

Abstract
Treatment of tuberculosis (TB) currently takes at least 6 months. Latent Mycobacterium tuberculosis (Mtb) are phenotypically tolerant to most anti-TB drugs. A key hypothesis is that drugs that kill non-replicating (NR) Mtb may shorten treatment when used in combination with conventional drugs. The Mtb proteasome (Mtb20S) could be such a target, because its pharmacological inhibition kills NR Mtb and its genetic deletion renders Mtb unable to persist in mice. Here we report a series of macrocyclic peptides that potently and selectively target the Mtb20S over human proteasomes, including macrocycle 6. The co-crystal structure of macrocycle 6 with Mtb20S revealed structural bases for the species selectivity. Inhibition of 20S within Mtb by 6 dose-dependently led to accumulation of Pup-tagged GFP that is degradable but resistant to depupylation., and death of non-replicating Mtb under nitrosative stress. These results suggest that compounds of this class have the potential to develop as anti-TB therapeutics.
Keywords: Mycobacterium tuberculosis, species selectivity, proteasome inhibitors, macrocyclic peptides, pupylated proteins
Graphical Abstract
INTRODUCTION
Host immune stresses can drive Mycobacterium tuberculosis (Mtb) into a state of non-replication, in which Mtb becomes phenotypically resistant to many of the drugs used to treat tuberculosis (TB).1–4 Drugs that kill non-replicating persisters are anticipated to shorten the duration of TB treatment.5 Mtb relies on specific enzymatic pathways to survive the host stresses that suppress its replication, including the generation of nitric oxide (NO) and reactive nitrogen intermediates derived from it (herein collectively called “NO” for simplicity).6 Among these defenses is the prokaryotic ubiquitin-like protein (Pup)–proteasome system (Figure 1).2, 7–13 Different from human 20S proteasome (hu20S) which contains 2 copies of 7 distinct α- and 7 distinct β-subunits among which only three β subunits (β1, β2, and β5) are enzymatically active, the Mtb 20S proteasome (Mtb20S) has 14 copies each of α and β subunits. Structures of proteasome are highly conserved in a four-layered, barrel shaped complex in the α1–7β1–7β1–7α1–7 fashion. Although the Mtb20S is dispensable under standard growth conditions in vitro, it is indispensable for Mtb to survive in the host, and it has been validated as a drug target both genetically and pharmacologically. A null mutant strain lacking prcBA genes that respectively encode the β and α subunits dies in the chronic phase of disease in mice.2, 3, 14 We have introduced several classes of Mtb proteasome inhibitors that kill NO-stressed or starved Mtb in vitro. These included the first inhibitors selective (>1000-fold) for the proteasome of a pathogen and not its host, and the first agents to kill bacteria by inhibiting protein breakdown rather than protein synthesis.2, 15, 16 We recently discovered a novel class of phenylimidazole based Mtb20S selective inhibitors.17 However, they were inactive against Mtb H37Rv under the conditions tested, which may be attributed to their poor uptake and/or intracellular biotransformation resulting in insufficient intracellular concentrations.
Figure 1.
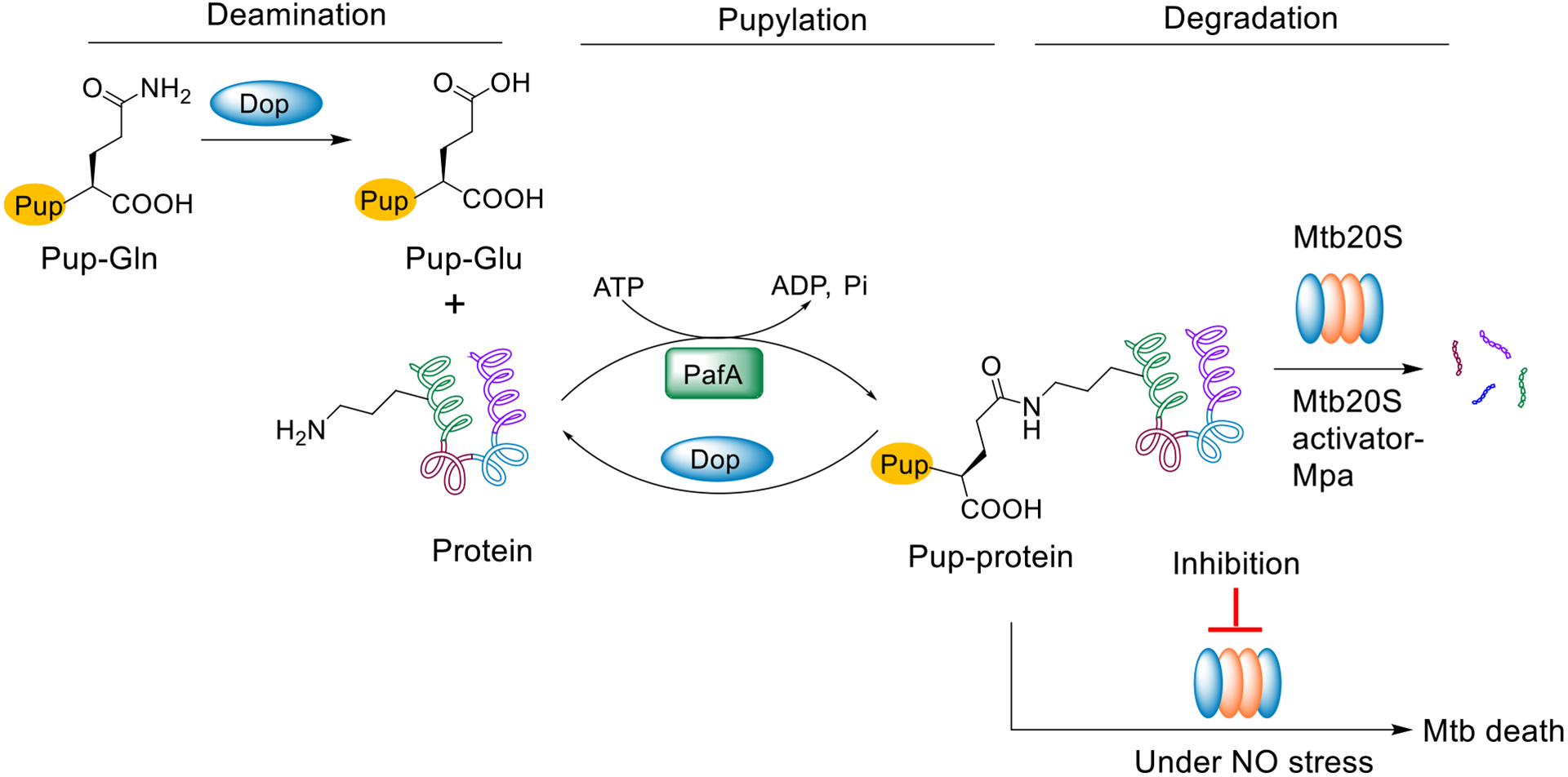
Schematic representation of Pup-proteasome system (PPS) in M. tuberculosis. Pup is expressed with a C-terminal glutamine (Pup-Gln), which is deamidated to glutamate (PupGlu) by Dop. PafA, a Pup ligase, then catalyzes the formation of an isopeptide bond between the C-terminal γ-carboxylate of Pup-Glu and the lysine ε-amino group of a protein substrate. The pupylated protein is recognized and unfolded by Mpa and degraded by the Mtb 20S proteasome. Alternatively, Dop can rescue the pupylated substrate by cleaving the specific isopeptide bond. Inhibition of the Mtb proteasome under NO stress (mimicking a host stress that can render Mtb non-replicating) results in cell death.
Macrocyclization is a structural optimization strategy that has been successfully applied in drug research and development.18 It can potentially improve potency, selectivity, and physicochemical properties through the restriction of conformation.19 Macrocycle-based inhibitors for eukaryotic proteasomes have been reported. For example, the natural tripeptide macrocycle TMZ-95a and its analogs have been extensively studied.20–23 A macrocyclic peptide inhibitor of the malaria proteasome was reported to possess potent antimalarial activity,24, 25 and a series of P1-P3 macrocyclic peptide aldehydes were reported to have anti-cancer activity.26 These results suggest the potential to cyclize either P1-P3 or P2-P4 of peptide-based proteasome inhibitors to tune them for specific activity. Here we describe our efforts to use this strategy to improve the activity and stability of a series of selective and potent Mtb proteasome inhibitors.
RESULTS AND DISCUSSION
Rationale.
The co-crystal structure of the acyclic N,C-capped dipeptide DPLG-2 and other N,C-capped dipeptides16, 27 bound to the Mtb20S open-gate, activated form (Mtb20SOG) shows that the ligand is occupying the non-prime S1, S2, S3, S4 binding pockets (Figure 2A). In the case of DPLG-2, the P1 naphthyl moiety inserts into the S1 binding pocket harboring the proteolytic residue Thr-1. The P2 p-F-benzyl group fills the shallow S2 pocket and has no interactions with the proteasome. The P3 N,N-diethyl Asn moiety occupies the S3 pocket, forming hydrogen bonds with Gln-22 and Ser-27 in addition to binding to the hydrophobic pocket via van der Waals interaction. The N-cap 3-phenylpropanoyl group occupies the S4 pocket, forming additional hydrophobic interactions with residues of neighboring subunits. Also, the peptide backbone forms several hydrogen bonds with substrate-binding cleft of Mtb20S. The close spatial proximity of the P2 and P4 substituents led us to hypothesize that it may be feasible to link them covalently into a macrocyclic ring, which may reduce the conformational freedom and increase the compounds’ potency. Such a macrocyclic strategy would also allow exploration of the region between P2 and P4 pockets.
Figure 2.
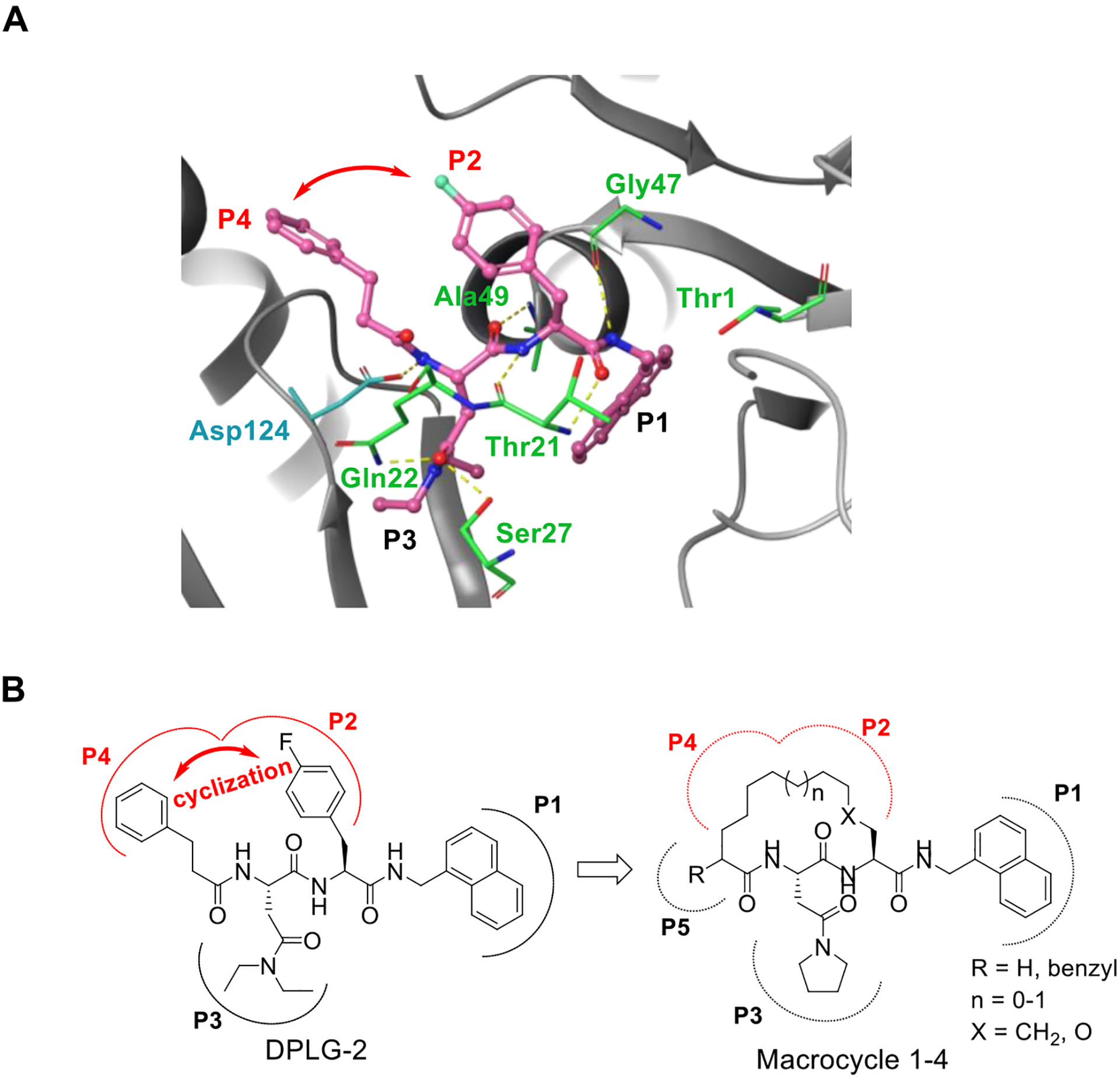
Rationale for introducing a tether between P2 and P4 in the DPLG-2 scaffold. A) X-ray crystal structure of acyclic N,C-capped dipeptide DPLG-2 in Mtb20SOG (PDB code 5TRG). DPLG-2 is shown in magenta. Hydrogen bonds are indicated by dashed yellow lines. The red arrow depicts the potential for macrocyclization between the P2 and P4 groups. B) Our initial macrocyclic strategy for the design of novel Mtb20S selective inhibitors. The P2 and P4 groups are linked with the conformationally flexible alkyl or ether linkers.
As shown in Figure 2B, our initial macrocyclic strategy involved removing the P2 4-fluorophenyl and connecting the P2 to the α-position of the carboxyl group of P4’s 3-phenylpropanoyl group via an alkyl or ether linker of varied length. The proposed linker would provide conformational flexibility and be suitable for verifying our strategy. A series of macrocycle 1 analogues ranging from 14- to 17-membered rings were evaluated with molecular docking using the Mtb20SOG co-crystal structure (PDB: 5TS0) as the docking template27. Cyclic peptides with a 15- or 16-membered ring showed good overlap with the parental N,C-capped dipeptide (Figure S1).
Structure-Activity Relationship (SAR) Studies.
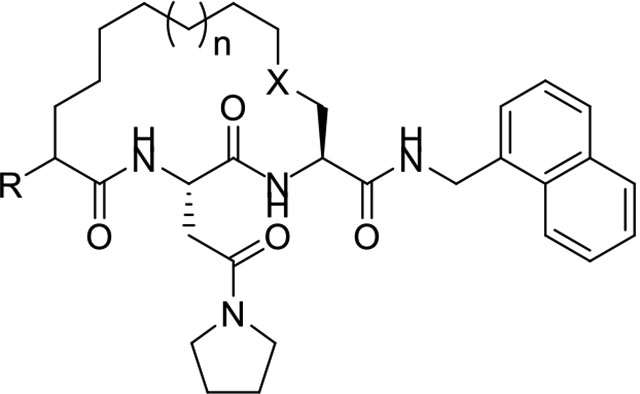
Our initial study of macrocyclic Mtb proteasome inhibitors focused on 15- and 16-membered alkyl- and ether-tethered macrocycles (Table 1). Considering the synthetic convenience, the 16-membered macrocycle 1 was synthesized as the racemic form and showed a significant loss in potency against Mtb20S compared with acyclic DPLG-2. Truncating the P5 benzyl substituent of macrocycle 1 gave the 16-membered macrocycle 2, which improved the potency by 33-fold. The 15-membered macrocycle 3 was 5-fold less potent than macrocycle 2, suggesting that 16-membered ring is preferrable. The potency of the 16-membered ether-linked macrocycle 4 was slightly improved over that of the corresponding alkyl-linked macrocycle 2.
Table 1.
IC50 values of macrocyclic peptides against Mtb20SOG and β5c and β5i of the human c-20S and i-20S, respectively.a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Proteasome inhibition IC50 (μM) | SIapp** | |||||||
| Macrocycle | R | n | X | Mtb20S | β5c | β5i | IC50–β5c | IC50–β5i |
| IC50–Mtb20S | IC50–Mtb20S | |||||||
| DPLG-2* | - | - | - | 0.015 | 70.0 | 54.7 | 4670 | 3650 |
| 1 | benzyl | 1 | CH2 | 5.0 ± 0.8 | >100 | >100 | >200 | >200 |
| 2 | H | 1 | CH2 | 0.15 ± 0.02 | >100 | >100 | >674 | >674 |
| 3 | H | 0 | CH2 | 0.71 ± 0.07 | >100 | >100 | >141 | >141 |
| 4 | H | 1 | O | 0.10 ± 0.01 | >100 | >100 | >984 | >984 |
Data shown are the mean of at least three independent experiments.
Data quoted from ref16 for comparison. LLVY: suc-LLVY-AMC, used for all three proteasomes. SIapp: apparent selectivity index.
SIapp values were rounded to three or fewer significant figures. β5c, β5 subunit of the human constitutive proteasome. β5i, β5 subunit of the human immunoproteasome.
Inspired by the recently reported CP124 and our N,C-capped dipeptide DPLG-2, we next sought to explore biphenyl ether–linked macrocycles. CP1 was developed to selectively target the Plasmodium falciparum proteasome (Pf20S) over human proteasomes. CP1 shares the same backbone with N,C-capped dipeptides but adds a 3,3’-biphenyl ether tether that links P2 and P4. We found that CP1 did not inhibit Mtb20S (Table 2), likely because the P3 homo-phenylalanine does not fit well in the shallow but wide S3 pocket. Our previous study showed that both 2-fluorobenzyl and naphthyl groups are well tolerated in the S1 pocket of Mtb20S.27 We chose 2-fluorobenzyl as P1 because of its lower molecular weight28 and better pharmacokinetic properties than naphthyl. We evaluated several macrocycle 5 analogues with 3,3’-, 3,4’-, or 4,3’-biphenyl ether tethers by computational docking in the Mtb20SOG (open gate) cocrystal structure (PDB code 5TS0) using Schrödinger’s Glide program.29, 30 Macrocycle 5 with a 3,4’-biphenyl ether tether had highest docking score. The overlay of macrocycle 5 and the parent N,C-capped dipeptide in the binding pocket indicated a similar binding pose (Figure 3A). The peptide backbone of the macrocycle forms four hydrogen bonds with residues Thr-21, Gly-47, Ala-49, and Asp-124 of the adjacent β-subunit. The P3 pyrrolidine Asn group forms two hydrogen bonds with residues Gln-22 and Ser-27. Macrocycle 5 had a sub-μM IC50 against Mtb20S and little inhibitory activity toward the human constitutive proteasome (c-20S) or the human immunoproteasome (i-20S) (Table 2).
Table 2.
IC50 of macrocyclic peptides against Mtb20SOG and β5c and β5i of the human c-20S and i-20Sa
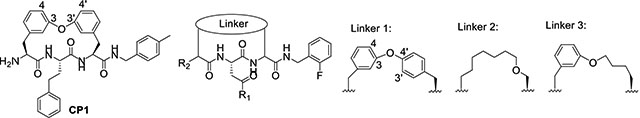 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Proteasome inhibition | ||||||||
| Macrocycle | Linker type | R1 | R2 | IC50 (μM) | SIapp* | |||
| Mtb20S | β5c | β5i | IC50–β5c | IC50–β5i | ||||
| IC50-Mtb20S | IC50-Mtb20S | |||||||
| CP1 | - | - | - | >100 | >100 | >100 | - | - |
| 5 | 1 | 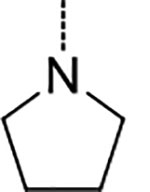 |
H | 0.20 ± 0.05 | >100 | 21.1 ± 5.2 | >500 | 108 |
| 6 | 1 | 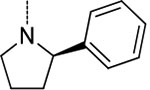 |
H | 0.0074 ± 0.0012 | >100 | >100 | >13,600 | >13,600 |
| 7 | 1 | 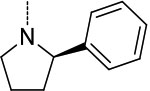 |
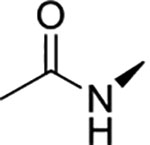 |
0.17 ± 0.02 | >100 | >100 | >594 | >594 |
| 8 | 1 | 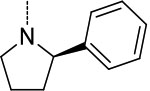 |
 |
0.84 ± 0.08 | >100 | >100 | >119 | >119 |
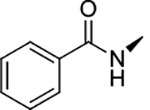
| 9 | 2 | 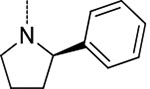 |
H | 0.007 ± 0.001 | >100 | >100 | >14,000 | >14,000 |
| 10 | 3 | 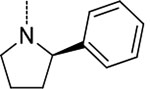 |
H | 0.007 ± 0.003 | >100 | >100 | >14,400 | >14,400 |
| 11 | 3 | 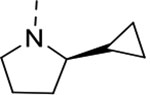 |
H | 0.007 ± 0.0004 | 0.073 ± 0.002 | 0.069 ± 0.028 | 11 | 11 |
| 12 | 3 | 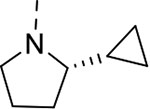 |
H | 0.079 ± 0.014 | 1.5 ± 0.1 | 0.87 ± 0.37 | 19 | 11 |
| 13 | 3 | 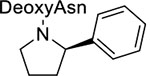 |
H | 1.5 ± 0.2 | 86.2± 12.1 | 47.3 ± 4.9 | 57 | 31 |
Data are the mean of at least three independent experiments. LLVY: suc-LLVY-AMC, used for all three proteasomes. SIapp: apparent selectivity index.
SI values were rounded to 3 or fewer significant figures.
Figure 3.
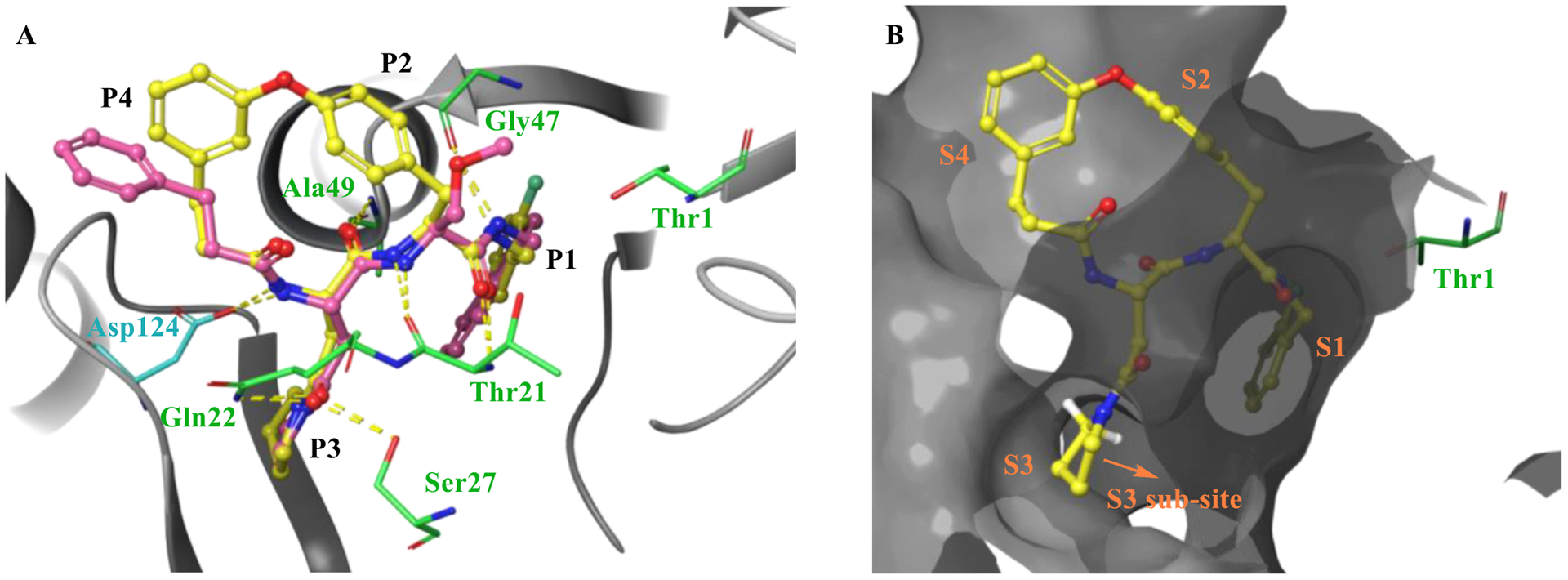
Molecular docking of macrocycle 5 to the co-crystal structure of Mtb20SOG (PDB 5TS0) executed with Schrödinger’s Glide. A) Overlay of macrocycle 5 (yellow) and parent N,C-capped dipeptide PKS-2208 suggests that the two have similar binding modes. Hydrogen bonds are indicated by dashed yellow lines. B) Molecular surface representation of the Mtb20SOG in complex with macrocycle 5. The binding pocket is depicted by a dark gray surface.
We next focused on modifying Asn P3 to further improve potency against Mtb20S based on macrocycle 5 (IC50 = 0.2 μM). As shown in Figure 3B, molecular docking analysis of macrocycle 5 revealed that the α-hydrogen atom of P3 pyrrolidine points to a large S3 sub-pocket, which is lined by residues Ala-49 and Val-31. We hypothesized that incorporation of a hydrophobic substituent to the α-position of pyrrolidine might provide valuable interactions with the S3 sub-pocket. Remarkably, incorporation of a bulky 2-phenyl substituent on the pyrrolidinyl ring of macrocycle 5 led to macrocycle 6 (TDI5575), which showed a 27-fold increase in potency against Mtb20S plus about a 5-fold reduced inhibition of human i-20S. The result was a >13,600-fold selectivity for Mtb20S over both human c-20S and human i-20S. We then extended the P5 amides CH3CONH or PhCONH on macrocycle 6, giving macrocycles 7 and 8, which inhibited Mtb20S with IC50 values of 0.17 μM and 0.84 μM, respectively, which were 23 and 114 times less potent than macrocycle 6. Structural analysis of the macrocycle 6–Mtb20SOG complex indicated that the space available for accommodating the P5 group in the active site was only 3.8 Å (Figure S2A). This suggested that P5 amides in the context of a rigid macrocyclic peptide were not well tolerated at the S5 pocket of Mtb20S.
The 16-membered alkyl ether–tethered macrocycle was identified as a promising scaffold in our initial SAR studies. We then synthesized a macrocycle 4 analogue, macrocycle 9, with 2-fluorobenzyl as P1 and 2-phenylpyrrolidinyl-Asn amide as P3. Macrocycle 9 had improved potency (Mtb20S IC50 = 7.1 nM) and maintained selectivity over human c-20S (hu-β5c IC50 >100 μM) and human i-20S (hu-β5i IC50 >100 μM) compared with macrocycle 4. Despite these favorable characteristics, both macrocycles 9 and 6 had extremely poor solubility (1.4 μg/mL and below the level of detection, respectively) (Table 3). To overcome the solubility issue, we further altered the tether by replacing the biphenyl ether in macrocycle 6 with a monophenyl ether, giving macrocycle 10, which showed improved solubility (33 μg/mL) while maintaining high potency (Mtb20S IC50 = 7.0 nM) for Mtb20S and selectivity over human c-20S (>14,400-fold) and i-20S (>14,400-fold).
Table 3.
Physicochemical properties and in vitro safety profile of macrocycles 6, 9, and 10
| Macrocycle | PAMPA permeability (nm/s) | Solubility pH 6.8 (μg/mL)a | Stability in human plasmab | Viability of HepG2 at 30 μMc | Microsome stability CLint (μL/min/mg) h/me |
|---|---|---|---|---|---|
| 6 | 227 | 1.4 | 94% | 98% | >768/>768 |
| 9 | 260 | –d | 86% | 91% | >768/>768 |
| 10 | 206 | 33 | 97% | 102% | >768/>768 |
Kinetic solubility.
Plasma stability was determined as the percentage remaining after incubation for 2 h with fresh human plasma.
HepG2 cells were treated with 30 μM test compounds for 72 h and viability was assessed.
Below the level of detection.
Stability of the macrocycles in human (h) and mouse (m) liver microsomes. Clint: intrinsic clearance.
To investigate the contributions of the P3 side chain 2-phenylpyrrolidinyl-Asn to potency and selectivity, we synthesized macrocycles 11 and 12, in which 2-(R)-cyclproylpyrrolidinyl-Asn or 2-(S)-cyclproylpyrrolidinyl-Asn, respectively, replaced 2-phenylpyrrolidinyl-Asn. Macrocycle 11 showed comparable potency to macrocycle 10 against Mtb20S (IC50 = 6.5 nM), but replacing the phenyl with the cyclopropyl markedly reduced the selectivity between the Mtb and human proteasomes. Compared with macrocycle 11, the inhibitory activity of macrocycle 12 was reduced for both Mtb20S and human proteasomes by 11- to 20-fold. These results indicate that the (R) conformation at the 2-position of the pyrrolidinyl is generally preferred over (S) by proteasomes, and the bulky phenyl group is responsible for species selectivity for Mtb20S over human c-20S and i-20S.
We also investigated whether the asparagine amide could be removed without affecting the binding affinity. We synthesized macrocycle 13 by replacing the C=O of P3 asparagine amide in macrocycle 10 with a CH2. The removal of this H-bond receptor reduced the potency against Mtb20S by 219-fold, suggesting that the carbonyl oxygen of the P3 amide is crucial for proteasome inhibition potency.
We further assessed inhibitory activity of macrocycles 1-12 against β1c or β2c subunits of the human c-20S or β1i or β2i of the human i-20S subunits (Table S1). Except macrocycle 11 exhibiting moderate inhibition against β2i (IC50 44.5 μM), all compounds showed <50% inhibition even at 100 μM. Therefore, this series of macrocyclic peptides are highly species-selective for Mtb20S over all active subunits of both human c-20S and i-20S.
Inhibition Kinetics.
By following the time-course of the peptide LLVY hydrolysis activity of Mtb20S in the presence of macrocycle 6 (Figure 4A), we found that the macrocyclic peptides were time-dependent inhibitors (Figure 4B). The plot of kobs values derived from each time-dependent curve versus macrocycle 6 yielded a k3 of 5.7 × 104 s−1M−1 and k4 of 0.7 × 10−3 s−1, resulting in a ki of 12.1 nM and t1/2 of 16.5 min for the Mtb20S-inhibitor complex (Figure 4C).
Figure 4.
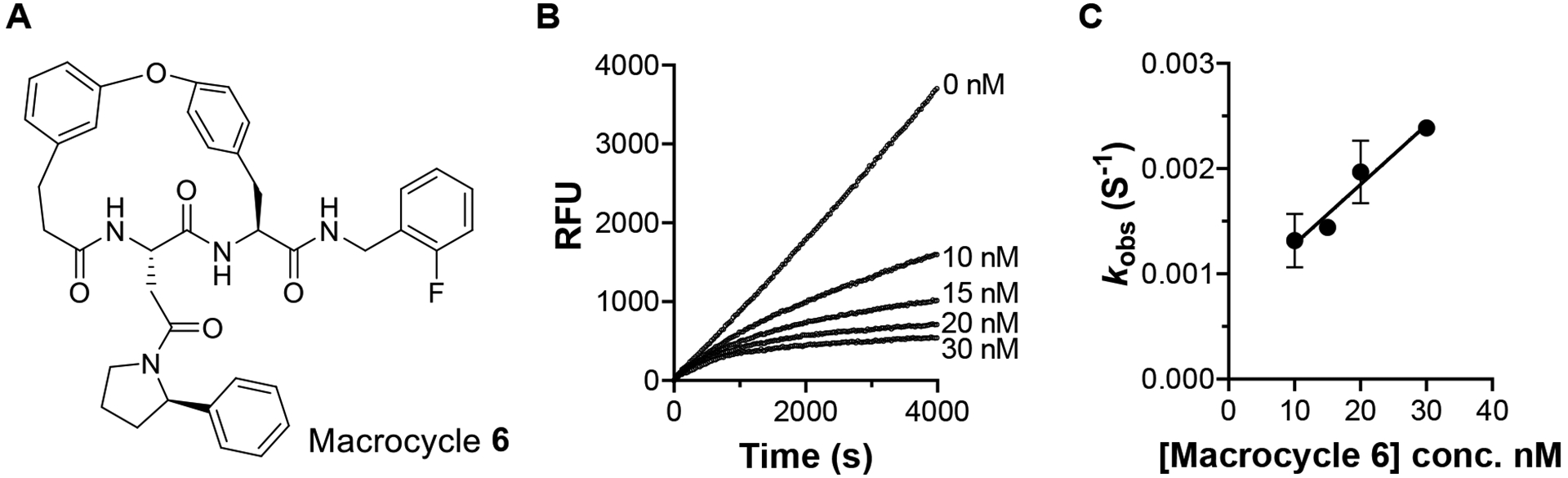
Inhibition of Mtb20S by macrocycle 6. A) Structure of macrocycle 6. B) Time-dependent inhibition of Mtb20S by macrocycle 6 at indicated concentration C) Plot of kobs vs concentration of macrocycle 6 yielded k3 of 5.7 × 104 s−1M−1 and k4 of 0.7 × 10−3 s−1.
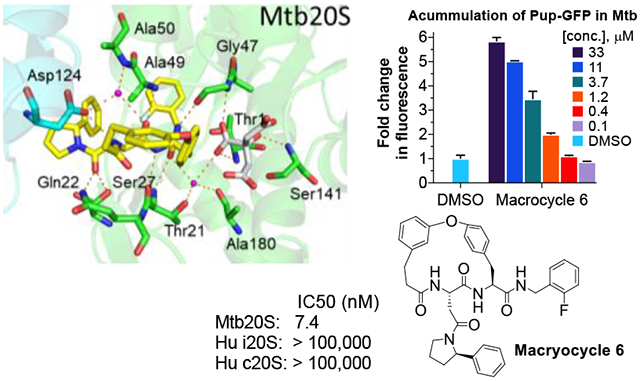
Crystal Structure of macrocycle 6 (TDI5575) in Complex with Mtb20SOG.
To understand the binding modes of the macrocyclic dipeptide inhibitors, macrocycle 6 was soaked into crystals of Mtb20SOG. The inhibitor-soaked crystals diffracted X-rays to 2.28 Å (Table S2). Molecular replacement was used to solve the inhibitor-bound Mtb20SOG structure using the available DPLG-2-bound Mtb20S structure (PDB ID 5TRG) as the search model. After refinement, both the Fo-Fc and 2Fo-Fc 3D maps had well-defined inhibitor density for modeling macrocycle 6 (Figure 5A). Each of the 14 β-subunits of the proteasome was occupied by one molecule of macrocycle 6, consistent with all β-subunits being active in the Mtb20S (Figure 5B–C). Like the reported N,C-capped dipeptides27 and phenylimidazole-based inhibitors,17 macrocycle 6 binds noncovalently to the Mtb20S active site and does not significantly alter the active site conformation, as compared with the apo Mtb20S structure (PDB ID 3HFA).
Figure 5.
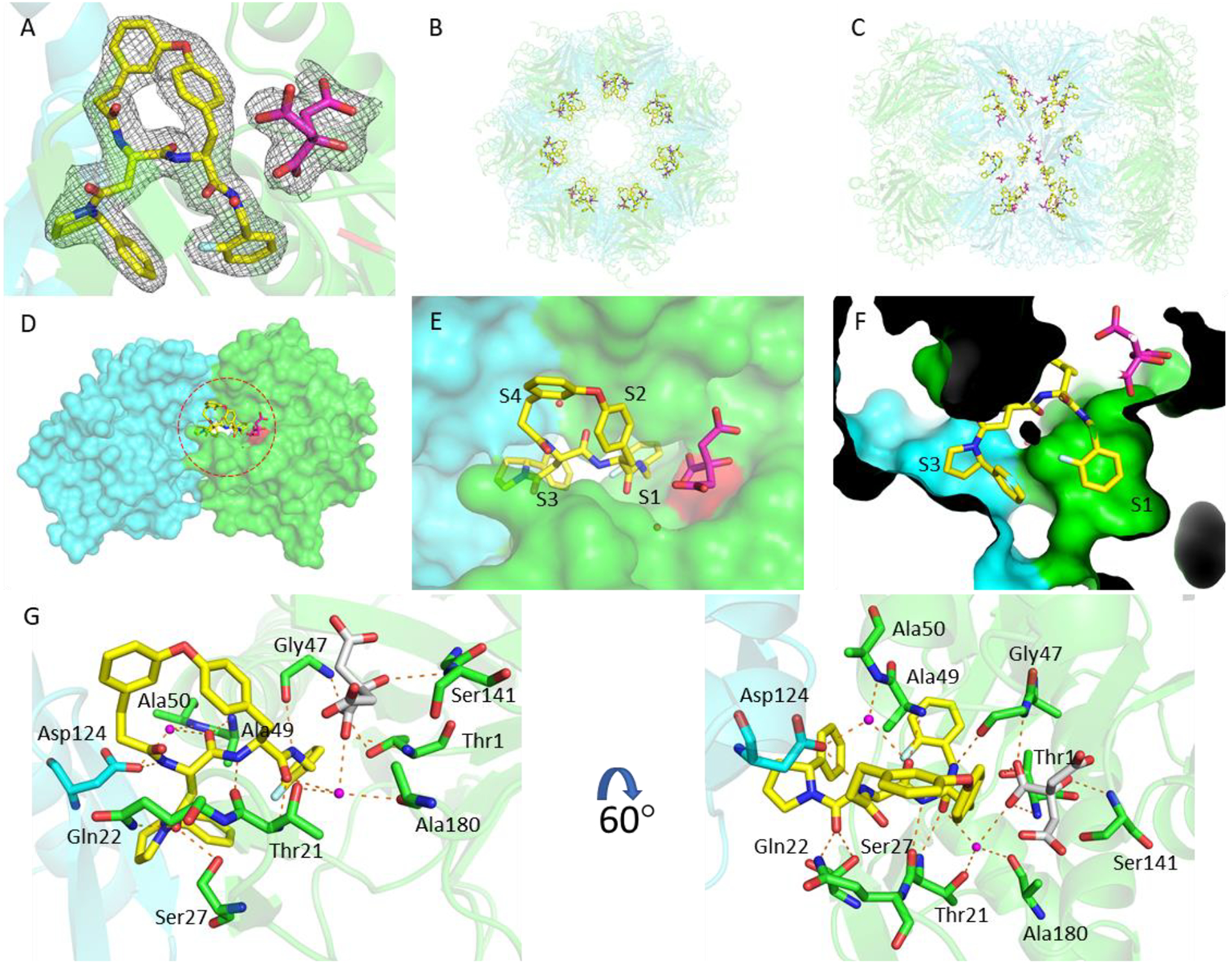
The binding mode of macrocycle 6 in the Mtb20SOG active site. A) The 2mFo–DFc electron density map is rendered at 1σ threshold and shown as gray meshes. Macrocycle 6 is in yellow and the citrate from solvent in magenta sticks. The two neighboring β-subunits are in green and cyan, respectively, and the catalytic residue Thr-1 is in red. B-C) A top and a side view of the Mtb20S structure, highlighting the 14 macrocycle 6 molecules in the active sites of the 14 catalytic β-subunits. D) A surface view of two neighboring β-subunits (green and cyan) as viewed from the interior chamber, demonstrating that the inhibitor in the substrate pocket of the green subunit also contacts the cyan subunit. E) A close-up view of the inhibitor-bound active site. F) P1 and P3 are inserted into the S1 and S3 binding pockets, respectively. G) The binding mode of macrocycle 6 in Mtb20SOG. Hydrogen bonds between macrocycle 6 and the β-subunits are shown as dashed orange lines. The two neighboring β-subunits are in green and cyan; water molecules are shown as magenta spheres; and citrate is in gray sticks.
The substrate specificity of a proteasome is mainly determined by the S1 and S3 binding pockets formed between two adjacent β-subunits. The P1 2-fluorobenzyl group and P3 2-phenylpyrrolindyl group are inserted into S1 and S3 pockets, respectively, with the 3,4’-tethered P2 and P4 phenyl groups facing the corresponding S2 and S4 sites (Figure 5D–F). No marked conformational changes were observed in the P1 and P3 pockets upon inhibitor binding, indicating that no conformational change in the Mtb20S catalytic pocket is required for the binding of the macrocyclic dipeptide inhibitor. macrocycle 6 binds to Mtb20S by forming a short antiparallel β-strand with the β-strands B3 and B6 of the β-subunits. The dipeptide backbone of the inhibitor forms hydrogen bonds with Thr-21 of B3, Gly-47 and Ala-49 of B6, and Asp-124 of the adjacent β-subunit (Figure 5G). The Mtb-specific residue Gln-22, along with Ser-27 (which is conserved in β5i), contributes an extra hydrogen bond with the carbonyl group of P3 2-phenylpyrrolidinyl-Asn, explaining the reduced inhibition activity of macrocycle 13. Macrocycle 6 does not interact directly with the catalytic Thr-1. However, a citrate molecule from the crystallization buffer was observed to interact with the OG1 of Thr-1 through a hydrogen bond. Two water molecules help to stabilize the inhibitor in the binding site: one water molecule is coordinated by Thr-21, Ala-180, the citrate, and the C-terminal carbonyl group of the dipeptide backbone. The other water molecule is coordinated by Ala-50, Asp-124 of the neighboring β-subunit, and the first carbonyl group of the dipeptide backbone. There is no hydrogen-bonding between the connected P2-P4 groups and the neighboring S2 and S4 sites.
To compare the binding modes of the 2-fluorobenzyl and naphthyl groups, we aligned the β-subunits of macrocycle 6-Mtb20SOG and DPLG-2-Mtb20SOG. The 2-fluorobenzyl group in macrocycle 6 and the naphthyl group in DPLG-2 had very similar binding modes in the S1 pockets (Figure S2B). In the P3 groups, incorporation of a 2-phenyl group (macrocycle 6, 9, and 10) or a 2-cyclopropyl group (11 and 12) on the pyrrolidinyl ring greatly increased the inhibitory potency against Mtb20S. The aligned macrocycle 6 and DPLG-2 showed similar binding modes of P3. The phenyl group of P3 in macrocycle 6 points toward the S1 site and the naphthyl group of P1 in DPLG-2 faces toward the S3 site. Thus, the bulky P3 group 2-phenylpyrrolindyl cannot coexist with the bulky naphthyl P1 group in the same inhibitor. From the activity and structural analyses, we observed the preference for bulky hydrophobic groups in the S1-S3 tunnel (Figure 5F). Although the P2 and P4 groups were linked to a cyclic form in this study, the binding mode of P2-P4 in macrocycle 6 still resembles that of DPLG-2, i.e., the P2 and P4 project into solvent and only partially interact with the S2 and S4 sites. As such, changing the linkers of P2 and P4 did not significantly change the inhibitory potency, as seen in macrocycles 6, 9 and 10.
Accumulation of Pup-GFP—a Novel Intracellular Reporter—by Macrocyclic Mtb20S Inhibitors.
One major obstacle to studying Mtb20S inhibition in Mtb is the dual function of Dop in the Pup–proteasome system.31, 32 Dop deamidates the C-terminal glutamine of Pup to convert it to glutamate, which is required for pupylation to take place. But Dop also removes Pup from pupylated proteins.31, 32 To test whether macrocycle 6 and other macrocyclic peptides can penetrate Mtb and engage the intrabacterial target, we used the Mtb strain MHD1441 (H37Rv WT + pSCK1). This strain produces Pup-GFP, in which Pup is linearly fused to a green fluorescent protein (GFP) at its C-terminus. Pup-GFP is therefore constitutively pupylated independently of Dop deamidation activity and cannot be depupylated by Dop, because it is linked to GFP by a peptide bond. Therefore, the inhibition of Mtb20S core particles in this Mtb strain causes Pup-GFP accumulation, which can be quantified by fluorescence (Ex495nm/Em510nm). Using this novel intracellular reporter strain, we screened 136 in-house synthesized macrocycles and identified 10 macrocycles whose addition to Mtb cultures resulted in the accumulation of Pup-GFP (Figure S3). Macrocycles 6, 9, and 10 further showed a dose-dependent increase of RFU (Figure 6A), strongly suggesting that these compounds penetrated the Mtb cell wall to inhibit intracellular proteasomal activity.
Figure 6.
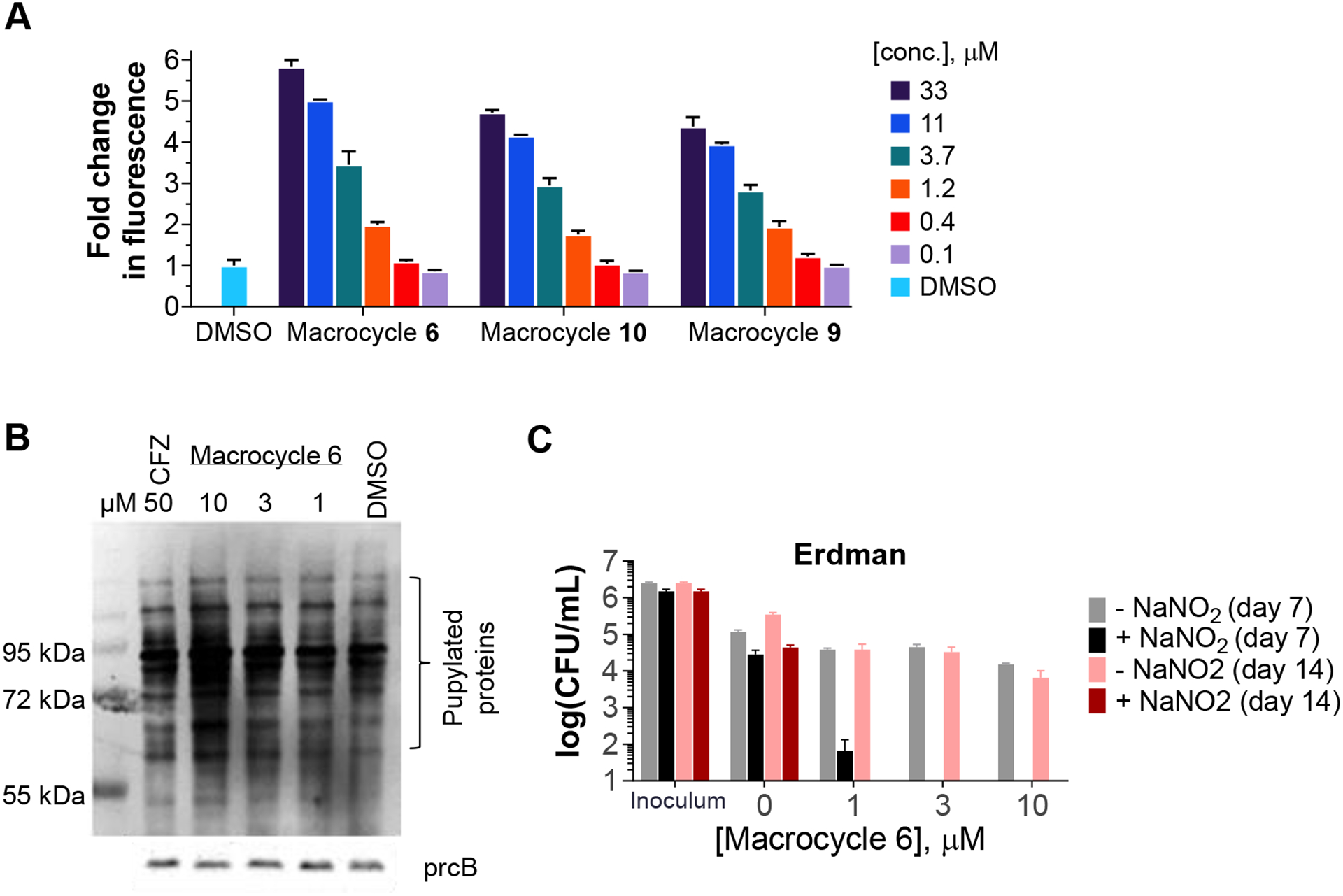
The macrocyclic inhibitors engage the intracellular target, and macrocycle 6 is mycobactericidal in the presence of nitric oxide. A) The Mtb strain MHD1441 (WT H37Rv producing Pup-GFP) was treated with macrocycles 6, 9, or 10 at the designated concentrations. Optical density at 580 nm and relative fluorescence units (RFU; at 495 nm excitation, 510 nm emission) were measured on day 4 post treatment. RFU values were normalized by optical density and fold changes were calculated by normalizing to the value of DMSO-treated MHD1441. Data shown are representative of at least 3 independent experiments. B) Mtb strains (Pup-Glu and dop–/–) were treated with macrocycle 6 at the indicated concentrations. Carfilzomib (CFZ) at 50 μM was used as a positive control. Pupylated proteins were probed with anti-Pup antibody and anti-PrcB was used as a loading control. C) Killing of Mtb Erdman in Sauton’s medium with 0.5 mM NaNO2 at pH 5.5, which generates NO.
Macrocycle 6 Induces Accumulation of Pupylated Proteins and Kills Mtb under Nitrosative Stress.
Host macrophages produce nitric oxide and other reactive nitrogen intermediates to control infection by M. tuberculosis. The Mtb proteasome serves as a defense for the pathogen against the nitrosative stress.2 We tested macrocycle 6 against non-replicating Mtb under nitrosative stress, as reported.16, 17 We found that macrocycle 6 showed marked, dose-dependent mycobactericidal activity (Figure 6C). To investigate whether the bacterial killing was due to inhibition of intracellular Mtb20S by macrocycle 6, we assayed the pupylome level in an Mtb strain in which proteins cannot be depupylated (CDC1551 dop::MycoMarT7 expressing pupGGE)33. This strain does not have the depupylase Dop but produces Pup-GGE, a C-terminal Pup variant that does not require Dop-mediated deamidation prior to substrate ligation by PafA.33 Without depupylation activity, the pupylome will only increase when Mtb20S core particles are inhibited. We treated the strain with macrocycle 6 at 1, 3, or 10 μM for 48 h. As shown in Figure 6B, macrocycle 6 dose-dependently promoted the accumulation of pupylated proteins, suggesting that macrocycle 6 penetrated Mtb and inhibited Mtb20S. Carfilzomib (CFZ), a proteasome inhibitor approved for treatment of multiple myeloma, also led to accumulation of pupylated proteins, at a concentration of 50 μM.
The Macrocycles Are Stable in Plasma and Well Tolerated by Human Cells.
The passive permeability of selected macrocycles (macrocycle 6, 9, and 10) was assessed using the parallel artificial membrane permeability assay (PAMPA). All three compounds showed high passive permeability (Table 3). Replacing the biphenyl ether of macrocycle 6 with the mono-phenyl tether of macrocycle 10 decreased the hydrophobicity (cLogP) from 6.0 to 4.9, which translated into dramatically improved solubility, while maintaining potency against Mtb20S. All three macrocycles were stable in human blood plasma, with ⩾ 86% of the parent remaining intact after 2 h incubation at 37 °C. We found no appreciable cytotoxicity toward human liver carcinoma cells (HepG2) for macrocycle 6, 9, and 10 at 30 μM, indicating a promising safety profile of this series of Mtb-selective proteasome inhibitors. However, macrocycles 6, 9, and 10 were evaluated in microsome stability assays, which showed all three compounds were rapidly metabolized by both human and mouse liver microsome (Clint > 768 μL/min/mg).
CONCLUSION
There have been only five broad classes of antibiotic targets: inhibition of synthesis of nucleic acids, proteins, cell walls and folate, and disruption of membranes. Following the success of proteasome inhibitor drugs for the treatment of hematopoietic malignancy, our recent work has validated a fifth class: inhibition of a protein degradation pathway.15 Our work has led to the discovery by others of pathogen-selective proteasome inhibitors that kill the protozoal agents of malaria, Chagas disease, and leishmaniasis.34, 35 Targeting another ancient, ATP-dependent compartmentalized protease, ClpP, in bacteria has also made tremendous progress.36–38
There is an intense effort to identify and target the Mtb genes that are essential for its survival in the host. Recent studies have shown that Mtb relies on several enzymatic pathways to survive host stresses while entering a non-replicative state. Among these is the Pup–proteasome system discovered over several years, beginning in 2003.2, 7–9, 11–13, 39, 40 In that system, Pup is deaminated by Dop, conjugated to a client protein by PafA (a Pup ligase)39–41 and fed by the Mtb20S activator Mpa39, 42, 43 into the Mtb proteasome for degradation.8, 15, 44 Although the Mtb proteasome is dispensable under standard growth conditions, it has been validated as a drug target both genetically and pharmacologically: the knock-out strain dies in the chronic phase of disease in mice. We have focused on targeting the Mtb proteasome because it is the most downstream of the five known enzymes in the system and we have succeeded in developing both irreversible and reversible Mtb 20S inhibitors that are species-selective and kill non-replicating Mtb.15, 16
In this study, we developed a series of macrocyclic Mtb proteasome inhibitors. A structure-guided study of their structure-activity relationships led to improved potency and species selectivity. We found that 1) targeting the S3 sub-pocket with a bulky phenyl group at the α-position of P3 pyrrolidine boosted both potency against Mtb20S and selectivity over human proteasomes; 2) a 16-membered alkyl ether linker, a 3,4’-biphenyl ether linker, and a monophenyl ether linker are accommodated in the P2 and P4 pockets, parts of which are exposed to solvent; 3) P5 amides of the rigid macrocyclic peptide were not well tolerated at the P5 pocket of Mtb20S.
The macrocycles 6, 9, and 10 have low-nanomolar potency against Mtb20S and >10,000-fold selectivity over human c-20S and i-20S. Macrocycle 6 is stable in plasma, non-toxic to human cells, and active against Mtb20S in a time-dependent manner. It enters Mtb and prevents protein degradation by Mtb20S, leading to the accumulation of pupylated proteins and bacterial death under nitrosative stress. Given these characteristics, further improvement on metabolic stability of the macrocycles identified in this report may lead to viable anti-TB drug candidates.
EXPERIMENTAL SECTION
The fluorogenic substrate Suc-LLVY-AMC, Z-LLE-AMC, and Z-VLR-AMC were from Boston Biochem (Cambridge, MA). Human immunoproteasomes (i-20S, isolated from peripheral blood mononuclear cells), human constitutive proteasomes (c-20S, isolated from red blood cells), and recombinant human PA28 activator alpha subunit (PA28α) were from Boston Biochem Inc. Mtb20SOG was expressed and purified as reported.27 Mouse and human liver microsomes were purchased from Sekisui XenoTech, LLC. (Kansas City, KS). Purity of the compounds was determined on a Waters Acquity™ UPLC / MS system equipped with a C18 column (100 × 2.1 mm, 1.7 μm), coupled with a PDA detector. The purity of all final compounds was > 95% except macrocycle 13. The purity of macrocycle 13 was 93%.
IC50 Determination.
The IC50 values of compounds against Mtb20SOG, β5i, β2i, β1i of i-20S and β5c, β2c, β1c of c-20S were determined from tests in a 96-well format as reported.16 One microliter of compound in a 3× series dilution in DMSO at concentrations from 100 μM to 0.0017 μM was spotted to the bottom of wells in a black 96-well plate. Reaction buffer (100 μL; 20 mM HEPES, 0.5 mM EDTA, pH 7.5) containing enzyme and substrate were dispensed into each well and the plate was then spun in a simple benchtop plate centrifuge at 1000 rpm for 1 min, followed by shaking on an orbital shaker for 1 min. The time course of hydrolysis in each well was plotted by recording the fluorescence of the product AMC (λex 360 nm and λem 460 nm) on a SpectraMax M5 plate reader over 1.5–2 h. The initial reaction velocity of each well was fit to a dose-dependent inhibition equation using PRISM to determine the IC50.
Suc-LLVY-AMC was used for Mtb20SOG activity at 50 μM, for c-20S β5c at 25 μM, and for i-20S β5i at 15 μM. Z-LLE-AMC was used for c-20S β1c and i-20S β1i at 50 μM. Z-VLR-AMC was used for c-20S β2c and i-20S β2i at 50 μM. Final concentrations of Mtb20S, β5i, and β5c were 2 nM, 0.4 nM, and 0.2 nM, respectively. Sodium dodecyl sulfate was used as an activator at 0.02% for human β1i, β1c, β5i, and β5c. PA28α was used as an activator at 6 nM for human β2i and β2c.
Protein Expression and Purification.
Cloning and expression of Mtb20SOG have been described.27 Briefly, a PACYCDuet-1 vector (Novagen, Madison, WI, USA) consisting of the Mtb prcAOG (encoding truncated-PrcA) and prcB genes was expressed in the BL21(DE3) strain of E. coli. Cells were grown at 37 °C to OD600 = 0.5–0.6 before being induced with 0.2 mM IPTG. The Mtb proteasome was expressed at 37 °C for 18 h to ensure full processing of β-subunits. Cells were harvested by centrifugation, resuspended in 10 mM potassium phosphate (pH 8.0), 10 mM imidazole, and 0.25 M NaCl, and lysed by passing through a Microfluidizer cell disruptor. The homogenate was clarified by spinning at 27,000 × g, and the supernatant was applied to a HiTrap-Ni column (GE Healthcare) pre-equilibrated with the lysis buffer. His-tagged proteins were eluted with a 10–300 mM imidazole gradient in 10 mM potassium phosphate (pH 8.0) and 0.25 M NaCl. Eluted fractions containing Mtb proteasomes were applied to a Hi-Trap Q column pre-equilibrated with 10 mM Tris (pH 8.0) and 50 mM NaCl and were eluted with a 50–500 mM NaCl gradient. Mtb20SOG was further purified with a Superdex 200 column (16 × 600 mm, GE Healthcare) in buffer containing 10 mM HEPES (pH 7.5), 1 mM dithiothreitol, and 0.15 M NaCl. For crystallization, the purified Mtb20SOG complex was concentrated to 10 mg/mL.
Kinetic Solubility.
2 μL 10 mM DMSO stock solution of test compounds were added to 98 μL PBS buffer at pH 6.8. After incubation for 24 h, the samples were filtered through filter plates to remove undissolved compound. The filtrates were then analyzed on an Acquity™ UPLC / MS system, equipped with a PDA detector, for solubility quantification. The kinetic solubility (in μg/mL) was calculated by dividing peak area of sample filtrate by peak area of standard (10 μM test compound in acetonitrile/water (1:1, v/v)) and multiplied by the target concentration (in μg/mL, 100 × MW / 1000 μg/mL).
Liver Microsomal Stability Assay.
Test compounds (1 μM) and the microsomes (0.2 mg protein/mL) were mixed in phosphate buffer at pH 7.4. The reactions were initiated by adding an NADPH generating system (a mixture of MgCl2, NADP+, glucose-6-phosphate, and glucose-6-phosphate dehydrogenase) to the mixtures in a final volume of 25 μL. The concentration of organic solvents was 1% (0.01% DMSO and 0.99% acetonitrile) in the final incubation system. The mixtures were incubated at 37 °C for 0, 15, 30 min in duplicates. The reactions were stopped at each time point by adding 75 μL acetonitrile containing internal standard. For the T0 min samples, 75 μL acetonitrile containing internal standard was added to mixtures before adding an NADPH generating system. After the samples were mixed and centrifuged, the supernatants were analyzed by LC/MS-MS. The intrinsic clearance (Clint, in μL/min/mg) was calculated by dividing rate constant (k, in min−1) by microsomal protein concentration (0.2 mg/mL). The rate constant (k, in min−1) calculated based on the exponential function of the percentage of compound remaining vs time curve on the assumption that it is the first order elimination. Flutamide was included as the positive control compound in each experiment.
Molecular Docking.
Molecular docking was performed in the Maestro (version 12.2) workspace using modules (Protein Preparation Wizard, LigPrep, and Glide) in the Schrödinger software package (release 2019-4). The X-ray crystal structures of Mtb20SOG (PDB ID 5TS0) were retrieved from the Protein Data Bank. The protein structures were prepared using the Protein Preparation Wizard by assigning bond orders, adding hydrogens, creating disulfide bonds, converting selenomethionines into methionines, and filling in missing side chains. Water molecules in the entire system were removed. PROPKA was used to assign H-bonds at pH 7.0. Restrained minimization was then performed by using OPLS3 force field. The docking grid centered on the co-crystallized ligand PKS2208 was created using the Receptor Grid Generation using default settings without any constraints. Ligands were built using the 2D Sketcher and prepared using LigPrep by generating ionization states at pH 7.0 ± 2 and tautomers to generate suitable 3D conformations for docking. The prepared ligands were docked using Glide in standard precision (SP) mode with flexible ligand sampling. The docked results were visualized and analyzed in Maestro. The best binding poses were chosen according to the glide docking scores.
Crystallization and Structure Determination.
Mtb20SOG was crystallized at 4 °C by the hanging-drop vapor diffusion method using 60 mM sodium citrate (pH 6.2) and 15% PEG-3350 as precipitant. To obtain the inhibitor–proteasome complex, Mtb20SOG crystals were transferred stepwise to the cryo-protectant containing 60 mM sodium citrate (pH 6.2), 15% PEG-3350, 35% dimethylformamide, and 1 mM macrocycle 6 inhibitor. After incubation at 4 °C for 20 h, crystals were flash-frozen in liquid nitrogen. Diffraction data were collected at the Life Sciences Collaborative Access Team (LS-CAT) beamline of APS and were processed with Mosflm. The space groups of inhibitor-soaked Mtb20SOG crystals were all P21. Molecular replacement was carried out to solve the macrocycle 6-bound Mtb20SOG structures using the program PHASER.45 The DPLG2-Mtb20SOG complex structure (PDB ID 5TRG) was used as initial search model. After building the model of macrocycle 6 in COOT,46 refinements were performed using Phenix-refine,47 and the refinement and modeling statistics are provided in Table S2.
Impact of Mtb20S Inhibitors on Pup-GFP in MHD1441.
Two microliters of Mtb20S inhibitor solution in DMSO were added to the inner wells of a 96-well black wall/clear bottom tissue culture plate. MHD1441 (H37Rv WT + pSCK1) was grown to OD 1.6 and adjusted to OD 0.05 with 7H9 medium. We dispensed 200 μL of MHD1441 in 7H9 into each well and incubated at 37 °C for 4 d. Optical density at 580 nm (OD580 nm) and relative fluorescence unit (RFU; excitation, 495nm and emission, 510 nm) values were measured on day 4. RFU values were then normalized by OD. We screened 255 compounds (phenylimidazoles, benzimidazoles and macrocyclic peptides) at final concentrations of 100 μM, 33 μM, and 11 μM, respectively. The dose-dependent impacts on Pup-GFP in Mtb of macrocycles 6, 9, and 10 were measured at final concentrations of 33, 11, 3.7, 1.2, 0.4, and 0.1 μM.
Human Plasma Stability Assay.
To 100 uL of human plasma (H3667, Sigma-Aldrich) was added 1μL of 10 mM test compound (6, 9, and 10). The mixtures were incubated at 37 °C for 0, 60, 120 min. The reactions were stopped at each time point by adding 200 μL ice-cold methanol containing 50 μM internal standard. For the T0 min samples, 200 μL ice-cold methanol containing internal standard was added to human plasma prior to addition of test compound. Precipitated proteins were pelleted by centrifuging at 13500 rpm for 20 min at 4 °C. The supernatants were collected and analyzed on an Acquity™ UPLC / MS sysem, coupled with a PDA detector. Column chromatography was carried out on a C18 Column, 130Å, 1.7 μm, 2.1 mm × 100 mm. The percentage of compounds remaining at each time points relative to starting concentration were calculated using integrated UV peak areas normalized to the internal control. Procaine was included as the positive control compound in each experiment.
Supplementary Material
ACKNOWLEDGMENTS
X-ray diffraction data were collected at the Life Sciences Collaborative Access Team (LS-CAT) beamline of the Advanced Photon Source (APS). LS-CAT was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (grant no. 085P1000817). The APS is a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation. We thank David Nadziejka (Van Andel Institute) for technical editing of this manuscript. The authors gratefully acknowledge the support to the project (not to the non-TDI labs) generously provided by the Tri-Institutional Therapeutics Discovery Institute (TDI), a 501(c)(3) organization. TDI receives financial support from Takeda Pharmaceutical Company, TDI’s parent institutes (Memorial Sloan Kettering Cancer Center, The Rockefeller University, and Weill Cornell Medicine), from a generous contribution from Mr. Lewis Sanders, and from other philanthropic sources.
Funding Sources
This work is supported by NIH grants R01 AI143714 (to G.L.), R21 AI144552 (to G.L.), and R01 AI070285 (to H.L.), the Tri-Institutional Therapeutics Discovery Institute and Weill Cornell Medicine Matching Fund (to G.L.), and by the Milstein Program in Translational Medicine. S.C.K. is supported in part by a Public Health Service Institutional Research Training Award T32 AI007180.
ABBREVIATIONS
- Mtb20S
M. tuberculosis 20S proteasome
- i-20S
immunoproteasome
- c-20S
constitutive proteasome
- three-letter codes
- amino acids
Footnotes
Supporting Information, including Figures S1, S2, and S3, Table S1 and S2, synthesis routes, and compound spectra, is available free of charge via the Internet at http://pubs.acs.org.
- Molecular formula strings (CSV)
- Synthesis and NMR spectra of final compounds and key intermediates and experimental procedures for the biological assays (PDF).
- Docking pose of macrocycle 5 at Mtb20SOG (PDB ID 5TS0)
- Overlay of macrocycle 5 to parent PKS-2208 at Mtb20SOG (PDB ID 5TS0)
Accession Codes
Atomic coordinates have been deposited in RCSB with PDB ID 6WNK.
The authors declare no competing financial interest.
REFERENCES
- 1.Russell DG; Barry CE; Flynn JL Tuberculosis: what we don’t know can, and does, hurt us. Science 2010, 328, 852–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Darwin KH; Ehrt S; Gutierrez-Ramos J-C; Weich N; Nathan CF The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 2003, 302, 1963–1966. [DOI] [PubMed] [Google Scholar]
- 3.Gandotra S; Schnappinger D; Monteleone M; Hillen W; Ehrt S In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat. Med 2007, 13, 1515–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin G; Tsu C; Dick L; Zhou XK; Nathan C Distinct specificities of Mycobacterium tuberculosis and mammalian proteasomes for N-acetyl tripeptide substrates. J. Biol. Chem 2008, 283, 34423–34431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gold B; Nathan C Targeting phenotypically tolerant Mycobacterium tuberculosis. Tuberculosis and the Tubercle Bacillus 2017, pp. 317–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nathan C; Gold B; Lin G; Stegman M; de Carvalho LPS; Vandal O; Venugopal A; Bryk R A philosophy of anti-infectives as a guide in the search for new drugs for tuberculosis. Tuberculosis 2008, 88, S25–S33. [DOI] [PubMed] [Google Scholar]
- 7.Darwin KH; Lin G; Chen Z; Li H; Nathan CF Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol. Microbiol 2005, 55, 561–571. [DOI] [PubMed] [Google Scholar]
- 8.Lin G; Hu G; Tsu C; Kunes YZ; Li H; Dick L; Parsons T; Li P; Chen Z; Zwickl P Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol. Microbiol 2006, 59, 1405–1416. [DOI] [PubMed] [Google Scholar]
- 9.Pearce MJ; Mintseris J; Ferreyra J; Gygi SP; Darwin KH Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 2008, 322, 1104–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imkamp F; Rosenberger T; Striebel F; Keller PM; Amstutz B; Sander P; Weber-Ban E Deletion of dop in Mycobacterium smegmatis abolishes pupylation of protein substrates in vivo. Mol. Microbiol 2010, 75, 744–754. [DOI] [PubMed] [Google Scholar]
- 11.Burns KE; Pearce MJ; Darwin KH Prokaryotic ubiquitin-like protein provides a two-part degron to Mycobacterium proteasome substrates. J. Bacteriol 2010, 192, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu K; Jastrab JB; Zhang S; Kovach A; Zhao G; Darwin KH; Li H Proteasome substrate capture and gate opening by the accessory factor PafE from Mycobacterium tuberculosis. J. Biol. Chem 2018, 293, 4713–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai L; Hu K; Wang T; Jastrab JB; Darwin KH; Li H Structural analysis of the dodecameric proteasome activator PafE in Mycobacterium tuberculosis. Proc. Nat. Acad. Sci 2016, 113, E1983–E1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gandotra S; Lebron MB; Ehrt S The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLOS Pathog 2010, 6, E1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin G; Li D; De Carvalho LPS; Deng H; Tao H; Vogt G; Wu K; Schneider J; Chidawanyika T; Warren JD Inhibitors selective for mycobacterial versus human proteasomes. Nature 2009, 461, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin G; Chidawanyika T; Tsu C; Warrier T; Vaubourgeix J; Blackburn C; Gigstad K; Sintchak M; Dick L; Nathan CN, C-Capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes: role of S3 and S1 binding pockets. J. Am. Chem. Soc 2013, 135, 9968–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhan W; Hsu H-C; Morgan T; Ouellette T; Burns-Huang K; Hara R; Wright AG; Imaeda T; Okamoto R; Sato K Selective phenylimidazole-based inhibitors of the Mycobacterium tuberculosis proteasome. J. Med. Chem 2019, 62, 9246–9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cummings MD; Sekharan S Structure-based macrocycle design in small-molecule drug discovery and simple metrics to identify opportunities for macrocyclization of small-molecule ligands. J. Med. Chem 2019, 62, 6843–6853. [DOI] [PubMed] [Google Scholar]
- 19.Driggers EM; Hale SP; Lee J; Terrett NK The exploration of macrocycles for drug discovery—an underexploited structural class. Nat. Rev. Drug Discov 2008, 7, 608–624. [DOI] [PubMed] [Google Scholar]
- 20.Berthelot A; Piguel S; Le Dour G; Vidal J Synthesis of macrocyclic peptide analogues of proteasome inhibitor TMC-95A. J. Org. Chem 2003, 68, 9835–9838. [DOI] [PubMed] [Google Scholar]
- 21.Kaiser M; Milbradt AG; Siciliano C; Assfalg-Machleidt I; Machleidt W; Groll M; Renner C; Moroder L TMC-95A analogues with endocyclic biphenyl ether group as proteasome inhibitors. Chem. Biodivers 2004, 1, 161–173. [DOI] [PubMed] [Google Scholar]
- 22.Groll M; Götz M; Kaiser M; Weyher E; Moroder L TMC-95-based inhibitor design provides evidence for the catalytic versatility of the proteasome. Chem. Biol 2006, 13, 607–614. [DOI] [PubMed] [Google Scholar]
- 23.Koguchi Y; Kohno J; Nishio M; Takahashi K; Okuda T; Ohnuki T; Komatsubara S TMC-95A, B, C, and D, novel proteasome inhibitors produced by Apiospora montagnei Sacc. TC 1093 taxonomy, production, isolation, and biological activities. J. Antibiot 2000, 53, 105–109. [DOI] [PubMed] [Google Scholar]
- 24.Li H; Tsu C; Blackburn C; Li G; Hales P; Dick L; Bogyo M Identification of potent and selective non-covalent inhibitors of the Plasmodium falciparum proteasome. J. Am. Chem. Soc 2014, 136, 13562–13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhan W; Zhang H; Ginn J; Leung A; Liu YJ; Michino M; Toita A; Wong TT; Imaeda T; Hara R; Yukawa T; Chelebieva S; Tumwebaze PK; Lafuente-Monasterio MJ; Martinez-Martinez MS; Vendome J; Beuming T; Sato K; Aso K; Rosenthal PJ; Cooper RA; Meinke PT; Nathan CF; Kirkman LA; Lin G Development of a highly selective Plasmodium falciparum proteasome inhibitor with anti-malaria activity in humanized mice. Angew. Chem. Int. Ed 2021. DOI: 10.1002/anie.202015845, accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neilsen PM; Pehere AD; Pishas KI; Callen DF; Abell AD New 26S proteasome inhibitors with high selectivity for chymotrypsin-like activity and p53-dependent cytotoxicity. ACS Chem. Biol 2013, 8, 353–359. [DOI] [PubMed] [Google Scholar]
- 27.Hsu H-C; Singh PK; Fan H; Wang R; Sukenick G; Nathan C; Lin G; Li H Structural basis for the species-selective binding of N, C-capped dipeptides to the Mycobacterium tuberculosis proteasome. Biochemistry 2017, 56, 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raymer B; Bhattacharya SK Lead-like Drugs: A Perspective: Miniperspective. J. Med. Chem 2018, 61, 10375–10384. [DOI] [PubMed] [Google Scholar]
- 29.Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 30.Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 31.Burns KE; Cerda-Maira FA; Wang T; Li H; Bishai WR; Darwin KH “Depupylation” of prokaryotic ubiquitin-like protein from mycobacterial proteasome substrates. Mol. Cell 2010, 39, 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imkamp F; Striebel F; Sutter M; Özcelik D; Zimmermann N; Sander P; Weber-Ban E Dop functions as a depupylase in the prokaryotic ubiquitin-like modification pathway. EMBO Rep 2010, 11, 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerda-Maira FA; Pearce MJ; Fuortes M; Bishai WR; Hubbard SR; Darwin KH Molecular analysis of the prokaryotic ubiquitin-like protein (Pup) conjugation pathway in Mycobacterium tuberculosis. Mol. Microbiol 2010, 77, 1123–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khare S; Nagle AS; Biggart A; Lai YH; Liang F; Davis LC; Barnes SW; Mathison CJ; Myburgh E; Gao M-Y Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H; O’Donoghue AJ; van der Linden WA; Xie SC; Yoo E; Foe IT; Tilley L; Craik CS; da Fonseca PC; Bogyo M Structure-and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gavrish E; Sit CS; Cao S; Kandror O; Spoering A; Peoples A; Ling L; Fetterman A; Hughes D; Bissell A Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem. Biol 2014, 21, 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitz KR; Handy EL; Compton CL; Gupta S; Bishai WR; Sauer RT; Sello JK Acyldepsipeptide antibiotics and a bioactive fragment thereof differentially perturb Mycobacterium tuberculosis ClpXP1P2 activity in vitro. ACS Chem. Biol 2020. DOI: 10.1021/acschembio.9b00454, accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conlon BP; Nakayasu ES; Fleck LE; LaFleur MD; Isabella VM; Coleman K; Leonard SN; Smith RD; Adkins JN; Lewis K Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Striebel F; Hunkeler M; Summer H; Weber-Ban E The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup’s N-terminus. EMBO J 2010, 29, 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Striebel F; Imkamp F; Sutter M; Steiner M; Mamedov A; Weber-Ban E Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat. Struct. Mol. Biol 2009, 16, 647–651. [DOI] [PubMed] [Google Scholar]
- 41.Sutter M; Damberger FF; Imkamp F; Allain FHT; Weber-Ban E Prokaryotic ubiquitin-like protein (Pup) is coupled to substrates via the side chain of its C-terminal glutamate. J. Am. Chem. Soc 2010, 132, 5610–5612. [DOI] [PubMed] [Google Scholar]
- 42.Wang T; Darwin KH; Li H Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat. Struct. Mol. Biol 2010, 17, 1352–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang T; Li H; Lin G; Tang C; Li D; Nathan C; Darwin KH; Li H Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure 2009, 17, 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu G; Lin G; Wang M; Dick L; Xu RM; Nathan C; Li H Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol. Microbiol 2006, 59, 1417–1428. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 47.Afonine PV; Grosse-Kunstleve RW; Echols N; Headd JJ; Moriarty NW; Mustyakimov M; Terwilliger TC; Urzhumtsev A; Zwart PH; Adams PD Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr., Sect. D: Biol. Crystallogr 2012, 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.