

Abstract
TAL1/SCL is one of the most prevalent oncogenes in T-cell acute lymphoblastic leukemia (T-ALL). TAL1 and its regulatory partners (GATA3, RUNX1, and MYB) positively regulate each other and coordinately regulate the expression of their downstream target genes in T-ALL cells. However, long non-coding RNAs (lncRNAs) regulated by these factors are largely unknown. Here we established a bioinformatics pipeline and analyzed RNA-seq datasets with deep coverage to identify lncRNAs regulated by TAL1 in T-ALL cells. Our analysis predicted 57 putative lncRNAs that are activated by TAL1. Many of these transcripts were regulated by GATA3, RUNX1, and MYB in a coordinated manner. We identified two novel transcripts that were activated in multiple T-ALL cell samples but were downregulated in normal thymocytes. One transcript near the ARID5B gene locus was specifically expressed in TAL1-positive T-ALL cases. The other transcript located between the FAM49A and MYCN gene locus was also expressed in normal hematopoietic stem cells and T-cell progenitor cells. In addition, we identified a subset of lncRNAs that were negatively regulated by TAL1 and positively regulated by E-proteins in T-ALL cells. This included a known lncRNA (lnc-OAZ3–2:7) located near the RORC gene, which was expressed in normal thymocytes but repressed in TAL1-positive T-ALL cells.
Introduction
Rapidly accumulating evidence demonstrates crucial roles for long non-coding RNAs (lncRNAs) in normal tissue development and cancer [1–6]. lncRNAs are generally defined as non-coding transcripts longer than 200 nucleotides that are encoded in the intergenic or intronic regions or overlapping with protein-coding genes [1]. lncRNAs are expressed in a tissue-specific manner and their expression levels are generally lower than those of protein-coding genes [7]. The lncRNA sequences are less conserved among species compared with protein-coding genes [8]. Importantly, a number of lncRNAs are functional and contribute to various aspects of cell homeostasis [5, 6]. The lncRNAs can regulate gene expression through interactions with genomic DNA, mRNA, or protein in the nucleus or cytoplasm [1, 3, 4], thus participating in the regulatory network in normal and malignant cells.
T-cell acute lymphoblastic leukemia (T-ALL) is a malignant disorder of thymic T-cell precursors [9]. One of the most prevalent oncogenes in T-ALL is the transcription factor TAL1/SCL, which is an essential regulator of hematopoiesis [10, 11]. TAL1 is normally expressed in hematopoietic stem cells (HSCs), progenitor cells, and erythro-megakaryocytic lineages, but it is silenced during T-cell development [12, 13]. In contrast, TAL1 is ectopically over-expressed in 40%–60% of T-ALL cases due to chromosomal translocation, intra-chromosomal rearrangement, or mutations in the non-coding element [14, 15]. In both normal hematopoietic cells and T-ALL cells, TAL1 forms a heterodimer complex with a class I bHLH protein, known as E-proteins (E2A, HEB), and makes a large transcriptional complex with LMO protein (LMO1 or LMO2), LDB1, and GATA protein (GATA1, GATA2, or GATA3). We previously reported that in T-ALL cells, TAL1 and several hematopoietic transcription factors (GATA3, RUNX1, and MYB) regulate the expression of downstream target genes in a coordinated manner [16]. These four factors also co-occupy their own regulatory elements and positively regulate each other, forming an interconnected auto-regulatory loop [15, 16]. The same structure has been reported in normal HSCs [17, 18].
In normal developing thymocytes, E-proteins form a homo- or heterodimer and regulate genes required for T-cell differentiation, such as RAG1, RAG2, and PTCRA [19, 20]. Deficiency in E-proteins leads to blocked differentiation and development of T-cell malignancies in mice [21]. TAL1 can inhibit the formation of E-protein dimers by forming a more stable transcriptional complex [22, 23], resulting in the deregulation of E-protein target genes. Together, TAL1 disrupts the transcriptional regulatory program in developing thymocytes by inducing stem cell-like transcriptional circuitry and by blocking T-cell differentiation program. Thus, it is important to identify downstream factors controlled by TAL1 and E-proteins. We have previously identified several protein-coding genes and microRNAs that are directly activated by the TAL1 complex and contribute to T-ALL pathogenesis [16, 24–27]. However, lncRNAs that are regulated by TAL1 and E-proteins in T-ALL cells are entirely unknown.
In this study, we established a bioinformatics pipeline for the prediction of lncRNAs and analyzed the RNA sequencing (RNA-seq) dataset with deep coverage after knockdown of each member of the TAL1 complex in T-ALL cells. This analysis identified 57 putative lncRNAs that are directly regulated by TAL1, including novel transcripts. Many were coordinately regulated by TAL1 and its regulatory partners. Some of them were repressed in normal thymocytes. In addition, we identified lncRNAs that are differentially controlled by TAL1 and E-proteins.
Materials and Methods
Cell culture and gene knockdown
All T-ALL cell lines were stocked in our laboratory [24, 25] and cultured in RPMI-1640 medium (BioWest) supplemented with 10% fetal bovine serum (FBS; BioWest). All cell lines were confirmed by DNA fingerprinting using the PowerPlex 1.2 system (Promega) in January 2013 and were used from the original stock. Cell lines were regularly tested for mycoplasma contamination. See Supplementary Table 1 for the expression of transcription factor genes defining subgroups of T-ALL. The mouse lymphohematopoietic progenitor cell line EML was cultured in Iscove’s modified Dulbecco’s medium supplemented with 20% FBS and 200 ng/ml Recombinant Murine SCF (Peprotech). Lentiviruses encoding short-hairpin RNAs (shRNAs) targeting each transcription factor were produced and infected the T-ALL cells, as described previously [24]. Detailed procedures are described in the Supplementary Information.
RNA-seq analysis
Total RNA was extracted using a miRNeasy kit (Qiagen) followed by DNase treatment (Turbo DNA-freeTM kit, Ambion). After the depletion of ribosomal RNAs, strand-specific library construction and sequencing of paired-end, 100 bp-long reads by the Illumina HiSeq4000 platform were performed at BGI Biotech Solutions Co., Ltd (Hong Kong). See Supplementary Table 2 for the number of sequencing reads and quality scores for each sample. All RNA-seq data have been deposited in the NCBI GEO database (GSE97514 and GSE103046) [27]. The RNA-seq datasets for human hematopoietic cells reported by Casero et al. [28] and for mouse hematopoietic cells reported by Sun et al. [29] were obtained from the GEO under accession numbers GSE69239 and GSE47819, respectively. Twenty-seven primary T-ALL cases from the Ma-Spore ALL 2003 study (DSRB ref number 2004/00275) were analyzed in this study (Supplementary Table 3). Primary cells were obtained with informed consent after approval by the institutional ethical committee. The RNA-seq dataset for primary samples have been reported [30] and deposited by Jun J. Yang in the EGA database (EGAD00001002151). Genomic sequence for the selected lncRNA retrieved from human (hg19) genome were aligned to mouse (mm10) genome using the UCSC BLAT tool. Details are described in the Supplementary Information.
ChIP-seq datasets
The Jurkat chromatin immunoprecipitation sequencing (ChIP-seq) datasets for TAL1, GATA3, RUNX1, MYB, and H3K4me1 have been reported by us and the Young lab [16, 26, 31], and can be downloaded from the GEO (GSE29181, GSE68976, and GSE59657). The Jurkat ChIP-seq dataset for H3K4me3 [32] was obtained from the GEO (GSM945267 and GSM945268). H3K27ac ChIP-seq datasets for Jurkat, RPMI-8402, CCRF-CEM, MOLT-4 [15], and normal thymus [33] were obtained from the GEO (GSM1296384, GSM1442003, GSM2037781, GSM2037790, and GSM1013125). H3K27ac ChIP-seq datasets for Loucy [34] and DND-41 [35] were downloaded from the GEO (GSE74312 and GSE54380). ChIP-seq data and super-enhancers were analyzed as described previously [24, 31, 36, 37]. Details are shown in the Supplementary Information.
Identification of lncRNAs
RNA-seq datasets of T-ALL cell lines were aligned to the hg19 human genome using STAR 2.5.2a and Ensembl database [38]. Alignment files were used for ab initio transcriptome assembly with Cufflinks v2.2.1 [39]. A RefSeq GTF file was applied for Cufflinks to guide the assembly. Transcriptome assemblies from each cell line were combined into one annotation using Cuffmerge from Cufflinks v2.2.1. The novel lncRNA transcripts were included when calculating the fragments per kilobase of transcript per million mapped read (FPKM) matrix. Details are described in the Supplementary Information.
Correlation analysis with neighboring gene
Two protein-coding genes or a pair of lncRNA and protein-coding gene as neighbors were defined by a minimal distance of less than 100 kb. To calculate the Pearson’s correlation of two neighbors, FPKM (cutoff 1 in at least one T-ALL cell line) was log2 transformed after addition of 0.05.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) [40] was performed for the normalized genes using the log2_Ratio-of-Classes as a metric for ranking genes. TAL1 target genes that were downregulated by knockdown were defined using the RNA-seq dataset and used as a gene set.
Quantitative reverse transcription PCR analysis
Total RNA was collected using a NucleoSpin RNA kit (Macherey-Nagel) and reverse-transcribed into cDNA using QuantiTect (QIAGEN). Quantitative PCR analysis was performed on a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) using Power SYBR Green PCR Master Mix (Roche). The PCR primer sequences can be found in the Supplementary Information.
Rapid amplification of cDNA ends
Rapid amplification of cDNA end (RACE) was performed using the SMARTer RACE 5’/3’ Kit (Takara) according to the manufacturer’s instructions. Briefly, total RNA was first extracted from Jurkat cells and subjected to the SMARTer first-strand cDNA synthesis to generate either 5′-RACE or 3′-RACE ready cDNA. The 5′-RACE and 3′-RACE PCR was then performed using touchdown PCR using specific gene primers (see Supplementary Information), and the generated PCR fragments were purified and cloned into the pRACE vector for subsequent Sanger sequencing using In-Fusion HD cloning provided by the kit.
In vitro transcription and translation assay
The in vitro transcription and translation assay was performed using a TNT Quick Coupled Transcription/Translation System (Promega) according to manufacturer’s instructions. Briefly, we first cloned the sense or antisense sequence of XLOC_005968, the full-length mRNA of C10ORF107/CABCOCO1 or GATA3 into the expression plasmid pCS2. See Supplementary Information for primer sequences. A total of 500 ng of plasmid DNA was mixed with reagents for the in vitro transcription and translation assay, and incubated at 30 °C for 90 min. Western immunoblotting was performed using the lysates to detect the presence of translated protein. Details are described in the Supplementary Information.
Statistical analysis
The Wald test was used to analyze differences in gene expression among groups by RNA-seq analysis. Two-tailed Student’s t-tests were used to analyze differences in gene expression by quantitative reverse transcription PCR (qRT-PCR) among groups. Adjusted P-values < 0.05 were considered statistically significant.
Results
Prediction of lncRNAs expressed in T-ALL cells
To identify lncRNAs expressed in T-ALL cells, we performed RNA-seq analysis in eight T-ALL cell lines, which represent several major subgroups of T-ALL (Supplementary Table 1). For RNA-seq analysis, we depleted ribosomal RNAs and included both polyA + and polyA – RNAs, because many lncRNAs have been known to lack polyA sequences. From this analysis, we obtained over 300 million reads of high quality for each sample (Supplementary Table 2). Using this dataset, we established an analytical pipeline to predict lncRNAs (Fig. 1a).
Fig. 1.

Identification of lncRNAs expressed in T-ALL cells. a Schematic diagram of the analytical pipeline. See Materials and Methods for details. b RNA-seq gene tracks showing the expression and genomic loci of three representative lncRNAs expressed in T-ALL cell lines: XLOC_040800/lnc-FAM160A1–6 (top), XLOC_012951/lnc-DUSP6–2 (middle), and XLOC_005308/lnc-TOMM20–2 (bottom). The x axis indicates the linear sequence of genomic DNA and the yaxis indicates the number of mapped reads per million. The black horizontal bar indicates the genomic scale in kilobases (kb). Black boxes in the gene map represent exons, and arrows indicate the location and direction of the transcriptional start site. c Violin plots showing expression levels of protein-coding genes and lncRNAs in T-ALL. The y axis indicates the maximum FPKM values (log10) of protein-coding genes and lncRNAs among eight T-ALL cell lines. Black boxes represent median and quartile expression levels. d Pie chart showing the percentage of each class of lncRNA. See Supplementary Figure 1A for details of classification. e Density distributions showing pairwise Pearson’s correlations between loci. Pairs were selected for lncRNAs or protein-coding genes (mRNA) with their neighboring protein-coding genes. As a control, 1000 mRNA–mRNA pairs were randomly selected. The y axis indicates density at each correlation value (x-axis).
We first mapped all sequence reads to the human genome (hg19), assembled them into transcripts, and defined the annotations (i.e., “XLOC_000001”). After removing short transcripts, we applied the Slncky pipeline developed by Chen et al. [41] to search for high-confidence lncRNAs (“putative lncRNAs 1”). As this tool potentially filters out lncRNAs that overlap with protein-coding genes, we included an additional pipeline to predict lncRNAs that share genomic DNA sequences with protein-coding genes. From transcripts overlapping with protein-coding genes, which were rejected from Slncky pipeline, we identified 1031 additional transcripts (“putative lncRNAs 2”). Finally, we filtered out low-expression lncRNAs for which the FKPM value was below the cutoff of 0.5. From this analysis, we identified a total of 2236 putative lncRNA loci including 4857 transcripts (Supplementary Table 4).
To confirm the validity of our pipeline, we compared our result with two public databases and the results for T-ALL samples reported by others. Out of 2236 putative lncRNA loci, we identified 1321 (59%) that were annotated in the GENCODE version 25 (total 15,875 loci); 1055 (47%) were annotated in the LNCipedia version 4.0 (total 48,044 loci). Importantly, several lncRNAs were previously reported to be differentially expressed among the different subgroups of T-ALL in the dataset by Wallaert et al. [34]. For example, lnc-FAM160A1–6 was shown to be highly expressed in the TAL1-positive subgroup, whereas lnc-DUSP6–2 and lnc-TOMM20–2 were reported to be highly expressed in the ETP/immature T-ALL and TLX-positive subgroups, respectively [34]. Consistently, our pipeline identified the lnc-FAM160A1–6 that was highly expressed in four TAL1-positive T-ALL cell lines (Jurkat, RPMI-8401, CCRF-CEM, and MOLT-4) (Fig. 1b, top; and Supplementary Table 1). In contrast, lnc-DUSP6–2 was specifically expressed in DND-41 cells (Fig. 1b, middle), which represents the TLX-positive subgroup. In addition, lnc-TOMM20–2 was specifically expressed in Loucy cells (Fig. 1b, bottom), which represents the ETP/immature T-ALL subgroup [42]. These results indicated that our pipeline accurately predicted lncRNAs known to be expressed in T-ALL.
Characterization of lncRNAs expressed in T-ALL cells
We next analyzed the expression levels of the predicted lncRNAs and positional overlap with protein-coding genes. As generally reported, the RNA expression level of putative lncRNAs was significantly lower than that of protein-coding genes, for which the median log10 maximum FPKM value was 0.17 (Fig. 1c). Classification of lncRNAs based on the positional relationship with annotated protein-coding genes indicated that approximately half were located in intergenic regions; the other half were found within the protein-coding genes (“sense”, “antisense,” or “intronic”) or in the opposite direction to nearby protein-coding genes ( < 1,000 bp) sharing the same transcriptional start site (“divergent”/bidirectional) (Fig. 1d and see Supplementary Figure 1A for the definitions).
We then analyzed the correlation in RNA expression levels between putative lncRNAs and mRNAs transcribed from neighboring protein-coding genes. This analysis demonstrated a higher positive correlation in the lncRNA–mRNA pairs (blue) than the random pairs (purple: Fig. 1e). This level was similar to that in the mRNA-mRNA pairs (red). These results support previous findings reported by others [7, 43, 44]. We did not observe notable differences between the “putative lncRNAs 1” and “putative lncRNAs 2” (Supplementary Figure 1B).
Regulation of lncRNAs by TAL1 and its regulatory partners in T-ALL cells
We next aimed to identify lncRNAs that are directly regulated by TAL1 and its regulatory partners (E2A, HEB, GATA3, RUNX1, and MYB) in T-ALL cells. For this purpose, we performed RNA-seq analysis after lentiviral shRNA knockdown of each of these transcription factors in one of the TAL1-positive cell lines (Jurkat). We independently validated the efficiency of knockdown using qRT-PCR analysis (Supplementary Figure 2A). We used the same annotations defined earlier (i.e., “XLOC_000001”) to compare the expression level of each lncRNA between the control and knockdown samples.
We first selected the putative lncRNAs for which the TAL1 protein binds near or at the lncRNA loci using our ChIP-seq dataset in Jurkat cells [16]. We applied the same cutoff (within 12 kb of the lncRNA loci) that we previously used for identifying protein-coding genes and microRNAs targeted by TAL1 [16, 26]. We then filtered transcripts that were significantly downregulated after shRNA knockdown of TAL1 in the same cells (adjusted p-value < 0.05 and log2-fold change < −0.2). A total of 57 putative lncRNAs were selected by these criteria (Fig. 2a and Supplementary Table 5). We next analyzed the effect of the regulatory partners of TAL1 (GATA3, RUNX1, and MYB) on the expression of TAL1-regulated lncRNAs in the same cells. The heatmap analysis illustrated that many of the transcripts were also downregulated after knockdown of GATA3, RUNX1, or MYB (Fig. 2b). The GSEA analysis demonstrated a positive correlation in the RNA expression changes after knockdown of each transcription factor (Fig. 2c). This trend was essentially similar to that observed for protein-coding genes using microarray analysis in our previous study [16] and by RNA-seq analysis in our current study (Supplementary Figures 2B–D). This indicated that TAL1 coordinately regulates lncRNAs with its regulatory partners in T-ALL cells.
Fig. 2.

Regulation of lncRNAs by TAL1 and its regulatory partners in Jurkat cells. a,b Expression of TAL1 target lncRNAs after knockdown (KD) of transcription factor genes in Jurkat cells. Jurkat cells were transduced with control shRNA (GFP or Luc) or shRNA targeting a transcription factor gene (TAL1, GATA3, RUNX1, or MYB) by lentivirus infection. Two different shRNAs (1 and 2) were used for each gene. Heatmap images represent expression levels of 57 selected lncRNAs in two controls and two knockdown samples. c GSEA was performed to determine the correlation of lncRNAs positively regulated by TAL1 (n = 57) and gene expression changes upon KD of GATA3, RUNX1, and MYB. GSEA plot indicates the degree to which TAL1 target lncRNAs are overrepresented at the extreme left (downregulated by KD) or right (upregulated by KD) of the entire ranked list. Each solid bar represents one lncRNA gene within the gene set. Normalized enrichment scores (NES) and p-values are shown. d Venn diagram represents the number of lncRNAs directly regulated by each transcription factor in Jurkat cells.
In addition, we evaluated the overlap of target lncRNAs that are directly regulated by each transcription factor (Fig. 2d). As expected, we observed a high level of overlap between TAL1 and other transcription factors, where 49 (86.0%) out of 57 putative lncRNAs were also directly regulated by at least one transcription factor (GATA3, RUNX1, or MYB). In contrast, RUNX1 possessed many other target lncRNAs that were not directly regulated by TAL1, GATA3, or MYB. This result was similar to the previous observation for protein-coding genes [16], suggesting that RUNX1 could also regulate lncRNA expression independent of the TAL1 complex in T-ALL cells.
Two novel lncRNAs aberrantly activated by the TAL1 complex in T-ALL cells
Next, we sought lncRNAs that are normally downregulated in thymocytes and are activated by TAL1 in T-ALL cells. Among 57 putative lncRNAs selected as direct targets of TAL1 (Fig. 2a), we filtered the ones that were also significantly downregulated after knockdown of GATA3, RUNX1, and MYB, and were bound by all these factors (Fig. 3a). We further narrowed down this list to select lncRNAs associated with super-enhancers, which are defined as clusters of enhancer that exhibit high levels of the H3K27ac active histone mark [31, 36, 37], in the same T-ALL cells but not normal thymus in ChIP-seq analysis (Fig. 3b,c and Supplementary Figures 3A and 3B). This selection fielded lncRNAs that might be abnormally activated in T-ALL cells. We independently validated the expression of each candidate by qRT-PCR in eight T-ALL cell lines (Supplementary Figures 3C and 3D) and after TAL1 knockdown in Jurkat cells (Supplementary Figure 3E). We excluded one transcript, which was not significantly downregulated by qRT-PCR. Lastly, we focused on the transcripts that have not been previously annotated. These criteria selected two putative lncRNAs (XLOC_030252 and XLOC_005968) (Fig. 3b,c).
Fig. 3.
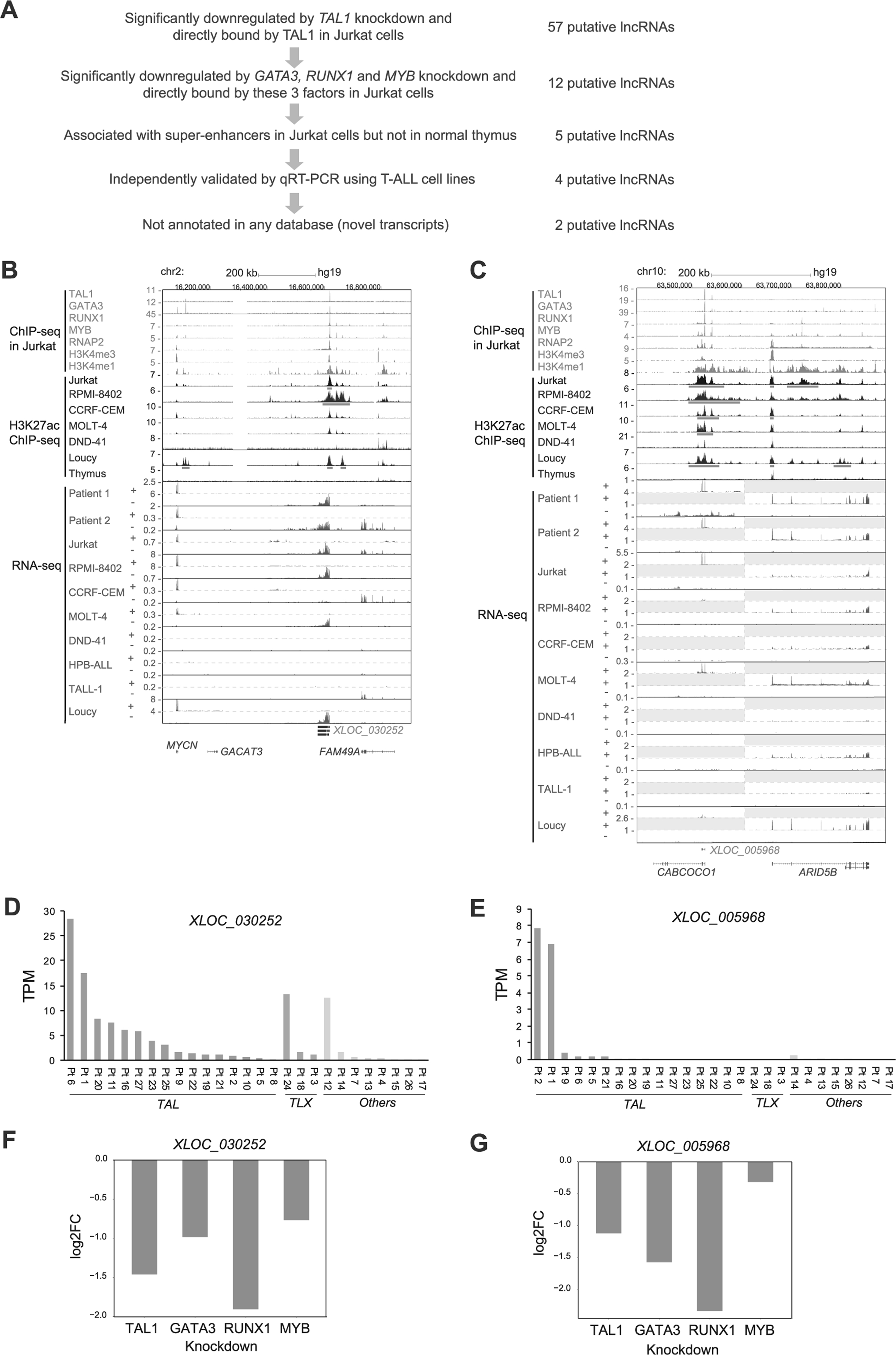
lncRNAs aberrantly activated by TAL1 in T-ALL cells. a Schematic diagram of selection criteria. b,c ChIP-seq gene tracks showing binding locations of TAL1, its regulatory partners (GATA3, RUNX1, and MYB) and RNA polymerase 2 (RNAP2) at XLOC_030252 b and XLOC_005968 c loci in Jurkat cells. ChIP-seq data for H3K27ac in six T-ALL cell lines and normal thymus (bulk) and RNA-seq data in eight T-ALL cell lines and two primary samples (Patients 1 and 2) are also shown. The y axis indicates the total number of mapped ChIP-seq reads per million. +, positive strand; –, negative strand, analyzed by strand-specific RNA-seq. See Fig. 1b legend for details of RNA-seq gene tracks. Two scales were included for the positive strand in Fig. 3c, because expression levels of XLOC_005968 and protein-coding genes were highly different. d,e RNA expressions of XLOC_030252 d and XLOC_005968 e in 27 primary T-ALL cases reported by Qian et al. [30]. T-ALL samples were classified into TAL (TAL1 or TAL2 positive), TLX (TLX1 or TLX3 positive), or others. Values are shown by the transcripts per kilobase million (TPM). There was no statistical significance observed due to small size of the cohort. f,g RNA expression changes of XLOC_030252 f and XLOC_005968 g after knockdown of TAL1, GATA3, RUNX1, and MYB in Jurkat cells analyzed by RNA-seq. Values are shown in log2 fold-change (FC).
XLOC_030252 was identified as a “putative lncRNAs 1” (Fig. 1a) and was located in the intergenic region between the MYCN and FAM49A gene locus (Fig. 3b). The expression was observed in four TAL1-positive T-ALL cell lines (Jurkat, RPMI-8402, CCRF-CRM, and MOLT-4) and one TAL1-negative cell line (Loucy) (Fig. 3b and Supplementary Table 3). This transcript was expressed in many of TAL-positive subgroup of primary T-ALL, whereas it was also found in TLX-positive or other cases (Fig. 3b,d and Supplementary Figure 3F). On the other hand, XLOC_005968 was identified as a “putative lncRNAs 2” in our pipeline (Fig. 1a), which could not be predicted by the Slncky model, because the genomic position of XLOC_005968 overlapped with the last 3 exons (exons 5–7) of a protein-coding gene CABCOCO1/C10ORF107 (Fig. 3c). We detected the expression of this particular transcript in two TAL1-positive T-ALL cell lines (Jurkat and MOLT-4) and two TAL1-positive primary leukemia samples (Patients 1 and 2) (Fig. 3c,e). This transcript was not expressed in TAL1-negative T-ALL cases above the cut-off value (Fig. 3e). There was no statistical significance observed due to small size of the cohort. Importantly, the full-length of CABCOCO1 was not expressed in any T-ALL samples based on the RNA-seq analysis (Fig. 3c). To verify this result, we designed specific primers targeting the exon 3 of CABCOCO1, which was not shared by XLOC_005968, or the exon 7, which are shared by XLOC_005968, and measured the expression using qRT-PCR analysis. This result revealed that only exon 7 was expressed in Jurkat and MOLT-4 cells (Supplementary Figures 3C and 3D).
Critically, XLOC_030252 and XLOC_005968 loci were bound by TAL1 and its regulatory partner proteins (GATA3, RUNX1, and MYB) (Fig. 3b,c). The expression of both transcripts was downregulated after knockdown of each of these factors (Fig. 3f,g). Those loci were associated with super-enhancers in TAL1-positive T-ALL cell lines but not normal thymus (Fig. 3b,c, red bars). Together, our results suggested that XLOC_030252 and XLOC_005968 are normally repressed in developing thymocytes but are highly activated by TAL1 under super-enhancers in T-ALL cells.
Expression of predicted lncRNAs in normal hematopoietic cells
We next examined whether XLOC_030252 and XLOC_005968 were expressed in normal human hematopoietic cells, using a dataset reported by Casero et al. [28]. Notably, XLOC_030252 was expressed in human HSCs and early double-negative stage thymocyte/progenitor cells (“thy1–2”: see figure legend for definition). However, it was downregulated in double-positive (DP) stage (“thy4”) and single-positive (SP) stage thymocytes (“thy5, 6”) (Fig. 4a, b), which consist the vast majority of thymocytes. This result was consistent with the H3K27ac ChIP-seq data (Fig. 3b). Interestingly, chromosomal position of XLOC_030252 was conserved between human and mouse (Fig. 4c). Using the RNA-seq dataset for mouse HSCs reported by Sun et al. [29], we were able to detect RNA expression between the Mycn and Fam49a gene locus (Fig. 4c), which corresponded to the human XLOC_030252 locus. We validated this result by qRT-PCR analysis using freshly isolated samples. Although the expression pattern was slightly different from human cells, we found that the transcript was expressed in mouse HSCs and progenitor cells and downregulated after DN1 stage thymocytes (Fig. 4d), which is similar to the expression pattern of mouse Tal1 (Supplementary Figure 4A). Notably, the expression of this transcript was significantly downregulated after knockdown of Tal1 in the mouse hematopoietic progenitor cell line (EML) (Fig. 4e). These results indicated that XLOC_030252 is expressed in normal HSCs and progenitor cells but is downregulated during T-cell development. It is noted that one TAL1-negative cell line (Loucy), which represents the ETP subtype of T-ALL [42], also expressed XLOC_030252 (Fig. 3b). As this transcript is expressed in T-cell progenitors (“thy1”) (Fig. 4a,b), which correspond to ETP that is characterized by the absence of CD1a and CD8 expression, activation of this lncRNA in Loucy cells may reflect the stage of normal T-cell development.
Fig. 4.

Expression of XLOC_030252 in human and mouse hematopoietic cells. a,b Expression of XLOC_030252 in different stages of human hematopoietic cells. RNA-seq gene tracks showing RNA expression in human hematopoietic cells at XLOC_030252 locus a. Each cell type was defined by Casero et al. [28] as follows: HSC, CD34+CD38neglinneg; lymphoid-primed multipotent progenitors (LMPP), CD34+CD38+CD10negCD45RA+CD62Lhighlinneg; common lymphoid progenitor (CLP), CD34+CD38+CD10+CD45RA+linneg; Thy1, CD34+CD7negCD1anegCD4negCD8neg; Thy2, CD34+CD7+CD1anegCD4negCD8neg; Thy3, CD34+CD7+CD1a+CD4negCD8neg; Thy4, CD4+CD8+; Thy5, CD3+CD4+CD8neg; and Thy6, CD3+CD4negCD8+. Two scales were included, because expression levels of XLOC_030252 and protein-coding genes were different. Expression level of XLOC_030252 in duplicate samples reported by Casero et al. [28] were shown by the TPM values. b,c RNA-seq dataset reported by Sun et al. [29] was used to analyze RNA expression in mouse HSCs collected at different ages (4 and 24 months) at the genomic locus conserved with human XLOC_030252 sequence. d The RNA expression of putative paralog of XLOC_030252 in different stages of mouse hematopoietic cells were measured by qRT-PCR and normalized to β2M expression. Long-term HSC (LT-SHC), short-term HSC (ST-HSC), multi potent progenitor (MPP), common lymphoid progenitor (CLP), DN1–4, DP, CD4 single-positive and CD8 single-positive cells. Fold-change values compared with LT-HSC are shown as the mean ±standard deviation (SD) of duplicate samples. See Supplementary Materials for the definition of each population. e Mouse lymphoid progenitor cell line EML was transduced with control shRNA (shLuc) or shRNA targeting mouse Tal1 (sh1 and sh2) by lentivirus infection. RNA expression of putative paralog of human XLOC_030252 was measured by qRT-PCR and normalized to β2M expression. Fold-change values compared with the control are shown as the mean ± SD of duplicate samples. **p < 0.01 by two-sample, two-tailed t-test.
In contrast, the expression of XLOC_005968 was found to be unique to human TAL1-positive T-ALL cells. This transcript was not detected in normal human hematopoietic cells in the dataset by Casero et al [28] (data not shown). The mouse genome possesses the Cabcoco1 gene near the Arid5b gene locus (Supplementary Figure 4B), similar to the human genome, where XLOC_005968 is located (Fig. 3c). However, RNA expression was detected in all exons of Cabcoco1, in marked contrast to human T-ALL cells where only XLOC_005968, but not the full-length CABCOCO1, was expressed (Fig. 3c). According to qRT-PCR analysis, we independently confirmed that exon 1 of the Cabcoco1 gene was expressed in mouse HSCs (Supplementary Figure 4C). Thus, these results indicated that the expression of XLOC_005968 was specific to TAL1-positive human T-ALL cells.
lncRNAs differentially controlled by TAL1 and E-proteins in T-ALL cells
We next analyzed the lncRNAs that are differentially controlled by TAL1 and E-proteins. TAL1 has been reported to inhibit E-protein function by sequestering E-protein dimers, leading to deregulation of E-protein target genes [22, 23]. Other group previously reported that the mutant form of TAL1, which cannot bind to DNA, could still induce T-cell leukemia/lymphoma in mice [45], indicating that TAL1 can also exert its oncogenic ability through inhibition of E-protein functions. Hence, it would be of interest to identify lncRNAs that are positively regulated by E-proteins and opposed by TAL1 in T-ALL cells. From our current RNA-seq analysis, we confirmed that RAG1, RAG2, and PTCRA, which are known E-protein targets [19, 20], were upregulated by knockdown of TAL1 and were further downregulated by knockdown of E2A and HEB in Jurkat cells (Supplementary Figures 5A–C). Although basal expression levels of these genes are low, this cell line is still useful for analysis of transcripts activated by E-proteins.
First, we selected putative lncRNAs (n = 199) that were upregulated by TAL1 knockdown (Fig. 5a and Supplementary Table 6). We next analyzed the expression changes of these transcripts after knockdown of E2A and HEB in the same cell line. Interestingly, 81 putative lncRNAs were downregulated by E2A and/or HEB knockdown (top), showing that these are positively regulated by E-proteins and are opposed by TAL1. In contrast, 49 putative lncRNAs were also upregulated by E2A and/or HEB knockdown (bottom), indicating that these transcripts are cooperatively regulated by TAL1 and E-proteins, possibly as the TAL1-E-protein heterodimer. GSEA showed a mixed pattern of gene regulation, including both positively and negatively correlated genes (Fig. 5b).
Fig. 5.

lncRNAs differentially controlled by TAL1 and E-proteins in T-ALL cells. a Jurkat cells were transduced with control shRNA (shGFP or shLuc) or shRNA targeting a transcription factor gene (TAL1, E2A, or HEB) by lentivirus infection. Two different shRNAs (1 and 2) were used for each gene. Heatmap images represent expression levels of 199 selected putative lncRNAs in two controls and two knockdown (KD) samples. b GSEA was performed to determine the correlation of lncRNAs negatively regulated by TAL1 (n = 199) and gene expression changes on KD of E2A and HEB. See Fig. 2c legend for details. c RNA-seq gene track showing expression and genomic loci of XLOC_001561/lnc-OAZ3–2:7 and RORC gene in T-ALL cell lines and normal hematopoietic cells. See Fig. 1b legend for details. d RNA expression changes of XLOC_001561/lnc-OAZ3–2:7 after knockdown of TAL1, E2A, and HEB in Jurkat cells. e RNA expression levels of XLOC_001561/lnc-OAZ3–2:7 in different stages of normal hematopoietic cells. See Fig. 4a legend for details.
For example, lncRNAs that were differentially controlled by TAL1 and E-proteins included the XLOC_001561, which is transcribed from the antisense strand encoding the RORC gene on chromosome 1 (Fig. 5c). This transcript has been annotated as lnc-OAZ3–2:7 in the LNCipedia. RORC has been reported as an E2A target [46]. Both XLOC_001561 and RORC expression were downregulated by E2A and HEB knockdown and were upregulated by TAL1 knockdown in Jurkat cells (Fig. 5d and Supplementary Figure 5D). We independently validated this result by qRT-PCR analysis in eight T-ALL cell lines (Supplementary Figure 5E) and after TAL1 knockdown in Jurkat cells (Supplementary Figure 5F).
Notably, DNA binding of TAL1 was not observed at this locus (Supplementary Figure 5G), suggesting that TAL1 negatively regulates the expression of XLOC_001561 and RORC in a DNA binding-independent manner, possibly through the inhibition of E-protein dimers. Both XLOC_001561 and RORC were more highly expressed in TAL1-negative cell lines (HPB-ALL and TALL-1) than in TAL1-positive cases (Fig. 5c). Importantly, this putative lncRNA was upregulated in the DP stage (“thy4”) and CD4+ or CD8+ SP stages of thymocytes (“thy5” and “thy6”) (Fig. 5e). A similar pattern was observed for the RORC gene (Supplementary Figure 5H). These results indicated that XLOC_001561 and RORC are activated by E-proteins in developing T-cells and are repressed by TAL1 in malignant T-cells.
Cloning of a novel lncRNA located near the ARID5B gene locus
Finally, we validated the protein-coding potential of the candidate lncRNA. We selected XLOC_005968 (Fig. 3c) for this purpose, because this transcript was uniquely identified by our pipeline and specifically expressed in human T-ALL cells. The expected size of the transcript was also < 1 kb, and thus it could be inserted into an expression vector, whereas the other novel lnRNA (XLOC_030252) was too large to be cloned.
We first tried to detect the full-length sequence of XLOC_005968 by the RACE method using specific primers (Fig. 6a and Supplementary Figure 6A). We successfully identified 802-bp nucleotide sequence for this transcript (Supplementary Figure 6B). We then performed an in vitro transcription assay followed by an in vitro translation analysis to analyze whether it can encode any proteins. We included full-length CABCOCO1 and GATA3 cDNA as positive controls. This analysis revealed that the sense or anti-sense strand of XLOC_005968 did not produce any proteins, while CABCOCO1 and GATA3 showed the expected sizes of translated protein (Fig. 6b). This result clearly indicated that XLOC_005968 is a lncRNA and validated our bioinformatics pipeline. Importantly, XLOC_005968 has not been previously annotated in public databases. Therefore, XLOC_005968 is a novel lncRNA.
Fig. 6.

XLOC_005968 is a novel lncRNA expressed in T-ALL cells. a The 5′ and 3′ RACE primers were designed to target exons 3 and 1 of the XLOC_005968 transcript, respectively. b In vitro transcription and translation assay was performed using expression vectors encoding sense or anti-sense strands of XLOC_005968. The cDNA for full-length CABCOCO1 and GATA3 were used as positive controls. Proteins were visualized by Western blot analysis using Strepavidin-HRP.
Discussion
Recent advances in RNA research have demonstrated pivotal roles of lncRNAs in normal tissue development and various cancers [1–5]. Several studies have implicated lncRNAs in T-ALL pathogenesis. Trimarchi et al. [44] showed that the lncRNA LUNAR is induced by NOTCH1, which is another prevalent oncogene in T-ALL, and is required for T-ALL growth due to its ability to enhance IGF1R mRNA expression. More recently, Wallaert et al. [34] demonstrated that primary T-ALL cases can be classified into four subgroups based on the lncRNA expression profiles, similar to classification based on expression of protein-coding genes. In addition, here we identified lncRNAs regulated by TAL1 in T-ALL cells.
For this purpose, we performed RNA-seq analysis with deep coverage. Using this dataset, we first established a pipeline for the prediction of lncRNAs. We used the Slncky model [41], which is less confounded by evolutional conservation than codon substitution models such as PhyloCSF [47] implemented in previous studies. However, this tool requires a cut-off for filtering out transcripts that overlap with protein-coding genes on the same strand (sense lncRNAs). Hence, we additionally applied four stringent steps to predict sense lncRNAs. For example, we identified a novel lncRNA (XLOC_005968) overlapping with the last 3 exons of CABCOCO1 gene as a sense lncRNA, which could not be found by the Slncky using various minimum overlap values. Importantly, we have shown that XLOC_005968 does not have the potential to encode a protein, whereas full-length CABCOCO1 can produce a protein, proving that XLOC_005968 is indeed a novel lncRNA. Hence, our pipeline accurately predicted lncRNA and is more comprehensive than previously reported approaches. In addition, we confirmed that expression levels of lncRNAs are lower than for protein-coding genes. Correlation analysis of the expression of neighboring protein-coding genes showed high correlations in these pairs. All these results support earlier findings reported for other cell types [7, 43, 44]. Notably, among 2236 putative lncRNA loci identified in our study, 59% and 47% have been already annotated in GENCODE version 25 and the LNCipedia version, respectively. In addition, 50% of lncRNAs were previously reported to be more highly expressed in TAL1-positive T-ALL subgroup than other subgroups [34]. Although these numbers are relatively low, this could possibly be due to differences in the analytical platform (microarray vs. RNA-seq), library preparation (e.g., depletion of ribosomal RNAs, inclusion of polyA-RNAs), sequencing depth, annotation, or tissue specificity.
Critically, we identified putative lncRNAs that are transcriptionally activated by TAL1 in T-ALL cells. Many were also positively regulated by GATA3, RUNX1, and MYB. Thus, our study demonstrates that TAL1 and its regulatory partners coordinately regulate the expression of both protein-coding genes and lncRNAs. In particular, we highlighted two novel lncRNAs, XLOC_030252 and XLOC_005968, in this study. Both loci were highly activated in T-ALL cells under super-enhancers but were downregulated in normal DP stage thymocytes. Interestingly, XLOC_030252 was also expressed in HSCs where TAL1 is physiologically expressed. Our result suggests that these transcripts are normally repressed in developing thymocytes but could be aberrantly activated in T-cells upon ectopic expression of TAL1. As XLOC_030252 is also expressed in normal HSCs and progenitor cells, this putative lncRNA may also play a role in normal hematopoiesis. Notably, both XLOC_030252 and XLOC_005968 were located near the TAL1-bound region, and the expression of XLOC_030252 and XLOC_005968 was significantly downregulated after TAL1 knockdown. Hence, a possible explanation is that those lncRNAs might be transcribed as a result of enhancer activation. Alternatively, those lncRNAs may act as enhancer RNAs that control the expression of neighboring genes, as reported for LUNAR, which controls IGF1R expression. In particular, XLOC_005968 is located near the ARID5B gene, which was recently reported by us as a critical downstream target of TAL1 in T-ALL [27]. ARID5B positively regulates the expression of the TAL1 complex and the MYC oncogene in T-ALL cells. ARID5B also promotes the growth and survival of T-ALL cells in vitro and in vivo. This study implicated ARID5B as a pro-oncogenic factor in the context of TAL1-positive T-ALL. Hence, XLOC_005968 is possibly involved in the regulatory mechanism and in T-cell leukemogenesis. Further investigation is necessary to elucidate their molecular functions.
In this study, we also focused on the regulation of lncRNAs by TAL1 and E-proteins (E2A and HEB). E-protein is a critical heterodimerization partner of TAL1 but also functions as an E-protein dimer to regulate genes required for lymphocyte development [19, 20]. A number of studies have shown that E-proteins serve as tumor suppressors in the context of T-ALL [21, 48]. TAL1 counteracts E-protein functions in T-ALL cells. Therefore, it is of great interest to identify lncRNAs that are differentially controlled by TAL1 and E-proteins. We found that many putative lncRNAs are negatively regulated by TAL1 and positively regulated by E2A and HEB. This included a putative lncRNA XLOC_001561 near the RORC locus. Both the XLOC_001561 and RORC genes were activated by E-proteins and repressed by TAL1 in T-ALL cells, whereas those transcripts are induced during thymocyte development. The RORC gene has been reported as an E2A target and has been implicated in normal thymus development [46, 49, 50]. Although further investigations are necessary, our study suggests that XLOC_001561 is involved in T-cell differentiation and/or may contribute to tumorigenesis when repressed by TAL1 overexpression.
Supplementary Material
Acknowledgements
We thank Toshio Suda’s laboratory for providing the EML cell line. The research is supported by the National Research Foundation (NRF) Singapore and the Singapore Ministry of Education (MOE) under its Research Centres of Excellence initiative. The research is also supported by the NRF under its Competitive Research Programme (NRF-NRFF2013–02) and the RNA Biology Center at CSI Singapore, NUS, from funding by the Singapore MOE’s Tier 3 grants (MOE2014-T3–1-006). A.E.J.Y. is supported by the National Medical Research Council, Singapore (NMRC/CSA/0053/2013). D.G.T. is supported by NIH grants R35CA197697 from the NCI and P01HL131477 from NHLBI.
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41375–018-0110–4) contains supplementary material, which is available to authorized users.
References
- 1.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9. [DOI] [PubMed] [Google Scholar]
- 4.Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. [DOI] [PubMed] [Google Scholar]
- 5.Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21:1253–61. [DOI] [PubMed] [Google Scholar]
- 6.Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29:452–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ulitsky I. Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat Rev Genet. 2016;17:601–14. [DOI] [PubMed] [Google Scholar]
- 9.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–43. [DOI] [PubMed] [Google Scholar]
- 10.Porcher C, Chagraoui H, Kristiansen MS. SCL/TAL1: a multi-faceted regulator from blood development to disease. Blood. 2017;129:2051–60. [DOI] [PubMed] [Google Scholar]
- 11.Sanda T, Leong WZ. TAL1 as a master oncogenic transcription factor in T-cell acute lymphoblastic leukemia. Exp Hematol. 2017;53:7–15. [DOI] [PubMed] [Google Scholar]
- 12.Mouthon MA, Bernard O, Mitjavila MT, Romeo PH, Vainchenker W, Mathieu-Mahul D. Expression of tal-1 and GATA-binding proteins during human hematopoiesis. Blood. 1993;81:647–55. [PubMed] [Google Scholar]
- 13.Herblot S, Steff AM, Hugo P, Aplan PD, Hoang T. SCL and LMO1 alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T alpha chain expression. Nat Immunol. 2000;1:138–44. [DOI] [PubMed] [Google Scholar]
- 14.Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–64. [DOI] [PubMed] [Google Scholar]
- 15.Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation: an oncogenic super-enhancer formed through somatic mutaton of a noncoding intergenic element. Science. 2014;346:1373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22:209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole MF, Young RA. Mapping key features of transcriptional regulatory circuitry in embryonic stem cells. Cold Spring Harb Symp Quant Biol. 2008;73:183–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moignard V, Woodhouse S, Fisher J, Gottgens B. Transcriptional hierarchies regulating early blood cell development. Blood Cells Mol Dis. 2013;51:239–47. [DOI] [PubMed] [Google Scholar]
- 19.Kee BL. E and ID proteins branch out. Nat Rev Immunol. 2009;9:175–84. [DOI] [PubMed] [Google Scholar]
- 20.Murre C. Helix-loop-helix proteins and lymphocyte development. Nat Immunol. 2005;6:1079–86. [DOI] [PubMed] [Google Scholar]
- 21.Bain G, Engel I, Robanus Maandag EC, te Riele HP, Voland JR, Sharp LL, et al. E2A deficiency leads to abnormalities in alpha-beta T-cell development and to rapid development of T-cell lymphomas. Mol Cell Biol. 1997;17:4782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldfarb AN, Lewandowska K. Inhibition of cellular differentiation by the SCL/tal oncoprotein: transcriptional repression by an Id-like mechanism. Blood. 1995;85:465–71. [PubMed] [Google Scholar]
- 23.El Omari K, Hoosdally SJ, Tuladhar K, Karia D, Hall-Ponsele E, Platonova O, et al. Structural basis for LMO2-driven recruitment of the SCL:E47bHLH heterodimer to hematopoietic-specific transcriptional targets. Cell Rep. 2013;4:135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liau WS, Tan SH, Ngoc PC, Wang CQ, Tergaonkar V, Feng H, et al. Aberrant activation of the GIMAP enhancer by oncogenic transcription factors in T-cell acute lymphoblastic leukemia. Leukemia. 2017;31:1798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan SH, Yam AW, Lawton LN, Wong RW, Young RA, Look AT, et al. TRIB2 reinforces the oncogenic transcriptional program controlled by the TAL1 complex in T-cell acute lymphoblastic leukemia. Leukemia. 2016;30:959–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mansour MR, Sanda T, Lawton LN, Li X, Kreslavsky T, Novina CD, et al. The TAL1 complex targets the FBXW7 tumor suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. J Exp Med. 2013;210:1545–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leong WZ, Tan SH, Ngoc PCT, Amanda S, Yam AWY, Liau WS, et al. ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 2017;31:2343–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casero D, Sandoval S, Seet CS, Scholes J, Zhu Y, Ha VL, et al. Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages. Nat Immunol. 2015;16:1282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian M, Zhang H, Kham SK, Liu S, Jiang C, Zhao X, et al. Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res. 2017;27:185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallaert A, Durinck K, Van Loocke W, Van de Walle I, Matthijssens F, Volders PJ, et al. Long noncoding RNA signatures define oncogenic subtypes in T-cell acute lymphoblastic leukemia. Leukemia. 2016;30:1927–30. [DOI] [PubMed] [Google Scholar]
- 35.Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J, Shishkin AA, Zhu X, Kadri S, Maza I, Guttman M, et al. Evolutionary analysis across mammals reveals distinct classes of long non-coding RNAs. Genome Biol. 2016;17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chonghaile TN, Roderick JE, Glenfield C, Ryan J, Sallan SE, Silverman LB, et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. 2014;4:1074–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kornienko AE, Dotter CP, Guenzl PM, Gisslinger H, Gisslinger B, Cleary C, et al. Long non-coding RNAs display higher natural expression variation than protein-coding genes in healthy humans. Genome Biol. 2016;17:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A, et al. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell. 2014;158:593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Neil J, Billa M, Oikemus S, Kelliher M. The DNA binding activity of TAL-1 is not required to induce leukemia/lymphoma in mice. Oncogene. 2001;20:3897–905. [DOI] [PubMed] [Google Scholar]
- 46.Miyazaki M, Rivera RR, Miyazaki K, Lin YC, Agata Y, Murre C. The opposing roles of the transcription factor E2A and its antagonist Id3 that orchestrate and enforce the naive fate of T cells. Nat Immunol. 2011;12:992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Neil J, Shank J, Cusson N, Murre C, Kelliher M. TAL1/SCL induces leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell. 2004;5:587–96. [DOI] [PubMed] [Google Scholar]
- 49.Kurebayashi S, Ueda E, Sakaue M, Patel DD, Medvedev A, Zhang F, et al. Retinoid-related orphan receptor gamma (ROR-gamma) is essential for lymphoid organogenesis and controls apoptosis during thymopoiesis. Proc Natl Acad Sci USA. 2000;97:10132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science. 2000;288:2369–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.