

Abstract
A reaction of aromatic halides bearing electron-withdrawing groups with tertiary amines in the presence of an iridium catalyst under blue light irradiation is described. Products of the aromatic substitution of the halide by the dialkylamino fragment are obtained. The interaction of aryl radicals with tertiary amines to generate zwitterionic radical species is believed to be the key factor responsible for the reaction efficiency.
Keywords: aromatic substitution, tertiary amines, photoredox catalysis, radical reactions
1. Introduction
The substitution of halogen by heteroatom-centered nucleophiles in the aromatic ring is an important class of processes leading to numerous industrial products [1,2,3,4]. Since direct nucleophilic substitution proceeds through the loss of aromaticity, these reactions may require harsh conditions. Alternatively, aryl halides may be involved in the transition of metal catalyzed reactions mainly based on the application of complexes of palladium, nickel, or copper [5,6,7,8,9]. In the last decade, photoredox catalysis has emerged as a powerful tool for performing chemical reactions [10,11] and it is also applied for aromatic substitution [12].
Several types of photoredox mediated substitution reactions were realized differing by the mode of action of the photocatalyst serving either as oxidant or reductant (Scheme 1). The oxidative pathway involves a single electron oxidation of the aromatic substrate by the light activated catalyst followed by nucleophilic attack and subsequent reduction [13,14,15,16,17]. This pathway works well for aromatics bearing electron donating groups. Another mode involves the application of strongly reducing catalysts, which can reduce the aromatic halide to generate aryl radical capable of interacting with a nucleophile with subsequent oxidation [18,19]. Reactions may also be performed via dual activation involving both photoredox and transition metal catalysis [20,21,22,23].
Scheme 1.

Modes of photocatalytic substitution.
The application of reductive photocatalysts toward generation of radicals is most appropriate for electron deficient substrates such as fluorinated or heteroaromatic compounds [24]. However, the resulting aryl radicals are typically trapped by π-systems (alkenes, alkynes, electron rich aromatics) or abstract a hydrogen atom from a suitable reagent [24,25,26,27,28].
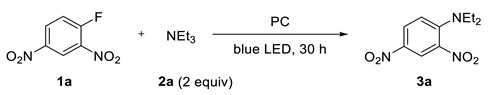
Herein, we report a substitution of aromatic halides by dialkylamine group in reaction with tertiary amines. The reaction proceeds via the photoredox generation of the aryl radical, which interacts with the amine followed by loss of electron and elimination of one group from the ammonium fragment. Classical uncatalyzed nucleophilic substitution reactions of chlorine or heavier halogens with tertiary amines are known but proceed under thermal conditions [29,30,31,32], while such reactions of fluorides have not been described.
2. Results and Discussion
2,4-Dinitrofluorobenzene (1a) and triethylamine were selected as model compounds and their reaction was evaluated (Table 1). In dimethylsulfoxide, in the absence of photocatalyst or in the dark, there was no product formation at room temperature within 30 h. Traces of amine 3a containing diethylamino fragment were observed when the reaction was heated at 50 °C for more than two days. Irradiation with blue LED at room temperature led to the 10% conversion (entry 3). Rewardingly, the use of photocatalysts gave noticeable improvement, with strongly reductive ones being more efficient. The best result was achieved using fac-Ir(ppy)3 affording product 3a in 84% isolated yield (entry 8). Though incomplete conversion of 1a was observed, increase of the reaction time did not give the increase of the yield of 3a. Other solvents were evaluated but gave inferior results (entries 10–12).
Table 1.
Optimization studies.
| Entry | PC 1 | Solv. | Ratio 1a:3a |
|---|---|---|---|
| 1 2 | – | DMSO | 100:0 |
| 2 2,3 | – | DMSO | 95:5 |
| 3 | – | DMSO | 90:10 |
| 4 | Ru(phen)3(PF6)2 (1%) | DMSO | 68:32 |
| 5 | 4CzIPN (2%) | DMSO | 33:67 |
| 6 | 3DPA2FBN (2%) | DMSO | 28:72 |
| 7 | Ir(ppy)2dtbbpy PF6 (0.5%) | DMSO | 29:71 |
| 8 | Ir(ppy)3 (0.3%) | DMSO | 15:85 (84) 4 |
| 9 | Ir(ppy)3 (0.3%), 1.1 equiv of NEt3 | DMSO | 61:39 |
| 10 | Ir(ppy)3 (0.3%) | DMF | 35:65 |
| 11 | Ir(ppy)3 (0.3%) | MeCN | 57:43 |
| 12 | Ir(ppy)3 (0.3%) | CH2Cl2 | 85:15 |
1 Abbreviations: 4CzIPN, 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; 3DPA2FBN, 2,4,6-tris(diphenylamino)-3,5-difluorobenzonitrile. 2 In the dark. 3 At 50 °C for 60 h. 4 Isolated yield.
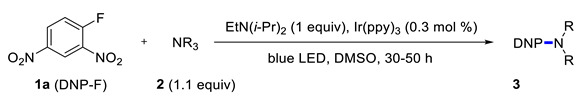
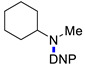
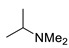
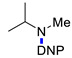
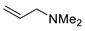
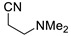
Under the above conditions, a series of tertiary amines were involved in the reaction with 2,4-dinitrofluorobenzene leading to products of the substitution of the fluoride by the dialkylamino group (Table 2). To reduce the amount of amine to 1.1 equiv, Hünig base (1 equiv) was added, which does not react with the substrate due to steric reasons but serves as a basic scavenger. Amines bearing non-identical substituents may afford various products. In the reaction of cyclic N-methylamines, the methyl group was selectively removed (products 3c–f). The reaction of 2-optimized cyanoethyldimethylamine unexpectedly furnished dimethyl-substituted amine 3g as a single product. The preferred removal of the 2-cyanoethyl compared to the methyl group may be associated with the presence of acidic proton at the α-position of the nitrile, thereby favoring E2 elimination with the formation of acrylonitrile. In case of unsymmetrical alkyldimethylamines, the methyl group is cleaved preferentially, though traces of the products arising from the removal of another alkyl group were formed, and the portion of the latter products increases along with the expected efficiency of the nucleophilic substitution at carbon connected to nitrogen. These data allow to propose the following row of substituents with respect to increasing propensity of being detached from the amine: Cy < i-Pr < primary alkyl < Bn < allyl.
Table 2.
Reaction of fluoride 1a with amines.
| Amine | Product | Y., % 1 | Amine | Major Product | Y., % 1 | Minor Product | Y., % 1 | |||
|---|---|---|---|---|---|---|---|---|---|---|

|
|
3a | 84 |
|
|
3h | 86 |
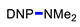
|
3g | 1 |
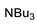
|
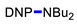
|
3b | 68 |
|
|
3i | 64 |
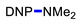
|
3g | 3 |
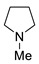
|
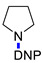
|
3c | 71 |
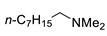
|
|
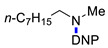
3j | 83 |
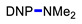
|
3g | 9 |
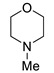
|
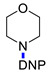
|
3d | 87 |
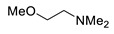
|
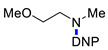
|
3k | 76 |
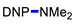
|
3g | 10 |
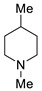
|
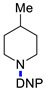
|
3e | 86 |
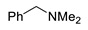
|
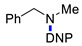
|
3l | 70 |
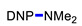
|
3g | 19 |
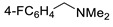
|
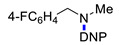
|
3m | 70 |
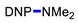
|
3g | 20 | ||||
|
|
3f | 88 |
|
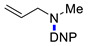
|
3n | 55 |
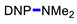
|
3g | 32 |
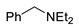
|
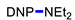
|
3a | 50 |
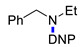
|
3o | 41 | ||||
|
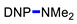
|
3g | 85 |
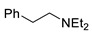
|
|
3p | 59 |
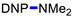
|
3g | 30 |
1 Isolated yield.
Then, reactions of various aromatic substrates 1 with 2 equiv of triethylamine were evaluated (Scheme 2). The fluoride can be displaced starting from pentafluoropyridine and 4-thio-substituted tetrafluorinated pyridines [33,34]. The reaction worked well with a series of heteroaromatic chlorides. Derivatives of benzoxazole, oxadiazole, and diazines gave good yields. At the same time, 2-chlorobenzothiazole, 1-chloro-2,4,6-trinitrobenzene, 4-nitrofluorobenzene, 4-bromofluorobenzne, ethyl 4-fluorobenzoate were unreactive.
Scheme 2.

Reaction of aryl halides with NEt3. Isolated yields are shown, reaction times are in parenthesis. 1 Performed in CH2Cl2.
The proposed mechanism is shown in Scheme 3. Starting aryl halide is reduced by the photoexcited iridium complex with elimination of the halide anion and generation of the aryl radical. The radical interacts with tertiary amine leading to zwitterionic species A, which is converted into the product 3 along with regeneration of the Ir(III) complex. The transformation of intermediate A into the product involves single electron oxidation of the aromatic π-system and removal of the alkyl group from nitrogen with the aid of the amine (Scheme 3, right). The detachment of the alkyl group may be realized either via nucleophilic substitution or through E2 elimination mechanism, especially if the group has acidic hydrogen at the β-position. The order of oxidation and alkyl group removal may depend on the nature of substituents in the aromatic ring and at the nitrogen atom. The radical character of the process was supported by an experiment with a radical trap. Thus, when the reaction of 2,4-dinitrofluorobenzene with triethylamine were carried out in the presence of TEMPO (1.5 equiv) under standard conditions, no product was formed, leaving the starting arylfluoride unconsumed.
Scheme 3.

Proposed mechanism.
The redox potential of 2,4-dinitrofluorobenzene (1a) in DMSO was measured by cyclic voltammetry affording a value of −0.93 V (vs. SCE) (see Supplementary Materials for CV curves). The photoexcited fac-Ir(ppy)3 has the potential of −1.73 V (from Ir(III)* to Ir(IV)) [10], thereby suggesting facile single electron reduction of 1a. It should also be pointed out that the photoexcited catalyst may be reductively quenched by tertiary amine generating Ir(II), which has even stronger reducing power (for Ir(II)/Ir(III), −2.19 V [10]). The use of iridium photocatalysts for the generation of aryl radicals by reductive cleavage of the C-F bond in aromatic aryl fluorides was described [24].
To gain insight into the alkyl detachment step from intermediate A, a photoreaction of pentafluoropyridine with two equivalents of N-methylpyrrolidine was evaluated (Scheme 4). The reaction was performed in acetonitrile, and after completion the mixture was directly analyzed by 1H, 13C, 19F NMR, and no alkyl fluorides were observed. The acetonitrile was evaporated and the residue was washed to remove all non-ionic organic compounds. When the residue was analyzed by NMR spectroscopy in DMSO-d6, N,N-dimethylpyrrolidinium cation was observed (the reference ammonium cation was formed by interaction of N-methylpyrrolidine with methyl iodide). The combined methyl tert-butyl ether phases were analyzed by GC-MS demonstrating that tetrafluorinated 4-pyrrolydinopyridine is a single substitution product.
Scheme 4.

Formation of ammonium salt.
To support the hypothesis of the formation of species A, the free energies of the interaction of aryl radicals with trimethylamine were derived by means of quantum chemical calculations. Energies were calculated at M06-2X/6-31+G(d) [35], and for the stationary points, energies were calculated by CPCM method in DMSO solution (Figure 1). In case of perfluorinated pyridinyl and benzoxazolyl radicals, in the resulting complexes A1 and A2, the amine nitrogen is located out of plane of the heteroaromatic ring having relatively long C,N bond of 1.595 Å and 1.559 Å, respectively. Such a geometry can be considered as a weak Meisenheimer complex between the amine and the aromatic system. On the other hand, for the complexes originated from 2,4-dinitrophenyl and 3-nitropyridinyl radicals, the amine fragment is located within the plane of the aromatic ring, with the C,N bonds in A3 and A4 being equal to 1.507 Å and 1.500 Å, respectively. Of special note, for the latter complexes, the free energies of their formation are notably more negative compared to those of A1 and A2.
Figure 1.

Calculated structures of complexes A formed upon interaction of aryl radicals with NMe3 (UM06-2X/6-31+G(d), DMSO as solvent). Hydrogen atoms are omitted. Bond length between the carbon atom of the aromatic ring and the nitrogen and free energies of complex formation are shown.
3. Materials and Methods
3.1. General Information
All reactions were performed under an argon atmosphere. DMSO was distilled from CaH2 and stored over MS 4Å. Column chromatography was carried out employing silica gel (230–400 mesh). High resolution mass-spectra (HRMS) were measured using electrospray ionization (ESI) and a time-of-flight (TOF) mass analyzer (Bruker MicrOTOF II, Bruker, Billerica, USA). The measurements were done in a positive-ion mode (interface capillary voltage −4500 V) or in a negative-ion mode (3200 V); the mass ranged from m/z 50 to m/z 3000. For irradiation, a strip of 455 nm light-emitting diodes (SMD 2835–120 LED 1 M Blue, 12 V, 24 W/m; 70 cm strip length) was used. The distance between the reaction vessel and diodes was about 30 mm, and the reaction vial was cooled with a fan.
4-(Cyclohexylthio)-2,3,5,6-tetrafluoropyridine [34] and 2,3,5,6-tetrafluoro-4-(phenylthio)pyridine [36] were obtained according to literature procedures.
2-[(2,3,5,6-Tetrafluoropyridin-4-yl)sulfanyl]-1,3-benzothiazole. Potassium carbonate (1.74 g, 12.8 mmol) was added to a solution of 2-mercaptobenzothiazole (2.1 g, 12.6 mmol) in DMF (15 mL). The mixture was stirred for 15 min, then added pentafluoropyridine (2.3 g, 13.8 mmol), and the mixture was stirred for one hour at room temperature. The mixture was poured into water (100 mL), the precipitate was filtered, washed with water, and dried in air. The obtained material was purified vie short silica gel column eluting with dichloromethane. Yield 3.2 g (80%). Colorless solid. Mp 69–71 °C (EtOH). 1H NMR (300 MHz, DMSO-d6) δ 8.10 (d, J = 7.4 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.47–7.53 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 149.4.7, 152.6, 143.4 (dm, J = 239 Hz), 142.5 (dm J = 258 Hz), 136.3, 127.2, 126.1, 125.2 (m), 122.6, 122.5. 19F NMR (282 MHz, DMSO-d6) δ −90.8 (d, J = 7 Hz), −134.1 (d, J = 7 Hz). HRMS (ESI): calcd for C12H5F4N2S2, 316.9825 [M + H]; found, 316.9814.
3.2. Reactions of Aryl Halides with Amines
3.2.1. Reaction of 2,4-Dinitrofluorobenzene with Tertiary Amines. Synthesis of Amines 3. General Procedure 1 (GP 1)
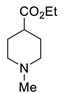
fac-Ir(ppy)3 (2 mg, 0.003 mmol), EtN(i-Pr)2 (174 μL, 1 mmol) and amine (1.1 mmol) were added to a solution of 2,4-dinitrofluorobenezene (186 mg, 1 mmol) in DMSO (2 mL). The mixture was irradiated at room temperature with blue LED [for 3c,g,j,n, 30 h; for 3a,b,d–f,h,i,k,m, 40 h; for 3l,o,p, 50 h]. For the work-up, the mixture was poured into 2% aqueous hydrochloric acid (20 mL) and extracted with EtOAc (4 × 15 mL). The combined organic phases were washed with water (10 mL), a 5% solution of potassium carbonate (10 mL), brine (10 mL), and dried over Na2SO4. The solvent was evaporated, and the residue was purified by column chromatography eluting with dichloromethane.
3.2.2. Reactions of Aryl Halides with Tertiary Amines. Synthesis of Amines 4. General Procedure 2 (GP 2)
fac-Ir(ppy)3 (2 mg, 0.003 mmol) and NEt3 (282 μL, 2 mmol) were added to a solution of aromatic halide (1 mmol) in DMSO (2 mL). The mixture was irradiated at room temperature with blue LED for 15–40 h (the reaction time is shown in Scheme 3). The work-up is the same as in General Procedure 1.
N,N-Diethyl-2,4-dinitroaniline (3a) [37]. According to GP 1; yield 201 mg (84%). According to GP 2 from BnNEt2, yield 120 mg (50%); from PhCH2CH2NEt2, yield 72 mg (30%); from NEt3, yield 203 mg (85%). Yellow solid. Mp 75–77 °C. 1H NMR (300 MHz, CDCl3) δ 8.64 (d, J = 2.8 Hz, 1H), 8.21 (dd, J = 9.6 Hz, 2.8 Hz 1H), 7.08 (d, J = 9.6 Hz, 1H), 3.38 (q, J = 7.2 Hz, 4H), 1.26 (t, J = 7.2 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 148.0, 137.2, 136.4, 127.5, 123.8, 118.4, 46.3, 12.3.
N,N-Dibutyl-2,4-dinitroaniline (3b) [38]. Yield 201 mg (68%). Orange liquid. 1H NMR (300 MHz, CDCl3) δ 8.65 (d, J = 2.8 Hz, 1H), 8.19 (dd, J = 9.5 Hz, 2.8 Hz 1H), 7.08 (d, J = 9.5 Hz, 1H), 3.28 (t, J = 7.3 Hz, 2H), 1.59 (m, 2H), 1.31 (m, 2H), 0.93 (t, J = 7.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 148.7, 137.7, 136.8, 127.6, 124.0, 118.8, 52.0, 29.4, 20.0, 13.7.
1-(2,4-Dinitrophenyl)pyrrolidine (3c) [39]. Yield 168 mg (71%). Yellow solid. Mp 97–98 °C. 1H NMR (300 MHz, CDCl3) δ 8.68 (d, J = 2.6 Hz, 1H), 8.21 (dd, J = 9.5 Hz, 2.6 Hz 1H), 6.92 (d, J = 9.5 Hz, 1H), 3.37 (m, 4H), 1.26 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 145.6, 135.3, 134.8, 127.5, 123.8, 115.6, 51.1, 25.6.
4-(2,4-Dinitrophenyl)morpholine (3d) [40]. Yield 220 mg (87%). Yellow solid. Mp 116–118 °C. 1H NMR (300 MHz, CDCl3) δ 8.70 (d, J = 2.8 Hz, 1H), 8.29 (dd, J = 9.4 Hz, 2.8 Hz 1H), 7.13 (d, J = 9.4 Hz, 1H), 3.88 (m, 4H), 3.29 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 149.3, 139.9, 138.6, 128.4, 123.6, 119.2, 66.2, 50.9.
1-(2,4-Dinitrophenyl)-4-methylpiperidine (3e) [41]. Yield 228 mg (86%). Yellow solid. Mp 84–86 °C. 1H NMR (300 MHz, CDCl3) δ 8.70 (d, J = 2.7 Hz, 1H), 8.23 (dd, J = 9.4 Hz, 2.7 Hz 1H), 7.09 (d, J = 9.4 Hz, 1H), 3.45 (m, 2H), 3.11 (m, 2H), 1.76–1.82 (m, 3H), 1.39 (m, 2H), 1.03 (d, J = 6.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 149.7, 137.3, 137.2, 128.0, 123.9, 119.2, 51.2, 33.6, 30.2, 21.6.
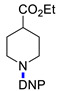
Ethyl 1-(2,4-dinitrophenyl)piperidine-4-carboxylate (3f). Yield 284 mg (88%). Yellow solid. Mp 94–96 °C (EtOH). 1H NMR (300 MHz, CDCl3) δ 8.72 (d, J = 2.6 Hz, 1H), 8.26 (dd, J = 9.3 Hz, 2.6 Hz 1H), 7.12 (d, J = 9.3 Hz, 1H), 4.21 (q, J = 7.1 Hz, 2H), 3.48 (m, 2H), 3.18 (m, 2H), 2.63 (m, 1H), 2.06–2.61 (m, 4H), 1.97 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.8, 149.6, 138.1, 137.9, 128.2, 123.8, 119.5, 60.8, 50.2, 39.9, 27.6, 14.2. HRMS (ESI): calcd for C14H18N3O6 [M + H], 324.1190; found, 324.1188.
N,N-Dimethyl-2,4-dinitroaniline (3g) [42]. Yield 179 mg (85%) from Me2NCH2CH2CN. Yield 68 mg (32%) from AllylNMe2. Yield 42 mg (20%) from 4-FPhCH2NMe2. Yield 2 mg (1%) from CyclohexylNMe2. Yield 19 mg (9%) from C8H17NMe2. Yield 40 mg (19%) from BnNMe2. Yield 21 mg (10%) from MeOCH2CH2NMe2. Yield 6 mg (3%) from i-PrNMe2. Yellow solid. Mp 84–86 °C. 1H NMR (300 MHz, CDCl3) δ 8.60 (d, J = 2.7 Hz, 1H), 8.15 (dd, J = 9.5 Hz, 2.7 Hz 1H), 7.01 (d, J = 9.5 Hz, 1H), 3.06 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 149.2, 136.2, 135.5, 127.7, 124.1, 116.7, 42.4.
N-Cyclohexyl-N-methyl-2,4-dinitroaniline (3h) [43]. Yield 240 mg (86%). Yellow solid. Mp 115–118 °C. 1H NMR (300 MHz, CDCl3) δ 8.64 (d, J = 2.7 Hz, 1H), 8.16 (dd, J = 9.6 Hz, 2.7 Hz 1H), 7.08 (d, J = 9.6 Hz, 1H), 3.57 (m, 1H), 2.77 (s, 3H), 1.91 (m, 4H), 1.21–1.76 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 149.4, 136.3, 136.1, 127.5, 124.3, 117.9, 62.1, 34.6, 29.8, 25.6, 25.4.
N-Methyl-N-(1-methylethyl)-2,4-dinitroaniline (3i). Yield 153 mg (64%). Yellow solid. Mp 73–75 °C (EtOH). 1H NMR (300 MHz, CDCl3) δ 8.66 (d, J = 2.7 Hz, 1H), 8.19 (dd, J = 9.6 Hz, 2.7 Hz 1H), 7.09 (d, J = 9.6 Hz, 1H), 4.03 (hept, J = 6.6 Hz, 1H), 2.75 (s, 3H), 1.33 (d, J = 6.6 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 149.3, 136.4, 136.3, 127.7, 124.3, 117.7, 53.3, 33.0, 19.4. HRMS (ESI): calcd for C10H14N3O4 [M + H], 240.0979; found, 240.0975.
N-Methyl-2,4-dinitro-N-octylaniline (3j). Yield 256 mg (83%). Orange liquid. 1H NMR (300 MHz, CDCl3) δ 8.65 (d, J = 2.7 Hz, 1H), 8.17 (dd, J = 9.5 Hz, 2.7 Hz 1H), 7.05 (d, J = 9.5 Hz, 1H), 3.38 (t, J = 7.6 Hz, 2H), 2.93 (s, 3H), 1.31 (m, 2H), 1.26–1.30 (m, 10H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 148.8, 136.2, 136.0, 127.6, 124.2, 117.2, 54.2, 40.5, 31.7, 29.2, 29.1, 26.9, 26.6, 22.6, 14.0. HRMS (ESI): calcd for C15H24N3O4 [M + H] 310.1761; found, 310.1758.
N-(2-Methoxyethyl)-N-methyl-2,4-dinitroaniline (3k). Yield 194 mg (76%). Orange liquid. 1H NMR (300 MHz, CDCl3) δ 8.62 (d, J = 2.6 Hz, 1H), 8.17 (dd, J = 9.5 Hz, 2.6 Hz 1H), 7.19 (d, J = 9.5 Hz, 1H), 3.65 (t, J = 5.1 Hz, 2H), 3.58 (t, J = 5.1 Hz, 2H), 3.35 (s, 3H), 3.00 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 149.2, 136.7, 136.4, 127.5, 123.9, 118.3, 69.5, 59.1, 54.0, 40.3. HRMS (ESI): calcd for C10H14N3O5 [M + H], 256.0928, found, 256.0926.
N-Benzyl-N-methyl-2,4-dinitroaniline (3l) [44]. Yield 201 mg (70%). Yellow solid. Mp 143–145 °C. 1H NMR (300 MHz, CDCl3) δ 8.70 (d, J = 2.6 Hz, 1H), 8.16 (dd, J = 9.5 Hz, 2.6 Hz 1H), 7.22–7.39 (m, 5H), 7.05 (d, J = 9.5 Hz, 1H), 4.66 (s, 2H), 2.97 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 149.1, 137.0, 136.4, 134.9, 129.2, 128.1, 127.7, 126.9, 124.0, 118.0, 57.8, 41.0.
N-(4-Fluorobenzyl)-N-methyl-2,4-dinitroaniline (3m). Yield 214 mg (70%). Orange liquid. 1H NMR (300 MHz, CDCl3) δ 8.68 (d, J = 2.7 Hz, 1H), 8.17 (dd, J = 9.5 Hz, 2.7 Hz 1H), 7.22 (m, 2H), 7.03–7.10 (m, 3H), 4.61 (s, 2H), 2.95 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 162.4 (d, J = 245.5 Hz), 149.0, 137.3, 136.7, 130.7.8, 128.9 (d, J = 22.7 Hz), 127.8, 123.9, 118.1, 116.1 (d, J = 7.6 Hz), 57.2, 40.9. 19F NMR (282 MHz, CDCl3) δ −114.54. HRMS (ESI): calcd for C14H12FN3O4Na [M + Na], 328.0704; found, 328.0693.
N-Methyl-2,4-dinitro-N-prop-2-en-1-ylaniline (3n) [45]. Yield 130 mg (55%). Yellow solid. Mp 60–62 °C. 1H NMR (300 MHz, CDCl3) δ 8.65 (d, J = 2.7 Hz, 1H), 8.18 (dd, J = 9.5 Hz, 2.7 Hz 1H), 7.04 (d, J = 9.5 Hz, 1H), 5.88 (m, 1H), 5.35 (dd, J = 17.1 Hz, 10.2 Hz, 2H), 4.02 (d, J = 5.1 Hz, 2H), 2.94 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 148.8, 136.7, 136.1, 130.7, 127.6, 124.0, 118.9, 117.7, 56.6, 40.1.
N-Benzyl-N-ethyl-2,4-dinitroaniline (3o) [46]. Yield 123 mg (41%). Yellow solid. Mp 72–73 °C. 1H NMR (300 MHz, CDCl3) δ 8.66 (d, J = 2.7 Hz, 1H), 8.18 (dd, J = 9.5 Hz, 2.7 Hz 1H), 7.24–7.36 (m, 5H), 7.09 (d, J = 9.5 Hz, 1H), 4.54 (s, 2H), 3.37 (q, J = 7.1 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 148.7, 135.6, 129.0, 128.0, 127.6, 127.4, 123.7, 119.5, 55.2, 47.8, 12.4.
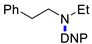
N-(2,4-Dinitrophenyl)-N-ethyl-N-(2-phenylethyl)amine (3p). Yield 178 mg (59%). Yellow solid. Mp 74–76 °C (EtOH). 1H NMR (300 MHz, CDCl3) δ 8.62 (d, J = 2.8 Hz, 1H), 8.17 (dd, J = 9.5 Hz, 2.8 Hz, 1H), 7.05–7.29 (m, 5H), 7.03 (d, J = 9.5 Hz, 1H), 3.54 (t, J = 7.4 Hz, 2H), 3.51 (q, J = 7.1 Hz, 2H), 3.41 (t, J = 7.4 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 148.0, 137.9, 137.8, 137.0, 128.8, 128.7, 127.5, 126.9, 123.8, 118.8, 53.4, 47.2, 33.7, 12.5. HRMS (ESI): calcd for C16H18N3O4 [M + H], 316.1292; found, 316.1288.
4-(Diethylamino)-2,3,5,6-tetrafluorobenzonitrile (4a) [47]. Yield 112 mg (45%). Colorless solid. Mp 47–49 °C. 1H NMR (300 MHz, CDCl3) δ 3.41 (qm, J = 7.1 Hz, 4H), 1.20 (t, J = 7.1 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 148.2 (ddm, J = 256 Hz, 19 Hz), 140.6 (dm, J = 247 Hz), 134.3 (m), 108.7 (t, J = 4 Hz), 82.5 (t, J = 18 Hz), 46.8 (t, J = 5 Hz), 13.6 (t, J = 2 Hz). 19F NMR (282 MHz, CDCl3) δ −136.5 (q, J = 14 Hz), −151.1 (q, J = 14 Hz).
N,N-Diethyl-2,3,5,6-tetrafluoropyridin-4-amine (4b) [47]. Yield 164 mg (74%). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 3.44 (q, J = 7.1 Hz, 4H), 1.24 (tm, J = 7.1 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 145.2 (ddd, J = 236 Hz, 32 Hz, 2 Hz), 139.4 (m), 134.3 (dm, J = 249 Hz), 46.6 (t, J = 5 Hz), 13.8. 19F NMR (282 MHz, CDCl3) δ −96.7 (m), −158.2 (m).
4-(Cyclohexylsulfanyl)-N,N-diethyl-3,5,6-trifluoropyridin-2-amine (4c). Yield 162 mg (51%). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 3.44 (dq, J = 7.0 Hz, 1.7 Hz, 4H), 1.63–1.95 (m, 4H), 1.30–1.59 (m, 7H), 1.17 (t, J = 7.0 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 144.5 (ddd, J = 247 Hz, 16 Hz, 4 Hz), 143.6 (dm, J = 239 Hz), 141.3 (m), 135.2 (dd, J = 240 Hz, 21 Hz), 124.9 (ddd, J = 21 Hz, 18 Hz, 4 Hz), 46.0 (d, J = 3 Hz), 44.2 (d, J = 6 Hz), 33.5, 25.8, 25.5, 13.6 (d, J = 1 Hz). 19F NMR (282 MHz, CDCl3) δ −93.3 (m), −130.7 (m), −152.6 (m). HRMS (ESI): calcd for C15H23F3N2S [M + H], 319.1450; found, 319.1462.
N,N-Diethyl-3,5,6-trifluoro-4-(phenylsulfanyl)pyridin-2-amine (4d). Yield 203 mg (65%). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.26–7.29 (m, 2H), 7.12–7.17 (m, 3H), 3.30 (dq, J = 7.1 Hz, 1.7 Hz, 4H), 1.04 (t, J = 7.1 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 144.8 (ddd, J = 232 Hz, 16 Hz, 3 Hz), 142.2 (dm, J = 241 Hz), 141.3 (m), 134.6 (ddd, J = 248 Hz, 30 Hz, 2 Hz), 133.0, 132.4, 130.8, 127.8, 125.0 (dm, J = 22 Hz), 44.2 (d, J = 6 Hz), 13.6 (d, J = 1 Hz). 19F NMR (282 MHz, CDCl3) δ −92.4 (m), -130.6 (m), −152.7 (m). HRMS (ESI): calcd for C15H16F3N2S [M + H], 313.0971; found, 313.0972.
4-(1,3-Benzothiazol-2-ylsulfanyl)-N,N-diethyl-3,5,6-trifluoropyridin-2-amine (4e). Yield 207 mg (56%). Colorless solid. Mp 87–88 °C (CCl4). 1H NMR (300 MHz, CDCl3) δ 7.89 (d, J = 8.1 Hz, 1H), 7.73 (d, J = 7.9 Hz, 1H), 7.41 (m, 1H), 7.33 (m, 1H), 3.47 (dq, J = 7.1 Hz, 1.8 Hz, 4H), 1.21 (t, J = 7.1 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 161.7, 153.2, 144.9 (ddd, J = 232 Hz, 16 Hz, 3 Hz), 142.6 (dm J = 259 Hz), 141.1 (m), 135.9, 134.2 (ddd, J = 253 Hz, 32 Hz, 2 Hz), 126.5, 125.2, 122.4, 121.0, 120.2 (dm, J = 26 Hz), 44.3 (d, J = 6 Hz), 13.6 (d, J = 2 Hz). 19F NMR (282 MHz, CDCl3) δ −90.0 (m), −126.9 (m), −148.9 (m). HRMS (ESI): calcd for C16H15F3N3S2 [M + H], 370.0654; found, 370.0649.
N,N-Diethyl-1,3-benzoxazol-2-amine (4f) [48]. Yield 179 mg (87%). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.39 (d, J = 7.8 Hz, 1H), 7.27 (d, J = 8.1 Hz, 1H), 7.15 (m, 1H), 7.03 (m, 1H), 3.62 (q, J = 7.0 Hz, 4H), 1.31 (t, J = 7.0 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 162.1, 148.8, 143.5, 123.8, 120.0, 115.7, 108.5, 42.9, 13.4.
N,N-Diethyl-5-methyl-1,3-benzoxazol-2-amine (4g) [49]. Yield 171 mg (84%). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.19 (d, J = 0.6 Hz, 1H), 7.13 (d, J = 8.1 Hz, 1H), 6.81 (dd, J = 8.1 Hz, 0.6 Hz, 1H), 3.61 (q, J = 7.1 Hz, 4H), 2.40 (s, 3H), 1.29 (t, J = 7.1 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 162.3, 146.9, 143.6, 133.4, 120.6, 116.2, 107.8, 42.9, 21.5, 13.6.
N,N-Diethyl-5-phenyl-1,3,4-thiadiazol-2-amine (4h) [50]. Yield 179 mg (77%). Colorless solid. Mp 40–42 °C. 1H NMR (300 MHz, CDCl3) δ 7.80–7.83 (m, 2H), 7.41–7.44 (m, 3H), 3.62 (q, J = 7.1 Hz, 4H), 1.33 (t, J = 7.0 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 170.6, 153.2, 131.7, 129.4, 129.3, 127.6, 46.2, 11.3.
2-Chloro-N,N-diethylpyrimidin-4-amine (4i) [51]. Yield 133 mg (72%). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.90 (d, J = 6.2 Hz, 1H), 6.19 (d, J = 6.2 Hz, 1H), 3.43 (m, 4H), 1.13 (t, J = 7.1 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 161.8, 160.7, 156.5, 101.0, 42.5, 12.5.
3-Chloro-N,N-diethylpyrazin-2-amine (4j). Yield 154 mg (83%). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 8.03 (d, J = 2.5 Hz, 1H), 7.73 (d, J = 2.5 Hz, 1H), 3.53 (q, J = 7.1 Hz, 4H), 1.22 (t, J = 7.1 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 162.4, 153.8, 139.4, 132.8, 44.3, 13.2. HRMS (ESI): calcd for C8H13ClN3 [M + H], 186.0793; found, 186.0783.
N,N-Diethyl-3-phenyl-1,2,4-oxadiazol-5-amine (4k) [52]. Yield 193 mg (89%). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 8.01–8.04 (m, 2H), 7.44–7.47 (m, 3H), 3.58 (q, J = 7.1 Hz, 4H), 1.31 (t, J = 7.0 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 171.0, 168.7, 130.5, 128.5, 128.0, 127.2, 43.5, 13.3.
N,N-Diethyl-2,6-dinitro-4-(trifluoromethyl)aniline (4l) [53]. Yield 236 mg (77%). Yellow solid. Mp 95–96 °C. 1H NMR (300 MHz, CDCl3) δ 8.09 (s, 2H), 3.13 (q, J = 7.0 Hz, 4H), 1.19 (t, J = 7.0 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 145.9, 141.2, 126.5, 123.5 (q, J = 271 Hz), 122.2, 46.2, 12.7. 19F NMR (282 MHz, CDCl3) δ −63.8.
N,N-Diethyl-5-nitropyridin-2-amine (4m) [54]. Yield 193 mg (99%). Yellow solid. Mp 72–75 °C. 1H NMR (300 MHz, CDCl3) δ 9.00 (d, 1.5 Hz, 1H), 8.12 (dd, J = 9.5 Hz, 1.5 Hz, 1H), 6.41 (d, J = 9.5 Hz, 1H), 3.60 (q, J = 7.1 Hz, 4H), 1.21 (t, J = 7.2 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 159.5, 146.9, 134.2, 132.6, 104.1, 43.5, 12.7.
N,N-Diethyl-3-nitropyridin-2-amine (4n) [55]. Yield 189 mg (97%). Orange oil. 1H NMR (300 MHz, CDCl3) δ 8.28 (dd, J = 4.5 Hz, 1.7 Hz, 1H), 8.01 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 6.63 (dd, J = 8.1 Hz, 4.5 Hz, 1H), 3.42 (q, J = 7.2 Hz, 4H), 1.20 (t, J = 7.2 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 152.1, 151.2, 135.2, 132.7, 117.8, 44.1, 12.6.
5-Bromo-N,N-diethyl-3-nitropyridin-2-amine (4o). Yield 262 mg (96%). Orange oil. 1H NMR (300 MHz, CDCl3) δ 8.31 (d, J = 2.2 Hz, 1H), 8.15 (d, J = 2.2 Hz, 1H), 3.42 (q, J = 7.1 Hz, 4H), 1.20 (t, J = 7.1 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 151.9, 150.6, 136.8, 104.3, 44.3, 12.5. HRMS (ESI): calcd for C9H13BrN3O2 [M + H], 274.0186; found, 274.0194.
N,N-Diethyl-3-(1-methylethyl)-1,2,4-oxadiazol-5-amine (4p). Yield 118 mg (86%). Colorless oil. 1H NMR (300 MHz, CDCl3) δ 3.43 (q, J = 7.1 Hz, 4H), 2.82 (h, J = 7.0 Hz, 1H), 1.23 (d, J = 7.0 Hz, 6H), 1.19 (t, J = 7.1 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 175.6, 170.6, 43.3, 27.0, 20.4, 13.2. HRMS (ESI): calcd for C9H18N2O [M + H], 184.1444; found, 184.1451.
3.2.3. Mechanistic Experiment
fac-Ir(ppy)3 (2 mg, 0.003 mmol) and N-methylpyrrolidine (170 mg, 2 mmol) were added to a solution of pentafluoropyridine (169 mg, 1 mmol) in acetonitrile (2 mL). The mixture was irradiated at room temperature with blue LED for 20 h. An aliquot was withdrawn and analyzed by 1H, 13C, 19F NMR. The solvent was evaporated under vacuum, the residue was washed with methyl tert-butyl ether (4 × 5 mL). The obtained solid was dried under vacuum and dissolved in DMSO-d6. NMR analysis indicated the presence of N,N-dimethylpyrrolidinium salt. 1H NMR (300 MHz, DMSO-d6) δ 3.47 (m, 4H), 3.11 (s, 6H), 2.10 (m, 4H). 13C NMR (75 MHz, DMSO-d6) δ 65.3, 51.6, 21.8. 19F NMR (282 MHz, DMSO-d6) δ −138.4 (br).
4. Conclusions
A method for the dealkylative arylation of tertiary amines by means of electron deficient aromatic and heteroaromatic halides under photoredox conditions is described. The reaction proceeds through the generation of aryl radicals, which interact with the amines followed by the dealkylation of the tryalkylamino fragment.
Acknowledgments
The authors thank Vladimir Kokorekin (Zelinsky Institute) for performing cyclic voltammetry measurements.
Supplementary Materials
The following are available online, Figures S1–S4: Cyclic voltammetry curves; Cartesian coordinates and energies; Copies of NMR spectra.
Author Contributions
D.L.L., A.E.F., A.Y.T., experiment; V.V.L., calculations and writing; A.D.D., writing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Scientific Schools Development Program by Zelinsky Institute of organic chemistry.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Terrier F. Modern Nucleophilic Aromatic Substitution. Wiley-VCH; Weinheim, Germany: 2013. [Google Scholar]
- 2.Caron S., McInturff E. Nucleophilic aromatic substitution. In: Caron S., editor. Practical Synthetic Organic Chemistry. John Wiley & Sons Inc.; Hoboken, NJ, USA: 2020. pp. 231–246. [Google Scholar]
- 3.Sandford G. Perfluoroheteroaromatic chemistry: Multifunctional systems from perfluorinated heterocycles by nucleophilic aromatic substitution processes. Top. Heterocycl. Chem. 2012;27:1–32. [Google Scholar]
- 4.Langlois B., Gilbert L., Forat G., Jean-Roger D., Serge R. Fluorination of aromatic compounds by halogen exchange with fluoride anions (‘‘halex’’ reaction) In: Desmurs J.-R., Ratton S., editors. Industrial Chemistry Library. Volume 8. Elsevier; Amsterdam, The Netherlands: 1996. pp. 244–292. [Google Scholar]
- 5.Hartwig J.F. Transition metal catalyzed synthesis of arylamines and aryl ethers from aryl halides and triflates: Scope and mechanism. Angew. Chem. Int. Ed. 1998;37:2047–2067. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 6.Yang B.H., Buchwald S.L. Palladium-catalyzed amination of aryl halides and sulfonates. J. Organomet. Chem. 1999;576:125–146. doi: 10.1016/S0022-328X(98)01054-7. [DOI] [Google Scholar]
- 7.Buchwald S.L., Mauger C., Mignani G., Scholz U. Industrial-scale palladium-catalyzed coupling of aryl halides and amines—A personal account. Adv. Synth. Catal. 2006;348:23–39. doi: 10.1002/adsc.200505158. [DOI] [Google Scholar]
- 8.Sambiagio C., Marsden S.P., Blacker A.J., McGowan P.C. Copper catalysed Ullmann type chemistry: From mechanistic aspects to modern development. Chem. Soc. Rev. 2014;43:3525–3550. doi: 10.1039/C3CS60289C. [DOI] [PubMed] [Google Scholar]
- 9.Walsh K., Sneddon H.F., Moody C.J. Amination of heteroaryl chlorides: Palladium catalysis or SNAr in green solvents? ChemSusChem. 2013;6:1455–1460. doi: 10.1002/cssc.201300239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prier C.K., Rankic D.A., MacMillan D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marzo L., Pagire S.K., Reiser O., König B. Visible-light photocatalysis: Does it make a difference in organic synthesis? Angew. Chem. Int. Ed. 2018;57:10034–10072. doi: 10.1002/anie.201709766. [DOI] [PubMed] [Google Scholar]
- 12.Majek M., Jacobi von Wangelin A. Mechanistic perspectives on organic photoredox catalysis for aromatic substitutions. Acc. Chem. Res. 2016;49:2316–2327. doi: 10.1021/acs.accounts.6b00293. [DOI] [PubMed] [Google Scholar]
- 13.Tay N.E.S., Nicewicz D.A. Cation radical accelerated nucleophilic aromatic substitution via organic photoredox catalysis. J. Am. Chem. Soc. 2017;139:16100–16104. doi: 10.1021/jacs.7b10076. [DOI] [PubMed] [Google Scholar]
- 14.Pistritto V.A., Schutzbach-Horton M.E., Nicewicz D.A. Nucleophilic aromatic substitution of unactivated fluoroarenes enabled by organic photoredox catalysis. J. Am. Chem. Soc. 2020;142:17187–17194. doi: 10.1021/jacs.0c09296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venditto N.J., Nicewicz D.A. Cation radical-accelerated nucleophilic aromatic substitution for amination of alkoxyarenes. Org. Lett. 2020;22:4817–4822. doi: 10.1021/acs.orglett.0c01621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holmberg-Douglas N., Nicewicz D.A. Arene cyanation via cation-radical accelerated-nucleophilic aromatic substitution. Org. Lett. 2019;21:7114–7118. doi: 10.1021/acs.orglett.9b02678. [DOI] [PubMed] [Google Scholar]
- 17.Tay N.E.S., Chen W., Levens A., Pistritto V.A., Huang Z., Wu Z., Li Z., Nicewicz D.A. 19F- and 18F-arene deoxyfluorination via organic photoredox-catalysed polarity-reversed nucleophilic aromatic substitution. Nat. Catal. 2020;3:734–742. doi: 10.1038/s41929-020-0495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W., Li J., Huang C.-Y., Li C.-J. Aromatic chemistry in the excited state: Facilitating metal-free substitutions and cross-couplings. Angew. Chem. Int. Ed. 2020;59:1786–1796. doi: 10.1002/anie.201909138. [DOI] [PubMed] [Google Scholar]
- 19.Cowper N.G.W., Chernowsky C.P., Williams O.P., Wickens Z.K. Potent reductants via electron-primed photoredox catalysis: Unlocking aryl chlorides for radical coupling. J. Am. Chem. Soc. 2020;142:2093–2099. doi: 10.1021/jacs.9b12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corcoran E.B., Pirnot M.T., Lin S., Dreher S.D., DiRocco D.A., Davies I.W., Buchwald S.L., MacMillan D.W.C. Aryl amination using ligand-free Ni(II) salts and photoredox catalysis. Science. 2016;353:279–283. doi: 10.1126/science.aag0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zuo Z., Cong H., Li W., Choi J., Fu G.C., MacMillan D.W.C. Enantioselective decarboxylative arylation of o±-amino acids via the merger of photoredox and nickel catalysis. J. Am. Chem. Soc. 2016;138:1832–1835. doi: 10.1021/jacs.5b13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavagnino M.N., Liang T., MacMillan D.W.C. HARC as an open-shell strategy to bypass oxidative addition in Ullmann–Goldberg couplings. Proc. Natl. Acad. Sci. USA. 2020;117:21058–21064. doi: 10.1073/pnas.2011831117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim K., Hong S.H. Photoinduced Copper(I)-catalyzed cyanation of aromatic halides at room temperature. Adv. Synth. Catal. 2017;359:2345–2351. doi: 10.1002/adsc.201700213. [DOI] [Google Scholar]
- 24.Arora A., Weaver J.D. Visible Light photocatalysis for the generation and use of reactive azolyl and polyfluoroaryl intermediates. Acc. Chem. Res. 2016;49:2273–2283. doi: 10.1021/acs.accounts.6b00259. [DOI] [PubMed] [Google Scholar]
- 25.Singh A., Fennell C.J., Weaver J.D. Photocatalyst size controls electron and energy transfer: Selectable E/Z isomer synthesis via C–F alkenylation. Chem. Sci. 2016;7:6796–6802. doi: 10.1039/C6SC02422J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer A.U., Slanina T., Yao C.-J., Koenig B. Metal-free perfluoroarylation by visible light photoredox catalysis. ACS Catal. 2016;6:369–375. doi: 10.1021/acscatal.5b02410. [DOI] [Google Scholar]
- 27.Senaweera S., Weaver J.D. Dual C–F, C–H Functionalization via photocatalysis: Access to multifluorinated biaryls. J. Am. Chem. Soc. 2016;138:2520–2523. doi: 10.1021/jacs.5b13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ou W., Zou R., Han M., Yu L., Su C. Tailorable carbazolyl cyanobenzene-based photocatalysts for visible light-induced reduction of aryl halides. Chin. Chem. Lett. 2020;31:1899–1902. doi: 10.1016/j.cclet.2019.12.017. [DOI] [Google Scholar]
- 29.Hamilton G.L., Backes B.J. Dealkylative functionalization of tertiary amines with electron deficient heteroaryl chlorides. Tetrahedron Lett. 2006;47:2229–2231. doi: 10.1016/j.tetlet.2006.01.104. [DOI] [Google Scholar]
- 30.Khalaf A.I., Alvarez R.G., Suckling C.J., Waigh R.D. Unexpected dealkylation during nucleophilic substitution: Synthesis of 2-N,N-dialkylamino benzoxazoles and benzothiazoles. Tetrahedron. 2000;56:8567–8571. doi: 10.1016/S0040-4020(00)00802-4. [DOI] [Google Scholar]
- 31.Kolesinska B., Kaminski Z.J. The umpolung of substituent effect in nucleophilic aromatic substitution. A new approach to the synthesis of N,N-disubstituted melamines (triazine triskelions) under mild reaction conditions. Tetrahedron. 2009;65:3573–3576. doi: 10.1016/j.tet.2009.03.017. [DOI] [Google Scholar]
- 32.Lee M., Rucil T., Hesek D., Oliver A.G., Fisher J.F., Mobashery S. Regioselective control of the SNAr amination of 5-substituted-2,4-dichloropyrimidines using tertiary amine nucleophiles. J. Org. Chem. 2015;80:7757–7763. doi: 10.1021/acs.joc.5b01044. [DOI] [PubMed] [Google Scholar]
- 33.Zubkov M.O., Kosobokov M.D., Levin V.V., Kokorekin V.A., Korlyukov A.A., Hu J., Dilman A.D. A novel photoredox-active group for the generation of fluorinated radicals from difluorostyrenes. Chem. Sci. 2020;11:737–741. doi: 10.1039/C9SC04643G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panferova L.I., Zubkov M.O., Kokorekin V.A., Levin V.V., Dilman A.D. Using the thiyl radical for aliphatic hydrogen-atom transfer: Thiolation of unactivated C–H bonds. Angew. Chem. Int. Ed. 2021;60:2849–2854. doi: 10.1002/anie.202011400. [DOI] [PubMed] [Google Scholar]
- 35.Ormazabal-Toledo R., Richter S., Robles-Navarro A., Maulen B., Matute R.A., Gallardo-Fuentes S. Meisenheimer complexes as hidden intermediates in the aza-SNAr mechanism. Org. Biomol. Chem. 2020;18:4238–4247. doi: 10.1039/D0OB00600A. [DOI] [PubMed] [Google Scholar]
- 36.Christopher J.A., Brophy L., Lynn S.M., Miller D.D., Sloan L.A., Sandford G. Synthetic utility of 4-bromo-2,3,5,6-tetrafluoropyridine. J. Fluorine Chem. 2008;129:447–454. doi: 10.1016/j.jfluchem.2008.01.004. [DOI] [Google Scholar]
- 37.Grossi L., Strazzari S. Aromatic radical anions as possible intermediates in the nucleophilic aromatic substitution (SNAr): An EPR study. J. Chem. Soc. Perkin Trans. 2. 1999:2141–2146. doi: 10.1039/a903407b. [DOI] [Google Scholar]
- 38.Imoto M., Matsui Y., Takeda M., Tamaki A., Taniguchi H., Mizuno K., Ikeda H. A probable hydrogen-bonded meisenheimer complex: An unusually high SNAr reactivity of nitroaniline derivatives with hydroxide ion in aqueous media. J. Org. Chem. 2011;76:6356–6361. doi: 10.1021/jo2007219. [DOI] [PubMed] [Google Scholar]
- 39.Kuehne M.E. The arylation of enamines. J. Am. Chem. Soc. 1962;84:837–847. doi: 10.1021/ja00864a032. [DOI] [Google Scholar]
- 40.Panahi F., Daneshgar F., Haghighi F., Khalafi-Nezhad A. Immobilized Pd nanoparticles on silica-starch substrate (PNP-SSS): Efficient heterogeneous catalyst in Buchwald–Hartwig C–N cross coupling reaction. J. Organomet. Chem. 2017;851:210–217. doi: 10.1016/j.jorganchem.2017.09.037. [DOI] [Google Scholar]
- 41.Al-Rawi J.M.A., Khuthier A.-H., Hanna S.Y. Carbon-13 nuclear magnetic resonance spectra of some N-(2,4-dinitrophenyl) Amines, The Corresponding N-Oxides, and Their Thermal Rearrangement Products. Spectrosc. Lett. 1988;21:249–259. doi: 10.1080/00387018808075715. [DOI] [Google Scholar]
- 42.Feng Y.-S., Mao L., Bu X.-S., Dai J.-J., Xu H.-J. Pd(OAc)2-catalyzed dinitration reaction of aromatic amines. Tetrahedron. 2015;71:3827–3832. doi: 10.1016/j.tet.2015.04.013. [DOI] [Google Scholar]
- 43.Bellucci G. Synthesis and determination of the configuration of the diastereoisomeric 4,N-dimethyl-N-phenylcyclohexylamines. Gazz. Chim. Ital. 1969;99:1208–1216. [Google Scholar]
- 44.Graymore J. 175. The reduction products of certain cyclic methyleneamines. Part II. J. Chem. Soc. 1932:1353–1357. doi: 10.1039/jr9320001353. [DOI] [Google Scholar]
- 45.Kleinschmidt R.F., Cope A.C. Rearrangement of allyl groups in dyad systems. amine oxides. J. Am. Chem. Soc. 1944;66:1929–1933. doi: 10.1021/ja01239a040. [DOI] [Google Scholar]
- 46.McIntire F.C., Clements L.M., Sproull M. 1-Fluoro-2,4-dinitrobenzene as quantitative reagent for primary and secondary amines. Anal. Chem. 1953;25:1757–1758. doi: 10.1021/ac60083a050. [DOI] [Google Scholar]
- 47.Matsuda N., Hirano K., Satoh T., Miura M. Copper-catalyzed direct amination of polyfluoroarenes and azoles with hydroxylamines and its application to the synthesis of 3-aminobenzoheteroles. Synthesis. 2012;44:1792–1797. doi: 10.1002/chin.201241037. [DOI] [Google Scholar]
- 48.Grube H., Suhr H. Nucleophile substitution, XI. reaktionen von chlorsubstituierten heteroaromaten mit aminen. Chem. Ber. 1969;102:1570–1579. doi: 10.1002/cber.19691020518. [DOI] [Google Scholar]
- 49.Qiu Y., Struwe J., Meyer T.H., Oliveira J.C.A., Ackermann L. Catalyst- and reagent-free electrochemical azole C–H amination. Chem. Eur. J. 2018;24:12784–12789. doi: 10.1002/chem.201802832. [DOI] [PubMed] [Google Scholar]
- 50.Yonemoto K., Shibuya I. Reaction of 1,4,2-dithiazolium salts with amino compounds. Bull. Chem. Soc. Jpn. 1988;61:4043–4049. doi: 10.1246/bcsj.61.4043. [DOI] [Google Scholar]
- 51.Jo J., Kim S.H., Kim H., Jeong M., Kwak J.-H., Taek Han Y., Jeong J.-Y., Jung Y.-S., Yun H. Discovery and SAR studies of novel 2-anilinopyrimidine-based selective inhibitors against triple-negative breast cancer cell line MDA-MB-468. Bioorg. Med. Chem. Lett. 2019;29:62–65. doi: 10.1016/j.bmcl.2018.11.010. [DOI] [PubMed] [Google Scholar]
- 52.Su D., Duan H., Wei Z., Cao J., Liang D., Lin Y. Condensation of Vilsmeier salts, derived from tetraalkylureas, with amidoximes: A novel approach to access N,N-dialkyl-1,2,4-oxadiazol-5-amines. Tetrahedron Lett. 2013;54:6959–6963. doi: 10.1016/j.tetlet.2013.10.061. [DOI] [Google Scholar]
- 53.Joardar S., Bhattacharyya A., Das S. A Palladium on carbon catalyzed one-pot synthesis of substituted benzimidazoles. Synthesis. 2014;46:3121–3132. doi: 10.1055/s-0034-1378454. [DOI] [Google Scholar]
- 54.Adamson A.J., Jondi W.J., Tipping A.E. Reaction of metal diethylnitroxides with pentafluoropyridine, pentafluorobenzene, octafluorotoluene and 2-chloro-3- or 5-nitropyridine. J. Fluorine Chem. 1996;76:67–78. doi: 10.1016/0022-1139(95)03355-6. [DOI] [Google Scholar]
- 55.Nandi D., Islam R.U., Devi N., Siwal S., Mallick K. A palladium nanoparticle-catalyzed aryl–amine coupling reaction: High performance of aryl and pyridyl chlorides as the coupling partner. New J. Chem. 2018;42:812–816. doi: 10.1039/C7NJ03447D. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available in this article.