

Abstract
The current review provides a historical perspective on the evolution of hypothesized mechanisms for senescent neurophysiology, focused on the CA1 region of the hippocampus, and the relationship of senescent neurophysiology to impaired hippocampal-dependent memory. Senescent neurophysiology involves processes linked to calcium (Ca2+) signaling including an increase in the Ca2+-dependent afterhyperpolarization (AHP), decreasing pyramidal cell excitability, hyporesponsiveness of N-methyl-D-aspartate (NMDA) receptor function, and a shift in Ca2+-dependent synaptic plasticity. Dysregulation of intracellular Ca2+ and downstream signaling of kinase and phosphatase activity lies at the core of senescent neurophysiology. Ca2+-dysregulation involves a decrease in Ca2+ influx through NMDA receptors and an increase release of Ca2+ from internal Ca2+ stores. Recent work has identified changes in redox signaling, arising in middle-age, as an initiating factor for senescent neurophysiology. The shift in redox state links processes of aging, oxidative stress and inflammation, with functional changes in mechanisms required for episodic memory. The link between age-related changes in Ca2+ signaling, epigenetics and gene expression is an exciting area of research. Pharmacological and behavioral intervention, initiated in middle-age, can promote memory function by initiating transcription of neuroprotective genes and rejuvenating neurophysiology. However, with more advanced age, or under conditions of neurodegenerative disease, epigenetic changes may weaken the link between environmental influences and transcription, decreasing resilience of memory function.
Keywords: Aging, Hippocampus, Afterhyperpolarization, Synaptic plasticity, N-methyl-D-aspartate receptor, Transcription, Epigenetics
1. Introduction
Aging is associated with a weakening of executive function, and processing speed; however, the most notable decline is observed as impaired episodic memory, including spatial memory (Foster, Defazio, & Bizon, 2012; Hughes, Agrigoroaei, Jeon, Bruzzese, & Lachman, 2018; Roberson et al., 2012). Episodic memories are flexible and rapidly acquired such that episodic information can be updated from moment to moment over the course of training. Impairment in flexible memory processes that depend on the hippocampus are a common complaint of aging, partly due to the emergence of deficits in middle-age. Thus, much of the research on neural mechanisms of cognitive aging has concentrated on the hippocampus and hippocampal-dependent episodic memory (Foster et al., 2012; Foster, 1999, 2012).
During the 1980s, fundamental research characterized hippocampal senescent neurophysiology, including decreased CA1 pyramidal cell excitability and altered synaptic plasticity (Barnes & McNaughton, 1985; Barnes, 1979; Disterhoft, Thompson, Moyer, & Mogul, 1996; Landfield & Pitler, 1984; Pitler & Landfield, 1990), processes linked to calcium (Ca2+) signaling. The results provided a foundation for the Ca2+ hypothesis of aging and Alzheimer’s disease, which suggested that long-term Ca2+ dysregulation and oxidative stress would, over time, result in neuronal death (Harman, 1981; Khachaturian, 1989, 1994; Siesjo, 1981). However, the revelation that normal aging is not associated with a loss of hippocampal neurons (West, Coleman, Flood, & Troncoso, 1994) and the accumulation of research demonstrating that age-related cognitive decline is less severe than that of patients with neurodegenerative disease that damage the hippocampus (Foster, 1999), prompted a reconsideration of the link between Ca2+ dysregulation and memory impairment. Similarly, ideas about the role of oxidative stress in brain aging have evolved over the past forty years to suggest that, rather than oxidative damage observed for neurodegenerative diseases, aging is associated with a shift in redox signaling involved in Ca2+ regulation (Kumar, Yegla, & Foster, 2017). This review is centered on neurons, although age-related changes have been noted for other senescent cell types (Yeoman, Scutt, & Faragher, 2012). The review describes Ca2+-dependent hippocampal senescent physiology, specifically an increase in the afterhyperpolarization (AHP) and hypofunction of N-methyl-D-aspartate (NMDA) glutamate receptors. Furthermore, these processes interact to alter synaptic plasticity. In addition, the review covers recent research that defines a role for redox state as a mechanism for Ca2+ dysregulation. Finally, with the development of molecular techniques, current research suggests that epigenetic mechanisms could provide the link between environmental influences on senescent physiology and variability in the trajectory of cognitive decline.
2. An increase in the AHP during aging
A major line of evidence for the original Ca2+ hypothesis of brain aging came from intracellular recordings of CA1 pyramidal cells by Landfield and associates (Landfield & Pitler, 1984; Thibault, Gant, & Landfield, 2007). These recordings revealed an age-related increase in the Ca2+ activated, and K+-mediated, AHP that follows a burst of action potentials (Fig. 1A). An increase in the amplitude of the AHP is now a well-established marker of aging in CA1 pyramidal neurons and has been observed in rabbits, mice, and rats, both male and female (Disterhoft et al., 1996; Kaczorowski & Disterhoft, 2009; Kumar and Foster, 2002, 2004; Landfield & Pitler, 1984; Murphy, Shah, Hell, & Silva, 2006; Power, Wu, Sametsky, Oh, & Disterhoft, 2002; Tombaugh, Rowe, & Rose, 2005). The larger hyperpolarization influences the relative refractory period of the action potential, reducing the number of action potentials evoked during depolarization (spike frequency accommodation) and shifts the discharge activity evoked by distinct patterns of afferent stimulation (Gant & Thibault, 2009). Reduced cell excitability may influence signal processing and input-output relationships, contributing to age-related differences in the stability or modifiability of environmentally evoked and behaviorally relevant hippocampal cell discharge activity (Barnes, Suster et al., 1997; Shen, Barnes, McNaughton, Skaggs, & Weaver, 1997; McEchron, Weible, & Disterhoft, 2001; Yan, Zhang, Roder, & McDonald, 2003).
Fig. 1.
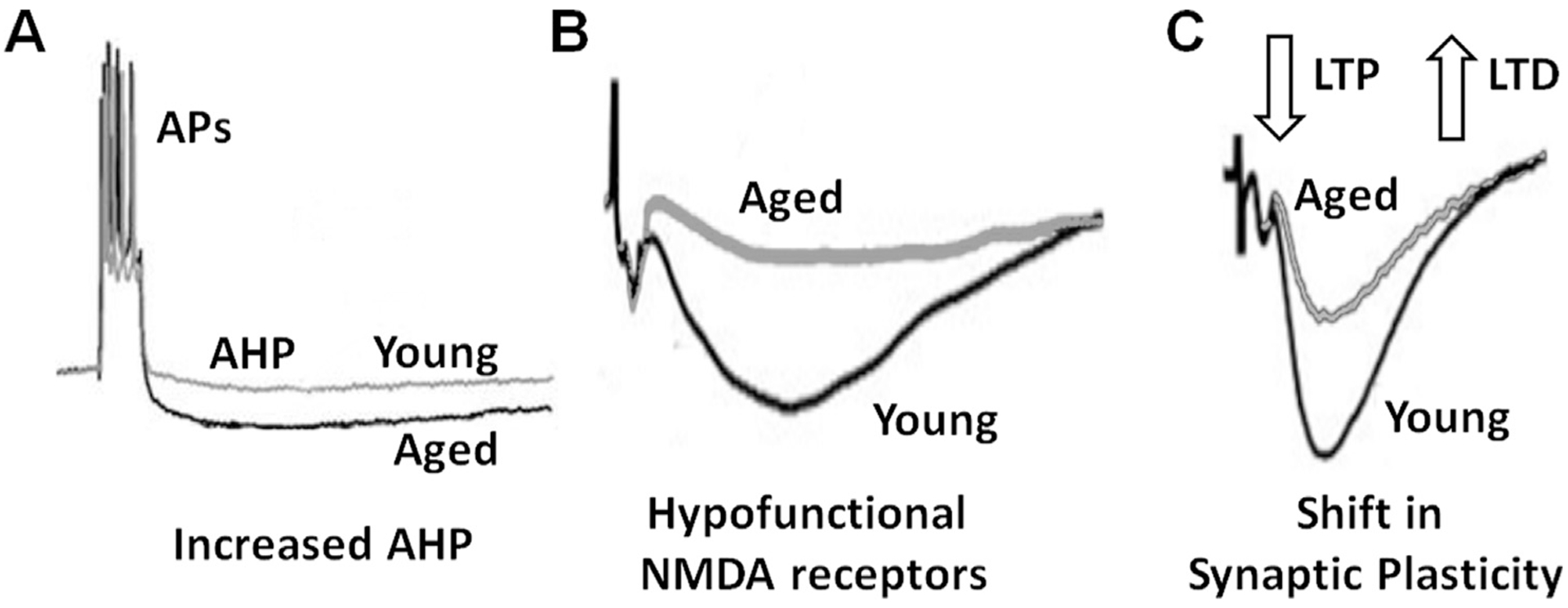
Senescent neurophysiology. Senescent neurophysiology in the CA1 region of the hippocampus includes an increase in the afterhyperpolarization (AHP), a decrease in the NMDA receptor component of synaptic transmission, and a shift in synaptic plasticity. (A) A burst of action potentials (APs) initiates Ca2+-dependent activation of K+ channels resulting in a subsequent AHP. The amplitude of the AHP increases during aging. (B) Illustration of isolated NMDA receptor synaptic transmission. The NMDA receptor component of synaptic transmission decreases during aging. (C) The larger AHP and decrease in NMDA receptor function underlie a shift in synaptic plasticity, decreasing the probability or amplitude of LTP and increasing susceptibility for LTD.
Importantly, the age-related increase in the AHP is specific to distinct neuronal populations. In the prefrontal cortex of aged monkeys, an increase in the AHP is observed, mainly in layer 3 relative to layer 5 pyramidal cells (Luebke & Amatrudo, 2012; Luebke & Chang, 2007) and in layer 3 neurons relative to layer 2 neurons of the medial entorhinal cortex of the aging rat (Gant, Kadish et al., 2018). In the hippocampus, an age-related increase in the AHP is unique to CA1 pyramidal cells, and is not observed in CA3 pyramidal cells (Simkin et al., 2015) or dentate gyrus granule cells (Baskys, Niesen, & Carlen, 1987). While the mechanism for neuronal specificity is unclear, several groups have suggest that specificity arises due to differences in vulnerability to oxidative stress, Ca2+ dysregulation, and excitotoxicity of the different regions (Du, Eid, Lothman, Kohler, & Schwarcz, 1995; Jackson, Rani, Kumar, & Foster, 2009; Burger, 2010; Wang & Michaelis, 2010; Zeier et al., 2011; Ianov, De Both et al., 2017; Datta & Arnsten, 2018).
In order to understand the role of an increase in the AHP amplitude in age-related memory impairment, it is important to visit ideas about the role of the AHP in hippocampal-dependent learning and memory. From a scientific and historical perspective, it is interesting to note that around the time that the AHP was reported to increase in region CA1 with age, Disterhoft and colleagues demonstrated an increase in cell excitability and decrease in the amplitude of the AHP in CA1 pyramidal cells, associated with behavioral conditioning (Coulter et al., 1989; de Jonge, Black, Deyo, & Disterhoft, 1990; Disterhoft, Coulter, & Alkon, 1986; Moyer, Thompson, & Disterhoft, 1996; Thompson, Moyer, & Disterhoft, 1996). The time course for the reduction in the AHP, which lasts several days following learning, suggested that increased excitability might enhance correlated neuronal activity, promoting the transfer of information from the hippocampus to the cortex during memory consolidation (Oh, Oliveira, & Disterhoft, 2010). In this case, it would be important to demonstrate that only those neurons involved in encoding the memory are the neurons that exhibit a decrease in the AHP and increase activity during consolidation. Alternatively, a generalized increase in cell excitability can contribute to learning. For example, learning and memory are facilitated by pharmacological treatments that reduce the AHP amplitude including neuromodulators (e.g. acetylcholine and norepinephrine), hormones (e.g. insulin and estrogen), and Ca2+ and K+ channel blockers (Disterhoft et al., 1999; Kumar & Foster, 2002; Maimaiti et al., 2017; Messier et al., 1991; Thomas, 2015; Thompson, Deyo, & Disterhoft, 1990).
Another indication that the AHP regulates learning comes from studies demonstrating that prior training on tasks that increase CA1 cell excitability can facilitate acquisition of other hippocampal-dependent behaviors (Kuo, Lee, & Disterhoft, 2006; Zelcer et al., 2006). Together, the data point to the AHP amplitude as a critical factor for successful learning. In the case of aged animals, the baseline AHP amplitude is larger; however, treatments to reduce the AHP also facilitate learning in older animals (Deyo, Straube, & Disterhoft, 1989; Oh et al., 2010). Similarly, genetic knockout of the potassium channel, Kvβ1.1, reduces the AHP of old animals to a level similar to that observed in young animals and improves learning (Giese, Peters, & Vernon, 2001). Thus, the larger AHP with advanced age likely contributes to impaired learning.
3. NMDA receptor hypofunction and synaptic plasticity during aging
One proposed mechanism for AHP involvement in memory involves the regulation of neuronal depolarization required for Ca2+-dependent synaptic plasticity, which is altered during aging (Fig. 1C) (Foster & Norris, 1997). Long-term potentiation (LTP) and long-term depression (LTD), two major forms of activity-dependent synaptic plasticity, are considered cellular correlates of learning and memory, and are studied extensively across various brain regions (Foster, 1999, 2012; Rosenzweig & Barnes, 2003). LTP is expressed as an increase in synaptic transmission mediated via α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors. LTP is induced by a large rise in intracellular Ca2+, mainly due to Ca2+ influx through NMDA receptor activation. In contrast, LTD is a decrease in AMPA receptor-mediated synaptic transmission and requires a modest rise in intracellular Ca2+. Carol Barnes first demonstrated that induction of LTP in aging rats was problematic, requiring an increase in the number of bouts of patterned stimulation in order to obtain the same level of LTP as that observed in young animals. Furthermore, the fast rate at which LTP decayed back to baseline correlated with increased forgetting of older animals (Barnes & McNaughton, 1985; Barnes, 1979). Later studies indicated that an age-related increase in susceptibility to induction of LTD and reversal of LTP contributed to the decay of LTP and rapid forgetting (Foster & Kumar, 2007; Kumar & Foster, 2005; Norris, Korol, & Foster, 1996). The results are consistent with the idea that LTP represents a memory mechanism, while LTD may contribute to increased forgetting and suggests that age-related impairment in learning and memory are due to decreased propensity for induction of LTP and increased susceptibility to induction of LTD (Foster, 2012).
An age-related decrease in the NMDA receptor component of synaptic transmission is another consistent marker of senescent neurophysiology (Barnes, Rao et al., 1997; Bodhinathan, Kumar, & Foster, 2010b; Kumar & Foster, 2013; Lee, Kumar, Rani, & Foster, 2014; Guidi, Kumar, & Foster, 2015; Kumar, Rani, Scheinert, Ormerod, & Foster, 2018) (Fig. 1B). Activation of postsynaptic NMDA receptors requires the binding of glutamate in conjunction with postsynaptic depolarization. Thus, NMDA receptor hypofunction and hyperpolarization due to the larger AHP, likely contribute to a shift in synaptic plasticity (Foster & Norris, 1997; Foster, 1999, 2007, 2012; Norris et al., 1996). Consequently, it may be no coincidence that these three characteristics of senescent neurophysiology, an increase in AHP, a decrease in NMDA receptor synaptic transmission, and altered synaptic plasticity, emerge at approximately the same time, in middle-age (Gant, Sama, Landfield, & Thibault, 2006; Guidi et al., 2015; Kaczorowski & Disterhoft, 2009; Kumar & Foster, 2013). Moreover, these senescent physiology characteristics correlate with the extent of memory impairment (Barnes, 1979; Foster & Kumar, 2007; Kaczorowski & Disterhoft, 2009; Kumar & Foster, 2013; Kumar, Rani, Tchigranova, Lee, & Foster, 2012; Tombaugh et al., 2005).
The amplitude of the AHP provides a constraint on spike generation, postsynaptic depolarization required for NMDA receptor activation, and may determine the threshold for induction of LTP. The larger AHP can inhibit action potential generation and reduces the time window for spike-timing-dependent plasticity (Fuenzalida, Fernandez de Sevilla, & Buno, 2007; Gant & Thibault, 2009). In their commentary on hippocampal synaptic senescence, Foster and Norris (1997) noted that an age-related impairment in the induction of LTP is particularly apparent for afferent stimulation that approximates the endogenous theta rhythm (~3–15 Hz) and the burst pattern of hippocampal neurons (i.e. prime burst or theta burst stimulation) observed in the rodent hippocampus. For aged animals, the interval between a pair of theta burst episodes would result in synaptic activity for the second burst occurring near the peak hyperpolarization initiated from the previous burst of action potentials (Foster & Norris, 1997). The model for the interaction of LTPAHP predicted that the window of the AHP would shift the threshold stimulation frequency for induction of LTP (Foster, 1999). Construction of frequency-response curves confirmed that the threshold frequency for induction of LTP was ~3–15 Hz for young animals. For aged animals, the threshold frequency was shifted, such that stimulation in the theta range resulted in a plateau region of no synaptic plasticity or induction of LTD (Hsu et al., 2002; Kumar and Foster, 2007a). The importance of the AHP window for synaptic plasticity is emphasized by studies that demonstrate that treatments to reduce the AHP enabled the induction of a robust LTP following 5 Hz stimulation (5 Hz-LTP), specifically for aged animals (Kumar & Foster, 2004; Norris et al., 1998b). Interestingly, impairment in low frequency induction of LTP is associated with unstable hippocampal place cells (Rotenberg, Mayford, Hawkins, Kandel, & Muller, 1996), possibly due to weaker synaptic modifications during processes for establishing the place field.
4. Dysregulation of Ca2+ signaling and redox state
The regulation of intracellular Ca2+ and downstream signaling through kinase and phosphatase activity lies at the core of altered cell excitability and synaptic function associated with memory and with senescent physiology. During aging, the hippocampus exhibits a shift in the balance of kinase/phosphatase activity, favoring activation of the protein phosphatases, protein phosphatase 1 and the Ca2+-dependent phosphatase, calcineurin (Foster, Sharrow, Masse, Norris, & Kumar, 2001; Jouvenceau & Dutar, 2006; Kumar, Bodhinathan, & Foster, 2009; Monti, Berteotti, & Contestabile, 2005; Norris et al., 1998a). This shift, with higher phosphatase activity, contributes to an age-related decrease in synaptic transmission (Bodhinathan et al., 2010b; Norris et al., 1998a). In addition, the balance of kinase/phosphatase activity may contribute to the larger AHP and an inability of behavioral training to reduce the AHP in aged animals (Oh, McKay, Power, & Disterhoft, 2009; Zhang, Ouyang, Ganellin, & Thomas, 2013). The amplitude of the AHP is reduced by activation of protein kinase A (PKA) (Madison & Nicoll, 1982; Pedarzani & Storm, 1993) or inhibition of the phosphatase, calcineurin (Pedarzani, Krause, Haug, Storm, & Stuhmer, 1998; Vogalis, Harvey, & Furness, 2004). Interestingly, PKA activation does not reduce the AHP in animals that exhibit learning on hippocampal-dependent tasks suggesting that kinase activity contributes to the maintenance of the decrease in the AHP amplitude (Oh et al., 2010). In this case, an increase in basal phosphatase activity during aging could preclude kinase activation, preventing a sustained decrease in the AHP.
The next question to address concerns the source of Ca2+ dysregulation. Ca2+ dysregulation may involve altered metabolism of aging (i.e. increased oxidative stress) and changes in the expression of genes/proteins involved in Ca2+ signaling. The major sources of the intracellular Ca2+ that contributes to the AHP is through influx from voltage gated Ca2+ channels and release of Ca2+ from intracellular Ca2+ stores (ICS). Initial evidence indicated involvement of L-type voltage gated channels, possibly due to increased expression of L-type channels near the site of the AHP generation (Nunez-Santana et al., 2014; Thibault & Landfield, 1996; Veng & Browning, 2002). Pharmacological blockade of L-channels reduces the AHP and promotes LTP in aged animals (Norris et al., 1998b). However, several studies suggest that the effects of L-channel antagonists are not age specific. First, L-channel antagonists enhance learning and memory in young and aged subjects (Foster, 2012). In addition, L-channel blockade decreases the AHP to the same extent (~30%) in young and aged animals (Power et al., 2002). The results suggest that other Ca2+ sources may underlie the age-related increase in the AHP.
Kumar, Foster, and associates established that the larger amplitude AHP involves an increase in release of Ca2+ from ICS. In this case, a small influx of Ca2+ is magnified through ryanodine receptor activation in a process known as Ca2+ induced Ca2+ release. The initial report demonstrated a greater reduction in the AHP amplitude in aged animals relative to young following ryanodine receptor blockade or depletion of Ca2+ from ICS (Kumar & Foster, 2004). Subsequent studies confirmed that removal of ICS as a source of Ca2+ reduced the AHP to a greater extent in aged animals (Bodhinathan et al., 2010a; Gant et al., 2006). Furthermore, the reduction in AHP amplitude permitted induction of 5 Hz-LTP (Kumar & Foster, 2004). The significance of ICS as a source for Ca2+-dysregulation for senescent physiology is emphasized by the fact that, LTP, a process that depends on a large rise in intracellular Ca2+, is facilitated in aged animals by decreasing intracellular Ca2+ coming from ICS.
The increase in Ca2+ from ICS likely involves altered function of ryanodine receptors. An age-related change in the expression of the ryanodine receptor regulatory protein, FK506-binding protein 1b, has been reported (Gant et al., 2011; Gant et al., 2015; Gant, Blalock et al., 2018). In addition, the function of the ryanodine receptor is redox sensitive (Bull et al., 2008; Kumar et al., 2017). The role of redox state in regulating the AHP through ryanodine receptor function and ICS was demonstrated by application of the reducing agent, dithiothreitol (DTT), which age-dependently decreased the AHP amplitude (Bodhinathan et al., 2010a) (Fig. 2A and B). For aged animals, the magnitude decrease (~50%) in the AHP was similar to that observed following ryanodine receptor blockade or depletion of Ca2+ from ICS. In fact, decreasing Ca2+ release from intracellular stores by application of thapsigargin or ryanodine prevented the DTT-mediated reduction of AHP (Fig. 2C). In contrast, blockade of L-type channels did not occlude the ability of DTT to reduce the AHP, indicating an explicit role for ICS in mediating DTT-effects. Further evidence for the idea that redox state underlies age-differences came from studies examining the effects of oxidizing agents, which mimicked the effects of aging, increasing AHP, specifically in young animals (Bodhinathan et al., 2010a) (Fig. 2D). Thus, the effects of redox manipulations on the AHP are age specific and suggest that aged animals are in a more oxidized redox state.
Fig. 2.
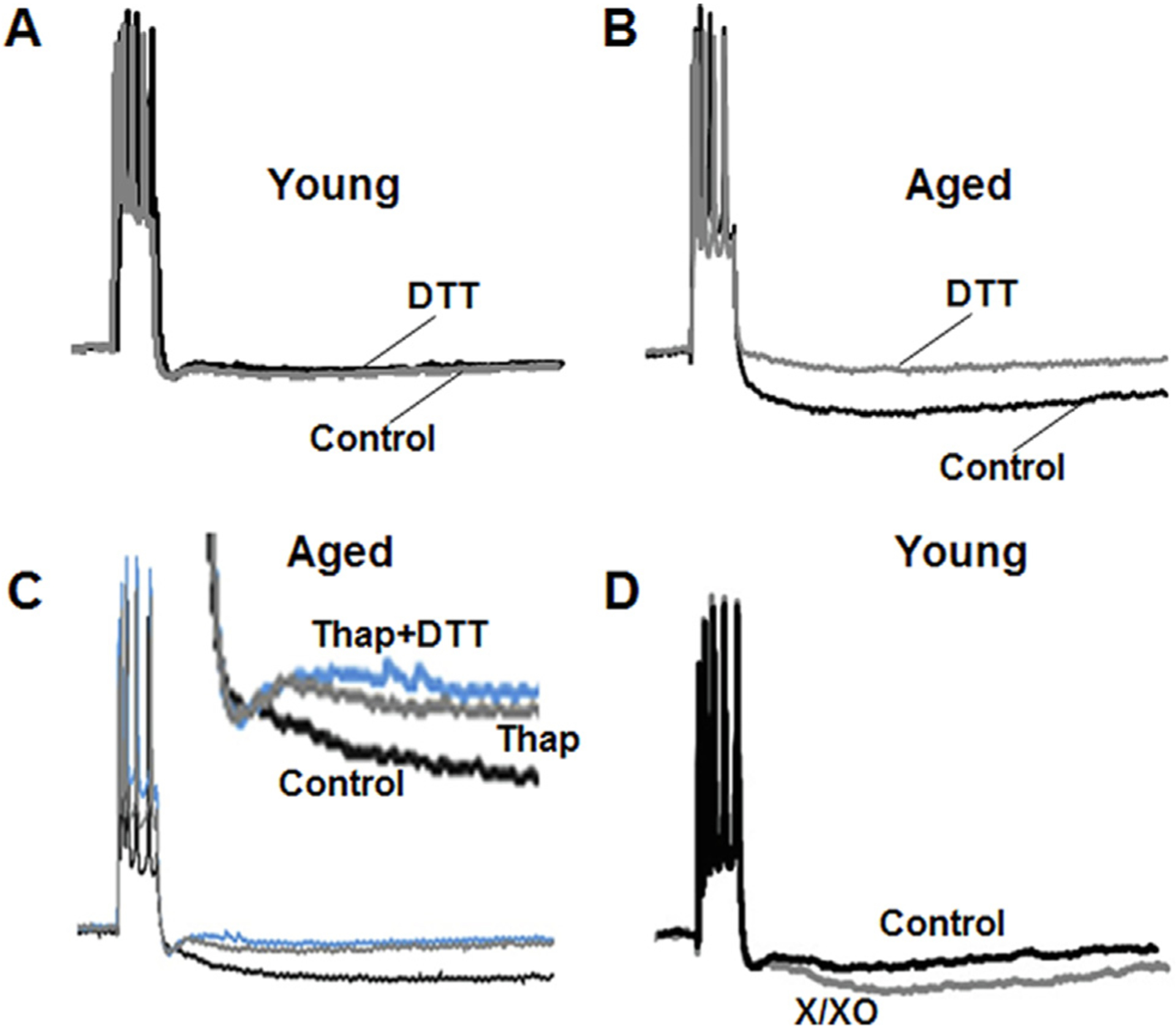
Age-dependent redox regulation of the AHP through intracellular Ca2+ stores (ICS). Representative traces illustrating the change in the AHP of (A) young and (B) aged animals under control conditions (black line) and following application of the reducing agent, DTT (gray line). (C) The large AHP of older animals under control condition (black line) is reduced by depletion of Ca2+ from ICS by thapsigargin (gray line). Following depletion of ICS, DTT no longer reduces the AHP (blue line). (D) In young animals, the smaller AHP recorded under control conditions (black line) increases under oxidizing conditions following application xanthine/xanthine oxidase (X/XO) (gray line). Figure adapted from Bodhinathan et al. (2010a).
In contrast to ICS, Ca2+ influx through NMDA receptors decreases with advancing age. However, like the AHP, NMDA receptor function is redox sensitive in an age-dependent manner. The initial study comparing age-dependent effects of oxidizing and reducing agents on the NMDA receptor synaptic response confirmed a more oxidized intracellular redox state during aging (Bodhinathan et al., 2010b). Thus, reducing agents produce a robust enhancement of NMDA receptor synaptic responses in older animals and oxidizing agents decrease the NMDA receptor synaptic response, specifically in young animals (Bodhinathan et al., 2010b; Guidi et al., 2015; Kumar & Foster, 2013; Kumar et al., 2018) (Fig. 3). The increase in NMDA receptor synaptic response following application of DTT is dependent on Ca2+-calmodulin-dependent kinase II (CaMKII), such that blockade of CaMKII prevents the DTT-mediated NMDA receptor potentiation (Bodhinathan et al., 2010b).
Fig. 3.
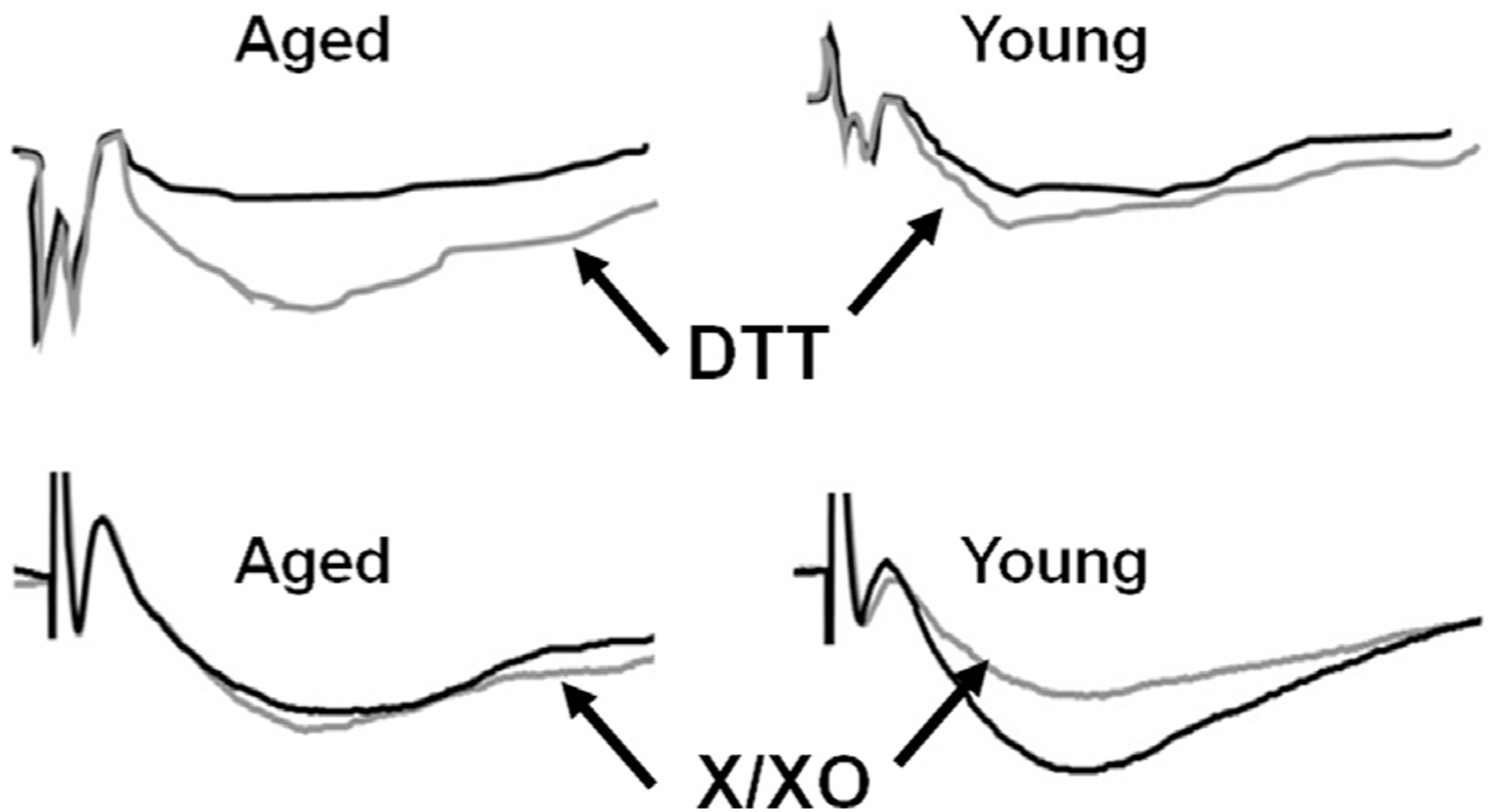
Age-dependent redox regulation of the NMDA receptor component of synaptic transmission. The top panels illustrates the response under control conditions (black line) for aged (left) and young (right) and following application of the reducing agent, DTT (gray line). DTT induces a large increase in the NMDA receptor response in older animals. The bottom panel illustrates the response under control conditions (black line) for aged (left) and young (right) and following application of the oxidizing agents, X/XO (gray line). Oxidizing conditions decrease the NMDA receptor response in young animals. Figure adapted from Bodhinathan et al. (2010b).
The redox regulation of CaMKII and NMDA receptor function likely contributes to impaired LTP of NMDA receptor synaptic transmission in older animals (Clayton, Grosshans, & Browning, 2002) and impaired LTP of NMDA receptor synaptic transmission under oxidizing conditions in young animals (Bernard, Hirsch, Khazipov, Ben-Ari, & Gozlan, 1997). Similar to LTP of NMDA receptors in young animals (Aniksztejn & Ben-Ari, 1995; Bashir, Alford, Davies, Randall, & Collingridge, 1991; Berretta et al., 1991; Muller & Lynch, 1988; Xie, Berger, & Barrionuevo, 1992), the DTT-mediate increase in NMDA receptor function in aged animals requires NMDA receptor activity and Ca2+ influx to initiate trafficking of NMDA receptors to the synapse (Kumar, Thinschmidt, & Foster, 2019). In young animals, NMDA receptor trafficking is mediated by CaMKII binding to the GluN2B NMDA receptor subunit, increasing the contribution of GluN2B to the synaptic response (Barcomb, Hell, Benke, & Bayer, 2016; Barria & Malinow, 2002; Yan et al., 2011). Likewise, the DTT-mediated NMDA receptor potentiation in aged animals involves an increased contribution of GluN2B to the synaptic response (Fig. 4). The ratio of GluN2A/GluN2B determines the decay of the synaptic response with increasing duration of the response as the contribution of GluN2B increases (Flint, Maisch, Weishaupt, Kriegstein, & Monyer, 1997; Stocca & Vicini, 1998). DTT increased the half time of the decay of excitatory postsynaptic current (EPSC) ~2 fold in young and ~4 fold in aged animals (Fig. 4A and B). The increased contribution of GluN2B to the synaptic response was also evident in the effectiveness of selective GluN2 antagonists in reducing the peak response under control conditions and following DTT application (Fig. 4C). These results suggest that redox state influences the contribution of GluN2B to the synaptic response (Kumar et al., 2019). The redox-mediated decline in the ability to traffic GluN2B to the synaptic may be critical for understanding how aging contributes to age-related neurodegenerative diseases. Specifically, the balance of synaptic and extrasynaptic GluN2B subunit activity is thought to contribute to amyloid beta-mediated toxicity (Hardingham & Bading, 2010; Karpova et al., 2013; Mota, Ferreira, Pereira, Oliveira, & Rego, 2012; Ronicke et al., 2011). Moreover, a shift in synaptic and extrasynaptic GluN2B activity can disrupt transcriptional signaling from the synapse to the nucleus, altering the expression of genes for neuroprotection and maintenance of synaptic connectivity (Bading, 2017).
Fig. 4.
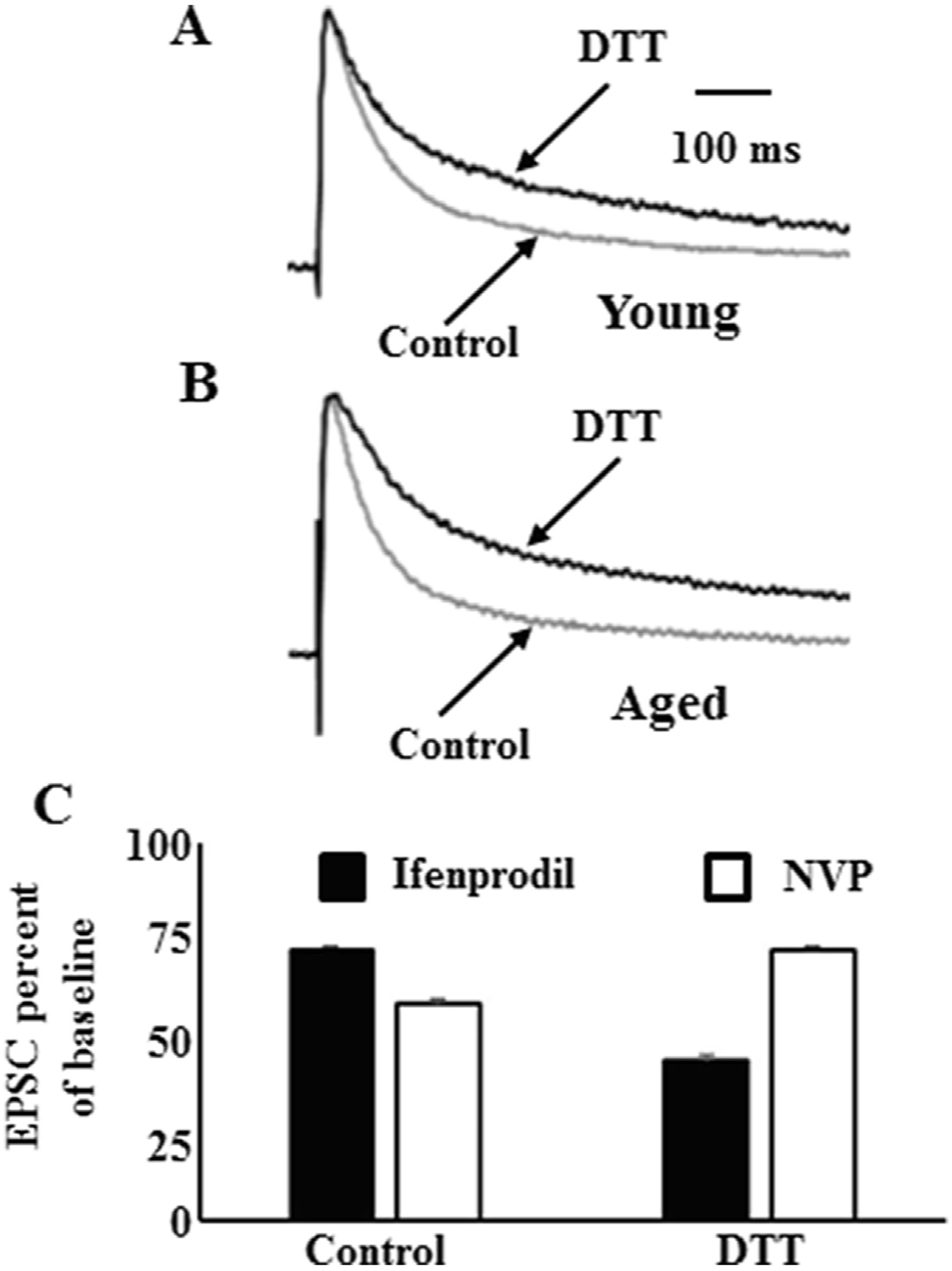
Redox state regulates the contribution of GluN2B to the synaptic response. Time course of EPSC response amplitude normalized to the peak of the response and averaged across all CA1 pyramidal cells in the control condition (gray trace) and in the presence of DTT (black trace). Recordings from (A) young animals and (B) aged animals. DTT increased the time to half decay ~2 fold in young and ~4 fold in aged animals. (C) Effect of subunit selective antagonists on the peak NMDA receptor EPSC in aged animals. In the control condition, the GluN2A selective antagonist, NVP, decreased the response more than the GluN2B selective antagonist, ifenprodil. In contrast, following the DTT mediated growth of the NMDA receptor synaptic response; ifenprodil decreased the response more than NVP. Figure adapted from Kumar et al. (2019).
5. Signaling to the nucleus and epigenetics
Senescent physiology is expected to involve altered gene expression. For example, the AHP is increased several hours after glucocorticoid treatment, suggesting a role for gene transcription (Joels & de Kloet, 1989; Kerr, Campbell, Hao, & Landfield, 1989; Pillai, Henckens, Fernandez, & Joels, 2014). Similarly, estrogen can have rapid and long-term effects that mitigate senescent neurophysiology (Foster, 2005). Ca2+ influx through synaptic NMDA receptors and due to neuronal activity induces the expression of pro-survival genes, transcription factors, and synaptic component genes (Hardingham & Bading, 2010; Papadia, Stevenson, Hardingham, Bading, & Hardingham, 2005; Tan, Zhang, Hoffmann, & Bading, 2012). Examination of age-related changes in transcription in region CA1 indicates altered expression of genes for Ca2+ signaling, ion channels, and synaptic plasticity (Blalock et al., 2003; Ianov, Rani, Beas, Kumar, & Foster, 2016; Ianov, De Both et al., 2017). In the case of Ca2+ signaling, the direction of altered transcription and protein expression suggests that, rather than acting as the cause of Ca2+-dysregulation, expression represents an attempt by CA1 neurons to compensate for a basal increase in intracellular Ca2+ (Ianov, De Both et al., 2017). Genes for proteins that facilitate Ca2+ entry or release from Ca2+ stores, as well as downstream Ca2+ signaling genes are decreased in animals that exhibit impaired episodic memory. For example, hippocalcin acts as Ca2+ sensors for the AHP, such that Ca2+ binding increases the AHP amplitude (Tzingounis, Kobayashi, Takamatsu, & Nicoll, 2007). Interestingly, decreased hippocalcin protein and mRNA has been reported in region CA1 during aging (Furuta, Kobayashi, Masaki, & Takamatsu, 1999; Ianov, De Both et al., 2017). A decrease in hippocalcin should decrease the AHP amplitude. Thus, while some transcriptional changes may contribute to senescent physiology, other modifications, such as the reduction in hippocalcin in memory-impaired animals, may represent a failed attempt to compensate for Ca2+-dysregulation and senescent physiology.
One mechanism for influencing transcription to promote cell excitability involves the activity of the cAMP-responsive element-binding protein (CREB) transcription factor. The shift in kinase/phosphatase activity during aging, favoring phosphatase activity, results in a decrease in CREB phosphorylation, which correlates with impaired memory (Foster et al., 2001). Overexpression of CREB decreases the AHP amplitude indicating that CREB activity can regulate cell excitability (Lopez de Armentia et al., 2007; Viosca, Lopez de Armentia, Jancic, & Barco, 2009; Zhou et al., 2009). Interestingly, overexpression of CREB had no apparent effect on the acquisition of a spatial reference memory; however, increasing CREB levels improved retention in older animals (Yu, Curlik, Oh, & Yin, 2017). In addition, enhanced expression of constitutively active CREB facilitates induction and maintenance of LTP induced by threshold stimulation (Barco, Alarcon, & Kandel, 2002). The molecular mechanism for CREB effects is unknown, but may involve the expression of brain derived neurotrophic factor (BDNF) (Finkbeiner et al., 1997), which also acts to decrease the AHP (Kramar et al., 2004) and increase NMDA receptor function (Caldeira et al., 2007; Levine, Crozier, Black, & Plummer, 1998; Marie, Morishita, Yu, Calakos, & Malenka, 2005). Moreover, CREB is involved in the transcriptional regulation of genes that are neuroprotective against oxidative stress (Lee et al., 2009; St-Pierre et al., 2006; Tan et al., 2012).
6. Treatments to rejuvenate senescent neurophysiology and improve cognition
In considering possible treatment for an age-related decline in episodic memory, pharmacological treatments that reduce the AHP (Disterhoft et al., 1999; Kumar & Foster, 2002; Maimaiti et al., 2017; Messier et al., 1991; Thomas, 2015; Thompson et al., 1990) or enhance NMDA receptor function (Burgdorf et al., 2011; Portero-Tresserra et al., 2018; Thompson & Disterhoft, 1997) can improve cognitive function. However, it is currently unclear whether adverse reactions might also occur from long-term treatments.
The increase in AHP amplitude and NMDA receptor hypofunction emerge in middle-age as a component of reversible redox signaling rather than irreversible oxidative damage, which may occur with more advanced age or in neurodegenerative disease (Kumar et al., 2017). Thus, it may be possible to prevent or reverse senescent physiology by approaches that reduce redox stress. It is important to emphasize that treatments may want to focus on redox signaling rather than oxidative damage. Indeed, increasing the activity of the antioxidant enzyme, superoxide dismutase 1 (SOD1), decreases oxidative damage by converting the damaging molecule, superoxide, to the redox-signaling molecule, hydrogen peroxide. In this case, the excess hydrogen peroxide promotes senescent neurophysiology (Lee et al., 2012, 2014). In contrast, treatments to decrease hydrogen peroxide, by increasing activity of catalase, glutathione peroxidase, or increasing the level of redox buffers may ameliorate senescent physiology (Billard, 2015; Bodhinathan et al., 2010b; Braidy et al., 2014; Clausen, Xu, Bi, & Baudry, 2012; Haxaire et al., 2012; Lee et al., 2012, 2014; Liu et al., 2003; Martin et al., 2016; More et al., 2018; Parihar, Kunz, & Brewer, 2008; Robillard, Gordon, Choi, Christie, & MacVicar, 2011).
Inflammation is a source of redox stress and anti-inflammatory treatments have been reported to decrease the AHP (Blalock et al., 2010), diminish the age-related and redox-mediated NMDA receptor hypofunction, and improve memory, suggesting that inflammation contributes to cognitive impairment through an increase in redox stress (Kumar et al., 2018; Mesches et al., 2004). However, older individuals may have problems with efficacy and adverse effects of anti-inflammatory drugs (Barkin et al., 2010). Alternatively, behavioral modification of diet, exercise, or cognitive stimulation can promote successful aging, reducing inflammation and oxidative stress, possibly regulating senescent physiology (Bettio, Rajendran, & Gil-Mohapel, 2017; Rowe & Kahn, 1987). For example, cognitive stimulation through environmental enrichment or physical activity rejuvenates the AHP and synaptic plasticity in older animals (Kumar and Foster, 2007b; Kumar et al., 2012; Bettio et al., 2017; Di Benedetto, Muller, Wenger, Duzel, & Pawelec, 2017).
The mechanism for the effects of exercise and environmental enrichment are far from clear. One proposed mechanism involves feedback due to coordinated hippocampal neural activity associated with locomotion or environmental stimulation, which regulates gene expression possibly through epigenetic mechanisms, including DNA methylation (Barter & Foster, 2018; Kumar et al., 2012). In general, DNA methylation is associated with decreased transcription and aging is associated increase DNA methylation for genes normally activated during learning and by environmental stimulation (Ianov, Riva et al., 2017; Barter & Foster, 2018). The DNA methylation marks may be reversible due to behavioral training, inducing neural activity, and activation of signaling cascades (Barter & Foster, 2018; Penner et al., 2011, 2016). However, with advanced age, senescent physiology may result in a prolonged decrease in activation of transcription factors that support synaptic plasticity and neural activity. For genes that exhibit reduced transcriptional activity, passive DNA methylation may increase within the promoter and the gene body (Barter & Foster, 2018; Thurman, 2012). In turn, the increase in DNA methylation could reduce neuronal resiliency to behavioral stimulation, inhibiting or delaying transcription in response to transcriptional signals. Thus, cognitively impaired animals are characterized by decreased transcription and increased DNA methylation of genes involved in synaptic plasticity and ion channel signaling a (Blalock et al., 2003; Ianov et al., 2016; Ianov, De Both et al., 2017; Rani et al., 2017).
In conclusion, in the CA1 region of the hippocampus, senescent neurophysiology is well-characterized and associated with impaired cognition. In particular, considerable research indicates that the larger AHP and hypofunction of NMDA receptors underlie a shift in synaptic plasticity, impairing the rate or extent of learning and increasing forgetting or impairing consolidation of episodic memory. We are at the early stages of understanding the molecular mechanisms for senescent physiology. Redox state provides a link between mechanisms of aging (oxidative stress and inflammation) and disruption of memory mechanisms (synaptic plasticity and NMDA receptor trafficking). Redox mediated senescent neurophysiology emerges in middle-age and may be rejuvenated by pharmacological or behavioral interventions. However, problems may compound with advanced aging due to the interaction of senescent physiology signaling to the nucleus and epigenetic mechanisms for the regulation of transcription. In addition, an understanding of how senescent physiology interacts with epigenetics and the environment to regulate neurodegenerative diseases of aging will be required in order to obtain optimal treatment plans.
Acknowledgements
Supported by National Institute of Aging grants AG037984, AG036800, AG049711, AG052258, P30AG028740 and the Evelyn F. McKnight Brain Research Foundation. Special thanks to Ashok Kumar for editing and comments.
References
- Aniksztejn L, & Ben-Ari Y (1995). Expression of LTP by AMPA and/or NMDA receptors is determined by the extent of NMDA receptors activation during the tetanus. Journal of Neurophysiology, 74, 2349–2357. [DOI] [PubMed] [Google Scholar]
- Bading H (2017). Therapeutic targeting of the pathological triad of extrasynaptic NMDA receptor signaling in neurodegenerations. Journal of Experimental Medicine, 214, 569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, & Kandel ER (2002). Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell, 108, 689–703. [DOI] [PubMed] [Google Scholar]
- Barcomb K, Hell JW, Benke TA, & Bayer KU (2016). The CaMKII/GluN2B protein interaction maintains synaptic strength. Journal of Biological Chemistry, 291, 16082–16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkin RL, Beckerman M, Blum SL, Clark FM, Koh EK, & Wu DS (2010). Should nonsteroidal anti-inflammatory drugs (NSAIDs) be prescribed to the older adult? Drugs and Aging, 27, 775–789. [DOI] [PubMed] [Google Scholar]
- Barnes CA (1979). Memory deficits associated with senescence: A neurophysiological and behavioral study in the rat. Journal of Comparative and Physiological Psychology, 93, 74–104. [DOI] [PubMed] [Google Scholar]
- Barnes CA, & McNaughton BL (1985). An age comparison of the rates of acquisition and forgetting of spatial information in relation to long-term enhancement of hippocampal synapses. Behavioral Neuroscience, 99, 1040–1048. [DOI] [PubMed] [Google Scholar]
- Barnes CA, Rao G, & Shen J (1997). Age-related decrease in the N-methyl-D-aspartateR-mediated excitatory postsynaptic potential in hippocampal region CA1. Neurobiology of Aging, 18, 445–452. [DOI] [PubMed] [Google Scholar]
- Barnes CA, Suster MS, Shen J, & McNaughton BL (1997). Multistability of cognitive maps in the hippocampus of old rats. Nature, 388, 272–275. [DOI] [PubMed] [Google Scholar]
- Barria A, & Malinow R (2002). Subunit-specific NMDA receptor trafficking to synapses. Neuron, 35, 345–353. [DOI] [PubMed] [Google Scholar]
- Barter JD, & Foster TC (2018). Aging in the brain: New roles of epigenetics in cognitive decline. Neuroscientist, 24, 516–525. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Alford S, Davies SN, Randall AD, & Collingridge GL (1991). Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature, 349, 156–158. [DOI] [PubMed] [Google Scholar]
- Baskys A, Niesen CE, & Carlen PL (1987). Altered modulatory actions of serotonin on dentate granule cells of aged rats. Brain Research, 419, 112–118. [DOI] [PubMed] [Google Scholar]
- Bernard CL, Hirsch JC, Khazipov R, Ben-Ari Y, & Gozlan H (1997). Redox modulation of synaptic responses and plasticity in rat CA1 hippocampal neurons. Experimental Brain Research, 113, 343–352. [DOI] [PubMed] [Google Scholar]
- Berretta N, Berton F, Bianchi R, Brunelli M, Capogna M, & Francesconi W (1991). Long-term potentiation of NMDA receptor-mediated EPSP in guinea-pig hippocampal slices. European Journal of Neuroscience, 3, 850–854. [DOI] [PubMed] [Google Scholar]
- Bettio LEB, Rajendran L, & Gil-Mohapel J (2017). The effects of aging in the hippocampus and cognitive decline. Neuroscience & Biobehavioral Reviews, 79, 66–86. [DOI] [PubMed] [Google Scholar]
- Billard JM (2015). D-Serine in the aging hippocampus. Journal of Pharmaceutical and Biomedical Analysis, 116, 18–24. [DOI] [PubMed] [Google Scholar]
- Blalock EM, Chen KC, Sharrow K, Herman JP, Porter NM, Foster TC, et al. (2003). Gene microarrays in hippocampal aging: Statistical profiling identifies novel processes correlated with cognitive impairment. Journal of Neuroscience, 23, 3807–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Phelps JT, Pancani T, Searcy JL, Anderson KL, Gant JC, et al. (2010). Effects of long-term pioglitazone treatment on peripheral and central markers of aging. PLoS ONE, 5, e10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodhinathan K, Kumar A, & Foster TC (2010b). Intracellular redox state alters NMDA receptor response during aging through Ca2+/calmodulin-dependent protein kinase II. Journal of Neuroscience, 30, 1914–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodhinathan K, Kumar A, & Foster TC (2010a). Redox sensitive calcium stores underlie enhanced after hyperpolarization of aged neurons: Role for ryanodine receptor mediated calcium signaling. Journal of Neurophysiology, 104, 2586–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Poljak A, Grant R, Jayasena T, Mansour H, Chan-Ling T, et al. (2014). Mapping NAD(+) metabolism in the brain of ageing Wistar rats: Potential targets for influencing brain senescence. Biogerontology, 15, 177–198. [DOI] [PubMed] [Google Scholar]
- Bull R, Finkelstein JP, Galvez J, Sanchez G, Donoso P, Behrens MI, et al. (2008). Ischemia enhances activation by Ca2+ and redox modification of ryanodine receptor channels from rat brain cortex. Journal of Neuroscience, 28, 9463–9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf J, Zhang XL, Weiss C, Matthews E, Disterhoft JF, Stanton PK, et al. (2011). The N-methyl-D-aspartate receptor modulator GLYX-13 enhances learning and memory, in young adult and learning impaired aging rats. Neurobiology of Aging, 32, 698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger C (2010). Region-specific genetic alterations in the aging hippocampus: Implications for cognitive aging. Frontiers in Aging Neuroscience, 2, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira MV, Melo CV, Pereira DB, Carvalho RF, Carvalho AL, & Duarte CB (2007). BDNF regulates the expression and traffic of NMDA receptors in cultured hippocampal neurons. Molecular and Cellular Neuroscience, 35, 208–219. [DOI] [PubMed] [Google Scholar]
- Clausen A, Xu X, Bi X, & Baudry M (2012). Effects of the superoxide dismutase/catalase mimetic EUK-207 in a mouse model of Alzheimer’s disease: Protection against and interruption of progression of amyloid and tau pathology and cognitive decline. Journal of Alzheimer’s Disease, 30, 183–208. [DOI] [PubMed] [Google Scholar]
- Clayton DA, Grosshans DR, & Browning MD (2002). Aging and surface expression of hippocampal NMDA receptors. Journal of Biological Chemistry, 277, 14367–14369. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Lo Turco JJ, Kubota M, Disterhoft JF, Moore JW, & Alkon DL (1989). Classical conditioning reduces amplitude and duration of calcium-dependent afterhyperpolarization in rabbit hippocampal pyramidal cells. Journal of Neurophysiology, 61, 971–981. [DOI] [PubMed] [Google Scholar]
- Datta D, & Arnsten AFT (2018). Unique molecular regulation of higher-order prefrontal cortical circuits: Insights into the neurobiology of schizophrenia. ACS Chemical Neuroscience, 9, 2127–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge MC, Black J, Deyo RA, & Disterhoft JF (1990). Learning-induced afterhyperpolarization reductions in hippocampus are specific for cell type and potassium conductance. Experimental Brain Research, 80, 456–462. [DOI] [PubMed] [Google Scholar]
- Deyo RA, Straube KT, & Disterhoft JF (1989). Nimodipine facilitates associative learning in aging rabbits. Science, 243, 809–811. [DOI] [PubMed] [Google Scholar]
- Di Benedetto S, Muller L, Wenger E, Duzel S, & Pawelec G (2017). Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neuroscience & Biobehavioral Reviews, 75, 114–128. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Coulter DA, & Alkon DL (1986). Conditioning-specific membrane changes of rabbit hippocampal neurons measured in vitro. Proceedings of the National Academy of Sciences of the United States of America, 83, 2733–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disterhoft JF, Kronforst-Collins M, Oh MM, Power JM, Preston AR, & Weiss C (1999). Cholinergic facilitation of trace eyeblink conditioning in aging rabbits. Life Sciences, 64, 541–548. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Thompson LT, Moyer JR Jr., & Mogul DJ (1996). Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life Sciences, 59, 413–420. [DOI] [PubMed] [Google Scholar]
- Du F, Eid T, Lothman EW, Kohler C, & Schwarcz R (1995). Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. Journal of Neuroscience, 15, 6301–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, & Greenberg ME (1997). CREB: A major mediator of neuronal neurotrophin responses. Neuron, 19, 1031–1047. [DOI] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, & Monyer H (1997). NR2A subunit expression shortens NMDA receptor synaptic currents in developing neo-cortex. Journal of Neuroscience, 17, 2469–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC (1999). Involvement of hippocampal synaptic plasticity in age-related memory decline. Brain Research. Brain Research Reviews, 30, 236–249. [DOI] [PubMed] [Google Scholar]
- Foster TC (2005). Interaction of rapid signal transduction cascades and gene expression in mediating estrogen effects on memory over the life span. Frontiers in Neuroendocrinology, 26, 51–64. [DOI] [PubMed] [Google Scholar]
- Foster TC (2007). Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell, 6, 319–325. [DOI] [PubMed] [Google Scholar]
- Foster TC (2012). Dissecting the age-related decline on spatial learning and memory tasks in rodent models: N-methyl-D-aspartate receptors and voltage-dependent Ca2+ channels in senescent synaptic plasticity. Progress in Neurobiology, 96, 283–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC, Defazio RA, & Bizon JL (2012). Characterizing cognitive aging of spatial and contextual memory in animal models. Frontiers in Aging Neuroscience, 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC, & Kumar A (2007). Susceptibility to induction of long-term depression is associated with impaired memory in aged Fischer 344 rats. Neurobiology of Learning and Memory, 87, 522–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC, & Norris CM (1997). Age-associated changes in Ca(2+)-dependent processes: Relation to hippocampal synaptic plasticity. Hippocampus, 7, 602–612. [DOI] [PubMed] [Google Scholar]
- Foster TC, Sharrow KM, Masse JR, Norris CM, & Kumar A (2001). Calcineurin links Ca2+ dysregulation with brain aging. Journal of Neuroscience, 21, 4066–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuenzalida M, Fernandez de Sevilla D, & Buno W (2007). Changes of the EPSP waveform regulate the temporal window for spike-timing-dependent plasticity. Journal of Neuroscience, 27, 11940–11948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y, Kobayashi M, Masaki T, & Takamatsu K (1999). Age-related changes in expression of hippocalcin and NVP2 in rat brain. Neurochemical Research, 24, 651–658. [DOI] [PubMed] [Google Scholar]
- Gant JC, Blalock EM, Chen KC, Kadish I, Thibault O, Porter NM, et al. (2018). FK506-Binding protein 12.6/1b, a negative regulator of [Ca(2+)], rescues memory and restores genomic regulation in the hippocampus of aging rats. Journal of Neuroscience, 38, 1030–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Chen KC, Kadish I, Blalock EM, Thibault O, Porter NM, et al. (2015). Reversal of aging-related neuronal Ca2+ dysregulation and cognitive impairment by delivery of a transgene encoding FK506-binding protein 12.6/1b to the hippocampus. Journal of Neuroscience, 35, 10878–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Chen KC, Norris CM, Kadish I, Thibault O, Blalock EM, et al. (2011). Disrupting function of FK506-binding protein 1b/12.6 induces the Ca(2)+-dysregulation aging phenotype in hippocampal neurons. Journal of Neuroscience, 31, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Kadish I, Chen KC, Thibault O, Blalock EM, Porter NM, & Landfield PW (2018). Aging-related calcium dysregulation in rat entorhinal neurons homologous with the human entorhinal neurons in which Alzheimer’s disease neurofibrillary tangles first appear. Journal of Alzheimer’s Disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Sama MM, Landfield PW, & Thibault O (2006). Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. Journal of Neuroscience, 26, 3482–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, & Thibault O (2009). Action potential throughput in aged rat hippocampal neurons: Regulation by selective forms of hyperpolarization. Neurobiology of Aging, 30, 2053–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Peters M, & Vernon J (2001). Modulation of excitability as a learning and memory mechanism: A molecular genetic perspective. Physiology & Behavior, 73, 803–810. [DOI] [PubMed] [Google Scholar]
- Guidi M, Kumar A, & Foster TC (2015). Impaired attention and synaptic senescence of the prefrontal cortex involves redox regulation of NMDA receptors. Journal of Neuroscience, 35, 3966–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, & Bading H (2010). Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nature Reviews Neuroscience, 11, 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D (1981). The aging process. Proceedings of the National Academy of Sciences of the United States of America, 78, 7124–7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haxaire C, Turpin FR, Potier B, Kervern M, Sinet PM, Barbanel G, et al. (2012). Reversal of age-related oxidative stress prevents hippocampal synaptic plasticity deficits by protecting D-serine-dependent NMDA receptor activation. Aging Cell, 11, 336–344. [DOI] [PubMed] [Google Scholar]
- Hsu KS, Huang CC, Liang YC, Wu HM, Chen YL, Lo SW, et al. (2002). Alterations in the balance of protein kinase and phosphatase activities and age-related impairments of synaptic transmission and long-term potentiation. Hippocampus, 12, 787–802. [DOI] [PubMed] [Google Scholar]
- Hughes ML, Agrigoroaei S, Jeon M, Bruzzese M, & Lachman ME (2018). Change in cognitive performance from midlife into old age: Findings from the Midlife in the United States (MIDUS) study. Journal of the International Neuropsychological Society, 24, 805–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianov L, De Both M, Chawla MK, Rani A, Kennedy AJ, Piras I, et al. (2017). Hippocampal transcriptomic profiles: Subfield vulnerability to age and cognitive impairment. Frontiers in Aging Neuroscience, 9, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianov L, Rani A, Beas BS, Kumar A, & Foster TC (2016). Transcription profile of aging and cognition-related genes in the medial prefrontal cortex. Frontiers in Aging Neuroscience, 8, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianov L, Riva A, Kumar A, & Foster TC (2017). DNA methylation of synaptic genes in the prefrontal cortex is associated with aging and age-related cognitive impairment. Frontiers in Aging Neuroscience, 9, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TC, Rani A, Kumar A, & Foster TC (2009). Regional hippocampal differences in AKT survival signaling across the lifespan: Implications for CA1 vulnerability with aging. Cell Death and Differentiation, 16, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joels M, & de Kloet ER (1989). Effects of glucocorticoids and norepinephrine on the excitability in the hippocampus. Science, 245, 1502–1505. [DOI] [PubMed] [Google Scholar]
- Jouvenceau A, & Dutar P (2006). A role for the protein phosphatase 2B in altered hippocampal synaptic plasticity in the aged rat. Journal of Physiology – Paris, 99, 154–161. [DOI] [PubMed] [Google Scholar]
- Kaczorowski CC, & Disterhoft JF (2009). Memory deficits are associated with impaired ability to modulate neuronal excitability in middle-aged mice. Learning & Memory, 16, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Bera S, Bar J, Reddy PP, Behnisch T, et al. (2013). Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell, 152, 1119–1133. [DOI] [PubMed] [Google Scholar]
- Kerr DS, Campbell LW, Hao SY, & Landfield PW (1989). Corticosteroid modulation of hippocampal potentials: Increased effect with aging. Science, 245, 1505–1509. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS (1989). The role of calcium regulation in brain aging: Reexamination of a hypothesis. Aging (Milano), 1, 17–34. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS (1994). Calcium hypothesis of Alzheimer’s disease and brain aging. Annals of the New York Academy of Sciences, 747, 1–11. [DOI] [PubMed] [Google Scholar]
- Kramar EA, Lin B, Lin CY, Arai AC, Gall CM, & Lynch G (2004). A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. Journal of Neuroscience, 24, 5151–5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, & Foster TC (2007a). Neurophysiology of old neurons and synapses. In Riddle DR (Ed.). Brain aging: Models, methods, and mechanisms. Boca Raton (FL) [PubMed] [Google Scholar]
- Kumar A, Bodhinathan K, & Foster TC (2009). Susceptibility to calcium dysregulation during brain aging. Frontiers in Aging Neuroscience, 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Thinschmidt JS, & Foster TC (2019). Subunit contribution to NMDA receptor hypofunction and redox sensitivity of hippocampal synaptic transmission during aging. Aging (Albany NY). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, & Foster TC (2002). 17beta-estradiol benzoate decreases the AHP amplitude in CA1 pyramidal neurons. Journal of Neurophysiology, 88, 621–626. [DOI] [PubMed] [Google Scholar]
- Kumar A, & Foster TC (2004). Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. Journal of Neurophysiology, 91, 2437–2444. [DOI] [PubMed] [Google Scholar]
- Kumar A, & Foster TC (2005). Intracellular calcium stores contribute to increased susceptibility to LTD induction during aging. Brain Research, 1031, 125–128. [DOI] [PubMed] [Google Scholar]
- Kumar A, & Foster T (2007b). Environmental enrichment decreases the after-hyperpolarization in senescent rats. Brain Research, 1130, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, & Foster TC (2013). Linking redox regulation of NMDAR synaptic function to cognitive decline during aging. Journal of Neuroscience, 33, 15710–15715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Rani A, Scheinert RB, Ormerod BK, & Foster TC (2018). Nonsteroidal anti-inflammatory drug, indomethacin improves spatial memory and NMDA receptor function in aged animals. Neurobiology of Aging, 70, 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Rani A, Tchigranova O, Lee WH, & Foster TC (2012). Influence of late-life exposure to environmental enrichment or exercise on hippocampal function and CA1 senescent physiology. Neurobiology of Aging, 33(828), e821–e1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Yegla B, & Foster TC (2017). Redox signaling in neurotransmission and cognition during aging. Antioxidants & Redox Signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AG, Lee G, & Disterhoft JF (2006). Simultaneous training on two hippocampus-dependent tasks facilitates acquisition of trace eyeblink conditioning. Learning & Memory, 13, 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfield PW, & Pitler TA (1984). Prolonged Ca2+-dependent after-hyperpolarizations in hippocampal neurons of aged rats. Science, 226, 1089–1092. [DOI] [PubMed] [Google Scholar]
- Lee B, Cao R, Choi YS, Cho HY, Rhee AD, Hah CK, et al. (2009). The CREB/CRE transcriptional pathway: Protection against oxidative stress-mediated neuronal cell death. Journal of Neurochemistry, 108, 1251–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WH, Kumar A, Rani A, & Foster TC (2014). Role of antioxidant enzymes in redox regulation of N-methyl-D-aspartate receptor function and memory in middle-aged rats. Neurobiology of Aging, 35, 1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WH, Kumar A, Rani A, Herrera J, Xu J, Someya S, et al. (2012). Influence of viral vector-mediated delivery of superoxide dismutase and catalase to the hippocampus on spatial learning and memory during aging. Antioxidants & Redox Signaling, 16, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, & Plummer MR (1998). Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proceedings of the National Academy of Sciences of the United States of America, 95, 10235–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Liu IY, Bi X, Thompson RF, Doctrow SR, Malfroy B, et al. (2003). Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proceedings of the National Academy of Sciences of the United States of America, 100, 8526–8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Armentia M, Jancic D, Olivares R, Alarcon JM, Kandel ER, & Barco A (2007). cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. Journal of Neuroscience, 27, 13909–13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebke JI, & Amatrudo JM (2012). Age-related increase of sI(AHP) in prefrontal pyramidal cells of monkeys: Relationship to cognition. Neurobiology of Aging, 33, 1085–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebke JI, & Chang YM (2007). Effects of aging on the electrophysiological properties of layer 5 pyramidal cells in the monkey prefrontal cortex. Neuroscience, 150, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, & Nicoll RA (1982). Noradrenaline blocks accommodation of pyramidal cell discharge in the hippocampus. Nature, 299, 636–638. [DOI] [PubMed] [Google Scholar]
- Maimaiti S, Frazier HN, Anderson KL, Ghoweri AO, Brewer LD, Porter NM, et al. (2017). Novel calcium-related targets of insulin in hippocampal neurons. Neuroscience, 364, 130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, & Malenka RC (2005). Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron, 45, 741–752. [DOI] [PubMed] [Google Scholar]
- Martin SA, DeMuth TM, Miller KN, Pugh TD, Polewski MA, Colman RJ, et al. (2016). Regional metabolic heterogeneity of the hippocampus is nonuniformly impacted by age and caloric restriction. Aging Cell, 15, 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEchron MD, Weible AP, & Disterhoft JF (2001). Aging and learning-specific changes in single-neuron activity in CA1 hippocampus during rabbit trace eyeblink conditioning. Journal of Neurophysiology, 86, 1839–1857. [DOI] [PubMed] [Google Scholar]
- Mesches MH, Gemma C, Veng LM, Allgeier C, Young DA, Browning MD, et al. (2004). Sulindac improves memory and increases NMDA receptor subunits in aged Fischer 344 rats. Neurobiology of Aging, 25, 315–324. [DOI] [PubMed] [Google Scholar]
- Messier C, Mourre C, Bontempi B, Sif J, Lazdunski M, & Destrade C (1991). Effect of apamin, a toxin that inhibits Ca(2+)-dependent K+ channels, on learning and memory processes. Brain Research, 551, 322–326. [DOI] [PubMed] [Google Scholar]
- Monti B, Berteotti C, & Contestabile A (2005). Dysregulation of memory-related proteins in the hippocampus of aged rats and their relation with cognitive impairment. Hippocampus, 15, 1041–1049. [DOI] [PubMed] [Google Scholar]
- More J, Galusso N, Veloso P, Montecinos L, Finkelstein JP, Sanchez G, et al. (2018). N-acetylcysteine prevents the spatial memory deficits and the redox-dependent RyR2 decrease displayed by an Alzheimer’s disease rat model. Frontiers in Aging Neuroscience, 10, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mota SI, Ferreira IL, Pereira C, Oliveira CR, & Rego AC (2012). Amyloid-beta peptide 1–42 causes microtubule deregulation through N-methyl-D-aspartate receptors in mature hippocampal cultures. Current Alzheimer Research, 9, 844–856. [DOI] [PubMed] [Google Scholar]
- Moyer JR Jr., Thompson LT, & Disterhoft JF (1996). Trace eyeblink conditioning increases CA1 excitability in a transient and learning-specific manner. Journal of Neuroscience, 16, 5536–5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, & Lynch G (1988). Long-term potentiation differentially affects two components of synaptic responses in hippocampus. Proceedings of the National Academy of Sciences of the United States of America, 85, 9346–9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy GG, Shah V, Hell JW, & Silva AJ (2006). Investigation of age-related cognitive decline using mice as a model system: Neurophysiological correlates. Am J Geriatr Psychiatry, 14, 1012–1021. [DOI] [PubMed] [Google Scholar]
- Norris CM, Halpain S, & Foster TC (1998b). Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. Journal of Neuroscience, 18, 3171–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Halpain S, & Foster TC (1998a). Alterations in the balance of protein kinase/phosphatase activities parallel reduced synaptic strength during aging. Journal of Neurophysiology, 80, 1567–1570. [DOI] [PubMed] [Google Scholar]
- Norris CM, Korol DL, & Foster TC (1996). Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. Journal of Neuroscience, 16, 5382–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez-Santana FL, Oh MM, Antion MD, Lee A, Hell JW, & Disterhoft JF (2014). Surface L-type Ca2+ channel expression levels are increased in aged hippocampus. Aging Cell, 13, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, McKay BM, Power JM, & Disterhoft JF (2009). Learning-related postburst afterhyperpolarization reduction in CA1 pyramidal neurons is mediated by protein kinase A. Proceedings of the National Academy of Sciences of the United States of America, 106, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Oliveira FA, & Disterhoft JF (2010). Learning and aging related changes in intrinsic neuronal excitability. Frontiers in Aging Neuroscience, 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H, & Hardingham GE (2005). Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. Journal of Neuroscience, 25, 4279–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar MS, Kunz EA, & Brewer GJ (2008). Age-related decreases in NAD(P)H and glutathione cause redox declines before ATP loss during glutamate treatment of hippocampal neurons. Journal of Neuroscience Research, 86, 2339–2352. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Krause M, Haug T, Storm JF, & Stuhmer W (1998). Modulation of the Ca2+-activated K+ current sIAHP by a phosphatase-kinase balance under basal conditions in rat CA1 pyramidal neurons. Journal of Neurophysiology, 79, 3252–3256. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, & Storm JF (1993). PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron, 11, 1023–1035. [DOI] [PubMed] [Google Scholar]
- Penner MR, Parrish RR, Hoang LT, Roth TL, Lubin FD, & Barnes CA (2016). Age-related changes in Egr1 transcription and DNA methylation within the hippocampus. Hippocampus, 26, 1008–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penner MR, Roth TL, Chawla MK, Hoang LT, Roth ED, Lubin FD, et al. (2011). Age-related changes in Arc transcription and DNA methylation within the hippocampus. Neurobiology of Aging, 32, 2198–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai AG, Henckens MJ, Fernandez G, & Joels M (2014). Delayed effects of corticosterone on slow after-hyperpolarization potentials in mouse hippocampal versus prefrontal cortical pyramidal neurons. PLoS ONE, 9, e99208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, & Landfield PW (1990). Aging-related prolongation of calcium spike duration in rat hippocampal slice neurons. Brain Research, 508, 1–6. [DOI] [PubMed] [Google Scholar]
- Portero-Tresserra M, Marti-Nicolovius M, Tarres-Gatius M, Candalija A, Guillazo-Blanch G, & Vale-Martinez A (2018). Intra-hippocampal D-cycloserine rescues decreased social memory, spatial learning reversal, and synaptophysin levels in aged rats. Psychopharmacology (Berl), 235, 1463–1477. [DOI] [PubMed] [Google Scholar]
- Power JM, Wu WW, Sametsky E, Oh MM, & Disterhoft JF (2002). Age-related enhancement of the slow outward calcium-activated potassium current in hippocampal CA1 pyramidal neurons in vitro. Journal of Neuroscience, 22, 7234–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani A, O’Shea A, Ianov L, Cohen RA, Woods AJ, & Foster TC (2017). miRNA in circulating microvesicles as biomarkers for age-related cognitive decline. Frontiers in Aging Neuroscience, 9, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Defazio RA, Barnes CA, Alexander GE, Bizon JL, Bowers D, et al. (2012). Challenges and opportunities for characterizing cognitive aging across species. Frontiers in Aging Neuroscience, 4, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robillard JM, Gordon GR, Choi HB, Christie BR, & MacVicar BA (2011). Glutathione restores the mechanism of synaptic plasticity in aged mice to that of the adult. PLoS ONE, 6, e20676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronicke R, Mikhaylova M, Ronicke S, Meinhardt J, Schroder UH, Fandrich M, et al. (2011). Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiology of Aging, 32, 2219–2228. [DOI] [PubMed] [Google Scholar]
- Rosenzweig ES, & Barnes CA (2003). Impact of aging on hippocampal function: Plasticity, network dynamics, and cognition. Progress in Neurobiology, 69, 143–179. [DOI] [PubMed] [Google Scholar]
- Rotenberg A, Mayford M, Hawkins RD, Kandel ER, & Muller RU (1996). Mice expressing activated CaMKII lack low frequency LTP and do not form stable place cells in the CA1 region of the hippocampus. Cell, 87, 1351–1361. [DOI] [PubMed] [Google Scholar]
- Rowe JW, & Kahn RL (1987). Human aging: Usual and successful. Science, 237, 143–149. [DOI] [PubMed] [Google Scholar]
- Shen J, Barnes CA, McNaughton BL, Skaggs WE, & Weaver KL (1997). The effect of aging on experience-dependent plasticity of hippocampal place cells. Journal of Neuroscience, 17, 6769–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjo BK (1981). Cell damage in the brain: A speculative synthesis. Journal of Cerebral Blood Flow & Metabolism, 1, 155–185. [DOI] [PubMed] [Google Scholar]
- Simkin D, Hattori S, Ybarra N, Musial TF, Buss EW, Richter H, et al. (2015). Aging-related hyperexcitability in CA3 pyramidal neurons is mediated by enhanced A-type K+ channel function and expression. Journal of Neuroscience, 35, 13206–13218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocca G, & Vicini S (1998). Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. Journal of Physiology, 507(Pt 1), 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, et al. (2006). Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell, 127, 397–408. [DOI] [PubMed] [Google Scholar]
- Tan YW, Zhang SJ, Hoffmann T, & Bading H (2012). Increasing levels of wild-type CREB up-regulates several activity-regulated inhibitor of death (AID) genes and promotes neuronal survival. BMC Neuroscience, 13, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Gant JC, & Landfield PW (2007). Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: Minding the store. Aging Cell, 6, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, & Landfield PW (1996). Increase in single L-type calcium channels in hippocampal neurons during aging. Science, 272, 1017–1020. [DOI] [PubMed] [Google Scholar]
- Thomas SA (2015). Neuromodulatory signaling in hippocampus-dependent memory retrieval. Hippocampus, 25, 415–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LT, Deyo RA, & Disterhoft JF (1990). Nimodipine enhances spontaneous activity of hippocampal pyramidal neurons in aging rabbits at a dose that facilitates associative learning. Brain Research, 535, 119–130. [DOI] [PubMed] [Google Scholar]
- Thompson LT, & Disterhoft JF (1997). Age- and dose-dependent facilitation of associative eyeblink conditioning by D-cycloserine in rabbits. Behavioral Neuroscience, 111, 1303–1312. [DOI] [PubMed] [Google Scholar]
- Thompson LT, Moyer JR Jr., & Disterhoft JF (1996). Transient changes in excitability of rabbit CA3 neurons with a time course appropriate to support memory consolidation. Journal of Neurophysiology, 76, 1836–1849. [DOI] [PubMed] [Google Scholar]
- Thurman RE, et al. (2012). The accessible chromatin landscape of the human genome. Nature, 489, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, & Rose GM (2005). The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. Journal of Neuroscience, 25, 2609–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Kobayashi M, Takamatsu K, & Nicoll RA (2007). Hippocalcin gates the calcium activation of the slow afterhyperpolarization in hippocampal pyramidal cells. Neuron, 53, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veng LM, & Browning MD (2002). Regionally selective alterations in expression of the alpha(1D) subunit (Ca(v)1.3) of L-type calcium channels in the hippocampus of aged rats. Brain Research Molecular Brain Research, 107, 120–127. [DOI] [PubMed] [Google Scholar]
- Viosca J, Lopez de Armentia M, Jancic D, & Barco A (2009). Enhanced CREB-dependent gene expression increases the excitability of neurons in the basal amygdala and primes the consolidation of contextual and cued fear memory. Learning & Memory, 16, 193–197. [DOI] [PubMed] [Google Scholar]
- Vogalis F, Harvey JR, & Furness JB (2004). Suppression of a slow post-spike afterhyperpolarization by calcineurin inhibitors. European Journal of Neuroscience, 19, 2650–2658. [DOI] [PubMed] [Google Scholar]
- Wang X, & Michaelis EK (2010). Selective neuronal vulnerability to oxidative stress in the brain. Frontiers in Aging Neuroscience, 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Coleman PD, Flood DG, & Troncoso JC (1994). Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet, 344, 769–772. [DOI] [PubMed] [Google Scholar]
- Xie X, Berger TW, & Barrionuevo G (1992). Isolated NMDA receptor-mediated synaptic responses express both LTP and LTD. Journal of Neurophysiology, 67, 1009–1013. [DOI] [PubMed] [Google Scholar]
- Yan JZ, Xu Z, Ren SQ, Hu B, Yao W, Wang SH, et al. (2011). Protein kinase C promotes N-methyl-D-aspartate (NMDA) receptor trafficking by indirectly triggering calcium/calmodulin-dependent protein kinase II (CaMKII) autophosphorylation. Journal of Biological Chemistry, 286, 25187–25200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Zhang Y, Roder J, & McDonald RJ (2003). Aging effects on spatial tuning of hippocampal place cells in mice. Experimental Brain Research, 150, 184–193. [DOI] [PubMed] [Google Scholar]
- Yeoman M, Scutt G, & Faragher R (2012). Insights into CNS ageing from animal models of senescence. Nature Reviews Neuroscience, 13, 435–445. [DOI] [PubMed] [Google Scholar]
- Yu XW, Curlik DM, Oh MM, & Yin JC (2017). CREB overexpression in dorsal CA1 ameliorates long-term memory deficits in aged rats. Elife 6. Disterhoft JF [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeier Z, Madorsky I, Xu Y, Ogle WO, Notterpek L, & Foster TC (2011). Gene expression in the hippocampus: Regionally specific effects of aging and caloric restriction. Mechanisms of Ageing and Development, 132, 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer I, Cohen H, Richter-Levin G, Lebiosn T, Grossberger T, & Barkai E (2006). A cellular correlate of learning-induced metaplasticity in the hippocampus. Cerebral Cortex, 16, 460–468. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ouyang M, Ganellin CR, & Thomas SA (2013). The slow afterhyperpolarization: A target of beta1-adrenergic signaling in hippocampus-dependent memory retrieval. Journal of Neuroscience, 33, 5006–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, et al. (2009). CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nature Neuroscience, 12, 1438–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]