

Generative deep learning and miniaturized bench-top synthesis were combined to automate the design of novel LXR agonists.
Abstract
Automating the molecular design-make-test-analyze cycle accelerates hit and lead finding for drug discovery. Using deep learning for molecular design and a microfluidics platform for on-chip chemical synthesis, liver X receptor (LXR) agonists were generated from scratch. The computational pipeline was tuned to explore the chemical space of known LXRα agonists and generate novel molecular candidates. To ensure compatibility with automated on-chip synthesis, the chemical space was confined to the virtual products obtainable from 17 one-step reactions. Twenty-five de novo designs were successfully synthesized in flow. In vitro screening of the crude reaction products revealed 17 (68%) hits, with up to 60-fold LXR activation. The batch resynthesis, purification, and retesting of 14 of these compounds confirmed that 12 of them were potent LXR agonists. These results support the suitability of the proposed design-make-test-analyze framework as a blueprint for automated drug design with artificial intelligence and miniaturized bench-top synthesis.
INTRODUCTION
Rapid iteration of the molecular design-make-test-analyze (DMTA) cycle has the potential for making “better decisions faster” (1, 2), with numerous applications in drug discovery and related fields (3, 4). Recent advances in chemical reaction monitoring and optimization, computing hardware, and algorithms have boosted the automation of several parts of the drug discovery process, such as robotic synthesis (5–8), computational molecular design (9–11), and synthesis planning (12–15). Standardized experimental procedures with robotic assistance increase the reproducibility of results, reduce errors, and decrease the consumption of materials, thereby contributing to “green chemistry” (16). Furthermore, reasoning with machine intelligence supports the discovery of novel drug-like molecules by freeing the molecular design and optimization process from personal biases (1). Pioneering studies combined microfluidics platforms with machine intelligence for synthesis planning (7, 17) as well as automated hit finding and hit-to-lead optimization in combinatorial libraries (8, 18). Computer-assisted molecular design is a critical element of this automation process. Molecular structure generation is often performed in a “rule-based” manner, i.e., by using algorithms for molecule assembly from predefined virtual reactions and reactants (19). Generative deep learning models extend the capabilities of rule-based de novo molecule generators by sampling new molecules from a latent chemical space representation (20–23), without the need for human-crafted molecule construction rules. Recently, the prospective applicability of “rule-free” generative deep learning for de novo molecular design has been demonstrated in combination with batch synthesis (9, 10, 24–26).
This study aims to pioneer the integration of generative molecular design with automated synthesis. Here, a recently published generative deep learning model (27) was adapted to generate compounds that are at the same time (i) bioactive on a selected macromolecular target and (ii) synthesizable on a bench-top microfluidic synthesis platform (16, 28). We challenged this automated DMTA pipeline to design liver X receptor (LXR) agonists from scratch, with minimal human interference. LXRs have emerged as promising drug targets because of their regulatory role in lipid metabolism and inflammation, thereby causing increased reverse cholesterol transport and reduction of atherosclerosis (29–32). With 28 molecules successfully synthesized and 12 fully validated for LXR activation in vitro, this present study pioneers the integration of generative artificial intelligence and automated synthesis by designing and experimentally testing the highest number of molecules reported thus far. The proposed modular framework has the potential to accelerate the DMTA cycle, thereby addressing one of the main bottlenecks of the preclinical drug discovery process (33).
RESULTS AND DISCUSSION
Modular DMTA platform
The automated molecular design pipeline was composed of three modules (Fig. 1):
Fig. 1. Schematic of the modular molecular design pipeline.
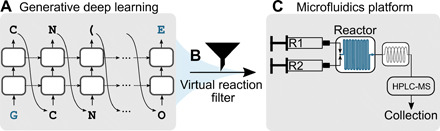
(A) A generative deep learning model (27) based on a long-short term memory network (34) was used to generate putative liver X receptor α agonists. The two-step network training procedure first trained the model on 656,070 compounds predicted as compatible with the microfluidics system and then fine-tuned this pretrained model with 40 known LXRα agonists. (B) The de novo generated molecules were filtered on the basis of their predicted synthesis route, using a set of 17 reactions specified in the SMARTS notation (“virtual reaction filter”). Molecular building blocks for synthesis were automatically retrieved from a commercial supplier catalog. (C) A total of 41 de novo designs were retained for synthesis on a microfluidics platform, which contained two syringes for the handling of reagents R1 and R2, a Cetoni Qmix element equipped with a Dean Flow microfluidic reactor chip, and a Rheodyne MRA splitter for automated sample transfer to an HPLC-MS system.
1) Module 1: A generative deep learning model (27) based on a recurrent neural network with long-short term memory (LSTM) cells (34). LSTM models were used for the design of new molecules represented as simplified molecular input line entry systems (SMILES) (35) strings (20, 21, 36). This LSTM-based “chemical language model” served as the de novo structure generator (Fig. 1A).
2) Module 2: A virtual reaction filter that captured 17 one-step reactions that were compatible with the microfluidics system (module 3). These reactions were encoded as SMILES arbitrary target specification (SMARTS) strings (37) (table S1). This filtering module selected those generated molecules that were synthetically compatible within the microfluidics platform (Fig. 1B).
3) Module 3: A microfluidics platform designed to minimize the amount of manual labor needed to optimize reaction conditions and synthesize focused compound libraries via one-step reactions. This compact bench-top system combined the automated retrieval of required reagents with the optimization of reaction conditions, online reaction product monitoring by high-performance liquid chromatography–mass spectrometry (HPLC-MS), and the collection of the reaction mixtures (Fig. 1C).
De novo molecular design with artificial intelligence
The deep learning model (Fig. 1A) was pretrained on the SMILES strings of 656,070 commercially available molecules from four compound vendors (for details, see Materials and Methods), which were predicted by the virtual reaction system to be suitable for the on-chip synthesis within our microfluidics platform. This pretraining step enabled the model to capture the syntax of the SMILES strings. After pretraining, 83 ± 2% of the generated SMILES strings were unique and chemically valid (average of three repetitions, 3000 SMILES strings sampled at each repetition) and corresponded to novel molecules that were not included in the pretraining set. Compared to model pretraining performed in previous studies using bioactive molecules from the ChEMBL database (9, 27, 38), the approach used here resulted in a significantly higher proportion of de novo molecules that were considered synthesizable in flow (P < 0.001, Kruskal-Wallis test; Fig. 2A). This result highlights the capability of the deep learning model to implicitly learn the desired molecular features (here, compound synthesizability as defined by the virtual reaction filter), without the need for explicit, rule-based design constraints.
Fig. 2. Automating de novo design with deep learning.
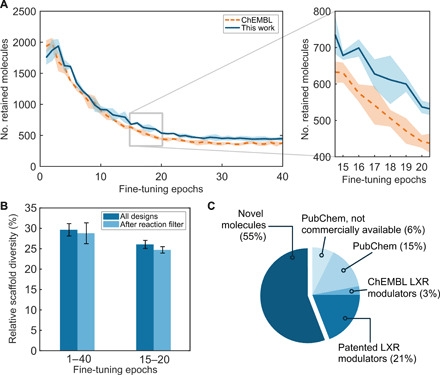
(A) Number of de novo designs retained by the virtual reaction filter depending on the pretraining (mean and SD over three replicates and 3000 sampled SMILES strings each). Compared to previous studies using bioactive molecules from ChEMBL (dashed lines) (9, 27, 38), this pretraining strategy (solid line) led to a larger number of compounds retained by the virtual reaction tool (P < 0.001, Kruskal-Wallis test), with up to 255 ± 97 more designs retained in each fine-tuning epoch. The epochs chosen for sampling are highlighted (epochs 15 to 20, gray rectangle). (B) Relative scaffold diversity (i.e., unique scaffolds/total number of scaffolds) of the de novo designs before and after applying the virtual reaction filter. No statistically significant difference in scaffold diversity was observed (Wilcoxon test, α = 0.05). (C) Analysis of 67 de novo designs retained for potential synthesis: 14 compounds were patented LXR modulators annotated in SureChEMBL or Reaxys (22%); 15 compounds existed in PubChem, of which 10 compounds are annotated as commercially available (15% of the total); 4 (6%) compounds lack vendor information; and 2 compounds (3%) were known LXR modulators annotated in ChEMBL27 [median inhibitory concentration (IC50)/EC50 ≤ 2 μM]. Thirty-seven compounds (55%) were not found in either PubChem, ChEMBL27, SciFinder, SureChEMBL, or Reaxys databases.
After pretraining, the model was fine-tuned with the SMILES strings of 40 LXRα agonists [median effective concentration (EC50) < 0.5 μM; tables S2 and S3] that were not included in the pretraining set. This fine-tuning step allowed us to focus the model on features that are shared by a chosen set of compounds (20, 39); therefore, it was used to bias the generation of new SMILES strings toward the chemical space of known LXRα agonists. Both the number of fine-tuning epochs for molecule design and the sampling temperature for SMILES string generation were automatically determined to optimize three parameters simultaneously (fig. S1), namely, the (i) predicted LXR activity (40), (ii) scaffold diversity (41), and (iii) pharmacophore similarity to the fine-tuning compounds (42). From the fine-tuned model (epochs 15 to 20), 3000 SMILES strings were sampled per epoch. Only those generated molecules that were not included in the pretraining and fine-tuning sets were retained, resulting in a total of 3626 de novo designs.
The retrosynthetic route of each generated molecule was predicted using the chosen set of 17 virtual reaction schemes (Fig. 1B). The compounds that could be decomposed into suitable reactants were kept (1911 designs). Notably, with a relative scaffold diversity equal to 23% both before and after reaction filtering, this filtering step did not markedly alter the scaffold diversity (41) of the designs. In general, no statistically significant decrease in the relative scaffold diversity due to the application of the virtual reaction filter was observed (α = 0.05, Wilcoxon test; Fig. 2B), rendering this method suitable for the design of reaction-focused compound libraries. Whenever the predicted reaction product was compatible with the microfluidics system (i.e., potentially synthesizable following 1 of the 17 selected reaction schemes), the predicted reactants were automatically retrieved from the Sigma-Aldrich catalog extracted from PubChem (43) (27 February 2019). For 67 designs, all of the required reactants were available.
A novelty check of the remaining 67 molecules was performed in PubChem (43), ChEMBL27 (44), SciFinder (45), SureChEMBL (46), and Reaxys compound databases (47). Of the 67 designs, 17 molecular structures corresponded to patented or otherwise known LXR agonists, with EC50 values ranging from 0.2 to 2 μM (Fig. 2C). This result indicated that the deep learning model correctly captured the relevant molecular features for LXR binding and activation. For the remaining 51 de novo designs (table S4), no information on bioactivity for LXR was available. Of this compound set, 37 molecules were novel, 10 compounds were commercially available, and 4 were described in the PubChem database but were unavailable for purchase (Fig. 2C). Overall, 41 compounds were selected for synthesis and 3 were purchased, while the remaining compounds were discarded because of unavailability or the high price of the respective building blocks.
Microfluidics-assisted synthesis “on-chip” and first-pass screening
The 41 selected de novo molecules were synthesized in flow using the computationally suggested reactions. Of these compounds, 21 were predicted to be synthesizable by sulfonamide formation, 19 by amide bond formation, and compound 21 by ester bond formation (table S5). These predictions were in agreement with the distribution of the pretraining set, in which these three reactions were the most frequently predicted ones (98% of the molecules; table S6). On the basis of the respective HPLC-MS mass peaks, a total of 25 compounds were successfully synthesized on the microfluidics platform (1 to 25; Fig. 3A and fig. S2 to S26), corresponding to a 61% success rate. Compounds 26 to 28 were purchased.
Fig. 3. Compound structures and first-pass in vitro screening results.
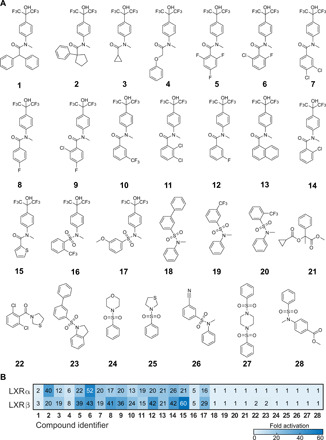
(A) Compounds 1 to 25 were synthesized in flow, and compounds 26 to 28 were purchased. (B) LXRα and LXRβ activation by compounds 1 to 28, as determined by the hybrid Gal4 reporter gene assays on the crude reaction products (test concentration ~10 μM, n = 2 with two technical replicates each). The numbers and color intensity indicate the fold activation of LXRα and LXRβ by each compound.
Compounds 1 to 28 were subjected to preliminary testing for LXRα and LXRβ activation in hybrid Gal4 reporter gene assays with human embryonic kidney (HEK) 293T cells (48). This assay relies on chimeric transcription factors composed of the respective human nuclear receptor ligand-binding domain and the DNA-binding domain of the yeast protein Gal4. Gal4-responsive firefly luciferase served as the reporter gene, and a constitutively expressed Renilla luciferase was used for normalization and to monitor the toxicity of the test compounds. As a cellular test system, this assay also captured the cell penetration and cytotoxicity of the test compounds. Crude reaction mixtures (49, 50) of all 28 test compounds were analyzed at a single concentration for LXRα and LXRβ activation, with two independent biological duplicates (Fig. 2B). The test compound concentration was roughly adjusted to 10 μM based on the HPLC traces of the samples.
Reaction mixtures of compounds 1 to 17 displayed ≥3-fold LXR activation in this preliminary screening, potentially corresponding to up to 68% actives among the 25 synthesized molecules. “Fold activation” refers to the fold LXR-induced reporter activity compared to untreated [dimethyl sulfoxide (DMSO)] cells. The reference LXR agonist T0901317 (51) achieved 81 ± 3–fold activation of LXRα and 129 ± 7–fold activation of LXRβ in this assay (at 1 μM). Compounds 6 (52-fold LXRα activation) and 15 (60-fold LXRβ activation) exhibited the strongest response in the primary screening. All compounds showing more than twofold LXR activation had a hexafluoro-2-phenyl-isopropanoyl moiety, suggesting particular relevance of this molecular feature for the observed bioactivity. This feature was also present in 12 of the fine-tuning compounds (29%), with an additional 4 (10%) and 8 (20%) fine-tuning compounds having a hexafluoro-2-aryl-isopropanoyl moiety and an aryl-trifluoromethyl motif, respectively. All compounds showing more than 10-fold LXRα activation in the preliminary screening were selected for full characterization of the dose-response curve. Compound 7 was excluded because of its cytotoxicity. Compound 1 (twofold LXRα activation) was included in the follow-up study because of its novel atomic scaffold (41), which is not present in any molecule annotated for LXRs in ChEMBL27 or in a repository for nuclear receptor bioactivity (52).
Bioactivity determination
The selected 14 compounds were prepared in-batch (scheme S1), purified, and fully characterized on LXRα and LXRβ (Table 1). Of these compounds, only compounds 2 and 3 were not confirmed to be active in the follow-up screening, suggesting that some other components in the crude reaction product mixture had activated LXRs in the primary screening. This finding indicates that the hexafluoro-2-phenyl-isopropanoyl moiety is not sufficient for LXR activation, despite its ubiquity among the de novo designs. The potencies of the remaining 12 LXR modulators were in the range of EC50 = 0.18 to 4.5 μM for LXRα and EC50 = 0.34 to 4.0 μM for LXRβ. In agreement with the primary screening data, compound 6 displayed the highest potency on LXRα, with an EC50 of 0.183 ± 0.006 μM and a 32-fold maximum activation. Compound 15 was confirmed as the most potent LXRβ agonist, with an EC50 of 0.34 ± 0.02 μM and 38-fold maximum receptor activation.
Table 1. LXRα and LXRβ modulatory potency of compounds selected for phase 2.
Potency was determined in cellular Gal4-based hybrid reporter gene assays. EC50 values and fold activation are reported as mean ± SE (n = 3). n.d., not determined.
| ID | LXRα | LXRβ | Selectivity for LXRα | ||
| EC50 (μM) | Fold activation | EC50 (μM) | Fold activation | (LXRβ/LXRα) | |
| 1 | 4.5 ± 0.1 | 20.5 ± 0.2 | >10 | n.d. | >2.2 |
| 2 | >10 | n.d. | >10 | n.d. | n.d. |
| 3 | >10 | n.d. | >10 | n.d. | n.d. |
| 5 | 0.26 ± 0.01 | 18.1 ± 0.1 | 1.30 ± 0.03 | 25.3 ± 0.3 | 5.0 ± 0.2 |
| 6 | 0.183 ± 0.006 | 32.4 ± 0.2 | 0.40 ± 0.01 | 23.3 ± 0.1 | 2.19 ± 0.09 |
| 8 | 1.05 ± 0.01 | 15.2 ± 0.1 | 1.72 ± 0.04 | 24.7 ± 0.3 | 1.64 ± 0.04 |
| 9 | 1.68 ± 0.03 | 20.3 ± 0.1 | 4.0 ± 0.1 | 22.2 ± 0.1 | 2.38 ± 0.07 |
| 10 | 1.19 ± 0.01 | 11.2 ± 0.1 | 3.1 ± 0.1 | 20.0 ± 0.4 | 2.61 ± 0.09 |
| 11 | 1.31 ± 0.03 | 23.0 ± 0.2 | 2.37 ± 0.02 | 19.2 ± 0.1 | 1.81 ± 0.04 |
| 12 | 0.8 ± 0.3 | 24.7 ± 0.7 | 1.08 ± 0.02 | 27.9 ± 0.3 | 1.4 ± 0.5 |
| 13 | 1.1 ± 0.1 | 13.5 ± 0.2 | 2.23 ± 0.02 | 19.0 ± 0.1 | 2.0 ± 0.2 |
| 14 | 0.30 ± 0.01 | 26.6 ± 0.1 | 1.41 ± 0.02 | 30.1 ± 0.2 | 4.7 ± 0.2 |
| 15 | 0.24 ± 0.04 | 22 ± 2 | 0.34 ± 0.02 | 38.3 ± 0.3 | 1.4 ± 0.3 |
| 17 | 0.21 ± 0.02 | 18.5 ± 0.1 | 1.25 ± 0.01 | 22.9 ± 0.1 | 6.0 ± 0.6 |
The bioactive hits had varying levels of fragment similarity to the fine-tuning compounds (25 ± 13% on average), in terms of their Tanimoto similarity on Morgan molecular fingerprints (53). Seven designs showed a fragment similarity <65% to all fine-tuning compounds (table S6). Compound 29 (Fig. 4A) was the nearest fine-tuning neighbor to several bioactive designs, with fragment similarity values ranging from 55 to 75%. Among those designs, bioactive compounds 1, 13, and 15 had novel atomic scaffolds (“Murcko scaffolds”) (41) compared to the LXRα and LXRβ agonists, with EC50 ≤ 50 μM annotated in the ChEMBL27 database (Fig. 4A). Furthermore, the scaffolds of compounds 1 and 13 were not present in any of the 15,247 molecules annotated in the Nuclear Receptor Activity (NURA) dataset (52). Compound 15, the most potent LXRβ agonist, had the lowest fragment similarity to the two closest fine-tuning molecules (table S6). These results corroborate the capacity of the computational pipeline to explore narrow regions of the chemical space defined by the known LXR agonists while, at the same time, providing hitherto unexplored molecular cores for further compound optimization.
Fig. 4. Analysis of selected de novo designs.
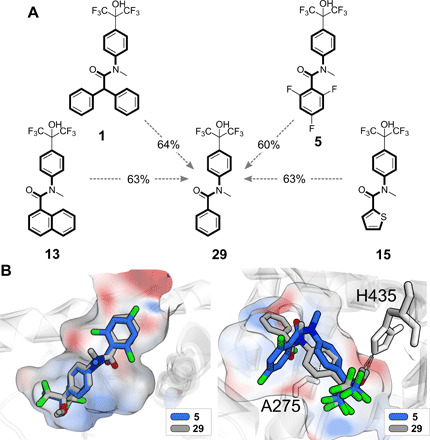
(A) Selected de novo designs (1, 5, 13, and 15; Table 1), with atomic scaffolds highlighted with a thicker line, in comparison with the most similar fine-tuning compound (29, ChEMBL-ID: 379225, LXRα: EC50 = 0.4 μM, LXRβ: EC50 = 0.18 μM). The corresponding fragment similarity (Morgan molecular fingerprints and Tanimoto index) is indicated in percentage values. Compounds 1, 13, and 15 have novel atomic scaffolds compared to the known LXR modulators annotated in the ChEMBL27 database. Compound 5 has a similar agonistic effect on LXRα to compound 29, and it is five times more selective for the α subtype. (B) Automated ligand docking of compounds 5 (de novo design, blue) and 29 (fine-tuning compound, light gray) to the binding pockets of LXRα [PDB ID: 3IPS (63), left] and LXRβ [PDB ID: 1PQC (64), right]. GOLD (65) docking software was used. The solvent-accessible surface of the binding pockets is colored according to the computed electrostatic potential; red: negatively charged and blue: positively charged.
All the confirmed active compounds had a preference for LXRα over the LXRβ subtype (Table 1). This observation reflects the desired effect of fine-tuning the artificial intelligence model with LXRα modulators. Compounds 17 and 5 showed the highest LXRα selectivity, with five to six times greater activity than on LXRβ (Table 1). Compound 17 activated LXRα less potently than its closest structural relative among the fine-tuning compounds (EC50 < 0.1 μM; table S6). Compound 5 (LXRα, EC50 = 0.26 ± 0.01 μM; LXRβ, EC50 = 1.30 ± 0.03 μM) constituted only a minor structural modification of fine-tuning compound 29, which had an EC50 of 0.4 μM on LXRα (54, 55) and an EC50 of 0.18 μM on LXRβ (54). While compounds 5 and 29 were comparable in their agonistic effects on LXRα, compound 5 was more than seven times less potent than 29 on the β subtype. As suggested by automated ligand-receptor docking, the preference for LXRα could be ascribed to the different positioning of compounds 5 and 29 in the LXRβ binding pocket (Fig. 4B). While compounds 5 and 29 are predicted to adopt similar binding poses within the LXRα pocket [root mean square deviation (RMSD), <1.8 Å], compound 29 was predicted to engage in an additional CH3-π interaction in the binding pocket of LXRβ. This hypothesis potentially explains the greater affinity of compound 29 for LXRβ as compared to that of compound 5.
Study significance and outlook
With 61% of the computational designs successfully synthesized and 12 novel LXR agonists with low-micromolar to sub-micromolar activity identified, the integrated de novo design platform shows promise for automating the DMTA cycle in drug discovery. By tailoring the model optimization to the available experimental pipeline, the benefits of rule-free de novo design, virtual reaction specification, and automated synthesis were combined. The results further validate the ability of generative molecular design approaches to capture desired molecular properties such as chemical synthesizability and on-target bioactivity, as well as their potential to support automation. The proposed DMTA framework offers the promise of fast iterations through the molecular design cycle and data-driven compound optimization. By relying on generative deep learning, the pipeline could be operated with minimal human interference, only requiring human input for the curation of the pretraining and fine-tuning compounds, and not using any human-crafted rules for molecule construction. Owing to its modular character, the approach can be tailored to other de novo design applications by replacing the computational molecule generator, reactions used for filtering, or synthesis technology. For the purpose of this proof-of-concept study, we successfully obtained molecules that were synthesizable within the microfluidics platform and had desired activity on LXR. These results indicate the usefulness of the pipeline for compound optimization and structure-activity relationship studies. Within the constraints of the two optimization goals (i.e., synthesizability and bioactivity on LXR), limited exploration of the chemical space was achieved. While conserving the overall structural similarity of the compounds in the fine-tuning set, novel structural features were introduced and new molecular scaffolds were identified. The three novel bioactive scaffolds (EC50 values ranging from 0.24 to 4.5 μM) highlight the capability of the computational pipeline to explore narrow regions of the relevant chemical space while, at the same time, providing access to unexplored molecular scaffolds.
The integrated de novo design platform can be further expanded by explicitly including the structural diversity of the molecular designs in the multiparameter optimization process. To achieve a broader exploration of the chemical space, several strategies can be adopted, for example, choosing different optimization criteria for the generative deep learning model or including other artificial intelligence models (15, 56) in the definition of compatible organic reactions. The proposed approach demonstrates the possibility to achieve closed-loop benchtop platforms for compound design and iterative optimization driven by artificial intelligence. Future work will be concerned with extending the microfluidic system to enable multistep synthesis, exploring automated batch synthesis as an alternative, as well as establishing active learning (57, 58) for improved process efficiency.
MATERIALS AND METHODS
Computational
Virtual reaction filter
A total of 17 decomposition reactions written in the SMARTS language (in the form “product >> reactant”) were adapted from a recent study (59) to capture feasible synthetic routes for the microfluidics-assisted platform (see table S1). The program code of the virtual reaction filter can be accessed at the following URL: https://github.com/ETHmodlab/ai-on-a-chip.
Training data
The pretraining library was obtained from a dataset of 3,383,942 commercially available synthetic compounds, assembled from four providers: Asinex (http://asinex.com/libraries-html/—Elite, Fragments, Gold, and Platinum collections), ChemBridge screening compound collection (http://chembridge.com), Enamine advanced and HTS collections (http://enamine.net), and Specs screening compounds (https://specs.net). The library was filtered using the respective reaction SMARTS. Only compounds that could be successfully decomposed into their corresponding reactants (656,689 molecules) were retained for model pretraining. Fine-tuning was performed on a set of 40 LXRα agonists (table S2), manually curated from ChEMBL26 (44) (target ID = CHEMBL2808), with cell-based activity data demonstrating LXRα agonism with EC50 < 500 nM.
Molecule preprocessing
The molecular structures were standardized with the Molecular Operating Environment (MOE) “wash” procedure (MOE v.2018.01, default settings) before computing molecular descriptors and performing target prediction. As in our previous study (27), before LSTM training, molecular structures were encoded as canonical SMILES (60) strings using the RDKit package (v.2018.03, www.rdkit.org); stereochemical information was removed and only SMILES strings with a length of up to 80 SMILES characters were retained (656,070 and 40 molecules for pretraining and fine-tuning, respectively). The preprocessed training set data can be accessed at the following URL: https://github.com/ETHmodlab/ai-on-a-chip.
Target prediction and molecular descriptors
Target prediction was performed with the in-house software SPiDER (40), using MOE2D and Chemically Advanced Template Search 2 (CATS2) descriptors (42) as input. Only predictions with P < 0.05 were considered. CATS2 descriptors were calculated using in-house software (settings: CorrelationDistance = 10, Scaling = Types, Distance = Euclidean). MOE2D descriptors were calculated with the “QSAR descriptors” node of MOE 2018.01 in a KNIME 3.7.0 environment (61) (charge calculation = MMFF94*).
Model architecture and settings
The chemical language model was implemented in Python (v. 3.6.5) using Keras (https://keras.io/, v2.2.0) with the TensorFlow GPU backend (www.tensorflow.org, v1.9.0) as a recurrent neural network with LSTM cells (34), as previously published (27). The neural network used consisted of four layers, for a total of 5,820,515 parameters: (i) BatchNormalization layer, (ii) LSTM layer with 1024 units, (iii) LSTM layer with 256 units, and (iv) BatchNormalization layer. The model was trained with SMILES strings encoded as one-hot vectors. SMILES randomization and 10-fold augmentation, as recently published (27), were used. We used the categorical cross-entropy loss and the Adam optimizer (62). The model was pretrained for 10 epochs with a learning rate equal to 0.001. We selected the pretraining epoch (epoch 2) as the one maximizing the number of designs that could be decomposed into compatible reactants (building blocks) available from the Sigma-Aldrich catalog. The selected model was fine-tuned for 40 epochs (learning rate = 0.002). The model code can be accessed at the following URL: https://github.com/ETHmodlab/virtual_libraries.
Temperature and sampling epoch choice
Sampling temperature and fine-tuning epochs were automatically determined to optimize three parameters simultaneously: (i) predicted LXR activity by SPiDER (40), (ii) scaffold diversity (41), and (iii) distance to the fine-tuning compounds [as encoded by CATS2 (42) descriptors with Euclidean distance]. We tested three sampling temperatures (T = 0.2, T = 0.7, and T = 1.2; fig. S1) and fine-tuning epochs 1 to 40 (fig. S3). Sampling temperature T = 0.70 and fine-tuning epochs in the range of 15 to 20 were chosen to generate the final designs, as they resulted in the best compromise between scaffold diversity, number of compounds predicted as LXR modulators (P < 0.05), and CATS2 similarity between the fine-tuning set and de novo design.
Novelty and scaffold analysis
The 67 de novo designs retained after reaction-based filtering were checked for their structural novelty on PubChem (43), ChEMBL27 (44), SciFinder (version 2019; Chemical Abstracts Service) (45), SureChEMBL (46), and Reaxys (47) (accessed 4 April 2019). Atomic scaffolds (41) were computed using RDKit in KNIME (v. 3.6.2).
Similarity analysis
ChEMBL27 compounds that were structurally similar to the bioactive hits [as determined by the Tanimoto similarity on RDKit Morgan (53) fingerprints with radius equal to 2 and 1024 bits; table S7] were retrieved using the ChEMBL web resource client “similarity filter” function (https://github.com/chembl/chembl_webresource_client, beta version, 6 November 2020, Python 3.7.7). The same strategy was used to report the two most similar fine-tuning compounds for each bioactive hit (table S6).
Automated ligand docking
The crystal structures of LXRα [Protein Data Bank (PDB) ID: 3IPS (63)] and LXRβ [PDB ID: 1PQC (64)] were retrieved from the PDB (https://www.rcsb.org/) and prepared with MOE v.2019.0102 (QuickPrep module: “Preserve Sequence and Neutralize”; “Use Protonate 3D for Protonation” = True; “Allow ASN/GLN/HIS ‘Flips’ in Protonate 3D” = True; “Delete Water Molecules Farther than 4.5 Å from Ligand or Receptor” = True; Tether Receptor: Strength = 10, Buffer = 0.25; Fix: “Atoms Farther than 8 Å from Ligands”, hydrogens close to ligands not fixed; Refine: “to RMS Gradient of 0.1 kcal/mol/Å”; “Retain QuickPrep Minimization Restraints” = True). Compounds 5 and 29 were docked with GOLD (65) within MOE v.2019.0102 (Efficiency = default, Score Efficiency = 100; Early Termination = [number:3, RMS = 1.5], PLP scoring, Rigid Receptor, 30 poses per compound), and poses were refined with MOE GBVI/WSA dG (10 refinement poses). Redocking of the crystalized ligand led to RMSD values of 0.8037 and 0.3775 Å for 3IPS and 1PQC, respectively.
Synthesis
Chemicals
All chemicals and solvents were reagent grade and used without further purification unless specified otherwise. The building block chemicals were purchased from Sigma-Aldrich (St. Louis, USA; www.sigmaaldrich.com), Apollo Scientific (Cheshire, UK; www.apolloscientific.co.uk), Alfa Aesar (Kandel, Germany; www.alfa.com), Fluorochem (Derbyshire, United Kingdom; www.fluorochem.co.uk), Acros Organics (Geel, Belgium; www.acros.com), Enamine (Riga, Latvia; www.enamine.net), Maybridge (Waltham, MA, USA; www.fishersci.com), ABCR (Karlsruhe, Germany; www.abcr.de), and ChemDiv (San Diego, CA, USA; www.chemdiv.com). Compounds 26 and 27 were purchased from Enamine (www.enamine.net; compounds Z45510435 and Z45410017, respectively; purity = 90%); 28 was purchased from ChemDiv (www.chemdiv.com; compound 8012-4386, purity = 90%).
Microfluidics platform
Instruments
Automated synthesis was performed on a Cetoni flow chemistry system (Cetoni GmbH, Korbussen, DE) using two gas-tight borosilicate glass syringes (SGE gas tight 2.5 ml, luer lock, Trajan Scientific), a reaction chip (Chip Type Dean Flow A, 16 × 12.5 mm, DFM-A1, 5 μl), and an 800-μl reaction coil of polytetrafluoroethylene tubing. The flow through the system was directed by three-way solenoid valves (100T3/S116, Bio-Chem Valve Inc., Chrom Tech, Apple Valley, MN, USA). The analysis block consisted of a Rheodyne MRA splitter (MRA100-000, Kinesis, Vernon Hills, IL, USA) coupled with an Advion Expression CMS (Advion, Ithaca, NY, USA) for in-line mass analysis. This equipment used L-216OU pumps from a VWR LaChrom ULTRA HPLC system (Radnor, PA, USA). An analytical HPLC system (Shimadzu, Kyoto, Japan) equipped with an analytical C18 reverse phase column (Macherey-Nagel, Nucleodur C18 HTec; 5 μm, 150 × 3 mm) was used for follow-up sample analysis. Mass signals were recorded using a Shimadzu LCMS-2020 system (Kyoto, Japan). The automated synthesis system was controlled using the QmixElements software supplied by Cetoni on an Aspire X3990 PC (i3 Intel Core 2120 CPU, 8GB, 1066 MHz DDR3 RAM, Windows 7 OS).
Protocol
The building blocks were loaded into two 96-well plates and sealed using adhesive slit seal sheets. User input was requested for the type of reaction, the number of reactions to be performed, the desired on-chip residence time, choice of reaction chip, and desired temperatures. Upon reaching the desired reaction conditions, aspiration of the reagent solutions was initiated. Using the Move-To-Container functions, the 360° rotAXYS arm moved to the location of the first building block and the dose-volume function was used to aspirate the dead volume between the syringe and the well plate. This volume was discarded, and the syringe was aspirated with a plug of dissolved building block consisting of 0.75 ml diluted with 0.25 ml of clean solvent. Before moving to the second building block container, the 360° rotAXYS arm moved to a clean solvent container using the “Move-To-Container” function to clean the needle. The same aspiration procedure was used for the second syringe. The volume contained in the syringes was then injected into the reaction chip using the dose-volume function at a flow rate calculated from the desired residence time. Once the syringes were empty, they were refilled with 2.5 ml of clean solvent using the dose-volume function and solvent was injected until the remaining part of the reaction plug had passed the reaction coil with the correct residence time. Once the reaction plug had passed the analysis unit and reached the end of the tubing, the Move-To-Container function was used to move the second rotAXYS arm to an empty container, where it was allowed to remain until the reaction plug was collected. The arm then moved back to the waste container position using the Move-XY function. HPLC-MS was used as an online monitoring tool. A stream splitting device coupled the ambient-pressure reactor system in an isolated fashion to the high-pressure side required by HPLC-MS.
Automated synthesis
General amide bond synthesis procedure (compounds 1 to 15 and 22)
Solutions (0.2 M) of the respective acid chloride building block in tetrahydrofuran (THF) and the amine building block in acetonitrile/N,N′-dimethylformamide (MeCN/DMF) (9:1 v/v) were prepared, and 1 ml of each solution was loaded into individual wells of a 96-well plate. For automated synthesis, 0.75 ml of each building block solution was used per reaction and diluted with the running solvent (MeCN/THF; 50:50, v/v) to a total volume of 1.0 ml. The residence time was set to 15 min, and the temperature was set to 55°C. After mixing both solutions in the reaction chip, the reaction mixture was pumped through the coil and 2 ml per sample was collected in another 96-well plate.
General ester bond synthesis procedure (compound 21)
Solutions (0.2 M) of the respective acid chloride building block in THF and the alcohol building block in MeCN/DMF (9:1, v/v) were prepared, and 1 ml of each solution was transferred into individual wells in a 96-well plate. The residence time was set to 5 min, and the temperature was set to 55°C. A 0.75-ml portion of each solution was aspirated into the syringe pumps and diluted to 1-ml total volume with the running solvent (MeCN/THF, 50:50, v/v). After mixing both solutions in the reaction chip, the reaction mixture was pumped through the coil and 2 ml was collected in a 96-well plate.
General sulfonamide synthesis procedure (compounds 16 to 20 and 23 to 25)
Solutions (0.2 M) of the sulfonyl chloride building block in THF and the amine building block with one equivalent of triethylamine in MeCN/THF (50:50 v/v) were prepared, and 1 ml of each solution was loaded into individual wells of a 96-well plate. For automated synthesis, 0.75 ml of each building block solution was used per reaction and diluted with the running solvent (MeCN/THF, 50:50, v/v) to a total volume of 1.0 ml. The residence time was set to 10 min, and the temperature was set to 55°C. After mixing both solutions in the reaction chip, the reaction mixture was pumped through the coil and 2 ml per sample was collected in another 96-well plate.
Batch synthesis
Fourteen compounds were synthesized in-batch for dose-response characterization (compounds 1 to 3, 5 to 6, 8 to 15, and 17). All compounds had a purity greater than 97% according to the HPLC-UV (ultraviolet) analysis (λ = 254 nm, λ = 290 nm). For details of the batch synthesis and analytical characterization, see the Supplementary Materials (figs. 29 and 30).
Biological characterization
The Gal4-fusion receptor plasmid [pFA-CMV-hLXRα-LBD (48) and pFA-CMV-hLXRβ-LBD (48)] coding for the hinge region and ligand-binding domain of the canonical isoform of the respective nuclear receptor have been reported previously. pFR-Luc (Stratagene) was used as the reporter plasmid along with pRL-SV40 (Promega) for normalization of the transfection efficiency and cell growth. HEK293T cells were grown in high-glucose Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal calf serum, sodium pyruvate (1 mM), penicillin (100 U ml−1), and streptomycin (100 μg ml−1) at 37°C and 5% CO2. HEK293T cells were seeded 1 day before transfection in 96-well plates (3.0 × 104 cells per well). Before transfection, the medium was changed to Opti-MEM without supplements. Transient transfection was carried out using Lipofectamine LTX reagent (Invitrogen) according to the manufacturer’s protocol, with pFR-Luc (Stratagene), pRL-SV40 (Promega), and the respective pFA-CMV-hLXR-LBD plasmid. Five hours after transfection, the medium was changed to Opti-MEM supplemented with penicillin (100 U ml−1), streptomycin (100 μg ml−1), 0.1% DMSO, and the respective test compounds or 0.1% DMSO alone as an untreated control. During primary screening, the concentration of each sample was roughly adjusted to 10 μM. Each sample was duplicated and tested in two biological repeats. For dose-response characterization of the purified compounds, each concentration was duplicated, and each experiment was repeated independently at least three times. Following overnight (12 to 14 hours) incubation with the test compounds, the cells were assayed for luciferase activity using the Dual-Glo luciferase assay system (Promega) according to the manufacturer’s protocol. Luminescence was measured using a Tecan Spark luminometer (Tecan Deutschland GmbH, Germany). Normalization of the transfection efficiency and cell growth was performed by dividing the firefly luciferase data by Renilla luciferase data and multiplying the value by 1000, resulting in relative light units (RLUs). Fold activation was obtained by dividing the mean RLU of the test compounds at a respective concentration by the mean RLU of the untreated control. T0901317 served as a reference agonist for assay validation and monitoring assay performance.
Acknowledgments
We thank S. Haller, B. Winkler, D. Gautschi, P. Schneider, and J. A. Hiss for their technical support. Funding: This work was financially supported by the Swiss National Science Foundation (grant no. 205321_182176 to G.S.) and by the RETHINK initiative at ETH Zurich. Author contributions: G.S. conceived the study, with contributions from F.G. and D.M.; F.G. designed the computational workflow and analysis, with contributions from A.L.B., M.M., and D.M.; B.J.H.H. curated the pretraining molecules; D.M. curated the fine-tuning molecules; M.M. trained the LSTM model and generated the designs; F.G. optimized the LSTM settings and performed post hoc analysis and docking; A.L.B. performed the retrosynthetic analysis; B.J.H.H. developed the microfluidic synthesis platform and performed the synthesis in flow; D.M. designed the in vitro screening procedure and performed the in vitro characterization; K.A. performed the batch synthesis and purification. F.G. drafted the manuscript, with contributions from D.M., B.J.H.H., M.M., and G.S. All authors contributed to manuscript revision. Competing interests: G.S. declares a potential financial conflict of interest as a cofounder of inSili.com LLC, Zurich, and in his role as a consultant to the pharmaceutical industry. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabg3338/DC1
REFERENCES AND NOTES
- 1.Schneider G., Automating drug discovery. Nat. Rev. Drug Discov. 17, 97–113 (2018). [DOI] [PubMed] [Google Scholar]
- 2.King R. D., Rowland J., Oliver S. G., Young M., Aubrey W., Byrne E., Liakata M., Markham M., Pir P., Soldatova L. N., Sparkes A., Whelan K. E., Clare A., The automation of science. Science 324, 85–89 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Chow S., Liver S., Nelson A., Streamlining bioactive molecular discovery through integration and automation. Nat. Rev. Chem. 2, 174–183 (2018). [Google Scholar]
- 4.Chapman T., Lab automation and robotics: Automation on the move. Nature 421, 661–663 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Granda J. M., Donina L., Dragone V., Long D.-L., Cronin L., Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature 559, 377–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steiner S., Wolf J., Glatzel S., Andreou A., Granda J. M., Keenan G., Hinkley T., Aragon-Camarasa G., Kitson P. J., Angelone D., Cronin L., Organic synthesis in a modular robotic system driven by a chemical programming language. Science 363, eaav2211 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Coley C. W., Thomas D. A. III, Lummiss J. A., Jaworski J. N., Breen C. P., Schultz V., Hart T., Fishman J. S., Rogers L., Gao H., Hicklin R. W., Plehiers P. P., Byington J., Piotti J. S., Green W. H., Hart A. J., Jamison T. F., Jensen K. F., A robotic platform for flow synthesis of organic compounds informed by AI planning. Science 365, eaax1566 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Desai B., Dixon K., Farrant E., Feng Q., Gibson K. R., van Hoorn W. P., Mills J., Morgan T., Parry D. M., Ramjee M. K., Selway C. N., Tarver G. J., Whitlock G., Wright A. G., Rapid discovery of a novel series of Abl kinase inhibitors by application of an integrated microfluidic synthesis and screening platform. J. Med. Chem. 56, 3033–3047 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Merk D., Friedrich L., Grisoni F., Schneider G., De novo design of bioactive small molecules by artificial intelligence. Mol. Inform. 37, 1700153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhavoronkov A., Ivanenkov Y. A., Aliper A., Veselov M. S., Aladinskiy V. A., Aladinskaya A. V., Terentiev V. A., Polykovskiy D. A., Kuznetsov M. D., Asadulaev A., Volkov Y., Zholus A., Shayakhmetov R. R., Zhebrak A., Minaeva L. I., Zagribelnyy B. A., Lee L. H., Soll R., Madge D., Xing L., Guo T., Aspuru-Guzik A., Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 37, 1038–1040 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Nagarajan D., Nagarajan T., Roy N., Kulkarni O., Ravichandran S., Mishra M., Chakravortty D., Chandra N., Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem. 293, 3492–3509 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies I. W., The digitization of organic synthesis. Nature 570, 175–181 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Shen Y., Borowski J. E., Hardy M. A., Sarpong R., Doyle A. G., Cernak T., Automation and computer-assisted planning for chemical synthesis. Nat. Rev. Meth. Primers 1, 23 (2021). [Google Scholar]
- 14.Segler M. H. S., Preuss M., Waller M. P., Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555, 604–610 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Schwaller P., Petraglia R., Zullo V., Nair V. H., Haeuselmann R. A., Pisoni R., Bekas C., Iuliano A., Laino T., Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy. Chem. Sci. 11, 3316–3325 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y., Jiang X., Why microfluidics? Merits and trends in chemical synthesis. Lab Chip 17, 3960–3978 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Kondo M., Wathsala H. D. P., Sako M., Hanatani Y., Ishikawa K., Hara S., Takaai T., Washio T., Takizawa S., Sasai H., Exploration of flow reaction conditions using machine-learning for enantioselective organocatalyzed Rauhut–Currier and [3+2] annulation sequence. Chem. Commun. 56, 1259–1262 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Pant S. M., Mukonoweshuro A., Desai B., Ramjee M. K., Selway C. N., Tarver G. J., Wright A. G., Birchall K., Chapman T. M., Tervonen T. A., Klefström J., Design, synthesis, and testing of potent, selective hepsin inhibitors via application of an automated closed-loop optimization platform. J. Med. Chem. 61, 4335–4347 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Reutlinger M., Rodrigues T., Schneider P., Schneider G., Multi-objective molecular de novo design by adaptive fragment prioritization. Angew. Chem. Int. Ed. 53, 4244–4248 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Segler M. H., Kogej T., Tyrchan C., Waller M. P., Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 4, 120–131 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olivecrona M., Blaschke T., Engkvist O., Chen H., Molecular de-novo design through deep reinforcement learning. J. Cheminform. 9, 48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez-Lengeling B., Aspuru-Guzik A., Inverse molecular design using machine learning: Generative models for matter engineering. Science 361, 360–365 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Ikebata H., Hongo K., Isomura T., Maezono R., Yoshida R., Bayesian molecular design with a chemical language model. J. Comput. Aided Mol. Des. 31, 379–391 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merk D., Grisoni F., Friedrich L., Schneider G., Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid X receptor modulators. Commun. Chem. 1, 68 (2018). [Google Scholar]
- 25.Bruns D., Merk D., Santhana Kumar K., Baumgartner M., Schneider G., Synthetic activators of cell migration designed by constructive machine learning. ChemistryOpen 8, 1303–1308 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan W., Jiang D., Nambiar D. K., Liew L. P., Hay M. P., Bloomstein J., Lu P., Turner B., Le Q.-T., Tibshirani R., Khatri P., Moloney M. G., Koong A. C., Chemical space mimicry for drug discovery. J. Chem. Inf. Model. 57, 875–882 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moret M., Friedrich L., Grisoni F., Merk D., Schneider G., Generative molecular design in low data regimes. Nat. Mach. Intell. 2, 171–180 (2020). [Google Scholar]
- 28.Rodrigues T., Schneider P., Schneider G., Accessing new chemical entities through microfluidic systems. Angew. Chem. Int. Ed. 53, 5750–5758 (2014). [DOI] [PubMed] [Google Scholar]
- 29.El-Gendy B. E.-D. M., Goher S. S., Hegazy L. S., Arief M. M. H., Burris T. P., Recent advances in the medicinal chemistry of liver X receptors. J. Med. Chem. 61, 10935–10956 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Collins J. L., Therapeutic opportunities for liver X receptor modulators. Curr. Opin. Drug Discov. Devel. 7, 692–702 (2004). [PubMed] [Google Scholar]
- 31.Li N., Wang X., Liu P., Lu D., Jiang W., Xu Y., Si S., E17110 promotes reverse cholesterol transport with liver X receptor β agonist activity in vitro. Acta Pharm. Sin. B. 6, 198–204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong C., Tontonoz P., Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 13, 433–444 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Plowright A. T., Johnstone C., Kihlberg J., Pettersson J., Robb G., Thompson R. A., Hypothesis driven drug design: Improving quality and effectiveness of the design-make-test-analyse cycle. Drug Discov. Today 17, 56–62 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Hochreiter S., Schmidhuber J., Long short-term memory. Neural Comput. 9, 1735–1780 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Weininger D., SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988). [Google Scholar]
- 36.Gupta A., Müller A. T., Huisman B. J., Fuchs J. A., Schneider P., Schneider G., Generative recurrent networks for de novo drug design. Mol. Inform. 37, 1700111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daylight Theory: SMARTS—A Language for Describing Molecular Patterns (Daylight Chemical Information System Inc., 2019); https://www.daylight.com/dayhtml/doc/theory/theory.smarts.html.
- 38.Grisoni F., Moret M., Lingwood R., Schneider G., Bidirectional molecule generation with recurrent neural networks. J. Chem. Inf. Model. 60, 1175–1183 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Awale M., Sirockin F., Stiefl N., Reymond J.-L., Drug analogs from fragment-based long short-term memory generative neural networks. J. Chem. Inf. Model. 59, 1347–1356 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Reker D., Rodrigues T., Schneider P., Schneider G., Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc. Natl. Acad. Sci. U.S.A. 111, 4067–4072 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bemis G. W., Murcko M. A., The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 39, 2887–2893 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Reutlinger M., Koch C. P., Reker D., Todoroff N., Schneider P., Rodrigues T., Schneider G., Chemically Advanced Template Search (CATS) for scaffold-hopping and prospective target prediction for ‘orphan’ molecules. Mol. Inform. 32, 133–138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B. A., Thiessen P. A., Yu B., Zaslavsky L., Zhang J., Bolton E. E., PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 47, D1102–D1109 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mendez D., Gaulton A., Bento A. P., Chambers J., De Veij M., Félix E., Magariños M. P., Mosquera J. F., Mutowo P., Nowotka M., Gordillo-Marañón M., Hunter F., Junco L., Mugumbate G., Rodriguez-Lopez M., Atkinson F., Bosc N., Radoux C. J., Segura-Cabrera A., Hersey A., Leach A. R., ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 47, D930–D940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chemical Abstract Service, SciFinder, v. 2019 (2019).
- 46.Papadatos G., Davies M., Dedman N., Chambers J., Gaulton A., Siddle J., Koks R., Irvine S. A., Pettersson J., Goncharoff N., Hersey A., Overington J. P., SureChEMBL: A large-scale, chemically annotated patent document database. Nucleic Acids Res. 44, D1220–D1228 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.A. J. Lawson, J. Swienty-Busch, T. Géoui, D. Evans, The making of Reaxys—Towards unobstructed access to relevant chemistry information, in The Future of the History of Chemical Information (ACS Symposium Series, American Chemical Society, 2014), vol. 1164, pp. 127–148. [Google Scholar]
- 48.Heitel P., Achenbach J., Moser D., Proschak E., Merk D., DrugBank screening revealed alitretinoin and bexarotene as liver X receptor modulators. Bioorg. Med. Chem. Lett. 27, 1193–1198 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Karageorgis G., Warriner S., Nelson A., Efficient discovery of bioactive scaffolds by activity-directed synthesis. Nat. Chem. 6, 872–876 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Baker L. M., Aimon A., Murray J. B., Surgenor A. E., Matassova N., Roughley S. D., Collins P. M., Krojer T., von Delft F., Hubbard R. E., Rapid optimisation of fragments and hits to lead compounds from screening of crude reaction mixtures. Commun. Chem. 3, 122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han S., Zhuang H., Shumyak S., Wu J., Xie C., Li H., Yang L.-J., Reeves W. H., Liver X receptor agonist therapy prevents diffuse alveolar hemorrhage in murine lupus by repolarizing macrophages. Front. Immunol. 9, 135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valsecchi C., Grisoni F., Motta S., Bonati L., Ballabio D., NURA: A curated dataset of nuclear receptor modulators. Toxicol. Appl. Pharmacol. 407, 115244 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Rogers D., Hahn M., Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Aoyama A., Endo-Umeda K., Kishida K., Ohgane K., Noguchi-Yachide T., Aoyama H., Ishikawa M., Miyachi H., Makishima M., Hashimoto Y., Design, synthesis, and biological evaluation of novel transrepression-selective liver X receptor (LXR) ligands with 5,11-Dihydro-5-methyl-11-methylene-6H-dibenz[b,e]azepin-6-one skeleton. J. Med. Chem. 55, 7360–7377 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Li L., Liu J., Zhu L., Cutler S., Hasegawa H., Shan B., Medina J. C., Discovery and optimization of a novel series of liver X receptor-α agonists. Bioorg. Med. Chem. Lett. 16, 1638–1642 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Johansson S., Thakkar A., Kogej T., Bjerrum E., Genheden S., Bastys T., Kannas C., Schliep A., Chen H., Engkvist O., AI-assisted synthesis prediction. Drug Discov. Today Technol. 32-33, 65–72 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Reker D., Schneider G., Active-learning strategies in computer-assisted drug discovery. Drug Discov. Today 20, 458–465 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Warmuth M. K., Liao J., Rätsch G., Mathieson M., Putta S., Lemmen C., Active learning with support vector machines in the drug discovery process. J. Chem. Inf. Comput. Sci. 43, 667–673 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Button A., Merk D., Hiss J. A., Schneider G., Automated de novo molecular design by hybrid machine intelligence and rule-driven chemical synthesis. Nat. Mach. Intell. 1, 307–315 (2019). [Google Scholar]
- 60.O’Boyle N. M., Towards a Universal SMILES representation - A standard method to generate canonical SMILES based on the InChI. J. Chem. 4, 22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berthold M. R., Cebron N., Dill F., Gabriel T. R., Kötter T., Meinl T., Ohl P., Thiel K., Wiswedel B., KNIME—The Konstanz information miner: Version 2.0 and beyond. SIGKDD Explor. Newsl. 11, 26–31 (2009). [Google Scholar]
- 62.D. P. Kingma, J. Ba, Adam: A Method for Stochastic Optimization (2014); arXiv:1412.6980 [cs.LG] (22 December 2014).
- 63.Fradera X., Vu D., Nimz O., Skene R., Hosfield D., Wynands R., Cooke A. J., Haunsø A., King A., Bennett D. J., McGuire R., Uitdehaag J. C. M., X-ray structures of the LXRα LBD in its homodimeric form and implications for heterodimer signaling. J. Mol. Biol. 399, 120–132 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Färnegårdh M., Bonn T., Sun S., Ljunggren J., Ahola H., Wilhelmsson A., Gustafsson J.-A., Carlquist M., The three-dimensional structure of the liver X receptor β reveals a flexible ligand-binding pocket that can accommodate fundamentally different ligands. J. Biol. Chem. 278, 38821–38828 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Verdonk M. L., Cole J. C., Hartshorn M. J., Murray C. W., Taylor R. D., Improved protein–ligand docking using GOLD. Proteins 52, 609–623 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabg3338/DC1