

Abstract
Mutations in the calreticulin (CALR) gene are seen in about 30% of essential thrombocythemia and primary myelofibrosis patients. To address the contribution of the human CALR mutants to the pathogenesis of myeloproliferative neoplasms (MPNs) in an endogenous context, we modeled the CALRdel52 and CALRins5 mutants by induced pluripotent stem cell (iPSC) technology using CD34+ progenitors from 4 patients. We describe here the generation of several clones of iPSC carrying heterozygous CALRdel52 or CALRins5 mutations. We showed that CALRdel52 induces a stronger increase in progenitors than CALRins5 and that both CALRdel52 and CALRins5 mutants favor an expansion of the megakaryocytic lineage. Moreover, we found that both CALRdel52 and CALRins5 mutants rendered colony forming unit–megakaryocyte (CFU-MK) independent from thrombopoietin (TPO), and promoted a mild constitutive activation level of signal transducer and activator of transcription 3 in megakaryocytes. Unexpectedly, a mild increase in the sensitivity of colony forming unit-granulocyte (CFU-G) to granulocyte-colony stimulating factor was also observed in iPSC CALRdel52 and CALRins5 compared with control iPSC. Moreover, CALRdel52-induced megakaryocytic spontaneous growth is more dependent on Janus kinase 2/phosphoinositide 3-kinase/extracellular signal-regulated kinase than TPO-mediated growth and opens a therapeutic window for treatments in CALR-mutated MPN. The iPSC models described here represent an interesting platform for testing newly developed inhibitors. Altogether, this study shows that CALR-mutated iPSC recapitulate MPN phenotypes in vitro and may be used for drug screening.
Introduction
Classical non–BCR-ABL1 myeloproliferative neoplasms (MPNs) include 3 main disorders: essential thrombocythemia (ET), polycythemia vera (PV), and primary myelofibrosis (PMF). They are due to disease-causing “driver mutations” affecting the cytokine receptor/Janus kinase 2 (JAK2)/signal transducer and activator of transcription (STAT) pathway. Mutations in JAK2 have been identified in 95% of PV and around 50% of ET and PMF. Mutations in the thrombopoietin receptor (myeloproliferative leukemia [MPL]) have been discovered in only 5% to 10% of ET and PMF. Finally, calreticulin (CALR) mutations were identified in 25% to 30% of patients harboring thrombocytosis such as ET and PMF.1,2 CALR mutations associated with MPNs are insertions and/or deletions that result in a +1 “frameshift” to a specific alternative reading frame, leading to the loss of the KDEL motif and the synthesis of a novel predominantly positively charged C-terminal peptide. Two mutations called CALRdel52 and CALRins5 have been shown to be the most prevalent in ET and PMF with CALRdel52 being more frequent than CALRins5 in PMF and are generally found heterozygous in MPN.3–5 Physiologically, CALR is a chaperone protein of the endoplasmic reticulum (ER) lumen that controls proper folding of neosynthesized glycoproteins and plays a role in calcium homeostasis with no evident role in cytokine signaling.6 Studies including from our group have shown that CALR mutants activate the MPL receptor after binding to its N-glycosylated residues in the ER and activate the JAK2/STAT signaling pathway.7–10 This activation required the positive charge of mutant CALR C-terminus peptide, the lectin-binding domain and the extracellular N-linked sugars of MPL. CALR mutants behave as an abnormal chaperone of MPL allowing its traffic to the cell membrane by the canonical pathway, but in an immature form.11,12 Induction of MPL signaling by CALR mutants through JAK2 activation mostly occurs at the cell surface.11,13,14 Moreover, CALR mutants are not able to activate other cytokine receptors than MPL except weakly granulocyte-colony stimulating factor receptor (G-CSF-R) in Ba/F3 and γ2A cell lines,7,15 where activation does not suffice for long-term autonomous growth.
The CALR mutations recapitulate MPN in mice mimicking an ET progressing to myelofibrosis only for CALRdel52 mice, whereas the phenotype for CALRins5 is milder. Disease development induced by CALR mutants was dependent on MPL.9,16–19
In addition to CALR drivers, a growing list of somatic mutations in genes affecting mainly epigenetic regulators and chromatin modifiers (TET2, DNMT3A, EZH2, ASXL1, IDH1/2), transcription factors (RUNX1, NFE2, P53, IKZF), and less frequently in splicing regulators (SF3B1, SRSF2, U2AF1) have been reported in MPN patients.20 They frequently occur after CALR mutations in hematopoietic stem cells.21,22 They are “modifying mutations” that change the phenotype or accelerate the disease.23
Induced pluripotent stem cells (iPSC) have been used not only to model hereditary disorders with germline mutations,24,25 but they were also successfully derived from acquired malignant disorders such as MPN or juvenile myelomonocytic leukemia.26–28 In previous studies, we and others have generated JAK2V617F and CALR-mutated iPSC29 and have shown that this technology can be used to model classical MPN and their response to small molecules.28,30,31 To understand and compare the role of endogenous CALRdel52 and ins5 mutants in the human setting, we have generated iPSC from CD34+ cells isolated from the blood of 4 MPN patients, with CALRdel52 or CALRins5 mutation associated or not with other mutations. Using hematopoietic differentiation, we showed that both CALRdel52 and CALRins5 iPSC induced a megakaryocytic phenotype since we found an increased generation of megakaryocyte (MK) and sensitivity of the MK progenitors to thrombopoietin (TPO) through spontaneous activation of STAT3. Unexpectedly, we found an increased sensitivity to granulocyte-colony stimulating factor (G-CSF) of both CALRdel52 and CALRins5 granulocytic progenitors. Finally, this study uncovers a more prominent impact of CALRdel52 on total number of human progenitors than CALRins5.
Methods
Patients, iPSC generation, and cultures
All participants to this study gave their written informed consent in accordance with the Declaration of Helsinki and the study was approved by the CNIL DR-2015-692 (Paris, France) and the Ethics Committee of the Brazilian National Cancer Institute under the number 062/08. MPN was defined following World Health Organization criteria for ET and PMF.32 Blood from a healthy donor was used as control. CD34+ cells were purified from blood mononuclear cells and cells were cultured in serum-free medium with cytokines for 5 days before being infected with Sendai viruses encoding Oct4, c-Myc, Klf4, and Sox2.33 Six days later, cells were seeded on irradiated murine embryonic fibroblasts in embryonic stem (ES) medium.34 Colonies with an ES-like morphology were picked from day 20 to 30 and expanded.
Antibodies and flow cytometry analysis
Directly conjugated monoclonal antibodies were used for iPSC characterization (Human Pluripotent Stem Cell Transcription Factor Analysis Kit, expression of stage-specific embryonic antigen [SSEA]3, eBioscience, San Diego, CA and tumor rejection antigen 1 [TRA-1]-81, Becton Dickinson, le Pont de Claix, France), and for sorting and characterization of hematopoietic cells (anti-CD34, Beckman, Villepinte, France; anti-CD43, CD42 and GPA, Invitrogen, Cergy-Pontoise, France; and anti-CD41 and CD15, Pharmingen, San Diego, CA). Cells were sorted on an Influx flow cytometer (Becton Dickinson) and analyzed on a FACS Canto II (Becton Dickinson).
Teratoma assays
IPSC (1 × 106) were scrapped and resuspended in 140 μL ES medium. Undiluted matrigel (60 μL) was added before subcutaneous injection into non-obese diabetic/severe combined immunodeficiency/γc−/− (NOD-SCID γc−/−) mice. After 8 to 12 weeks, tumors were isolated and fixed in formalin (10%). Sections were stained for germ layers analysis. Spontaneous differentiation was generated by embryoid body formation.35
Hematopoietic differentiation and quantification of progenitors in semisolid cultures
IPSC were seeded on geltrex (Thermofisher, Les Ulis, France) in StemMACS iPS-Brew (Miltenyi Biotec, Paris, France) or in Essential 8 medium (Thermofisher) the day before differentiation. On the first day of differentiation, iPSC were cultured in StemPro-34 (Thermofisher) in the presence of vascular endothelial growth factor (VEGF) (50 ng/mL) (Peprotech, Neuilly-sur-Seine, France), CHIR99021 (2 μM) (Sigma, Saint Quentin Fallavier, France), and bone morphogenetic protein (BMP)-4 (5 ng/mL) (Peprotech). On day 2, CHIR99021 was removed and basic fibroblast growth factor (bFGF) (20 ng/mL) (Miltenyi Biotec) was added. On day 4, BMP-4 was removed and VEGF and bFGF were added at lower concentrations: 15 and 5 ng/mL, respectively. On day 6, VEGF (50 ng/mL), bFGF (50 ng/mL), human stem cell factor (SCF) (50 ng/mL) (Biovitrum AB, Stockholm, Sweden), and fms like tyrosine kinase 3 (5 ng/mL) were added to the medium. From day 7 to the end, different cytokines were added to the cocktail: TPO (50 ng/mL) (Kirin Brewery, Tokyo, Japan) and interleukin (IL)-6 (10 ng/mL) were added to obtain MK, erythropoietin (EPO) (1 U/mL) (Amgen, Thousand Oaks, CA) was added to obtain erythroblasts, and G-CSF (20 ng/mL) and IL-3 (10 ng/mL) (Miltenyi Biotec) were added to get granulocytes. At day 10, cells were dissociated and sorted for CD34- and CD43-positive markers. The recovered cells were cultured or sorted on the expression of CD34, CD43, CD41, GPA, and CD15.
Cells were plated either in methylcellulose with SCF and EPO or G-CSF or GM-CSF to quantify erythroid (erythroid progenitor [EryP]) and granulo or granulo-monocytic (colony forming unit-granulocyte [CFU-G] or colony forming unit-granulocyte monocyte [CFU-GM]) progenitors or in serum-free fibrin clot assays with SCF and TPO to quantify colony forming unit–megakaryocyte (CFU-MK) (30). Cultures were scored after 12 to 14 days for EryP and CFU-GM–derived colonies (29). MK colonies were enumerated at day 10, after labeling by an indirect immunoalkaline phosphatase staining technique using an anti-CD41a monoclonal antibody (Becton Dickinson, clone HIP8), as previously described.36
Western blot analysis
Signaling studies were performed on cultured megakaryoblasts (CD41+) after overnight cytokine deprivation in serum-free medium. Restimulation by TPO (20 ng/mL) for 15 minutes was done as positive control. Samples were subjected to Western blot analysis using polyclonal antibodies against the phosphorylated forms of STAT3 (Tyr705), extracellular signal-regulated kinase (ERK)1/2 (Thr202/Tyr204), and AKT (Ser473) and against the pan proteins (Cell Signaling Technology, Ozyme, Saint-Cyr-L’École, France). Monoclonal anti-β-actin antibody from Sigma was used as loading control.
Small molecules
Increasing concentrations of small molecules inhibitors were tested in CFU-MK colony assays in the presence of SCF and with or without TPO. JAK2 inhibitors (ruxolitinib), a pan phosphoinositide 3-kinase (PI3K) inhibitor (LY294002), a heat shock protein 90 (HSP90) inhibitor (AUY922), and a mitogen-activated protein kinase kinase (MEK) inhibitor (UO126) were tested. All inhibitors were from Selleck chemicals (Euromedex, Souffelweyersheim, France).
Results
Derivation of human iPSC from CD34+ cells of MPN patients and a healthy donor
Using next generation sequencing (whole exome sequencing for patients number 11, 38, and 35 or targeted for 34), we selected 4 MPN patients with heterozygous CALRdel52 or CALRins5 mutation associated or not with other acquired mutations. Patient 38 presented a PMF and harbored a CALRdel52 mutation and a SETD1B mutation in nearly all the granulocytes. Patient 35 presented an ET and harbored CALRdel52 mutation (50% variant allele frequency [VAF]) and a TET2 mutation (21% VAF). Patient 11 presented an ET and harbored a CALRins5 mutation (43% VAF) and a PIK3CD mutation (41% VAF). Finally, patient 34 presented a PMF and harbored a CALRins5 (50% VAF) mutation associated with TET2 (81%) and ASXL1 (16%) mutations (Table 1).
Table 1.
Genotypes of Patients and iPSC.
| Patient | Patient Mutations | Patient Allele Burden | iPSC Genotype | Name of iPS Cell Line | Performed Experiments |
|---|---|---|---|---|---|
| Control (healthy donor) (by retrovirus) | - | - | WT | Alpha | CD34+CD43+ and CD41+ and GPA+ cell generation |
| Beta | Sensitivity of progenitors to TPO and EPO | ||||
| Control (healthy donor) (by Sendai virus) | - | - | WT | WT-A | All experiments with 2 iPS clones |
| WT-B | |||||
| No. 38 (by Sendai virus) | CALRdel52 | 50% | CALRdel52 + SETD1B | P38-A | All experiments with 2 iPS clones |
| SETD1B | 50% | P38-B | |||
| No. 35 (by Sendai virus) | CALRdel52 | 50% | CALRdel52 | P35 | All experiments with 1 iPS clone |
| TET2 | 21% | ||||
| No. 11 (by Sendai virus) | CALRins5 | 43% | CALRins5 + PIK3CD | P11-A | All experiments with 2 iPS clones |
| PIK3CD | 41% | P11-B | |||
| No. 34 (by Sendai virus) | CALRins5 | 50% | CALRins5 | P34 | All experiments with 1 iPS clone |
| ASXL1 | 16% | ||||
| TET2 | 81% |
EPO = erythropoietin; GPA = glycophorin A; iPSC = induced pluripotent stem cell; TPO = thrombopoietin; WT = wild-type.
Cells were reprogrammed according to the protocol of Yamanaka by using commercial nonintegrating Sendai viruses33 (Table 1). We generated iPSC from these 4 MPN patients and from 1 healthy donor as a control. In all cases, ES-like colonies were individually expanded. Only 2 iPS clones (P38-A and P38-B) were obtained from patient number 38 and both were positive for CALRdel52 and for SETD1B mutations after Sanger sequencing. More than 10 CALRdel52 iPS clones were obtained from patient number 35, but only 1 was negative for TET2 mutation and was selected for further studies (see Supplemental Digital Content, Figure 1, http://links.lww.com/HS/A162). Hundreds of iPS clones were obtained from patient number 11 and all were positive for CALRins5 but also for a PIK3CD mutation after Sanger sequencing. Two clones (P11-A and P11-B) were chosen for experiments (see Supplemental Digital Content, Figure 2, http://links.lww.com/HS/A162). Thirty-two iPS clones were obtained from patient number 34 and 15 clones carried CALRins5 alone without TET2 and ASXL1 mutations. One IPS clone (P34) was selected for experiments (see Supplemental Digital Content, Figure 3, http://links.lww.com/HS/A162). Two iPS clones generated from the healthy donor (wild-type A and wild-type B) were used in further studies as controls.
The iPSC showed normal karyotypes (WT), comparative genomic hybridization array (P35, P38, P11), or ploidy by LG-WGS (P34) that did not identify other significant differences in the iPSC compared to the starting cells (see Supplemental Digital Content, Figure 4, http://links.lww.com/HS/A162, Figure 5, http://links.lww.com/HS/A162). We verified the pluripotency of the 8 undifferentiated iPSC that expressed high levels of cell surface pluripotency markers including TRA-1-81 and SSEA-3/4 and endogenous pluripotency-associated genes such as POU5F1, NANOG, and SRY-box transcription factor 2 (SOX2) (Figure 1A, B). Absence of the Sendai viruses was verified by reverse transcription polymerase chain reaction after 10 passages (data not shown). Pluripotency functions were further demonstrated using biological assays including in vivo teratoma formation with the generation of the 3 germ layer tissues (Figure 1C).
Figure 1.
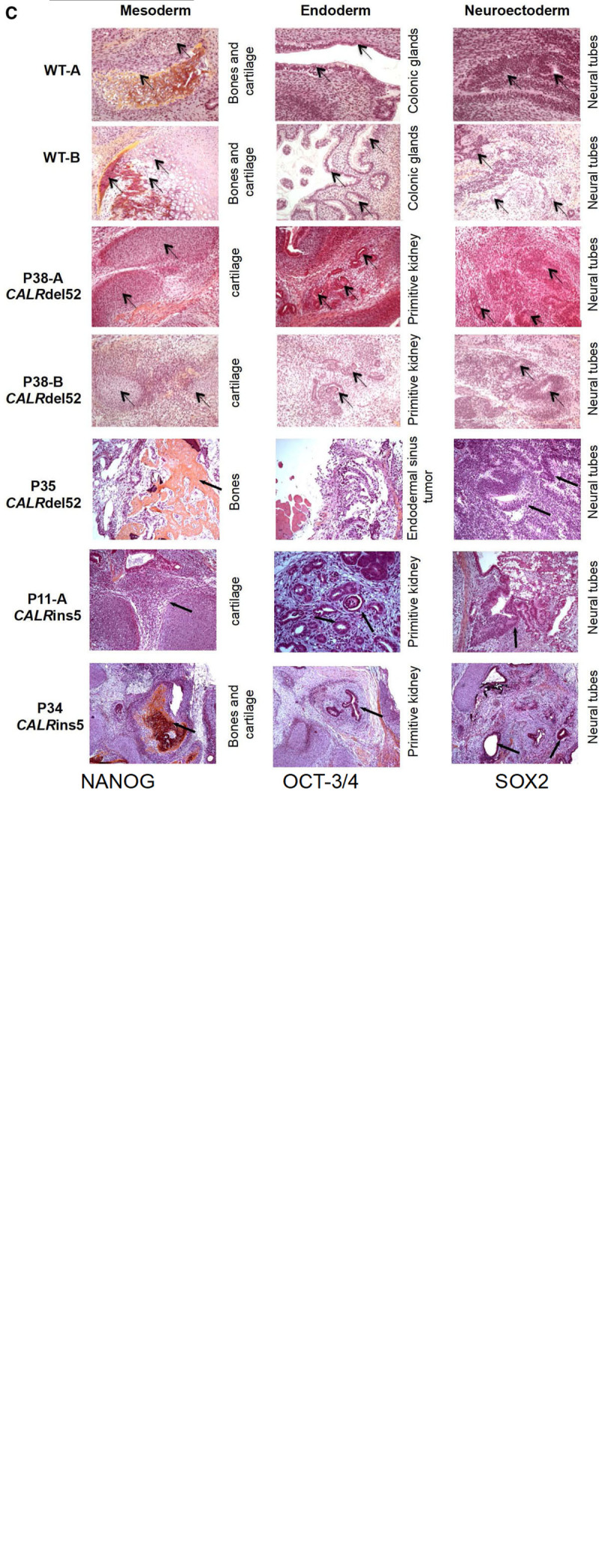
Biological characterization of iPSC (continued on next page). (A/B), Flow cytometry analysis of pluripotency cell surface markers TRA1-81 and SSEA-3/4 and endogenous pluripotency transcription factors NANOG, OCT3/4, and SOX2 in CALRdel52 clones (A) and CALRins5 clones (B). (C - next page), Functional characterization of iPSC in vivo. Analysis of spontaneous differentiation of iPSC in vivo by teratoma formation assay. Teratomas were analyzed after hematoxylin eosin saffron staining. The arrows point the tissue of interest (named on the right side of each pictures). IGG = immunoglobuline G; iPSC = induced pluripotent stem cell; OCT-3/4 = octamer-binding transcription factor 3/4; SOX2 = SRY-box transcription factor 2; SSE4-3/4 = expression of stage-specific embryonic antigen-3/4; TRA1-81 = tumor rejection antigen 1-81; WT = wild-type.
Altogether, these results show that we have generated bona fide CALRdel52 and CALRins5 iPSC from MPN patients. To evaluate the impact of these mutations on MPN phenotypes, we analyzed their roles in hematopoietic differentiation potential.
Effect of CALR mutations on hematopoiesis differentiation, progenitor cytokine sensitivity, and JAK2/STAT signaling pathway
Hematopoietic differentiation was performed on feeder-free culture (Figure 2A–C). All the experiments were done with the 8 iPS clones (2 WT, 3 CALRdel52, and 3 CALRins5). To avoid heterogeneity between donors, we also compared 2 iPS WT clones generated by Sendai virus with 2 other previously published iPS WT clones generated by expressing octamer-binding transcription factor 4, SOX2, Kruppel-like factor 4, and c-myelocytomatosis from Moloney-derived retroviruses vectors.25 The analysis of the main phenotypes did not show any significant difference between these 2 types of control iPSC (Supplemental Digital Content, Figure 6, http://links.lww.com/HS/A162). All the iPSC were evaluated for cell differentiation kinetics between days 10 and 20 by flow cytometry (Figure 3). We observed that P35 iPSC gave rise to significantly more CD41+ cells at day 20 and GPA+ cells at day 15 compared with controls iPSC (Figure 3A, D). We also observed that P11 iPSC and P34 iPSC led to a significant increase in CD41+ cells from day 15 to 17 compared to control iPSC. Moreover, the P11 iPSC led to decreased GPA+ cells and increased CD15+ cells at day 20 compared with controls (Figure 3B, D). Finally, when we combined the data of the different CALRdel52 iPSC or the different CALRins5 iPSC, we found that both CALRdel52 and CALRins5 were able to significantly increase the CD41+ cells compared with controls. Of note, CALRdel52 favored a transient GPA+ cell generation at day 15 (Figure 3C, E).
Figure 2.
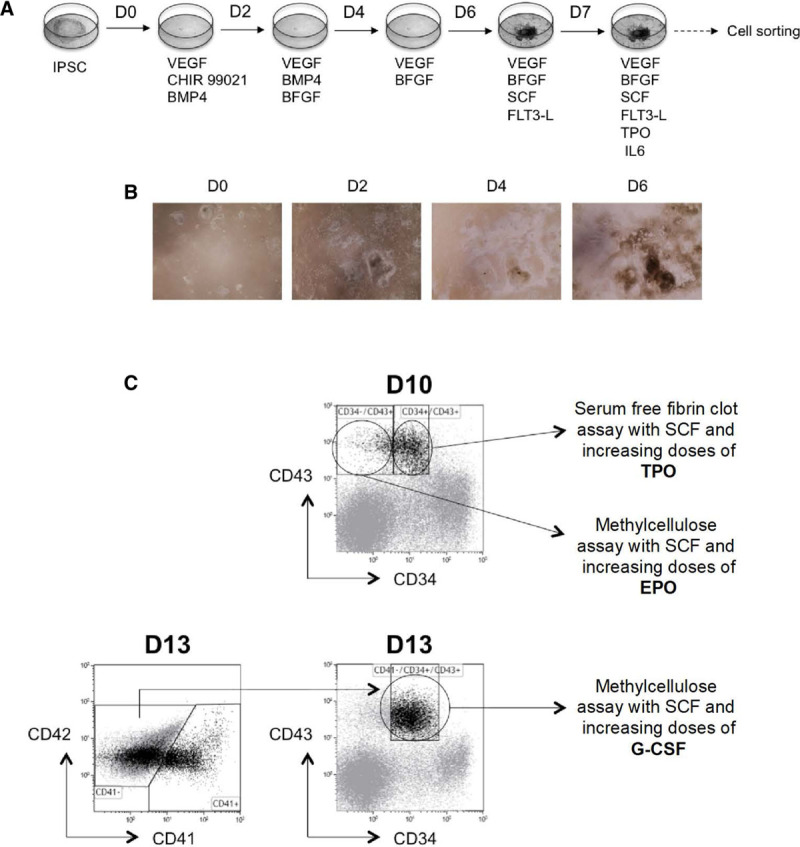
Hematopoietic differentiation of iPSC. (A), Feeder-free differentiation protocol. (B), Pictures of iPSC differentiation in feeder-free culture from day 0 to 6. (C), Scheme for characterization of hematopoietic progenitor sensitivity to cytokines. CD34−CD43+ progenitors were sorted on day 10 and cultured in semisolid condition with SCF and increasing doses of EPO for 14 d. Alternatively, CD41−CD34+CD43+ progenitors were sorted on day 10 and cultured in fibrin-clot assay with SCF and increasing doses of TPO for 10 d. At day 13, CD41−CD34+CD43+ progenitors were sorted and cultured in semisolid condition with SCF and increasing doses of G-CSF for 14 d. bFGF = basic fibroblast growth factor; BMP = bone morphogenetic protein; EPO = erythropoietin; FLT-3 = fms like tyrosine kinase 3; G-CSF = granulocyte-colony stimulating factor; GM-CSF = granulocyte macrophage-colony stimulating factor; IL-6 = interleukin 6; iPSC = induced pluripotent stem cell; SCF = stem cell factor; TPO = thrombopoietin; VEGF = vascular endothelial growth factor.
Figure 3.
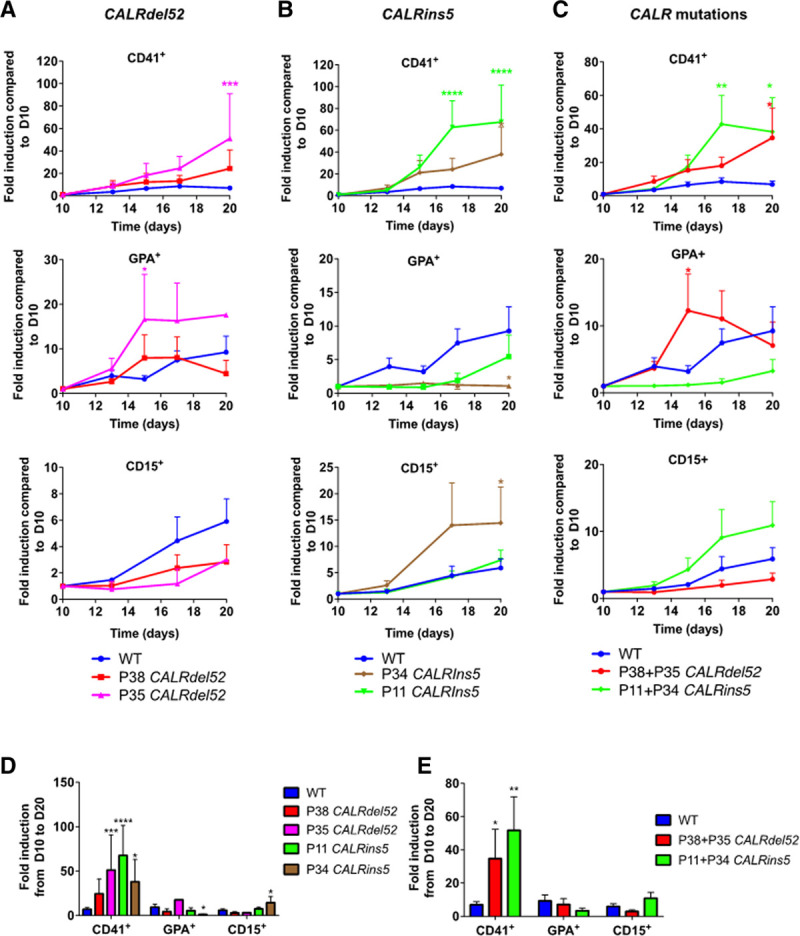
CALRdel52/ins5 increase the generation of hematopoietic cells enriched in megakaryocytes. Kinetics of hematopoietic cells (CD41+, GPA+, CD15+) from day 10 to 20 of (A) CALRdel52, (B) CALRins5, or (C) CALR-mutated iPSC. Fold induction from day 10 to 20 for (D) for all iPSCs and (E) for CALR-mutated iPSC. Results are expressed as fold induction compared to day 10 (mean ± SEM, WT: n = 10–18, P38: n = 7–11, P35: n = 5–8, P11 = 3–7; P34 = 3–7). *P < 0.05; **P < 0.01, ***P < 0.001, ***P < 0.0001, 2-way ANOVA, Tukey’s multiple comparison test. ANOVA = analysis of variance; GPA = glycophorin A; iPSC = induced pluripotent stem cell; SEM = standard error of the mean; WT = wild-type.
We next evaluated the effect of CALR mutations on the number of hematopoietic progenitors and their sensitivity to cytokines by seeding day 10-CD34+CD43+ progenitors in fibrin-clot assay for megakaryocyte progenitors (CFU-MK) or day 10-CD34−CD43+ progenitors in methylcellulose for erythroid colonies (EryP). Alternatively, day 13-CD41−CD34+CD43+ progenitors were plated in methylcellulose to check the CFU-G (Figure 2C). CFU-MK colonies were enumerated at days 10 to 12 and no marked differences were observed compared with controls (Figure 4A). EryP and CFU-G were counted after 14 days of culture in the presence of cytokines (SCF + EPO or SCF + G-CSFG), and CALRdel52 or CALRins5 had no significant impact on total number of colonies compared to controls (Figure 4B, C). However, we found a significantly higher number of any types of progenitors with CALRdel52 compared with CALRins5 (Figure 4A–C).
Figure 4.
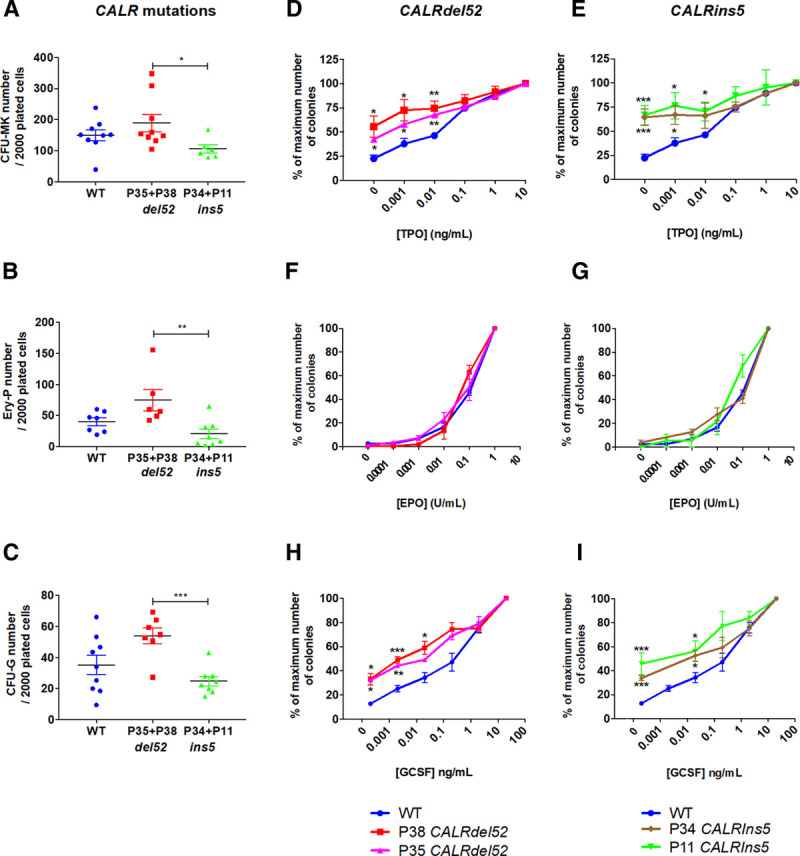
CALRdel52/ins5 increase sensitivity to TPO and G-CSF. (A), Cloning efficiency of CD34+CD43+ progenitors in the presence of SCF and TPO. (B), Cloning efficiency of CD34−CD43+ progenitors in the presence of SCF and EPO. (C), Cloning efficiency of CD41−CD34+CD43+ progenitors in the presence of SCF and G-CSF. (D, E), Sensitivity of CFU-MK to TPO (mean ± SEM, WT: n = 7; P38: n = 6, P35: n = 3, P11: n = 3, P34: n = 3). (F, G), Sensitivity of EryP to EPO (mean ± SEM, WT: n = 7; P38: n = 4, P35: n = 3, P11: n = 3, P34: n = 4). (H, I), Sensitivity of CFU-G to G-CSF (mean ± SEM, WT: n = 6; P38: n = 4, P35: n = 3, P11: n = 3, P34: n = 3). *P < 0.05; **P < 0.01; ***P < 0.001. Student t test. CFU-G = colony forming unit-granulocyte; CFU-MK = colony forming unit-megakaryocyte; EPO = erythropoietin; EryP = erythroid progenitor; G-CSF = granulocyte-colony stimulating factor; SCF = stem cell factor; SEM = standard error of the mean; TPO = thrombopoietin; WT = wild-type.
Since a hallmark of MPN is independency or hypersensitivity of progenitors to cytokines,20,33 particularly to EPO and TPO, we investigated if the CALR-mutant iPSC recapitulated this property by evaluating the sensitivity of progenitors for their responses to cytokines (Figure 4D–I). The CD34+CD43+ progenitor cell fraction was sorted and grown in fibrin clot assay, in the presence of SCF and increasing concentrations of TPO. In these conditions, we observed a 40% to 65% independent growth of CFU-MK progenitors, as well as a hypersensitivity to TPO in all CALR-mutated iPSC from the 4 patients compared with control iPSC (Figure 4D, E). The CD34−CD43+ progenitor cell fraction was sorted and grown in methylcellulose in the presence of SCF and increasing concentrations of EPO. CALRdel52 and CALRins5 did not induce any hypersensitivity of EryP to EPO (Figure 4F, G). The CD41−CD34+CD43+ progenitor cell fraction was sorted and grown in methylcellulose in the presence of SCF and increasing concentrations of G-CSF. Unexpectedly, we also observed a mild 30% to 40% independent growth of CFU-G progenitors as well as a hypersensitivity to G-CSF both for CALRdel52 and CALRins5 (Figure 4H, I). Together, our data demonstrate that CALRdel52 and CALRins5 profoundly modify the response to TPO and to G-CSF, but not to EPO. Moreover, CALRdel52 and CALRins5 iPSC showed larger CFU-MK colonies than the control (Figure 5A, B), suggesting that either CALRdel52 and CALRins5 stimulate MK precursor proliferation or that they generate more immature MK progenitors capable of increased proliferation than in control conditions. Overall, CALR mutations may modify the MK differentiation at multiple steps.
Figure 5.
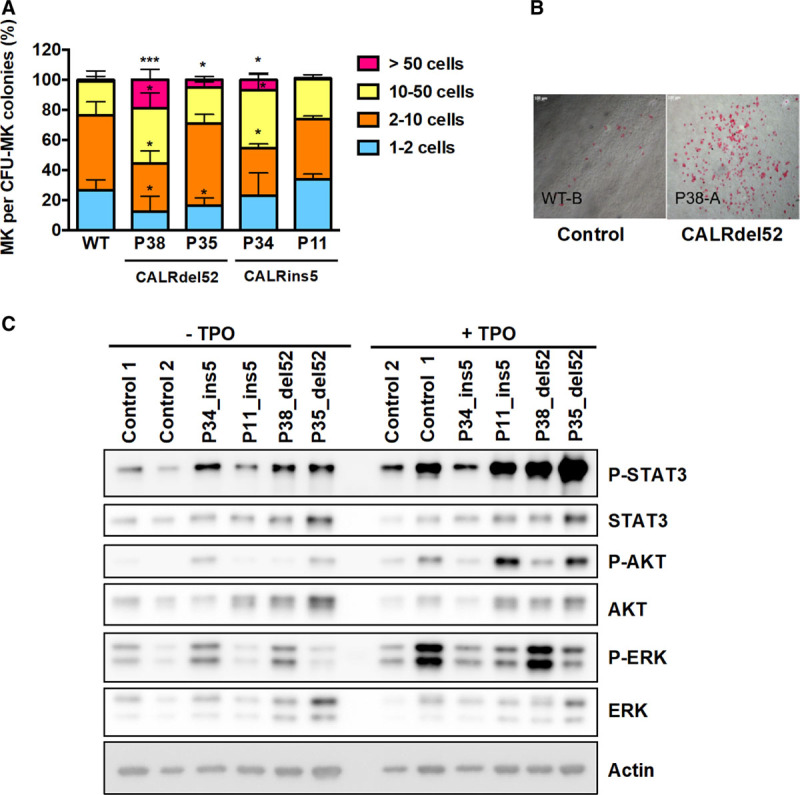
CALRdel52/ins5 induce a constitutive activation of JAK2/STAT pathway in megakaryoblasts. (A, B), CD34+CD43+ progenitors were plated in fibrin-clot assay in presence of SCF and TPO (10 ng/mL) and the number of megakaryocytes were counted in each colony. (A), 50 colonies were counted per condition. (mean ± SEM, WT: n = 7; P38: n = 4, P35: n = 3, P11: n = 3, P34: n = 3). *P < 0.05; ***P < 0.001. Student t test. (B), Representative pictures of CALRdel52/ins5-induced larger CFU-MK colonies compared with control cells. (C), Day 12-megakaryoblasts (CD41+) from all the iPSC were sorted, cytokine-deprived for 24 h and then restimulated or not with 20 ng/mL of TPO and subjected to western blot analysis using specific antibodies. A representative experiment is shown out of 2. CFU-MK = colony forming unit-megakaryocyte; iPSC = induced pluripotent stem cell; JAK2 = Janus kinase 2; MEK = mitogen-activated protein kinase; MK = megakaryocyte; SCF = stem cell factor; SEM = standard error of the mean; STAT = signal transducer and activator of transcription; TPO = thrombopoietin; WT = wild-type.
Overexpression of CALR mutant has been shown to induce a constitutive activation of the JAK2/STAT pathway in cell lines7–10 and we tested whether endogenous CALRdel52 or CALRins5 mutants were able to spontaneously activate this signaling pathway. Day 12-megakaryoblasts (CD41+) were sorted, deprived of cytokines for 24 hours and stimulated or not by TPO before Western-blot analysis (Figure 5C and Supplemental Digital Content, Figure S7, http://links.lww.com/HS/A162). We detected significant spontaneous phosphorylation of STAT3 for all CALR-mutated compared with control iPSC-derived MKs, but a variable phosphorylation of ERK and AKT.
Taken together, these results show that both CALRdel52 and CALRins5 induce an increase in the MK lineage due to an independence/hypersensitivity to TPO with activation of STAT3. Moreover, both CALR mutants induce hypersensitivity of progenitors to G-CSF.
Effect of JAK2, HSP90, PI3K, and MEK signaling inhibitors on megakaryocytic colonies
Finally, we wondered if CALRdel52 iPSC could be used for testing small molecules targeting JAK2, PI3K, or ERK signaling or a chaperone protein of JAK2, HSP90. For this purpose, we seeded day 10-CD34+CD43+ progenitors in fibrin-clot assay and tested the effect of different inhibitors such as the JAK1/2 inhibitor ruxolitinib (Figure 6A, B), a HSP90 inhibitor, AUY922 (Figure 6C, D), a pan PI3K inhibitor, LY294002 (Figure 6E, F), and a MEK inhibitor, UO126 (Figure 6G, H). Dose-response studies were performed on the growth of CFU-MK in the presence or absence of TPO. All these 4 small molecules inhibited TPO-dependent megakaryocytic growth in a dose-dependent manner in all iPSC (WT and CALRdel52; Figure 6A, C, E, G). A trend to increased sensitivity to ruxolitinib and LY294002 were found for CALRdel52 progenitors compared with WT progenitors (Figure 6I). Spontaneous CALRdel52-induced CFU-MK were twice as more sensitive to JAK1/2, PI3K, and MEK inhibitors than the TPO-dependent CFU-MK colonies (Figure 6B, D, F, H, J).
Figure 6.
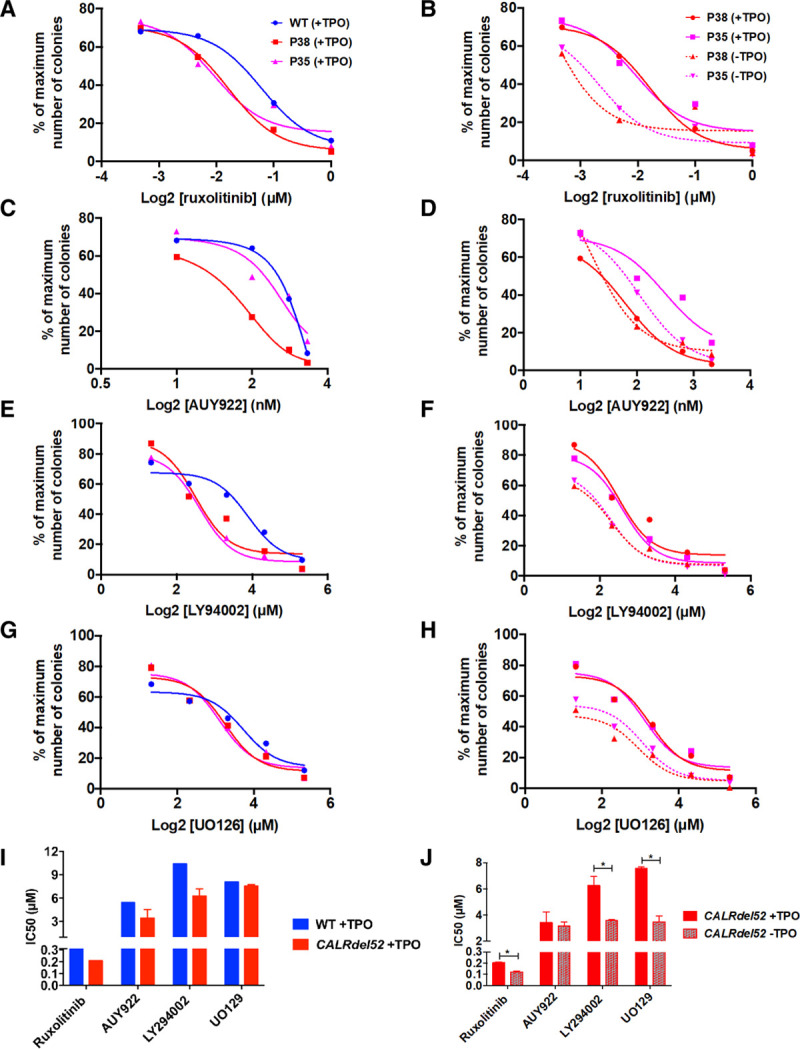
Impact of JAK2, HSP90, PI3K, and MEK signaling inhibitors on CFU-MK. Day 10-CD34+/CD43+ cells were seeded in fibrin clot assay with SCF and with or without TPO in presence of various inhibitors. (A), Ruxolitinib +TPO (mean ± SEM, WT, P38 and P35: n = 3). (B), Ruxolitinib ±TPO (mean ± SEM, P38 and P35: n = 3). (C), AUY922 + TPO (mean ± SEM, WT, P38 and P35: n = 3). (D), AU922 ±TPO (mean ± SEM, P38 and P35: n = 3). (E), LY94002 ±TPO (mean ± SEM, WT, P38 and P35: n = 3). (F), LY94002 ± TPO (mean ± SEM, P38 and P35: n = 3). (G), UO126 +TPO (mean ± SEM, WT, P38 and P35: n = 3). (H), UO126 ±TPO (mean ± SEM, P38 and P35: n = 3). (I, J), IC50 were calculated for each curve. (I), The graph represents the IC50 for WT and CALRdel52 (P35 and P38) in the presence of TPO. (J), The graph represents the IC50 for CALRdel52 (P35 and P38) in the presence or absence of TPO. *P < 0.05. Student t test. CFU-MK = colony forming unit-megakaryocyte; HSP90 = heat shock protein 90; IC50 = half-maximal inhibitory concentration; JAK2 = Janus Kinase 2; MEK = mitogen-activated protein kinase; PI3K = phosphoinositide 3-kinase; SCF = stem cell factor; SEM = standard error of the mean; TPO = thrombopoietin; WT = wild-type.
Altogether, this study brings the proof of concept that iPSC can be used for studying drug efficacy and shed light on JAK2/PI3K/ERK as specific druggable targets.
Discussion
IPSC have been proven to be a useful and powerful tool to model acquired hematological diseases. In this study, we have generated iPSC from 2 patients harboring CALRdel52 mutation either alone or associated with SETD1B mutation or from 2 patients harboring CALRins5 mutation alone or associated with PIK3CD mutation. Of note, we got a very high number of iPSC associated with PIK3CD mutation suggesting a potential positive impact of this mutation on reprogramming. Based on this technology, we studied the role of the endogenous driver mutations CALRdel52 and CALRins5 in a human hematopoietic context.
Here, we provide evidence that CALRdel52 and CALRins5 iPSC reproduce the MK hyperplasia observed in patients and in mouse models.7–9,15,22,37 We observed an increase in the generation of MK, a TPO hypersensitivity of the CFU-MK progenitors as well as an increase in their size in CALR-mutated iPSC derived from 4 different patients compared with control iPSC, suggesting a stronger MK precursor proliferation and/or a generation of more immature MK progenitors. Thus, CALR mutations modify the MK differentiation at multiple steps. Accordingly, we found a constitutive phosphorylation of STAT3 in MK obtained from CALRdel52 and CALRins5 iPSC. These phenotypes were not much impacted by the presence of PIK3CD or SETD1B mutation. Previously, similar effects on megakaryopoiesis were found for iPSCs with heterozygous and homozygous JAK2V617F28 and with the CALRins5 iPSC generated by the group of Komatsu.31 They are also in line with the MPL-dependent activation of MK hyperplasia induced by CALR mutants in primary cells, in iPSC, and in retroviral mouse models.7–10,15,16,18,37 We also show that CALRdel52 and CALRins5 impact the G-CSF sensitivity of progenitors. Unfortunately, we were not able to specifically test the effect of CALR mutants on granulocytic differentiation and signaling since the CD15 is also present on macrophages, which represents the majority of the cells in this type of iPSC-derived culture. These results are also in accordance with the fact that CALRdel52 does induce a weak activation of G-CSF-R and a transient independent growth of the Ba/F3 cell lines expressing G-CSF-R,7,15 but are more unexpected for CALRins5. However, the impact of this phenotype on the clinical feature is still elusive since JAK2V617F-mutated patients exhibit higher leukocytosis than CALR-mutated patients.3,4,38 Further studies should be conducted to precisely compare the effect of JAK2V617F and CALRdel52/ins5 on G-CSF-R signaling including STAT3/5, ERK, and AKT. Of note, iPSC differentiate into an embryonic type of hematopoiesis and it is possible that G-CSF-R activation by CALR mutants impact differently embryonic versus adult hematopoiesis. In contrast to homozygous JAK2V617F iPSC, we observe no or a moderate effect on the erythroid lineage and no EPO hypersensitivity of progenitors with CALRdel52 and even a tendency to a decreased generation of erythroblasts with CALRins5 previously observed in the work of Takei et al.28,30,31
A very interesting finding is the difference found in the generation of progenitors between CALRdel52 and CALRins5, suggesting that CALRdel52 may have a higher impact on the total number of myeloid progenitors than CALRins5. This result is comparable to what is observed in the CALRdel52 and CALRins5 KI mouse models or in the bone marrow model after retroviral transfer, and probably explains the lower severity of the disease in the case of CALRins5.9,16 Altogether, iPSC represent a valuable tool for MPN modeling, mimicking the 2 different diseases driven either by JAK2V617F or CALRdel52/ins5. Indeed, iPSC showed phenotypes that correlate with clinical features in patients: a MK phenotype with heterozygous JAK2V617F or CALRdel52/ins5 and an additive erythroid phenotype with homozygous JAK2V617F.10,28,30,31,39 They also fit well with the mouse modeling giving an ET/PV-like disease progressing to myelofibrosis for JAK2V617F and an ET phenotype progressing to myelofibrosis for CALRdel52.16,17,40,41
Previous studies have shown that overexpression of CALR mutants including CALRdel52 and CALRins5 activate the JAK2/STAT signaling pathway in cellular models and primary cells via MPL.7–9,15,22,37 Here, we confirm that endogenous CALRdel52/ins5 activates JAK2/STAT pathway in MKs by observing a constitutive phosphorylation of STAT3. The constitutive phosphorylation of ERK was not reproducible for all CALR-mutated iPSC unlike a previous study using primary cells from patients or transduced with lentiviral vectors encoding CALR mutations,42 although MEK inhibitor could inhibit the CALRdel52-induced spontaneous CFU-MK growth.
As previously shown, iPSC are a good model to test the efficacy and to screen molecules that are used in clinic or clinical trials.28,30 Dose-response studies performed on the growth of MK colonies with a JAK1/2 inhibitor (ruxolitinib), a PI3K inhibitor (LY294002), a HSP90 inhibitor (AUY922), and a MEK inhibitor (UO126) showed a lack of specificity for CALRdel52 progenitors from iPSC in the presence of cytokines. However, we found that the CALRdel52-mutated CFU-MK progenitors are more sensitive to ruxolitinib, LY294002, and UO126 in the absence than in the presence of TPO. This result indicates that the CALRdel52-induced MK spontaneous growth is more dependent on JAK2/PI3K/ERK than TPO-mediated growth and opens a therapeutic window for treatments of CALR-mutated MPN. It would be interesting to test others inhibitors, particularly STAT3 inhibitors for which we pointed a rationale to use, or newly developed inhibitors in future studies using these iPSC.
In aggregate, this study brings the proof-of-concept that iPSC can be used for studying CALRdel52/ins5-mutated MPN pathogenesis and drug screening.
Acknowledgments
We thank P. Rameau and Y. Lecluse for flow cytometry and cell sorting. We thank D. Muller for technical mouse support, O. Bawa for histology, and the staff of the animal facilities of Gustave Roussy directed by P. Gonin.
Sources of funding
This work was supported by grants from Institut National du Cancer (PLBIO2015 and PLBIO2017 to IP); Ligue Nationale Contre le Cancer (“Equipe labellisée 2013, 2016, 2019,” HR); MPN research foundation (IP), and Institut National de la Santé et de la Recherche Médicale (Inserm), CAPES-COFECUB (IP and MHB), PRONON (BM-M and MHB) and FAPERJ (BM-M). The program was supported by molecular medicine in oncology MMO (Gustave Roussy, Villejuif). LS was supported by PhD grants from the Region Ile de France (Cancéropôle and DIM cellule souche) and then Association pour la Recherche sur le Cancer. ME-K was funded by Société Française d’Hématologie. Support to SNC is acknowledged from Fondation contre le cancer Belgium, Salus Sanguinis, projects Action de recherché concertée (ARC) 16/21-073 and WelBio F 44/8/5 - MCF/UIG – 10955. CGL and SdS-B were supported by PhD grants from the Brazilian National Institute of Cancer and CAPES-COFECUB. Laboratory of Excellence Globule Rouge-Excellence (IP, WV) is funded by the program “Investissements d’avenir.”
Disclosures
The authors have no conflicts of interest to disclose.
Supplementary Material
Footnotes
The authors LS, CGL, ADS, and IP equally contributed to the study.
Supplemental digital content is available for this article.
References
- 1.Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013; 369:2379–2390. [DOI] [PubMed] [Google Scholar]
- 2.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013; 369:2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cabagnols X, Defour JP, Ugo V, et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: relevance for disease evolution. Leukemia. 2015; 29:249–252. [DOI] [PubMed] [Google Scholar]
- 4.Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia. 2016; 30:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tefferi A, Wassie EA, Guglielmelli P, et al. Type 1 versus type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol. 2014; 89:E121–E124. [DOI] [PubMed] [Google Scholar]
- 6.Michalak M, Groenendyk J, Szabo E, et al. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J. 2009; 417:651–666. [DOI] [PubMed] [Google Scholar]
- 7.Chachoua I, Pecquet C, El-Khoury M, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. 2016; 127:1325–1335. [DOI] [PubMed] [Google Scholar]
- 8.Elf S, Abdelfattah NS, Chen E, et al. Mutant calreticulin requires both its mutant c-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 2016; 6:368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marty C, Pecquet C, Nivarthi H, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016; 127:1317–1324. [DOI] [PubMed] [Google Scholar]
- 10.Araki M, Yang Y, Masubuchi N, et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood. 2016; 127:1307–1316. [DOI] [PubMed] [Google Scholar]
- 11.Pecquet C, Chachoua I, Roy A, et al. Calreticulin mutants as oncogenic rogue chaperones for TpoR and traffic-defective pathogenic TpoR mutants. Blood. 2019; 133:2669–2681. [DOI] [PubMed] [Google Scholar]
- 12.Elf S, Abdelfattah NS, Baral AJ, et al. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood. 2018; 131:782–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Araki M, Yang Y, Imai M, et al. Homomultimerization of mutant calreticulin is a prerequisite for MPL binding and activation. Leukemia. 2019; 33:122–131. [DOI] [PubMed] [Google Scholar]
- 14.Masubuchi N, Araki M, Yang Y, et al. Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia. 2020; 34:499–509. [DOI] [PubMed] [Google Scholar]
- 15.Balligand T, Achouri Y, Pecquet C, et al. Pathologic activation of thrombopoietin receptor and JAK2-STAT5 pathway by frameshift mutants of mouse calreticulin. Leukemia. 2016; 30:1775–1778. [DOI] [PubMed] [Google Scholar]
- 16.Benlabiod C, Cacemiro MDC, Nédélec A, et al. Calreticulin del52 and ins5 knock-in mice recapitulate different myeloproliferative phenotypes observed in patients with MPN. Nat Commun. 2020; 11:4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Prins D, Park HJ, et al. Mutant calreticulin knockin mice develop thrombocytosis and myelofibrosis without a stem cell self-renewal advantage. Blood. 2018; 131:649–661. [DOI] [PubMed] [Google Scholar]
- 18.Balligand T, Achouri Y, Pecquet C, et al. Knock-in of murine Calr del52 induces essential thrombocythemia with slow-rising dominance in mice and reveals key role of Calr exon 9 in cardiac development. Leukemia. 2020; 34:510–521. [DOI] [PubMed] [Google Scholar]
- 19.Shide K, Kameda T, Yamaji T, et al. Calreticulin mutant mice develop essential thrombocythemia that is ameliorated by the JAK inhibitor ruxolitinib. Leukemia. 2017; 31:1136–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Courtier F, Carbuccia N, Garnier S, et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica. 2017; 102:e11–e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundberg P, Nienhold R, Ambrosetti A, et al. Somatic mutations in calreticulin can be found in pedigrees with familial predisposition to myeloproliferative neoplasms. Blood. 2014; 123:2744–2745. [DOI] [PubMed] [Google Scholar]
- 22.El-Khoury M, Cabagnols X, Mosca M, et al. Different impact of calreticulin mutations on human hematopoiesis in myeloproliferative neoplasms. Oncogene. 2020; 39:5323–5337. [DOI] [PubMed] [Google Scholar]
- 23.Pasquier F, Cabagnols X, Secardin L, et al. Myeloproliferative neoplasms: JAK2 signaling pathway as a central target for therapy. Clin Lymphoma Myeloma Leuk. 2014; 14(suppl):S23–S35. [DOI] [PubMed] [Google Scholar]
- 24.Antony-Debré I, Manchev VT, Balayn N, et al. Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood. 2015; 125:930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saliba J, Saint-Martin C, Di Stefano A, et al. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat Genet. 2015; 47:1131–1140. [DOI] [PubMed] [Google Scholar]
- 26.Gandre-Babbe S, Paluru P, Aribeana C, et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood. 2013; 121:4925–4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumano K, Arai S, Hosoi M, et al. Generation of induced pluripotent stem cells from primary chronic myelogenous leukemia patient samples. Blood. 2012; 119:6234–6242. [DOI] [PubMed] [Google Scholar]
- 28.Saliba J, Hamidi S, Lenglet G, et al. Heterozygous and homozygous JAK2(V617F) states modeled by induced pluripotent stem cells from myeloproliferative neoplasm patients. PLoS One. 2013; 8:e74257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gomez Limia CE, Devalle S, Reis M, et al. Generation and characterization of a human induced pluripotent stem (iPS) cell line derived from an acute myeloid leukemia patient evolving from primary myelofibrosis carrying the CALR 52bp deletion and the ASXL1 p.R693X mutation. Stem Cell Res. 2017; 24:16–20. [DOI] [PubMed] [Google Scholar]
- 30.Ye Z, Liu CF, Lanikova L, et al. Differential sensitivity to JAK inhibitory drugs by isogenic human erythroblasts and hematopoietic progenitors generated from patient-specific induced pluripotent stem cells. Stem Cells. 2014; 32:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takei H, Edahiro Y, Mano S, et al. Skewed megakaryopoiesis in human induced pluripotent stem cell-derived haematopoietic progenitor cells harbouring calreticulin mutations. Br J Haematol. 2018; 181:791–802. [DOI] [PubMed] [Google Scholar]
- 32.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127:2391–2405. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131:861–872. [DOI] [PubMed] [Google Scholar]
- 34.Mali P, Ye Z, Hommond HH, et al. Improved efficiency and pace of generating induced pluripotent stem cells from human adult and fetal fibroblasts. Stem Cells. 2008; 26:1998–2005. [DOI] [PubMed] [Google Scholar]
- 35.Klimchenko O, Mori M, Distefano A, et al. A common bipotent progenitor generates the erythroid and megakaryocyte lineages in embryonic stem cell-derived primitive hematopoiesis. Blood. 2009; 114:1506–1517. [DOI] [PubMed] [Google Scholar]
- 36.Debili N, Coulombel L, Croisille L, et al. Characterization of a bipotent erythro-megakaryocytic progenitor in human bone marrow. Blood. 1996; 88:1284–1296. [PubMed] [Google Scholar]
- 37.Nivarthi H, Chen D, Cleary C, et al. Thrombopoietin receptor is required for the oncogenic function of CALR mutants. Leukemia. 2016; 30:1759–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tefferi A, Lasho TL, Finke CM, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014; 28:1472–1477. [DOI] [PubMed] [Google Scholar]
- 39.Ye Z, Zhan H, Mali P, et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009; 114:5473–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hasan S, Lacout C, Marty C, et al. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNα. Blood. 2013; 122:1464–1477. [DOI] [PubMed] [Google Scholar]
- 41.Tiedt R, Hao-Shen H, Sobas MA, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008; 111:3931–3940. [DOI] [PubMed] [Google Scholar]
- 42.Kollmann K, Warsch W, Gonzalez-Arias C, et al. A novel signalling screen demonstrates that CALR mutations activate essential MAPK signalling and facilitate megakaryocyte differentiation. Leukemia. 2017; 31:934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.