

Abstract
Rodents are the most common models in studies of alcoholic liver disease (ALD). Although several rodents ALD models have been established and multiple mechanisms have been elucidated based on them, these models have some non-negligible shortcomings, specifically only inducing early stage (mainly steatosis, slight to moderate steatohepatitis) but not the whole spectrum of human ALD. The resistance of rodents to advanced ALD has been suggested to be due to the physiological differences between rodents and human beings. Previous studies have reported significant interstrain differences in the susceptibility to ethanol-induced liver injury and in the manifestation of ALD (such as different alteration of lipid profiles). Therefore, it would be interesting to characterize the manifestation of ethanol-induced liver damage in various rodents, which may provide a recommendation to investigators of ALD. Furthermore, more severe ALD models need to be established for the study of serious ALD forms, which may be achieved by using genetic modified rodents.
Keywords: ethanol, alcoholic liver disease, animal model, rodents
Introduction
Alcoholic liver disease (ALD), a progressively aggravated liver disease with a wide spectrum ranging from reversible steatosis to irreversible fibrosis and cirrhosis, remains to be one of the leading causes of alcohol-related death in both developed and developing countries [1–3]. The investigation of the hepatotoxicity of ethanol can date back to 60 years ago, when the natural aversion of rodents to ethanol was overcome and high blood alcohol concentration (BAC) was achieved by incorporating ethanol into the liquid diet [4]. Since then, a huge number of studies have revealed multiple mechanisms underlying the onset of ALD such as the toxicity of acetaldehyde, reactive oxygen species (ROS), mitochondrial dysfunction, activation of cytochrome P4502E1 (CYP2E1), disturbance of lipid homeostasis, endoplasmic reticulum stress, and the gut/adipose-liver axes [5–14]. Unfortunately, effective therapeutic strategy for ALD is still lacking, which is at least partially due to the absence of ideal animal models. Although primary hepatocytes and various liver cell lines have been used as in vitro models, they could not mimic the crosstalk between hepatocytes and other intrahepatic cells (such as Kupffer cells, KCs) and the communication between liver and gut/adipose. Currently, rodents are the most commonly used animals in ALD study. However, the available rodent models mainly produce early stage of ALD (mainly steatosis, slight to moderate steatohepatitis). This mini-review focuses on the current available rodent ALD models in which ethanol is delivered via gastrointestinal tract, and discusses the limitation and application of these rodent models. Although some studies achieve high BAC by utilizing other methods such as inhalation, intravenous and intraperitoneal injection, these models are not discussed here as they fail to mimic the natural alcohol drinking pattern of human ALD patients.
ALD: a progressive liver disease with complicated mechanisms
Ethanol is primarily metabolized into acetaldehyde and then to acetate in hepatocytes by cytosolic alcohol dehydrogenase (ADH) and mitochondrial acetaldehyde dehydrogenase (ALDH), respectively. Acetaldehyde, a well-known hepatotoxicant, is considered as one of the principal culprits of ALD, as it could binds nonenzymatically to free amino groups in the proteins of the liver cells, leading to the functional impairments of key proteins [15–17]. ALD is also closely related with the inducible CYP2E1, which has been demonstrated by using CYP2E1 inhibitors and Cyp2e1 ablated/knockin mice [14, 18–21]. In addition to the direct effects on hepatocytes, the activation of KCs and hepatic stellate cells (HSCs) contribute to the inflammation and fibrosis/cirrhosis in ALD. Specifically, ethanol abuse leads to the disturbance of intestine microbiome and impairment of the gastrointestinal mucosa, favoring the translocation of gut lipopolysaccharide (LPS) to liver followed by the activation of KCs and the release of proinflammatory cytokines and chemokines [22]. Elimination of KCs or blocking the Toll-like receptor (TLR)-mediated signaling suppressed ethanol-induced liver damage, demonstrating the crucial roles of innate immune mechanism in ALD [23–28]. HSCs, the major producers of extracellular matrix in fibrotic liver, could be activated by proinflammatory cytokines, profibrotic cytokines, danger-associated molecular patterns (DAMPs) released by injured hepatocytes, and ROS [6, 9, 12]. The accumulation of collagen deposition will lead to the portal tract-septal fibrosis and eventually result in the formation of fibrous septate and scar tissue [29]. Furthermore, other intrahepatic cells such as invariant natural killer T (iNKT) and extrahepatic cells including adipocytes may be also involved in the development of ALD [30, 31]. A brief scheme of pathogenesis of ALD is presented in Figure 1.
Figure 1.
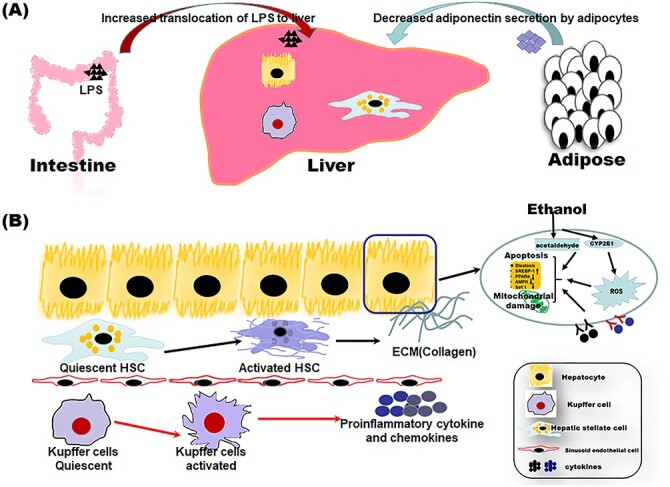
A mechanism scheme of alcoholic liver disease (ALD). The onset of ALD is initiated by the metabolism of ethanol in hepatocytes, which could lead to production of toxic intermediate acetaldehyde, the overproduction of reactive oxygen species (ROS), and mitochondrial dysfunction. The impairment of the intestinal mucosa and disturbance of gut microbiome lead to translocation of gut lipopolysaccharide (LPS) to liver, resulting in the activation of hepatic resident macrophage, Kupffer cells. The activated Kupffer cells release a great amount of proinflammatory cytokines and chemokines, inducing the infiltration of peripheral macrophages and neutrophiles. The activated hepatic stellate cells (HSCs) produce a great amount of extracellular matrix (ECM). Furthermore, ethanol consumption results in the decline of adiponectin secretion, further aggravating liver damage.
Rodents utilized in ALD study: species, strains, and gender differences
Rats and mice are the most popular rodents in ALD studies, whereas hamsters are used in some earlier studies [32, 33]. Almost all the available strains of rats (including Sprague–Dawley (SD) rats, Wistar rats, Long–Evans rats, and Fischer-344 rats, Brown Norway rats, and Lewis rats) and mice (such as C56BL/6, BALB/c, ICR, 129S1/SvlmJ, C3H) have been used, and significant interstrain differences in sensitivity to ethanol-induced liver injury has been reported. One study showed that Long-Evans rats were more susceptible to ALD compared with SD and Fischer-344 rats after receiving same volume of liquid diet for 8 weeks, which was thought to be due to the differences in expression of ethanol metabolizing enzymes [34]. Interestingly, another study found that ethanol induced mainly pericentral lipid deposition pattern in Fisher-344 rats (similar to human ALD) but hepatic midzonal steatosis in Wistar rats, suggesting that Fischer-344 rats were better suited for lipid studies in early stage of ALD [35]. In regard with mice, C57BL/6 mice were found to consume highest volume of ethanol voluntarily compared with other 21 inbred mice under normal circumstances (>10 g/kg/d) [36]. Another study compared the sensitivity of 14 inbred strains of mice (129S1/SvImJ, AKR/J, BALB/cJ, BALB/cByJ, BTBR T + tf/J, C3H/HeJ, C57BL/10J, DBA/2J, FVB/NJ, KK/HIJ, MOLF/EiJ, NZW/LacJ, PWD/PhJ, and WSB/EiJ) using an intragastric intubation model, and found profound interstrain differences in ethanol-induced steatohepatitis in spite of consistently high urine ethanol level [37]. This study revealed that NZW/Lacj was most susceptive to ethanol-induced liver injury, whereas Wsb/Eij was most resistant [37]. However, C57BL/6 strain is not involved in this study, although it is the most popular strain used in ALD studies. Apparently, much more works are needed to identify the optimal strains of rodents for the study of ALD.
Epidemiological studies and animal studies have demonstrated that females are more susceptible to ethanol-induced liver injuries than males [38–41]. Although the exact mechanisms remain to be elucidated, available evidences suggest that gender differences in ALD susceptibility may be related with the differences in bioavailability of ethanol, the sex hormone levels, and the activation status of Kupffer cells between male and female drinkers [42–45]. Although female rats are more susceptible to ALD, both male and female rodents have been used in previous studies. Interestingly, female C57BL/6 mice were found to be less sensitive to the high fat-plus-binge-induced liver injury than the male counterparts, probably because female mice gained much less body weight post high-fat feeding [46].
Binge drinking model
One “binge” is defined as consumption of five and four drinks for men and women, respectively, in 2 h to achieve BAC over 80 mg/dl by the National Institute on Alcohol Abuse and Alcoholism (Bethesda, MD, USA) [47, 48]. One single binge model (4–6 g/kg, by gavage) developed by Carson and Stephen could mimic blood alcohol levels, behavioral effects, and physiological changes in human binge drinkers, and has been wildly used [49–53]. In a more commonly used multiple binge model, three doses of ethanol bolus (usually 5 g/kg) lead to marked increase of serum aminotransferase activities, hepatocyte apoptosis, liver steatosis, liver lipid peroxidation, induction of CYP2E1, and increased production of hepatic tumor necrosis factor α (TNF-α) [54–58]. In addition to the dose of ethanol and the number of binges, several other critical factors may influence the effects of ethanol such as the concentration of ethanol solution and the time points of sample collection [48]. Ethanol of higher concentration and fasting before ethanol gavage would lead to damage of gastrointestinal tract, more severe liver damage, and even mortality of mice [49, 59]. Interestingly, previous studies have reported biphasic effect of acute ethanol/binge drinking on the activation of KCs and the innate immune response, which might be associated with the time of examination [22, 60, 61]. Therefore, the time point of sampling is very important, especially when the aim of study is to investigate the effects of ethanol on innate immune system. Furthermore, maltodextrin is usually selected as a control solution which could not be neglected as it could provide isocaloric intake in control animals.
Chronic rodent ALD models
Voluntary drinking model
Rodents are allowed free access to chow diet and ethanol-containing drinking water (0–40% (v/v)) to accurately simulate human drinking pattern. However, a pioneer study showed that rats received 15% (v/v) ethanol-containing water for 177 days had no obvious liver injury [62]. Similarly, mice exposed to 10–20% ethanol-containing drinking water only developed slight steatosis [63, 64]. Interestingly, moderate ethanol treatment through drinking water exerted beneficial effects on nonalcoholic fatty liver in mice fed a high-fat diet [65]. The absence of liver toxicity of ethanol is attributed to the lower BAC due to the natural aversion of rodents to ethanol and the higher rate of ethanol metabolism compared with human being [66–68]. Although this “voluntary drinking” mode could induce damage after long time treatment with high ethanol concentration [69–71], it cannot exclude the contribution of malnutrition in the toxicity observed in ethanol group.
Lieber–DeCarli liquid diet model
The liquid diet model established by Lieber and Decarli is a breakthrough in the study of ALD. This elegant model is designed to overcome the aversion characteristic of rodents to ethanol by feeding animals only with ethanol-containing liquid diet. Typically, the control liquid diet is composed of protein (18% of total of calories), fat (35% of total calories), carbohydrate (47% of total calories), vitamins and salts mixes, whereas 36% of total energy provided by carbohydrate is replaced by ethanol in ethanol-containing liquid diet [72]. The animals are fed with gradually increased ethanol-containing diet (from 1% to 5%, w/v) during 1-week period, and then with 5% ethanol diet for 4–8 weeks. The daily ethanol intake could reach to 12–18 g/d for rats and 24–34 g/d for mice [72, 73]. The amount of liquid diet given to animals in control group is equal to those ingested by the ethanol-treated littermate, and thus accurately control energy balance between ethanol-exposed rodents and the control animals [72, 74–76]. The advantage of this model is easy handling, inexpensive, short time-consuming, low mortality of animals, and convenient (liquid diet commercially available), and thus applicable for all laboratories. However, no serious liver injury was induced in this model, which might be attributed to the low BAC levels possibly due to the long time needed to consume the ethanol-containing diets (eating slowing) [77]. Anyway, this model provides a useful tool for studying the early stage of ALD.
Tsukamoto–French intragastric infusion model
Tsukamoto and French developed a rat ALD model by directly injecting alcohol and nutrients via a cannula inserted in the stomach [78, 79]. This model achieves high BAC level, as rodents passively receive ethanol-containing liquid diets and investigators can exactly control the diet balance between control and ethanol-fed animals [80]. Ethanol delivered using this method could induce steatosis, megamitochondria, apoptosis, central lobular and pericellular fibrosis, portal fibrosis, bridging fibrosis, central necrosis, and mixed inflammatory infiltrate, which closely resembled human ALD [77]. Although initially developed in rats, the intragastric infusion model has been successfully established in mice [81, 82]. The advantage of this model is that, for the first time, the control and ethanol-fed animals were in exactly the same state of nutrition, as the diet and ethanol intake are completely under the control of investigator [83]. As higher BAC level and more severe liver injuries can be achieved, this model can serve as a useful tool for studying advanced ALD. Furthermore, this model provides a method for the study of multifactorial liver disease, such as the synergy or antagonism between the environment/nutrients and alcohol. However, the application of this model is restricted due to complicated operating techniques, difficulty in postoperative animal health maintenance, and expensive equipment.
Chronic-plus-binge model
Binge drinking after chronic ethanol consumption is one of the important factors contributing to the progression of steatosis to steatohepatitis. Chronic-plus-binge model simulates the “long-term drinking history and recent alcoholism” drinking pattern observed in ALD patients. Aroor et al. developed a chronic-plus-binge rat model in which chronic ethanol-containing (5%, w/v) liquid diet feeding rats were gavaged with single dose of ethanol (5 g/kg) [84]. A similar model was established in mice by Gao group, and named as NIAAA model or Gao–Binge model. In the Gao–Binge model, the ethanol group mice receive an alcoholic liquid diet for 10 days followed by an acute ethanol gavage (5 g/kg), and sacrificed 9 h later for liver injury examination [85]. In both models, single binge significantly increased BAC level (from 100 mg/dl to 175 mg/dl in rats, and from 180 mg/dl to 400 mg/dl in mice) and augmented liver injuries. Extension of chronic ethanol feeding period or multiple binges resulted in more serious neutrophile infiltration and aggravated liver damage [84, 85]. The advantage of this model is flexible and easy to operation, suitable for exploring the pathological mechanism of hepatitis.
High fat-plus-binge/chronic drinking models
High fat-plus-binge/chronic drinking models were developed based on the concept that obesity could exacerbate the hepatotoxicity of ethanol. Tsukamoto et al. established a high-fat diet feeding and intragastric ethanol infusion model by feeding mice 2 weeks of high-fat diet (1% cholesterol, 20% calories from lard, 17% calories from corn oil) followed by intragastric infusion of ethanol (27 g/kg/d) and high-fat liquid diet (60% of total calories) for 8 weeks. The results showed that high-fat diet dramatically enhanced BAC level in ethanol-fed mice (90 mg/dl and 340 mg/dl in chow diet plus ethanol group mice and high-fat diet plus ethanol-fed mice, respectively) and significantly augmented liver injury (15-fold increase of alanine aminotransferase levels and severe steatohepatitis, perisinusoidal and pericellular fibrosis) [86, 87]. High fat-plus binge drinking model was established and characterized in a following study, in which mice were fed with a high-fat diet before exposure to a single binge or multiple binge (5 g/kg). It was found that as little as 3 days of high-fat feeding could significantly exacerbate single binge-induced neutrophilia, hepatic neutrophil infiltration, and liver injury [88]. The deleterious effects of ethanol on liver could be aggravated if the feeding time of high-fat diet was extended to 3 months [46, 88]. These models serve as useful tools to study the synergistic effect of a high-fat diet and alcohol on liver injury. The detailed factors affecting chronic-plus-binge model have been reviewed recently [46]. One specific point needed to pay attention is that binge drinking (5 g/kg bw) will lead to high mortality in mice of higher weight, which can be avoided by reducing ethanol dose or shortening the period of chronic high-fat feeding (reducing the weight of mice) [46].
“Second or multiple hit” model
Ethanol with a “second hit” (another hepatotoxicant such as LPS, carbon tetrachloride, diethyl nitrosamine) could achieve more severe liver damage, providing a model for the study of serious lesions in the end-stage ALD [89]. However, it is obvious that certain differences exist in the pathological mechanisms between liver damage induced by ethanol per se and those by combination of ethanol and a second hepatotoxicant.
The shortcomings of available rodent ALD models
The primary shortcoming of the above rodent ALD models is that they all fail to cover the whole spectrum of human ALD. Even the aversion of rodents can be overcome by incorporating ethanol into liquid diet or by gavage, however, most of these models only induce early stage of ALD. For example, none of the above ALD model could develop hepatocellular carcinoma (HCC), although ethanol is classified as group 1 carcinogen (known to be carcinogenic to humans) by the International Agency for Research on Cancer (IARC) [90]. Although early HCC could be induced by combing ethanol and other hepatotoxicants (such as diethyl nitrosamine, carbon tetrachloride, and urethane) [91–94], the molecular and cellular mechanisms observed in these models cannot be directly ascribed to ethanol per se [95].
The resistance of rodents to advanced ALD may be related with the physiological differences between human beings and rodents. Firstly, the catabolic rate of ethanol in rodents is significantly higher than that in human beings, suggesting that higher BAC level is needed to produce similar liver injuries in rodents as observed in human beings [96]. Second, the innate immune system, which plays critical roles in pathogenesis of ALD, has been demonstrated to vary significantly between mice and human [96]. Specifically, rodents are resistant to LPS challenge, which might be due to the lower proportion of neutrophiles in blood leukocytes [97, 98]. Thirdly, genetic factors has been suggested to be involved in the development of human ALD, as only about 30% of continued drinkers develop fibrosis or cirrhosis and about 5–15% of abstainers develop fibrosis [99].
Conclusion and future research perspective
Several rodent ALD models have been established for the studies of ALD mechanisms and the evaluation of potential medicines. The versatile Liber–DeCarli liquid diet model is convenient, flexible, and applicable for most laboratories, whereas the NIAAA model could mimic the “chronic-plus-binge” pattern of human ALD. However, all ALD models have drawbacks and specifically only induce early stage of ALD, which may be related with the physiological differences between human beings and rodents. Although the Tsukamoto–French intragastric infusion model can cause aggravated liver damage such as fibrosis, its application is limited due to complicate operation, expensive equipment, time-consuming characteristic and difficulty in postoperative animal health maintenance. The “second hit” model can induce more severe liver damage (liver fibers, cirrhosis, and liver cancer), but the addition of another hit makes it hard to ascertain the contribution of ethanol per se in the onset of liver injury (Table 1).
Table 1.
Summary of common rodent models of alcoholic liver disease
| Models | Characteristics and application | Special attention and disadvantage | Phenotypes of ALD |
|---|---|---|---|
| Binge drinking model | Mimic the binge drinking pattern of human | The time point of sampling; control solution. | Elevation of ALT and AST; steatosis; mild inflammation |
| Voluntary drinking model | [1] Simulation of human drinking pattern [2] Simple with low mortality rate [3] Inexpensive |
Natural aversion of rodents cannot be overcome; unbalanced nutrition status between control and ethanol group rodents | Mild elevation of ALT and AST; usually no severe liver injuries |
| Lieber-DeCarli liquid diet model | [1] Simulation of human drinking pattern [2] Easy to control effect of nutrients [3] Overcoming the aversion of rodents to alcohol [4] Flexible application [5] Time and cost efficient [6] High blood alcohol concentration |
No fibrosis and end-stage injuries | Various degrees of steatosis; mild inflammation |
| Tsukamoto–French intragastric infusion model | [1] Nutrition balance between pair-feed animals [2] Overcoming the aversion of experimental animals to alcohol [3] Flexible application |
Complicated operating technique; difficulty in postoperative animal health maintenance; expensive equipment | Steatosis; inflammation; mild fibrosis; focal liver necrosis |
| Chronic-plus-binge model | [1] Mimicking the chronic-plus-binge drinking pattern [2] Flexible application [3] Time and cost efficient [4] High blood alcohol concentration |
No fibrosis and end-stage injuries | Steatosis; inflammation |
| High fat-plus-binge model | Mimicing the deleterious effects of ethanol in obesity population | High mortality in overweight mice | Steatosis; inflammation |
| “Second hit” model | Induction of end-stage liver injuries | Difficulty in analysis of experiment result | Advanced liver injury (cirrhosis, hepatocellular carcinoma) |
Future researches on ALD models may focus on two aspects: mapping the manifestation of ethanol-induced liver damage in various rodents and establishing models of advanced ALD. Previous studies have suggested that rodents of different strains may have different sensitivity to ALD and discrepant alteration of lipid profiles after ethanol exposure [34, 35]. Thus, it would be interesting to map the manifestation of ethanol-induced liver damage in various rodents, which may finally provide a recommendation to investigators of ALD. Besides, more severe ALD models need to be established for the study of serious form of human ALD, which may be achieved by using genetic modified rodents. Mechanisms studies have suggested that CYP2E1 was responsible for oxidative stress, hepatotoxicity, and carcinogenic ethno-DNA lesions in ALD [11, 21, 100], whereas Aldh2 deficiency promoted alcohol-associated liver cancer [91]. Interestingly, people with a homozygous c2c2 genotype of Cyp2e1 (higher CYP2E1 activity) or with *2 allele of Aldh2 gene (decreased ALDH2 activity) were suggested to have increased susceptibility to ALD [91, 101]. Results of these studies suggest that genetic modified mice may serve as invaluable tools to explore novel mechanisms, develop diagnostic biomarkers, and screen potential medicines of advanced ALD.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (Grant No. 81872653 and 81473004).
Contributor Information
Shi-Xuan Liu, Institute of Toxicology, School of Public Health, Cheeloo College of Medicine, Shandong University, 44 Wenhua Xi Road, Jinan, Shandong, 250012, China.
Yan-Chao Du, Jinan Institute for Product Quality Inspection, 1311 Longao Bei Road, Jinan, Shandong, 250102, China.
Tao Zeng, Institute of Toxicology, School of Public Health, Cheeloo College of Medicine, Shandong University, 44 Wenhua Xi Road, Jinan, Shandong, 250012, China.
Conflict of interest statement
None declared.
References
- 1. Sheron N. Alcohol and liver disease in Europe--simple measures have the potential to prevent tens of thousands of premature deaths. J Hepatol 2016;64:957–67. [DOI] [PubMed] [Google Scholar]
- 2. Fan JG. Epidemiology of alcoholic and nonalcoholic fatty liver disease in China. J Gastroenterol Hepatol 2013;28:11–7. [DOI] [PubMed] [Google Scholar]
- 3. Asrani SK, Mellinger J, Arab JP et al. Reducing the global burden of alcohol-associated liver disease: a blueprint for action. Hepatology 2020. doi: 10.1002/hep.31583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lieber CS. The discovery of the microsomal ethanol oxidizing system and its physiologic and pathologic role. Drug Metab Rev 2004;36:511–29. [DOI] [PubMed] [Google Scholar]
- 5. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: pathophysiological basis for therapy. J Hepatol 2020;72:558–77. [DOI] [PubMed] [Google Scholar]
- 6. Nagy LE, Ding WX, Cresci G et al. Linking pathogenic mechanisms of alcoholic liver disease with clinical phenotypes. Gastroenterology 2016;150:1756–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhu H, Jia Z, Misra H et al. Oxidative stress and redox signaling mechanisms of alcoholic liver disease: updated experimental and clinical evidence. J Dig Dis 2012;13:133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. An L, Wang X, Cederbaum AI. Cytokines in alcoholic liver disease. Arch Toxicol 2012;86:1337–48. [DOI] [PubMed] [Google Scholar]
- 9. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu Y, Wu D, Wang X et al. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radic Biol Med 2010;49:1406–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cederbaum AI. Role of CYP2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Dig Dis 2010;28:802–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meng FG, Zhang XN, Liu SX et al. Roles of peroxisome proliferator-activated receptor alpha in the pathogenesis of ethanol-induced liver disease. Chem Biol Interact 2020;327:109176. [DOI] [PubMed] [Google Scholar]
- 13. Zhao N, Guo FF, Xie KQ et al. Targeting Nrf-2 is a promising intervention approach for the prevention of ethanol-induced liver disease. Cell Mol Life Sci 2018;75:3143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zeng T, Zhang CL, Zhao N et al. Impairment of Akt activity by CYP2E1 mediated oxidative stress is involved in chronic ethanol-induced fatty liver. Redox Biol 2018;14:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longev 2010;3:178–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barry RE. Role of acetaldehyde in the pathogenesis of alcoholic liver disease. Br J Addict 1988;83:1381–6. [DOI] [PubMed] [Google Scholar]
- 17. You M, Fischer M, Deeg MA et al. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J Biol Chem 2002;277:29342–7. [DOI] [PubMed] [Google Scholar]
- 18. Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J Hepatol 2013;58:395–8. [DOI] [PubMed] [Google Scholar]
- 19. Abdelmegeed MA, Banerjee A, Yoo SH et al. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J Hepatol 2012;57:860–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu Y, Cederbaum AI. CYP2E1 potentiation of LPS and TNFalpha-induced hepatotoxicity by mechanisms involving enhanced oxidative and nitrosative stress, activation of MAP kinases, and mitochondrial dysfunction. Genes Nutr 2010;5:149–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu Y, Zhuge J, Wang X et al. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology 2008;47:1483–94. [DOI] [PubMed] [Google Scholar]
- 22. Zeng T, Zhang C-L, Xiao M et al. Critical roles of Kupffer cells in the pathogenesis of alcoholic liver disease: from basic science to clinical trials. Front Immunol 2016;7:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koop DR, Klopfenstein B, Iimuro Y et al. Gadolinium chloride blocks alcohol-dependent liver toxicity in rats treated chronically with intragastric alcohol despite the induction of CYP2E1. Mol Pharmacol 1997;51:944–50. [DOI] [PubMed] [Google Scholar]
- 24. Adachi Y, Bradford BU, Gao W et al. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology 1994;20:453–60. [PubMed] [Google Scholar]
- 25. Uesugi T, Froh M, Arteel GE et al. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol 2002;168:2963–9. [DOI] [PubMed] [Google Scholar]
- 26. Uesugi T, Froh M, Arteel GE et al. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001;34:101–8. [DOI] [PubMed] [Google Scholar]
- 27. Yin M, Wheeler MD, Kono H et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology 1999;117:942–52. [DOI] [PubMed] [Google Scholar]
- 28. Yin M, Bradford BU, Wheeler MD et al. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol 2001;166:4737–42. [DOI] [PubMed] [Google Scholar]
- 29. Brandon-Warner E, Schrum LW, Schmidt CM et al. Rodent models of alcoholic liver disease: of mice and men. Alcohol 2012;46:715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. You M, Rogers CQ. Adiponectin: a key adipokine in alcoholic fatty liver. Exp Biol Med (Maywood) 2009;234:850–9. [DOI] [PubMed] [Google Scholar]
- 31. Marrero I, Maricic I, Morgan TR et al. Differential activation of unconventional T cells, including iNKT cells, in alcohol-related liver disease. Alcohol Clin Exp Res 2020;44:1061–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Keung WM, Klyosov AA, Vallee BL. Daidzin inhibits mitochondrial aldehyde dehydrogenase and suppresses ethanol intake of Syrian golden hamsters. Proc Natl Acad Sci USA 1997;94:1675–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fujii H, Ohmachi T, Sagami I et al. Liver microsomal drug metabolism in ethanol-treated hamsters. Biochem Pharmacol 1985;34:3881–4. [DOI] [PubMed] [Google Scholar]
- 34. Denucci SM, Tong M, Longato L et al. Rat strain differences in susceptibility to alcohol-induced chronic liver injury and hepatic insulin resistance. Gastroenterol Res Pract 2010;2010:312790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bhopale KK, Kondraganti S, Fernando H et al. Alcoholic steatosis in different strains of rat: a comparative study. J Drug Alcohol Res 2015;4:235912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoneyama N, Crabbe JC, Ford MM et al. Voluntary ethanol consumption in 22 inbred mouse strains. Alcohol 2008;42:149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsuchiya M, Ji C, Kosyk O et al. Interstrain differences in liver injury and one-carbon metabolism in alcohol-fed mice. Hepatology 2012;56:130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loft S, Olesen KL, Dossing M. Increased susceptibility to liver disease in relation to alcohol consumption in women. Scand J Gastroenterol 1987;22:1251–6. [DOI] [PubMed] [Google Scholar]
- 39. Becker U, Deis A, Sorensen TI et al. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology 1996;23:1025–9. [DOI] [PubMed] [Google Scholar]
- 40. Pares A, Caballeria J, Bruguera M et al. Histological course of alcoholic hepatitis. Influence of abstinence, sex and extent of hepatic damage. J Hepatol 1986;2:33–42. [DOI] [PubMed] [Google Scholar]
- 41. Iimuro Y, Frankenberg MV, Arteel GE et al. Female rats exhibit greater susceptibility to early alcohol-induced liver injury than males. Am J Physiol 1997;272:G1186–94. [DOI] [PubMed] [Google Scholar]
- 42. Kono H, Wheeler MD, Rusyn I et al. Gender differences in early alcohol-induced liver injury: role of CD14, NF-κB, and TNF-α. Am J Physiol Gastrointest Liver Physiol 2000;278:G652–61. [DOI] [PubMed] [Google Scholar]
- 43. Eagon PK. Alcoholic liver injury: influence of gender and hormones. World J Gastroenterol 2010;16:1377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Müller C. Liver, alcohol and gender. Wien Med Wochenschr 2006;156:523–6. [DOI] [PubMed] [Google Scholar]
- 45. Frezza M, di Padova C, Pozzato G et al. High blood alcohol levels in women. The role of decreased gastric alcohol dehydrogenase activity and first-pass metabolism. N Engl J Med 1990;322:95–9. [DOI] [PubMed] [Google Scholar]
- 46. Gao B, Xu MJ, Bertola A et al. Animal models of alcoholic liver disease: pathogenesis and clinical relevance. Gene Expr 2017;17:173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. National Institute on Alcohol Abuse and Alcoholism . NIAAA council approves definition of binge drinking. NIAAA Newsletter 2004;3:3. [Google Scholar]
- 48. Ghosh Dastidar S, Warner JB, Warner DR et al. Rodent models of alcoholic liver disease: role of binge ethanol administration. Biomolecules 2018;8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carson EJ, Pruett SB. Development and characterization of a binge drinking model in mice for evaluation of the immunological effects of ethanol. Alcohol Clin Exp Res 1996;20:132–8. [DOI] [PubMed] [Google Scholar]
- 50. Kanuri G, Weber S, Volynets V et al. Cinnamon extract protects against acute alcohol-induced liver steatosis in mice. J Nutr 2009;139:482–7. [DOI] [PubMed] [Google Scholar]
- 51. Lu ZM, Tao WY, Zou XL et al. Protective effects of mycelia of Antrodia camphorata and Armillariella tabescens in submerged culture against ethanol-induced hepatic toxicity in rats. J Ethnopharmacol 2007;110:160–4. [DOI] [PubMed] [Google Scholar]
- 52. Zeng T, Zhang CL, Pan GB et al. The protective effects of garlic oil on acute ethanol-induced oxidative stress in the liver of mice. J Sci Food Agric 2008;88:2238–43. [Google Scholar]
- 53. Lambert JC, Zhou Z, Wang L et al. Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. J Pharmacol Exp Ther 2003;305:880–6. [DOI] [PubMed] [Google Scholar]
- 54. Song Z, Deaciuc I, Song M et al. Silymarin protects against acute ethanol-induced hepatotoxicity in mice. Alcohol Clin Exp Res 2006;30:407–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xiong Y, Yang Y, Yang J et al. Tectoridin, an isoflavone glycoside from the flower of Pueraria lobata, prevents acute ethanol-induced liver steatosis in mice. Toxicology 2010;276:64–72. [DOI] [PubMed] [Google Scholar]
- 56. Huang LL, Wan JB, Wang B et al. Suppression of acute ethanol-induced hepatic steatosis by docosahexaenoic acid is associated with downregulation of stearoyl-CoA desaturase 1 and inflammatory cytokines. Prostaglandins Leukot Essent Fatty Acids 2013;88:347–53. [DOI] [PubMed] [Google Scholar]
- 57. Abdelmegeed MA, Banerjee A, Jang S et al. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic Biol Med 2013;65:1238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zeng T, Zhang CL, Song FY et al. The activation of HO-1/Nrf-2 contributes to the protective effects of diallyl disulfide (DADS) against ethanol-induced oxidative stress. Biochim Biophys Acta 2013;1830:4848–59. [DOI] [PubMed] [Google Scholar]
- 59. Shukla SD, Pruett SB, Szabo G et al. Binge ethanol and liver: new molecular developments. Alcohol Clin Exp Res 2013;37:550–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Enomoto N, Ikejima K, Bradford B et al. Alcohol causes both tolerance and sensitization of rat Kupffer cells via mechanisms dependent on endotoxin. Gastroenterology 1998;115:443–51. [DOI] [PubMed] [Google Scholar]
- 61. Yamashina S, Wheeler MD, Rusyn I et al. Tolerance and sensitization to endotoxin in Kupffer cells caused by acute ethanol involve interleukin-1 receptor-associated kinase. Biochem Biophys Res Commun 2000;277:686–90. [DOI] [PubMed] [Google Scholar]
- 62. Best CH, Hartroft WS et al. Liver damage produced by feeding alcohol or sugar and its prevention by choline. Br Med J 1949;2:1002–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Larosche I, Choumar A, Fromenty B et al. Prolonged ethanol administration depletes mitochondrial DNA in MnSOD-overexpressing transgenic mice, but not in their wild type littermates. Toxicol Appl Pharmacol 2009;234:326–38. [DOI] [PubMed] [Google Scholar]
- 64. Harrison-Findik DD, Lu S. The effect of alcohol and hydrogen peroxide on liver hepcidin gene expression in mice lacking antioxidant enzymes, glutathione peroxidase-1 or catalase. Biomolecules 2015;5:793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bucher S, Begriche K, Catheline D et al. Moderate chronic ethanol consumption exerts beneficial effects on nonalcoholic fatty liver in mice fed a high-fat diet: possible role of higher formation of triglycerides enriched in monounsaturated fatty acids. Eur J Nutr 2020;59:1619–32. [DOI] [PubMed] [Google Scholar]
- 66. Rodd-Henricks ZA, Bell RL, Kuc KA et al. Effects of ethanol exposure on subsequent acquisition and extinction of ethanol self-administration and expression of alcohol-seeking behavior in adult alcohol-preferring (P) rats: II. Adult exposure. Alcohol Clin Exp Res 2002;26:1642–52. [DOI] [PubMed] [Google Scholar]
- 67. Smith DG, Learn JE, McBride WJ et al. Long-term effects of alcohol drinking on cerebral glucose utilization in alcohol-preferring rats. Pharmacol Biochem Behav 2001;69:543–53. [DOI] [PubMed] [Google Scholar]
- 68. Bloomer SA, Broadhurst KA, Maleah Mathahs M et al. Effects of long-term ethanol ingestion on hepatic iron metabolism in two mouse strains. Clin Exp Pharmacol Physiol 2020. doi: 10.1111/1440-1681.13445. [DOI] [PubMed] [Google Scholar]
- 69. Keegan A, Martini R, Batey R. Ethanol-related liver injury in the rat: a model of steatosis, inflammation and pericentral fibrosis. J Hepatol 1995;23:591–600. [DOI] [PubMed] [Google Scholar]
- 70. Kanbagli O, Balkan J, Aykac-Toker G et al. Hepatic mitochondrial prooxidant and antioxidant status in ethanol-induced liver injury in rats. Biol Pharm Bull 2002;25:1482–4. [DOI] [PubMed] [Google Scholar]
- 71. Robin MA, Sauvage I, Grandperret T et al. Ethanol increases mitochondrial cytochrome P450 2E1 in mouse liver and rat hepatocytes. FEBS Lett 2005;579:6895–902. [DOI] [PubMed] [Google Scholar]
- 72. Lieber CS, DeCarli LM, Sorrell MF. Experimental methods of ethanol administration. Hepatology 1989;10:501–10. [DOI] [PubMed] [Google Scholar]
- 73. He L, Marecki JC, Serrero G et al. Dose-dependent effects of alcohol on insulin signaling: partial explanation for biphasic alcohol impact on human health. Mol Endocrinol 2007;21:2541–50. [DOI] [PubMed] [Google Scholar]
- 74. DeCarli LM, Lieber CS. Fatty liver in the rat after prolonged intake of ethanol with a nutritionally adequate new liquid diet. J Nutr 1967;91:331–6. [DOI] [PubMed] [Google Scholar]
- 75. Lieber CS, DeCarli LM. The feeding of alcohol in liquid diets: two decades of applications and 1982 update. Alcohol Clin Exp Res 1982;6:523–31. [DOI] [PubMed] [Google Scholar]
- 76. Lieber CS, DeCarli LM. Liquid diet technique of ethanol administration: 1989 update. Alcohol Alcohol 1989;24:197–211. [PubMed] [Google Scholar]
- 77. French SW. Intragastric ethanol infusion model for cellular and molecular studies of alcoholic liver disease. J Biomed Sci 2001;8:20–7. [DOI] [PubMed] [Google Scholar]
- 78. Tsukamoto H, French SW, Reidelberger RD et al. Cyclical pattern of blood alcohol levels during continuous intragastric ethanol infusion in rats. Alcohol Clin Exp Res 1985;9:31–7. [DOI] [PubMed] [Google Scholar]
- 79. Tsukamoto H, Reidelberger RD, French SW et al. Long-term cannulation model for blood sampling and intragastric infusion in the rat. Am J Physiol 1984;247:R595–9. [DOI] [PubMed] [Google Scholar]
- 80. Tsukamoto H, French SW, Benson N et al. Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology 1985;5:224–32. [DOI] [PubMed] [Google Scholar]
- 81. Kono H, Rusyn I, Yin M et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest 2000;106:867–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yan AW, Fouts DE, Brandl J et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011;53:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tsukamoto H, Horne W, Kamimura S et al. Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest 1995;96:620–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Aroor AR, Jackson DE, Shukla SD. Elevated activation of ERK1 and ERK2 accompany enhanced liver injury following alcohol binge in chronically ethanol-fed rats. Alcohol Clin Exp Res 2011;35:2128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bertola A, Mathews S, Ki SH et al. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kisseleva T, Cong M, Paik Y et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA 2012;109:9448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lazaro R, Wu R, Lee S et al. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology 2015;61:129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chang B, Xu MJ, Zhou Z et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology 2015;62:1070–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tsukamoto H, Machida K, Dynnyk A et al. "Second hit" models of alcoholic liver disease. Semin Liver Dis 2009;29:178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ganne-Carrie N, Nahon P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J Hepatol 2019;70:284–93. [DOI] [PubMed] [Google Scholar]
- 91. Seo W, Gao Y, He Y et al. ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J Hepatol 2019;71:1000–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ambade A, Satishchandran A, Gyongyosi B et al. Adult mouse model of early hepatocellular carcinoma promoted by alcoholic liver disease. World J Gastroenterol 2016;22:4091–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rafacho BP, Stice CP, Liu C et al. Inhibition of diethylnitrosamine-initiated alcohol-promoted hepatic inflammation and precancerous lesions by flavonoid luteolin is associated with increased sirtuin 1 activity in mice. Hepatobiliary Surg Nutr 2015;4:124–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Beland FA, Benson RW, Mellick PW et al. Effect of ethanol on the tumorigenicity of urethane (ethyl carbamate) in B6C3F1 mice. Food Chem Toxicol 2005;43:1–19. [DOI] [PubMed] [Google Scholar]
- 95. Zhang XN, Meng FG, Wang YR et al. Transformed ALDH2(−/−) hepatocytes by ethanol could serve as a useful tool for studying alcoholic hepatocarcinogenesis. Med Hypotheses 2020;146:110366. [DOI] [PubMed] [Google Scholar]
- 96. Holmes RS, Duley JA, Algar EM et al. Biochemical and genetic studies on enzymes of alcohol metabolism: the mouse as a model organism for human studies. Alcohol Alcohol 1986;21:41–56. [PubMed] [Google Scholar]
- 97. Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol 2007;35:757–66. [DOI] [PubMed] [Google Scholar]
- 98. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol 2004;172:2731–8. [DOI] [PubMed] [Google Scholar]
- 99. O'Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology 2010;51:307–28. [DOI] [PubMed] [Google Scholar]
- 100. Wang Y, Millonig G, Nair J et al. Ethanol-induced cytochrome P4502E1 causes carcinogenic etheno-DNA lesions in alcoholic liver disease. Hepatology 2009;50:453–61. [DOI] [PubMed] [Google Scholar]
- 101. Zeng T, Guo FF, Zhang CL et al. Roles of cytochrome P4502E1 gene polymorphisms and the risks of alcoholic liver disease: a meta-analysis. PLoS One 2013;8:e54188. [DOI] [PMC free article] [PubMed] [Google Scholar]