediated cAMP generation but did not affect responses to PGE2 (Deshpande, et al., 2008). Accordingly, while tracheal rings from β2arr−−/− mice contracted to an equivalent extent as those from WT mice in response to methacholine, β2 adrenergic receptor-mediated (but not PGE2-mediated) relaxation was increased (Deshpande, et al., 2008). These results suggest that β-arrestin-2 regulates AHR by promoting airway relaxation to certain (but not all) bronchodilators without directly affecting airway contraction.
3. Conclusions/therapeutic implications
Here we have emphasized the primary GPCR signaling pathways inducing ASM contraction and relaxation and discussed regulation of these pathways by G-proteins, RGS proteins, and GRK/β-arrestin-2. These mechanisms have important implications for the treatment of asthma and other respiratory diseases. While current prophylactic therapies are aimed at alleviating allergic inflammation (e.g. corticosteroids, anti-cytokine or anti-IgE antibodies), these approaches may only be effective in specific asthma endotypes and/or are limited by serious side effects or cost (McGregor, et al., 2019). Current therapies directly targeting ASM are squarely focused on promoting bronchodilation (e.g. β2-agonists), a process that is subject to “tachyphylaxis”, or resistance to therapy, due to receptor downregulation (Amrani & Bradding, 2017). Overuse of β2-agonists has been linked to fatalities in patients with asthma, and LABA monotherapy for asthma is currently contraindicated (Pera & Penn, 2016).
Given the breadth of contraction-inducing mediators acting on GPCRs, targeting G-proteins or downstream effectors, or indirectly by increasing activity and/or expression of RGS proteins/GRKs, represents a new alternative strategy. The efficacy of Gq inhibitors in preclinical models demonstrates the feasibility and promise of this approach. Although bronchoprotective RGS proteins expressed in ASM such as RGS2 and RGS5 represent potential therapeutic targets, both proteins are subject to rapid turnover due to proteasome-mediated degradation (Alqinyah & Hooks, 2018). Preliminary studies have suggested that increasing steady state RGS protein levels might be a potential strategy to enhance their activity. A natural compound screen revealed that indolactam V, which is a known activator of PKC, induced sustained upregulation of RGS2 in a PKCβ-dependent manner (Raveh, et al., 2014). In a separate study, RGS2 degradation was shown to be mediated by the F-box Only Protein 44 (FBXO44), which recruits RGS2 to the proteasome complex for ubiquitination (Sjogren, et al., 2015). RGS2 interacts with FBXO44 through a stretch of residues in the amino terminus (McNabb, et al., 2020). Small molecule inhibitors of RGS2-FBXO44 interactions, which would be predicted to stabilize steady-state levels of RGS2, might be developed. Systemic effects of such drugs, or other compounds predicted to increase RGS levels such as proteasome inhibitors bortezomib or carfilzomib, which are approved for the treatment of cancers including multiple myeloma (Okazuka & Ishida, 2018), might be limited by inhalational administration for the treatment of respiratory diseases. Finally, RGS4 represents a unique target for treatment of AHR. The small molecule CCG203769, which is a thiol-reactive compound that covalently binds to RGS4 and irreversibly inhibits its activity (Blazer, et al., 2015), reduced AHR in experimental models of allergen- and aspirin-associated asthma (Wong, et al., 2020). Further studies of the effects of RGS4 inhibition in models of chronic, established asthma will be needed to determine feasibility of this approach.
Acknowledgements
Funding: This research was funded in part by NIAID/NIH Intramural Program (Z01-AI-000746) (K.M.D); National Center for Translational Science (UL1TR003017) and National Heart, Lung and Blood Institute (1P01HL114471) grants (R.A.P). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- AC
adenylyl cyclase
- Af
Aspergillus fumigatus
- AHR
airways hyperresponsiveness
- ANS
autonomic nervous system
- AR
adrenergic receptor
- ASM
airway smooth muscle
- BALF
bronchoalveolar lavage fluid
- cAMP
cyclic adenosine monophosphate
- CPI-17
C-potentiated inhibitor protein of 17 kDa (protein phosphatase 1)
- DAG
diacylglycerol
- DEP
disheveled-EGL10-Pleckstrin homology domain
- Epac
exchange protein directly activated by cAMP
- FcεR1
high-affinity IgE receptor
- GAPs
GTPase activating proteins
- GDP
guanosine diphosphate
- GEFs
guanine nucleotide exchange factors
- GPCRs
G protein-coupled receptors
- GRKs
G protein-coupled receptor kinases
- GTP
guanosine triphosphate
- HDM
house dust mite
- ILC2
type 2 innate lymphoid cells
- IP3
inositol-1,4,5-trisphosphate
- LABA
long-acting β2-adrenoreceptor agonists
- MLCK
myosin light chain kinase
- MYPT-1
myosin light chain phosphatase 1
- Orai1
Ca2+-activated Ca2+ membrane channel protein
- OVA
ovalbumin
- PARs
protease-activated receptors
- PLCβ
phospholipase C beta
- PCLS
precision cut lung slices
- PDEs
phosphodiesterases
- PGE2
prostaglandin E2
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKA
protein kinase A
- PKC
protein kinase C
- PLC
phospholipase C
- PTB
phosphotyrosine binding domain
- R9AP
RGS9 Anchoring Protein
- R7BP
R7 Binding Protein
- RBD
Ras binding domain
- RGS
Regulator of G protein signaling
- RyR
ryanodine receptor
- ROCC
receptor- operated Ca2+ channels
- ROCK
RhoA-associated coiled-coil forming kinase
- SEA
Schistosoma mansoni egg antigen
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+-ATPase
- SABA
short-acting β2-adrenoreceptor agonists
- SOCC
store-operated Ca2+ channels
- SOCE
store-operated Ca2+ entry
- SR
sarcoplasmic reticulum
- STIM1
SR protein stromal interaction molecule-1
- Th2
T helper type 2
- TSLP
thymic stromal lymphopoietin
- VOCC
voltage-operated Ca2+ channels
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
References
- Alqinyah M, & Hooks SB (2018). Regulating the regulators: Epigenetic, transcriptional, and post-translational regulation of RGS proteins. Cell Signal, 42, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani Y, & Bradding P (2017). beta2-Adrenoceptor Function in Asthma. Adv Immunol, 136, 1–28. [DOI] [PubMed] [Google Scholar]
- An SS, Mitzner W, Tang WY, Ahn K, Yoon AR, Huang J, et al. B. (2016). An inflammation-independent contraction mechanophenotype of airway smooth muscle in asthma. J Allergy Clin Immunol, 138, 294–297 e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthracopoulos MB, & Everard ML (2020). Asthma: A Loss of Post-natal Homeostatic Control of Airways Smooth Muscle With Regression Toward a Pre-natal State. Front Pediatr, 8, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balenga NA, Jester W, Jiang M, Panettieri RA Jr., & Druey KM (2014). Loss of regulator of G protein signaling 5 promotes airway hyperresponsiveness in the absence of allergic inflammation. J Allergy Clin Immunol, 134, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal G, Xie Z, Rao S, Nocka KH, & Druey KM (2008a). Suppression of immunoglobulin E-mediated allergic responses by regulator of G protein signaling 13. Nat Immunol, 9, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal G, DiVietro JA, Kuehn HS, Rao S, Nocka KH, Gilfillan AM, et al. (2008b). RGS13 controls g protein-coupled receptor-evoked responses of human mast cells. J Immunol, 181, 7882–7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (1992). Effect of bradykinin on airway function. Agents Actions Suppl, 38 ( Pt 3), 432–438. [PubMed] [Google Scholar]
- Barnes PJ (2013). Theophylline. Am J Respir Crit Care Med, 188, 901–906. [DOI] [PubMed] [Google Scholar]
- Bastin G, Singh K, Dissanayake K, Mighiu AS, Nurmohamed A, & Heximer SP (2012). Amino-terminal cysteine residues differentially influence RGS4 protein plasma membrane targeting, intracellular trafficking, and function. J Biol Chem, 287, 28966–28974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beadling C, Druey KM, Richter G, Kehrl JH, & Smith KA (1999). Regulators of G protein signaling exhibit distinct patterns of gene expression and target G protein specificity in human lymphocytes. J Immunol, 162, 2677–2682. [PubMed] [Google Scholar]
- Berman DM, Kozasa T, & Gilman AG (1996). The GTPase-activating protein RGS4 stabilizes the transition state for nucleotide hydrolysis. J Biol Chem, 271, 27209–27212. [DOI] [PubMed] [Google Scholar]
- Berman DM, Wilkie TM, & Gilman AG (1996). GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell, 86, 445–452. [DOI] [PubMed] [Google Scholar]
- Billington CK, Ojo OO, Penn RB, & Ito S (2013). cAMP regulation of airway smooth muscle function. Pulm Pharmacol Ther, 26, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billington CK, & Penn RB (2003). Signaling and regulation of G protein-coupled receptors in airway smooth muscle. Respir Res, 4, 2. [PMC free article] [PubMed] [Google Scholar]
- Blazer LL, Storaska AJ, Jutkiewicz EM, Turner EM, Calcagno M, Wade SM, et al. (2015). Selectivity and anti-Parkinson’s potential of thiadiazolidinone RGS4 inhibitors. ACS Chem Neurosci, 6, 911–919. [DOI] [PubMed] [Google Scholar]
- Boonpiyathad T, Sozener ZC, Satitsuksanoa P, & Akdis CA (2019). Immunologic mechanisms in asthma. Semin Immunol, 46, 101333. [DOI] [PubMed] [Google Scholar]
- Bosse Y, Rousseau E, Amrani Y, & Grunstein MM (2013). Smooth muscle hypercontractility in airway hyperresponsiveness: innate, acquired, or nonexistent? J Allergy (Cairo), 2013, 938046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, et al. , T. (1999). Selective regulation of Galpha(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J Biol Chem, 274, 34483–34492. [DOI] [PubMed] [Google Scholar]
- Cervantes-Villagrana RD, Adame-Garcia SR, Garcia-Jimenez I, Color-Aparicio VM, Beltran-Navarro YM, Konig GM, et al. (2019). Gbetagamma signaling to the chemotactic effector P-REX1 and mammalian cell migration is directly regulated by Galphaq and Galpha13 proteins. J Biol Chem, 294, 531–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Guo L, Hadas J, Gutowski S, Sprang SR, & Sternweis PC (2012). Activation of p115-RhoGEF requires direct association of Galpha13 and the Dbl homology domain. J Biol Chem, 287, 25490–25500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuinas A, Garcia-Morales V, Vina D, Gil-Longo J, & Campos-Toimil M (2016). Activation of PKA and Epac proteins by cyclic AMP depletes intracellular calcium stores and reduces calcium availability for vasoconstriction. Life Sci, 155, 102–109. [DOI] [PubMed] [Google Scholar]
- D’Urzo KA, Mok F, & D’Urzo AD (2020). Variation Among Spirometry Interpretation Algorithms. Respir Care. [DOI] [PubMed] [Google Scholar]
- Damera G, Druey KM, Cooper PR, Krymskaya VP, Soberman RJ, Amrani Y, et al. (2012). An RGS4-mediated phenotypic switch of bronchial smooth muscle cells promotes fixed airway obstruction in asthma. PLoS One, 7, e28504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BE, Blais CM, & Cockcroft DW (2018). Methacholine challenge testing: comparative pharmacology. J Asthma Allergy, 11, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries L, Mousli M, Wurmser A, & Farquhar MG (1995). GAIP, a protein that specifically interacts with the trimeric G protein G alpha i3, is a member of a protein family with a highly conserved core domain. Proc Natl Acad Sci U S A, 92, 11916–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande DA, Theriot BS, Penn RB, & Walker JK (2008). Beta-arrestins specifically constrain beta2-adrenergic receptor signaling and function in airway smooth muscle. FASEB J, 22, 2134–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman HG, Song J, Ma D, Courchesne WE, & Thorner J (1996). Sst2, a negative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae: expression, localization, and genetic interaction and physical association with Gpa1 (the G-protein alpha subunit). Mol Cell Biol, 16, 5194–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druey KM, Blumer KJ, Kang VH, & Kehrl JH (1996). Inhibition of G-protein-mediated MAP kinase activation by a new mammalian gene family. Nature, 379, 742–746. [DOI] [PubMed] [Google Scholar]
- Eto M (2009). Regulation of cellular protein phosphatase-1 (PP1) by phosphorylation of the CPI-17 family, C-kinase-activated PP1 inhibitors. J Biol Chem, 284, 35273–35277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics, 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Sun Q, & Roth M (2020). Immunologic and Non-Immunologic Mechanisms Leading to Airway Remodeling in Asthma. Int J Mol Sci, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Huang X, Piao L, Lopatin AN, & Neubig RR (2007). Endogenous RGS proteins modulate SA and AV nodal functions in isolated heart: implications for sick sinus syndrome and AV block. Am J Physiol Heart Circ Physiol, 292, H2532–2539. [DOI] [PubMed] [Google Scholar]
- Gauvreau GM, Watson RM, & O’Byrne PM (1999). Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am J Respir Crit Care Med, 159, 31–36. [DOI] [PubMed] [Google Scholar]
- George T, Bell M, Chakraborty M, Siderovski DP, Giembycz MA, & Newton R (2017). Protective Roles for RGS2 in a Mouse Model of House Dust Mite-Induced Airway Inflammation. PLoS One, 12, e0170269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George T, Chakraborty M, Giembycz MA, & Newton R (2018). A bronchoprotective role for Rgs2 in a murine model of lipopolysaccharide-induced airways inflammation. Allergy Asthma Clin Immunol, 14, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghil S, McCoy KL, & Hepler JR (2014). Regulator of G protein signaling 2 (RGS2) and RGS4 form distinct G protein-dependent complexes with protease activated-receptor 1 (PAR1) in live cells. PLoS One, 9, e95355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Chiba Y, Matsusue K, Hattori Y, Maitani Y, Sakai H, et al. (2010). The proximal STAT6 and NF-kappaB sites are responsible for IL-13- and TNF-alpha-induced RhoA transcriptions in human bronchial smooth muscle cells. Pharmacol Res, 61, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan R, Xu X, Chen M, Hu H, Ge H, Wen S, et al. (2013). Advances in the studies of roles of Rho/Rho-kinase in diseases and the development of its inhibitors. Eur J Med Chem, 70, 613–622. [DOI] [PubMed] [Google Scholar]
- Guida G, & Riccio AM (2019). Immune induction of airway remodeling. Semin Immunol, 46, 101346. [DOI] [PubMed] [Google Scholar]
- Guo Q, Subramanian H, Gupta K, & Ali H (2011). Regulation of C3a receptor signaling in human mast cells by G protein coupled receptor kinases. PLoS One, 6, e22559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, & Gurevich EV (2019). GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front Pharmacol, 10, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, & Gurevich EV (2020). Biased GPCR signaling: Possible mechanisms and inherent limitations. Pharmacol Ther, 211, 107540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler JR, & Gilman AG (1992). G proteins. Trends Biochem Sci, 17, 383–387. [DOI] [PubMed] [Google Scholar]
- Holden NS, Bell MJ, Rider CF, King EM, Gaunt DD, Leigh R, et al. (2011). beta2-Adrenoceptor agonist-induced RGS2 expression is a genomic mechanism of bronchoprotection that is enhanced by glucocorticoids. Proc Natl Acad Sci U S A, 108, 19713–19718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden NS, George T, Rider CF, Chandrasekhar A, Shah S, Kaur M, et al. (2014). Induction of regulator of G-protein signaling 2 expression by long-acting beta2-adrenoceptor agonists and glucocorticoids in human airway epithelial cells. J Pharmacol Exp Ther, 348, 12–24. [DOI] [PubMed] [Google Scholar]
- Hollingsworth JW, Theriot BS, Li Z, Lawson BL, Sunday M, Schwartz DA, et al. (2010). Both hematopoietic-derived and non-hematopoietic-derived {beta}-arrestin-2 regulates murine allergic airway disease. Am J Respir Cell Mol Biol, 43, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F, Haldeman BD, Jackson D, Carter M, Baker JE, & Cremo CR (2011). Biochemistry of smooth muscle myosin light chain kinase. Arch Biochem Biophys, 510, 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Charbeneau RA, Fu Y, Kaur K, Gerin I, MacDougald OA, & Neubig RR (2008). Resistance to diet-induced obesity and improved insulin sensitivity in mice with a regulator of G protein signaling-insensitive G184S Gnai2 allele. Diabetes, 57, 77–85. [DOI] [PubMed] [Google Scholar]
- Hwang IY, Park C, Harrison K, Boularan C, Gales C, & Kehrl JH (2015). An essential role for RGS protein/Galphai2 interactions in B lymphocyte-directed cell migration and trafficking. J Immunol, 194, 2128–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang IY, Park C, Harrison K, & Kehrl JH (2017). Normal Thymocyte Egress, T Cell Trafficking, and CD4(+) T Cell Homeostasis Require Interactions between RGS Proteins and Galphai2. J Immunol, 198, 2721–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AL, Elliot JG, Jones RL, Carroll ML, Mauad T, Bai TR, et al. (2012). Airway smooth muscle hypertrophy and hyperplasia in asthma. Am J Respir Crit Care Med, 185, 1058–1064. [DOI] [PubMed] [Google Scholar]
- Jayaraman M, Zhou H, Jia L, Cain MD, & Blumer KJ (2009). R9AP and R7BP: traffic cops for the RGS7 family in phototransduction and neuronal GPCR signaling. Trends Pharmacol Sci, 30, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Delmotte P, Aravamudan B, Pabelick CM, Prakash YS, & Sieck GC (2013). Effects of the inflammatory cytokines TNF-alpha and IL-13 on stromal interaction molecule-1 aggregation in human airway smooth muscle intracellular Ca(2+) regulation. Am J Respir Cell Mol Biol, 49, 601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Xie Y, Abel PW, Wolff DW, Toews ML, Panettieri RA, et al. (2015). Regulator of G-protein signaling 2 repression exacerbates airway hyper-responsiveness and remodeling in asthma. Am J Respir Cell Mol Biol, 53, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, & Ghil S (2020). Regulators of G-protein signaling, RGS2 and RGS4, inhibit protease-activated receptor 4-mediated signaling by forming a complex with the receptor and Galpha in live cells. Cell Commun Signal, 18, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimple AJ, Soundararajan M, Hutsell SQ, Roos AK, Urban DJ, Setola V, et al. (2009). Structural determinants of G-protein alpha subunit selectivity by regulator of G-protein signaling 2 (RGS2). J Biol Chem, 284, 19402–19411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelle MR, & Horvitz HR (1996). EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell, 84, 115–125. [DOI] [PubMed] [Google Scholar]
- Komi DEA, Mortaz E, Amani S, Tiotiu A, Folkerts G, & Adcock IM (2020). The Role of Mast Cells in IgE-Independent Lung Diseases. Clin Rev Allergy Immunol, 58, 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong KC, Gandhi U, Martin TJ, Anz CB, Yan H, Misior AM, et al. (2008). Endogenous Gs-coupled receptors in smooth muscle exhibit differential susceptibility to GRK2/3-mediated desensitization. Biochemistry, 47, 9279–9288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziol-White C, Johnstone TB, Corpuz ML, Cao G, Orfanos S, Parikh V, et al. (2020). Budesonide enhances agonist-induced bronchodilation in human small airways by increasing cAMP production in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol, 318, L345–L355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam M, Lamanna E, & Bourke JE (2019). Regulation of Airway Smooth Muscle Contraction in Health and Disease. Adv Exp Med Biol, 1124, 381–422. [DOI] [PubMed] [Google Scholar]
- Lambrecht BN, & Hammad H (2015). The immunology of asthma. Nat Immunol, 16, 45–56. [DOI] [PubMed] [Google Scholar]
- Lambrecht BN, Hammad H, & Fahy JV (2019). The Cytokines of Asthma. Immunity, 50, 975–991. [DOI] [PubMed] [Google Scholar]
- Lee K, Lee SH, & Kim TH (2020). The Biology of Prostaglandins and Their Role as a Target for Allergic Airway Disease Therapy. Int J Mol Sci, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LY, & Yu J (2014). Sensory nerves in lung and airways. Compr Physiol, 4, 287–324. [DOI] [PubMed] [Google Scholar]
- Liang G, Bansal G, Xie Z, & Druey KM (2009). RGS16 inhibits breast cancer cell growth by mitigating phosphatidylinositol 3-kinase signaling. J Biol Chem, 284, 21719–21727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert E, Yowe DL, Gonzalo JA, Justice JP, Webster JM, Fedyk ER, et al. (2003). Role of regulator of G protein signaling 16 in inflammation-induced T lymphocyte migration and activation. J Immunol, 171, 1542–1555. [DOI] [PubMed] [Google Scholar]
- Liu C, Li Q, Zhou X, Kolosov VP, & Perelman JM (2013). Regulator of G-protein signaling 2 inhibits acid-induced mucin5AC hypersecretion in human airway epithelial cells. Respir Physiol Neurobiol, 185, 265–271. [DOI] [PubMed] [Google Scholar]
- Liu C, Weng J, Wang D, Yang M, Jia M, & Wang W (2018). A Residue outside the Binding Site Determines the Galpha Binding Specificity of GoLoco Motifs. Biochemistry, 57, 6562–6569. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, & Lefkowitz RJ (1990). beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science, 248, 1547–1550. [DOI] [PubMed] [Google Scholar]
- Madigan LA, Wong GS, Gordon EM, Chen WS, Balenga N, Koziol-White CJ, et al. (2018). RGS4 Overexpression in Lung Attenuates Airway Hyperresponsiveness in Mice. Am J Respir Cell Mol Biol, 58, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahn K, Hirst SJ, Ying S, Holt MR, Lavender P, Ojo OO, et al. (2009). Diminished sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) expression contributes to airway remodelling in bronchial asthma. Proc Natl Acad Sci U S A, 106, 10775–10780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahn K, Ojo OO, Chadwick G, Aaronson PI, Ward JP, & Lee TH (2010). Ca(2+) homeostasis and structural and functional remodelling of airway smooth muscle in asthma. Thorax, 65, 547–552. [DOI] [PubMed] [Google Scholar]
- Mansfield PJ, Shayman JA, & Boxer LA (2000). Regulation of polymorphonuclear leukocyte phagocytosis by myosin light chain kinase after activation of mitogen-activated protein kinase. Blood, 95, 2407–2412. [PubMed] [Google Scholar]
- Masuho I, Balaji S, Muntean BS, Skamangas NK, Chavali S, Tesmer JJG, et al. (2020). A Global Map of G Protein Signaling Regulation by RGS Proteins. Cell, 183, 503–521 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthey M, Roberts R, Seidinger A, Simon A, Schroder R, Kuschak M, et al. (2017). Targeted inhibition of Gq signaling induces airway relaxation in mouse models of asthma. Sci Transl Med, 9. [DOI] [PubMed] [Google Scholar]
- McGraw DW, & Liggett SB (1997). Heterogeneity in beta-adrenergic receptor kinase expression in the lung accounts for cell-specific desensitization of the beta2-adrenergic receptor. J Biol Chem, 272, 7338–7344. [DOI] [PubMed] [Google Scholar]
- McGregor MC, Krings JG, Nair P, & Castro M (2019). Role of Biologics in Asthma. Am J Respir Crit Care Med, 199, 433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNabb HJ, Gonzalez S, Muli CS, & Sjogren B (2020). N-Terminal Targeting of Regulator of G Protein Signaling Protein 2 for F-Box Only Protein 44-Mediated Proteasomal Degradation. Mol Pharmacol, 98, 677–685. [DOI] [PubMed] [Google Scholar]
- Momotani K, & Somlyo AV (2012). p63RhoGEF: a new switch for G(q)-mediated activation of smooth muscle. Trends Cardiovasc Med, 22, 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance MR, Kreutz B, Tesmer VM, Sterne-Marr R, Kozasa T, & Tesmer JJ (2013). Structural and functional analysis of the regulator of G protein signaling 2-galphaq complex. Structure, 21, 438–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak AP, & Penn RB (2020). The proton-sensing receptor ovarian cancer G-protein coupled receptor 1 (OGR1) in airway physiology and disease. Curr Opin Pharmacol, 51, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig RR (2015). RGS-Insensitive G Proteins as In Vivo Probes of RGS Function. Prog Mol Biol Transl Sci, 133, 13–30. [DOI] [PubMed] [Google Scholar]
- Nichols HL, Saffeddine M, Theriot BS, Hegde A, Polley D, El-Mays T, et al. (2012). beta-Arrestin-2 mediates the proinflammatory effects of proteinase-activated receptor-2 in the airway. Proc Natl Acad Sci U S A, 109, 16660–16665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi KM, Sandberg MA, Kooijman AC, Martemyanov KA, Pott JW, Hagstrom SA, et al. (2004). Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature, 427, 75–78. [DOI] [PubMed] [Google Scholar]
- Nlend MC, Bookman RJ, Conner GE, & Salathe M (2002). Regulator of G-protein signaling protein 2 modulates purinergic calcium and ciliary beat frequency responses in airway epithelia. Am J Respir Cell Mol Biol, 27, 436–445. [DOI] [PubMed] [Google Scholar]
- Nunn C, Mao H, Chidiac P, & Albert PR (2006). RGS17/RGSZ2 and the RZ/A family of regulators of G-protein signaling. Semin Cell Dev Biol, 17, 390–399. [DOI] [PubMed] [Google Scholar]
- Okazuka K, & Ishida T (2018). Proteasome inhibitors for multiple myeloma. Jpn J Clin Oncol, 48, 785–793. [DOI] [PubMed] [Google Scholar]
- Oliveira-Dos-Santos AJ, Matsumoto G, Snow BE, Bai D, Houston FP, Whishaw IQ, et al. , (2000). Regulation of T cell activation, anxiety, and male aggression by RGS2. Proc Natl Acad Sci U S A, 97, 12272–12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peebles RS Jr. (2019). Prostaglandins in asthma and allergic diseases. Pharmacol Ther, 193, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn RB, Panettieri RA Jr., & Benovic JL (1998). Mechanisms of acute desensitization of the beta2AR-adenylyl cyclase pathway in human airway smooth muscle. Am J Respir Cell Mol Biol, 19, 338–348. [DOI] [PubMed] [Google Scholar]
- Pera T, & Penn RB (2016). Bronchoprotection and bronchorelaxation in asthma: New targets, and new ways to target the old ones. Pharmacol Ther, 164, 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan HTN, Jackson WF, Shaw VS, Watts SW, & Neubig RR (2019). Loss-of-Function Mutations in Human Regulator of G Protein Signaling RGS2 Differentially Regulate Pharmacological Reactivity of Resistance Vasculature. Mol Pharmacol, 96, 826–834. [DOI] [PubMed] [Google Scholar]
- Raedler D, Ballenberger N, Klucker E, Bock A, Otto R, Prazeres da Costa O, et al. (2015). Identification of novel immune phenotypes for allergic and nonallergic childhood asthma. J Allergy Clin Immunol, 135, 81–91. [DOI] [PubMed] [Google Scholar]
- Raveh A, Schultz PJ, Aschermann L, Carpenter C, Tamayo-Castillo G, Cao S, et al. (2014). Identification of protein kinase C activation as a novel mechanism for RGS2 protein upregulation through phenotypic screening of natural product extracts. Mol Pharmacol, 86, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Garcia J, Flores-Soto E, Carbajal-Garcia A, Sommer B, & Montano LM (2018). Maintenance of intracellular Ca2+ basal concentration in airway smooth muscle (Review). Int J Mol Med, 42, 2998–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman RW, Strock J, Hains MD, Cabanilla NJ, Lau KK, Siderovski DP, & Diverse-Pierluissi M (2005). RGS12 interacts with the SNARE-binding region of the Cav2.2 calcium channel. J Biol Chem, 280, 1521–1528. [DOI] [PubMed] [Google Scholar]
- Roscioni SS, Maarsingh H, Elzinga CR, Schuur J, Menzen M, Halayko AJ, et al. (2011). Epac as a novel effector of airway smooth muscle relaxation. J Cell Mol Med, 15, 1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross EM, & Wilkie TM (2000). GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem, 69, 795–827. [DOI] [PubMed] [Google Scholar]
- Sakai H, Suto W, Kai Y, & Chiba Y (2017). Mechanisms underlying the pathogenesis of hyper-contractility of bronchial smooth muscle in allergic asthma. J Smooth Muscle Res, 53, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki F, & Yokomizo T (2019). The leukotriene receptors as therapeutic targets of inflammatory diseases. Int Immunol, 31, 607–615. [DOI] [PubMed] [Google Scholar]
- Shankar SP, Wilson MS, DiVietro JA, Mentink-Kane MM, Xie Z, Wynn TA, et al. (2012). RGS16 attenuates pulmonary Th2/Th17 inflammatory responses. J Immunol, 188, 6347–6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siderovski DP, Hessel A, Chung S, Mak TW, & Tyers M (1996). A new family of regulators of G-protein-coupled receptors? Curr Biol, 6, 211–212. [DOI] [PubMed] [Google Scholar]
- Siderovski DP, & Willard FS (2005). The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci, 1, 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren B, Swaney S, & Neubig RR (2015). FBXO44-Mediated Degradation of RGS2 Protein Uniquely Depends on a Cullin 4B/DDB1 Complex. PLoS One, 10, e0123581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow BE, Brothers GM, & Siderovski DP (2002). Molecular cloning of regulators of G-protein signaling family members and characterization of binding specificity of RGS12 PDZ domain. Methods Enzymol, 344, 740–761. [DOI] [PubMed] [Google Scholar]
- Snow BE, Krumins AM, Brothers GM, Lee SF, Wall MA, Chung S, et al. (1998). A G protein gamma subunit-like domain shared between RGS11 and other RGS proteins specifies binding to Gbeta5 subunits. Proc Natl Acad Sci U S A, 95, 13307–13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song KS, Choi YH, Kim JM, Lee H, Lee TJ, & Yoon JH (2009a). Suppression of prostaglandin E2-induced MUC5AC overproduction by RGS4 in the airway. Am J Physiol Lung Cell Mol Physiol, 296, L684–692. [DOI] [PubMed] [Google Scholar]
- Song KS, Kim HJ, Kim K, Lee JG, & Yoon JH (2009b). Regulator of G-protein signaling 4 suppresses LPS-induced MUC5AC overproduction in the airway. Am J Respir Cell Mol Biol, 41, 40–49. [DOI] [PubMed] [Google Scholar]
- Soundararajan M, Willard FS, Kimple AJ, Turnbull AP, Ball LJ, Schoch GA, et al. (2008). Structural diversity in the RGS domain and its interaction with heterotrimeric G protein alpha-subunits. Proc Natl Acad Sci U S A, 105, 6457–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterne-Marr R, Dhami GK, Tesmer JJ, & Ferguson SS (2004). Characterization of GRK2 RH domain-dependent regulation of GPCR coupling to heterotrimeric G proteins. Methods Enzymol, 390, 310–336. [DOI] [PubMed] [Google Scholar]
- Steury MD, Kang HJ, Lee T, Lucas PC, McCabe LR, & Parameswaran N (2018). G protein-coupled receptor kinase-2-deficient mice are protected from dextran sodium sulfate-induced acute colitis. Physiol Genomics, 50, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassheim D, Gerasimovskaya E, Irwin D, Dempsey EC, Stenmark K, & Karoor V (2019). RhoGTPase in Vascular Disease. Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian H, Gupta K, Parameswaran N, & Ali H (2014). Regulation of FcinRI signaling in mast cells by G protein-coupled receptor kinase 2 and its RH domain. J Biol Chem, 289, 20917–20927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney D, Hollins F, Gomez E, Mistry R, Saunders R, Challiss RA, et al. (2015). No evidence for altered intracellular calcium-handling in airway smooth muscle cells from human subjects with asthma. BMC Pulm Med, 15, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, et al. (2004). A novel Galphaq/11-selective inhibitor. J Biol Chem, 279, 47438–47445. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Berman DM, Gilman AG, & Sprang SR (1997). Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell, 89, 251–261. [DOI] [PubMed] [Google Scholar]
- Upham JW, & Chung LP (2018). Optimising treatment for severe asthma. Med J Aust, 209, S22–S27. [DOI] [PubMed] [Google Scholar]
- Vural A, Nabar NR, Hwang IY, Sohn S, Park C, Karlsson MCI, et al. (2019). Galphai2 Signaling Regulates Inflammasome Priming and Cytokine Production by Biasing Macrophage Phenotype Determination. J Immunol, 202, 1510–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JK, Fong AM, Lawson BL, Savov JD, Patel DD, Schwartz DA, & Lefkowitz RJ (2003). Beta-arrestin-2 regulates the development of allergic asthma. J Clin Invest, 112, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WC, Mihlbachler KA, Bleecker ER, Weiss ST, & Liggett SB (2008). A polymorphism of G-protein coupled receptor kinase5 alters agonist-promoted desensitization of beta2-adrenergic receptors. Pharmacogenet Genomics, 18, 729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Wang L, & Zheng YM (2020). Canonical Transient Potential Receptor-3 Channels in Normal and Diseased Airway Smooth Muscle Cells. Adv Exp Med Biol, 1131, 471–487. [DOI] [PubMed] [Google Scholar]
- Watson N, Linder ME, Druey KM, Kehrl JH, & Blumer KJ (1996). RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature, 383, 172–175. [DOI] [PubMed] [Google Scholar]
- Wendell SG, Fan H, & Zhang C (2020). G Protein-Coupled Receptors in Asthma Therapy: Pharmacology and Drug Action. Pharmacol Rev, 72, 1–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JW, Yau D, Sethakorn N, Kach J, Reed EB, Moore TV, et al. (2013). RGS3 controls T lymphocyte migration in a model of Th2-mediated airway inflammation. Am J Physiol Lung Cell Mol Physiol, 305, L693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong GS, Redes JL, Balenga N, McCullough M, Fuentes N, Gokhale A, et al. (2020). RGS4 promotes allergen- and aspirin-associated airway hyperresponsiveness by inhibiting PGE2 biosynthesis. J Allergy Clin Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Jiang H, Nguyen H, Jia S, Berro A, Panettieri RA Jr., W., et al. (2012). Regulator of G protein signaling 2 is a key modulator of airway hyperresponsiveness. J Allergy Clin Immunol, 130, 968–976 e963. [DOI] [PubMed] [Google Scholar]
- Yamada Y, & Matsumoto T (2017). House Dust Mites Induce Production of Endothelin-1 and Matrix Metalloproteinase-9 in Keratinocytes via Proteinase-Activated Receptor-2 Activation. Int Arch Allergy Immunol, 173, 84–92. [DOI] [PubMed] [Google Scholar]
- Yamauchi K, & Ogasawara M (2019). The Role of Histamine in the Pathophysiology of Asthma and the Clinical Efficacy of Antihistamines in Asthma Therapy. Int J Mol Sci, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Balenga N, Cooper PR, Damera G, Edwards R, Brightling CE, Panettieri RA Jr., & Druey KM (2012). Regulator of G-protein signaling-5 inhibits bronchial smooth muscle contraction in severe asthma. Am J Respir Cell Mol Biol, 46, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Cooper PR, Damera G, Mukhopadhyay I, Cho H, Kehrl JH, et al. (2011). Beta-agonist-associated reduction in RGS5 expression promotes airway smooth muscle hyper-responsiveness. J Biol Chem, 286, 11444–11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu OM, & Brown JH (2015). G Protein-Coupled Receptor and RhoA-Stimulated Transcriptional Responses: Links to Inflammation, Differentiation, and Cell Proliferation. Mol Pharmacol, 88, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wang L, Liao Q, Zhang L, Xu L, Chen C, et al. (2013). Genetic associations with hypertension: meta-analyses of six candidate genetic variants. Genet Test Mol Biomarkers, 17, 736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Saradna A, Ratan R, Ke X, Tu W, Do DC, et al. (2020). RhoA/Rho-kinases in asthma: from pathogenesis to therapeutic targets. Clin Transl Immunology, 9, e01134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Nunn C, Ramineni S, Hepler JR, & Chidiac P (2013). The Ras-binding domain region of RGS14 regulates its functional interactions with heterotrimeric G proteins. J Cell Biochem, 114, 1414–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Ma YC, Ostrom RS, Lavoie C, Gill GN, Insel PA, et al. , (2001). RGS-PX1, a GAP for GalphaS and sorting nexin in vesicular trafficking. Science, 294, 1939–1942. [DOI] [PubMed] [Google Scholar]