

Abstract
Bipolar disorder (BD) is a heritable mental illness with complex etiology. We performed a genome-wide association study (GWAS) of 41,917 BD cases and 371,549 controls of European ancestry, which identified 64 associated genomic loci. BD risk alleles were enriched in genes in synaptic signaling pathways and brain-expressed genes, particularly those with high specificity of expression in neurons of the prefrontal cortex and hippocampus. Significant signal enrichment was found in genes encoding targets of antipsychotics, calcium channel blockers, antiepileptics, and anesthetics. Integrating eQTL data implicated 15 genes robustly linked to BD via gene expression, encoding druggable targets such as HTR6, MCHR1, DCLK3 and FURIN. Analyses of BD subtypes indicated high but imperfect genetic correlation between BD type I and II and identified additional associated loci. Together, these results advance our understanding of the biological etiology of BD, identify novel therapeutic leads, and prioritize genes for functional follow-up studies.
Bipolar disorder (BD) is a complex mental disorder characterized by recurrent episodes of (hypo)mania and depression. It is a common condition affecting an estimated 40 to 50 million people worldwide1. This, combined with the typical onset in young adulthood, an often chronic course, and increased risk of suicide2, make BD a major public health concern and a major cause of global disability1. Clinically, BD is classified into two main subtypes: bipolar I disorder, in which manic episodes typically alternate with depressive episodes, and bipolar II disorder, characterized by the occurrence of at least one hypomanic and one depressive episode3. These subtypes have a lifetime prevalence of ~1% each in the population4,5.
Family and molecular genetic studies provide convincing evidence that BD is a multifactorial disorder, with genetic and environmental factors contributing to its development6. On the basis of twin and family studies, the heritability of BD is estimated at 60–85%7,8. Genome-wide association studies (GWAS)9–23 have led to valuable insights into the genetic etiology of BD. The largest such study has been conducted by the Psychiatric Genomics Consortium (PGC), in which genome-wide SNP data from 29,764 BD patients and 169,118 controls were analyzed and 30 genome-wide significant loci were identified (PGC2)24. SNP-based heritability () estimation using the same data suggested that common genetic variants genome-wide explain ~20% of BD’s phenotypic variance24. Polygenic risk scores generated from the results of this study explained ~4% of phenotypic variance in independent samples. Across the genome, genetic associations with BD converged on specific biological pathways including regulation of insulin secretion25,26, retrograde endocannabinoid signaling24, glutamate receptor signaling27 and calcium channel activity9.
Despite this considerable progress, only a fraction of the genetic etiology of BD has been identified, and the specific biological mechanisms underlying the development of the disorder are still unknown. In the present study, we report the results of the third GWAS meta-analysis of the PGC Bipolar Disorder Working Group, comprising 41,917 individuals with BD and 371,549 controls. These results confirm and expand on many previously reported findings, identify novel therapeutic leads, and prioritize genes for functional follow-up studies28,29. Thus, our results further illuminate the biological etiology of BD.
Results
GWAS results.
We conducted a GWAS meta-analysis of 57 BD cohorts collected in Europe, North America and Australia (Supplementary Table 1), totaling 41,917 BD cases and 371,549 controls of European descent (effective n = 101,962, see Online Methods). For 52 cohorts, individual-level genotype and phenotype data were shared with the PGC and cases met international consensus criteria (DSM-IV, ICD-9 or ICD-10) for lifetime BD, established using structured diagnostic interviews, clinician-administered checklists or medical record review. BD GWAS summary statistics were received for five external cohorts (iPSYCH30, deCODE genetics31, Estonian Biobank32, Trøndelag Health Study (HUNT)33 and UK Biobank34), in which most cases were ascertained using ICD codes. The GWAS meta-analysis identified 64 independent loci associated with BD at genome-wide significance (P < 5 × 10−8) (Fig. 1, Table 1, and Supplementary Table 2). Using LD Score regression (LDSC)35, the of BD was estimated to be 18.6% (s.e. = 0.008, P = 5.1 × 10−132) on the liability scale, assuming a BD population prevalence of 2%, and 15.6% (s.e. = 0.006, P = 5.0 × 10−132) assuming a population prevalence of 1% (Supplementary Table 3). The genomic inflation factor (λGC) was 1.38 and the LD Score regression (LDSC) intercept was 1.04 (s.e. = 0.01, P = 2.5 × 10−4) (Supplementary Fig. 1). While the intercept has frequently been used as an indicator of confounding from population stratification, it can rise above 1 with increased sample size and heritability. The attenuation ratio—(LDSC intercept - 1)/(mean of association chi-square statistics - 1)—which is not subject to these limitations, was 0.06 (s.e. = 0.02), indicating that the majority of inflation of the GWAS test statistics was due to polygenicity35,36. Of the 64 genome-wide significant loci, 33 are novel discoveries (i.e. loci not overlapping with any locus previously reported as genome-wide significant for BD). Novel loci include the major histocompatibility complex (MHC) and loci previously reaching genome-wide significance for other psychiatric disorders, including 10 for schizophrenia, 4 for major depression, and 3 for childhood-onset psychiatric disorders or problematic alcohol use (Table 1).
Figure 1 |. Manhattan plot of genome-wide association meta-analysis of 41,917 bipolar disorder cases and 371,549 controls.
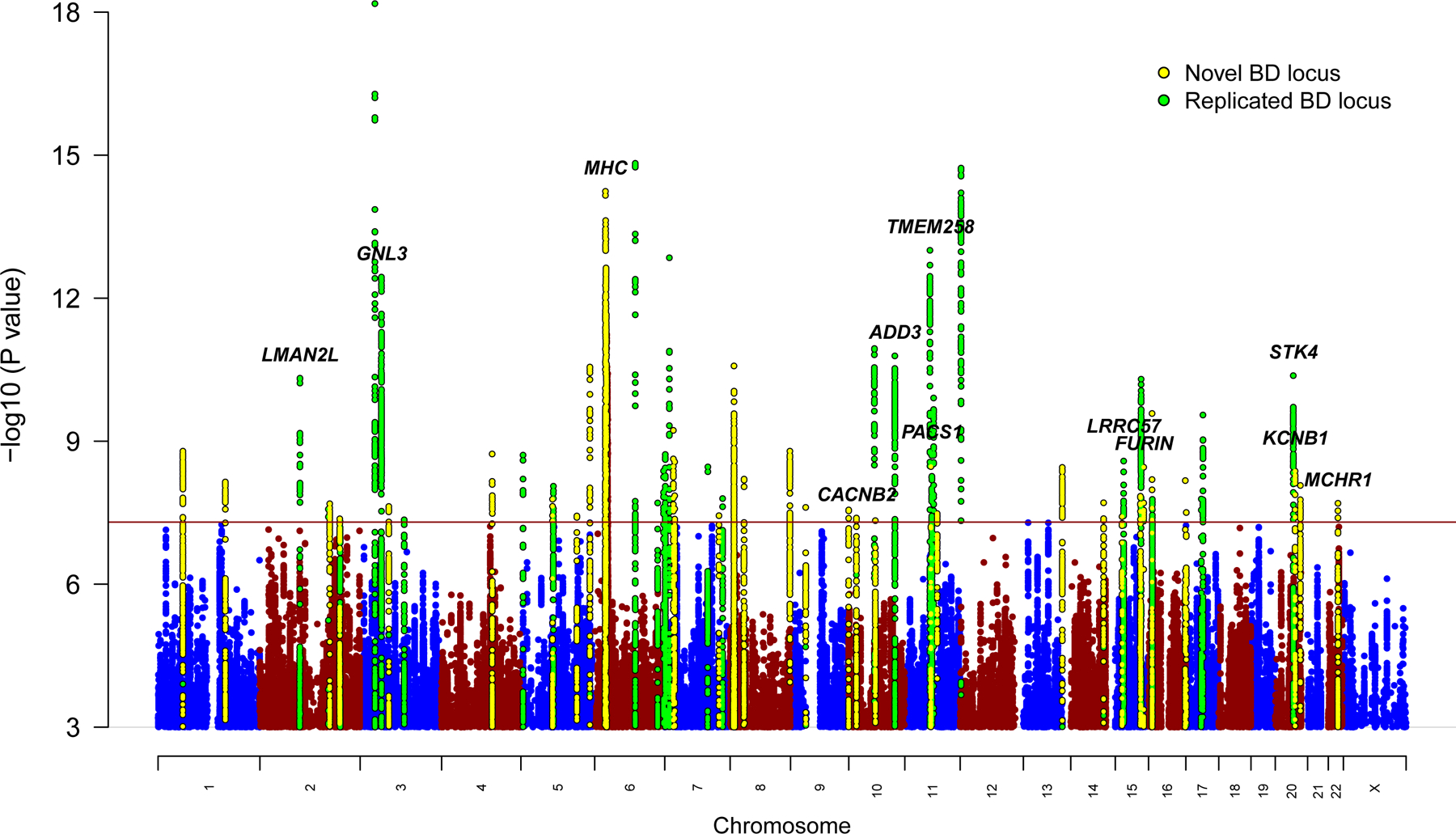
The x-axis shows genomic position (chromosomes 1–22 and X), and the y-axis shows statistical significance as −log10(P value). P values are two-sided and based on an inverse variance weighted fixed effects meta-analysis. The red line shows the genome-wide significance threshold (P < 5 × 10−8). SNPs in genome-wide significant loci are colored green for loci previously associated with bipolar disorder (BD) and yellow for novel associations from this study. The genes labeled are those prioritized by integrative eQTL analyses or notable genes in novel loci (MHC, CACNB2, KCNB1).
Table 1 |.
Genome-wide significant loci for bipolar disorder from meta-analysis of 41,917 cases and 371,549 controls
| Locus | CHR | BP | SNP | P | OR | s.e. | A1/A2 | A1 freq in controls | Previous report^ for BD (citation) | Name for novel locus+ | Previous report^ for psychiatric disorders |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 61105668 | rs2126180 | 1.6E-09 | 1.058 | 0.009 | A/G | 0.457 | LINC01748 | ||
| 2 | 1 | 163745389 | rs10737496 | 7.2E-09 | 1.056 | 0.009 | C/T | 0.444 | NUF2 | CDG | |
| 3* | 2 | 97416153 | rs4619651 | 4.8E-11 | 1.068 | 0.010 | G/A | 0.670 | LMAN2L (PGC2) | CDG | |
| 4 | 2 | 166152389 | rs17183814 | 2.7E-08 | 1.108 | 0.019 | G/A | 0.924 | SCN2A (PGC2) | ||
| 5 | 2 | 169481837 | rs13417268 | 2.1E-08 | 1.064 | 0.011 | C/G | 0.758 | CERS6 | ||
| 6 | 2 | 193738336 | rs2011302 | 4.3E-08 | 1.055 | 0.010 | A/T | 0.377 | PCGEM1 | CDG | |
| 7 | 2 | 194437889 | rs2719164 | 4.9E-08 | 1.053 | 0.010 | A/G | 0.564 | intergenic (PGC2) | CDG | |
| 8* | 3 | 36856030 | rs9834970 | 6.6E-19 | 1.087 | 0.009 | C/T | 0.481 | TRANK1 (PGC2) | SCZ, CDG | |
| 9* | 3 | 52626443 | rs2336147 | 3.6E-13 | 1.070 | 0.009 | T/C | 0.498 | ITIH1 (PGC2) | SCZ, CDG | |
| 10 | 3 | 70488788 | rs115694474 | 2.4E-08 | 1.068 | 0.012 | T/A | 0.799 | MDFIC2 | ||
| 11 | 3 | 107757060 | rs696366 | 4.5E-08 | 1.053 | 0.009 | C/A | 0.550 | CD47 (PGC2) | ||
| 12* | 4 | 123076007 | rs112481526 | 1.9E-09 | 1.065 | 0.011 | G/A | 0.256 | KIAA1109 | MD | |
| 13* | 5 | 7542911 | rs28565152 | 2.0E-09 | 1.070 | 0.011 | A/G | 0.238 | ADCY2 (PGC2) | ||
| 14* | 5 | 78849505 | rs6865469 | 1.7E-08 | 1.060 | 0.010 | T/G | 0.274 | HOMER1 | ||
| 15 | 5 | 80961069 | rs6887473 | 8.8E-09 | 1.062 | 0.011 | G/A | 0.739 | SSBP2 (PGC2) | ||
| 16* | 5 | 137712121 | rs10043984 | 3.7E-08 | 1.062 | 0.011 | T/C | 0.236 | KDM3B | CDG | |
| 17 | 5 | 169289206 | rs10866641 | 2.8E-11 | 1.065 | 0.009 | T/C | 0.575 | DOCK2 | ||
| 18* | 6 | 26463575 | rs13195402 | 5.8E-15 | 1.146 | 0.018 | G/T | 0.919 | MHC | MD, SCZ, CDG, MOOD | |
| 19* | 6 | 98565211 | rs1487445 | 1.5E-15 | 1.078 | 0.009 | T/C | 0.487 | POU3F2 (PGC2) | CDG | |
| 20 | 6 | 152793572 | rs4331993 | 2.0E-08 | 1.056 | 0.010 | A/T | 0.382 | SYNE1 (Green 2013) | ||
| 21* | 6 | 166995260 | rs10455979 | 4.2E-09 | 1.057 | 0.010 | G/C | 0.500 | RPS6KA2 (PGC2) | ||
| 22* | 7 | 2020995 | rs12668848 | 1.9E-09 | 1.059 | 0.010 | G/A | 0.575 | MAD1L1 (Hou 2016, Ikeda 2017) | MD, SCZ, CDG | |
| 23* | 7 | 11871787 | rs113779084 | 1.4E-13 | 1.079 | 0.010 | A/G | 0.299 | THSD7A (PGC2) | ||
| 24* | 7 | 21492589 | rs6954854 | 5.9E-10 | 1.060 | 0.009 | G/A | 0.425 | SP4 | ||
| 25 | 7 | 24647222 | rs12672003 | 2.7E-09 | 1.096 | 0.016 | G/A | 0.113 | MPP6 | SCZ, CDG, MOOD | |
| 26 | 7 | 105043229 | rs11764361 | 3.5E-09 | 1.063 | 0.010 | A/G | 0.668 | SRPK2 (PGC2) | SCZ, ASD, CDG | |
| 27 | 7 | 131870597 | rs6946056 | 3.7E-08 | 1.055 | 0.010 | C/A | 0.623 | PLXNA4 | ||
| 28 | 7 | 140676153 | rs10255167 | 1.6E-08 | 1.068 | 0.012 | A/G | 0.778 | MRPS33 (PGC2) | CDG | |
| 29* | 8 | 9763581 | rs62489493 | 2.6E-11 | 1.094 | 0.014 | G/C | 0.128 | miR124–1 | SCZ, ALC, ASD | |
| 30* | 8 | 10226355 | rs3088186 | 2.1E-08 | 1.058 | 0.010 | T/C | 0.287 | MSRA | SCZ, ALC, ASD | |
| 31 | 8 | 34152492 | rs2953928 | 6.3E-09 | 1.124 | 0.020 | A/G | 0.067 | RP1–84O15.2 (lincRNA) | SCZ, ADHD, CDG | |
| 32* | 8 | 144993377 | rs6992333 | 1.6E-09 | 1.062 | 0.010 | G/A | 0.410 | PLEC | ||
| 33 | 9 | 37090538 | rs10973201 | 2.5E-08 | 1.101 | 0.017 | C/T | 0.110 | ZCCHC7 | MD, CDG, MOOD | |
| 34* | 9 | 141066490 | rs62581014 | 2.8E-08 | 1.067 | 0.012 | T/C | 0.366 | TUBBP5 | ||
| 35* | 10 | 18751103 | rs1998820 | 4.1E-08 | 1.087 | 0.015 | T/A | 0.886 | CACNB2 | SCZ, CDG | |
| 36* | 10 | 62322034 | rs10994415 | 1.1E-11 | 1.125 | 0.017 | C/T | 0.082 | ANK3 (PGC2) | ||
| 37 | 10 | 64525135 | rs10761661 | 4.7E-08 | 1.053 | 0.009 | T/C | 0.472 | ADO | ||
| 38* | 10 | 111648659 | rs2273738 | 1.6E-11 | 1.096 | 0.014 | T/C | 0.135 | ADD3 (Charney 2017, PGC2) | ||
| 39* | 11 | 61618608 | rs174592 | 9.9E-14 | 1.074 | 0.010 | G/A | 0.395 | FADS2 (PGC2) | MD, CDG, MOOD | |
| 40 | 11 | 64009879 | rs4672 | 3.4E-09 | 1.107 | 0.017 | A/G | 0.083 | FKBP2 | ||
| 41* | 11 | 65848738 | rs475805 | 2.0E-09 | 1.070 | 0.011 | A/G | 0.767 | PACS1 (PGC2) | ||
| 42* | 11 | 66324583 | rs678397 | 5.5E-09 | 1.056 | 0.009 | T/C | 0.457 | PC (PGC1, PGC2) | ||
| 43* | 11 | 70517927 | rs12575685 | 1.2E-10 | 1.067 | 0.010 | A/G | 0.327 | SHANK2 (PGC2) | MD | |
| 44 | 11 | 79092527 | rs12289486 | 3.3E-08 | 1.086 | 0.015 | T/C | 0.115 | ODZ4 (PGC1) | ||
| 45* | 12 | 2348844 | rs11062170 | 1.9E-15 | 1.081 | 0.010 | C/G | 0.333 | CACNA1C (PGC2) | SCZ, CDG, MOOD | |
| 46 | 13 | 113869045 | rs35306827 | 3.6E-09 | 1.068 | 0.011 | G/A | 0.775 | CUL4A | ||
| 47 | 14 | 99719219 | rs2693698 | 2.0E-08 | 1.055 | 0.009 | G/A | 0.551 | BCL11B | SCZ, CDG | |
| 48* | 15 | 38973793 | rs35958438 | 3.8E-08 | 1.066 | 0.012 | G/A | 0.772 | C15orf53 | CDG | |
| 49* | 15 | 42904904 | rs4447398 | 2.6E-09 | 1.086 | 0.014 | A/C | 0.131 | STARD9 (PGC2) | ||
| 50 | 15 | 83531774 | rs62011709 | 1.4E-08 | 1.064 | 0.011 | T/A | 0.747 | HOMER2 | SCZ | |
| 51* | 15 | 85149575 | rs748455 | 5.0E-11 | 1.070 | 0.010 | T/C | 0.719 | ZNF592 (PGC2) | SCZ, CDG | |
| 52 | 15 | 91426560 | rs4702 | 3.5E-09 | 1.059 | 0.010 | G/A | 0.446 | FURIN | SCZ, CDG | |
| 53 | 16 | 9230816 | rs28455634 | 2.6E-10 | 1.065 | 0.010 | G/A | 0.620 | C16orf72 | ||
| 54 | 16 | 9926348 | rs7199910 | 1.7E-08 | 1.057 | 0.010 | G/T | 0.312 | GRIN2A (PGC2) | SCZ, CDG | |
| 55 | 16 | 89632725 | rs12932628 | 6.7E-09 | 1.058 | 0.010 | T/G | 0.487 | RPL13 | ||
| 56 | 17 | 1835482 | rs4790841 | 3.1E-08 | 1.075 | 0.013 | T/C | 0.151 | RTN4RL1 | ||
| 57 | 17 | 38129841 | rs11870683 | 2.8E-08 | 1.059 | 0.010 | T/A | 0.650 | ERBB2 (Hou 2016) | ||
| 58 | 17 | 38220432 | rs61554907 | 1.6E-08 | 1.091 | 0.015 | T/G | 0.124 | ERBB2 (Hou 2016) | ||
| 59* | 17 | 42191893 | rs228768 | 2.8E-10 | 1.067 | 0.010 | G/T | 0.294 | HDAC5 (PGC2) | ||
| 60* | 20 | 43682551 | rs67712855 | 4.2E-11 | 1.070 | 0.010 | T/G | 0.687 | STK4 (PGC2) | ||
| 61* | 20 | 43944323 | rs6032110 | 1.0E-09 | 1.059 | 0.009 | A/G | 0.512 | WFDC12 (PGC2) | ||
| 62* | 20 | 48033127 | rs237460 | 4.3E-09 | 1.057 | 0.009 | T/C | 0.412 | KCNB1 | CDG | |
| 63 | 20 | 60865815 | rs13044225 | 8.5E-09 | 1.056 | 0.010 | G/A | 0.440 | OSBPL2 | ||
| 64 | 22 | 41153879 | rs5758064 | 2.0E-08 | 1.054 | 0.009 | T/C | 0.523 | SLC25A17 | MD, SCZ, CDG, MOOD |
CHR, chromosome; BP, GRCh37 base pair position; SNP, single nucleotide polymorphism; OR, odds ratio; s.e., standard error, A1, tested allele; A2, other allele; freq, frequency; BD, bipolar disorder; CDG, Cross-disorder GWAS of the Psychiatric Genomics Consortium; MD, major depression; SCZ, schizophrenia; MOOD, mood disorders; ASD, autism spectrum disorder; ALC, alcohol use disorder or problematic alcohol use; ADHD, attention deficit/hyperactivity disorder.
Locus overlaps with genome-wide significant locus for bipolar I disorder.
Previous report refers to previous association of a SNP in the locus with the psychiatric disorder at genome-wide significance. PGC1 = PMID 21926972; PGC2 = PMID 31043756; Hou 2016 = PMID 27329760; Ikeda 2017 = PMID 28115744; Green 2013 = PMID 22565781; Charney 2017 = PMID 28072414.
Novel loci are named using the nearest gene to the index SNP. P values are two-sided and based on an inverse variance weighted fixed effects meta-analysis.
Enrichment analyses.
Genome-wide analyses using MAGMA37 indicated significant enrichment of BD associations in 161 genes (Supplementary Table 4) and 4 gene sets related to synaptic signaling (Supplementary Table 5). The BD association signal was enriched amongst genes expressed in different brain tissues (Supplementary Table 6), especially genes with high specificity of gene expression in neurons (both excitatory and inhibitory) versus other cell types, within cortical and subcortical brain regions in mice (Supplementary Fig. 2)38. In human brain samples, signal enrichment was also observed in hippocampal pyramidal neurons and interneurons of the prefrontal cortex and hippocampus, compared with other cell types (Supplementary Fig. 2).
In a gene-set analysis of the targets of individual drugs (from the Drug-Gene Interaction Database DGIdb v.239 and the Psychoactive Drug Screening Database Ki DB40), the targets of the calcium channel blockers mibefradil and nisoldipine were significantly enriched (Supplementary Table 7). Grouping drugs according to their Anatomical Therapeutic Chemical (ATC) classes41, there was significant enrichment in the targets of four broad drug classes (Supplementary Table 8): psycholeptics (drugs with a calming effect on behavior) (especially hypnotics and sedatives, antipsychotics and anxiolytics), calcium channel blockers, antiepileptics, and (general) anesthetics (Supplementary Table 8).
eQTL integrative analyses.
We conducted a transcriptome-wide association study (TWAS) using FUSION42 and eQTL data from the PsychENCODE Consortium (1,321 brain samples)43. BD-associated alleles significantly influenced expression of 77 genes in the brain (Supplementary Table 9 and Supplementary Fig. 3). These genes encompassed 40 distinct regions. We performed TWAS fine-mapping using FOCUS44 to model the correlation among the TWAS signals and prioritize the most likely causal gene(s) in each region. Within the 90%-credible set, FOCUS prioritized 22 genes with a posterior inclusion probability (PIP) > 0.9 (encompassing 20 distinct regions) and 32 genes with a PIP > 0.7 (29 distinct regions) (Supplementary Table 10).
We used summary data-based Mendelian randomization (SMR)45,46 to identify putative causal relationships between SNPs and BD via gene expression by integrating the BD GWAS results with brain eQTL summary statistics from the PsychENCODE43 Consortium and blood eQTL summary statistics from the eQTLGen Consortium (31,684 whole blood samples)47. The eQTLGen results represent the largest existing eQTL study and provide independent eQTL data. Of the 32 genes fine-mapped with PIP > 0.7, 15 were significantly associated with BD in the SMR analyses and passed the HEIDI (heterogeneity in dependent instruments) test45,46, suggesting that their effect on BD is mediated via gene expression in the brain and/or blood (Supplementary Table 11). The genes located in genome-wide significant loci are labeled in Figure 1. Other significant genes included HTR6, DCLK3, HAPLN4 and PACSIN2.
MHC locus.
Variants within and distal to the major histocompatibility complex (MHC) locus were associated with BD at genome-wide significance. The most highly associated SNP was rs13195402, 3.2 Mb distal to any HLA gene or the complement component 4 (C4) genes (Supplementary Fig. 4). Imputation of C4 alleles using SNP data uncovered no association between the five most common structural forms of the C4A/C4B locus (BS, AL, AL-BS, AL-BL, and AL-AL) and BD, either before or after conditioning on rs13195402 (Supplementary Fig. 5). While genetically predicted C4A expression initially showed a weak association with BD, this association was non-significant after controlling for rs13195402 (Supplementary Fig. 6).
Polygenic risk scoring.
The performance of polygenic risk scores (PRS) based on these GWAS results was assessed by excluding cohorts in turn from the meta-analysis to create independent test samples. PRS explained ~4.57% of phenotypic variance in BD on the liability scale (at GWAS P-value threshold (PT) < 0.1, BD population prevalence 2%), based on the weighted mean R2 across cohorts (Fig. 2 and Supplementary Table 12). This corresponds to a weighted mean area under the curve (AUC) of 65%. Results per cohort and per wave of recruitment to the PGC are in Supplementary Tables 12 and 13 and Supplementary Figure 7. At PT < 0.1, individuals in the top 10% of BD PRS had an odds ratio of 3.5 (95% CI 1.7–7.3) of being affected with the disorder compared with individuals in the middle decile (based on the weighted mean OR across PGC cohorts), and an odds ratio of 9.3 (95% CI 1.7–49.3) compared with individuals in the lowest decile. The generalizability of PRS from this meta-analysis was examined in several non-European cohorts. PRS explained up to 2.3% and 1.9% of variance in BD in two East Asian samples, and 1.2% and 0.4% in two admixed African American samples (Fig. 2 and Supplementary Table 14). The variance explained by the PRS increased in every cohort with increasing sample size of the PGC BD European discovery sample (Supplementary Fig. 8 and Supplementary Table 14).
Figure 2 |. Phenotypic variance in bipolar disorder explained by polygenic risk scores.
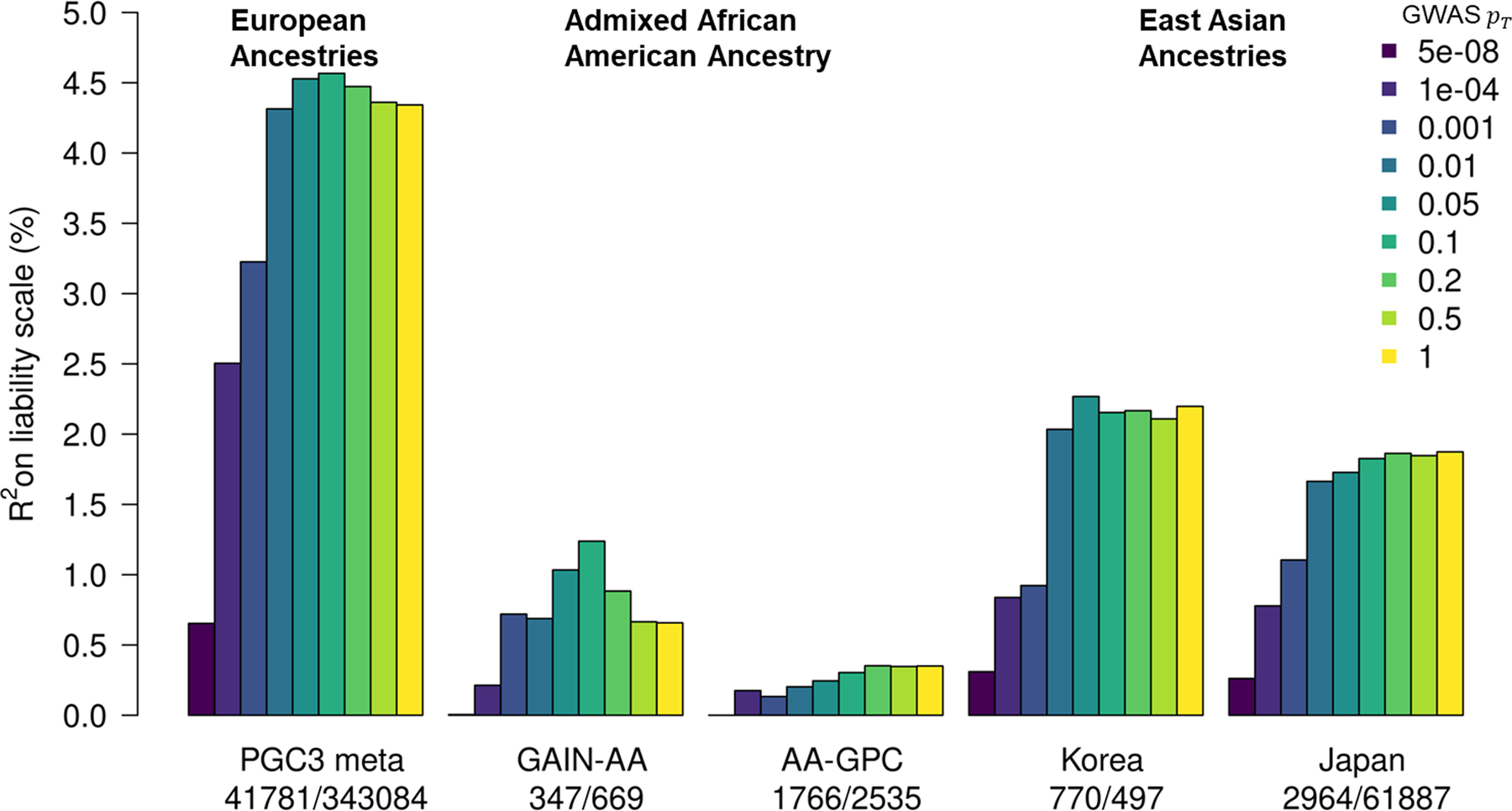
Variance explained is presented on the liability scale, assuming a 2% population prevalence of bipolar disorder. For European ancestries, the results shown are the weighted mean R2 values across all 57 cohorts in the PGC3 meta-analysis, weighted by the effective n per cohort. The numbers of cases and controls are shown from left to right under the barplot for each study. GWAS PT, the color of the bars represents the P value threshold used to select SNPs from the discovery GWAS; GAIN-AA, Genetic Association Information Network African American cohort; AA-GPC, African American Genomic Psychiatry Cohort.
Genetic architecture of BD and other traits.
The genome-wide genetic correlation (rg) of BD with a range of diseases and traits was assessed on LD Hub48. After correction for multiple testing, BD showed significant rg with 16 traits among 255 tested from published GWAS (Supplementary Table 15). Genetic correlation was positive with all psychiatric disorders assessed, particularly schizophrenia (rg = 0.68) and major depression (rg = 0.44), and to a lesser degree anorexia, attention deficit/hyperactivity disorder, and autism spectrum disorder (rg ≈ 0.2). We found evidence of positive rg between BD and smoking initiation, cigarettes per day, problematic alcohol use, and drinks per week (Fig. 3). BD was also positively genetically correlated with measures of sleep quality (daytime sleepiness, insomnia, sleep duration) (Fig. 3). Among 514 traits measured in the general population of the UK Biobank, there was significant rg between BD and many psychiatric-relevant traits or symptoms, dissatisfaction with interpersonal relationships, poorer overall health rating, and feelings of loneliness or isolation (Supplementary Table 16).
Figure 3 |. Relationships between bipolar disorder and modifiable risk factors based on genetic correlations, generalized summary statistics-based Mendelian randomization, and bivariate gaussian mixture modeling.
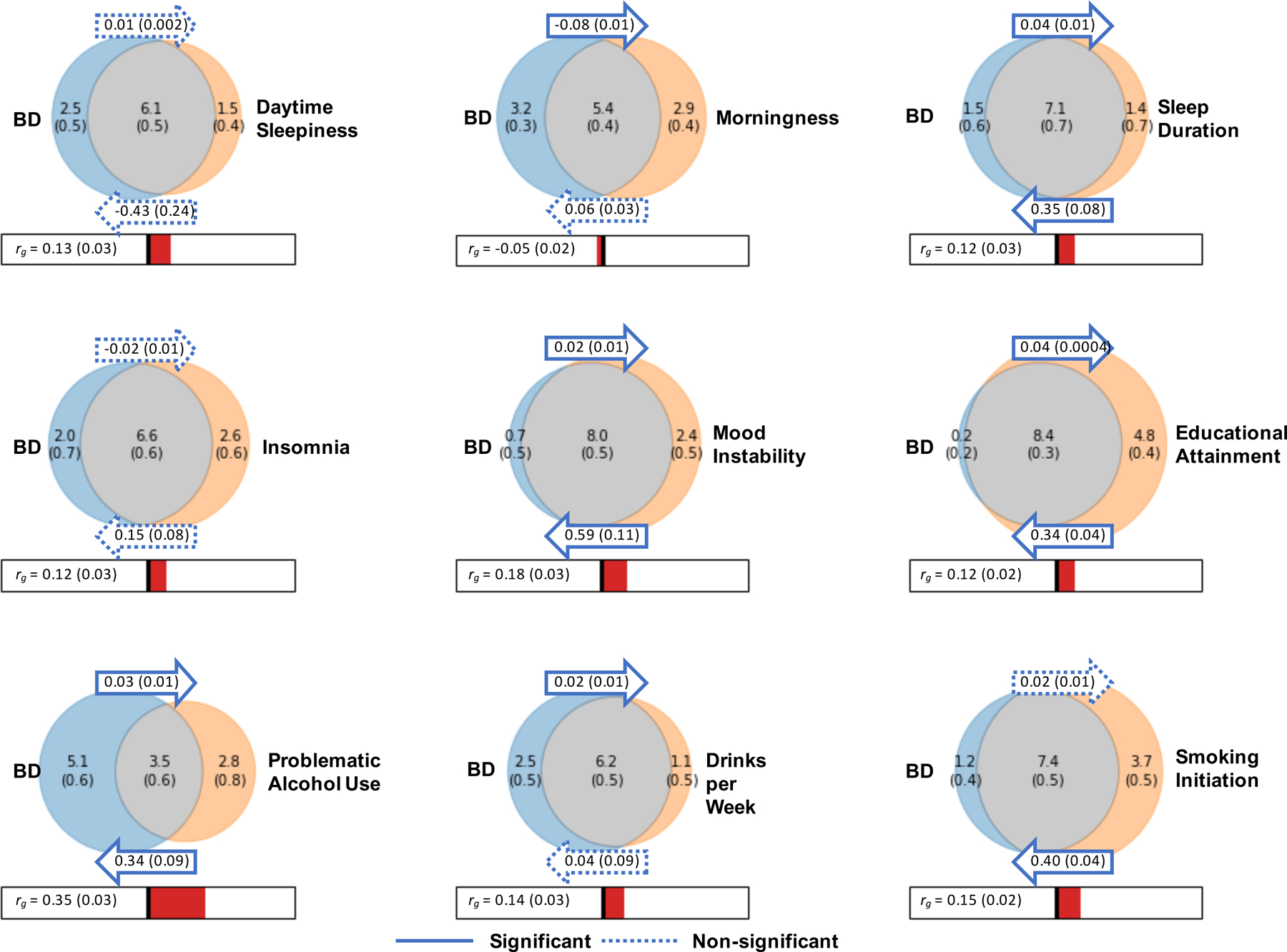
Venn diagrams depict MiXeR results of the estimated number of influencing variants shared between bipolar disorder (BD) and each trait of interest (grey), unique to BD (blue) and unique to the trait of interest (orange). The number of influencing variants and standard error are shown in thousands. The size of the circles reflects the polygenicity of each trait, with larger circles corresponding to greater polygenicity. The estimated genetic correlation (rg) between BD and each trait of interest and standard error from LD Score regression is shown below the corresponding Venn diagram, with an accompanying scale (-1 to +1). The arrows above and below the Venn diagrams indicate the results of generalized summary statistics-based Mendelian randomization (GSMR) of BD on the trait of interest, and the trait of interest on BD, respectively. The GSMR effect size and standard error is shown inside the corresponding arrow. Solid arrows indicate a significant relationship between the exposure and the outcome, after correction for multiple comparisons (P < 1.47 × 10−3), and dashed arrows indicate a non-significant relationship.
Bivariate gaussian mixture models were applied to the GWAS summary statistics for BD and other complex traits using the MiXeR tool49,50 to estimate the number of variants influencing each trait that explain 90% of and their overlap between traits. MiXeR estimated that approximately 8.6 k (s.e. = 0.2 k) variants influence BD, which is similar to the estimate for schizophrenia (9.7 k, s.e. = 0.2 k) and somewhat lower than that for major depression (12.3 k, s.e. = 0.6 k) (Supplementary Table 17 and Supplementary Fig. 9). When considering the number of shared loci as a proportion of the total polygenicity of each trait, the vast majority of loci influencing BD were also estimated to influence major depression (97%) and schizophrenia (96%) (Supplementary Table 17 and Supplementary Fig. 9). Interestingly, within these shared components, the variants that influenced both BD and schizophrenia had high concordance in direction of effect (80%, s.e. = 2%), while the portion of concordant variants between BD and MDD was only 69% (s.e. = 1%) (Supplementary Table 17).
Genetic and causal relationships between BD and modifiable risk factors.
Ten traits associated with BD from clinical and epidemiological studies were investigated in detail for genetic and potentially causal relationships with BD via LDSC35, generalized summary statistics-based Mendelian randomization (GSMR)51, and bivariate gaussian mixture modeling49. BD has been strongly linked with sleep disturbances52, alcohol use53 and smoking54, higher educational attainment55,56, and mood instability57. Most of these traits had modest but significant genetic correlations with BD (rg = −0.05–0.35) (Fig. 3). Examining the effects of these traits on BD via GSMR, smoking initiation was associated with BD, corresponding to an OR of 1.49 (95% CI 1.38–1.61) for developing the disorder (P = 1.74 × 10−22) (Fig. 3). Testing the effect of BD on the traits, BD was significantly associated with reduced likelihood of being a morning person and increased number of drinks per week (P < 1.47 × 10−3) (Fig. 3). Positive bi-directional relationships were identified between BD and longer sleep duration, problematic alcohol use, educational attainment (EA), and mood instability (Fig. 3). Notably, the instrumental variables for mood instability were selected from a GWAS conducted in the general population, excluding individuals with psychiatric disorders58. For all of the aforementioned BD-trait relationships, the effect size estimates from GSMR were consistent with those calculated using the inverse variance weighted regression method, and there was no evidence of bias from horizontal pleiotropy. Full MR results are in Supplementary Tables 18 and 19. Bivariate gaussian mixture modeling using MiXeR indicated large proportions of variants influencing both BD and all other traits tested, particularly educational attainment, where approximately 98% of variants influencing BD were estimated to also influence EA. While cigarettes per day was a trait of interest, MiXeR could not model these data due to low polygenicity and heritability, and the effect of cigarettes per day on BD was inconsistent between MR methods, suggesting a violation of MR assumptions (Supplementary Tables 18–20).
BD subtypes.
We conducted GWAS meta-analyses of bipolar I disorder (BD I) (25,060 cases, 449,978 controls) and bipolar II disorder (BD II) (6,781 cases, 364,075 controls). The BD I analysis identified 44 genome-wide significant loci, 31 of which overlapped with genome-wide significant loci from the main BD GWAS (Table 1 and Supplementary Table 21). The remaining 13 genome-wide significant loci for BD I all had P < 4.0 × 10−5 in the main BD GWAS. One genome-wide significant locus was identified in the GWAS meta-analysis of BD II and had a P < 1.1 × 10−4 in the main GWAS of BD (Supplementary Table 21). The estimates on the liability scale for BD I and BD II were 20.9% (s.e. = 0.009, P = 1.0 × 10−111) and 11.6% (s.e. = 0.01, P = 3.9 × 10−15), respectively, assuming a 1% population prevalence of each subtype. These heritability values are significantly different from each other (P = 2.4 × 10−25, block jackknife). The genetic correlation between BD I and BD II was 0.85 (s.e. = 0.05, P = 2.88 × 10−54), which is significantly different from 1 (P = 1.6 × 10−3). The genetic correlation of BD I with schizophrenia (rg = 0.66, s.e. = 0.02) was higher than that of BD II (rg = 0.54, s.e. = 0.05), whereas major depression was more strongly genetically correlated with BD II (rg = 0.66, s.e. = 0.05) than with BD I (rg = 0.34, s.e. = 0.03) (Supplementary Table 22).
Discussion
In a GWAS of 41,917 BD cases, we identified 64 associated genomic loci, 33 of which are novel discoveries. With a 1.5-fold increase in effective sample size compared with the PGC2 BD GWAS, this study more than doubled the number of associated loci, representing an inflection point in the rate of risk variant discovery. We observed consistent replication of known BD loci, including 28/30 loci from the PGC2 GWAS24 and several implicated by other BD GWAS15,16,17, including a study of East Asian cases59.
The 33 novel loci discovered here encompass genes of expected biological relevance to BD, such as the ion channels CACNB2 and KCNB1. Amongst the 64 BD loci, 17 have previously been implicated in GWAS of schizophrenia60, and seven in GWAS of major depression61, representing the first overlap of genome-wide significant loci between the mood disorders. For these genome-wide significant loci shared across disorders, 17/17 and 5/7 of the BD index SNPs had the same direction of effect on schizophrenia and major depression, respectively (Supplementary Table 23). More generally, 50/64 and 62/64 BD loci had a consistent direction of effect on major depression and schizophrenia, respectively, considerably greater than chance (P < 1 × 10−5, binomial test). Bivariate gaussian mixture modeling estimated that across the entire genome, almost all variants influencing BD also influence schizophrenia and major depression, albeit with variable effects62. SNPs in and around the MHC locus reached genome-wide significance for BD for the first time. However, unlike in schizophrenia, we found no influence of C4 structural alleles or gene expression63. Rather the association was driven by variation outside the classical MHC locus, with the index SNP (rs13195402) being a missense variant in BTN2A1, a brain-expressed gene64 encoding a plasma membrane protein.
The genetic correlation of BD with other psychiatric disorders was consistent with previous reports65,66. Our results also corroborate previous genetic and clinical evidence of associations between BD and sleep disturbances67, problematic alcohol use68, and smoking69. While the genome-wide genetic correlations with these traits were modest (rg = −0.05–0.35), MiXeR estimated that, for all traits, more than 55% of trait-influencing variants also influence BD (Fig. 3). Taken together, these results point to shared biology as one possible explanation for the high prevalence of substance use in BD. However, excluding genetic variants associated with both traits, MR analyses suggested that smoking is also a putatively “causal” risk factor for BD, while BD has no effect on smoking, consistent with a previous report70. (We use the word “causal” with caution here as we consider MR an exploratory analysis to identify potentially modifiable risk factors that warrant more detailed investigations to understand their complex relationship with BD.) In contrast, MR indicated that BD had bi-directional “causal” relationships with problematic alcohol use, longer sleep duration, and mood instability. Insights into the relationship of such behavioral correlates with BD may have future impact on clinical decision making in the prophylaxis or management of the disorder. Higher educational attainment has previously been associated with BD in epidemiological studies55,56, while lower educational attainment has been associated with schizophrenia and major depression71,72. Here, educational attainment had a significant positive effect on risk of BD and vice versa. Interestingly, MiXeR estimated that almost all variants that influence BD also influence educational attainment. The substantial genetic overlap observed between BD and the other phenotypes suggests that many variants likely influence multiple phenotypes, which may be differentiated by phenotype-specific effect size distributions among the shared influencing variants.
The integration of eQTL data with our GWAS results yielded 15 high-confidence genes for which there was converging evidence that their association with BD is mediated via gene expression. Amongst these were HTR6, encoding a serotonin receptor targeted by antipsychotics and antidepressants73, and MCHR1 (melanin-concentrating hormone receptor 1), encoding a target of the antipsychotic haloperidol73. We note that, for both of these genes, their top eQTLs have opposite directions of effect on gene expression in the brain and blood, possibly playing a role in the tissue-specific gene regulation influencing BD74. BD was associated with decreased expression of FURIN, a gene with a neurodevelopmental role that has already been the subject of functional genomics experiments in neuronal cells following its association with schizophrenia in GWAS75. The top association in our GWAS was in the TRANK1 locus on chromosome 3, which has previously been implicated in BD12,18,59. Although BD-associated SNPs in this locus are known to regulate TRANK1 expression76, our eQTL analyses support a stronger but correlated regulation of DCLK3, located 87 kb upstream of TRANK143,77. Both FURIN and DCLK3 also encode druggable proteins (although they are not targets for any current psychiatric medications)73,78. These eQTL results provide promising BD candidate genes for functional follow-up experiments29. While several of these are in genome-wide significant loci, many are not the closest gene to the index SNP, highlighting the value of probing underlying molecular mechanisms to prioritize the most likely causal genes in the loci.
GWAS signals were enriched in the gene targets of existing BD pharmacological agents, such as antipsychotics, mood stabilizers, and antiepileptics. However, enrichment was also found in the targets of calcium channel blockers used to treat hypertension and GABA-receptor targeting anesthetics (Supplementary Table 8). Calcium channel antagonists have long been investigated for the treatment of BD, without becoming an established therapeutic approach, and there is evidence that some antiepileptics have calcium channel-inhibiting effects79,80. These results underscore the opportunity for repurposing some classes of drugs, particularly calcium channel antagonists, as potential BD treatments81.
BD associations were enriched in gene sets involving neuronal parts and synaptic signaling. Neuronal and synaptic pathways have been described in cross-disorder GWAS of multiple psychiatric disorders including BD82–84. Dysregulation of such pathways has also been suggested by previous functional and animal studies85. Analysis of single-cell gene expression data revealed enrichment in genes with high specificity of gene expression in neurons (both excitatory and inhibitory) of many brain regions, in particular the cortex and hippocampus. These findings are similar to those reported in GWAS data of schizophrenia86 and major depressive disorder38.
PRS for BD explained on average 4.57% of phenotypic variance (liability scale) across European cohorts, although this varied in different waves of the BD GWAS, ranging from 6.6% in the PGC1 cohorts to 2.9% in the External biobank studies (Supplementary Fig. 7 and Supplementary Table 12). These results are in line with the of BD per wave, which ranged from 24.6% (s.e. =0.01) in PGC1 to 11.9% (s.e. = 0.01) in External studies (Supplementary Table 3). Some variability in estimates may arise from the inclusion of cases from population biobanks, who may have more heterogeneous clinical presentations or less severe illness than BD patients ascertained via inpatient or outpatient psychiatric clinics. Across the waves of clinically ascertained samples within the PGC, and the R2 of PRS also varied, likely reflecting clinical and genetic heterogeneity in the type of BD cases ascertained; the PGC1 cohorts consisted mostly of BD I cases9, known to be the most heritable of the BD subtypes11,24, while later waves included more individuals with BD II24. Overall, the of BD calculated from the meta-analysis summary statistics was 18% on the liability scale, a decrease of ~2% compared with the PGC2 GWAS24, which may be due to the addition of cohorts with lower estimates and heterogeneity between cohorts (Supplementary Table 3). However, despite differences in and R2 of PRS per wave, the genetic correlation of BD between all waves was high (weighted mean rg = 0.94, s.e. = 0.03), supporting our rationale for combining cases with different BD subtypes or ascertainment to increase power for discovery of risk variants. In Europeans, individuals in the top 10% of PRS had an OR of 3.5 for BD, compared with individuals with average PRS (middle decile), which translates into a modest absolute lifetime risk of the disorder (7% based on PRS alone). While PRS are invaluable tools in research settings, the current BD PRS lack sufficient power to separate individuals into clinically meaningful risk categories, and therefore have no clinical utility at present87,88. PRS from this European BD meta-analysis yield higher R2 values in diverse ancestry samples than PRS based on any currently available BD GWAS within the same ancestry59. However, performance still greatly lags behind that in Europeans, with ~2% variance explained in East Asian samples and substantially less in admixed African American samples, likely due to differences in allele frequencies and LD structures, consistent with previous studies89,90. There is a pressing need for more and larger studies in other ancestry groups to ensure that any future clinical utility is broadly applicable. Exploiting the differences in LD structure between diverse ancestry samples will also assist in the fine-mapping of risk loci for BD.
Our analyses confirmed that BD is a highly polygenic disorder, with an estimated 8.6 k variants explaining 90% of its . Hence, many more SNPs than those identified here are expected to account for the common variant architecture underlying BD. This GWAS marks an inflection point in risk variant discovery, and we expect that, from this point forward, the addition of more samples will lead to a dramatic increase in genetic findings. Nevertheless, fewer genome-wide significant loci have been identified in BD than in a schizophrenia GWAS of comparable sample size60. This may be due to the clinical and genetic heterogeneity that exists in BD.
Our GWAS of subtypes BD I and BD II identified additional associated loci. Consistent with previous findings24, our analysis showed that the two subtypes were highly but imperfectly genetically correlated (rg = 0.85), and that BD I is more genetically correlated with schizophrenia, while BD II has stronger genetic correlation with major depression. The subtypes are sufficiently similar to justify joint analysis as BD, but are not identical in their genetic composition, and as such contribute to the genetic heterogeneity of BD91. We identified 13 loci passing genome-wide significance for BD I, and one for BD II, which did not reach significance in the main BD GWAS, further illustrating the partially differing genetic composition of the two subtypes. Understanding the shared and distinct genetic components of BD subtypes and symptoms requires detailed phenotyping efforts in large cohorts and is an important area for future psychiatric genetics research.
In summary, these new data advance our understanding of the biological etiology of BD and prioritize a set of candidate genes for functional follow-up experiments. Several lines of evidence converge on the involvement of calcium channel signaling, providing a promising avenue for future therapeutic development.
Methods
Sample description.
The meta-analysis sample comprises 57 cohorts collected in Europe, North America and Australia, totaling 41,917 BD cases and 371,549 controls of European descent (Supplementary Table 1). The total effective n, equivalent to an equal number of cases and controls in each cohort (4*ncases*ncontrols/(ncases + ncontrols)), is 101,962. For 52 cohorts, individual-level genotype and phenotype data were shared with the PGC. Cohorts have been added to the PGC in five waves (PGC19, PGC224, PGC PsychChip, PGC3 and External Studies); all cohorts from previous PGC BD GWAS were included. The source and inclusion/exclusion criteria for cases and controls for each cohort are described in the Supplementary Note. Cases were required to meet international consensus criteria (DSM-IV, ICD-9, or ICD-10) for a lifetime diagnosis of BD, established using structured diagnostic instruments from assessments by trained interviewers, clinician-administered checklists, or medical record review. In most cohorts, controls were screened for the absence of lifetime psychiatric disorders and randomly selected from the population. For five cohorts (iPSYCH30, deCODE genetics31, Estonian Biobank32, Trøndelag Health Study (HUNT)33 and UK Biobank34), GWAS summary statistics for BD were shared with the PGC. In these cohorts, BD cases were ascertained using ICD codes or self-report during a nurse interview, and the majority of controls were screened for the absence of psychiatric disorders via ICD codes. Follow-up analyses included four non-European BD case-control cohorts, two from East Asia (Japan59 and Korea92), and two admixed African American cohorts22,93, providing a total of 5,847 cases and 65,588 controls. These BD cases were ascertained using international consensus criteria (DSM-IV)22,93 through psychiatric interviews (Supplementary Note).
Genotyping, quality control and imputation.
For 52 cohorts internal to the PGC, genotyping was performed following local protocols and genotypes were called using standard genotype calling softwares from commercial sources (Affymetrix and Illumina). Subsequently, standardized quality control, imputation and statistical analyses were performed centrally using RICOPILI (Rapid Imputation for COnsortias PIpeLIne) (version 2018_Nov_23.001)94, separately for each cohort. Briefly, the quality control parameters for retaining SNPs and subjects were: SNP missingness < 0.05 (before sample removal), subject missingness < 0.02, autosomal heterozygosity deviation (Fhet < 0.2), SNP missingness < 0.02 (after sample removal), difference in SNP missingness between cases and controls < 0.02, SNP Hardy-Weinberg equilibrium (P > 10 × 10−10 in psychiatric cases and P > 10 × 10−6 in controls). Relatedness was calculated across cohorts using identity by descent and one of each pair of related individuals (pi_hat > 0.2) was excluded. Principal components (PCs) were generated using genotyped SNPs in each cohort separately using EIGENSTRAT v6.1.495. Based on visual inspection of plots of PCs for each dataset (which were all of European descent according to self-report/clinical data), we excluded samples to obtain more clearly homogeneous datasets. Genotype imputation was performed using the pre-phasing/ imputation stepwise approach implemented in Eagle v2.3.596 and Minimac397 to the Haplotype Reference Consortium (HRC) reference panel v1.098. Data on the X chromosome were available for 50 cohorts internal to the PGC and one external cohort (HUNT), and the X chromosome was imputed to the HRC reference panel in males and females separately within each cohort. The five external cohorts were processed by the collaborating research teams using comparable procedures and imputed to the HRC or a custom reference panel as appropriate. Full details of the genotyping, quality control and imputation for each of these cohorts are available in the Supplementary Note. Identical individuals between PGC cohorts and the Estonian Biobank and UK Biobank cohorts were detected using genotype-based checksums (https://personal.broadinstitute.org/sripke/share_links/zpXkV8INxUg9bayDpLToG4g58TMtjN_PGC_SCZ_w3.0718d.76) and removed from PGC cohorts.
Genome-wide association study.
For PGC cohorts, GWAS were conducted within each cohort using an additive logistic regression model in PLINK v1.9099, covarying for PCs 1–5 and any others as required. Association analyses of the X chromosome were conducted in males and females separately using the same procedures, with males coded as 0 or 2 for 0 or 1 copies of the reference allele. Results from males and females were then meta-analyzed within each cohort. For external cohorts, GWAS were conducted by the collaborating research teams using comparable procedures (Supplementary Note). To control test statistic inflation at SNPs with low minor allele frequency (MAF) in small cohorts, SNPs were retained only if cohort MAF was > 1% and minor allele count was > 10 in either cases or controls (whichever had smaller n). There was no evidence of stratification artifacts or uncontrolled inflation of test statistics in the results from any cohort (λGC = 0.97–1.05) (Supplementary Table 1). Meta-analysis of GWAS summary statistics was conducted using an inverse variance-weighted fixed effects model in METAL (version 2011-03-25)100 across 57 cohorts for the autosomes (41,917 BD cases and 371,549 controls) and 51 cohorts for the X chromosome (35,691 BD cases and 96,731 controls). A genome-wide significant locus was defined as the region around a SNP with P < 5 × 10−8, with linkage disequilibrium (LD) r2 > 0.1, within a 3,000 kilobase (kb) window. Regional association plots and forest plots of the index SNP for all genome-wide significant loci are presented in Supplementary Data 1 and 2, respectively.
Overlap of loci with other psychiatric disorders.
Genome-wide significant loci for BD were assessed for overlap with genome-wide significant loci for other psychiatric disorders, using the largest available GWAS results for major depression61, schizophrenia60, attention deficit/hyperactivity disorder101, post-traumatic stress disorder102, lifetime anxiety disorder103, Tourette’s Syndrome104, anorexia nervosa105, alcohol use disorder or problematic alcohol use68, autism spectrum disorder106, mood disorders91 and the cross-disorder GWAS of the Psychiatric Genomics Consortium66. The boundaries of the genome-wide significant loci were calculated in the original publications. Overlap of loci was calculated using bedtools v2.29.2107.
Enrichment analyses.
P values quantifying the degree of association of genes and gene sets with BD were calculated using MAGMA v1.0837, implemented in FUMA v1.3.6a64,108. Gene-based tests were performed for 19,576 genes (Bonferroni-corrected P-value threshold = 2.55 × 10−6). A total of 11,858 curated gene sets including at least 10 genes from MSigDB V7.0 were tested for association with BD (Bonferroni-corrected P-value threshold = 4.22 × 10−6). Competitive gene-set tests were conducted correcting for gene size, variant density and LD within and between genes. Tissue-set enrichment analyses were also performed using MAGMA implemented in FUMA, to test for enrichment of association signal in genes expressed in 54 tissue types from GTEx V8 (Bonferroni-corrected P-value threshold = 9.26 × 10−4)64,108.
For single-cell enrichment analyses, publicly available single-cell RNA-seq data were compiled from five studies of the adult human and mouse brain86,109–112. The mean expression for each gene in each cell type was computed from the single-cell expression data (if not provided). For the Zeisel dataset109, we used the mean expression at level 4 (39 cell types from 19 regions for the mouse nervous system). For the Saunders dataset110, we computed the mean expression of the different classes in each of the 9 different brain regions sampled (88 cell types in total). We filtered out any genes with non-unique names, genes not expressed in any cell types, non-protein coding genes, and, for mouse datasets, genes that had no expert curated 1:1 orthologs between mouse and human (Mouse Genome Informatics, The Jackson Laboratory, version 11/22/2016, http://www.informatics.jax.org/downloads/reports/index.html#homology), resulting in 16,472 genes. Gene expression was then scaled to a total of 1 million UMIs (unique molecular identifiers) (or transcript per million (TPM)) for each cell type/tissue. Using a previously described method38, a metric of gene expression specificity was calculated by dividing the expression of each gene in each cell type by the total expression of that gene in all cell types, leading to values ranging from 0 to 1 for each gene (0 meaning that the gene is not expressed in that cell type and 1 meaning that all of the expression of the gene is in that cell type). We then selected the top 10% most specific genes for each cell type/tissue for enrichment analysis. MAGMA v1.0837 was used to test gene-set enrichment using GWAS summary statistics, covarying for gene size, gene density, mean sample size for tested SNPs per gene, the inverse of the minor allele counts per gene and the log of these metrics. We excluded any SNPs with INFO score < 0.6, with MAF < 1% or with estimated odds ratio > 25 or smaller than 1/25, as well as SNPs located in the MHC region (chr6:25–34 Mb). We set a window of 35 kb upstream to 10 kb downstream of the gene coordinates to compute gene-level association statistics and used the European reference panel from the phase 3 of the 1000 Genomes Project as the reference population113. We then used MAGMA to test whether the 10% most specific genes (with an expression of at least 1 TPM or 1 UMI per million) for each cell type/tissue were associated with BD. The P-value threshold for significance was P < 9.1 × 10−3, representing a 5% false discovery rate (FDR) across datasets.
Further gene-set analyses were performed restricted to genes targeted by drugs, assessing individual drugs and grouping drugs with similar actions. This approach has been described previously41. Gene-level and gene-set analyses were performed in MAGMA v1.0837. Gene boundaries were defined using build 37 reference data from the NCBI, available on the MAGMA website (https://ctg.cncr.nl/software/magma), extended 35 kb upstream and 10 kb downstream to include regulatory regions outside of the transcribed region. Gene-level association statistics were defined as the aggregate of the mean and the lowest variant-level P value within the gene boundary, converted to a Z-value. Gene sets were defined comprising the targets of each drug in the Drug-Gene Interaction database DGIdb v.239 and in the Psychoactive Drug Screening Database Ki DB40, both downloaded in June 201641. Analyses were performed using competitive gene-set analyses in MAGMA. Results from the drug-set analysis were then grouped according to the Anatomical Therapeutic Chemical class of the drug41. Only drug classes with at least 10 valid drug gene sets within them were analyzed. Drug-class analysis was performed using enrichment curves. All drug gene sets were ranked by their association in the drug set analysis, and then for a given drug class an enrichment curve was drawn scoring a “hit” if the drug gene set was within the class, or a “miss” if it was outside of the class. The area under the curve was calculated, and a P-value for this calculated as the Wilcoxon Mann-Whitney test comparing drug gene sets within the class to drug gene sets outside of the class41. Multiple testing was controlled using a Bonferroni-corrected significance threshold of P < 5.60 × 10−5 for drug-set analysis and P < 7.93 × 10−4 for drug-class analysis, accounting for 893 drug-sets and 63 drug classes tested.
eQTL integrative analysis.
A transcriptome-wide association study (TWAS) was conducted using the precomputed gene expression weights from PsychENCODE data (1,321 brain samples)43, available online with the FUSION software42. For genes with significant cis-SNP heritability (13,435 genes), FUSION software (vOct 1, 2019) was used to test whether SNPs influencing gene expression are also associated with BD (Bonferroni-corrected P-value threshold < 3.72 × 10−6). For regions including a TWAS significant gene, TWAS fine-mapping of the region was conducted using FOCUS (fine-mapping of causal gene sets, v0.6.10)44. Regions were defined using the correlation matrix of predicted effects on gene expression around TWAS significant genes44. A posterior inclusion probability (PIP) was assigned to each gene for being causal for the observed TWAS association signal. Based on the PIP of each gene and a null model, whereby no gene in the region is causal for the TWAS signal, the 90%-credible gene set for each region was computed44.
Summary data-based Mendelian randomization (SMR) (v1.03)45,46 was applied to further investigate putative causal relationships between SNPs and BD via gene expression. SMR was performed using eQTL summary statistics from the eQTLGen (31,684 blood samples)47 and PsychENCODE43 consortia. SMR analysis is limited to transcripts with at least one significant cis-eQTL (P < 5 × 10−8) in each dataset (15,610 in eQTLGen; 10,871 in PsychENCODE). The Bonferroni-corrected significance threshold was P < 3.20 × 10−6 and P < 4.60 × 10−6 for eQTLGen and PsychENCODE, respectively. The significance threshold for the HEIDI test (heterogeneity in dependent instruments) was PHEIDI ≥ 0.0146. While the results of TWAS and SMR indicate an association between BD and gene expression, a non-significant HEIDI test additionally indicates either a direct causal role or a pleiotropic effect of the BD-associated SNPs on gene expression.
Complement component 4 (C4) imputation.
To investigate the major histocompatibility complex (MHC; chr6:24–34 Mb on hg19), the alleles of complement component 4 genes (C4A and C4B) were imputed in 47 PGC cohorts for which individual-level genotype data were accessible, totaling 32,749 BD cases and 53,370 controls. The imputation reference panel comprised 2,530 reference haplotypes of MHC SNPs and C4 alleles, generated using a sample of 1,265 individuals with whole-genome sequence data, from the Genomic Psychiatry cohort114. Briefly, imputation of C4 as a multi-allelic variant was performed using Beagle v4.1115,116, using SNPs from the MHC region that were also in the haplotype reference panel. Within the Beagle pipeline, the reference panel was first converted to bref format. We used the conform-gt tool to perform strand-flipping and filtering of specific SNPs for which strand remained ambiguous. Beagle was run using default parameters with two key exceptions: we used the GRCh37 PLINK recombination map, and we set the output to include genotype probability (i.e., GP field in VCF) for correct downstream probabilistic estimation of C4A and C4B joint dosages. The output consisted of dosage estimates for each of the common C4 structural haplotypes for each individual. The five most common structural forms of the C4A/C4B locus (BS, AL, AL-BS, AL-BL, and AL-AL) could be inferred with reasonably high accuracy (generally 0.70 < r2 < 1.00). The imputed C4 alleles were tested for association with BD in a joint logistic regression that included (i) terms for dosages of the five most common C4 structural haplotypes (AL-BS, AL-BL, AL-AL, BS, and AL), (ii) rs13195402 genotype (top lead SNP in the MHC) and (iii) PCs as per the GWAS. The genetically regulated expression of C4A was predicted from the imputed C4 alleles using a model previously described63. Predicted C4A expression was tested for association with BD in a joint logistic regression that included (i) predicted C4A expression, (ii) rs13195402 genotype (top lead SNP in the MHC) and (iii) PCs as per the GWAS.
Polygenic risk scoring.
PRS from our GWAS meta-analysis were tested for association with BD in individual cohorts, using a discovery GWAS where the target cohort was left out of the meta-analysis. Briefly, the GWAS results from each discovery GWAS were pruned for LD using the P-value informed clumping method in PLINK v1.9099 (r2 0.1 within a 500-kb window) based on the LD structure of the HRC reference panel98. Subsets of SNPs were selected from the results below nine increasingly liberal P-value thresholds (PT) (5 × 10−8, 1 × 10−4, 1 × 10−3, 0.01, 0.05, 0.1, 0.2, 0.5, 1). Sets of alleles, weighted by their log odds ratios from the discovery GWAS, were summed into PRS for each individual in the target datasets, using PLINK v1.90 implemented via RICOPILI94,99. PRS were tested for association with BD in the target dataset using logistic regression, covarying for PCs as per the GWAS in each cohort. PRS were tested in the external cohorts by the collaborating research teams using comparable procedures. The variance explained by the PRS (R2) was converted to the liability scale to account for the proportion of cases in each target dataset, using a BD population prevalence of 2% and 1%117. The weighted average R2 values were calculated using the effective n for each cohort. The odds ratios for BD for individuals in the top decile of PRS compared with those in the lowest decile and middle decile were calculated in the 52 datasets internal to the PGC. To assess cross-ancestry performance, PRS generated from the meta-analysis results were tested for association with BD using similar methods in a Japanese sample59, a Korean sample92, and two admixed African American samples. Full details of the QC, imputation, and analysis of these samples are in the Supplementary Note.
LD score regression.
LD Score regression (LDSC)35 was used to estimate the of BD from GWAS summary statistics. was converted to the liability scale, using a lifetime BD prevalence of 2% and 1%. LDSC bivariate genetic correlations attributable to genome-wide SNPs (rg) were estimated with 255 human diseases and traits from published GWAS and 514 GWAS of phenotypes in the UK Biobank from LD Hub48. Adjusting for the number of traits tested, the Bonferroni-corrected P-value thresholds were P < 1.96 × 10−4 and P < 9.73 × 10−5, respectively.
MiXeR.
We applied causal mixture models49,118 to the GWAS summary statistics, using MiXeR v1.3. MiXeR provides univariate estimates of the proportion of non-null SNPs (“polygenicity”) and the variance of effect sizes of non-null SNPs (“discoverability”) in each phenotype. For each SNP, i, univariate MiXeR models its additive genetic effect of allele substitution, βi, as a point-normal mixture, , where π1 represents the proportion of non-null SNPs (`polygenicity`) and represents variance of effect sizes of non-null SNPs (`discoverability`). Then, for each SNP, j, MiXeR incorporates LD information and allele frequencies for M = 9,997,231 SNPs extracted from 1000 Genomes Phase 3 data to estimate the expected probability distribution of the signed test statistic, , where N is sample size, Hi indicates heterozygosity of i-th SNP, rij indicates allelic correlation between i-th and j-th SNPs, and is the residual variance. Further, the three parameters, π1, , , are fitted by direct maximization of the likelihood function. The optimization is based on a set of approximately 600,000 SNPs, obtained by selecting a random set of 2,000,000 SNPs with minor allele frequency of 5% or higher, followed by LD pruning procedure at LD r2 = 0.8 threshold. The random SNP selection and full optimization procedure are repeated 20 times to obtain mean and standard errors of model parameters. The log-likelihood figures show individual curves for each of the 20 runs, each shifted vertically so that best log-likelihood point is shown at zero ordinate.
The total number of trait influencing variants is estimated as Mπ1, where M = 9,997,231 gives the number of SNPs in the reference panel. MiXeR Venn diagrams report the effective number of influencing variants, ηMπ_1, where η is a fixed number, η = 0.319, which gives the fraction of influencing variants contributing to 90% of trait’s heritability (with rationale for this adjustment being that the remaining 68.1% of influencing variants are small and cumulatively explain only 10% of trait’s heritability). Phenotypic variance explained on average by an influencing genetic variant is calculated as , where is the average heterozygosity across SNPs in the reference panel. Under the assumptions of the MiXeR model, SNP-heritability is then calculated as .
In the cross-trait analysis, MiXeR models additive genetic effects as a mixture of four components, representing null SNPs in both traits (π0); SNPs with a specific effect on the first and on the second trait (π1 and π2, respectively); and SNPs with non-zero effect on both traits (π12). In the last component, MiXeR models variance-covariance matrix as where ρ12 indicates correlation of effect sizes within the shared component, and and correspond to the discoverability parameter estimated in the univariate analysis of the two traits. These components are then plotted in Venn diagrams. After fitting parameters of the model, the Dice coefficient of polygenic overlap is then calculated as , and genetic correlation is calculated as . Fraction of influencing variants with concordant effect direction is calculated as twice the multivariate normal CDF at point (0, 0) for the bivariate normal distribution with zero mean and variance-covariance matrix Σ12. All code is available online (https://github.com/precimed/mixer).
Mendelian randomization.
We selected 17 traits associated with BD in clinical or epidemiological studies for Mendelian randomization (MR) to dissect their relationship with BD (Supplementary Note). Bi-directional generalized summary statistics-based MR (GSMR)51 analyses were performed between BD and the traits of interest using GWAS summary statistics, implemented in GCTA software (v1.93.1f beta). The instrumental variables (IVs) were selected by a clumping procedure internal to the GSMR software with parameters: --gwas-thresh 5e-8 --clump-r2 0.01. Traits with less than 10 IVs available were excluded from the GSMR analyses to avoid conducting underpowered tests51, resulting in 10 traits tested (Bonferroni-corrected P-value threshold < 2.5 × 10−3). The HEIDI-outlier test (heterogeneity in dependent instruments) was applied to test for horizontal pleiotropy (PHEIDI < 0.01)51. For comparison, the MR analyses were also performed using the inverse variance weighted regression method, implemented via the TwoSampleMR R package, using the IVs selected by GSMR119,120. To further investigate horizontal pleiotropy, the MR Egger intercept test was conducted using the TwoSampleMR package119,120 and MR-PRESSO software was used to perform the Global Test and Distortion Test121.
BD subtypes.
GWAS meta-analyses were conducted for BD I (25,060 cases, 449,978 controls from 55 cohorts, effective n = 64,802) and BD II (6,781 cases, 364,075 controls from 31 cohorts, effective n = 22,560) (Supplementary Table 1) using the same procedures described for the main GWAS. BD subtypes were defined based on international consensus criteria (DSM-IV, ICD-9, or ICD-10), established using structured diagnostic instruments from assessments by trained interviewers, clinician-administered checklists or medical record review. In the external biobank cohorts, BD subtypes were defined using ICD codes (Supplementary Note). LDSC35 was used to estimate the of each subtype, and the genetic correlation between the subtypes. The difference between the LDSC estimates for BD I and BD II was tested for deviation from 0 using the block jackknife122. The LDSC genetic correlation (rg) was tested for difference from 1 by calculating a chi-square statistic corresponding to the estimated rg as ((rg − 1)/ SE)2.
Supplementary Material
Acknowledgements
We thank the participants who donated their time, life experiences and DNA to this research and the clinical and scientific teams that worked with them. We are deeply indebted to the investigators who comprise the PGC. The PGC has received major funding from the US National Institute of Mental Health (PGC3: U01 MH109528; PGC2: U01 MH094421; PGC1: U01 MH085520). Statistical analyses were carried out on the NL Genetic Cluster Computer (http://www.geneticcluster.org) hosted by SURFsara and the Mount Sinai high performance computing cluster (http://hpc.mssm.edu), which is supported by the Office of Research Infrastructure of the National Institutes of Health under award numbers S10OD018522 and S10OD026880. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Full acknowledgements are included in the Supplementary Note.
Footnotes
Data availability
GWAS summary statistics are publicly available on the PGC website (https://www.med.unc.edu/pgc/results-and-downloads). Individual-level data are accessible through collaborative analysis proposals to the Bipolar Disorder Working Group of the PGC (https://www.med.unc.edu/pgc/shared-methods/how-to/). This study included some publicly available datasets accessed through dbGaP (PGC bundle phs001254.v1.p1) and the Haplotype Reference Consortium reference panel v1.0 (http://www.haplotype-reference-consortium.org/home). Databases used: Drug-Gene Interaction Database DGIdb v.2 (https://www.dgidb.org); Psychoactive Drug Screening Database Ki DB (https://pdsp.unc.edu/databases/kidb.php); DrugBank 5.0 (www.drugbank.ca); LDHub (http://ldsc.broadinstitute.org); FUMA (https://fuma.ctglab.nl).
Code availability
All software used is publicly available at the URLs or references cited.
Consortia
HUNT All-In Psychiatry
Bendik S. Winsvold19,33, Eystein Stordal221,222, Gunnar Morken21,198, John-Anker Zwart18,19,33, Ole Kristian Drange21,22, Sigrid Børte17,18,19
A full list of consortium members and affiliations appears in the Supplementary Note.
Competing interests
T.E. Thorgeirsson, S. Steinberg, H. Stefansson and K. Stefansson are employed by deCODE Genetics/Amgen. Multiple additional authors work for pharmaceutical or biotechnology companies in a manner directly analogous to academic co-authors and collaborators. A.H. Young has given paid lectures and advisory boards for the following companies with drugs used in affective and related disorders:
Astrazenaca, Eli Lilly, Lundbeck, Sunovion, Servier, Livanova, Janssen, Allegan, Bionomics, Sumitomo Dainippon Pharma. A.H. Young was Lead Investigator for Embolden Study (Astra Zeneca), BCI Neuroplasticity study and Aripiprazole Mania Study. Also an investigator for Janssen, Lundbeck, Livanova, Compass. J. Nurnberger is an investigator for Janssen. P.F. Sullivan reports the following potentially competing financial interests: Lundbeck (advisory committee), Pfizer (Scientific Advisory Board member), and Roche (grant recipient, speaker reimbursement). G. Breen reports consultancy and speaker fees from Eli Lilly and Illumina and grant funding from Eli Lilly. M. Landén has received speaker fees from Lundbeck. O.A. Andreassen has received speaker fees from Lundbeck. J.Antoni Ramos-Quiroga was on the speakers bureau and/or acted as consultant for Eli-Lilly, Janssen-Cilag, Novartis, Shire, Lundbeck, Almirall, Braingaze, Sincrolab, and Rubió in the last 5 years. He also received travel awards (air tickets + hotel) for taking part in psychiatric meetings from Janssen-Cilag, Rubió, Shire, and Eli- Lilly. The Department of Psychiatry chaired by him received unrestricted educational and research support from the following companies in the last 5 years: Eli-Lilly, Lundbeck, Janssen- Cilag, Actelion, Shire, Ferrer, Oryzon, Roche, Psious, and Rubió. Dr. E. Vieta has received grants and served as consultant, advisor or CME speaker for the following entities: AB-Biotics, Abbott, Allergan, Angelini, AstraZeneca, Bristol-Myers Squibb, Dainippon Sumitomo Pharma, Farmindustria, Ferrer, Forest Research Institute, Gedeon Richter, Glaxo-Smith-Kline, Janssen, Lundbeck, Otsuka, Pfizer, Roche, SAGE, Sanofi-Aventis, Servier, Shire, Sunovion, Takeda, the Brain and Behaviour Foundation, the Catalan Government (AGAUR and PERIS), the Spanish Ministry of Science, Innovation, and Universities (AES and CIBERSAM), the Seventh European Framework Programme and Horizon 2020, and the Stanley Medical Research Institute. T. Elvsåshagen has received speaker fees from Lundbeck. S. Kittel-Schneider received author’s and consultant honoraria from Medice Arzneimittel Pütter GmbH and Shire/Takeda. Prof. Serretti is or has been consultant/speaker for: Abbott, Abbvie, Angelini, Astra Zeneca, Clinical Data, Boheringer, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Innovapharma, Italfarmaco, Janssen, Lundbeck, Naurex, Pfizer, Polifarma, Sanofi, Servier. J.R. DePaulo has served as an unpaid consultant to Myriad – Neuroscience (formerly Assurex Health) in 2017 and 2019 and owns stock in CVS Health. H.R. Kranzler serves as an advisory board member for Dicerna Pharmaceuticals, is a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was sponsored in the past three years by AbbVie, Alkermes, Amygdala Neurosciences, Arbor Pharmaceuticals, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, and Pfizer. HRK is named as an inventor on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed January 24, 2018. ALM and CL are employees of Indivior, Inc. AA was an employee of Indivior, Inc. at the time the work was performed.
All other authors declare no financial interests or potential conflicts of interest.
References
- 1.GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1211–1259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plans L et al. Association between completed suicide and bipolar disorder: A systematic review of the literature. J. Affect. Disord 242, 111–122 (2019). [DOI] [PubMed] [Google Scholar]
- 3.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders 5th edn. (American Psychiatric Association Publishing, 2013). [Google Scholar]
- 4.Merikangas KR et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey replication. Arch. Gen. Psychiatry 64, 543–552 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merikangas KR et al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch. Gen. Psychiatry 68, 241–251 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craddock N & Sklar P Genetics of bipolar disorder. Lancet 381, 1654–1662 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Song J et al. Bipolar disorder and its relation to major psychiatric disorders: a family-based study in the Swedish population. Bipolar Disord 17, 184–193 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Bienvenu OJ, Davydow DS & Kendler KS Psychiatric ‘diseases’ versus behavioral disorders and degree of genetic influence. Psychol. Med 41, 33–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet 43, 977–983 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baum AE et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol. Psychiatry 13, 197–207 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charney AW et al. Evidence for genetic heterogeneity between clinical subtypes of bipolar disorder. Transl. Psychiatry 7, e993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen DT et al. Genome-wide association study meta-analysis of European and Asian-ancestry samples identifies three novel loci associated with bipolar disorder. Mol. Psychiatry 18, 195–205 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Cichon S et al. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am. J. Hum. Genet 88, 372–381 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreira MAR et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet 40, 1056–1058 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green EK et al. Association at SYNE1 in both bipolar disorder and recurrent major depression. Mol. Psychiatry 18, 614–617 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Green EK et al. Replication of bipolar disorder susceptibility alleles and identification of two novel genome-wide significant associations in a new bipolar disorder case-control sample. Mol. Psychiatry 18, 1302–1307 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou L et al. Genome-wide association study of 40,000 individuals identifies two novel loci associated with bipolar disorder. Hum. Mol. Genet 25, 3383–3394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mühleisen TW et al. Genome-wide association study reveals two new risk loci for bipolar disorder. Nat. Commun 5, 3339 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Schulze TG et al. Two variants in Ankyrin 3 (ANK3) are independent genetic risk factors for bipolar disorder. Mol. Psychiatry 14, 487–491 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott LJ et al. Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc. Natl. Acad. Sci. USA 106, 7501–7506 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sklar P et al. Whole-genome association study of bipolar disorder. Mol. Psychiatry 13, 558–569 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith EN et al. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol. Psychiatry 14, 755–763 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stahl EA et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet 51, 793–803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee S-H, Zabolotny JM, Huang H, Lee H & Kim Y-B Insulin in the nervous system and the mind: Functions in metabolism, memory, and mood. Mol. Metab 5, 589–601 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McIntyre RS et al. A randomized, double-blind, controlled trial evaluating the effect of intranasal insulin on neurocognitive function in euthymic patients with bipolar disorder. Bipolar Disord 14, 697–706 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Nurnberger JI Jr et al. Identification of pathways for bipolar disorder: a meta-analysis. JAMA Psychiatry 71, 657–664 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gordovez FJA & McMahon FJ The genetics of bipolar disorder. Mol. Psychiatry 25, 544–559 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Zhang C, Xiao X, Li T & Li M Translational genomics and beyond in bipolar disorder. Mol. Psychiatry 26, 186–202 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Pedersen CB et al. The iPSYCH2012 case-cohort sample: New directions for unravelling genetic and environmental architectures of severe mental disorders. Mol. Psychiatry 1, 6–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gudbjartsson DF et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet 47, 435–444 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Leitsalu L et al. Cohort Profile: Estonian Biobank of the Estonian Genome Center, University of Tartu. Int. J. Epidemiol 44, 1137–1147 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Krokstad S et al. Cohort Profile: the HUNT Study, Norway. Int. J. Epidemiol 42, 968–977 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Sudlow C et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loh P-R, Kichaev G, Gazal S, Schoech AP & Price AL Mixed-model association for biobank-scale datasets. Nat. Genet 50, 906–908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bryois J et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat. Genet 52, 482–493 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner AH et al. DGIdb 2.0: mining clinically relevant drug-gene interactions. Nucleic Acids Res 44, D1036–D1044 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roth BL, Lopez E, Patel S & Kroeze WK The multiplicity of serotonin receptors: uselessly diverse molecules or an embarrassment of riches? Neuroscientist 6, 252–262 (2000). [Google Scholar]
- 41.Gaspar HA & Breen G Drug enrichment and discovery from schizophrenia genome-wide association results: an analysis and visualisation approach. Sci. Rep 7, 12460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gandal MJ et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mancuso N et al. Probabilistic fine-mapping of transcriptome-wide association studies. Nat. Genet 51, 675–682 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Z et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet 48, 481–487 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Wu Y et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun 9, 918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Võsa U et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. Preprint at bioRxiv (2018) doi: 10.1101/447367. [DOI] [Google Scholar]
- 48.Zheng J et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frei O et al. Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat. Commun 10, 2417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holland D et al. Beyond SNP heritability: polygenicity and discoverability of phenotypes estimated with a univariate Gaussian mixture model. PLoS Genet 16, e1008612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Z et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun 9, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steardo L Jr et al. Sleep disturbance in bipolar disorder: neuroglia and circadian rhythms. Front. Psychiatry 10, 501 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hunt GE, Malhi GS, Cleary M, Lai HMX & Sitharthan T Prevalence of comorbid bipolar and substance use disorders in clinical settings, 1990–2015: Systematic review and meta-analysis. J. Affect. Disord 206, 331–349 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Heffner JL, Strawn JR, DelBello MP, Strakowski SM & Anthenelli RM The co-occurrence of cigarette smoking and bipolar disorder: phenomenology and treatment considerations. Bipolar Disord 13, 439–453 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vreeker A et al. High educational performance is a distinctive feature of bipolar disorder: a study on cognition in bipolar disorder, schizophrenia patients, relatives and controls. Psychol. Med 46, 807–818 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.MacCabe JH et al. Excellent school performance at age 16 and risk of adult bipolar disorder: national cohort study. Br. J. Psychiatry 196, 109–115 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Broome MR, Saunders KEA, Harrison PJ & Marwaha S Mood instability: significance, definition and measurement. Br. J. Psychiatry 207, 283–285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ward J et al. The genomic basis of mood instability: identification of 46 loci in 363,705 UK Biobank participants, genetic correlation with psychiatric disorders, and association with gene expression and function. Mol. Psychiatry 25, 3091–3099 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ikeda M et al. A genome-wide association study identifies two novel susceptibility loci and trans population polygenicity associated with bipolar disorder. Mol. Psychiatry 23, 639–647 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pardiñas AF et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet 50, 381–389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Howard DM et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci 22, 343–352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smeland OB, Frei O, Dale AM & Andreassen OA The polygenic architecture of schizophrenia - rethinking pathogenesis and nosology. Nat. Rev. Neurol 16, 366–379 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Sekar A et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.GTEx Consortium et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brainstorm Consortium et al. Analysis of shared heritability in common disorders of the brain. Science 360, eaap8757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 179, 1469–1482.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lewis KJS et al. Comparison of genetic liability for sleep traits among individuals with bipolar disorder I or II and control participants. JAMA Psychiatry 77, 303–310 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou H et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat. Neurosci 23, 809–818 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Okbay A et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vermeulen JM et al. Smoking and the risk for bipolar disorder: evidence from a bidirectional Mendelian randomisation study. Br. J. Psychiatry 218, 88–94 (2021). [DOI] [PubMed] [Google Scholar]
- 71.Peyrot WJ et al. The association between lower educational attainment and depression owing to shared genetic effects? Results in ~25 000 subjects. Mol. Psychiatry 20, 735–743 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swanson CL Jr, Gur RC, Bilker W, Petty RG & Gur RE Premorbid educational attainment in schizophrenia: association with symptoms, functioning, and neurobehavioral measures. Biol. Psychiatry 44, 739–747 (1998). [DOI] [PubMed] [Google Scholar]
- 73.Wishart DS et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46, D1074–D1082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mizuno A & Okada Y Biological characterization of expression quantitative trait loci (eQTLs) showing tissue-specific opposite directional effects. Eur. J. Hum. Genet 27, 1745–1756 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schrode N et al. Synergistic effects of common schizophrenia risk variants. Nat. Genet 51, 1475–1485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang X et al. Sodium valproate rescues expression of TRANK1 in iPSC-derived neural cells that carry a genetic variant associated with serious mental illness. Mol. Psychiatry 24, 613–624 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huckins LM et al. Transcriptomic imputation of bipolar disorder and bipolar subtypes reveals 29 novel associated genes. Preprint at bioRxiv (2017) doi: 10.1101/222786. [DOI] [Google Scholar]
- 78.Finan C et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med 9, eaag1166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.von Wegerer J, Hesslinger B, Berger M & Walden J A calcium antagonistic effect of the new antiepileptic drug lamotrigine. Eur. Neuropsychopharmacol 7, 77–81 (1997). [DOI] [PubMed] [Google Scholar]
- 80.Cipriani A et al. A systematic review of calcium channel antagonists in bipolar disorder and some considerations for their future development. Mol. Psychiatry 21, 1324–1332 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harrison PJ, Tunbridge EM, Dolphin AC & Hall J Voltage-gated calcium channel blockers for psychiatric disorders: genomic reappraisal. Br. J. Psychiatry 216, 250–253 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci 18, 199–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Forstner AJ et al. Identification of shared risk loci and pathways for bipolar disorder and schizophrenia. PLoS One 12, e0171595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium. Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell 173, 1705–1715.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee Y, Zhang Y, Kim S & Han K Excitatory and inhibitory synaptic dysfunction in mania: an emerging hypothesis from animal model studies. Exp. Mol. Med 50, 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Skene NG et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet 50, 825–833 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lewis CM & Vassos E Polygenic risk scores: from research tools to clinical instruments. Genome Med 12, 44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Torkamani A, Wineinger NE & Topol EJ The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet 19, 581–590 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Duncan L et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun 10, 3328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martin AR et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet 51, 584–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Coleman JRI et al. The genetics of the mood disorder spectrum: genome-wide association analyses of more than 185,000 cases and 439,000 controls. Biol. Psychiatry 88, 169–184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 92.Moon S et al. The Korea Biobank Array: design and identification of coding variants associated with blood biochemical traits. Sci. Rep 9, 1382 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bigdeli TB et al. Contributions of common genetic variants to risk of schizophrenia among individuals of African and Latino ancestry. Mol. Psychiatry 25, 2455–2467 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lam M et al. RICOPILI: Rapid Imputation for COnsortias PIpeLIne. Bioinformatics 36, 930–933 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Price AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet 38, 904–909 (2006). [DOI] [PubMed] [Google Scholar]
- 96.Loh P-R et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet 48, 1443–1448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Das S et al. Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McCarthy S et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Demontis D et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet 51, 63–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nievergelt CM et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat. Commun 10, 4558 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Purves KL et al. A major role for common genetic variation in anxiety disorders. Mol. Psychiatry 25, 3292–3303 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yu D et al. Interrogating the genetic determinants of Tourette’s syndrome and other tic disorders through genome-wide association studies. Am. J. Psychiatry 176, 217–227 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Watson HJ et al. Genome-wide association study identifies eight risk loci and implicates metabo-psychiatric origins for anorexia nervosa. Nat. Genet 51, 1207–1214 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grove J et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat. Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zeisel A et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Saunders A et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Habib N et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lake BB et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol 36, 70–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kamitaki N et al. Complement genes contribute sex-biased vulnerability in diverse disorders. Nature 582, 577–581 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Browning SR & Browning BL Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet 81, 1084–1097 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Browning BL & Browning SR Genotype imputation with millions of reference samples. Am. J. Hum. Genet 98, 116–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee SH, Goddard ME, Wray NR & Visscher PM A better coefficient of determination for genetic profile analysis. Genet. Epidemiol 36, 214–224 (2012). [DOI] [PubMed] [Google Scholar]
- 118.Holland D et al. Beyond SNP heritability: Polygenicity and discoverability of phenotypes estimated with a univariate Gaussian mixture model. PLoS Genet 16, e1008612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hemani G, Tilling K & Davey Smith G Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 13, e1007081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hemani G et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, e34408 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Verbanck M, Chen C-Y, Neale B & Do R Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet 50, 693–698 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hübel C et al. Genomics of body fat percentage may contribute to sex bias in anorexia nervosa. Am. J. Med. Genet. B Neuropsychiatr. Genet 180, 428–438 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.