

Abstract
Pathogenic bacteria demonstrate incredible abilities to evade conventional antibiotics through the development of resistance and formation of dormant, surface-attached biofilms. Therefore, agents that target and eradicate planktonic and biofilm bacteria are of significant interest. We explored a new series of halogenated phenazines (HP) through the use of N-aryl-2-nitrosoaniline synthetic intermediates that enabled functionalization of the 3-position of this scaffold. Several HPs demonstrated potent antibacterial and biofilm-killing activities (e.g., HP 29, against methicillin-resistant Staphylococcus aureus: MIC = 0.075 μM; MBEC = 2.35 μM), and transcriptional analysis revealed that HPs 3, 28, and 29 induce rapid iron starvation in MRSA biofilms. Several HPs demonstrated excellent activities against Mycobacterium tuberculosis (HP 34, MIC = 0.80 μM against CDC1551). This work established new SAR insights, and HP 29 demonstrated efficacy in dorsal wound infection models in mice. Encouraged by these findings, we believe that HPs could lead to significant advances in the treatment of challenging infections.
Graphical Abstract
1. INTRODUCTION
Novel agents that target multidrug-resistant bacteria are of critical importance due to significant clinical challenges posed by these important human pathogens.1–5 During infection, bacteria can rapidly develop resistance to conventional antibiotic therapies using a multitude of mechanisms, which include (1) target mutation to impede antibiotic binding, (2) alterations in membrane chemistry to reduce drug penetration, (3) increased efflux pump activity to reduce intracellular concentrations of the antibiotic, (4) overproduction of the antibacterial target, and (5) enzymatic deactivation of antibiotics.1,5–10 In contrast to resistance, free-floating planktonic bacteria communicate through quorum sensing to coordinate virulent behaviors,11–13 including the formation of surface-attached biofilm communities composed of enriched populations of dormant persistent cells innately tolerant to antibiotics.14–20 Bacterial biofilms are credited as the primary cause of chronic and recurring infections.1,20–23 As such, new agents capable of targeting antibiotic-resistant bacteria through mechanisms that eradicate surface-attached biofilms are of considerable interest to human health.
Despite extraordinary chemical diversity, conventional antibiotics operate through relatively few modes of action.5,6 Multiple classes of antibiotics inhibit bacterial ribosomes to impede protein synthesis (e.g., macrolides, tetracyclines, and aminoglycosides), while other classes inhibit cell wall synthesis (e.g., beta-lactams and glycopeptides).1,5,6 In addition, select antibiotic therapies inhibit DNA synthesis (e.g., quinolones), RNA polymerase (e.g., rifamycin), and folate synthesis (e.g., sulfonamides), while polymyxins target and disrupt bacterial membranes.1,5,6
Significant efforts to identify novel antibiotics that operate through unique modes of action have been made to overcome resistant and tolerant bacterial infections. A few recent discoveries in the antibiotic arena include the identification of teixobactin (targeting lipid II),24 darobactin (inhibits BamA, an essential chaperone and translocator that folds outer membrane proteins),25 and G0775 (synthetic arylomycin that covalently modifies and inhibits LepB, a membrane-bound protease that cleaves signal sequences from preproteins),26 the development of eNTRy rules to guide synthetic conversion of Gram-positive antibacterials into broad-spectrum agents,27–29 and new group A streptogramin antibiotic analogues that overcome virginiamycin acetyltransferase (Vat) resistance.30 In addition, recent progress has been made to identify agents that can effectively treat persister/biofilm infections in mouse models, including the ClpP protease-activating agent ADEP-4 (synthetic acyldepsipeptide)31 and membrane-disrupting retinoid CD437.32
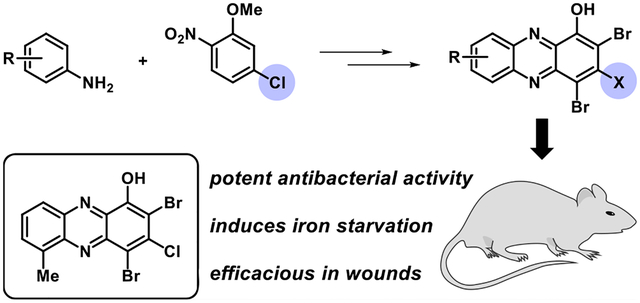
Our group has identified a series of halogenated phenazines (HP) that demonstrate antibacterial and biofilm eradication activities through a unique mechanism (Figure 1).33–39 We were initially interested in phenazine antibiotics (e.g., pyocyanin and phenazine-1-carboxylic acid) utilized by Pseudomonas aeruginosa to eradicate established Staphylococcus aureus infections during Cystic Fibrosis (CF) disease progression.40–43 Inspired by the interspecies competition between P. aeruginosa and S. aureus,40,42 we synthesized a series of phenazine antibiotics and non-natural phenazines, which were tested for antibacterial activities against S. aureus and Staphylococcus epidermidis.33 We discovered that 2-bromo-1-hydroxyphenazine 1, initially isolated from a marine Streptomyces strain, displayed the most potent antibacterial activities against S. aureus among the naturally occurring phenazines in our collection (1, MIC = 6.25 μM against S. aureus and S. epidermidis). Since our initially discovery that 1 exhibits good antibacterial activities against Gram-positive pathogens, we have synthesized a diversity of HPs 2 that have shown incredible potency and utility as probes to better understand biofilm viability.
Figure 1.
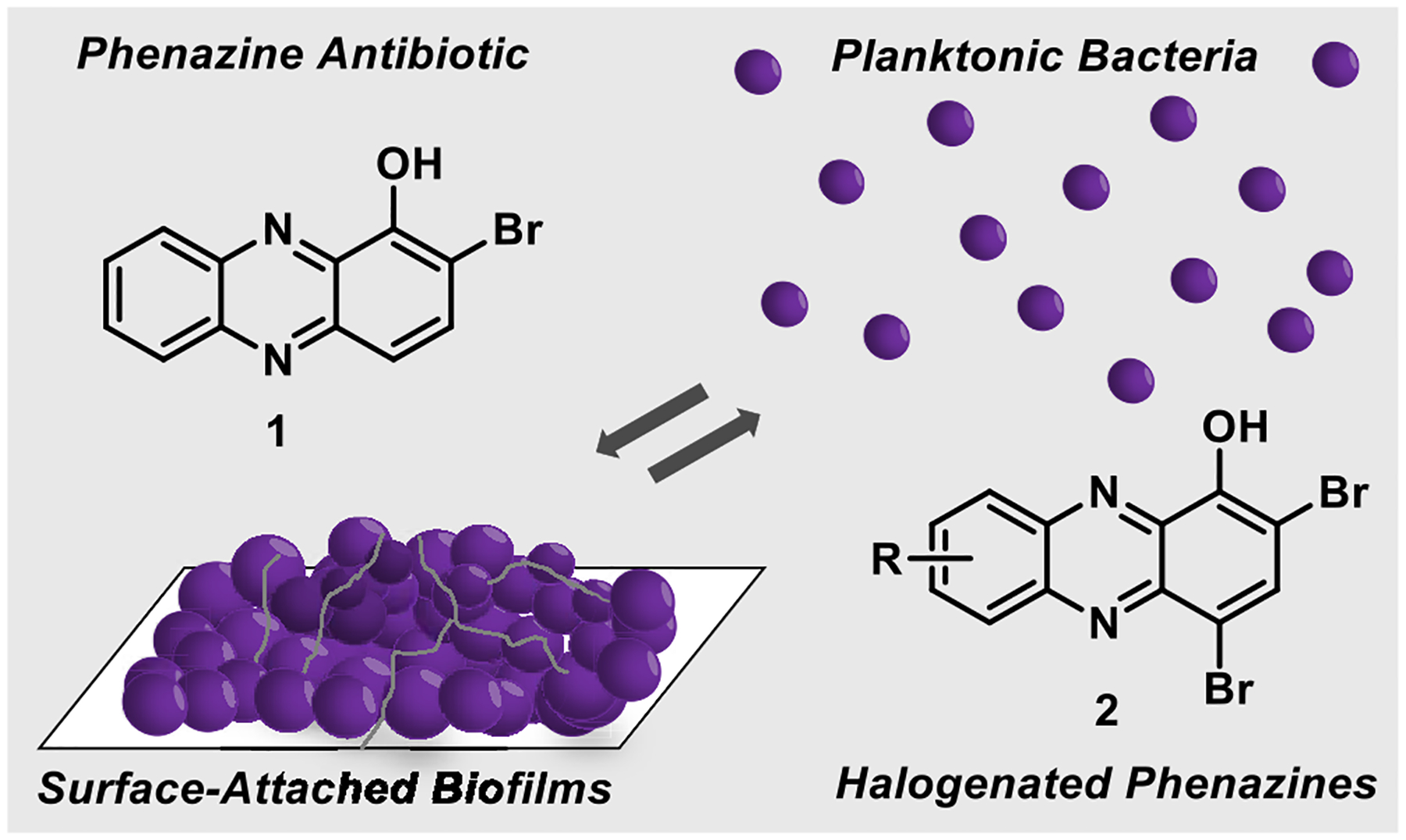
Marine phenazine 2-bromo-1-hydroxyphenazine 1 demonstrates antibacterial activities, and synthetic analogues 2 can eradicate both planktonic cells and established biofilms.
Our previous studies have focused on the (1) synthesis of diverse HPs to explore structure–activity relationships (SAR, see HPs 3–6; Figure 2)36–38 and (2) understanding HP-14’s mode of action by transcript profiling using RNA-seq technology.39 Through the synthesis and microbiological testing of >100 HP analogues, we have shown that several substituents in the 6-, 7-, and 8-positions of the HP scaffold significantly improve antibacterial/biofilm eradication potency, while substitution in the 9-position abolishes antibacterial activity. Transcript profiling of established MRSA BAA-1707 biofilms demonstrated that HP-14 rapidly induces the transcription of several gene clusters involved in iron acquisition (isd, iron-regulated surface determinant; sbn, staphyloferrin B, siderophore; sfa, staphyloferrin A, siderophore; and MW0695, ferrichrome ABC transporter),39 which aligned with our previous findings that HPs bind metal(II) cations, including iron(II).36–38 Overall, we have identified a synthetically tunable series of HPs that demonstrates potent antibacterial activity against planktonic cells and eradicates biofilms through a rapid iron starvation mode of action.
Figure 2.
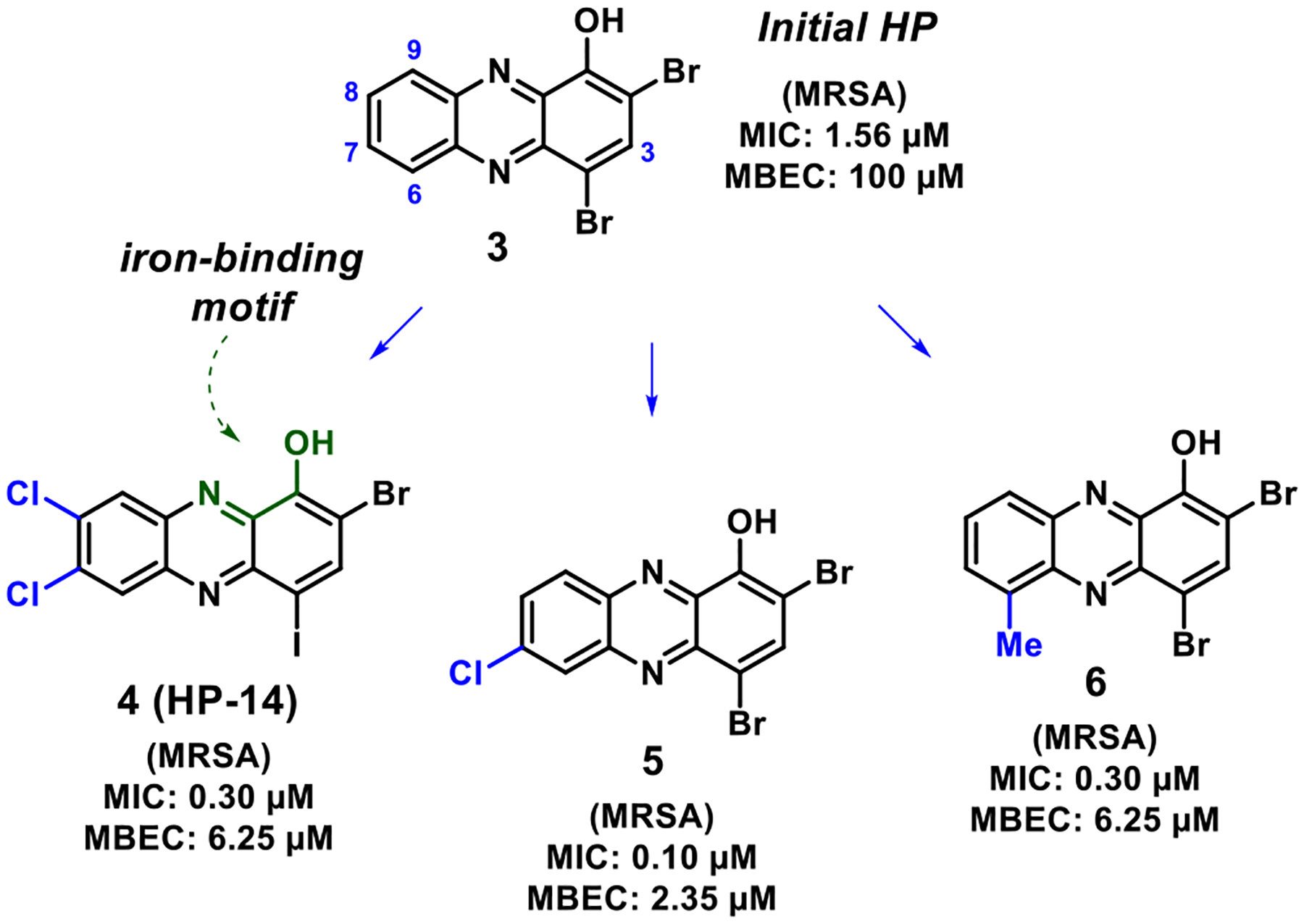
Select halogenated phenazine analogues that demonstrate potent antibacterial and biofilm eradication activities.
Our efforts toward exploring the HP scaffold began with the condensation of 4,5-disubstutied o-phenylenediamines 8 and quinone 9 to yield 1-methoxyphenazines, which were then subjected to (1) boron tribromide (BBr3) demethylation and (2) N-bromosuccinimide (NBS)-mediated bromination to final HPs 7 (Figure 3).35,36 In later work, we fused a series of diverse aniline building blocks 10 with 2-nitroanisole 11 through a Wohl–Aue reaction to access new 1-methoxyphenazine compounds, which were transformed to target HPs for biological investigation.37 Most recently, our lab reported the use of 2-bromo-3-nitroanisole 12 in a modular Buchwald–Hartwig cross-coupling reaction with diverse anilines 10 followed by a reductive cyclization under basic conditions to generate new 1-methoxyphenazines that were advanced to new HPs.38 Here, we report the utilization of N-aryl-2-nitrosoaniline intermediates to enable rapid access to HPs functionalized at the 3-position from anilines 10 and 2-nitro-5-chloroanisole 13 to further explore structure–activity relationships of this potent antibacterial scaffold.
Figure 3.
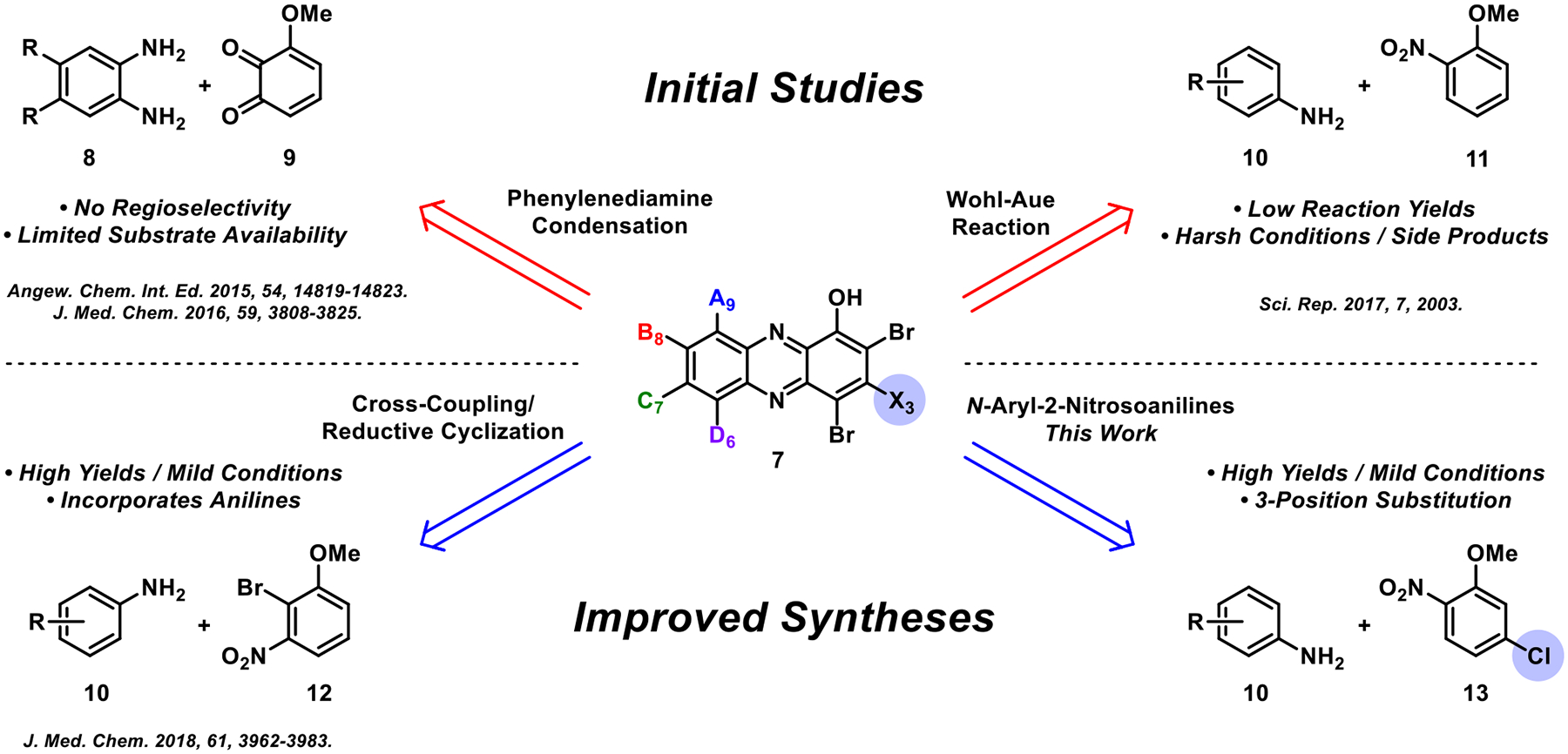
Overview of synthesis strategies utilized to access new HPs for microbiological investigations.
1.1. Design and Synthesis of New Halogenated Phenazines Using a Modular Route.
Our previous efforts to develop HP agents have not included synthetic chemistry that enables exploration at the 3-position of this scaffold. We came across an interesting method reported by Wróbel and co-workers utilizing aniline starting materials to access N-aryl-2-nitrosoaniline intermediates (e.g., 14, Scheme 1) which were then cyclized to phenazines upon treatment with the silylating agent N,O-bis(trimethylsilyl)acetamide (BSA) in N,N-dimethylformamide.44,45 In Wróbel’s approach, 3-chloro-1-methoxyphenazines (15, R = H) were accessed and we envisioned this as an entry point to investigate novel 3-subsituted HPs (e.g., 16).
Scheme 1.
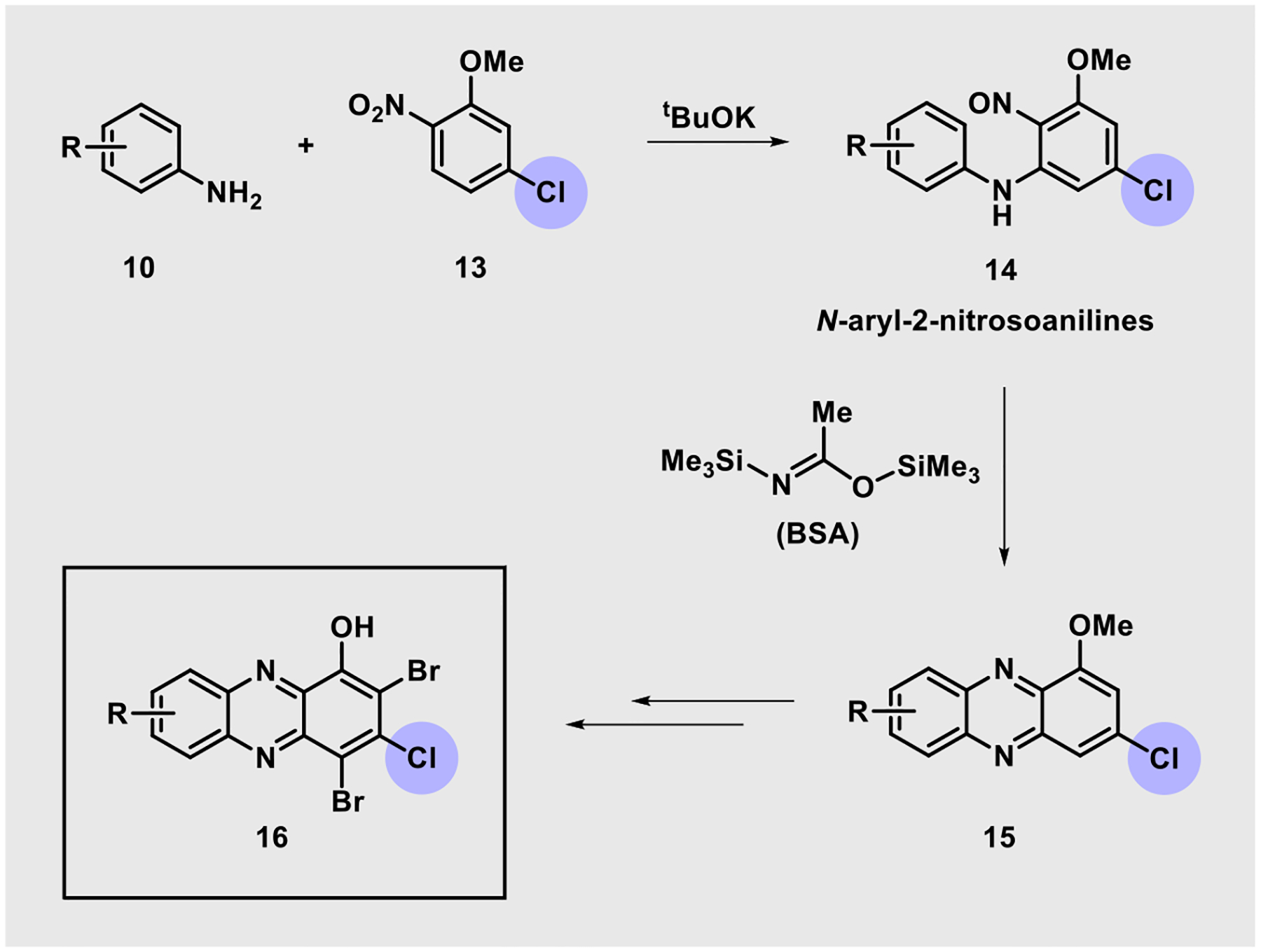
Utilization of N-Aryl-2-nitrosoaniline Intermediates for the Synthesis of New HPs
During the course of these investigations, 10 diverse, commercially available anilines 10 were reacted with potassium tert-butoxide (tBuOK) in the presence of 2-nitro-5-chloroanisole 13 to yield N-aryl-2-nitrosoaniline intermediates (Scheme 2). We had concerns regarding the stability of the corresponding N-aryl-2-nitrosoaniline intermediates, so they were each taken on crude and immediately treated with BSA to yield diverse 3-chloro-1-methoxyphenazines 15. This pathway proved to be fruitful as all 10 aniline starting materials were transformed to target phenazines in 34–82% yield (average yield = 66%) using this synthetic approach. Select N-aryl-2-nitrosoanilines were confirmed through MS analysis during these investigations; however, they were not fully characterized (see the Supporting Information). Following synthesis of the phenazine nucleus (e.g., 16), each 1-methoxyphenazine was subjected to (1) boron tribromide (BBr3) demethylation to the corresponding 1-hydroxyphenazine (18–27, average yield = 98%) and (2) bromination using N-bromosuccinimide (NBS) to generate target HP analogues (28–37, average yield = 59%).
Scheme 2.
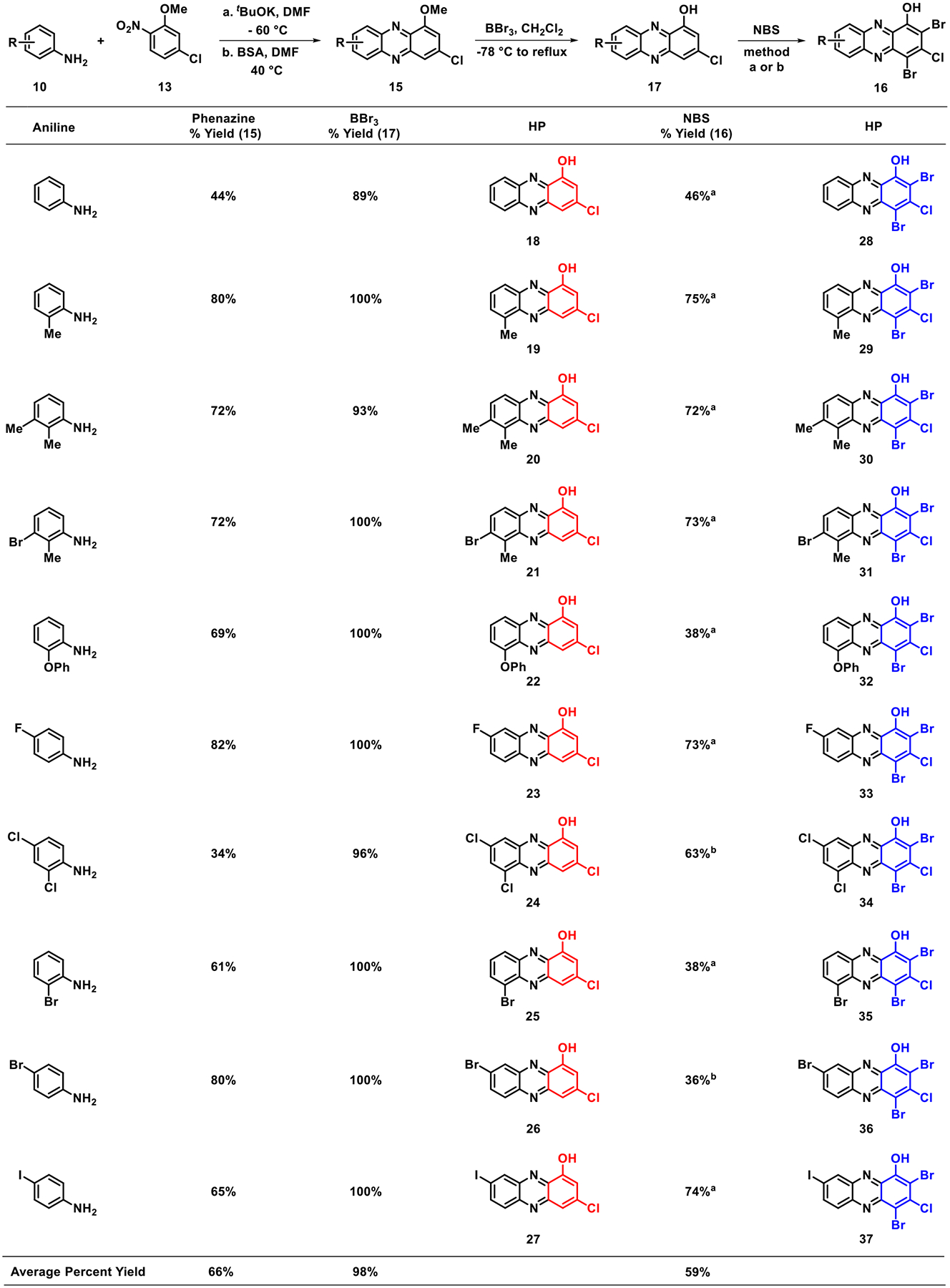
Modular Synthesis of 3-Substituted HPs from Diverse Anilines 10 and 2-Nitro-5-chloroanisole 13
Note: a NBS, CH2Cl2, rt. b NBS, PhMe, 50 °C. NBS: N-bromosuccinimide.
In addition to accessing 3-chloro-HP analogues 18–37, we utilized the chlorine atom as a synthetic handle in SNAr reactions with three thiol nucleophiles to generate 3-thiolated HP analogues 45–47. During these studies, thiols were reacted with 3-chlorophenazine compounds 19 and 38 to yield the corresponding 3-thiolated phenazine 39–44 (Scheme 3). Despite this reaction working well with potassium carbonate (K2CO3) in N,N-dimethylformamide (DMF) under oil bath heating, this reaction required 7 days to complete. Encouragingly, we found that microwave conditions allowed the desired SNAr reaction to occur in 1–3.5 min with good yields (47–100%, Scheme 3A). Interestingly, the 3-methoxy group of phenazine 38 was labile in the presence of the thiol nucleophile under these reaction conditions, yielding a mixture of 1-methoxy and 1-hydroxyphenazine (from demethylation) products that were readily separated via column chromatography. To circumvent problems with product mixtures, we performed the SNAr reaction on 1-hydroxy-3-chloro-6-methylphenazine 19 to yield the desired 3-thiolated products in 75–89% yields (Scheme 3A, entry 2 & 5). Each of the 3-thiol-1-hydroxyphenazines (42–44) was then subjected to NBS bromination to yield target 3-thiolated HP analogues 45–47 in 27–73% yield (Scheme 3B).
Scheme 3.
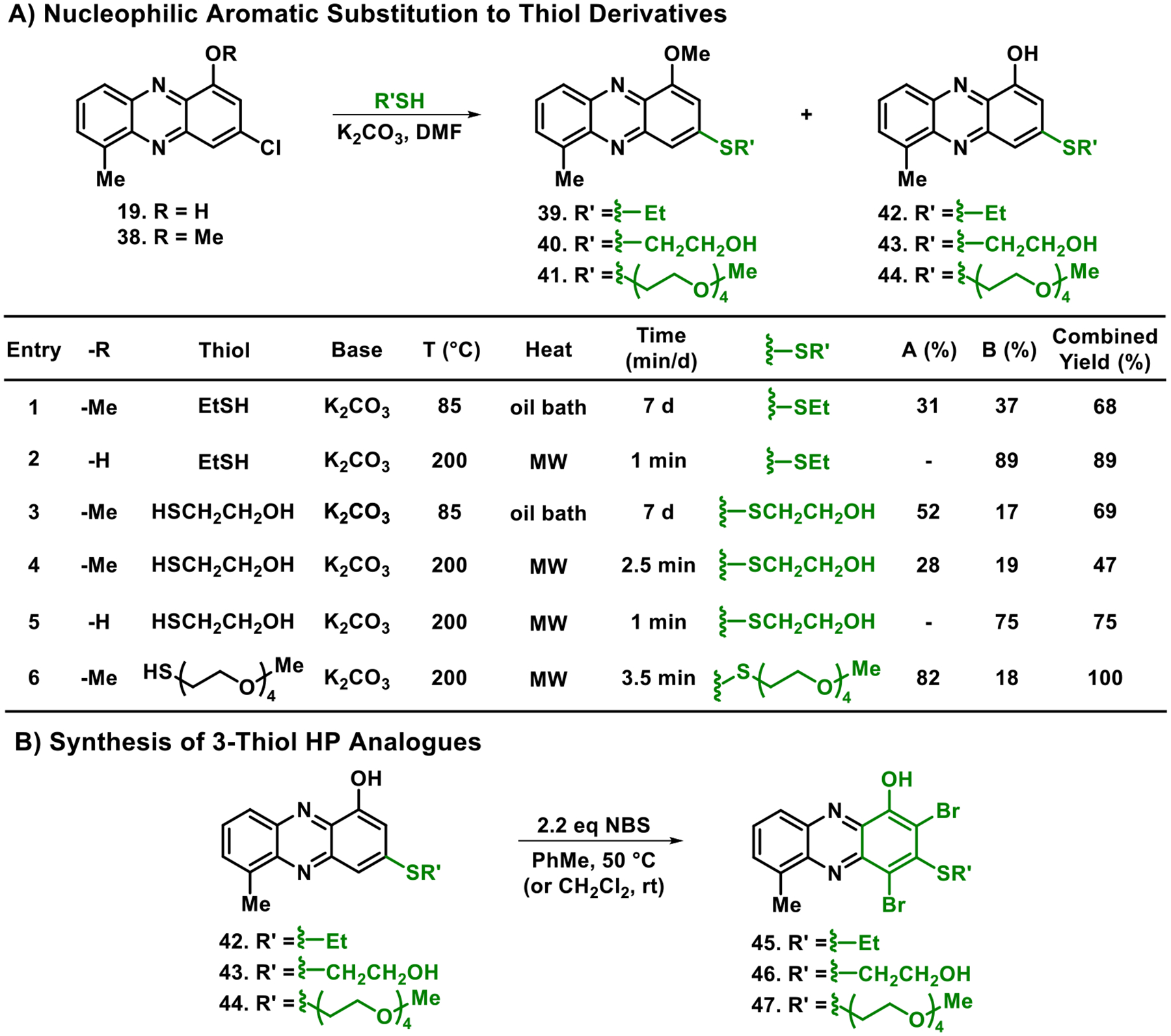
Chemical Synthesis of 3-Thiolated HPs Synthesized via Nucleophilic Aromatic Substitutiona
aMethod A: NBS, CH2Cl2, rt. Method B: NBS, PhMe, 50 °C.
1.2. In Vitro Antibacterial Studies.
We evaluated the new series of 23 HP analogues functionalized at the 3-position in antibacterial assays against a panel of pathogens, including several antibiotic-resistant strains (methicillin-resistant S. aureus strains MRSA-1707 and MRSA-44; methicillin-resistant S. epidermidis MRSE 35984; vancomycin-resistant Enterococcus faecium VRE 700221 and Enterococcus faecalis OG1RF; and Mycobacterium tuberculosis, Mtb strains H37Ra and CDC1551). These new HPs were categorized into three subseries to explore and define new structure–activity relationships regarding the HP scaffold, including subseries: (A) 3-chloro-1-hydroxyphenazines (10 nonbrominated HP analogues, 18–27), (B) 2,4-dibromo-3-chloro-1-hydroxyphenazines (10 dibrominated HPs, 28–37), and (C) 2,4-dibromo-3-thio-1-hydroxyphenazines (3 thiolated HPs, 45–47). Each of the three HP subseries produced active, submicromolar potent antibacterial agents against MRSA strains, while demonstrating outstanding activity profiles against all bacterial pathogens investigated (Table 1).
Table 1.
Summary of MIC Values Obtained during Antibacterial Assessment for 3-Functionalized HP Analogues and Comparator Compounds, Including Several Antibioticsa
| compound | MRSA-1707 | MRSA-44 | MRSE 35984 | VRE 700221 | E.faecalis OG1RF | S. pneumoniae 6303 | Mtb H37Ra | Mtb CDC1551 |
|---|---|---|---|---|---|---|---|---|
| 1 | 3.13 | 3.13 | 3.13 | 6.25 | 18.8b | |||
| 3 | 1.56c | 1.56c | 2.35b | 4.69b | 12.5c | 25 | 13.7 | |
| 18 | 25 | 18.8b | >50 | |||||
| 19 | 1.56 | 1.56 | 2.35b | 3.13 | ||||
| 20 | 2.35b | 2.35b | 2.35b | 3.13 | 12.5 | |||
| 21 | 0.30b | 0.30b | 0.78 | 1.17b | 2.35b | 6.25 | 3.27 | |
| 22 | 3.13 | 3.13 | 3.13 | 4.69b | ||||
| 23 | 6.25 | 18.8b | 18.8b | |||||
| 24 | 0.30b | 0.30b | 1.17b | 2.35b | 9.38b | 3.13 | 2.31 | |
| 25 | 4.69b | 9.38b | 18.8b | 18.8b | ||||
| 26 | 9.38b | >50 | >50 | |||||
| 27 | 0.78 | 1.56 | 3.13 | 3.13 | ||||
| 28 | 0.15b | 0.59b | 0.59b | 1.17b | 3.13 | 6.25 | 4.37 | |
| 29 | 0.08b | 0.08b | 0.08b | 0.10 | 0.30b | 0.50 | 6.25 | 1.59 |
| 30 | 0.08b | 0.08b | ≤0.05d | 0.08b | 0.20 | 3.62 | ||
| 31 | 0.59b | 1.17b | 0.10 | ≤0.05d | 0.08b | 0.88 | ||
| 32 | 0.15b | 0.10 | 0.20 | 0.15b | 0.78 | 3.13 | 3.01 | |
| 33 | 0.15b | 0.20 | 0.78 | 0.78 | 1.17b | 1.71 | ||
| 34 | 0.59b | 0.15b | ≤0.05d | 0.08b | 0.30b | 6.25 | 0.80 | |
| 35 | 0.30b | 0.20 | 0.10 | 0.15b | 0.39 | 3.91 | ||
| 36 | 0.59b | 1.56 | 0.10 | 0.30b | 0.78 | |||
| 37 | 0.15b | 0.78 | 0.30b | 0.59b | 1.56 | |||
| 45 | 0.30b | 0.30b | 0.30b | 0.39 | 0.78 | 1.69 | ||
| 46 | 6.25 | 6.25 | 4.69b | 12.5 | ||||
| 47 | 2.35b | 4.69b | 2.35b | 3.13 | ||||
| 48 (1-OHP) | 50 | 50 | 37.5b | >50 | >50 | |||
| 49 (QAC-10) | 4.69b | 2.35b | 2.35b | 2.35b | 3.13 | |||
| EDTA | 25 | 125 | 500 | |||||
| TPEN | 46.9b | 46.9b | 46.9b | |||||
| vancomycin | 0.39 | 0.78 | >100 | 0.78 | ||||
| daptomycin | 3.13 | |||||||
| linezolid | 12.5 | |||||||
| streptomycin | 1.32 |
All MIC values are reported in micromolar (μM) concentrations.
Midpoint of a twofold range in observed MIC values.
Midpoint of a fourfold range in observed MIC values.
Lowest test concentration.
The test range was 0.05–50 μM for all HPs and higher (e.g., 100 μM) for comparators. EDTA and TPEN are metal-binding agent comparators. Each MIC was determined from three to nine independent experiments.
This was the first time that we observed potent antibacterial activities regarding HP analogues not containing both bromine atoms at the 2- and 4-position of the HP scaffold, suggesting that the 3-chlorine atom dramatically impacts the SAR profile of these agents. Analogue 21 (subseries A) demonstrated potent activities against Staphylococcus strains MRSA-1707, MRSA-44, and MRSE 35984 (MIC = 0.30–0.78 μM, Table 1), while demonstrating good antibacterial potency against the other pathogens in the panel (VRE 700221, MIC = 1.17 μM; E. faecalis OG1RF, MIC = 2.35 μM; Mtb H37Ra, MIC = 6.25 μM; Mtb CDC1551, MIC = 3.27 μM). HP analogue 24 also demonstrated a similar antibacterial profile with some reduced potency against the Enterococcus strains when compared to 21. The remaining 3-chloro-1-hydroxyphenazines 18–20, 22, 23, and 25–27 demonstrated a range of reduced antibacterial activities; however, we found these profiles to be incredibly insightful to our SAR investigations.
The 2,4-dibromo-3-chloro-1-hydroxyphenazine subseries (B, 10 analogues; Table 1) demonstrated incredible potency against the panel of pathogenic bacteria. As an overview of subseries B activities, seven HPs demonstrated MIC = 0.08–0.30 μM against MRSA-1707 (vancomycin, MIC = 0.39 μM; comparator), six HPs reported MIC = 0.08–0.20 μM against MRSA-44 (vancomycin, MIC = 0.39 μM), eight HPs gave MIC = 0.05–0.30 μM against MRSE 35984 (vancomycin, MIC = 0.78 μM), seven HPs showed MIC = 0.05–0.30 μM against VRE 700221 (vancomycin, MIC >100 μM), and five HPs proved MIC = 0.08–0.39 μM against E. faecalis OG1RF (vancomycin, MIC = 0.78 μM). HPs 29, 30, 32, 34, and 35 each recorded submicromolar MICs against all Gram-positive strains in the panel (see representative MIC assays, Figure 4). Select analogues from subseries B were also evaluated against M. tuberculosis strains and found to have good to excellent anti-TB activities, including HPs 29 (H37Ra, MIC = 6.25 μM; CDC1551, MIC = 1.59 μM), 31 (CDC1551, MIC = 0.88 μM), 32 (H37Ra, MIC = 3.13 μM; CDC1551, MIC = 3.01 μM), and 34 (H37Ra, MIC = 6.25 μM; CDC1551, MIC = 0.80 μM). In addition, HP 29 displayed excellent antibacterial activity against Streptococcus pneumoniae 6303 (MIC = 0.50 μM), which is the first time we have shown HP activity against this pathogen.
Figure 4.
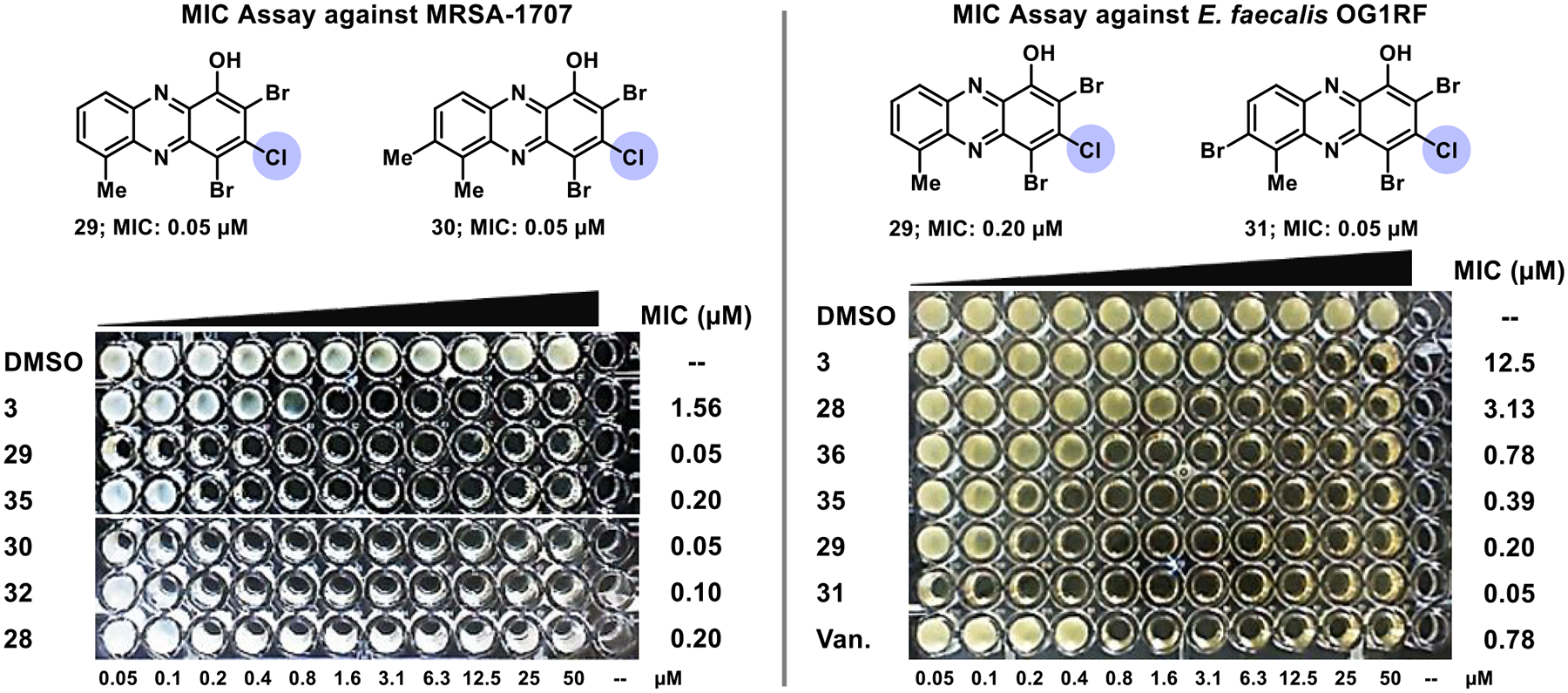
MIC assay results focused on selected 3-chloro HP analogues against MRSA-1707 and E. faecalis OG1RF. Note: “Van.” is for vancomycin.
In addition to HPs bearing a chlorine atom in the 3-position (subseries A and B), three 3-thiol HP analogues were tested for antibacterial activities. We also wanted to utilize the SNAr chemistry to explore water-solubilizing side chains (e.g., PEG in HP 47). We found HP 45 bearing an ethyl thiol moiety to have potent antibacterial activities against all Gram-positive strains tested (MRSA, MIC = 0.30 μM; MRSE 35984, MIC = 0.30 μM; VRE 700221, MIC = 0.39 μM); however, this was a reduction in activity compared to HP 29, which has a single group change from 3-ethyl thiol to 3-chlorine (29 and 45 possess a 6-methyl group on the HP scaffold). The two 3-thiol HP analogues aimed at improving water solubility (HP 46 bearing a 2-hydroxylethyl thiol moiety at the 3-position; HP 47 containing a four PEG unit thiol at the 3-position) demonstrated significant reductions in antibacterial activities (MICs = 2.35–12.5 μM against MRSA, MRSE, and VRE; Table 1).
HP analogues 3, 29, 34, and 45 were evaluated in MIC assays against six MRSA clinical isolates (Supporting Information, Table S2) obtained from patients treated at Shands Health Hospital (Gainesville, FL). Investigating new agents against clinical isolates is an important early step when working to translate interesting leads. MIC results against MRSA clinical isolates aligned with findings in lab strains as new HPs (29, 34, and 45) were significantly more active than parent HP 3 (MIC = 1.25–2.5 μM). HP 29 demonstrated outstanding antibacterial potency with MIC values between 0.06 and 0.12 μM against the MRSA isolates, which was considerably more active than vancomycin (MIC = 0.39–0.59 μM). In addition, HP 45 (MIC = 0.31–0.63 μM) and 34 (MIC = 0.47–1.25 μM) were found to have good to excellent activity profiles. Overall, HP analogues 29, 34, and 45 showed remarkable activities as three of six clinical isolates were multidrug resistant (methicillin, MIC ≥37.5 μM; ciprofloxacin, MIC >100 μM for MDR strains).
1.3. Cytotoxicity Assessment.
Following antibacterial investigations, we evaluated a select panel of active HP analogues against multiple mammalian cell types to determine cytotoxicity and bacterial targeting. From this series, 12 new HPs and parent HP 3 were evaluated against mammalian cell lines, including (1) three nonbrominated 3-chloro HPs (subseries A; 21, 24, and 27), (2) eight 2,4-dibromo-3-chloro HPs (subseries B; 28–35), and (3) one 2,4-dibromo-3-thiol HP (subseries C; 45). The cell lines used to determine cytotoxicity of HPs included HeLa (cervical cancer cell line; 24 h LDH release assay,46 HPs were evaluated at 25, 50, and 100 μM), J774 MΦ (macrophages; 24 h Alamar Blue assay,47 HPs tested in twofold dilutions up to 200 μM), HepG2 (hepatocellular carcinoma; 24 h Alamar Blue assay, HPs tested in twofold dilutions up to 200 μM), and HEK-293 (human embryonic kidney cells; 72 h MTT assay48 only HP 29 was evaluated in twofold dilutions up to 200 μM).
Results from the cytotoxicity assessment of new HPs were very encouraging (Table 2). The majority of new HPs reported IC50 > 100 μM against HeLa cells (six of nine new HPs evaluated) with HP 33 reporting the most cytotoxicity against HeLa cells with an IC50 > 25 μM. When tested against J774 MΦ and HepG2 cells, nearly all new HPs reported IC50 > 200 μM, resulting in a selectivity index (SI) of >2000 when considering the MIC values of HP analogues 29 and 30 against MRSA, MRSE, and VRE strains (MIC = 0.05–0.10 μM). HEK-293 cells demonstrated increased sensitivity toward HP 29 (IC50 = 18.3 ± 5.8 μM), however, gave a very good selectivity index (SI = 244) when comparing its IC50 against HEK-293 to MIC values against MRSA/MRSE strains (MIC = 0.075 μM). Overall, this new series of HPs demonstrates very good to outstanding bacterial targeting results based on relative cytotoxicity profiles to antibacterial activities.
Table 2.
| compound | MRSA-1707 MIC | HeLa cytotox. (IC50) | J774 MΦ cytotox. (IC50) | Hep G2 cytotox. (IC50) | HEK-293 cytotox. (IC50) |
|---|---|---|---|---|---|
| 3 | 1.56 | >100 | >100 | >100 | |
| 21 | 0.30 | >100 | >200 | >200 | |
| 24 | 0.30 | >100 | >200 | >200 | |
| 27 | 0.78 | 100 | |||
| 28 | 0.15 | >100 | >200 | >200 | |
| 29 | 0.08 | >100 | >200 | >200 | 18.3 ± 5.8 |
| 30 | 0.08 | >200 | >200 | ||
| 31 | 0.59 | >200 | >200 | ||
| 32 | 0.15 | >100 | >200 | >200 | |
| 33 | 0.15 | >25 | >200 | >200 | |
| 34 | 0.59 | >100 | >200 | >200 | |
| 35 | 0.30 | 100 | >200 | >200 | |
| 45 | 0.30 | >100 | >200 |
All results are reported in micromolar (μM) concentrations.
Assay information: HeLa cytotoxicity (24 h, LDH release assay), J774 MΦ cytotoxicity (24 h, Alamar Blue assay), HepG2 cytotoxicity (24 h, Alamar Blue assay), HEK-293 cytotoxicity (72 h, MTT assay).
Note: All experimental results are reported from a minimum of three independent cytotoxicity experiments.
1.4. Biofilm Eradication and Iron Starvation.
Initial MIC assays allowed us to determine planktonic growth inhibition activities of new HP analogues; however, HPs eradicate surface-attached bacterial biofilms with excellent potency, which require different microbiological assays. To investigate the biofilm-eradication activities of select compounds, our group has utilized calgary biofilm device (CBD) assays to determine minimum biofilm eradication concentration (MBEC) values.35–38 The CBD is a 96-well assay that has a specialized lid with anchored pegs that are submerged into microtiter wells, allowing biofilm formation and subsequent transfer to fresh 96-well plates as the assay progresses.49,50 Biofilm eradication assays have three phases, including the (1) biofilm establishment phase (inoculated media in a 96-well plate with CBD pegs submerged into microtiter wells providing a surface for biofilms to establish; 24 h incubation under static conditions), (2) compound treatment phase (pegs with established biofilms are gently rinsed to remove planktonic cells and transferred to a new 96-well plate containing a twofold serially diluted test compound in fresh media; 24 h incubation), and (3) recovery phase (lid bearing compound-treated biofilms on CBD pegs is transferred to a final 96-well plate with fresh media only; 24 h incubation). Upon completion of CBD assays, biofilm eradication is determined by a turbidity readout. Microtiter wells that are turbid result from viable biofilms on CBD peg surfaces (live biofilms will disperse planktonic bacteria into fresh media and undergo replication/bacterial growth), whereas nonturbid microtiter wells result from completely eradicated biofilms. The lowest concentration at which biofilm eradication occurs is referred to as the minimum biofilm eradication concentration (MBEC) value.
The CBD allows one to determine planktonic versus biofilm cell killing dynamics of small molecules as these assays allow for the determination of minimum bactericidal concentration (MBC) and MBEC values simultaneously. This is a significant advantage over comparing MBEC to MIC values obtained from significantly different microbiological assays. In the past, we have found HPs to report MBC/MBEC ratios of 1:1 to 1:3 from CBD assays, demonstrating near equipotent planktonic- and biofilm-killing properties.35–38
Since biofilm eradication assays are more sophisticated than MIC assays and HP’s antibacterial potency typically correlates to biofilm-killing activities, we advanced a subset of 14 new HPs to MBEC assays against MRSA-1707 (Table 3). We investigated three nonbrominated HPs from subseries A (21, 24, and 27) and found these analogues to have good activities with HPs 21 and 24 demonstrating the highest levels of biofilm killing (MBEC = 37.5 μM, Table 3). We evaluated all subseries B (dibrominated HP) analogues bearing a 3-chloro substituent in MBEC assays since these compounds demonstrated MIC values ≤0.59 μM against MRSA-1707. Each of the 10 dibrominated HP analogues (subseries B) reported MBEC values ≤75 μM with five analogues demonstrating outstanding biofilm killing with MBECs at 2.35–4.69 μM (HPs 29, 30, 32, 34, and 35, see Figure 5A). Finally, 3-thiolated HP 45 demonstrated relatively weak biofilm-killing activities against MRSA-1707 (MBEC = 200 μM).
Table 3.
Summary of Biofilm Eradication Studies against MRSA, MRSE, VRE, and E. faecalis Biofilmsa
| compound | MRSA-1707 MBC/MBEC | MRSA 44 MBC/MBEC | MRSE 35984 MBC/MBEC | VRE 700221 MBC/MBEC | E. faecalis OG1RF MBC/MBEC | % hemolysis at 200 μM |
|---|---|---|---|---|---|---|
| 3 | 50c/100c | 37.5b/75b | 25c/50c | 18.8b/12.5c | 25c/>200d | ≤1 |
| 21 | 18.8b/37.5b | ≤1 | ||||
| 24 | 18.8b/75b | ≤1 | ||||
| 27 | 37.5b/37.5b | 1.9 | ||||
| 28 | 50/50 | 37.5b/75b | 12.5c/25c | 4.69b/2.35b | 75b/>200d | ≤1 |
| 29 | 1.56c/2.35b | 4.69b/6.25c | 1.17b/0.59b | 0.59b/0.59b | 6.25/18.8b | ≤1 |
| 30 | 18.8b/4.69b | 3.13c/2.35b | 0.78c/0.30b | ≤1 | ||
| 31 | 75b/75b | 4.69b/1.17b | 2.35b/0.20 | 1.56c/0.78c | 27 | |
| 32 | 9.38b/2.35b | 4.69b/6.25c | 1.56c/1.56c | 0.78c/0.39 | 18.8b/75b | 2.6 |
| 33 | 12.5c/18.8b | 12.5/12.5 | 3.13c/1.17b | ≤1 | ||
| 34 | 2.35b/3.13c | 6.25c/6.25c | 0.59b/0.30b | 0.39c/0.20 | 3.13c/1.17b | ≤1 |
| 35 | 4.69b/4.69b | 6.25c/18.8b | 2.35b/2.35b | 0.59b/0.30b | 6.25/6.25 | ≤1 |
| 36 | 12.5/12.5 | 6.25/6.25 | 4.69b/0.59b | 5.1 | ||
| 37 | 12.5/9.38b | 6.25/6.25 | 4.69b/3.13c | 3.0 | ||
| 45 | 50c/200 | 12.5c/4.69b | 9.38b/3.13c | 5.7 | ||
| 49 (QAC-10) | 93.8b/93.8b | 3.13/3.13 | 3.0b/3.0b | >99 | ||
| TPEN | 375b/>2000 | 250/>2000 | 188c/>2000 | 500/>2000 | ≤1 | |
| EDTA | >2000/>2000 | 1000/>2000 | >2000/>2000 | ≤1 | ||
| Vancomycin | 7.8/>2000 | 7.8/>2000 | 3.0c/>2000 | 750b/>2000 | 11.7b/>2000 | |
| Daptomycin | 125/>2000 | 375/93.8b | ||||
| Linezolid | 31.3/>2000 | 4.69b/1.56 |
All biological results are reported in micromolar (μM) concentrations.
Midpoint of a twofold range in observed values.
Midpoint of a fourfold range in values.
Partial turbidity observed at the highest test concentration.
All values in this table resulted from a minimum of three independent experiments. Halogenated phenazines and QAC-10 were tested at concentrations of up to 200 μM. Conventional antibiotics (e.g., vancomycin), EDTA, and TPEN (metal-binding agents) were tested at concentrations of up to 2000 μM in CBD assays.
Figure 5.
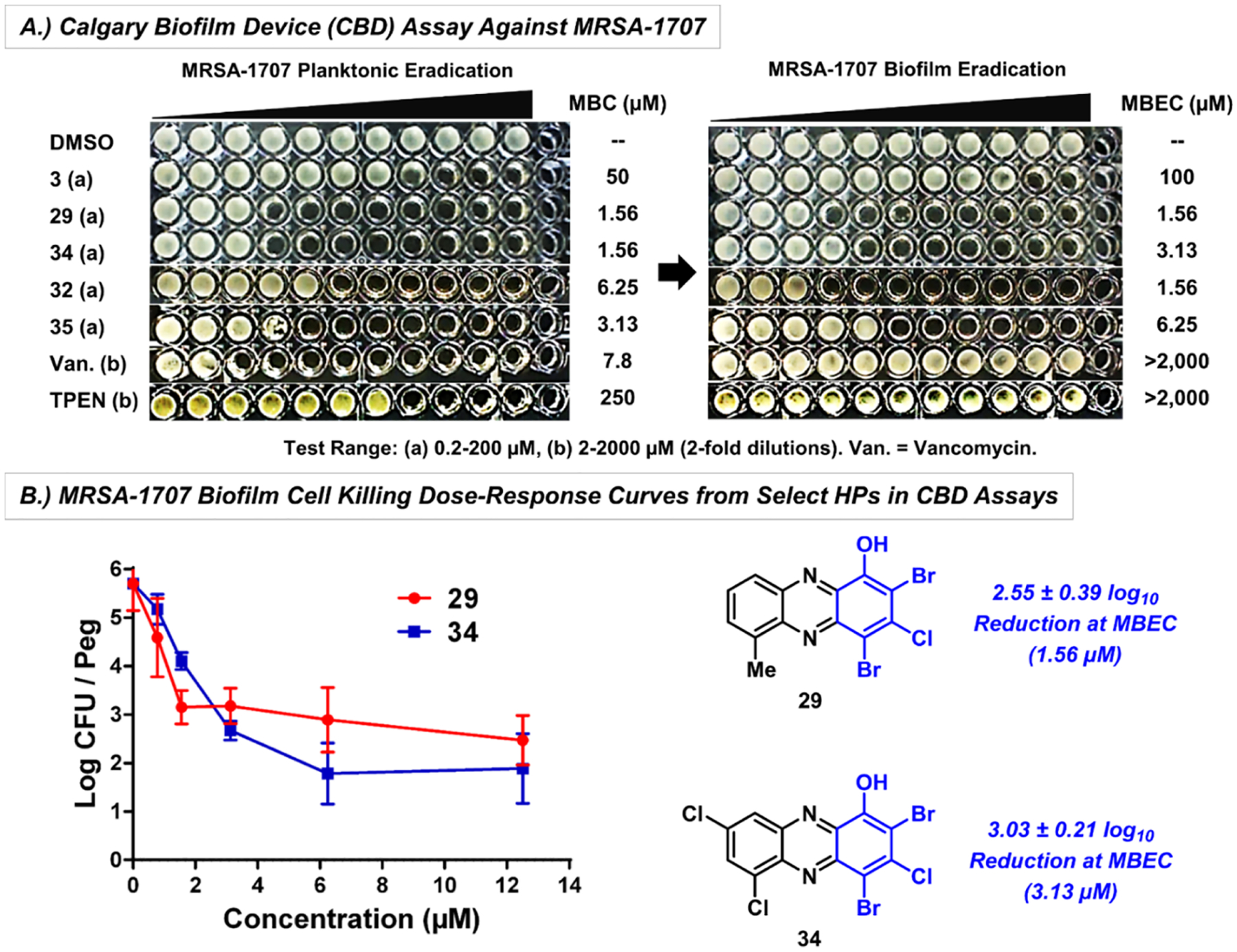
(A) CBD assay of a panel of HPs, vancomycin and TPEN against MRSA-1707. (B) MRSA-1707 biofilm cell killing (CFU/peg) for HPs 29 and 34 obtained from CBD pegs.
In addition, dose–response of biofilm killing was determined from CFU counts from CBD pegs against MRSA-1707 treated with HPs 29 (2.55 ± 0.39 log10 reduction of viable biofilm cells at the MBEC value, Figure 5B) and 34 (3.03 ± 0.21 log10 reduction of viable MRSA-1707 biofilm cells). Together, HPs 29 and 34 demonstrated ~3 log10 reduction or ~99.9% kill of MRSA-1707 biofilm cells at the corresponding MBEC value, similar to previous findings.35–38 Subseries B analogues 29, 32, 34, and 35 were also evaluated for biofilm eradication activities against MRSA BAA-44 and demonstrated excellent biofilm-killing potencies with MBEC values between 6.25 and 18.8 μM (Table 3).
In addition to MBEC assays, we showed that HPs 3, 28, and 29 rapidly induce iron starvation in MRSA-1707 biofilms using RT-qPCR experiments (Figure 6), which is in line with previous findings regarding 4 (HP-14).39 Briefly, established MRSA-1707 biofilms were treated for 4 h with 3, 28, and 29 with 1/10 × MBEC and 1 μM of each compound before RNA was extracted from HP- and vehicle-treated biofilm samples. RT-qPCR was then performed to investigate transcript levels from four different MRSA genes involved in iron uptake: isdB (iron regulated surface determinant; heme iron acquisition), sfaA (staphyloferrin A; siderophore), sbnC (staphyloferrin B; siderophore), and MW0695 (ferrichrome ABC transporter). Results from these experiments show that HPs induce a rapid upregulation of gene clusters involved in iron uptake (Figure 6B), and we conclude that biofilm eradication occurs due to iron starvation. HPs 18, 28, 29, and 34 were confirmed to directly bind iron(II) via UV–vis spectroscopy (see Figure 6C and Supporting Information), aligning with our RT-qPCR results related to the iron starvation of MRSA biofilms.
Figure 6.
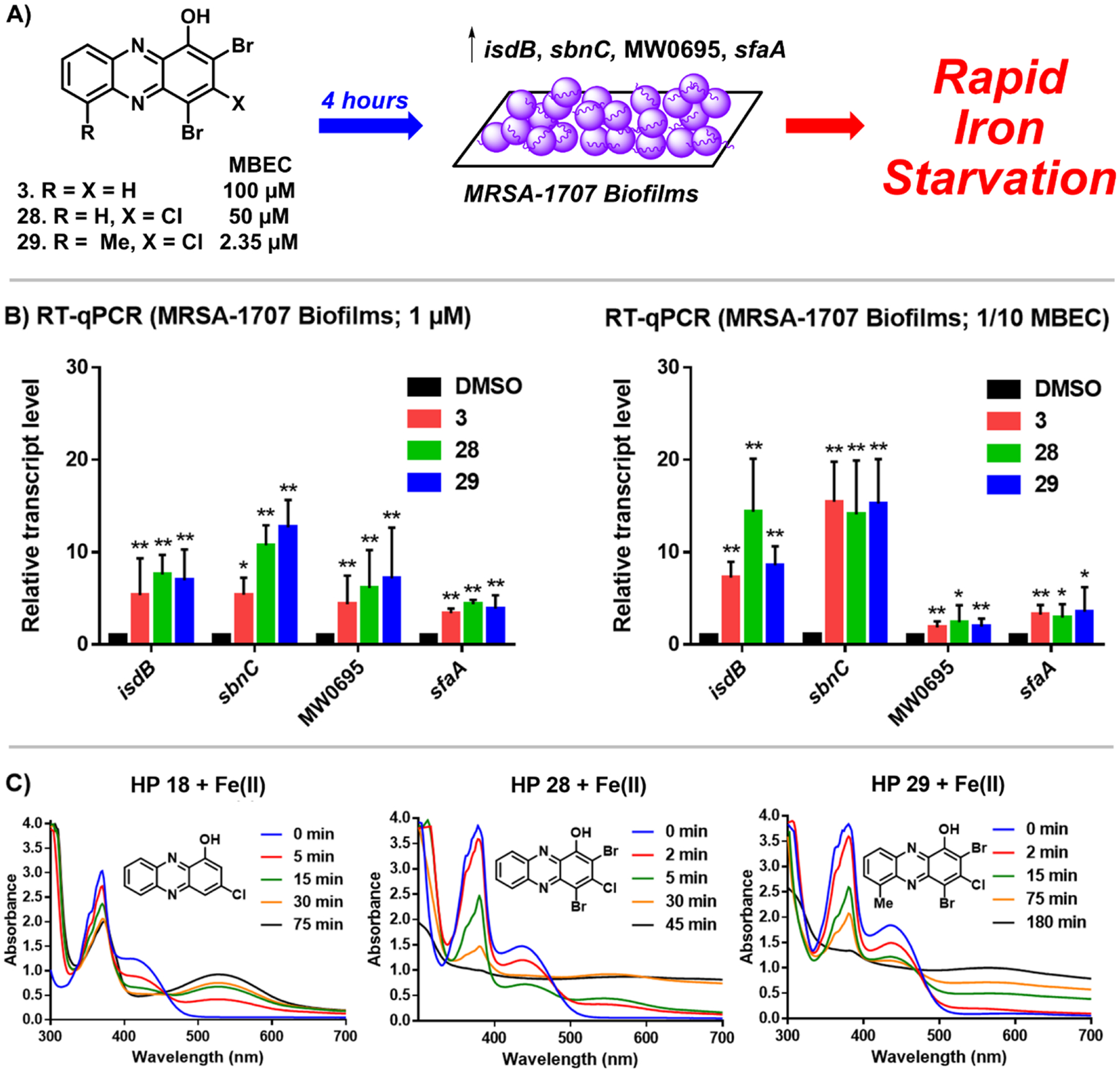
(A) Treating established MRSA-1707 biofilms with HPs 3, 28, and 29 leads to a rapid induction of iron starvation. RT-qPCR results of HPs 3, 28, and 29 upregulating iron uptake genes in MRSA-1707 biofilms following 4 h treatment at (B) 1 μM or 1/10 × MBEC; *p value ≤ 0.05, **p ≤ 0.01 (Student’s T-test). (C) UV–vis spectroscopy of HPs 18, 28, and 29 binding iron(II). Note: In previous work, we show that EDTA and TPEN do not upregulate this panel of iron uptake genes at 5 μM.39
Following initial biofilm-killing studies in MRSA, select HPs were evaluated against other Gram-positive pathogens in CBD assays (Table 3). Several HPs demonstrated good (MBEC = 12.5–25 μM) to outstanding (MBEC = 0.30–6.25 μM) biofilm-killing potencies against MRSE 35984 with 29 (MBEC = 0.59 μM), 31 (MBEC = 1.17 μM), 32 (MBEC = 1.56 μM), and 34 (MBEC = 0.30 μM) proving to be the most active. Dose-dependent killing of MRSE 35984 biofilm bacteria was determined by CFU counts from CBD pegs treated with HPs 29 (3.65 ± 1.33 log10 reduction of viable MRSE biofilm cells at the MBEC value; Figure 7B) and 34 (3.18 ± 0.40 log10 reduction of viable MRSE biofilm cells at the MBEC value). In addition, these dose–response experiments show that HPs 29 and 34 eradicate ~99.999% MRSE 35984 biofilm cells at 3.13 μM (~5 log10 reduction of viable CFUs).
Figure 7.
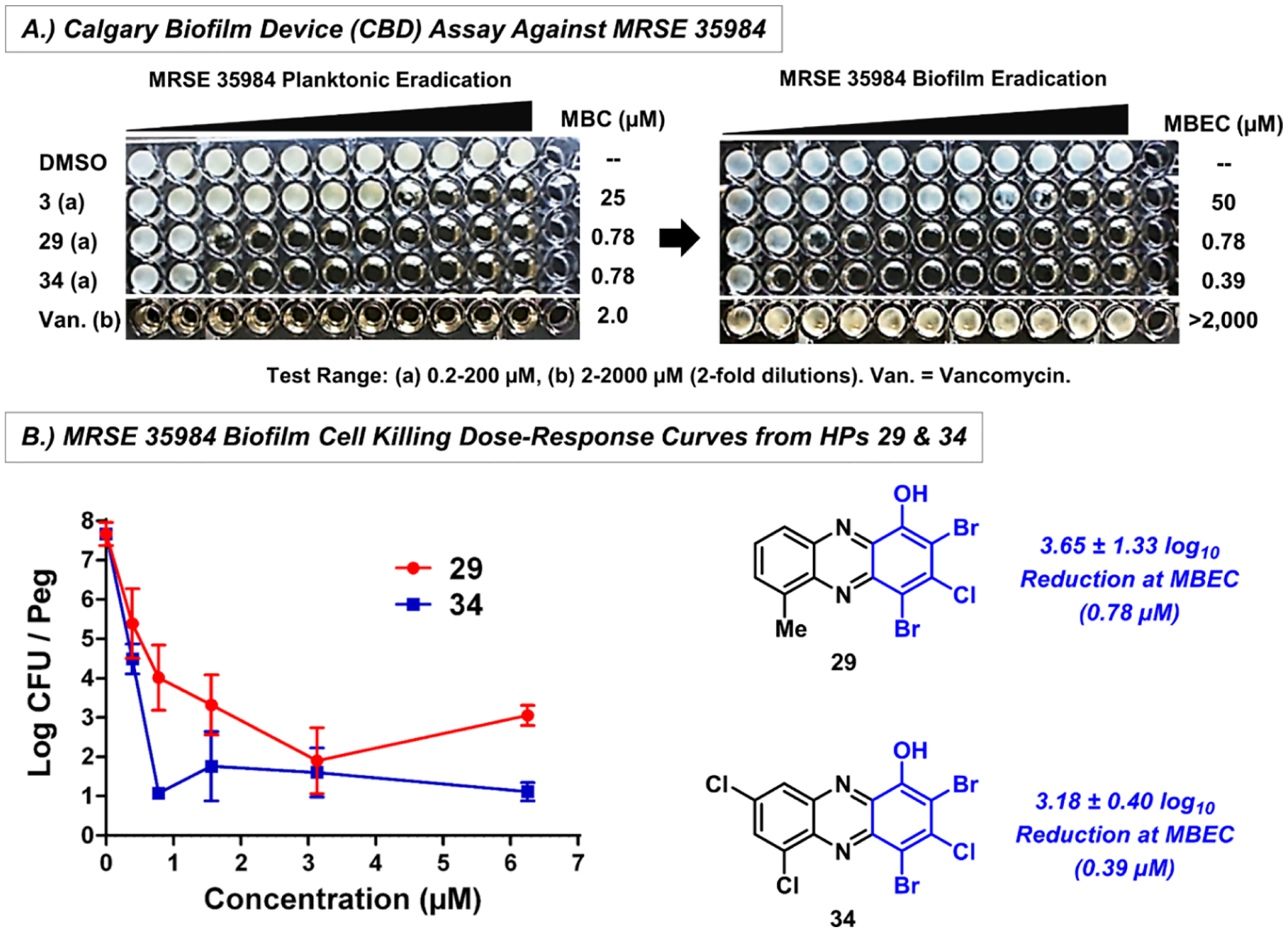
MRSE 35984 biofilm eradication results with HPs 29 and 34 from CBD assays. (A) Turbidity results to determine planktonic and biofilm eradication (MBC/MBEC values) and (B) biofilm killing from viable cell counts on CBD pegs.
In addition, HP analogues from subseries B demonstrated good to excellent biofilm eradicating activities against enterococci strains (Table 3). Seven HPs showed remarkable biofilm-killing activity against E. faecium 700221 with MBEC values 0.20–0.59 μM (HP 31 and 34, MBEC = 0.20 μM). Select HPs were evaluated against E. faecalis OG1RF biofilms with 31 (MBEC = 0.78 μM), 34 (MBEC = 1.17 μM), and 35 (MBEC = 6.25 μM) proving to have the highest killing activities.
Vancomycin, daptomycin, and linezolid are frontline antibiotics used to treat MRSA infections and, in our CBD assays, failed to eradicate MRSA-1707 biofilms despite effective planktonic killing (e.g., vancomycin, MBC = 7.8 μM against planktonic cells, MBEC >2000 μM against MRSA biofilms; Table 3). In addition, QAC-10 is a membrane-lysing quaternary ammonium cation that displays good biofilm-eradicating activities51 and serves as a valuable comparator in these investigations. QAC-10 eradicated MRSA-1707 biofilms with an MBEC of 93.8 μM in CBD assays, which is 20-fold less potent than our most active HPs in subseries B. In addition, EDTA and TPEN (membrane-permeable agent) are general metal-chelating agents and are unable to eradicate biofilms at the highest test concentration (MBECs >2000 μM; Table 3).
We then assessed the ability of our most active HP analogues to lyse red blood cells (RBCs). Hemolysis assays are used to determine the membrane-lysis activity of a compound, which is of particular interest to this work as membrane-lysing agents (e.g., QACs) can eradicate biofilms. During these investigations, we observed minimal hemolytic activity for nearly all new HP compounds at 200 μM (≤5% hemolysis; Table 3), with the exception of 31 which reported 27% hemolysis of RBCs. QAC-10 was tested alongside HP analogues as an active membrane-lysing agent (comparator) and caused >99% hemolysis of RBCs at 200 μM.
1.5. In Vivo Assessment of HP 29 in Dorsal Wound Infection Models.
Based on its potent antibacterial activities, rapid induction of iron starvation in MRSA biofilms, and excellent cytotoxicity profile, HP 29 was evaluated for in vivo efficacy against S. aureus and E. faecalis using dorsal wound infection models in mice. S. aureus and E. faecalis are highly prevalent in both hospital-acquired and wound infections.52,53 In addition, these pathogens are notorious for their antibiotic-resistant phenotypes and propensity to form tolerant biofilms.54,55
For the in vivo experiments, HP 29 was formulated in a PEG-based ointment52 for topical application. In separate experiments, wounds were created on the mouse’s dorsal mid-section using a biopsy punch to remove the dermal layer. The resulting wound was inoculated with either S. aureus UAMS-1 (1 × 107 CFU/mL) or E. faecalis OG1RF (6 × 1010 CFU/mL) to establish infection. Following infection, PEG ointment containing HP 29 or PEG ointment alone (vehicle control) was directly applied to the infected wounds twice (S. aureus infection) or once (E. faecalis infection) for three days before mice were sacrificed, and the bacterial load in each wound was determined as CFU per lesion (Figure 8).
Figure 8.
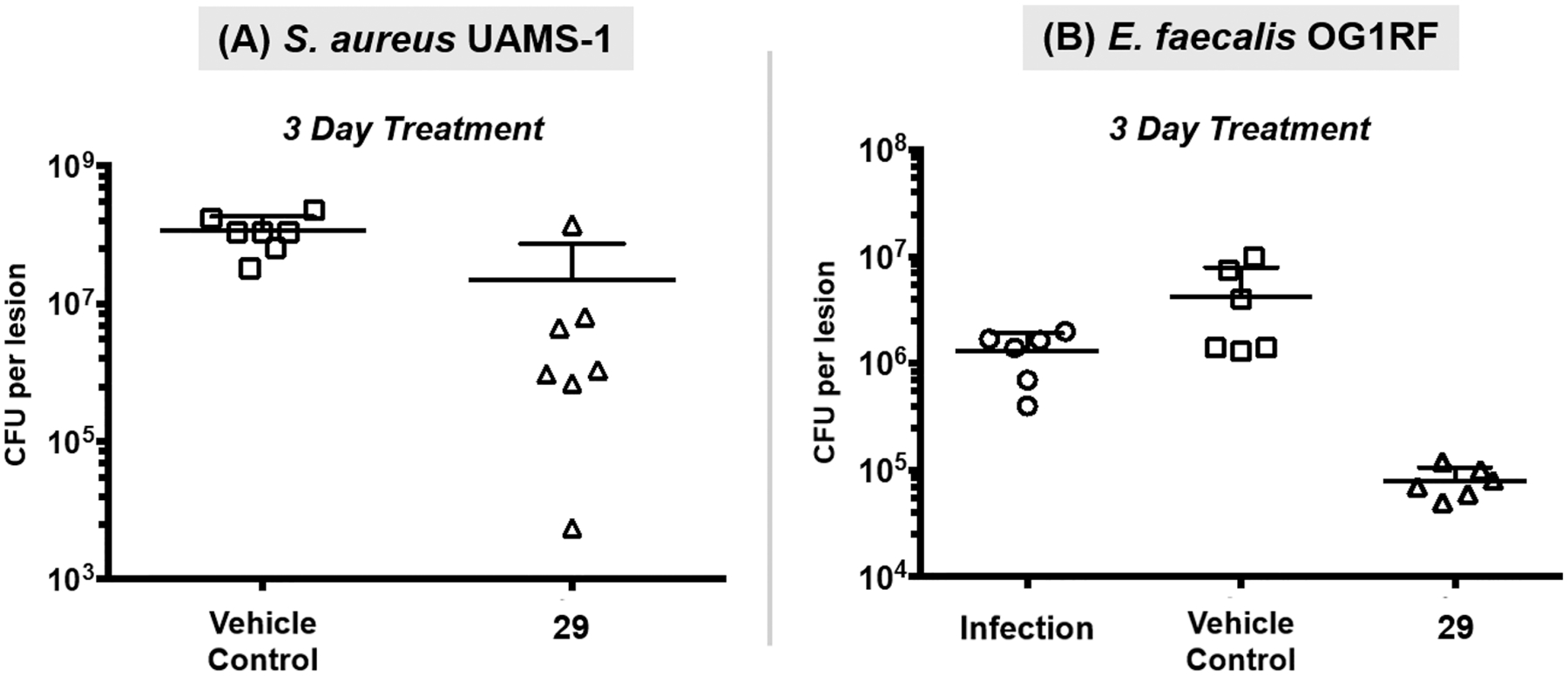
In vivo assessment of HP 29 in dorsal wound infections of S. aureus and E. faecalis in mice. (A) HP 29 reduces S. aureus UAMS-1 bacterial load in BALB/c mice (Student’s T-test: p value = 0.010; seven mice per group). (B) HP 29 reduces E. faecalis OG1RF bacterial load in C57BL/6J mice (comparing mice treated with 29 and infection control; ANOVA: day 3, p value = 0.068).
Results from the dorsal wound infection experiments demonstrated that HP 29 treatment led to a significant decolonization of both S. aureus and E. faecalis. HP 29-treated mice led to a 0.82-log10 reduction in CFU per lesion of S. aureus UAMS-1 compared to vehicle-treated mice following 3 consecutive days of treatment (Figure 8A). Against E. faecalis OG1RF, HP 29 showed more efficacious decolonization with 1.73-log10 reduction compared to vehicle control after treatment (Figure 8B). Collectively, these results indicate that HP agents are a useful alternative for the topical treatment of wounds infected by Gram-positive pathogens.
1.6. Structure–Activity Relationship Analysis.
This new collection of HPs has significantly expanded structure–activity relationship profiles for these antibacterial agents (Figure 9). In addition to the synthesis and biological assessment of 3-substituted HPs, we determined pKa and CLogP values (ChemDraw) of select analogues for further analysis.
Figure 9.
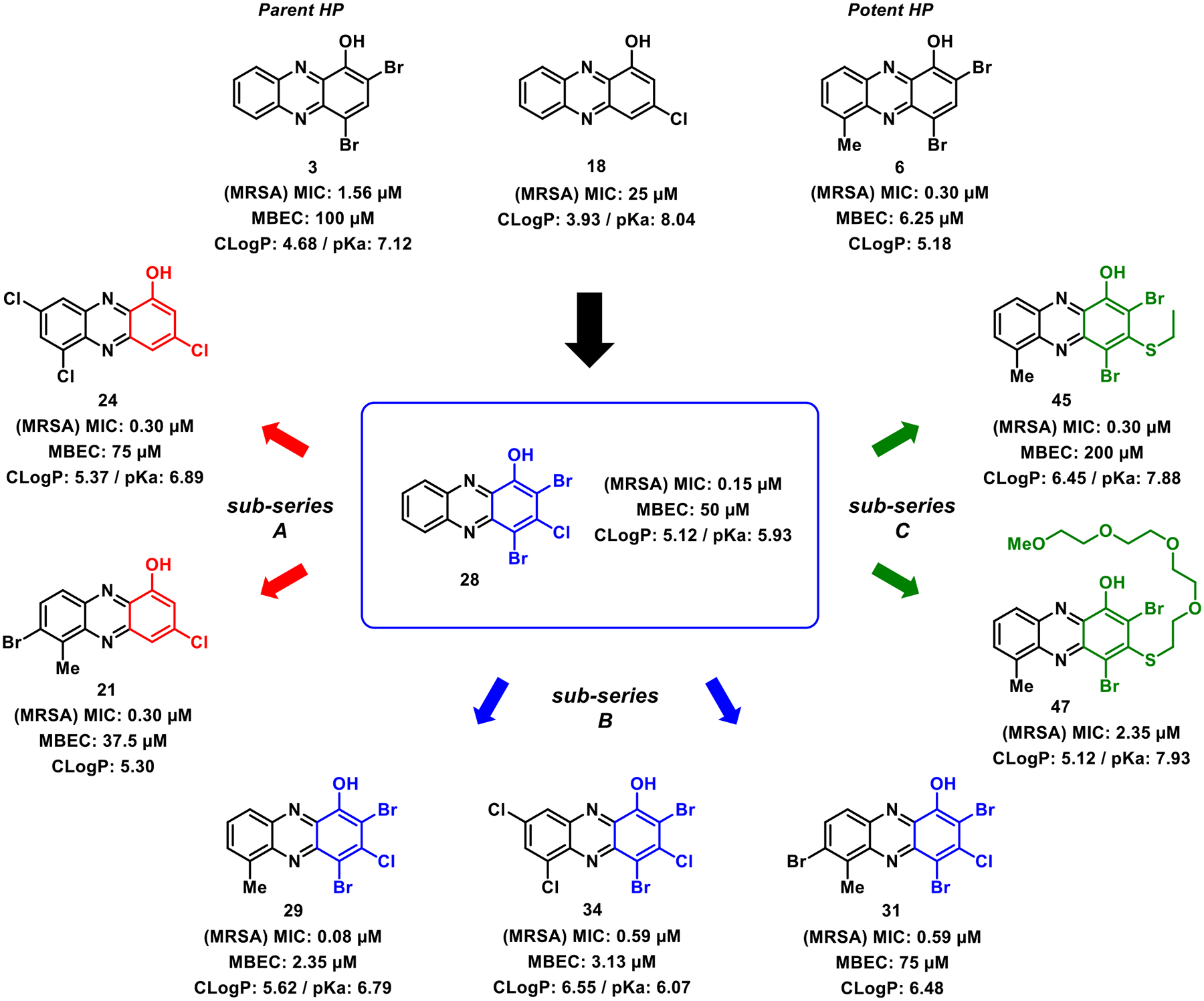
Structure–activity relationship profiles for new analogues functionalized at the 3-position of the HP scaffold. Note: MRSA data refer to MRSA-1707 findings in this SAR figure.
New key insights into HP antibacterial agents from these studies include the following: (1) a 3-chlorine atom on the HP scaffold increases the antibacterial/biofilm eradication activities, acidity of the phenolic proton, and CLogP values (see subseries B, Figure 9), (2) bromine/chlorine/iodine atoms at the 2- and 4-positions of the HPs are not critical for antibacterial activities, as previously determined,33,35–38 based on new findings with subseries A analogues 21 and 24 that demonstrate potent antibacterial activities and very good biofilm eradication activities, and (3) thiol substituents at the 3-position diminished antibacterial activities compared to 3-chlorinated HP analogues (comparing subseries C to subseries B analogues) and reduced acidity of the phenolic proton.
As noted, the 3-chlorine atom has a significant impact on the acidity of the phenolic proton of HP analogues, and we believe that this primes the HP scaffold for iron binding as the alkoxy anion. In addition, the 3-chlorine atom increases the CLogP value of HP analogues (that are largely anionic in biological assays performed at pH ~ 7), which could lead to rapid diffusion through bacterial membranes to bind intracellular iron(II). For instance, parent HP 3 is without a 3-chlorine atom and has an MIC = 1.56 μM (MRSA), MBEC = 100 μM (MRSA), a pKa of 7.12, and a CLogP value of 4.68, whereas HP 28 has the addition of a chlorine atom in the 3-position of the HP and has an MIC = 0.15 μM (MRSA), MBEC = 50 μM (MRSA), a pKa of 5.93, and a CLogP of 5.12 (Figure 9). A similar profile can be observed when comparing HPs 6 (MIC = 0.30 μM, MRSA; MBEC = 6.25 μM, MRSA; CLogP = 5.18; pKa not determined) and 29 (MIC = 0.08 μM, MRSA; MBEC = 2.35 μM, MRSA; CLogP = 5.62; pKa = 6.79; Figure 9).
Several HPs from subseries B demonstrate high levels of planktonic and biofilm-killing activities against Gram-positive pathogens, including multidrug-resistant strains. HP 29 demonstrated outstanding in vitro activities in addition to encouraging in vivo efficacy in dorsal wound infections against S. aureus UAMS-1 and E. faecalis OGRF1. Multiple HPs demonstrated rapid iron starvation in MRSA biofilms as a result of their ability to bind iron between the hydroxyl oxygen at the 1-position of the HP scaffold and nitrogen at the 10-position. Active HPs identified during these studies are ideal candidates for prodrug development through functionalization of the 1-hydroxyl group aimed to enhance physicochemical properties, prevent rapid metabolism (phenol conjugation), mitigate off-target metal binding that could lead to toxicity, and release via bacteria-specific mechanisms.37,38,56,57 Prodrug efforts related to new HP scaffolds from this study are underway, and findings will be reported in due course.
2. CONCLUSIONS
In conclusion, we utilized aniline building blocks to rapidly access a diverse series of N-aryl-2-nitrosoaniline intermediates en route to new halogenated phenazine antibacterial agents. This chemistry enabled the first study of HPs functionalized at the 3-position with chlorine or thiol substituents. This collection of >20 HP analogues demonstrated highly potent antibacterial and biofilm eradication activities against Gram-positive pathogens (e.g., HP 29, MIC = 0.075 μM, MBEC = 2.35 μM against MRSA-1707), and multiple HPs were shown to induce rapid iron starvation in MRSA biofilms. In addition, several new HPs reported good to excellent activities against M. tuberculosis (e.g., HP 34, MIC = 0.80 μM against M. tuberculosis CDC1551). Three diverse subseries of HPs provided significant SAR insights into this antibacterial scaffold, and HP 29 demonstrated in vivo efficacy against S. aureus and E. faecalis in wound infection models in mice. These findings could lead to significantly improved treatment options for antibiotic-resistant and -tolerant bacterial infections, including wound and chronic biofilm-associated infections.
3. EXPERIMENTAL SECTION
3.1. General Information.
All reagents for chemical synthesis were purchased at ≥95% purity from commercial sources and used without further purification. All microwave reactions were carried out using an Anton Paar Monowave 300 microwave synthesis reactor. A constant power was applied to ensure reproducibility regarding microwave reactions as temperature control was automated via the IR sensor and all indicated temperatures correspond to the maximal temperature reached during each experiment. Analytical thin layer chromatography (TLC) was performed using 250 μm silica gel 60 F254 precoated plates (EMD Chemicals Inc.) and used to monitor all reactions. Flash column chromatography was performed using 230–400 Mesh 60Å silica gel from Sorbent Technologies. Melting points were obtained, uncorrected, using a Mel-Temp capillary melting point apparatus from Laboratory Services, Inc.
NMR experiments were recorded using broadband probes on a Varian Mercury-Plus-400 spectrometer via VNMR-J software (400 MHz for 1H and 101 MHz for 13C), Varian Mercury-Plus-500 spectrometer via VNMR-J software (500 MHz for 1H and 126 MHz for 13C), Bruker Avance III (500 MHz for 1H; 126 MHz for 13C), and Bruker Avance II (600 MHz for 1H; 151 MHz for 13C). All spectra are presented using MestReNova 11.0 (Mnova) software and are displayed without the use of the signal suppression function. Spectra were obtained in the following solvents (reference peaks for 1H and 13C NMRs are included): CDCl3 (1H NMR, 7.26 ppm; 13C NMR, 77.23 ppm) and DMSO-d6 (1H NMR, 2.50 ppm; 13C NMR, 39.52 ppm). All NMR experiments were performed at room temperature. Chemical shift values (δ) are reported in parts per million (ppm) for all 1H NMR and 13C NMR spectra. 1H NMR multiplicities are reported as follows: s = singlet, br. s = broad singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. HSQC was used to identify a few challenging 13C signals, and these spectra are reported in the Supporting Information. High-resolution mass spectrometry (HRMS) was obtained for all new compounds from the Chemistry Department at the University of Florida.
Multiple controls and comparator agents (e.g., antibiotics and EDTA) were purchased and used in biological assays. All synthesized compounds evaluated in biological assays were determined to be ≥95% pure using a Shimadzu Prominence HPLC system, an AB Sciex 3200 QTRAP spectrometer, and a Kinetex C18 column (50 mm × 2.1 mm × 2.6 μm) with a 21 min linear gradient from 10 to 80% acetonitrile in 0.1% formic acid at a flow rate of 0.25 mL/min (traces and purity analysis can be viewed in the Supporting Information). All compounds were stored as DMSO stocks at room temperature in the absence of light for several months at a time without observing losses in biological activity. To ensure the integrity of DMSO stock solutions of test compounds, they were not subjected to freeze–thaw cycles. Bacterial strains used during these investigations include S. aureus BAA-1707 (“MRSA-1707”), BAA-44 and UAMS-1, S. epidermidis ATCC 35984, E. faecium ATCC 700221, E. faecalis OG1RF (ATCC 47077), S. pneumoniae ATCC 6303, M. tuberculosis H37Ra (ATCC 25177), and CDC1551. All animal experiments performed were conducted in compliance with institutional guidelines.
3.2. Chemistry.
This chemistry section includes the following items: (a) synthetic procedures and compound characterization (ordered by the synthetic route; note: most 1-methoxyphenazines synthesized during these studies are not presented in the manuscript; however, their full structures can be viewed in the Supporting Information Figure 2 and spectra section), (b) UV–vis for HPs binding iron(II), and (c) pKa determination of select HP analogues.
3.2.1. Synthetic Procedures and Compound Characterization.
3.2.1.1. General Two-Step Procedure for the Synthesis of 3-Chloro-1-methoxyphenazines (38, 50–58).44,45
Step 1. 4-Bromoaniline (303 mg, 1.76 mmol) dissolved in 2 mL of N,N-dimethylformamide was added dropwise to a stirring solution of potassium tert-butoxide (416 mg, 4.80 mmol) in N,N-dimethylformamide (12 mL) at −60 °C. Then, a solution of 5-choloro-2-nitroanisole 13 (300 mg, 1.60 mmol) in N,N-dimethylformamide (2 mL) was added to the mixture which continued to stir at −60 °C for 7 h. Upon completion of this reaction, the resulting mixture was transferred to a separatory funnel containing saturated aqueous ammonium chloride (80 mL) and the crude product was extracted with ethyl acetate (3 × 30 mL). The organic layers were then combined, washed with brine, and dried with sodium sulfate. The resulting organic layer was then filtered and concentrated in vacuo to obtain the crude nitroso intermediate that was used directly in the next step. Step 2. The crude nitroso intermediate (oil) was dissolved in N,N-dimethylformamide (10 mL) before N,O-bis(trimethylsilyl)acetamide (1.97 mL, 8.00 mmol) was added to the solution. The resulting mixture was allowed to stir at 50 °C for 16 h until completion. After this time, 2 mL of water was added to the mixture and stirring continued at room temperature for an additional 10 min before the precipitate was filtered and washed with cold ethyl acetate (this solid was a batch of the desired product). The filtrate was then transferred to a separatory funnel containing brine (50 mL) and extracted with ethyl acetate (3 × 30 mL) to isolate additional product. After extraction of the filtrate, the organic layers were combined and washed with water (3 × 50 mL), dried with sodium sulfate, filtered, and concentrated in vacuo. The resulting crude solid was purified via silica gel chromatography using hexanes/ethyl acetate (99:1 to 85:15) to afford a yellow solid as a second product batch, which was combined with the solid obtained above to afford 57 (413 mg, 80% yield).
3.2.1.2. 3-Chloro-1-methoxy-6-methylphenazine (38).
Yield: 80%; 578 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.18 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 2.0 Hz, 1H), 7.74−7.62 (m, 2H), 6.98 (d, J = 2.0 Hz, 1H), 4.16 (s, 3H), 2.87 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 155.6, 143.8, 143.3, 142.4, 137.8, 136.3, 135.2, 130.7, 130.6, 128.2, 120.7, 108.7, 57.0, 17.8. HRMS (ESI): calcd for C14H12ClN2O [M + H]+, 259.0633; found, 259.0642. mp 178–180 °C.
3.2.1.3. 3-Chloro-1-methoxyphenazine (50).
Yield: 44%; 105 mg was isolated as a yellow solid. Note: 1H NMR and 13C NMR spectral data match those previously reported for this compound (CAS: 1346682–87-8).44
3.2.1.4. 8-Chloro-6-methoxy-1,2-dimethylphenazine (51).
Yield: 72%; 211 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.10 (d, J = 9.0 Hz, 1H), 7.86 (d, J = 1.5, 1H), 7.64 (d, J = 9.0 Hz, 1H), 6.98 (d, J = 1.5 Hz, 1H), 4.17 (s, 3H), 2.82 (s, 3H), 2.57 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 155.7, 143.7, 143.3, 141.2, 139.0, 136.1, 134.6, 134.5, 134.2, 127.1, 120.7, 108.3, 57.0, 21.0, 13.3. HRMS (ESI): calcd for C15H14ClN2O [M + H]+, 273.0789; found, 273.0799. mp 174–176 °C.
3.2.1.5. 2-Bromo-8-chloro-6-methoxy-1-methylphenazine (52).
Yield: 72%; 388 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.04 (d, J = 9.3 Hz, 1H), 7.90 (d, J = 9.3 Hz, 1H), 7.84 (d, J = 2.0 Hz, 1H), 7.00 (d, J = 2.0 Hz, 1H), 4.17 (s, 3H), 2.95 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 155.6, 143.6, 143.4, 141.2, 137.5, 137.2, 135.1, 135.0, 128.7, 127.4, 120.6, 109.0, 57.1, 17.6. HRMS (ESI): calcd for C14H11BrClN2O [M + H]+, 336.9738; found, 336.9734. mp 218–220 °C.
3.2.1.6. 3-Chloro-1-methoxy-6-phenoxyphenazine (53).
Yield: 69%; 445 mg was isolated as a yellow solid. 1H NMR (600 MHz, CDCl3): δ 8.08 (d, J = 8.8 Hz, 1H), 8.00 (d, J = 2.0 Hz, 1H), 7.68 (dd, J = 8.8, 7.6 Hz, 1H), 7.47−7.41 (m, 2H), 7.26−7.21 (m, 3H), 7.08 (d, J = 7.6 Hz, 1H), 7.05 (d, J = 2.0 Hz, 1H), 4.20 (s, 3H). 13C NMR (151 MHz, CDCl3): δ 156.4, 155.5, 154.1, 143.2, 143.0, 137.7, 137.0, 135.9, 130.2, 130.1, 124.8, 124.3, 120.8, 120.8, 114.7, 109.3, 57.0. HRMS (ESI): calcd for C19H14ClN2O2 [M + H]+, 337.0738; found, 337.0752. mp 178–180 °C. Note: TMS was used as the reference in this 1H NMR spectrum at 0.00 pm due to the chloroform peak being buried in proton signals.
3.2.1.7. 3-Chloro-8-fluoro-1-methoxyphenazine (54).
Yield: 82%; 344 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.21 (dd, J = 9.5, 5.9 Hz, 1H), 7.96 (dd, J = 9.4, 2.8 Hz, 1H), 7.82 (d, J = 2.0 Hz, 1H), 7.68 (ddd, J = 9.5, 7.9, 2.8 Hz, 1H), 7.03 (d, J = 2.0 Hz, 1H), 4.18 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 163.2 (d, J = 255.7 Hz), 155.5, 143.6 (d, J = 2.8 Hz), 142.7 (d, J = 13.7 Hz), 141.7, 136.8 (d, J = 1.8 Hz), 136.0, 131.9 (d, J = 10.2 Hz), 123.6 (d, J = 28.3 Hz), 120.3 (d, J = 1.2 Hz), 112.6 (d, J = 21.5 Hz), 109.5, 57.1. HRMS (ESI): calcd for C13H9ClFN2O [M + H]+, 263.0382; found, 263.0394. mp 221–223 °C.
3.2.1.8. 1,3,8-Trichloro-6-methoxyphenazine (55).
Yield: 34%; 175 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.26 (s, 1H), 7.98−7.86 (m, 2H), 7.03 (s, 1H), 4.17 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 155.5, 143.7, 142.2, 139.4, 138.2, 136.3, 135.7, 134.2, 132.0, 127.9, 120.6, 110.1, 57.2. HRMS (DART): calcd for C13H8Cl3N2O [M + H]+, 312.9697; found, 312.9696. mp 221–223 °C.
3.2.1.9. 6-Bromo-3-chloro-1-methoxyphenazine (56).
Yield: 61%; 183 mg was isolated as a yellow solid. 1H NMR (500 MHz, CDCl3): δ 8.34 (dd, J = 8.8, 1.3 Hz, 1H), 8.20 (dd, J = 7.3, 1.3 Hz, 1H), 7.99 (d, J = 2.0 Hz, 1H), 7.68 (dd, J = 8.8, 7.3 Hz, 1H), 7.05 (d, J = 2.0 Hz, 1H), 4.19 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 155.5, 144.2, 142.6, 141.5, 137.9, 136.0, 134.7, 130.5, 130.3, 124.1, 120.6, 109.6, 57.1. HRMS (ESI): calcd for C13H9BrClN2O [M + H]+, 322.9581; found, 322.9578. mp 137–139 °C.
3.2.1.10. 8-Bromo-3-chloro-1-methoxyphenazine (57).
Yield: 80%; 413 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.55 (d, J = 2.1 Hz, 1H), 8.04 (d, J = 9.2 Hz, 1H), 7.89 (dd, J = 9.2, 2.1 Hz, 1H), 7.78 (d, J = 2.0 Hz, 1H), 7.01 (d, J = 2.0 Hz, 1H), 4.17 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 155.7, 144.1, 142.7, 142.3, 137.5, 135.9, 135.3, 132.3, 130.7, 125.1, 120.3, 109.5, 57.1. HRMS (ESI): calcd for C13H9BrClN2O [M + H]+, 322.9581; found, 322.9580. mp 225–227 °C.
3.2.1.11. 3-Chloro-8-iodo-1-methoxyphenazine (58).
Yield: 65%; 388 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.83 (d, J = 1.9 Hz, 1H), 8.06 (dd, J = 9.1, 1.9 Hz, 1H), 7.89 (d, J = 9.1 Hz, 1H), 7.80 (d, J = 2.0 Hz, 1H), 7.02 (d, J = 2.0 Hz, 1H), 4.17 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 155.8, 144.1, 143.0, 142.5, 140.2, 139.3, 137.6, 135.7, 130.4, 120.2, 109.4, 97.2, 57.1. HRMS (ESI): calcd for C13H9ClIN2O [M + H]+, 370.9443; found, 370.9448. mp 224–226 °C.
3.2.1.12. Boron Tribromide Demethylation of 3-Chloro-1-methoxyphenazines to 3-Chloro-1-hydroxyphenazines (18–27).
Compound 38 (200 mg, 0.77 mmol) was dissolved in anhydrous dichloromethane (50 mL) in a round bottom flask. The solution was then cooled to −78 °C before a 1 M solution of boron tribromide (4.6 mL, 4.6 mmol in dichloromethane) was added dropwise. The resulting reaction mixture was allowed to stir at −78 °C for 1 h before being warmed to room temperature overnight. After this time, the reaction was heated to reflux until completion (monitored by TLC). Upon completion, brine (50 mL) was added to the mixture to quench the reaction. The resulting mixture was then transferred to a separatory funnel and extracted with dichloromethane. The combined organic layers were dried with sodium sulfate, filtered, and concentrated in vacuo. The resulting solid was purified via column chromatography using dichloromethane to elute compound 19 as a yellow solid (191 mg, 100%).
3.2.1.13. 3-Chlorophenazin-1-ol (18).
Yield: 89%; 84.0 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.27 (br s, 1H), 8.26−8.18 (m, 2H), 7.92−7.83 (m, 2H), 7.79 (d, J = 2.0 Hz, 1H), 7.22 (d, J = 2.0 Hz, 1H). 13C NMR (101 MHz, CDCl3): δ 152.4, 145.0, 143.8, 141.2, 138.3, 133.5, 131.6, 131.0, 129.9, 129.4, 118.9, 111.0. HRMS (ESI): calcd for C12H8ClN2O [M + H]+, 231.0320; found, 231.0329. mp 213–215 °C. HPLC purity: 98.7%.
3.2.1.14. 3-Chloro-6-methylphenazin-1-ol (19).
Yield: 100%; 191 mg was isolated as a yellow solid. 1H NMR (600 MHz, DMSO-d6): δ 11.16 (br s, 1H), 8.08 (dd, J = 8.5, 1.6 Hz, 1H), 7.86−7.76 (m, 2H), 7.72 (d, J = 2.2 Hz, 1H), 7.15 (d, J = 2.2 Hz, 1H), 2.79 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 154.8, 142.8, 142.7, 141.4, 137.0, 136.2, 134.4, 130.9, 130.7, 127.4, 117.8, 111.3, 17.3. HRMS (ESI): calcd for C13H10ClN2O [M + H]+, 245.0476; found, 245.0464. mp 185–187 °C. HPLC purity: 97.8%.
3.2.1.15. 3-Chloro-6,7-dimethylphenazin-1-ol (20).
Yield: 93%; 123 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.19 (br s, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.74 (d, J = 2.0 Hz, 1H), 7.60 (d, J = 8.9 Hz, 1H), 7.12 (d, J = 2.0 Hz, 1H), 2.79 (s, 3H), 2.56 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 152.2, 144.3, 142.8, 140.0, 138.9, 137.2, 134.8, 134.5, 132.2, 126.0, 119.2, 110.3, 21.0, 13.4. HRMS (ESI): calcd for C14H12ClN2O [M + H]+, 259.0633; found, 259.0642. mp 183–185 °C. HPLC purity: 99.1%.
3.2.1.16. 7-Bromo-3-chloro-6-methylphenazin-1-ol (21).
Yield: 100%; 122 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.14 (s, 1H), 7.97−7.89 (m, 2H), 7.83 (d, J = 2.0 Hz, 1H), 7.22 (d, J = 2.1 Hz, 1H), 3.00 (s, 3H). 13C NMR (151 MHz, CDCl3): δ 152.3, 144.4, 143.1, 140.2, 138.5, 138.0, 135.4, 132.8, 127.7, 127.4, 119.3, 111.3, 17.8. HRMS (ESI): calcd for C13H7BrClN2O [M − H]−, 320.9436; found, 320.9421. mp 210–212 °C. HPLC purity: 99.3%.
3.2.1.17. 3-Chloro-6-phenoxyphenazin-1-ol (22).
Yield: 100%; 118 mg was isolated as a yellow solid. 1H NMR (500 MHz, CDCl3): δ 8.23 (s, 1H), 7.94 (d, J = 2.0 Hz, 1H), 7.92 (dd, J = 8.8, 1.1 Hz, 1H), 7.69 (dd, J = 8.8, 7.6 Hz, 1H), 7.48−7.41 (m, 2H), 7.28−7.20 (m, 4H), 7.08 (dd, J = 7.6, 1.1 Hz, 1H). 13C NMR (101 MHz, CDCl3): δ 156.3, 154.5, 152.2, 142.8, 141.9, 138.4, 138.2, 133.7, 130.5, 130.3, 125.1, 123.2, 120.9, 119.5, 114.5, 111.5. HRMS (ESI): calcd for C18H12ClN2O2 [M + H]+, 323.0582; found, 323.0570. mp 168–170 °C. HPLC purity: 97.4%.
3.2.1.18. 3-Chloro-8-fluorophenazin-1-ol (23).
Yield: 100%; 95 mg was isolated as a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 11.32 (br s, 1H), 8.29 (ddd, J = 9.4, 6.1, 0.7 Hz, 1H), 8.01−7.91 (m, 2H), 7.73 (d, J = 2.2 Hz, 1H), 7.17 (d, J = 2.2 Hz, 1H). 13C NMR (101 MHz, DMSO-d6): δ 162.5 (d, J = 253.0 Hz), 154.6, 143.0 (d, J = 2.5 Hz), 141.6 (d, J = 13.9 Hz), 141.0, 136.2 (d, J = 1.7 Hz), 135.0, 131.9 (d, J = 10.5 Hz), 123.6 (d, J = 28.4 Hz), 117.6, 112.0, 111.7 (d, J = 21.1 Hz). HRMS (ESI): calcd C12H5FClN2O for [M − H]−, 247.0080; found, 247.0078. mp 214–216 °C. HPLC purity: >99.9%.
3.2.1.19. 3,6,8-Trichlorophenazin-1-ol (24).
Yield: 96%; 116 mg was isolated as a yellow solid. 1H NMR (600 MHz, CDCl3): δ 8.15 (d, J = 2.2 Hz, 1H), 8.06 (s, 1H), 7.96 (d, J = 2.2 Hz, 1H), 7.92 (d, J = 2.0 Hz, 1H), 7.27 (d, J = 2.0 Hz, 1H). 13C NMR (101 MHz, CDCl3): δ 152.1, 143.4, 141.2, 140.2, 139.5, 136.1, 134.7, 134.1, 132.1, 127.0, 119.4, 112.6. HRMS (ESI): calcd for C12H4Cl3N2O [M − H]−, 296.9395; found, 296.9403. mp 234–236 °C. HPLC purity: 97.6%.
3.2.1.20. 6-Bromo-3-chlorophenazin-1-ol (25).
Yield: 100%; 141 mg was isolated as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.26−8.18 (m, 2H), 8.15 (s, 1H), 7.96 (d, J = 2.0 Hz, 1H), 7.71 (dd, J = 8.7, 7.3 Hz, 1H), 7.26 (m, 1H, partially buried in the reference signal). 13C NMR (151 MHz, CDCl3): δ 152.2, 143.9, 142.3, 141.7, 139.1, 134.8, 133.9, 130.8, 129.3, 124.7, 119.4, 111.9. HRMS (ESI): calcd for C12H5BrClN2O [M − H]−, 306.9279; found, 306.9277. mp 223–225 °C. HPLC purity: 98.2%.
3.2.1.21. 8-Bromo-3-chlorophenazin-1-ol (26).
Yield: 100%; 103 mg was isolated as a yellow solid. 1H NMR (600 MHz, DMSO-d6): δ 11.30 (br s, 1H), 8.45 (d, J = 2.1 Hz, 1H), 8.11 (d, J = 9.2 Hz, 1H), 8.05 (dd, J = 9.2, 2.1 Hz, 1H), 7.70 (d, J = 2.1 Hz, 1H), 7.16 (d, J = 2.1 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 154.8, 143.5, 142.0, 141.4, 136.9, 135.0, 134.9, 131.2, 130.8, 124.1, 117.5, 111.9. HRMS (ESI): calcd for C12H5BrClN2O [M − H]−, 306.9279; found, 306.9274. mp 251–253 °C. HPLC purity: 98.1%.
3.2.1.22. 3-Chloro-8-iodophenazin-1-ol (27).
Yield: 100%; 110 mg was isolated as a yellow solid. 1H NMR (600 MHz, DMSO-d6): δ 11.28 (br s, 1H), 8.69 (d, J = 1.8 Hz, 1H), 8.19 (dd, J = 9.1, 1.8 Hz, 1H), 7.96 (d, J = 9.1 Hz, 1H), 7.73 (d, J = 2.1 Hz, 1H), 7.18 (d, J = 2.1 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 154.9, 143.6, 142.3, 141.8, 140.0, 137.9, 136.9, 134.8, 130.4, 117.5, 111.8, 98.3. HRMS (ESI): calcd for C12H5ClIN2O [M − H]−, 354.9141; found, 354.9145. mp 233–235 °C. HPLC purity: 97.9%.
3.3. General Procedures for Nucleophilic Aromatic Substitution (NAS).
3.3.1. NAS Reaction Method A (39, 40, and 42).
3-Chloro-1-methoxy-6-methylphenazine 38 (160 mg, 0.62 mmol) and potassium carbonate were suspended in anhydrous N,N-dimethylformamide (6 mL) in a sealed tube. The resulting mixture was then purged with argon for 30 min before 2-mercaptoethanol (350 μL, 4.94 mmol) was added. The sealed tube was then closed, and the reaction was allowed to stir at 85 °C for 7 days. Upon completion, the reaction mixture was transferred to a separatory funnel containing brine (100 mL) and extracted with ethyl acetate (3 × 50 mL). The resulting organic extracts were then combined, washed with water, dried with anhydrous sodium sulfate, filtered, and concentrated in vacuo. The resulting crude solid was then purified via flash column chromatography using hexanes/ethyl acetate (4:1 to 1:1) to afford 43 as a yellow solid (30 mg, 17%) and 40 as a yellow solid (96 mg, 52%).
3.3.2. NAS Reaction Method B (41, 43 and 44).
3-Chloro-1-methoxy-6-methylphenazine 38 (147 mg, 0.57 mmol) was dissolved in N,N-dimethylformamide (6 mL) in a microwave reaction vessel. Then, potassium carbonate (392 mg, 2.84 mmol) and 2,5,8,11-tetraoxatridecane-13-thiol 61 (956 mg, 4.54 mmol) were added to the reaction vessel, which was then purged with argon for 3 min before being sealed and heated to 200 °C for 152 s. Upon completion, the reaction mixture was then transferred to a separatory funnel containing ethyl acetate (50 mL) and brine (50 mL). The product was extracted, and the organic layer was collected, washed with brine (3 × 50 mL), dried with anhydrous sodium sulfate, filtered, and concentrated in vacuo to give a crude red oil. The crude products were purified via column chromatography using hexane/ethyl acetate (1:1 to 1:99) to afford 44 (43 mg, 18%) as red oil and 41 as red oil (210 mg, 82%).
3.3.2.1. 3-(Ethylthio)-1-methoxy-6-methylphenazine (39).
Yield: 31%; 51 mg was isolated as a yellow solid (NAS method A). 1H NMR (600 MHz, CDCl3): δ 8.14 (dd, J = 8.4, 1.8 Hz, 1H), 7.63−7.55 (m, 2H), 7.50 (d, J = 1.8 Hz, 1H), 6.81 (d, J = 1.8 Hz, 1H), 4.11 (s, 3H), 3.16 (q, J = 7.4 Hz, 2H), 2.84 (s, 3H), 1.46 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3): δ 154.4, 143.8, 143.3, 142.1, 141.7, 137.3, 135.6, 130.3, 129.4, 128.2, 115.1, 107.0, 56.6, 26.3, 17.8, 13.8. HRMS (ESI): calcd for C16H17N2OS [M + H]+, 285.1056; found, 285.1069. mp 89–91 °C.
3.3.2.2. 2-((4-Methoxy-9-methylphenazin-2-yl)thio)ethan-1-ol (40).
Yield: 52%; 96 mg was isolated as a yellow solid (NAS method A). 1H NMR (500 MHz, CDCl3): δ 8.20 (m, 1H), 7.83 (s, 1H), 7.74−7.65 (m, 2H), 6.91 (d, J = 2.0 Hz, 1H), 4.16 (s, 3H), 4.01 (t, J = 6.2 Hz, 2H), 3.42 (t, J = 6.2 Hz, 2H), 2.92 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 154.8, 143.6, 143.4, 142.0, 140.6, 137.4, 135.7, 130.6, 129.9, 128.3, 116.3, 107.4, 60.6, 56.8, 35.4, 17.9. HRMS (ESI): calcd for C16H17N2O2S [M + H]+, 301.1005; found, 301.1016. mp 158–160 °C.
3.3.2.3. 3-((2,5,8,11-Tetraoxatridecan-13-yl)thio)-1-methoxy-6-methylphenazine (41).
Yield: 82%; 210 mg was isolated as a red residue (NAS method B). 1H NMR (600 MHz, CDCl3): δ 8.18 (m, 1H), 7.72−7.63 (m, 3H), 6.89 (d, J = 1.7 Hz, 1H), 4.14 (s, 3H), 3.88 (t, J = 6.6 Hz, 2H), 3.72−3.66 (m, 6H), 3.66−3.60 (m, 4H), 3.54−3.51 (m, 2H), 3.40 (t, J = 6.6 Hz, 2H), 3.36 (s, 3H), 2.90 (s, 3H).13C NMR (151 MHz, CDCl3): δ 154.6, 143.3, 142.9, 142.2, 141.9, 137.1, 135.8, 130.8, 129.7, 128.3, 115.2, 107.3, 72.1, 70.9, 70.8, 70.8, 70.7, 69.4, 59.2, 56.8, 32.1, 18.0. Note: one carbon signal is missing in our spectra due to overlap at 70.8 ppm (determined by HSQC; see spectra for details). HRMS (ESI): calcd for C23H31N2O5S [M + H]+, 447.1948; found, 447.1940.
3.3.2.4. 3-(Ethylthio)-6-methylphenazin-1-ol (42).
Yield: 37%; 58 mg was isolated as a yellow solid (NAS method A). 1H NMR (500 MHz, CDCl3): δ 8.13 (s, 1H), 7.93 (dd, J = 7.4, 2.5 Hz, 1H), 7.64−7.58 (m, 2H), 7.46 (d, J = 1.9 Hz, 1H), 7.04 (d, J = 1.9 Hz, 1H), 3.16 (q, J = 7.4 Hz, 2H), 2.86 (s, 3H), 1.48 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 151.1, 144.1, 143.6, 143.4, 140.6, 137.8, 133.5, 130.2, 129.7, 127.1, 114.0, 108.9, 26.3, 18.0, 13.9. HRMS (ESI): calcd C15H15N2OS for [M + H]+, 271.0900; found, 271.0896. mp 119–121 °C.
3.3.2.5. 3-((2-Hydroxyethyl)thio)-6-methylphenazin-1-ol (43).
Yield: 75%; 44 mg was isolated as a yellow solid (NAS method B). 1H NMR (600 MHz, DMSO-d6): 10.72 (br s, 1H), 8.06 (m, 1H), 7.79−7.73 (m, 2H), 7.46 (d, J = 2.0 Hz, 1H), 7.02 (d, J = 2.0 Hz, 1H), 5.13 (t, J = 5.6 Hz, 1H), 3.73 (q, J = 5.7 Hz, 2H), 3.28 (t, J = 6.6 Hz, 2H), 2.81 (s, 3H). 13C NMR (151 MHz, DMSO-d6): δ 153.0, 143.2, 142.8, 142.5, 140.7, 136.7, 134.6, 130.4, 129.6, 127.3, 112.6, 109.8, 59.5, 33.7, 17.4. HRMS (ESI): calcd for C15H15N2O2S [M +H]+, 287.0849; found, 287.0855. mp 154–156 °C.
3.3.2.6. 3-((2,5,8,11-Tetraoxatridecan-13-yl)thio)-6-methylphenazin-1-ol (44).
Yield: 18%; 43 mg was isolated as a red residue (NAS method B). 1H NMR (600 MHz, DMSO-d6): δ 10.73 (br s, 1H), 8.06 (m, 1H), 7.78−7.74 (m, 2H), 7.46 (d, J = 1.8 Hz, 1H), 7.03 (d, J = 1.8 Hz, 1H), 3.76 (t, J = 6.3 Hz, 2H), 3.59 (dd, J = 5.9, 3.5 Hz, 2H), 3.54 (dd, J = 5.9, 3.5 Hz, 2H), 3.51 (dd, J = 5.9, 3.5 Hz, 2H), 3.49−3.45 (m, 4H), 3.40−3.38 (m, 4H), 3.20 (s, 3H), 2.80 (s, 3H). 13C NMR (151 MHz, DMSO-d6): δ 153.1, 143.1, 142.5, 142.5, 140.7, 136.7, 134.6, 130.3, 129.6, 127.3, 112.9, 109.8, 71.3, 69.8, 69.8, 69.6, 68.6, 58.0, 30.9, 17.4. Note: two carbon signals from the PEG chain could not be viewed in our 13C NMR spectra due to signal overlap at 70 ppm, which is supported by HSQC (see the spectra section of the Supporting Information for details). HRMS (ESI): calcd for C22H29N2O5S [M + H]+, 433.1792; found, 433.1800.
3.4. General Procedures for Bromination of 3-Chloro-1-hydroxyphenazines to Target HPs.
3.4.1. Bromination Reaction Method A (Synthesis of 28–33, 35, 37, and 45).
Compound 27 (100 mg, 0.28 mmol) and N-bromosuccinimide (104 mg, 0.59 mmol) were dissolved with dichloromethane (75 mL) in a round bottom flask. The resulting reaction mixture was then allowed to stir at room temperature for 3 h until completion. After this time, the contents of the reaction were transferred to a separatory funnel with brine and extracted with dichloromethane. The organic layer extracts were collected, dried with sodium sulfate, filtered, and concentrated in vacuo. The resulting solid was purified via column chromatography using 100% dichloromethane to elute 37 (107 mg, 74%) as a yellow solid.
3.4.2. Bromination Reaction Method B (Synthesis of 34, 36, 46, and 47).
Compound 24 (102 mg, 0.34 mmol) was dissolved in toluene (7 mL) before N-bromosuccinimide (133 mg, 0.75 mmol) was added to the solution. The resulting reaction mixture was then heated to 50 °C for 6 h until completion. After this time, the reaction mixture was cooled to room temperature and concentrated via rotavap. The crude material was then absorbed onto silica gel (dry loaded) and purified via column chromatography using 100% dichloromethane to elute 34 (98 mg, 63%) as a yellow solid.
3.4.2.1. 2,4-Dibromo-3-chlorophenazin-1-ol (28).
Yield: 46%; 31 mg was isolated as a yellow solid (method A). 1H NMR (400 MHz, DMSO-d6): δ 8.37 (m, 1H), 8.32 (m, 1H), 8.11−8.03 (m, 2H). 13C NMR (101 MHz, DMSO-d6): δ 152.2, 143.4, 141.2, 139.7, 137.2, 133.9, 132.7, 132.3, 129.4, 128.9, 112.5, 106.1. HRMS (DART): calcd for C12H6Br2ClN2O [M + H]+, 388.8509; found, 388.8525. mp 218–220 °C. HPLC purity: 99.5%.
3.4.2.2. 2,4-Dibromo-3-chloro-6-methylphenazin-1-ol (29).
Yield: 75%; 434 mg was isolated as a yellow solid (method A). 1H NMR (400 MHz, DMSO-d6): δ 11.91 (s, 1H), 8.16 (d, J = 8.4 Hz, 1H), 7.95−7.85 (m, 2H), 2.85 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 152.0, 142.7, 141.4, 138.6, 137.4, 136.7, 133.4, 132.1, 131.4, 126.6, 112.9, 106.1, 16.9. HRMS (DART): calcd for C13H8Br2ClN2O [M + H]+, 402.8665; found, 402.8671. mp 216–218 °C. HPLC purity: 98.8%.
3.4.2.3. 2,4-Dibromo-3-chloro-6,7-dimethylphenazin-1-ol (30).
Yield: 72%; 112 mg was isolated as a yellow solid (method A). 1H NMR (400 MHz, DMSO-d6): δ 11.66 (br s, 1H), 8.06 (d, J = 8.9 Hz, 1H), 7.84 (d, J = 8.9 Hz, 1H), 2.80 (s, 3H), 2.57 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 151.8, 142.4, 140.0, 139.7, 138.3, 136.3, 135.6, 133.7, 132.4, 125.3, 112.8, 105.3, 20.1, 12.6. HRMS (DART): calcd for C14H10Br2ClN2O [M + H]+, 416.8822; found, 416.8816. mp 223–225 °C. HPLC purity: 95.5%.
3.4.2.4. 2,4,7-Tribromo-3-chloro-6-methylphenazin-1-ol (31).
Yield: 33%; 51 mg was isolated as a yellow solid (method A). 1H NMR (500 MHz, DMSO-d6): δ 8.12 (d, J = 9.3 Hz, 1H), 8.07 (d, J = 9.3 Hz, 1H), 2.92 (s, 3H). 13C NMR (101 MHz, DMSO-d6): δ 151.9, 142.5, 140.2, 138.8, 137.5, 136.9, 135.7, 133.2, 127.6, 127.1, 112.7, 106.4, 16.9. HRMS (DART): calcd for C13H7Br3ClN2O [M + H]+, 478.7792; found, 478.7801. mp 243–245 °C. HPLC purity: >99.9%.
3.4.2.5. 2,4-Dibromo-3-chloro-6-phenoxyphenazin-1-ol (32).
Yield: 38%; 47 mg was isolated as a yellow solid (method A). 1H NMR (500 MHz, DMSO-d6): δ 8.11 (d, J = 8.7 Hz, 1H), 7.96 (t, J = 8.2 Hz, 1H), 7.44 (t, J = 7.8 Hz, 2H), 7.40 (d, J = 7.6 Hz, 1H), 7.26−7.16 (m, 3H). 13C NMR (151 MHz, DMSO-d6): δ 156.8, 152.7, 152.1, 142.2, 138.9, 137.2, 133.9, 132.1, 130.1, 124.1, 123.6, 119.3, 117.5, 112.9, 106.6. Note: despite multiple experiments, one carbon signal is missing likely due to overlap. HRMS (DART): calcd for C18H10Br2ClN2O2 [M + H]+, 478.8792; found, 478.8813. mp 239–241 °C. HPLC purity: 98.1%.
3.4.2.6. 2,4-Dibromo-3-chloro-8-fluorophenazin-1-ol (33).
Yield: 73%; 89 mg was isolated as a yellow solid (method A). 1H NMR (500 MHz, DMSO-d6): δ 12.02 (br s, 1H), 8.39 (dd, J = 9.5, 5.9 Hz, 1H), 8.04 (ddd, J = 9.5, 8.2, 2.7 Hz, 1H), 8.00 (dd, J = 9.5, 2.7 Hz, 1H). 13C NMR (126 MHz, DMSO-d6): δ 163.2 (d, J = 255.6 Hz), 151.9, 141.7 (d, J = 14.2 Hz), 141.1, 139.2 (d, J = 1.8 Hz), 137.0, 134.1, 132.4 (d, J = 10.7 Hz), 124.3 (d, J = 28.4 Hz), 112.6, 111.1 (d, J = 21.5 Hz), 106.9. HRMS (ESI): calcd C12H3Br2ClFN2O for [M − H]−, 402.8290; found, 402.8286. mp 236–238 °C. HPLC purity: 99.6%.
3.4.2.7. 2,4-Dibromo-3,6,8-trichlorophenazin-1-ol (34).
Yield: 63%; 98 mg was isolated as a yellow solid (method B). 1H NMR (500 MHz, DMSO-d6): δ 8.39 (d, J = 2.1 Hz, 1H), 8.28 (d, J = 2.1 Hz, 1H). 13C NMR (126 MHz, DMSO-d6): δ 152.2, 141.4, 139.5, 138.7, 138.4, 135.8, 134.7, 133.7, 132.1, 126.6, 112.6, 107.8. HRMS (DART): calcd for C12H4Br2Cl3N2O [M + H]+, 458.7703; found, 458.7696. mp 246–248 °C. HPLC purity: 99.9%.
3.4.2.8. 2,4,6-Tribromo-3-chlorophenazin-1-ol (35).
Yield: 38%; 30 mg was isolated as an orange solid (method A). 1H NMR (500 MHz, DMSO-d6): δ 8.45 (dd, J = 7.3, 0.7 Hz, 1H), 8.35 (dd, J = 8.8, 0.7 Hz, 1H), 7.93 (dd, J = 8.6, 7.3 Hz, 1H). 13C NMR (101 MHz, DMSO-d6): δ 152.1, 141.9, 140.6, 139.8, 138.0, 135.5, 134.3, 132.4, 129.0, 123.5, 112.7, 107.1. HRMS (ESI): calcd for C12H3Br3ClN2O [M − H]−, 462.7490; found, 462.7486. mp 193–195 °C. HPLC purity: >99.9%.
3.4.2.9. 2,4,8-Tribromo-3-chlorophenazin-1-ol (36).
Yield: 36%; 46 mg was isolated as a yellow solid (method A). 1H NMR (400 MHz, DMSO-d6): δ 8.55 (d, J = 2.1 Hz, 1H), 8.26 (d, J = 9.3 Hz, 1H), 8.17 (dd, J = 9.3, 2.1 Hz, 1H). 13C NMR (101 MHz, DMSO-d6): δ 151.9, 142.0, 141.3, 139.7, 137.5, 135.5, 134.1, 131.1, 130.4, 125.5, 112.5, 106.8. HRMS (ESI): calcd for C12H3Br3ClN2O [M − H]−, 462.7490; found, 462.7469. mp 250 °C (decomp). HPLC purity: 98.9%.
3.4.2.10. 2,4-Dibromo-3-chloro-8-iodophenazin-1-ol (37).
Yield: 74%; 107 mg was isolated as an orange solid (method A). 1H NMR (600 MHz, DMSO-d6): δ 8.76 (d, J = 1.8 Hz, 1H), 8.28 (dd, J = 9.1, 1.8 Hz, 1H), 8.07 (d, J = 9.1 Hz, 1H). 13C NMR (101 MHz, DMSO-d6): δ 152.2, 142.4, 141.7, 140.7, 139.8, 137.6, 137.2, 133.9, 130.7, 112.7, 106.8, 100.4. HRMS (ESI): calcd for C12H3Br2ClIN2O [M − H]−, 510.7351; found, 510.7351. mp 258–260 °C. HPLC purity: 99.7%.
3.4.2.11. 2,4-Dibromo-3-(ethylthio)-6-methylphenazin-1-ol (45).
Yield: 73%; 35 mg was isolated as a yellow solid (method A). 1H NMR (500 MHz, CDCl3): δ 8.62 (s, 1H), 8.09 (d, J = 8.5 Hz, 1H), 7.81 (dd, J = 8.5, 7.0 Hz, 1H), 7.76 (d, J = 7.0 Hz, 1H), 3.17 (q, J = 7.4 Hz, 2H), 2.99 (s, 3H), 1.32 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 149.1, 144.5, 141.9, 140.2, 139.3, 139.3, 133.2, 132.5, 131.1, 126.6, 124.2, 111.5, 31.4, 17.7, 14.9. HRMS (DART): calcd for C15H13Br2N2OS [M + H]+, 428.9090; found, 428.9094. mp 163–165 °C. HPLC purity: 97.8%.
3.4.2.12. 2,4-Dibromo-3-((2-hydroxyethyl)thio)-6-methylphenazin-1-ol (46).
Yield: 28%; 25 mg was isolated as a yellow solid (method B). 1H NMR (600 MHz, DMSO-d6): δ 11.52 (br s, 1H), 8.16 (d, J = 8.6 Hz, 1H), 7.91 (dd, J = 8.6, 6.8 Hz, 1H), 7.87 (d, J = 6.8 Hz, 1H), 4.88 (br s, 1H), 3.59 (t, J = 7.1 Hz, 2H), 3.17 (t, J = 7.1 Hz, 2H), 2.87 (s, 3H). 13C NMR (151 MHz, DMSO-d6): δ 150.8, 142.8, 141.5, 139.7, 138.6, 137.6, 134.2, 132.3, 131.1, 126.5, 122.0, 112.3, 60.5, 38.9, 17.0. The 13C signal at 38.9 ppm was determined by HSQC and can be viewed in the spectra section. HRMS (ESI): calcd for C15H11Br2N2O2S [M − H]−, 442.8893; found, 442.8905. mp 178–180 °C. HPLC purity: 95.7%.
3.4.2.13. 3-((2,5,8,11-Tetraoxatridecan-13-yl)thio)-2,4-dibromo-6-methylphenazin-1-ol (47).
Yield: 27%; 16 mg was isolated as a yellow residue (method B). 1H NMR (600 MHz, CDCl3): δ 8.66 (br s, 1H), 8.09 (d, J = 8.6 Hz, 1H), 7.82 (dd, J = 8.6, 6.7 Hz, 1H), 7.76 (d, J = 6.7 Hz, 1H), 3.71 (t, J = 6.8 Hz, 2H), 3.62−3.54 (m, 10H), 3.51 (dd, J = 5.8, 3.6 Hz, 2H), 3.36 (s, 3H), 3.32 (t, J = 6.8 Hz, 2H), 2.98 (s, 3H). 13C NMR (151 MHz, CDCl3): δ 149.2, 144.5, 141.9, 140.2, 139.3, 139.3, 133.2, 132.6, 131.1, 126.6, 124.2, 111.3, 72.1, 70.8, 70.8, 70.7, 70.7, 70.6, 70.5, 59.2, 36.6, 17.7. HRMS (ESI): calcd for C22H27Br2N2O5S [M + H]+, 590.9983; found, 590.9992. HPLC purity: >99.9%.
3.4.3. UV–Vis for HPs Binding Iron(II).36–38
Halogenated phenazine–iron(II) complex formation was determined using UV–vis spectrometry. Ammonium iron(II) sulfate hexahydrate (0.5 equiv) was added to a stirring solution of an HP analogue (10 mM for HPs 18, 28, and 29; 5 mM for HP 34) in dimethyl sulfoxide. Aliquots of 50 μL (HPs 18, 28, and 29) or 100 μL (HP 34) were then removed from the resulting mixture and added to 1 mL of dimethyl sulfoxide in a cuvette. Spectral scanning was performed from 200 to 800 nm in 2 nm increments and a loss of absorbance at λmax (free HP) in the UV–vis spectrum, and apparent formation of a halogenated phenazine–iron(II) complex was observed over time. The disappearance of HPs 28, 29, and 34 was observed over the indicated time points, and the halogenated phenazine–iron(II) complex formation [in a 2:1 HP/iron(II) ratio] yielded a loss in absorbance due to precipitation.
3.4.4. pKa Determination of Select HP Analogues.36
Dissociation constants (pKa) for select HP analogues were determined using UV–vis spectroscopy and the Henderson–Hasselbalch equation. Buffers were prepared using potassium phosphate monobasic (KH2PO4) and sodium phosphate dibasic (Na2HPO4) in a 1:1 solution of water/methanol to achieve a pH range of 4.07–9.66 (ammonium hydroxide was added to prepare buffer pH over 9.80). Compounds were added from 10 mM stock solutions in dimethyl sulfoxide (25 μL) to 1975 μL of each buffer to evaluate each analogue to yield a final compound concentration of 125 μM. Full spectral scans were performed from 200 to 800 nm in 2 nm increments to determine λmax values for the protonated phenol (HA) and the deprotonated phenolate species (A−). The change in absorption at each determined λmax in relation to pH was monitored in each buffer and plotted as absorbance versus pH for each species. The pKa was first estimated by determining the pH of the point of intersection of the two linear curves (see the Supporting Information). The visual estimation was confirmed by plotting pH versus log[A−/HA]. The resulting plot yielded a linear regression line with a Y-intercept corresponding to a calculated pKa value. As a method validation, the pKa of 4-nitrophenol (lit. pKa = 7.15) was determined to be 7.52 under these experimental parameters.
3.5. Biological Studies.
This section includes (a) in vitro testing of HP analogues and (b) in vivo testing of HP 29.
3.5.1. In Vitro Testing of HP Analogues.
3.5.1.1. Minimum Inhibitory Concentration Susceptibility Assay for MRSA-1707, MRSA-44, MRSE 35984, VRE 700221, and E. faecalis OG1RF.33–38
The minimum inhibitory concentration (MIC) for each test compound was determined by the broth microdilution method as recommended by the Clinical and Laboratory Standards Institute (CLSI).58 In a 96-well plate, 11 twofold serial dilutions of each compound were made in a final volume of 100 μL of lysogeny broth (LB, MRSA-1707, MRSA-44, and MRSE 35984; brain heart infusion, BHI, VRE 700221, and E. faecalis OG1RF). Each well was inoculated with ~105 bacterial cells at the initial time of incubation, prepared from a fresh log phase culture (OD600 of 0.5–1.0 depending on bacterial strain). The MIC was defined as the lowest concentration of the compound that prevented bacterial growth after incubating for 16 h at 37 °C (MIC values were further supported by spectrophotometric readings at OD600). The concentration range tested for each HP compound during this study was 0.05–50 μM. DMSO served as our vehicle and negative control in each microdilution MIC assay. DMSO was serially diluted with a top concentration of 0.5% v/v. All compounds were tested in a minimum of three independent experiments.
3.5.1.2. MIC Assay for Mycobacterium tuberculosis (Mtb) H37Ra (ATCC 25177).35–38
M. tuberculosis H37Ra (ATCC 25177) was inoculated in 10 mL of Middlebrook 7H9 medium and allowed to grow for 2 weeks. The culture was then diluted with fresh medium to an OD600 = 0.01. Aliquots of 200 μL were then added to each well of a 96-well plate starting from the second column. Test compounds were dissolved in DMSO at a final concentration of 10 mM. Each compound (7.5 μL) along with DMSO (negative control) and streptomycin (positive control-40 mg/mL stock solution) was added to 1.5 mL of the diluted cultures, resulting in 50 μM final concentration of each halogenated phenazine analogues and 340 μM for streptomycin. The final DMSO concentration was maintained at 0.5%. Aliquots of 400 μL were added to wells of the first column of the 96-well plate and serially diluted twofold (200 μL) per well across the plate to obtain final concentrations that range from 0.024 to 50 μM for the test compounds and 0.16–340 μM for streptomycin. The plates were then incubated at 37 °C for 7 days. Minimum inhibitory concentrations are reported as the lowest concentration at which no bacterial growth was observed. OD600 absorbance was recorded using SpectraMax M5 (Molecular Devices). All compounds were tested in a minimum of three independent experiments.
3.5.1.3. MIC Assay for Mtb CDC1551.
A bioluminescent M. tuberculosis CDC1551 reporter strain (Mtb-lux) was grown in Middlebrook 7H9 media supplemented with 0.05% Tween 80 and 10% oleic acid-albumin-dextrose-catalase (OADC) under kanamycin (50 μg/mL) selection until an optical density (OD600) of 0.4–0.8. A twofold serial drug dilution series (0.003–200 μM) was prepared at 2× in media with 2% dimethyl sulfoxide (DMSO) in 15 μL in white 384-well plates (white with solid bottom, Corning #3570). Mtb-lux was diluted to OD600 0.02 and added to the drug dilution plate (15 μL/well). Luminescence was measured with a Synergy H4 plate reader after 5 days of treatment (37 °C, 5% CO2) and compared to the negative control (1% DMSO) and positive control (10 μM rifampicin) used to calculate % inhibition of growth. Dose–response curves were generated using GraphPad Prism, and MIC values were determined using a modified Gompertz model.
3.5.1.4. MIC Assay for S. pneumoniae (ATCC 6303).
The MIC of 29 against S. pneumoniae ATCC 6303 was determined according to the following procedure. Fresh overnight cultures were inoculated at 1% in microtiter wells containing tryptic soy broth with 5% sheep blood in the presence of 29 ranging from 0.5 to 1000 nM. The plate was incubated at 37 °C with 5% CO2 for 16–20 h. After this time, the lowest test concentration of 29 that resulted in a complete lack of turbidity (bacterial growth) was determined to be the MIC. All tests were performed in at least three independent experiments.
3.5.1.5. CBD Experiments.36–38
Biofilm eradication experiments were performed using the CBD to determine MBC/MBEC values for various compounds of interest (Innovotech, product code: 19111). The CBD (96-well plate with a lid containing pegs to establish biofilms) was inoculated with 125 μL of a mid-log phase culture diluted 1000-fold in tryptic soy broth with 0.5% glucose (TSBG; BHI for E. faecalis OG1RF) to establish bacterial biofilms after incubation at 37 °C for 24 h. The lid was then removed, washed, and transferred to another 96-well plate containing twofold serial dilutions of the test compounds (the “challenge plate”). The total volume of media with the compound in each well in the challenge plate is 150 μL. The CBD device was incubated at 37 °C for 24 h, the lid was then removed from the challenge plate, and MBC/MBEC values were determined using different experimental pathways. To determine MBC values, 20 μL of the challenge plate was transferred into a fresh 96-well plate containing 180 μL of TSBG (BHI for E. faecalis OG1RF) and incubated overnight at 37 °C. The MBC values were determined as the concentration, giving a lack of visible bacterial growth (i.e., turbidity). For determination of MBEC values, the CBD lid (with attached pegs/treated biofilms) was transferred to a new 96-well plate containing 150 μL of fresh TSBG (BHI for E. faecalis OG1RF) media in each well and incubated for 24 h at 37 °C to allow viable biofilms to disperse and grow, resulting in turbidity after the incubation period. MBEC values were determined as the lowest test concentration that resulted in eradicated biofilms (i.e., wells that had no turbidity after the final incubation period). In selected experiments, both treated and untreated CBD pegs were removed from the lid after final incubation, sonicated for 30 min in phosphate-buffered saline (PBS), and plated out to determine biofilm cell killing in colony-forming units per peg (CFU/peg). All data were obtained from a minimum of three independent experiments.
3.5.1.6. RT-qPCR Protocol to Determine Iron Starvation in MRSA Biofilms.39
Biofilm formation: MRSA BAA-1707 was grown in TSBG to an OD600 ~ 0.8–1.0. Then, 1 mL of this culture was added to a 24-well plate coated with 0.1% gelatin. The plate was then incubated for 20 h at 37 °C under static conditions to form biofilms. Following biofilm formation, the contents of the well were discarded, leaving only the biofilm. Treating the established biofilms with compounds: HPs 3, 28, and 29 were added to the established MRSA BAA-1707 biofilms in TSBG at the desired concentration (1 μM or 1/10 MBEC value). In addition, the same volume of DMSO (vehicle) was added as a negative control. The plate was then incubated under static conditions for 4 h at 37 °C. After the incubation period, the liquid culture was discarded, leaving only the surface-attached biofilm. Extraction of total RNA from MRSA BAA-1707 biofilms: 0.5 mL of the RNAprotect bacteria reagent (Qiagen) was added for 5 min to the plate, and the biofilm suspension was scraped and transferred into 2 mL tubes. The bacterial cells were then centrifuged for 1 min at 15,000g; then, the supernatant was removed. Total RNA was extracted using the RiboPure RNA purification kit, bacteria (Invitrogen, cat# AM1925) according to the manufacturer’s protocols. Genomic DNA was digested using the materials supplied by the kit. Each experiment was performed in three replicates. RNA quality control information: RNA concentration was determined on a Qubit 2.0 fluorometer (ThermoFisher/Invitrogen, Grand Island, NY), and RNA quality was assessed using the Agilent 2100 bioanalyzer (Agilent Technologies, Inc.). Total RNA with RNA integrity numbers (RIN) ≥ 7 were used for RT-qPCR validation. Quantitative real-time PCR (qPCR) for select gene transcripts (validation): Total RNA was isolated from MRSA BAA-1707 biofilms treated and untreated with HPs (see the Supporting Information for RNA quality control data). Real-time PCR reactions were performed using the Power SYBR Green RNA-to-CT 1-Step kit (Applied Biosystems 4389986) using the manufacturer’s guidelines. SYBR, primers Rt enzyme RNA, and water were added to a 1.5 mL Eppendorf tube on ice. After all contents were added to Eppendorf tubes, they were mixed by centrifugation for 1 min at 10,000g. 20 μL was then removed from the reaction tubes and was added to each well of a MicroAmp optical 96-well reaction plate with barcode (Applied Biosystems 4306737) on ice. The plate was then sealed with the MicoAmp optical adhesive film (Applied Biosystems 4311971). The plate was centrifuged for 2 min at 1200g. qPCR was carried out on an ABI 7300 sequence detection system using the thermocycler program: 30 min at 50 °C, 10 min at 95 °C, 15 s at 95 °C (40 cycles), and 1 min at 60 °C. Relative gene expression changes were calculated using the ΔΔCT method. For each experiment, the CT values of each gene tested were normalized to the CT values of the housekeeping gene ptaA. Graphs were plotted and data analysis was performed using GraphPad Prism 6. All primers used during these studies are listed in the Supporting Information. All qPCR data were generated from three independent experiments.
3.5.1.7. Lactate Dehydrogenate (LDH) Release Assay for HeLa Cytotoxicity Assessment.46
HeLa cytotoxicity was assessed using the LDH release assay described by CytoTox96 (Promega G1780). HeLa cells were grown in DMEM (Gibco) supplemented with 10% FBS at 37 °C with 5% CO2. When the HeLa cultures exhibited 70–80% confluence, halogenated phenazines were then diluted by DMEM (10% FBS) at concentrations of 25, 50, and 100 μM and added to HeLa cells. Triton X-100 (at 2% v/v) was used as the positive control for maximum LDH activity in this assay (i.e., complete cell death), while “medium only” lanes served as negative control lanes (i.e., no cell death). DMSO was used as our vehicle control. HeLa cells were treated with compounds for 24 h, and then 50 μL of the supernatant was transferred into a fresh 96-well plate where 50 μL of the reaction mixture was added to the 96-well plate and incubated at room temperature for 30 min. Finally, stop solution (50 μL) was added to the incubating plates, and the absorbance was measured at 490 nm. Results are from three independent experiments.
3.5.1.8. Cytotoxicity Assay against J774 Macrophages and HepG2 Cells.47
J774 macrophages and HepG2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% heat-inactivated fetal calf serum (Atlanta Biologicals), 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/mL penicillin, and 100 mg/mL streptomycin in T75 flasks until confluent. J774 macrophages were scraped in PBS, pelleted at 1000 rpm for 10 min, and resuspended in macrophage infection media (DMEM + 10% fetal bovine serum, FBS, 1% L-glutamine, and 1% sodium pyruvate, no phenol red). HepG2 cells were lifted with 0.25% trypsin, pelleted at 1000 rpm for 10 min, and resuspended in macrophage infection media. Both cell types were seeded into 384-well plates (black with clear bottom, Corning #3712) at 25,000 cells/well (in 24 μL total volume). After 4 h of attachment (37 °C, 5% CO2), 6 μL of 5× drugs (twofold dilution series spanning 0.003–200 μM test concentrations, prepared in water) was added to each well. Cells were treated overnight (16–18 h), followed by the addition of 6 μL/well Alamar Blue (0.02% resazurin in water). After 4 h, fluorescence was measured using a Synergy H4 plate reader (λex = 530 nm/λem = 590 nm). Viability was calculated as a percent of DMSO control. 2% Triton X-100 was used as a positive control.
3.5.1.9. Cytotoxicity Assay against HEK-293 Cells.48
The viability of HEK-293 cells was assessed by the MTT assay. The cells were cultured in DMEM medium containing 10% fetal bovine serum and 100 U/mL penicillin and streptomycin. The cells (104 cells/well) were seeded onto a 96-well plate and maintained at 37 °C in a humidified incubator under 5% CO2 overnight. Serial concentrations of 29 (final concentrations at 0, 1.56, 3.13, 6.25, 12.5, 25, 50, 100, and 200 μM) were then added to the wells (n = 6). After incubation at 37 °C for 72 h, 10 μL of MTT (5 mg/mL) in PBS was added to each well and incubated for 4 h, followed by the aspiration of the medium. Dimethyl sulfoxide (DMSO, 100 μL) was then added to each well to dissolve the MTT in the wells, and the plate was agitated for 1 h. The optical density (OD) was measured at 570 nm using a UV/vis microplate spectrophotometer (BioTek). The percent inhibition was calculated as follows: inhibition (%) = (1 − test OD570/nontreated OD570) × 100%. The data were analyzed using Origin.
3.5.1.10. Hemolysis Assay with Red Blood Cells.
Freshly drawn human red blood cells [hRBC with ethylenediaminetetraacetic acid (EDTA) as an anticoagulant] were washed with Tris-buffered saline [0.01 M Tris-base, 0.155 M sodium chloride (NaCl), pH 7.2] and centrifuged for 5 min at 3500 rpm. The washing was repeated three times with the buffer. In a 96-well plate, test compounds were added to the buffer from DMSO stocks. Then, 2% hRBCs (50 μL) in buffer were added to test compounds to give a final concentration of 200 μM. The plate was then incubated for 1 h at 37 °C. After incubation, the plate was centrifuged for 5 min at 3500 rpm before 80 μL of the supernatant was transferred to another 96-well plate to obtain an optical density (OD) read at 405 nm. DMSO served as the negative control (0% hemolysis), while Triton X served as the positive control (100% hemolysis) in these experiments. The percent hemolysis was calculated as (OD405 of the compound treated RBCs – OD405 DMSO control)/(OD405 Triton X treated RBCs – OD405 buffer) from three independent experiments.
3.5.2. In Vivo Testing of HP 29 for Efficacy.
3.5.2.1. Preparation of Polyethylene Glycol (PEG) Ointment.
PEG ointments were prepared based on reported procedures with minor modifications.52 For these investigations, the PEG ointment base was prepared by mixing PEG 400 (70%, wt/vol) with PEG 3350 (30%, wt/vol) as described by the U.S. Pharmacopeia and The National Formulary (USP 24-NF 19). PEG 400 (7 mL) was added to a flask containing PEG 3550 (3 g), and the resulting mixture was heated at 60 °C until the mixture liquified. Then, 1 mL of the resulting PEG liquid was transferred to a vial containing HP 29 (20 mg) to create a 2% suspension. The mixture was then allowed to stir at 60 °C for 30 min before being cooled to room temperature where the suspension solidified. A similar procedure was used to create vehicle control (without 29).
3.5.2.2. Bacterial Strains Used in Animal Models of Infection.
S. aureus strain UAMS-1, a well-characterized antibiotic-susceptible clinical isolate commonly used to study the organism’s biofilm formation and colonization properties, was used in animal experiments during these studies.59 Female BALB/c mice 4–6 weeks of age were obtained from Charles River Laboratories International, Inc. (Wilmington, MA) and housed individually according to approved University of Rochester Medical Center Council on Animal Research (UCAR) protocol UCAR-101864/2017–022. The UAMS-1 wound infection experiments were performed by the Dunman lab at the University of Rochester Medical Center.
E. faecalis strain OG1RF was used in animal experiments during these investigations. Seven week-old female C57BL/6J mice were purchased from Jackson Laboratories Ltd. and housed according to the University of Florida’s approved IACUC protocol 201709769. The OG1RF wound infection experiments were performed by the Lemos lab at the University of Florida.
3.5.2.3. S. aureus Dermal Wound Infection Model.
The effects of ointment compilations were evaluated for in vivo antimicrobial activity using a dermal wound infection mouse model, as previously described.52 Briefly, mice were anesthetized by intraperitoneal injection with a mixture of 100 mg kg−1 ketamine (Hospira Inc., Lake Forest IL) and 10 mg kg−1 xylazine (Lloyd Laboratories, Shenandoah IA) in PBS. Meloxicam (2 mg kg−1, Henry Schein Animal Health, Portland, ME) was administered prior to dermal wounding. The dorsal mid-section of the mouse was shaved and cleaned with a series of betadine scrub (Fisher Scientific), povidoneiodine pads (Professional Disposables International Inc; Orangeburg, NY), and isopropyl alcohol pads (Fisher Scientific) for a total contact time of 2 min. A wound was created in this sterile field on the mouse with a 4.5 mm biopsy punch (Fisher Scientific) to remove only the dermal layer and not disrupt the underlying musculature. The wounds of the mice were inoculated with 1 × 107 S. aureus strain UAMS-1 by pipetting 10 μL of culture directly onto the wound. Mice were then treated with ointment formulations (50 μL) containing either vehicle alone or test compound 45 min post inoculation; treatments were repeated every 12 h for three consecutive days Mice were then euthanized via CO2 asphyxiation and cervical dislocation. The wound and underlying muscle were excised with a 7 mm biopsy punch and placed in microcentrifuge tubes containing 1.4 mm ceramic beads (Fisher Scientific) and 1 mL of freshly made PBS. Samples were homogenized for 1 min using the Fisherbrand bead mill homogenizer, serially diluted, and plated on ChromoAgar plates. Plates were incubated for 16 h at 37 °C, and the number of S. aureus was enumerated.
3.5.2.4. E. faecalis Dorsal Wound Infection Model.
To test the efficacy of HP 29 for the treatment of wounds infected with E. faecalis, a 3 mm incision dorsal wound was created using a biopsy punch on 7 week-old female C57BL/6J mice and the wound was infected with 20 μL of 6 × 1010 CFU/mL of E. faecalis OG1RF, and the infected wound was covered with Tegaderm (Tegaderm, 3 M, St Paul Minnesota). 24 h after infection, the dressing was removed and the wounds were treated topically with HP 29 ointment or with the PEG vehicle control once a day. On day 3 (2 days of treatment) postinfection, animals were euthanized, the wounds were aseptically excised using a surgical blade, and the tissues were homogenized in 1 mL of sterile PBS. The homogenates were serially diluted and plated on trypticase soy (TSA) agar containing rifampicin and fusidic acid for bacterial enumeration.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the National Institute of General Medical Sciences and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health for providing support for this work (R35GM128621 to R.W.H.; R35GM128742 to Y.D.; R21AI37446 to J.A.L.). All high-resolution mass spectra were obtained for new compounds from the University of Florida’s Mass Spectrometry Research and Education Center and supported by NIH S10 OD021758-01A1.
ABBREVIATIONS
- 1-OHP
1-hydroxyphenazine
- BHI
brain heart infusion
- BSA
bis(trimethylsilyl)acetamide
- CBD
calgary biofilm device
- CFU
colony-forming unit
- CLogP
calculated logarithm of partition coefficient between n-octanol and water
- CO2
carbon dioxide
- d
day
- DMEM
Dulbecco’s modified Eagle’s medium
- DMF
N,N-dimethylformamide
- EDTA
ethylenediaminetetraacetic acid
- FBS
fetal bovine serum
- h
hour
- HP
halogenated phenazine
- hRBCs
human red blood cells
- LB
lysogeny broth
- LDH
lactate dehydrogenase
- MBC
minimum bactericidal concentration
- MBEC
minimum biofilm eradication concentration
- MDR
multidrug resistant
- MIC
minimum inhibitory concentration
- μM
micromolar
- min
minute
- MRSA
methicillin-resistant Staphylococcus aureus
- MRSE
methicillin-resistant Staphylococcus epidermidis
- Mtb
Mycobacterium tuberculosis
- NAS
nucleophilic aromatic substitution
- NBS
N-bromosuccinimide
- OD
optical density
- PBS
phosphate-buffered saline
- PEG
polyethylene glycol
- pKa
dissociation constant
- QAC-10
quaternary ammonium cation-10
- rt
room temperature
- RT-qPCR
real-time quantitative polymerase chain reaction
- SI
selectivity index
- TPEN
N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine
- TSA
trypticase soy agar
- TSB
tryptic soy broth
- TSBG
tryptic soy broth supplemented with 0.5% glucose
- VRE
vancomycin-resistant Enterococcus faecium
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00168.
Supporting figures, 1H/13C NMR spectra, dose–response curves, UV–vis results for iron(II) binding, pKa determination, and purity analysis (HPLC traces) for HPs evaluated in biological assays (PDF)
Molecular formula strings (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c00168
The authors declare no competing financial interest.
Contributor Information
Hongfen Yang, Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States;.
Shivani Kundra, Department of Oral Biology, College of Dentistry, University of Florida, Gainesville, Florida 32610, United States;.
Michaelle Chojnacki, Department of Microbiology and Immunology, University of Rochester, Rochester, New York 14642, United States;.
Ke Liu, Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States;.
Marisa A. Fuse, Burnett School of Biomedical Sciences, College of Medicine, University of Central Florida, Orlando, Florida 32827, United States
Yasmeen Abouelhassan, Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Dimitris Kallifidas, Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States.
Peilan Zhang, Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States;.
Guangtao Huang, Department of Molecular Genetics & Microbiology, College of Medicine, University of Florida, Gainesville, Florida 32610, United States.
Shouguang Jin, Department of Molecular Genetics & Microbiology, College of Medicine, University of Florida, Gainesville, Florida 32610, United States;.
Yousong Ding, Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States;.
Hendrik Luesch, Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States;.
Kyle H. Rohde, Burnett School of Biomedical Sciences, College of Medicine, University of Central Florida, Orlando, Florida 32827, United States;.
Paul M. Dunman, Department of Microbiology and Immunology, University of Rochester, Rochester, New York 14642, United States
José A. Lemos, Department of Oral Biology, College of Dentistry, University of Florida, Gainesville, Florida 32610, United States;.
Robert W. Huigens, III, Department of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), College of Pharmacy, University of Florida, Gainesville, Florida 32610, United States; Phone: (352) 273-7718;.
REFERENCES
- (1).Abouelhassan Y; Garrison AT; Yang H; Chávez-Riveros A; Burch GM; Huigens RW III Recent Progress in Natural-Product-Inspired Programs Aimed To Address Antibiotic Resistance and Tolerance. J. Med. Chem 2019, 62, 7618–7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lewis K The Science of Antibiotic Discovery. Cell 2020, 181, 29–45. [DOI] [PubMed] [Google Scholar]
- (3).Wood TK Combatting Bacterial Persister Cells. Biotechnol. Bioeng 2016, 113, 476–483. [DOI] [PubMed] [Google Scholar]
- (4).Fitzpatrick MA Real-World Antibiotic Needs for Resistant Gram-Negative Infections. Lancet Infect. Dis 2020, 20, 1108–1109. [DOI] [PubMed] [Google Scholar]
- (5).Liu K; Huigens RW III Instructive Advances in Chemical Microbiology Inspired by Nature’s Diverse Inventory of Molecules. ACS Infect. Dis 2020, 6, 541–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lewis K Platforms for Antibiotic Discovery. Nat. Rev. Drug Discovery 2013, 12, 371–387. [DOI] [PubMed] [Google Scholar]
- (7).Brown ED; Wright GD Antibacterial Drug Discovery in the Resistance Era. Nature 2016, 529, 336–343. [DOI] [PubMed] [Google Scholar]
- (8).Blair JMA; Webber MA; Baylay AJ; Ogbolu DO; Piddock LJV Molecular Mechanisms of Antibiotic Resistance. Nat. Rev. Microbiol 2015, 13, 42–51. [DOI] [PubMed] [Google Scholar]
- (9).Munita JM; Arias CA Mechanisms of Antibiotic Resistance. Microbiol. Spectr 2016, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wright GD Molecular Mechanisms of Antibiotic Resistance. Chem. Commun 2011, 47, 4055–4061. [DOI] [PubMed] [Google Scholar]
- (11).Miller MB; Bassler BL Quorum Sensing in Bacteria. Annu. Rev. Microbiol 2001, 55, 165–199. [DOI] [PubMed] [Google Scholar]
- (12).Waters CM; Bassler BL Quorum Sensing: Cell-to-Cell Communication in Bacteria. Annu. Dev. Biol 2005, 21, 319–346. [DOI] [PubMed] [Google Scholar]
- (13).Mukherjee S; Bassler BL Bacterial Quorum Sensing in Complex and Dynamically Changing Environments. Nat. Rev. Microbiol 2019, 17, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lewis K Persister Cells. Annu. Rev. Microbiol 2010, 64, 357–372. [DOI] [PubMed] [Google Scholar]
- (15).Donlan RM; Costerton JW; Donlan RM; Costerton JW Biofilms: Survival Mechanisms of Clinically Relevant Micro-organisms. Clin. Microbiol 2002, 15, 167–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hall-Stoodley L; Costerton JW; Stoodley P Bacterial Biofilms: From the Natural Environment to Infectious Diseases. Nat. Rev. Microbiol 2004, 2, 95–108. [DOI] [PubMed] [Google Scholar]
- (17).Flemming H-C; Wingender J; Szewzyk U; Steinberg P; Rice SA; Kjelleberg S Biofilms: An Emergent Form of Bacterial Life. Nat. Rev. Microbiol 2016, 14, 563–575. [DOI] [PubMed] [Google Scholar]
- (18).Yan J; Bassler BL Surviving as a Community: Antibiotic Tolerance and Persistence in Bacterial Biofilms. Cell Host Microbe 2019, 26, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lewis K Persister Cells: Molecular Mechanisms Related to Antibiotic Tolerance. Handb. Exp. Pharmacol 2012, 211, 121–133. [DOI] [PubMed] [Google Scholar]
- (20).Garrison AT; Huigens RW III Eradicating Bacterial Biofilms with Natural Products and Their Inspired Analogues That Operate Through Unique Mechanisms. Curr. Top. Med. Chem 2017, 17, 1954–1964. [PubMed] [Google Scholar]
- (21).Wolcott R; Dowd S The Role of Biofilms: Are We Hitting the Right Target? Plast. Reconstr. Surg 2011, 127, 28S–35S. [DOI] [PubMed] [Google Scholar]
- (22).Bjarnsholt T The Role of Bacterial Biofilms in Chronic Infections. APMIS Suppl 2013, 121, 1–58. [DOI] [PubMed] [Google Scholar]
- (23).Wu Y-K; Cheng N-C; Cheng C-M Biofilms in Chronic Wounds: Pathogenesis and Diagnosis. Trends Biotechnol 2019, 37, 505–517. [DOI] [PubMed] [Google Scholar]
- (24).Ling LL; Schneider T; Peoples AJ; Spoering AL; Engels I; Conlon BP; Mueller A; Schäberle TF; Hughes DE; Epstein S; Jones M; Lazarides L; Steadman VA; Cohen DR; Felix CR; Fetterman KA; Millett WP; Nitti AG; Zullo AM; Chen C; Lewis K A New Antibiotic Kills Pathogens without Detectable Resistance. Nature 2015, 517, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Imai Y; Meyer KJ; Iinishi A; Favre-Godal Q; Green R; Manuse S; Caboni M; Mori M; Niles S; Ghiglieri M; Honrao C; Ma X; Guo JJ; Makriyannis A; Linares-Otoya L; Böhringer N; Wuisan ZG; Kaur H; Wu R; Mateus A; Typas A; Savitski MM; Espinoza JL; O’Rourke A; Nelson KE; Hiller S; Noinaj N; Schäberle TF; D’Onofrio A; Lewis K A New Antibiotic Selectively Kills Gram-Negative Pathogens. Nature 2019, 576, 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Smith PA; Koehler MFT; Girgis HS; Yan D; Chen Y; Chen Y; Crawford JJ; Durk MR; Higuchi RI; Kang J; Murray J; Paraselli P; Park S; Phung W; Quinn JG; Roberts TC; Rougé L; Schwarz JB; Skippington E; Wai J; Xu M; Yu Z; Zhang H; Tan M-W; Heise CE Optimized Arylomycins are a New Class of Gram-Negative Antibiotics. Nature 2018, 561, 189–194. [DOI] [PubMed] [Google Scholar]
- (27).Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ Predictive Compound Accumulation Rules Yield a Broad-Spectrum Antibiotic. Nature 2017, 545, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Parker EN; Drown BS; Geddes EJ; Lee HY; Ismail N; Lau GW; Hergenrother PJ Implementation of Permeation Rules Leads to a Fabi Inhibitor with Activity against Gram-Negative Pathogens. Nat. Microbiol 2020, 5, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Motika SE; Ulrich RJ; Geddes EJ; Lee HY; Lau GW; Hergenrother PJ Gram-Negative Antibiotic Active Through Inhibition of an Essential Riboswitch. J. Am. Chem. Soc 2020, 142, 10856–10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li Q; Pellegrino J; Lee DJ; Tran AA; Chaires HA; Wang R; Park JE; Ji K; Chow D; Zhang N; Brilot AF; Biel JT; van Zundert G; Borrelli K; Shinabarger D; Wolfe C; Murray B; Jacobson MP; Mühle E; Chesneau O; Fraser JS; Seiple IB Synthetic Group A Streptogramin Antibiotics that Overcome Vat Resistance. Nature 2020, 586, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Conlon BP; Nakayasu ES; Fleck LE; LaFleur MD; Isabella VM; Coleman K; Leonard SN; Smith RD; Adkins JN; Lewis K Activated ClpP Kills Persisters and Eradicates a Chronic Biofilm Infection. Nature 2013, 503, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kim W; Zhu W; Hendricks GL; Van Tyne D; Steele AD; Keohane CE; Fricke N; Conery AL; Shen S; Pan W; Lee K; Rajamuthiah R; Fuchs BB; Vlahovska PM; Wuest WM; Gilmore MS; Gao H; Ausubel FM; Mylonakis E A New Class of Synthetic Retinoid Antibiotics Effective against Bacterial Persisters. Nature 2018, 556, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Borrero NV; Bai F; Perez C; Duong BQ; Rocca JR; Jin S; Huigens RW III Phenazine Antibiotic Inspired Discovery of Potent Bromophenazine Antibacterial Agents against Staphylococcus aureus and Staphylococcus epidermidis. Org. Biomol. Chem 2014, 12, 881–886. [DOI] [PubMed] [Google Scholar]
- (34).Garrison AT; Bai F; Abouelhassan Y; Paciaroni NG; Jin S; Huigens RW III Bromophenazine Derivatives with Potent Inhibition, Dispersion and Eradication Activities against Staphylococcus aureus Biofilms. RSC Adv 2015, 5, 1120–1124. [Google Scholar]
- (35).Garrison AT; Abouelhassan Y; Kallifidas D; Bai F; Ukhanova M; Mai V; Jin S; Luesch H; Huigens RW III Halogenated Phenazines that Potently Eradicate Biofilms, MRSA Persister Cells in Non-Biofilm Cultures, and Mycobacterium tuberculosis. Angew. Chem., Int. Ed 2015, 54, 14819–14823. [DOI] [PubMed] [Google Scholar]
- (36).Garrison AT; Abouelhassan Y; Norwood VM; Kallifidas D; Bai F; Nguyen MT; Rolfe M; Burch GM; Jin S; Luesch H; Huigens RW III Structure-Activity Relationships of a Diverse Class of Halogenated Phenazines That Targets Persistent, Antibiotic-Tolerant Bacterial Biofilms and Mycobacterium tuberculosis. J. Med. Chem 2016, 59, 3808–3825. [DOI] [PubMed] [Google Scholar]
- (37).Yang H; Abouelhassan Y; Burch GM; Kallifidas D; Huang G; Yousaf H; Jin S; Luesch H; Huigens RW III A Highly Potent Class of Halogenated Phenazine Antibacterial and Biofilm-Eradicating Agents Accessed Through a Modular Wohl-Aue Synthesis. Sci. Rep 2017, 7, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Garrison AT; Abouelhassan Y; Kallifidas D; Tan H; Kim YS; Jin S; Luesch H; Huigens RW III An Efficient Buchwald-Hartwig/Reductive Cyclization for the Scaffold Diversification of Halogenated Phenazines: Potent Antibacterial Targeting, Biofilm Eradication, and Prodrug Exploration. J. Med. Chem 2018, 61, 3962–3983. [DOI] [PubMed] [Google Scholar]
- (39).Abouelhassan Y; Zhang Y; Jin S; Huigens RW III Transcript Profiling of MRSA Biofilms Treated with a Halogenated Phenazine Eradicating Agent: A Platform for Defining Cellular Targets and Pathways Critical to Biofilm Survival. Angew. Chem., Int. Ed 2018, 57, 15523–15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Machan ZA; Pitt TL; White W; Watson D; Taylor GW; Cole PJ; Wilson R Interaction Between Pseudomonas aeruginosa and Staphylococcus aureus: Description of an Anti-staphylococcal Substance. J. Med. Microbiol 1991, 34, 213–217. [DOI] [PubMed] [Google Scholar]
- (41).Laursen JB; Nielsen J Phenazine Natural Products: Biosynthesis, Synthetic Analogues, and Biological Activity. Chem. Rev 2004, 104, 1663–1686. [DOI] [PubMed] [Google Scholar]
- (42).Price-Whelan A; Dietrich LEP; Newman DK Rethinking “Secondary” Metabolism: Physiological Roles for Phenazine Antibiotics. Nat. Chem. Biol 2006, 2, 71–78. [DOI] [PubMed] [Google Scholar]
- (43).Guttenberger N; Blankenfeldt W; Breinbauer R Recent Developments in the Isolation, Biological Function, Biosynthesis, and Synthesis of Phenazine Natural Products. Bioorg. Med. Chem 2017, 25, 6149–6166. [DOI] [PubMed] [Google Scholar]
- (44).Kwast A; Stachowska K; Trawczyński A; Wróbel Z N-Aryl-2-Nitrosoanilines as Intermediates in the Synthesis of Substituted Phenazines from Nitroarenes. Tetrahedron Lett 2011, 52, 6484–6488. [Google Scholar]
- (45).Wróbel Z; Plichta K; Kwast A Reactivity and Substituent Effects in the Cyclization of N-aryl-2-nitrosoanilines to Phenazines. Tetrahedron 2017, 73, 3147–3152. [Google Scholar]
- (46).Weidmann E; Brieger J; Jahn B; Hoelzer D; Bergmann L; Mitrou PS Lactate Dehydrogenase-Release Assay: A Reliable, Nonradioactive Technique for Analysis of Cytotoxic Lymphocyte-Mediated Lytic Activity against Blasts from Acute Myelocytic Leukemia. Annu. Hematol 1995, 70, 153–158. [DOI] [PubMed] [Google Scholar]
- (47).Gupta R; Rodrigues Felix C; Akerman MP; Akerman KJ; Slabber CA; Wang W; Adams J; Shaw LN; Tse-Dinh Y-C; Munro OQ; Rohde KH Evidence for Inhibition of Topoisomerase 1A by Gold(III) Macrocycles and Chelates Targeting Mycobacterium tuberculosis and Mycobacterium abscessus. Antimicrob. Agents Chemother 2018, 62, No. e01696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Abouelhassan Y; Zhang P; Ding Y; Huigens RW III Rapid kill assessment of an N-arylated NH125 analogue against drug-resistant microorganisms. Med. Chem. Commun 2019, 10, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ceri H; Olson ME; Stremick C; Read RR; Morck D; Buret A The Calgary Biofilm Device: New Technology for Rapid Determination of Antibiotic Susceptibilities of Bacterial Biofilms. J. Clin. Microbiol 1999, 37, 1771–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Harrison JJ; Stremick CA; Turner RJ; Allan ND; Olson ME; Ceri H Microtiter Susceptibility Testing of Microbes Growing on Peg Lids: A Miniaturized Biofilm Model for High-Throughput Screening. Nat. Protoc 2010, 5, 1236–1254. [DOI] [PubMed] [Google Scholar]
- (51).Jennings MC; Ator LE; Paniak TJ; Minbiole KPC; Wuest WM Biofilm-Eradicating Properties of Quaternary Ammonium Amphiphiles: Simple Mimics of Antimicrobial Peptides. ChemBioChem 2014, 15, 2211–2215. [DOI] [PubMed] [Google Scholar]
- (52).Blanchard C; Brooks L; Beckley A; Colquhoun J; Dewhurst S; Dunman PM Neomycin Sulfate Improves the Antimicrobial Activity of Mupirocin-Based Antibacterial Ointments. Antimicrob. Agents Chemother 2016, 60, 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Colomer-Winter C; Flores-Mireles AL; Baker SP; Frank KL; Lynch AJL; Hultgren SJ; Kitten T; Lemos JA Manganese Aquisition is Essential for Virulence of Enterococcus Faecalis. PLoS Pathog 2018, 14, No. e1007102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bhattacharya M; Wozniak DJ; Stoodley P; Hall-Stoodley L Prevention and treatment of Staphylococcus aureus biofilms. Expert Rev. Anti Infect. Ther 2015, 13, 1499–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ch’ng J-H; Chong KKL; Lam LN; Wong JJ; Kline KA Biofilm-associated Infection by Enterococci. Nat. Rev. Microbiol 2019, 17, 82–94. [DOI] [PubMed] [Google Scholar]
- (56).Xiao T; Liu K; Huigens RW III Progress Towards a Stable Cephalosporin-Halogenated Phenazine Conjugate for Antibacterial Prodrug Applications. Bioorg. Med. Chem. Lett 2020, 30, 127515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Yang H; Liu K; Jin S; Huigens RW III Design, Synthesis and Biological Evaluation of a Halogenated Phenazine-Erythromycin Conjugate Prodrug for Antibacterial Applications. Org. Biomol. Chem 2021, 19, 1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standards, 8th ed. (M7-M8); Clinical and Laboratory Standards Institute: Wayne, PA, 2009. [Google Scholar]
- (59).Gillaspy AF; Hickmon SG; Skinner RA; Thomas JR; Nelson CL; Smeltzer MS Role of the Accessory Gene Regulator (agr) in Pathogenesis of Staphylococcal Osteomyelitis. Infect. Immun 1995, 63, 3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.