

Abstract
Traumatic brain injury (TBI) is a chronic and progressive disease, and management requires an understanding of both the primary neurological injury and the secondary sequelae that affect peripheral organs, including the gastrointestinal (GI) tract. The brain-gut axis is composed of bidirectional pathways through which TBI-induced neuroinflammation and neurodegeneration impact gut function. The resulting TBI-induced dysautonomia and systemic inflammation contribute to the secondary GI events, including dysmotility and increased mucosal permeability. These effects shape, and are shaped by, changes in microbiota composition and activation of resident and recruited immune cells. Microbial products and immune cell mediators in turn modulate brain-gut activity. Importantly, secondary enteric inflammatory challenges prolong systemic inflammation and worsen TBI-induced neuropathology and neurobehavioral deficits. The importance of brain-gut communication in maintaining GI homeostasis highlights it as a viable therapeutic target for TBI. Currently, treatments directed toward dysautonomia, dysbiosis, and/or systemic inflammation offer the most promise.
Introduction
Traumatic brain injury (TBI) is a major cause of death and disability in adults across all age groups, with more than 50 million TBIs occurring worldwide each year (1). The social and economic burden of TBI has placed it at the forefront of public attention, yet decades of costly research has yielded limited clinical success, owing to the poor understanding of TBI’s heterogeneity and complexity. There are currently an estimated 3.17 to 5 million people in the United States living with a chronic TBI-related disability (2). Management of TBI involves both treatment of the acute insult and prevention and treatment of secondary sequelae, which are potential druggable targets. The lack of FDA-approved clinical interventions emphasizes the urgent need to identify and develop novel therapeutic targets for TBI patients.
TBI has profound effects on the gastrointestinal (GI) tract. Notably, TBI survivors are more likely to succumb to digestive disorders than healthy age-matched cohorts (3). The brain-gut axis is a bidirectional pathway that is critical for central nervous system (CNS) and GI homeostasis and regulates diverse functions including visceral pain, intestinal barrier function, gut motility, and neurobehavior (4). TBI induces a stress response that impacts the well-documented autonomic nervous system (ANS) control of GI function (5). Activation of the systemic immune system following TBI could play a major role in subsequent GI dysfunction, and the effects of TBI may be amplified further by psychological distress, which is known to exacerbate GI symptoms (6). Gut-to-brain communication is equally important, as sensory information arising from the gut lumen coordinates function across different regions of the gut. Changes in the gut microbiome are common in neurodegenerative disorders, spurring mechanistic studies on the role of specific microbiota and their microbial products (7). There is also an expanding appreciation of the impact of secondary GI challenges on chronically injured TBI patients that worsen long-term morbidity and mortality.
This article reviews the clinical and translational studies that define our understanding of the role of the brain-gut axis in TBI-induced effects on the gut and delineates current gaps in our knowledge. There is a need for an integrated assessment of how changes in the gut affect the progression of acute to chronic TBI. Thus, the mechanisms proposed to subserve brain-gut communications are discussed here with consideration of their therapeutic potential.
TBI pathophysiology and long-term consequences
TBI is defined as a blow to the head or body resulting in a disruption of normal brain function. Major causes of TBI include falls, motor vehicle accidents, domestic violence (e.g., intimate partner violence), sports-related injuries, and explosive blasts in combat zones (2). TBI is classified as mild, moderate, or severe, based on the Glasgow Coma Scale, which assigns scores to eye, verbal, and motor responses to yield a clinical score from 3 to 15 (mild: 13–15; moderate: 9–12; severe: 3–8) (8, 9). The majority of diagnosed TBIs are estimated to be mild (10), comprising approximately 70% of cases. Even when mild, TBI often results in chronic disabilities that have long-lasting effects on patient health and quality of life (11, 12). Long-term TBI-induced impairments in motor and cognitive function and social behavior, development of mood disorders, abnormal sleep patterns, and personality changes contribute to decreased quality of life and overall life expectancy, resulting in an estimated annual global financial burden of over US$400 billion in health care costs (1). Severe TBI is a major risk factor for neurodegeneration, dementia, stroke, and epilepsy (13), and is associated with an increased long-term mortality rate compared with rates for the general population (12). These neurodegenerative risks also occur in milder forms of TBI, especially after repetitive injuries (e.g., concussions) (13). Thus, TBI is not a single static injury event, but should be considered a chronic and progressive disease with serious long-term consequences.
The neurological consequences of TBI depend on injury severity, location, frequency, sex, age, genetics, and individual comorbidities. Thus, the heterogeneity of TBI injury reflects both the direct mechanical damage that results from the injury itself (primary injury) and the delayed molecular cascades that ensue (secondary injury) (Figure 1). Primary injury is characterized by the displacement of, and mechanical damage to, brain tissue, including contusion, vasculature damage, hemorrhages, alterations in cerebral blood flow and blood-brain barrier (BBB) permeability, and disruptions in metabolism. The ensuing secondary injury involves complex, multifactorial biochemical processes that are initiated within minutes of the primary mechanical injury and persist for days, months, and even years following the primary insult, contributing to and exacerbating neuroinflammation, neurodegeneration, and neurological deficits. Secondary injury processes include the continued flux of intracellular ions (Ca2+, Na+, K+) and release of excitotoxic neurotransmitters (e.g., glutamate). Additionally, cytoplasmic and nuclear proteins released from injured cells act as damage-associated molecular patterns (DAMPs), which are potent stimulators of central and systemic immune responses. The disruption of the BBB allows for the infiltration of peripheral immune cells, including leukocytes, into brain parenchyma. These infiltrating immune cells secrete chemokines and cytokines that mobilize and activate resident glial cells such as microglia and astrocytes, and perpetuate the infiltration of peripheral immune cells (14).
Figure 1. Innate immune responses in brain and periphery following traumatic brain injury.

(i) Depending on the severity of traumatic brain injury (TBI), the primary mechanical injury consists of meningeal contusion, axonal shearing, and cerebrovascular injury that leads to meningeal and neuronal cell death and activation of microglia and astrocytes. (ii) Neuronal injury and glial activation generate chemokines, cytokines, and reactive oxygen species (ROS) and release of damage-associated molecular patterns (DAMPs), eliciting an inflammatory response. (iii) When exposed to DAMPs, phagocytic microglia clear debris and produce neurotrophic factors. (iv) Chronic stimulation of these pathways induces secondary injury via recruitment of leukocytes, which initially facilitate clearance of tissue debris but then contribute to progression of inflammation and blood-brain barrier (BBB) breakdown. (v) The ensuing cytotoxic edema and impaired BBB function increase intracranial pressure (ICP) and lead to reduced cerebral blood flow (CBF), amplifying hypoxia to disrupt energy supply in the brain. This causes further neuronal loss and a feed-forward cycle of neuroinflammation and neurodegeneration. (vi) These progressive pathological changes lead to neurological dysfunction and deficits in motor, cognitive, and affective function. TBI also alters the autonomic nervous system (ANS), which is hard-wired to monitor and modulate DAMPs, thereby producing extracerebral and peripheral innate immune responses. (vii) Sympathetic ANS activation results in peripheral release of catecholamines (epinephrine/norepinephrine; E/NE), which suppress systemic immune responses. The vagus stimulates splenic T lymphocytes and inhibits proinflammatory responses of macrophages, via the cholinergic antiinflammatory pathway (CAP) that dampens systemic inflammation. (viii) The release of catecholamines and glucocorticoids via the hypothalamic-pituitary-adrenal (HPA) axis modulates systemic immune cell function after TBI. (ix) TBI can also disrupt systemic cellular defense mechanisms.
Activation of microglia and reactive astrogliosis are initially beneficial during the acute phase following TBI, supporting phagocytosis of cellular debris and promoting tissue repair and remodeling. However, prolonged propagation of this neuroinflammatory response is deleterious (15). Sustained neuroinflammation results in further tissue damage and neurodegeneration, thereby impairing, or potentially worsening, neurological recovery from TBI. Chronic activation of microglia results in the persistent production and release of proinflammatory molecules including reactive oxygen species (ROS) and cytokines (16). Sustained reactive astrogliosis progresses to the formation of a glial scar, which acts to inhibit axonal regeneration, resulting in dysfunction of axonal end bulbs that impairs functional recovery. Chronic astrogliosis also contributes to BBB breakdown by excessive production and release of matrix metalloproteinases (17–19). These chronic secondary injury processes in TBI patients potentially extend the therapeutic window for clinical intervention (20, 21). Ongoing secondary injury processes may also contribute to the development of comorbidities. TBI-related comorbidities and/or subsequent peripheral challenges can, in turn, exacerbate these secondary injury processes, worsening TBI-related long-term outcomes. Experimental models of TBI have been developed with the goal of investigating the mechanisms of primary and secondary injury sequelae (Tables 1 and 2 and refs. 22–27).
Table 1. Rodent models of TBI.

Table 2. Brain injury parameters for selected referenced preclinical papers.

TBI impacts gut physiology and function
TBI has critical effects on other organ systems, including the GI tract (28, 29), that impact morbidity and mortality (30–32). The few studies in patients that chronicle GI symptoms following TBI are focused primarily on inpatient complications involving food intolerance (33) and GI dysmotility (34) that affect the processes of nutrient and drug absorption essential for recovery (Figure 2). About 50% of severe TBI patients experience feeding intolerance, particularly in the first week after injury, which is correlated with injury severity (33). While the incidence of diarrhea in TBI patients in intensive care units (ICUs) ranged from 10.5% to 74% (33, 35, 36) and positively contributed to length of hospitalization (35), diarrhea may not be a specific result of TBI, as it has a high incidence in critically ill patients receiving enteral nutrition. Other GI-related outcomes experienced by TBI patients include gastroparesis (34, 37) and impaired lower esophageal function (38), which also contribute to food intolerance. Consistent with clinical findings, experimental models of TBI showed decreased intestinal smooth muscle contractility and delayed GI transit between 1 and 7 days after injury (39). The hierarchical neural control of GI motility, particularly of the upper GI tract, involves modulation by the ANS and enteric nervous system (ENS). Both systems are negatively impacted by TBI and likely contribute to the early GI dysmotility in hospitalized TBI patients.
Figure 2. TBI induces significant changes in gut function.

The secondary sequelae of TBI in the gut include (i) mucosal damage associated with increased permeability and (ii) malabsorption of nutrients and electrolytes. Enhanced mucosal permeability mobilizes gut defenses, which include increased numbers of activated enteric glial cells culminating in (iii) reactive gliosis and generation of products that promote barrier function and epithelial repair. TBI-induced dysautonomia is characterized by sympathetic dominance, which in combination with the (iv) local release of proinflammatory mediators from resident and recruited immune cells inhibits smooth muscle contraction. These early TBI-induced effects on GI motility include (v) gastroparesis leading to food intolerance. Dysmotility also promotes (vi) changes in microbial composition and (vii) microbial products and metabolites. Compromised barrier function facilitates their passage across the mucosa, leading to activation of (vii) vagal and (ix) spinal afferents that are fundamental to (x) gut-brain communication. A secondary gut challenge such as enteric infection or inflammation prolongs the effects of i–xi and contributes to (xi) levels of circulating inflammatory mediators. Long-lasting systemic inflammation, dysautonomia, and dysbiosis contribute to the chronicity of TBI-induced effects on the gut as well as the increased susceptibility of TBI patients to GI disorders. IECs, intestinal epithelial cells; TJ, tight junctions; AJ, adherens junctions; DS, desmosomes; GJ, gap junctions; GC, goblet cells; EEC, enteroendocrine cells; EGC, enteric glial cells; rEGC, reactive EGC; MM, muscularis mucosae; Th1, T helper cells; CM, circular muscle; EN, enteric neurons; LM, longitudinal muscle.
Mucosal barrier dysfunction is often cited as a major outcome of TBI that contributes to long-term sequelae (Figure 2). An effective barrier requires an intact epithelial cell layer connected by intercellular junctional protein complexes composed of tight junctions, adherens junctions, and desmosomes that allow selective passage of luminal contents (40). Regulation of barrier homeostasis entails a complex interaction among epithelial cells, immune cells, and enteric glial cells (41). Mucus is also a component of the barrier, as its close association with gut epithelial cells provides a physical barrier to limit direct contact with pathogenic microbiota or other deleterious agents (42). Clinical data on mucosal barrier function following TBI are sparse and limited to ICU patients. Using lactulose/mannitol absorption ratios to assess gut permeability in TBI patients, impaired barrier function was observed at 4 days after injury, but not earlier, and was correlated with severity of disease and long-term prognosis (43). Data from experimental rodent models show that TBI results in increased mucosal permeability concomitant with microscopic changes that are variable and depend, in part, on the experimental model, the species, and the time after the initial injury (44–49). In rats, impaired barrier function was associated with microscopic changes within hours after injury, including edema, villous shortening, epithelial shedding, and inflammatory infiltrate (45–47, 50). In murine models of TBI, enhanced ileal permeability was observed at 6 hours (44) and at 3 and 7 days after injury and was associated with villous shortening (48). Others reported no change in jejunal permeability or morphology at 24 hours or 28 days after TBI (49), suggesting there may be differences in the effects of TBI along the length of the intestine.
There is less information on the impact of TBI on colonic function. Moreover, the extent to which early changes in gut function facilitate progression from acute to chronic TBI is unclear. TBI had no effect on colonic mucosal injury or permeability at 6 hours after injury (45). In contrast, at 28 days after injury there were changes in morphology consistent with a colonic response to an inflammatory challenge (49). Coincidently, at 28 days after TBI, paracellular permeability was enhanced in the colon, but not in the small intestine (49), supporting both temporal and regional differences in the gut response to TBI. The importance of these chronic effects, particularly in the colon with its concentration of microbiota, is underscored by the observation that TBI patients who survived longer than a year after injury were 12 times more likely to die of complications of sepsis and 2.5 times more likely to die of digestive disease–related conditions compared with healthy, age-matched cohorts of the general population (3). Thus, the GI tract likely plays a key role in the long-lasting sequelae of TBI that impact patient health and quality of life.
Mechanisms of TBI-induced alterations in brain-gut communication
Systemic immune dysfunction.
TBI initiates a systemic stress response associated with activation of the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic branch of the ANS, resulting in the release of glucocorticoids and catecholamines, respectively (51–55). Catecholamine release into the circulation remained elevated for 14 days after injury and was proportional to the severity of injury (56). While this response may acutely compensate for TBI effects, a prolonged stress response is associated with numerous deleterious effects, leading to organ dysfunction including GI dysmotility, hypertension, and cardiac dysfunction, among others (52, 53, 57, 58).
In addition to the increased release of corticoids and catecholamines, TBI induces a systemic immune response, which can progress to systemic immune response syndrome (SIRS), releasing immune mediators of inflammation (cytokines, chemokines) into the circulation (59–63). Systemic inflammation can persist for months in patients suffering mild (59) and severe TBI (63–65). In prospective cohort studies, TBI patients had elevated serum IL-1β, IL-6, IL-8, IL-10, and TNF-α levels over the first year after injury when compared with age-matched healthy controls (64). A subacute systemic cytokine load score identified individuals at risk for unfavorable outcomes following TBI, and was predictive of global outcomes at 6 and 12 months (65). Higher proinflammatory burden with IL-6, relative to the antiinflammatory marker IL-10, was shown to be relevant to global TBI recovery. Moreover, the acute-phase cytokine load score predicted cognitive performance in moderate-to-severe TBI patients (66), such that IL-1β, TNF-α, sIL-6R, RANTES, and MIP-1β were negatively associated with cognition. Thus, there is a significant correlation between circulating cytokines and chemokines and worsened patient outcomes. Once in circulation, these immune mediators exert deleterious effects on other peripheral organ systems (61, 67, 68). Although not identical to the cytokine storm induced during TBI and SIRS, cytokine release syndrome, which is associated with the sudden release of large amounts of cytokines, results in peripheral organ dysfunction (69).
Preclinical studies indicate that TBI results in acute and chronic changes in peripheral immune function, prominently skewing toward widespread immunosuppression in the innate and adaptive arms of systemic immunity acutely after TBI, and then transforming to a hyperinflammatory and dysfunctional state during chronic recovery periods (70–72). Notably, neuroinflammation has been linked also to activation of systemic immune responses. Preclinical studies show that peripheral injections triggering a systemic immune response (e.g., LPS, IL-1β) at acute and chronic time points following experimental TBI induced microglial activation and exacerbated TBI-related neuropathology and neurological outcomes (73–76). It is proposed that intestinal inflammation promotes neuroinflammation through an activated systemic immune response. Proinflammatory factors released from circulating, activated immune cells in response to an enteric challenge could increase BBB permeability, allowing access of circulating immune cells and mediators to the brain that serve to activate microglia and trigger a neuroinflammatory response (77). Additionally, deficits in the cholinergic system in the brain following TBI have been reported (78, 79), which may be affected further by the sustained systemic immune response.
Autonomic and enteric nervous system dysfunction.
The GI tract is innervated by all three divisions of the ANS: the parasympathetic nervous system, the sympathetic nervous system, and the intrinsic ENS that is embedded in the wall of the gut. The afferents for these systems are located strategically all along the gut, including mucosal surfaces, and fibers project to dorsal root ganglia (vagal) or terminate in the spinal cord (spinal), transmitting high- and low-threshold mechano- and chemosensitive information (80). Sensory information from the gut is also transmitted by visceral afferents, which are associated with, but distinct from, the ANS. Importantly, visceral afferents can be sensitized by gut inflammation (81) and may be affected by elevated levels of intestinal TNF-α and IL-6 in the first week following experimental TBI (82). The incidence of ANS dysfunction at 14 days after injury was reported to be 8% in patients with moderate to severe TBI (83). Dysautonomia plays a critical role in both the acute and the long-term pathology of TBI (84), and is associated with worse outcomes and increased mortality (83).
The parasympathetic vagus nerve innervates almost the entire length of the gut from the esophagus to the mid colon and plays a key role in coordinating function between different regions of the gut. Preganglionic efferent fibers originate from the nucleus ambiguus and the dorsal motor nucleus of the vagus and pass uninterrupted to synapse on target cells such as the postganglionic neurons in the ENS plexuses located in the muscularis externa (Auerbach’s plexus) and submucosa (Meissner’s plexus). Although stimulation of the parasympathetic nerves has both excitatory and inhibitory actions on secretomotor gut function, the vagus is primarily a sensory nerve composed of approximately 80%–90% afferent fibers. Information carried from the gut to the CNS is critical to orchestration of the reflex activity in response to physiological meal-related mechano- and chemosensory stimuli, as well as to neuropeptides released from enteroendocrine cells in the gut. The vagus can also respond to nociceptive signals (81). Following TBI, the vagal afferents provide a mechanistic pathway for inflammatory mediators, microbiota, or microbial products to modulate CNS function via the brain-gut axis (Figure 2).
Sympathetic nerves comprise about 50% afferent fibers. Spinal afferents primarily transmit nociceptive information from the GI tract and are activated by gut inflammation and injury (81). Preganglionic efferent fibers synapse first in one of three prevertebral ganglia (celiac, superior, or inferior mesenteric ganglia), and postganglionic adrenergic fibers synapse on a variety of cells, including ENS neurons, sphincters, and blood vessels. Stimulation of sympathetic nerves plays a critical role in the regulation of blood flow to the gut and generally inhibits GI secretomotor function. TBI increases sympathetic activity early after injury, instigating a stress response leading to a sympathetic storm (51, 52, 54, 84, 85). The exaggerated catecholamine release likely contributes to GI dysmotility including gastroparesis and food intolerance.
Both the parasympathetic and sympathetic systems are tonically active, and imbalance of the input motivates the dysautonomia that impacts GI dysfunction (86). Heart rate variability (HRV) is a noninvasive determinant of baseline autonomic function (tone). It is used to ascertain the balance of sympathetic and vagal tone as an expression of the total amount of variations of both instantaneous heart rate and RR (beat-to-beat) intervals, which are the intervals between normal QRS complexes. The principal components of HRV are a high frequency (HF), reflective of basal vagal tone, and a low frequency (LF) related to sympathetic tone. The ratio of these frequencies (LF/HF) is considered a clinical index of ANS regulation of target organs. Decreased HRV, indicating sympathetic dominance, is reported in patients with all levels of TBI (87). Importantly, correction of dysautonomia in TBI patients is an emerging therapeutic target with early administration of propranolol having beneficial effects on TBI in a prospective randomized clinical trial (88). Propranolol’s potential beneficial effects on secondary GI sequelae are untested.
The vagus plays a key role in the intestinal mucosal response to injury and inflammation via the excitatory cholinergic reflex. The vagal antiinflammatory pathway is initiated by intestinal inflammation and involves vagal efferent activation of the ENS close to resident macrophages in the gut (89) or the spleen (90). In the gut, acetylcholine released by the ENS binds to the cholinergic nicotinic acetylcholine α7 receptors located on macrophages, leading to inhibition of proinflammatory cytokine release (91). Vagotomized mice exhibit a worsening of injury and inflammation, demonstrating a tonic vagal activation in response to intestinal damage (92). The severity of TBI-induced secondary injury including BBB permeability, systemic inflammation, and microglial activation was exacerbated in α7 receptor–knockout mice (93). In this same study, systemic administration of the proinflammatory cytokines IL-1β and TNF-α aggravated BBB permeability, thereby linking peripheral inflammation to TBI outcomes. Vagal nerve stimulation reduces systemic inflammation in preclinical TBI models (94, 95) and is currently undergoing clinical testing in patients with mild TBI and posttraumatic stress disorder (96, 97). Thus, restoration of vagal tone to improve TBI-induced dysautonomia, through direct electrical stimulation or pharmacological modulation using vagomimetic agents (98, 99), reversible cholinesterase inhibitors (99–101), or drugs targeting activity or expression of muscarinic/nicotinic acetylcholine receptors or β-adrenergic receptors (99, 102, 103), is a viable therapeutic target.
The ENS is the “little brain in the gut” that independently integrates intrinsic input from the gut lumen with extrinsic signals from the parasympathetic and sympathetic nerves (104). The extrinsic nerve fibers have little direct access to individual secretomotor cells in the gut, so the ENS acts as a “gatekeeper.” Synaptic connections between sensory neurons, interneurons, and motor neurons in the ENS coordinate input to provide effector functions appropriate for each region of the gut. The bidirectional communication between the CNS and ENS is the foundation of the brain-gut axis, and the ANS is central to this interaction. The CNS and ENS share a similar rich repertoire of neurotransmitters such as acetylcholine and norepinephrine that are implicated in acute and chronic dysfunction after TBI (105). Norepinephrine is the postganglionic neurotransmitter in the sympathetic nervous system and inhibits ENS neuron release of acetylcholine. Acetylcholine is the major neurotransmitter at preganglionic neurons in both the parasympathetic and sympathetic systems and plays a major role in the control of cognitive function including learning, memory (106), and attention (107). It is a viable therapeutic target, as decreased cholinergic activity in CNS regions was observed in both TBI patients (108) and preclinical models with impaired cognitive behavior associated with the loss of cholinergic neurotransmission (78).
A major component of the ENS are enteric glial cells (EGCs), which outnumber neurons by at least 4:1 (109). EGCs resemble their CNS counterparts, astrocytes, in morphology and expression of common markers such as S100B (110, 111), the precursor protein SOX10 (112), and the activation marker glial fibrillary acidic protein (GFAP). EGCs also have unique markers like proteolipid protein 1 (113) and lack the astrocyte marker aldehyde dehydrogenase 1 family member L1 (114). Marker expression changes with their location in the GI tract, and they respond strongly to environmental cues, such that EGCs remain glial cells in the gut but, under certain in vitro conditions, can be rederived into ENS neurons (115) and can function as astrocytes when transplanted into the CNS (116, 117). EGCs occupy two somewhat discrete areas in the GI tract, one within the myenteric plexus and one beneath the epithelium. The role of myenteric EGCs has expanded beyond providing a support for neurons, with EGC-derived products playing an active role in neurotransmission (118). The location of the mucosal EGC population impacts neuro-epithelial interactions, and mucosal EGC production of S-nitrosoglutathione directly affects epithelial permeability (119). Interestingly, vagal nerve stimulation activates α7-nicotinic receptors on EGCs and improves intestinal barrier integrity following severe injury (120).
In experimental models, TBI induces astrogliosis that provides a physical support for the cells within the injured area as well as a source of trophic factors and immune mediators that promote tissue remodeling and neurogenesis (121, 122). While astrogliosis is beneficial in repairing local damage and disruption of the BBB, chronic reactivity is associated with TBI-induced neuroinflammation leading to persistent deficits in patients attributable to impaired neural recovery (123). Like astrocytes in the CNS, EGCs can have both beneficial and deleterious effects. Glial cell–derived neurotrophic factor (GDNF) is necessary for maintenance of enteric neurons (124), and upregulation of GDNF has an antiinflammatory role supporting epithelial proliferation and turnover in inflammatory bowel disease (IBD) patients as well as preservation of mucosal integrity (125). In contrast, production of TGF-β1 results in inhibition of cell proliferation, thereby impairing restitution of barrier function after injury (125). In ulcerative colitis, upregulation of S100B and increased numbers of activated EGCs contribute to the increased levels of nitric oxide (126) that drive chronic changes in gut secretomotor function (127). TBI transiently decreases activated glial cells in the colon, but not the small intestine, at 24 hours after injury, followed at 28 days after injury by an increased number of activated mucosal EGCs (Figure 2 and ref. 49). Despite the anatomical connection between the CNS and ENS, the nature and extent of any communication between CNS astrocytes and ENS glial cells are unknown. It is also unclear whether astrocytes preferentially influence myenteric or mucosal EGCs. As inflammation alters the number and activity of EGCs in the gut, TBI-induced GI gliosis could be mediated by local events; however, TBI-induced EGC proliferation and activation in the colon occurred in the presence of increased colonic permeability, but in the absence of overt inflammation (49). Thus, TBI induces a region-specific reactive gliosis in the gut that may be part of a local neuroprotective response to impaired mucosal barrier function and may also contribute to the long-term secondary sequelae in the gut (49).
Gut microbiota in the brain-gut axis.
The GI tract, especially the colon, contains the largest concentration of microbiota, a diverse mixture of primarily bacteria, but also viruses, fungi, and yeasts. The human intestinal microbiota contains approximately 1000 bacterial species dominated by four phyla: Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria, comprising greater than 95% of resident bacteria in healthy adults (128). The microbiota influence the CNS via neural, vascular, and even lymphatic pathways, as part of the bidirectional brain-gut axis (28, 129–131). The primarily sensory vagus nerve serves as a direct, rapid pathway through which the gut microbiota can affect brain function (132–134). In the gut, microbial products bind to receptors on vagal afferents in the gut mucosa, providing information about changes in the composition and pathogenicity of the microbiome (135). CNS input influences ENS activity to affect gut microbiome composition and diversity by modulating intestinal motility, transit, barrier integrity, and secretion of factors into the lumen that influence microbial gene expression and function (136–141). Alterations in the gut microbiome composition favoring pathogenic bacteria over commensal bacteria have deleterious effects on both the CNS and the GI tract. Abnormal motility induces changes in the microbiota, setting up a positive-feedback loop between dysbiosis and dysmotility, and the detrimental impact on clinical outcomes is evident in chronic GI inflammatory pathologies such as IBD (142, 143).
The gut is the largest immune organ in the body and contains an abundance of resident immune cells, including innate lymphoid cells, macrophages, and dendritic cells, that, with enterocytes, provide the first line of defense in detecting alterations in the microbiome. These cells generate cytokines and chemokines to recruit and activate additional immune cells as part of innate and adaptive immune responses. The immune system in the gut plays an important role in maintaining the symbiotic relationship between the host and commensal bacteria (144). Commensal microbial metabolites such as methane and short-chain fatty acids, as well as secondary metabolites like tryptamine and bile acid metabolites, modulate GI functions. TBI significantly alters the gut microbiome (Table 3), decreasing commensal bacteria and increasing the presence of pathogenic bacteria, thereby promoting disease progression (145–148). TBI-induced paracellular permeability likely facilitates translocation of pathogenic products that disrupt normal brain-gut communication (Figure 2). Dysbiosis can persist in patients for years after the initial brain trauma (146), and reduced diversity in severely injured TBI patients is predictive of survival (149). The microbiome is exquisitely sensitive to changes in diet (150). As TBI patients often require nutritional support while in the ICU (151), these critically injured patients likely experience acute changes in microbiome as a result of food intolerance and dysmotility. Long-term follow-up studies indicate that TBI patients may continue to exhibit altered eating behaviors, including anorexia as well as hyperphagia (152), and overall are at greater risk of obesity (153). There is evidence of a causal link between microbiota composition and microbial metabolites associated with lean and obese body weights (154, 155). Thus, the dysbiosis induced during the primary TBI injury is compounded by the multifactorial processes during the secondary injury, all of which may be exacerbated further by changes in nutrition or food psychology.
Table 3. Effect of TBI on the microbiome.

In preclinical TBI studies (145, 146, 156–158), acute changes at the species and genus levels (158), as well as alterations in α-diversity with increased numbers of Proteobacteria and decreased Firmicutes, were correlated significantly with the severity of TBI (159). There is avid interest in the impact of changes in gut microbiota composition on neuroinflammation, neurological development, cognition, affect, and behavior (160–165). An early manifestation of experimental-TBI-induced gut injury is translocation of bacteria-derived endotoxin, which may promote systemic inflammation, leading to CNS neuroinflammation and neurodegeneration (46). Indeed, neuroinflammation is exacerbated by systemic administration of LPS following experimental TBI (74), contributing to a more pronounced TBI-induced cognitive decline (75). Notably, depletion of the gut microbiota following experimental TBI with an antibiotic cocktail ameliorated TBI-induced neurological outcomes, improving fear-based cue learning and reducing lesion volume (157). Additionally, dietary short-chain fatty acids are rapidly absorbed and have antiinflammatory effects on microglia (166) and improve neurological recovery after ischemic brain injury (167).
Thus, early changes in motility in TBI patients are likely accompanied by changes in microbiota. The dysbiosis, in turn, directly and indirectly contributes to TBI-induced outcomes including loss of barrier integrity, chronic activation of local and systemic immune responses, and dysmotility. These effects increase susceptibility to sepsis (168–171), impair cognitive function, worsen neuropathology and neuroinflammation, and increase the risk of neurodegenerative diseases (135, 160, 161, 172–175). Preclinical studies, however, demonstrated that antibiotic-induced depletion of gut microbiota prior to TBI aggravated outcomes, implicating a protective effect of specific microbiota or microbial products in TBI (148). The benefits of manipulating the microbiota in TBI are supported by studies showing that pretreatment with Lactobacillus acidophilus or Clostridium butyricum (176) attenuated TBI-induced neurological effects, neuroinflammation, and neuropathology (48). The therapeutic potential of gut microbiota in TBI is the focus of several recent reviews (177–179).
Secondary GI challenges and TBI outcomes.
GI infections and inflammation activate both local and systemic immune responses that become chronic as a result of persistent barrier dysfunction and intestinal inflammation (180). There is heightened attention to the brain-gut axis in an effort to unravel the impact of secondary challenges on CNS function, particularly in TBI patients. Of interest, then, is that in preclinical models, mild-to-moderate TBI did not alter the efficacy or efficiency of the host immune response to enteric pathogenic bacterial infection (49) nor did it aggravate the severity of injury or delay mucosal repair following acute colitis (181). Enteric challenge following experimental TBI, however, dramatically influenced gut-to-brain communication. A proposed scenario involves increased circulating microbial products and proinflammatory cytokines released from activated enteric and systemic immune cells in response to infection, increasing BBB permeability (77). This in turn can facilitate access of circulating immune cells and mediators to the CNS, activating microglia and triggering neuroinflammation (77). Severe TBI patients, however, exhibit repressed host immune response such that normally nonpathogenic strains of E. coli bacteria can trigger infections (182). This weakened cell-mediated immune response after TBI may contribute to the high incidence of infections in long-term TBI patients (183, 184).
Enteric pathogen-induced inflammation is implicated in the development of cognitive dysfunction and increased risk of neurodegenerative diseases in patients (185–188). Infection with E. coli resulted in deficits in working memory and fear-based retention memory for up to 7 days after enteric infection in rodents (189). Preclinical data thus support the association between clinical intestinal inflammation and neurodegenerative diseases reported in IBD patients, who have twice the risk of developing dementia (190). Similarly, TBI patients with persistent posttraumatic neuroinflammation and progressive neurodegeneration have increased risk of dementia (191, 192). Recent studies showed that brain-injured mice subjected to acute colonic inflammation exhibited neurobehavioral deficits initiated at the time of inflammation that were maintained throughout the recovery period (181). Indeed, colonic inflammation significantly exacerbated TBI-induced chronic deficits in fine motor coordination, declarative memory, spatial learning and memory, and social behavior (181). Importantly, both the magnitude and the severity of these inflammation-induced changes were similar in mild and moderate-to-severe TBI (181). Network-level gene analysis of the hippocampus of brain-injured mice with and without enteric inflammation strongly implicated genes associated with disease and neuroinflammation, and a downregulation of genes associated with neuronal structure, plasticity, transmission, and connectivity (181).
The mechanisms of these brain-gut pathological interactions may be linked to the effects of systemic inflammation on TBI neuropathology and ANS activity that are compounded by the impact of gut inflammation on TBI-induced dysautonomia. Experimental TBI resulted in an acute and transient increase in HRV in moderate-to-severe TBI, but subsequent enteric inflammation led to a persistent dominance of sympathetic tone regardless of TBI severity (181). This dysautonomia arises perhaps by the simultaneous inputs from the damaged brain and gut and is maintained by systemic inflammation and other unidentified mechanisms, impairing the conditions needed to resolve TBI-related neurological outcomes. These preclinical findings are consistent with literature regarding sickness behavior (193, 194), in which changes in brain energy metabolism and neuroinflammation induced by systemic inflammation result in alterations in cognitive function and affect (195). The enhanced risk of neurodegenerative and neuropsychiatric diseases following enteric challenge in TBI patients has yet to be determined and remains critically unexplored.
Conclusion
Substantial advances have been made in the understanding of bidirectional communication along the brain-gut axis that contributes to the development and progression of pathological responses in each organ following TBI. Dysfunction in neural and systemic pathways of the brain-gut axis — including brain injury and inflammation, changes in ANS, sympathetic stress, and immune cell function — and indirect communication via gut microbiota serve as mechanistic links that drive chronic TBI-related pathologies (Figure 3). Recognition of such brain-gut dysfunction will lead to a better appreciation of how other organ systems contribute to chronic TBI progression and recovery, which is essential to comprehend the full etiology of TBI. Furthermore, targeting mechanisms of brain-gut communication could facilitate development of novel therapeutic interventions for TBI. Emerging therapeutic strategies being developed for clinical translation include those that target systemic inflammation and the cholinergic antiinflammatory pathway by vagal stimulation, those that correct dysautonomia, and those that modulate the gut microbiome and microbial metabolites. Given that long-term TBI survivors are more likely to die of digestive disease–related conditions (3), the mechanisms by which secondary enteric challenges or injuries contribute to the long-lasting sequelae of TBI are an important area of future investigation and should be taken into consideration when addressing patient care.
Figure 3. Bidirectional interactions of the brain-gut axis and therapeutic targets.
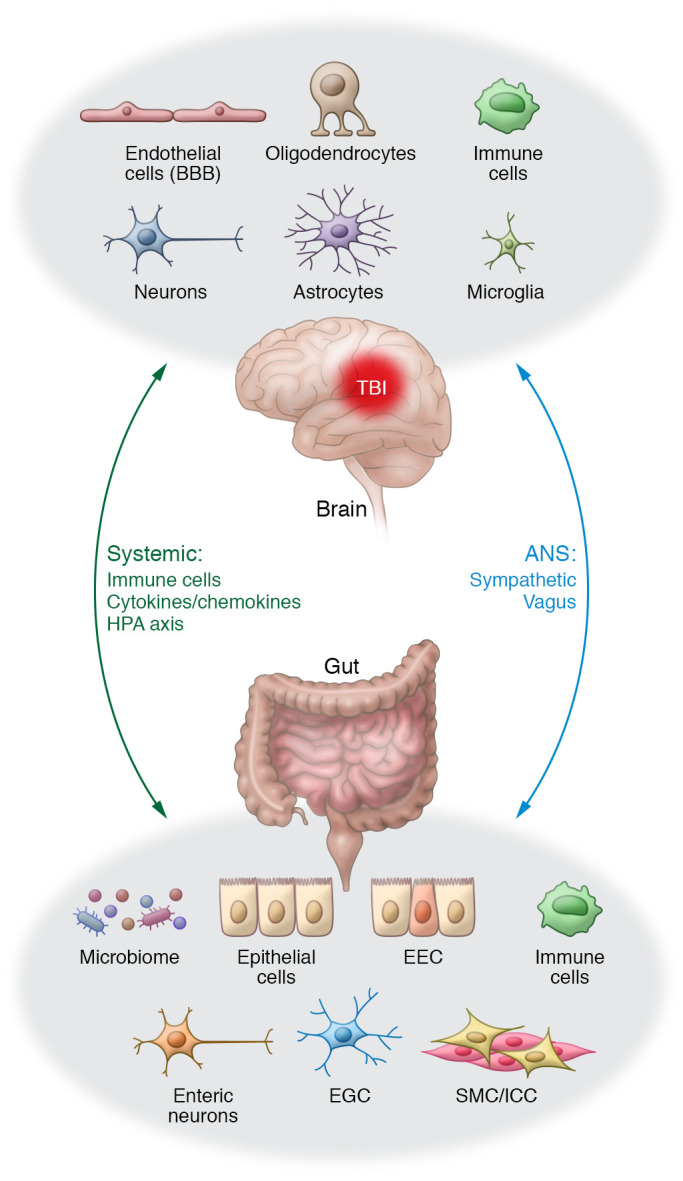
The brain and gut communicate through direct (neural) and indirect (systemic) bidirectional pathways. The brain influences GI function through the ANS (sympathetic and vagal efferents), systemic circulation (blood vessels and lymph), and HPA axis. Signals from the gut, including nutrients, mechanical stimuli, and microbiota and their products and metabolites (e.g., short-chain fatty acids), modulate brain function via neural (ENS neurons, glial cells, vagal afferents, spinal afferents), immune (resident and recruited immune cells), and endocrine (hormones released by enteroendocrine cells) mechanisms. TBI-induced GI dysfunction or secondary enteric challenges worsen neurological outcomes by activating local and systemic immune responses that increase BBB permeability and infiltration of activated circulating immune cells and exacerbate ongoing astrocyte- and microglia-mediated neuropathology. The cells and pathways involved in these bidirectional signaling pathways provide viable targets for therapeutic intervention in TBI patients, particularly those with GI comorbidities. Treatments that potentially benefit both the brain and the gut in TBI patients include reduction of sympathetic activation or restoration of vagal tone; use of pre-/probiotics or fecal microbial transplant to correct gut dysbiosis; and suppression of local and systemic proinflammatory immune responses through approved immunosuppressors or emerging nanotherapeutics. EEC, enteroendocrine cells; EGC, enteric glial cells; ICC, interstitial cells of Cajal; SMC, smooth muscle cells.
Acknowledgments
This work was supported by an NIH grant (R01NS110756 to DJL) and a Science Foundation Ireland grant (17/FRL/4860 to DJL). MH was supported by an NIH (National Institute of Diabetes and Digestive and Kidney Diseases) T32 award (DK067872; principal investigator, Jean-Pierre Raufman) and a Brain Injury Association of America Dissertation Award (Brain Injury Association of America’s Brain Injury Research Fund).
Version 1. 06/15/2021
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(12):e143777.https://doi.org/10.1172/JCI143777.
Contributor Information
Marie Hanscom, Email: mhanscom@som.umaryland.edu.
David J. Loane, Email: dloane@som.umaryland.edu.
Terez Shea-Donohue, Email: terez.shea-donohue@nih.gov.
References
- 1.Maas AIR, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16(12):987–1048. doi: 10.1016/S1474-4422(17)30371-X. [DOI] [PubMed] [Google Scholar]
- 2. Faul M, et al. CDC. Traumatic brain injury in the United States; emergency department visits, hospitalizations, and deaths, 2002–2006. https://stacks.cdc.gov/view/cdc/5571 Updated March 2010. Accessed August 10, 2020.
- 3.Harrison-Felix C, et al. Causes of death following 1 year postinjury among individuals with traumatic brain injury. J Head Trauma Rehabil. 2006;21(1):22–33. doi: 10.1097/00001199-200601000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Mayer EA, Tillisch K. The brain-gut axis in abdominal pain syndromes. Annu Rev Med. 2011;62:381–396. doi: 10.1146/annurev-med-012309-103958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayer EA. The neurobiology of stress and gastrointestinal disease. Gut. 2000;47(6):861–869. doi: 10.1136/gut.47.6.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. doi: 10.1007/s10620-020-06798-y. Glynn H, et al. Prevalence and impact of post-traumatic stress disorder in gastrointestinal conditions: a systematic review [published January 12, 2021]. Dig Dis Sci . [DOI] [PubMed]
- 7.Carabotti M, et al. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28(2):203–209. [PMC free article] [PubMed] [Google Scholar]
- 8.Iankova A. The Glasgow Coma Scale: clinical application in emergency departments. Emerg Nurse. 2006;14(8):30–35. doi: 10.7748/en2006.12.14.8.30.c4221. [DOI] [PubMed] [Google Scholar]
- 9.Teasdale G, Jennett B. Assessment and prognosis of coma after head injury. Acta Neurochir (Wien) 1976;34(1–4):45–55. doi: 10.1007/BF01405862. [DOI] [PubMed] [Google Scholar]
- 10.Faul M, Coronado V. Epidemiology of traumatic brain injury. Handb Clin Neurol. 2015;127:3–13. doi: 10.1016/B978-0-444-52892-6.00001-5. [DOI] [PubMed] [Google Scholar]
- 11.Carroll EL, et al. Mild traumatic brain injury recovery: a growth curve modelling analysis over 2 years. J Neurol. 2020;267(11):3223–3234. doi: 10.1007/s00415-020-09979-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stocchetti N, Zanier ER. Chronic impact of traumatic brain injury on outcome and quality of life: a narrative review. Crit Care. 2016;20(1):148. doi: 10.1186/s13054-016-1318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson L, et al. The chronic and evolving neurological consequences of traumatic brain injury. Lancet Neurol. 2017;16(10):813–825. doi: 10.1016/S1474-4422(17)30279-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das M, et al. Lateral fluid percussion injury of the brain induces CCL20 inflammatory chemokine expression in rats. J Neuroinflammation. 2011;8(1):148. doi: 10.1186/1742-2094-8-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simon DW, et al. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13(3):171–191. doi: 10.1038/nrneurol.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loane DJ, et al. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73(1):14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ekmark-Lewén S, et al. Traumatic axonal injury in the mouse is accompanied by a dynamic inflammatory response, astroglial reactivity and complex behavioral changes. J Neuroinflammation. 2013;10:44. doi: 10.1186/1742-2094-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menzel L, et al. Progranulin protects against exaggerated axonal injury and astrogliosis following traumatic brain injury. Glia. 2017;65(2):278–292. doi: 10.1002/glia.23091. [DOI] [PubMed] [Google Scholar]
- 19.Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5(2):146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 20.Henry RJ, et al. Microglial depletion with CSF1R inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J Neurosci. 2020;40(14):2960–2974. doi: 10.1523/JNEUROSCI.2402-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31(12):596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma X, et al. Animal models of traumatic brain injury and assessment of injury severity. Mol Neurobiol. 2019;56(8):5332–5345. doi: 10.1007/s12035-018-1454-5. [DOI] [PubMed] [Google Scholar]
- 23.Aravind A, et al. Behavioral deficits in animal models of blast traumatic brain injury. Front Neurol. 2020;11:990. doi: 10.3389/fneur.2020.00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiong Y, et al. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14(2):128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shultz SR, et al. The potential for animal models to provide insight into mild traumatic brain injury: translational challenges and strategies. Neurosci Biobehav Rev. 2017;76(pt B):396–414. doi: 10.1016/j.neubiorev.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 26.Namjoshi DR, et al. Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Mol Neurodegener. 2014;9:55. doi: 10.1186/1750-1326-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McNamara EH, et al. The closed-head impact model of engineered rotational acceleration (CHIMERA) as an application for traumatic brain injury pre-clinical research: a status report. Exp Neurol. 2020;333:113409. doi: 10.1016/j.expneurol.2020.113409. [DOI] [PubMed] [Google Scholar]
- 28.Sundman MH, et al. The bidirectional gut-brain-microbiota axis as a potential nexus between traumatic brain injury, inflammation, and disease. Brain Behav Immun. 2017;66:31–44. doi: 10.1016/j.bbi.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Iftikhar PM, et al. Traumatic brain injury causing intestinal dysfunction: a review. J Clin Neurosci. 2020;79:237–240. doi: 10.1016/j.jocn.2020.07.019. [DOI] [PubMed] [Google Scholar]
- 30.Lim HB, Smith M. Systemic complications after head injury: a clinical review. Anaesthesia. 2007;62(5):474–482. doi: 10.1111/j.1365-2044.2007.04998.x. [DOI] [PubMed] [Google Scholar]
- 31.Harrison-Felix CL, et al. Mortality over four decades after traumatic brain injury rehabilitation: a retrospective cohort study. Arch Phys Med Rehabil. 2009;90(9):1506–1513. doi: 10.1016/j.apmr.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 32.Anthony DC, Couch Y. The systemic response to CNS injury. Exp Neurol. 2014;258:105–111. doi: 10.1016/j.expneurol.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 33.Norton JA, et al. Intolerance to enteral feeding in the brain-injured patient. J Neurosurg. 1988;68(1):62–66. doi: 10.3171/jns.1988.68.1.0062. [DOI] [PubMed] [Google Scholar]
- 34.Kao CH, et al. Gastric emptying in head-injured patients. Am J Gastroenterol. 1998;93(7):1108–1112. doi: 10.1111/j.1572-0241.1998.00338.x. [DOI] [PubMed] [Google Scholar]
- 35.Vieira LV, et al. Incidence of diarrhea and associated risk factors in patients with traumatic brain injury and enteral nutrition. Metab Brain Dis. 2018;33(5):1755–1760. doi: 10.1007/s11011-018-0287-2. [DOI] [PubMed] [Google Scholar]
- 36.Li X, et al. Enteral combined with parenteral nutrition improves clinical outcomes in patients with traumatic brain injury. Nutr Neurosci. doi: 10.1080/1028415x.2020.1765114. [published online May 20, 2020]. [DOI] [PubMed] [Google Scholar]
- 37.Jackson MD, Davidoff G. Gastroparesis following traumatic brain injury and response to metoclopramide therapy. Arch Phys Med Rehabil. 1989;70(7):553–555. [PubMed] [Google Scholar]
- 38.Saxe JM, et al. Lower esophageal sphincter dysfunction precludes safe gastric feeding after head injury. J Trauma. 1994;37(4):581–584. doi: 10.1097/00005373-199410000-00010. [DOI] [PubMed] [Google Scholar]
- 39.Sun B, et al. The effects of lactobacillus acidophilus on the intestinal smooth muscle contraction through PKC/MLCK/MLC signaling pathway in TBI mouse model. PLoS One. 2015;10(6):e0128214. doi: 10.1371/journal.pone.0128214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fasano A, Shea-Donohue T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Pract Gastroenterol Hepatol. 2005;2(9):416–422. doi: 10.1038/ncpgasthep0259. [DOI] [PubMed] [Google Scholar]
- 41.Sharkey KA. Emerging roles for enteric glia in gastrointestinal disorders. J Clin Invest. 2015;125(3):918–925. doi: 10.1172/JCI76303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGuckin MA, et al. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. 2011;9(4):265–278. doi: 10.1038/nrmicro2538. [DOI] [PubMed] [Google Scholar]
- 43.Faries PL, et al. Intestinal permeability correlates with severity of injury in trauma patients. J Trauma. 1998;44(6):1031–1035. doi: 10.1097/00005373-199806000-00016. [DOI] [PubMed] [Google Scholar]
- 44.Bansal V, et al. Stimulating the central nervous system to prevent intestinal dysfunction after traumatic brain injury. J Trauma. 2010;68(5):1059–1064. doi: 10.1097/TA.0b013e3181d87373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feighery L, et al. Increased intestinal permeability in rats subjected to traumatic frontal lobe percussion brain injury. J Trauma. 2008;64(1):131–137. doi: 10.1097/TA.0b013e3181568d9f. [DOI] [PubMed] [Google Scholar]
- 46.Hang CH, et al. Alterations of intestinal mucosa structure and barrier function following traumatic brain injury in rats. World J Gastroenterol. 2003;9(12):2776–2781. doi: 10.3748/wjg.v9.i12.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Jiang X. Effects of enteral nutrition on the barrier function of the intestinal mucosa and dopamine receptor expression in rats with traumatic brain injury. JPEN J Parenter Enteral Nutr. 2015;39(1):114–123. doi: 10.1177/0148607113501881. [DOI] [PubMed] [Google Scholar]
- 48.Ma Y, et al. Lactobacillus acidophilus exerts neuroprotective effects in mice with traumatic brain injury. J Nutr. 2019;149(9):1543–1552. doi: 10.1093/jn/nxz105. [DOI] [PubMed] [Google Scholar]
- 49.Ma EL, et al. Bidirectional brain-gut interactions and chronic pathological changes after traumatic brain injury in mice. Brain Behav Immun. 2017;66:56–69. doi: 10.1016/j.bbi.2017.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Y, et al. Rosuvastatin alleviates intestinal injury by down-regulating the CD40 pathway in the intestines of rats following traumatic brain injury. Front Neurol. 2020;11:816. doi: 10.3389/fneur.2020.00816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kôiv L, et al. Changes of sympatho-adrenal and hypothalamo-pituitary-adrenocortical system in patients with head injury. Acta Neurol Scand. 1997;96(1):52–58. doi: 10.1111/j.1600-0404.1997.tb00238.x. [DOI] [PubMed] [Google Scholar]
- 52.Lemke DM. Riding out the storm: sympathetic storming after traumatic brain injury. J Neurosci Nurs. 2004;36(1):4–9. doi: 10.1097/01376517-200402000-00002. [DOI] [PubMed] [Google Scholar]
- 53.McDonald SJ, et al. Beyond the brain: peripheral interactions after traumatic brain injury. J Neurotrauma. 2020;37(5):770–781. doi: 10.1089/neu.2019.6885. [DOI] [PubMed] [Google Scholar]
- 54.Rosner M, et al. Mechanical brain injury: the sympathoadrenal response. J Neurosurg. 1984;61(1):76–86. doi: 10.3171/jns.1984.61.1.0076. [DOI] [PubMed] [Google Scholar]
- 55.Taylor AN, et al. Injury severity differentially affects short- and long-term neuroendocrine outcomes of traumatic brain injury. J Neurotrauma. 2008;25(4):311–323. doi: 10.1089/neu.2007.0486. [DOI] [PubMed] [Google Scholar]
- 56.Schroeppel TJ, et al. Beta-adrenergic blockade for attenuation of catecholamine surge after traumatic brain injury: a randomized pilot trial. Trauma Surg Acute Care Open. 2019;4(1):e000307. doi: 10.1136/tsaco-2019-000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 1984;5(1):1–24. doi: 10.1210/edrv-5-1-1. [DOI] [PubMed] [Google Scholar]
- 58.Stanford GG. The stress response to trauma and critical illness. Crit Care Nurs Clin North Am. 1994;6(4):693–702. doi: 10.1016/S0899-5885(18)30443-X. [DOI] [PubMed] [Google Scholar]
- 59.Chaban V, et al. Systemic inflammation persists the first year after mild traumatic brain injury: results from the prospective trondheim mild traumatic brain injury study. J Neurotrauma. 2020;37(19):2120–2130. doi: 10.1089/neu.2019.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jaffer U, et al. Cytokines in the systemic inflammatory response syndrome: a review. HSR Proc Intensive Care Cardiovasc Anesth. 2010;2(3):161–175. [PMC free article] [PubMed] [Google Scholar]
- 61.Ott L, et al. Cytokines and metabolic dysfunction after severe head injury. J Neurotrauma. 1994;11(5):447–472. doi: 10.1089/neu.1994.11.447. [DOI] [PubMed] [Google Scholar]
- 62.Zhang W, et al. Systemic inflammatory response syndrome in Sepsis-3: a retrospective study. BMC Infect Dis. 2019;19(1):139. doi: 10.1186/s12879-019-3790-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vijapur SM, et al. Treelet transform analysis to identify clusters of systemic inflammatory variance in a population with moderate-to-severe traumatic brain injury. Brain Behav Immun. doi: 10.1016/j.bbi.2021.01.026. [published online January 29, 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Di Battista AP, et al. Inflammatory cytokine and chemokine profiles are associated with patient outcome and the hyperadrenergic state following acute brain injury. J Neuroinflammation. 2016;13:40. doi: 10.1186/s12974-016-0500-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kumar RG, et al. Chronic inflammation after severe traumatic brain injury: characterization and associations with outcome at 6 and 12 months postinjury. J Head Trauma Rehabil. 2015;30(6):369–381. doi: 10.1097/HTR.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 66.Milleville KA, et al. Early chronic systemic inflammation and associations with cognitive performance after moderate to severe TBI. Brain Behav Immun. 2021;11(3):100185. doi: 10.1016/j.bbih.2020.100185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Syed-Ahmed M, Narayanan M. Immune dysfunction and risk of infection in chronic kidney disease. Adv Chronic Kidney Dis. 2019;26(1):8–15. doi: 10.1053/j.ackd.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 68.Zhao Q, et al. Immune response mediates cardiac dysfunction after traumatic brain injury. J Neurotrauma. 2018;36(4):619–629. doi: 10.1089/neu.2018.5766. [DOI] [PubMed] [Google Scholar]
- 69.Shimabukuro-Vornhagen A, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56. doi: 10.1186/s40425-018-0343-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hazeldine J, et al. Traumatic brain injury and peripheral immune suppression: primer and prospectus. Front Neurol. 2015;6:235. doi: 10.3389/fneur.2015.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ritzel RM, et al. Chronic alterations in systemic immune function after traumatic brain injury. J Neurotrauma. 2018;35(13):1419–1436. doi: 10.1089/neu.2017.5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwulst SJ, et al. Traumatic brain injury-induced alterations in peripheral immunity. J Trauma Acute Care Surg. 2013;75(5):780–788. doi: 10.1097/TA.0b013e318299616a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fenn AM, et al. Immune activation promotes depression one month after diffuse brain injury: a role for primed microglia. Biol Psychiatry. 2014;76(7):575–584. doi: 10.1016/j.biopsych.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hang C, et al. Effect of systemic LPS injection on cortical NF-κB activity and inflammatory response following traumatic brain injury in rats. Brain Res. 2004;1026(1):23–32. doi: 10.1016/j.brainres.2004.07.090. [DOI] [PubMed] [Google Scholar]
- 75.Muccigrosso MM, et al. Cognitive deficits develop 1 month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun. 2016;54:95–109. doi: 10.1016/j.bbi.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Utagawa A, et al. Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Exp Neurol. 2008;211(1):283–291. doi: 10.1016/j.expneurol.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oriá RB, et al. Early-life enteric infections: relation between chronic systemic inflammation and poor cognition in children. Nutr Rev. 2016;74(6):374–386. doi: 10.1093/nutrit/nuw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shin SS, Dixon CE. Alterations in cholinergic pathways and therapeutic strategies targeting cholinergic system after traumatic brain injury. J Neurotrauma. 2015;32(19):1429–1440. doi: 10.1089/neu.2014.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Valiyaveettil M, et al. Regional specific alterations in brain acetylcholinesterase activity after repeated blast exposures in mice. Neurosci Lett. 2012;506(1):141–145. doi: 10.1016/j.neulet.2011.10.067. [DOI] [PubMed] [Google Scholar]
- 80.Robinson DR, Gebhart GF. Inside information: the unique features of visceral sensation. Mol Interv. 2008;8(5):242–253. doi: 10.1124/mi.8.5.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Holzer P. Role of visceral afferent neurons in mucosal inflammation and defense. Curr Opin Pharmacol. 2007;7(6):563–569. doi: 10.1016/j.coph.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hang CH, et al. Expressions of intestinal NF-κB, TNF-α, and IL-6 following traumatic brain injury in rats. J Surg Res. 2005;123(2):188–193. doi: 10.1016/j.jss.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 83.Baguley IJ, et al. The incidence of dysautonomia and its relationship with autonomic arousal following traumatic brain injury. Brain Inj. 2007;21(11):1175–1181. doi: 10.1080/02699050701687375. [DOI] [PubMed] [Google Scholar]
- 84.Esterov D, Greenwald B. Autonomic dysfunction after mild traumatic brain injury. Brain Sci. 2017;7(12):100. doi: 10.3390/brainsci7080100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Clifton GL, et al. Circulating catecholamines and sympathetic activity after head injury. Neurosurgery. 1981;8(1):10–14. doi: 10.1227/00006123-198101000-00003. [DOI] [PubMed] [Google Scholar]
- 86.Cain KC, et al. Heart rate variability is related to pain severity and predominant bowel pattern in women with irritable bowel syndrome. Neurogastroenterol Motil. 2007;19(2):110–118. doi: 10.1111/j.1365-2982.2006.00877.x. [DOI] [PubMed] [Google Scholar]
- 87.Conder RL, Conder AA. Heart rate variability interventions for concussion and rehabilitation. Front Psychol. 2014;5:890. doi: 10.3389/fpsyg.2014.00890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ammar MA, Hussein NS. Using propranolol in traumatic brain injury to reduce sympathetic storm phenomenon: a prospective randomized clinical trial. Saudi J Anaesth. 2018;12(4):514–520. doi: 10.4103/sja.SJA_33_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Matteoli G, et al. A distinct vagal anti-inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut. 2014;63(6):938–948. doi: 10.1136/gutjnl-2013-304676. [DOI] [PubMed] [Google Scholar]
- 90.Rosas-Ballina M, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334(6052):98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cailotto C, et al. Neuro-anatomical evidence indicating indirect modulation of macrophages by vagal efferents in the intestine but not in the spleen. PLoS One. 2014;9(1):e87785. doi: 10.1371/journal.pone.0087785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ghia JE, et al. The vagus nerve: a tonic inhibitory influence associated with inflammatory bowel disease in a murine model. Gastroenterology. 2006;131(4):1122–1130. doi: 10.1053/j.gastro.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 93.Dash PK, et al. Activation of alpha 7 cholinergic nicotinic receptors reduce blood-brain barrier permeability following experimental traumatic brain injury. J Neurosci. 2016;36(9):2809–2818. doi: 10.1523/JNEUROSCI.3197-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou L, et al. Neuroprotective effects of vagus nerve stimulation on traumatic brain injury. Neural Regen Res. 2014;9(17):1585–1591. doi: 10.4103/1673-5374.141783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bansal V, et al. Vagal stimulation modulates inflammation through a ghrelin mediated mechanism in traumatic brain injury. Inflammation. 2012;35(1):214–220. doi: 10.1007/s10753-011-9307-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. University of Florida. Non-invasive Vagal Nerve Stimulation and Sleep. https://clinicaltrials.gov/ct2/show/NCT03170440 Updated July 14, 2020. Accessed April 22, 2021.
- 97. VA Office of Research and Development. Non-invasive Vagal Nerve Stimulation in Veterans With Mild Traumatic Brain Injury (mTBI). https://clinicaltrials.gov/ct2/show/NCT04437498 Updated March 30, 2021. Accessed April 22, 2021.
- 98.Liu L, et al. Choline ameliorates cardiovascular damage by improving vagal activity and inhibiting the inflammatory response in spontaneously hypertensive rats. Sci Rep. 2017;7(1):42553. doi: 10.1038/srep42553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu L, et al. Pharmacological modulation of vagal nerve activity in cardiovascular diseases. Neurosci Bull. 2018;35(1):156–166. doi: 10.1007/s12264-018-0286-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nóbrega AC, et al. Enhancement of heart rate variability by cholinergic stimulation with pyridostigmine in healthy subjects. Clin Auton Res. 2001;11(1):11–17. doi: 10.1007/BF02317797. [DOI] [PubMed] [Google Scholar]
- 101.Lataro RM, et al. Increase in parasympathetic tone by pyridostigmine prevents ventricular dysfunction during the onset of heart failure. Am J Physiol Regul Integr Comp Physiol. 2013;305(8):R908–R916. doi: 10.1152/ajpregu.00102.2013. [DOI] [PubMed] [Google Scholar]
- 102.Lampert R, et al. Effects of propranolol on recovery of heart rate variability following acute myocardial infarction and relation to outcome in the Beta-Blocker Heart Attack Trial. Am J Cardiol. 2003;91(2):137–142. doi: 10.1016/S0002-9149(02)03098-9. [DOI] [PubMed] [Google Scholar]
- 103.Bloom HL, et al. Differential effects of adrenergic antagonists (Carvedilol vs Metoprolol) on parasympathetic and sympathetic activity: a comparison of clinical results. Heart Int. 2014;9(1):15–21. [PMC free article] [PubMed] [Google Scholar]
- 104.Wood JD. Enteric neuroimmunophysiology and pathophysiology. Gastroenterology. 2004;127(2):635–657. doi: 10.1053/j.gastro.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 105.Bonnelle V, et al. Default mode network connectivity predicts sustained attention deficits after traumatic brain injury. J Neurosci. 2011;31(38):13442–13451. doi: 10.1523/JNEUROSCI.1163-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Aigner TG. Pharmacology of memory: cholinergic-glutamatergic interactions. Curr Opin Neurobiol. 1995;5(2):155–160. doi: 10.1016/0959-4388(95)80021-2. [DOI] [PubMed] [Google Scholar]
- 107.Blokland A. Acetylcholine: a neurotransmitter for learning and memory? Brain Res Brain Res Rev. 1995;21(3):285–300. doi: 10.1016/0165-0173(95)00016-X. [DOI] [PubMed] [Google Scholar]
- 108.Östberg A, et al. Cholinergic dysfunction after traumatic brain injury: preliminary findings from a PET study. Neurology. 2011;76(12):1046–1050. doi: 10.1212/WNL.0b013e318211c1c4. [DOI] [PubMed] [Google Scholar]
- 109.Gulbransen BD, Sharkey KA. Novel functional roles for enteric glia in the gastrointestinal tract. Nat Rev Gastroenterol Hepatol. 2012;9(11):625–632. doi: 10.1038/nrgastro.2012.138. [DOI] [PubMed] [Google Scholar]
- 110.Jessen KR, Mirsky R. Astrocyte-like glia in the peripheral nervous system: an immunohistochemical study of enteric glia. J Neurosci. 1983;3(11):2206–2218. doi: 10.1523/JNEUROSCI.03-11-02206.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ferri GL, et al. Evidence for the presence of S-100 protein in the glial component of the human enteric nervous system. Nature. 1982;297(5865):409–410. doi: 10.1038/297409a0. [DOI] [PubMed] [Google Scholar]
- 112.Young HM, et al. The projections of early enteric neurons are influenced by the direction of neural crest cell migration. J Neurosci. 2002;22(14):6005–6018. doi: 10.1523/JNEUROSCI.22-14-06005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rao M, et al. Enteric glia express proteolipid protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia. 2015;63(11):2040–2057. doi: 10.1002/glia.22876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boesmans W, et al. The astrocyte marker Aldh1L1 does not reliably label enteric glial cells. Neurosci Lett. 2014;566:102–105. doi: 10.1016/j.neulet.2014.02.042. [DOI] [PubMed] [Google Scholar]
- 115.Joseph NM, et al. Enteric glia are multipotent in culture but primarily form glia in the adult rodent gut. J Clin Invest. 2011;121(9):3398–3411. doi: 10.1172/JCI58186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jiang S, et al. Acceleration of blood-brain barrier formation after transplantation of enteric glia into spinal cords of rats. Exp Brain Res. 2005;162(1):56–62. doi: 10.1007/s00221-004-2119-3. [DOI] [PubMed] [Google Scholar]
- 117.Jiang S, et al. Enteric glia promote regeneration of transected dorsal root axons into spinal cord of adult rats. Exp Neurol. 2003;181(1):79–83. doi: 10.1016/S0014-4886(02)00030-4. [DOI] [PubMed] [Google Scholar]
- 118.Rühl A. Glial regulation of neuronal plasticity in the gut: implications for clinicians. Gut. 2006;55(5):600–602. doi: 10.1136/gut.2005.084426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Savidge TC, et al. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology. 2007;132(4):1344–1358. doi: 10.1053/j.gastro.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 120.Costantini TW, et al. Vagal nerve stimulation protects against burn-induced intestinal injury through activation of enteric glia cells. Am J Physiol Gastrointest Liver Physiol. 2010;299(6):G1308–G1318. doi: 10.1152/ajpgi.00156.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gyoneva S, Ransohoff RM. Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol Sci. 2015;36(7):471–480. doi: 10.1016/j.tips.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wanner IB, et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. 2013;33(31):12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lau LW, et al. Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat Rev Neurosci. 2013;14(10):722–729. doi: 10.1038/nrn3550. [DOI] [PubMed] [Google Scholar]
- 124.Brun P, et al. Toll-like receptor 2 regulates intestinal inflammation by controlling integrity of the enteric nervous system. Gastroenterology. 2013;145(6):1323–1333. doi: 10.1053/j.gastro.2013.08.047. [DOI] [PubMed] [Google Scholar]
- 125.Neunlist M, et al. Enteric glia inhibit intestinal epithelial cell proliferation partly through a TGF-beta1-dependent pathway. Am J Physiol Gastrointest Liver Physiol. 2007;292(1):G231–G241. doi: 10.1152/ajpgi.00276.2005. [DOI] [PubMed] [Google Scholar]
- 126.Cirillo C, et al. Increased mucosal nitric oxide production in ulcerative colitis is mediated in part by the enteroglial-derived S100B protein. Neurogastroenterol Motil. 2009;21(11):1209–e112. doi: 10.1111/j.1365-2982.2009.01346.x. [DOI] [PubMed] [Google Scholar]
- 127.Capoccia E, et al. Enteric glia: a new player in inflammatory bowel diseases. Int J Immunopathol Pharmacol. 2015;28(4):443–451. doi: 10.1177/0394632015599707. [DOI] [PubMed] [Google Scholar]
- 128.Dethlefsen L, et al. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449(7164):811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Foster JA, et al. Gut microbiota and brain function: an evolving field in neuroscience. Int J Neuropsychopharmacol. 2016;19(5):pyv114. doi: 10.1093/ijnp/pyv114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cryan JF, et al. The microbiota-gut-brain axis. Physiol Rev. 2019;99(4):1877–2013. doi: 10.1152/physrev.00018.2018. [DOI] [PubMed] [Google Scholar]
- 131.Ma Q, et al. Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J Neuroinflammation. 2019;16(1):53. doi: 10.1186/s12974-019-1434-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bonaz B, et al. The vagus nerve at the interface of the microbiota-gut-brain axis. Front Neurosci. 2018;12:49. doi: 10.3389/fnins.2018.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Forsythe P, Kunze WA. Voices from within: gut microbes and the CNS. Cell Mol Life Sci. 2013;70(1):55–69. doi: 10.1007/s00018-012-1028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Quigley EMM. Microbiota-brain-gut axis and neurodegenerative diseases. Curr Neurol Neurosci Rep. 2017;17(12):94. doi: 10.1007/s11910-017-0802-6. [DOI] [PubMed] [Google Scholar]
- 135. Forsythe P, et al. Vagal pathways for microbiome-brain-gut axis communication. In: Lyte M, Cryan JF, eds. Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease. Springer; 2014:115–133. [DOI] [PubMed] [Google Scholar]
- 136.Lyte JM, et al. Gut-brain axis serotonergic responses to acute stress exposure are microbiome-dependent. Neurogastroenterol Motil. 2020;32(11):e13881. doi: 10.1111/nmo.13881. [DOI] [PubMed] [Google Scholar]
- 137.Martin CR, et al. The brain-gut-microbiome axis. Cell Mol Gastroenterol Hepatol. 2018;6(2):133–148. doi: 10.1016/j.jcmgh.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sudo N. Microbiome, HPA axis and production of endocrine hormones in the gut. In: Lyte M, Cryan JF, eds. Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease. Springer; 2014:177–194. [DOI] [PubMed] [Google Scholar]
- 139.Taylor WR, Takemiya K. Hypertension opens the flood gates to the gut microbiota. Circ Res. 2017;120(2):249–251. doi: 10.1161/CIRCRESAHA.116.310339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wall R, et al. Bacterial neuroactive compounds produced by psychobiotics. In: Lyte M, Cryan JF, eds. Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease. Springer; 2014:221–239. [Google Scholar]
- 141.Yang T, Zubcevic J. Gut–brain axis in regulation of blood pressure. Front Physiol. 2017;8:845. doi: 10.3389/fphys.2017.00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Halfvarson J, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. 2017;2(5):1–7. doi: 10.1038/nmicrobiol.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kashyap PC, et al. Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology. 2013;144(5):967–977. doi: 10.1053/j.gastro.2013.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gensollen T, et al. How colonization by microbiota in early life shapes the immune system. Science. 2016;352(6285):539–544. doi: 10.1126/science.aad9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pathare N, et al. The impact of traumatic brain injury on microbiome composition: a systematic review. Biol Res Nurs. 2020;22(4):495–505. doi: 10.1177/1099800420943961. [DOI] [PubMed] [Google Scholar]
- 146.Urban RJ, et al. Altered fecal microbiome years after traumatic brain injury. J Neurotrauma. 2019;37(8):1037–1051. doi: 10.1089/neu.2019.6688. [DOI] [PubMed] [Google Scholar]
- 147.Zhu CS, et al. A review of traumatic brain injury and the gut microbiome: insights into novel mechanisms of secondary brain injury and promising targets for neuroprotection. Brain Sci. 2018;8(6):113. doi: 10.3390/brainsci8060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Celorrio M, et al. Gut microbial dysbiosis after traumatic brain injury modulates the immune response and impairs neurogenesis. Acta Neuropathol Commun. 2021;9(1):40. doi: 10.1186/s40478-021-01137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Burmeister DM, et al. The gut microbiome distinguishes mortality in trauma patients upon admission to the emergency department. J Trauma Acute Care Surg. 2020;88(5):579–587. doi: 10.1097/TA.0000000000002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.David LA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Costello LAS, et al. Nutrition therapy in the optimisation of health outcomes in adult patients with moderate to severe traumatic brain injury: findings from a scoping review. Injury. 2014;45(12):1834–1841. doi: 10.1016/j.injury.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 152.Crenn P, et al. Changes in weight after traumatic brain injury in adult patients: a longitudinal study. Clin Nutr. 2014;33(2):348–353. doi: 10.1016/j.clnu.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 153.McGlennon TW, et al. Bypassing TBI: metabolic surgery and the link between obesity and traumatic brain injury-a review. Obes Surg. 2020;30(12):4704–4714. doi: 10.1007/s11695-020-05065-3. [DOI] [PubMed] [Google Scholar]
- 154.Lee CJ, et al. Gut microbiome and its role in obesity and insulin resistance. Ann N Y Acad Sci. 2020;1461(1):37–52. doi: 10.1111/nyas.14107. [DOI] [PubMed] [Google Scholar]
- 155.Maruvada P, et al. The human microbiome and obesity: moving beyond associations. Cell Host Microbe. 2017;22(5):589–599. doi: 10.1016/j.chom.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 156.Angoa-Pérez M, et al. Repetitive, mild traumatic brain injury results in a progressive white matter pathology, cognitive deterioration, and a transient gut microbiota dysbiosis. Sci Rep. 2020;10(1):8949. doi: 10.1038/s41598-020-65972-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Simon DW, et al. Depletion of gut microbiota is associated with improved neurologic outcome following traumatic brain injury. Brain Res. 2020;1747:147056. doi: 10.1016/j.brainres.2020.147056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Treangen TJ, et al. Traumatic brain injury in mice induces acute bacterial dysbiosis within the fecal microbiome. Front Immunol. 2018;9:2757. doi: 10.3389/fimmu.2018.02757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nicholson SE, et al. Moderate traumatic brain injury alters the gastrointestinal microbiome in a time-dependent manner. Shock. 2019;52(2):240–248. doi: 10.1097/SHK.0000000000001211. [DOI] [PubMed] [Google Scholar]
- 160.Levy M, et al. Dysbiosis and the immune system. Nat Rev Immunol. 2017;17(4):219–232. doi: 10.1038/nri.2017.7. [DOI] [PubMed] [Google Scholar]
- 161.Liang S, et al. Gut-brain psychology: rethinking psychology from the microbiota–gut–brain axis. Front Integr Neurosci. 2018;12:33. doi: 10.3389/fnint.2018.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Freestone PPE, et al. Microbial endocrinology: how stress influences susceptibility to infection. Trends Microbiol. 2008;16(2):55–64. doi: 10.1016/j.tim.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 163.Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science. 2010;330(6012):1768–1773. doi: 10.1126/science.1195568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Neuman H, et al. Microbial endocrinology: the interplay between the microbiota and the endocrine system. FEMS Microbiol Rev. 2015;39(4):509–521. doi: 10.1093/femsre/fuu010. [DOI] [PubMed] [Google Scholar]
- 165.Lyte M. Microbial endocrinology and the microbiota-gut-brain axis. Adv Exp Med Biol. 2014;817:3–24. doi: 10.1007/978-1-4939-0897-4_1. [DOI] [PubMed] [Google Scholar]
- 166.Wenzel TJ, et al. Short-chain fatty acids (SCFAs) alone or in combination regulate select immune functions of microglia-like cells. Mol Cell Neurosci. 2020;105:103493. doi: 10.1016/j.mcn.2020.103493. [DOI] [PubMed] [Google Scholar]
- 167.Sadler R, et al. Short-chain fatty acids improve poststroke recovery via immunological mechanisms. J Neurosci. 2020;40(5):1162–1173. doi: 10.1523/JNEUROSCI.1359-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Adelman MW, et al. The gut microbiome’s role in the development, maintenance, and outcomes of sepsis. Crit Care. 2020;24(1):278. doi: 10.1186/s13054-020-02989-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fay KT, et al. The gut microbiome alters immunophenotype and survival from sepsis. FASEB J. 2019;33(10):11258–11269. doi: 10.1096/fj.201802188R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Haak BW, Wiersinga WJ. The role of the gut microbiota in sepsis. Lancet Gastroenterol Hepatol. 2017;2(2):135–143. doi: 10.1016/S2468-1253(16)30119-4. [DOI] [PubMed] [Google Scholar]
- 171.Ruan W, et al. Healthy human gastrointestinal microbiome: composition and function after a decade of exploration. Dig Dis Sci. 2020;65(3):695–705. doi: 10.1007/s10620-020-06118-4. [DOI] [PubMed] [Google Scholar]
- 172.Ogbonnaya ES, et al. Adult hippocampal neurogenesis is regulated by the microbiome. Biol Psychiatry. 2015;78(4):7–9. doi: 10.1016/j.biopsych.2014.12.023. [DOI] [PubMed] [Google Scholar]
- 173.Rogers GB, et al. From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol Psychiatry. 2016;21(6):738–748. doi: 10.1038/mp.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Thion MS, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. 2018;172(3):500–516. doi: 10.1016/j.cell.2017.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Westfall S, et al. Microbiome, probiotics and neurodegenerative diseases: deciphering the gut brain axis. Cell Mol Life Sci. 2017;74(20):3769–3787. doi: 10.1007/s00018-017-2550-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Li H, et al. Clostridium butyricum exerts a neuroprotective effect in a mouse model of traumatic brain injury via the gut-brain axis. Neurogastroenterol Motil. 2018;30(5):e13260. doi: 10.1111/nmo.13260. [DOI] [PubMed] [Google Scholar]
- 177.Patterson TT, et al. Complex feed-forward and feedback mechanisms underlie the relationship between traumatic brain injury and the gut-microbiota-brain axis. Shock. 2019;52(3):318–325. doi: 10.1097/SHK.0000000000001278. [DOI] [PubMed] [Google Scholar]
- 178.Zhang Y, et al. Gut microbiota-brain interaction: an emerging immunotherapy for traumatic brain injury. Exp Neurol. 2020;337:113585. doi: 10.1016/j.expneurol.2020.113585. [DOI] [PubMed] [Google Scholar]
- 179.Rice MW, et al. Gut microbiota as a therapeutic target to ameliorate the biochemical, neuroanatomical, and behavioral effects of traumatic brain injuries. Front Neurol. 2019;10:875. doi: 10.3389/fneur.2019.00875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rivera ED, et al. The mesentery, systemic inflammation, and Crohn’s disease. Inflamm Bowel Dis. 2019;25(2):226–234. doi: 10.1093/ibd/izy201. [DOI] [PubMed] [Google Scholar]
- 181.Hanscom M, et al. Acute colitis during chronic experimental traumatic brain injury in mice induces dysautonomia and persistent extraintestinal, systemic, and CNS inflammation with exacerbated neurological deficits. J Neuroinflammation. 2021;18(1):24. doi: 10.1186/s12974-020-02067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Liao Y, et al. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS One. 2013;8(7):e68963. doi: 10.1371/journal.pone.0068963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Meisel C, et al. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6(10):775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- 184.Esnault P, et al. Early-onset ventilator-associated pneumonia in patients with severe traumatic brain injury: incidence, risk factors, and consequences in cerebral oxygenation and outcome. Neurocrit Care. 2017;27(2):187–198. doi: 10.1007/s12028-017-0397-4. [DOI] [PubMed] [Google Scholar]
- 185.Ghezzi A, Zaffaroni M. Neurological manifestations of gastrointestinal disorders, with particular reference to the differential diagnosis of multiple sclerosis. Neurol Sci. 2001;22(8):S117–S122. doi: 10.1007/s100720100048. [DOI] [PubMed] [Google Scholar]
- 186.Franceschi F, et al. Microbes and Alzheimer’ disease: lessons from H. pylori and GUT microbiota. Eur Rev Med Pharmacol Sci. 2019;23(1):426–430. doi: 10.26355/eurrev_201901_16791. [DOI] [PubMed] [Google Scholar]
- 187.Archbald-Pannone LR, et al. Delirium and other clinical factors with Clostridium difficile infection that predict mortality in hospitalized patients. Am J Infect Control. 2015;43(7):690–693. doi: 10.1016/j.ajic.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ferro JM, et al. Neurologic manifestations of inflammatory bowel diseases. Handb Clin Neurol. 2014;120:595–605. doi: 10.1016/B978-0-7020-4087-0.00040-1. [DOI] [PubMed] [Google Scholar]
- 189.Jang SE, et al. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal Immunol. 2018;11(2):369–379. doi: 10.1038/mi.2017.49. [DOI] [PubMed] [Google Scholar]
- 190.Zhang B, et al. Inflammatory bowel disease is associated with higher dementia risk: a nationwide longitudinal study. Gut. 2021;70(1):85–91. doi: 10.1136/gutjnl-2020-320789. [DOI] [PubMed] [Google Scholar]
- 191.Nordström P, et al. Traumatic brain injury and young onset dementia: a nationwide cohort study. Ann Neurol. 2014;75(3):374–381. doi: 10.1002/ana.24101. [DOI] [PubMed] [Google Scholar]
- 192.Schaffert J, et al. Traumatic brain injury history is associated with an earlier age of dementia onset in autopsy-confirmed Alzheimer’s disease. Neuropsychology. 2018;32(4):410–416. doi: 10.1037/neu0000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dantzer R. Cytokine-induced sickness behaviour: a neuroimmune response to activation of innate immunity. Eur J Pharmacol. 2004;500(1):399–411. doi: 10.1016/j.ejphar.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 194.Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21(2):153–160. doi: 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kealy J, et al. Acute inflammation alters brain energy metabolism in mice and humans: role in suppressed spontaneous activity, impaired cognition, and delirium. J Neurosci. 2020;40(29):5681–5696. doi: 10.1523/JNEUROSCI.2876-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]