

ABSTRACT
Mitochondria respond to many cellular functions and act as central hubs in innate immunity against viruses. This response is notably due to their role in the activation of interferon (IFN) signaling pathways through the activity of MAVS (mitochondrial antiviral signaling protein) present at the mitochondrial surface. Here, we report that the BHRF1 protein, a BCL2 homolog encoded by Epstein-Barr virus (EBV), inhibits IFNB/IFN-β induction by targeting the mitochondria. Indeed, we have demonstrated that BHRF1 expression modifies mitochondrial dynamics and stimulates DNM1L/Drp1-mediated mitochondrial fission. Concomitantly, we have shown that BHRF1 is pro-autophagic because it stimulates the autophagic flux by interacting with BECN1/Beclin 1. In response to the BHRF1-induced mitochondrial fission and macroautophagy/autophagy stimulation, BHRF1 drives mitochondrial network reorganization to form juxtanuclear mitochondrial aggregates known as mito-aggresomes. Mitophagy is a cellular process, which can specifically sequester and degrade mitochondria. Our confocal studies uncovered that numerous mitochondria are present in autophagosomes and acidic compartments using BHRF1-expressing cells. Moreover, mito-aggresome formation allows the induction of mitophagy and the accumulation of PINK1 at the mitochondria. As BHRF1 modulates the mitochondrial fate, we explored the effect of BHRF1 on innate immunity and showed that BHRF1 expression could prevent IFNB induction. Indeed, BHRF1 inhibits the IFNB promoter activation and blocks the nuclear translocation of IRF3 (interferon regulatory factor 3). Thus, we concluded that BHRF1 can counteract innate immunity activation by inducing fission of the mitochondria to facilitate their sequestration in mitophagosomes for degradation.
Abbreviations: 3-MA: 3-methyladenine; ACTB: actin beta; BCL2: BCL2 apoptosis regulator; CARD: caspase recruitment domain; CCCP: carbonyl cyanide 3-chlorophenylhydrazone; CI: compaction index; CQ: chloroquine; DAPI: 4ʹ,6-diamidino-2-phenylindole, dihydrochloride; DDX58/RIG-I: DExD/H-box helicase 58; DNM1L/Drp1: dynamin 1 like; EBSS: Earle’s balanced salt solution; EBV: Epstein-Barr virus; ER: endoplasmic reticulum; EV: empty vector; GFP: green fluorescent protein; HEK: human embryonic kidney; IFN: interferon; IgG: immunoglobulin G; IRF3: interferon regulatory factor 3; LDHA: lactate dehydrogenase A; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MAVS: mitochondrial antiviral signaling protein; MMP: mitochondrial membrane potential; MOM: mitochondrial outer membrane; PINK1: PTEN induced kinase 1; RFP: red fluorescent protein; ROS: reactive oxygen species; SQSTM1/p62: sequestosome 1; STING1: stimulator of interferon response cGAMP interactor 1; TOMM20: translocase of outer mitochondrial membrane 20; VDAC: voltage dependent anion channel.
KEYWORDS: Autophagy, BCL2, BECN1, BHRF1, DNM1L, EBV, IFN, MAVS, mitochondrial dynamics, mitophagy
Introduction
The Epstein-Barr virus (EBV) is a ubiquitous virus belonging to the herpesviruses family. Although its prevalence is high in populations worldwide, EBV primary infection is asymptomatic when occurring in children, whereas it may be responsible for infectious mononucleosis in young adults. EBV essentially infects epithelial cells and B cells, in which it establishes a latent lifelong persistence. Several latency programs exist, corresponding to the expression of different viral proteins without the production of infectious viral particles [1,2]. Some forms of latency promote cell transformation, and EBV infection has, therefore, been associated with cancers, such as lymphomas and carcinomas [3]. It has been reported that EBV can block programmed cell death, thus prolonging the lifespan of infected cells, maximizing the production of progeny viruses, and facilitating the establishment of virus persistence [4,5].
EBV has a large 172-kbp DNA genome that can encode for more than 100 different proteins together with noncoding RNAs [4]. Among these proteins, BHRF1 is expressed during the viral productive cycle but also during some latency programs, and its only known function is to protect cells against apoptosis [6], notably during primary infection [7]. BHRF1 is a 17 kDa putative transmembrane protein, which is highly conserved among different EBV isolates and shows strong functional homology with the human BCL2 [8]. This 191-amino-acid viral protein owns two motifs referred to as BCL2 homology domains 1 and 2 (BH1 and BH2). Cellular BCL2 belongs to a family of more than 20 members with anti-apoptotic properties, such as BCL2 or BCL2L1/Bcl-xL, and pro-apoptotic proteins, such as BAX, BAK1, BAD or BCL2L11/Bim [9]. BHRF1 anti-apoptotic activity may at least in part rely on its ability to interact with pro-apoptotic BH3-only proteins (especially BCL2L11) and with the executioner BAK1 [10,11]. In addition, BHRF1, like BCL2, seems to reside mainly in the mitochondrial membrane [12,13].
Mitochondria fulfill multiple cellular functions, such as energy production, maintenance of calcium homeostasis, reactive oxygen species (ROS) generation, and apoptosis initiation. In addition, mitochondria also actively participate in innate immunity in order to limit viral infections. Indeed, the innate immune signaling receptor MAVS (mitochondrial antiviral signaling protein) is located at the mitochondrial outer membrane (MOM) and plays a pivotal role in signaling cascades that lead to type I interferon (IFN) and pro-inflammatory cytokine production. MAVS is activated following interaction with DDX58/RIG-I (DExD/H-box helicase 58) or IFIH1/MDA5 (interferon induced with helicase C domain 1), two cytoplasmic pattern recognition receptors that detect viral genome during infection. Functions of mitochondria are intertwined with the morphology and the number of mitochondria. Indeed, mitochondria are dynamic and mobile organelles that constantly undergo membrane remodeling through repeated cycles of fusion and fission, and their length is determined by the balance between fission/fusion rates. Maintenance of mitochondrial homeostasis also includes the control of the number of mitochondria through the stimulation of their biogenesis, and removal of damaged mitochondria by selective autophagy, a process called mitophagy. Selective autophagy begins in the recognition of a specific cargo by a molecular receptor that links the cargo to the autophagosome membrane, leading to its sequestration into this double-membrane vesicle [14]. Macroautophagy/autophagy can also be nonselective and can randomly sequester bulk cytosol and other cytoplasmic components into the autophagosomes [15]. In both cases, the maturation of autophagosomes ends with their fusion with the lysosomes, creating the autolysosomes, leading to the digestion of their contents by proteases.
Whereas the ability of BHRF1 to block apoptosis is well documented, no information is available so far regarding its putative role on mitochondrial dynamics and autophagy. Interestingly, several studies reported that EBV could subvert autophagy to improve viral replication and the final envelopment of viral particles [16–18]. Here, we demonstrate that ectopic expression of BHRF1 leads to a DNM1L/Drp1 (dynamin related protein 1)-dependent mitochondrial fission. Subsequently, BHRF1 induces the accumulation of autophagosomes by interacting with BECN1/Beclin 1, an autophagy machinery protein, which is known to interact with BCL2. These cellular modifications result in the formation of a mito-aggresome, a perinuclear mitochondrial clustering that precedes mitophagy. Given the central role of mitochondria in innate immunity, we explored the contribution of BHRF1 in the control of innate immunity and demonstrated that BHRF1-mediated mitochondrial degradation resulted in the inhibition of type I IFN induction.
Results
BHRF1 expression induces mitochondrial fission and the formation of mito-aggresomes
BHRF1 subcellular localization was investigated by confocal microscopy in HeLa cells following transfection with a BHRF1-HA expression vector for 24 h. Mitochondria were counterstained by MitoTracker Red CMXRos dye. In accordance with previous reports, we observed an important colocalization of BHRF1 with mitochondria (Figure 1A), corresponding to a perinuclear staining of BHRF1 and a mitochondrial distribution pattern similar to BCL2 [12,19]. Moreover, BHRF1 seemed to induce a modification on the shape of the nucleus. More interestingly, mitochondrial labeling with an antibody directed against the mitochondrial import receptor subunit TOMM20 confirmed that the mitochondrial network morphology was dramatically altered in BHRF1-expressing cells (Figure 1B). The mitochondrial average length was measured (Figure 1C), and it confirmed that the BHRF1 expression induced a marked reduction in mitochondrial length, which was indicative of mitochondrial fission. While the mitochondrial population exhibited a tubular network or an intermediate phenotype in the majority of control cells, BHRF1 caused fragmentation of the mitochondria, with almost 75% of them displaying a size under 1 µm (Figure 1C). Interestingly, no change in mitochondrial membrane potential (MMP) was noted upon BHRF1 expression (Figure S1A and B). BHRF1-expressing cells also showed abnormal and juxtanuclear mitochondrial aggregates, while these organelles were homogenously distributed as a network in the cytoplasm of control cells (Figure 1B). These rough aggregates were strongly reminiscent of structures previously described and classically called mito-aggresomes [20]. Based on a mitochondrial compaction index (CI) above 0.4 [21], used herein as the criterion for judging mitochondrion clustering, virtually 80% of BHRF1-expressing cells showed a mito-aggresome (Figures 1D and S1C). To confirm this observation in the context of native EBV infection, we first analyzed the mitochondrial morphology in EBV-positive Akata B cells during latency or following viral reactivation. The mitochondria were homogenously distributed in the cytoplasm of latent (non-reactivated) Akata cells (Figure 1E and Figure 1F). However, after the reactivation of the viral lytic cycle by anti-IgG treatment, they formed mito-aggresomes in reactivated cells that were characterized by the expression of immediate early protein BZLF1 (Figure 1E). Although BHRF1 was not expressed in latent Akata cells, BHRF1 accumulated in reactivated cells where it colocalized with the mito-aggresomes (Figure 1F). Based on the CI scoring, more than 90% of BHRF1-positive cells displayed mito-aggresomes (Figure 1F). In order to confirm the role of BHRF1 in the mito-aggresome formation in infected cells, we assessed alterations of mitochondrial network in the HEK293/EBV+ epithelial cell line. These cells contain the EBV genome of the B95-8 strain and can be reactivated by the expression of the trans-activator protein BZFL1 via transfection of an expression plasmid [22,23] (Figure 1I). We explored in parallel a similar cell line harboring the EBV genome deleted of the BHRF1 gene (∆BHRF1) [7]. As observed in Figure 1G, reactivation (visualized by Ea-D viral protein expression) led to the high compaction of mitochondria in WT EBV cells. However, the lack of BHRF1 (∆BHRF1) dramatically reduced the percentage of cells presenting a mito-aggresome, demonstrating that BHRF1 is required for the mitochondrial alterations during reactivation (Figure 1H).
Figure 1.
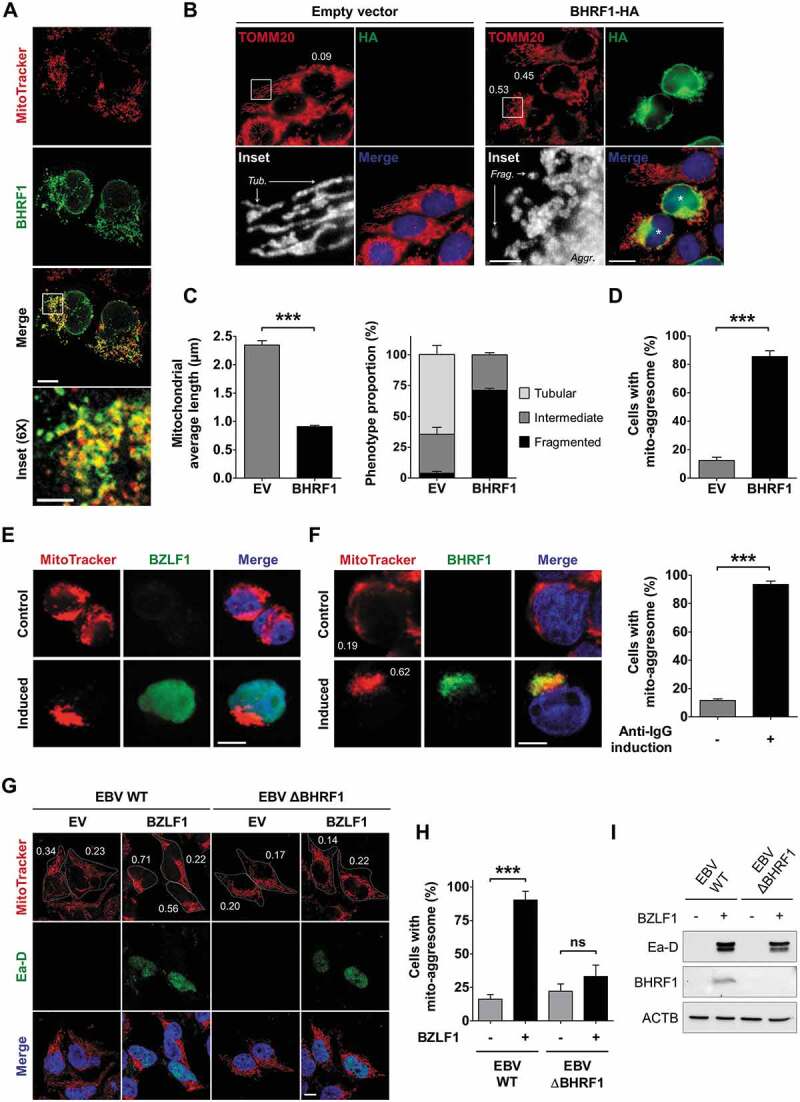
BHRF1 induces the formation of mito-aggresomes in HeLa cells and EBV-reactivated cells. (A) BHRF1 localizes to the mitochondria. Confocal images of BHRF1-HA-transfected HeLa cells immunostained for BHRF1 and the mitochondria (MitoTracker). Scale bars: 10 µm, and 3 µm for inset. (B-D) BHRF1 induces mitochondrial fission in HeLa cells. Cells were transfected with an empty vector (EV) or a plasmid encoding BHRF1-HA for 24 h. Cells were fixed and immunostained for TOMM20 to visualize the mitochondrial network, and HA to visualize BHRF1-expressing cells. Nuclei were subsequently stained with DAPI. (B) Representative images. Scale bars: 20 µm and 4 µm for insets. Tub., tubular mitochondria; Frag., fragmented mitochondria; Aggr., aggregated mitochondria. Values of mitochondrial compaction index (CI) are indicated on representative cells; cells indicated by a star (*) present a mito-aggresome (CI above 0.4). For details on CI scoring, see Figure S1C. (C) Left: mitochondrial average length. Right: mitochondrial phenotype. (D) Percentage of cells presenting a mito-aggresome. (E and F) EBV reactivation in B cells induces mito-aggresome formation. EBV-reactivated Akata cells were labeled with MitoTracker to visualize the mitochondrial phenotype and reactivated cells were detected by labeling EBV antigens. Nuclei were stained with Hoechst. Scale bar: 10 µm. (E) Confocal images of MitoTracker and BZLF1 co-staining. (F) Left: confocal images of MitoTracker and BHRF1 co-staining. Values of mitochondrial CI are indicated on representative cells. Right: percentage of EBV-reactivated Akata cells presenting a mito-aggresome. (G-I) EBV reactivation in the absence of BHRF1 blocks mito-aggresome formation in epithelial cells. (G) Confocal images of MitoTracker and Ea-D viral protein co-staining of HEK293 cells without reactivation (EV) or after reactivation (BZLF1 transfection). Nuclei were stained with DAPI. Scale bar: 10 µm. CI values are indicated on representative cells. (H) Percentage of cells presenting a mito-aggresome (CI above 0.4). (I) Immunoblot analysis of Ea-D and BHRF1 after BZLF1 transfection, confirming EBV reactivation in both cell lines and the lack of BHRF1 expression with the mutant virus. ACTB was used as a loading control. Data represent the mean ± SEM of three independent experiments. ns = non-significant; *** P < 0.001 (Student’s t-test)
DNM1L is required for BHRF1-induced mitochondrial fission and mito-aggresome formation
Mitochondria change their overall morphology by fusion and fission in response to cellular stress. Damaged mitochondria can trigger their fission to get rid of the unhealthy parts. DNM1L is a GTPase that plays a critical role in mitochondrial fission [24]. DNM1L mostly localizes in the cytoplasm and is translocated to the MOM to regulate mitochondrial fission [25]. Since BHRF1 expression was associated with increased mitochondrial fission, we monitored DNM1L localization in BHRF1-expressing cells by confocal microscopy (Figure 2A). Whereas DNM1L was diffusely distributed in the cytoplasm in control cells, DNM1L relocated upon BHRF1 expression into BHRF1-positive mito-aggresomes. Cell fractionation experiments were performed to separate the mitochondria-associated proteins [26]. This experiment confirmed that BHRF1 is specifically present at the mitochondria and demonstrated that DNM1L was slightly more abundant in the mitochondrial fraction in BHRF1-expressing cells than in control cells (Figure 2B). Overall DNM1L accumulation increased by 40% in the mitochondrial fraction in BHRF1-expressing cells (Figure 2B). Post-translational modifications of DNM1L, such as phosphorylation, sumoylation, ubiquitination, and S-nitrosylation, regulate mitochondrial fission in response to diverse cellular stimuli [25]. We analyzed the level of phosphorylation of DNM1L in position Ser637 in BHRF1-expressing cells (Figure 2C). Indeed, this phosphorylation site depends on the cAMP-dependent PRKA (protein kinase A) activation and impedes mitochondrial fission. Conversely, dephosphorylation of DNM1L by the phosphatase calcineurin, for example, induces the translocation of DNM1L and fragmentation of the organelle [27]. Whereas no modification of the total level of DNM1L occurred in BHRF1-expressing cells compared to control cells, a strong downregulation of DNM1L Ser637 phosphorylation was observed following BHRF1 expression (Figure 2C), which corroborates the translocation of DNM1L to the mitochondria and the mitochondrial fragmentation that was previously observed (Figures 2B and Figures 1C). As to confirm that DNM1L was indeed required for BHRF1-mediated mitochondrial fission, we generated HeLa cells DNM1L knockdown using lentiviruses, which delivered the sh-RNA that specifically targets the human DNM1L mRNA. We characterized them in the absence of BHRF1 using known mitochondrial fission inducers (Figure S2). In HeLa cells expressing sh-DNM1L, mitochondria formed a highly connected network (Figure S2A) along with an 83% decrease in DNM1L protein expression (Figure 2D). Moreover, BHRF1 did not induce mitochondrial reorganization in sh-DNM1L-expressing cells (Figure 2E). Accordingly, the mitochondrial length was unaffected by BHRF1 expression and DNM1L deficiency mostly abrogated the BHRF1 ability to induce the formation of mito-aggresomes (Figure 2F and Figure 2G). During the viral lytic cycle, DNM1L was similarly required for the formation of mito-aggresomes since the treatment of Akata cells by Mdivi-1, a chemical inhibitor of DNM1L, totally impeded the formation of mito-aggresomes (Figure 2H and Figure 2I). Altogether, our results demonstrated that BHRF1 expression induces DNM1L-dependent mitochondrial fission, which is required for mitochondrial clustering.
Figure 2.
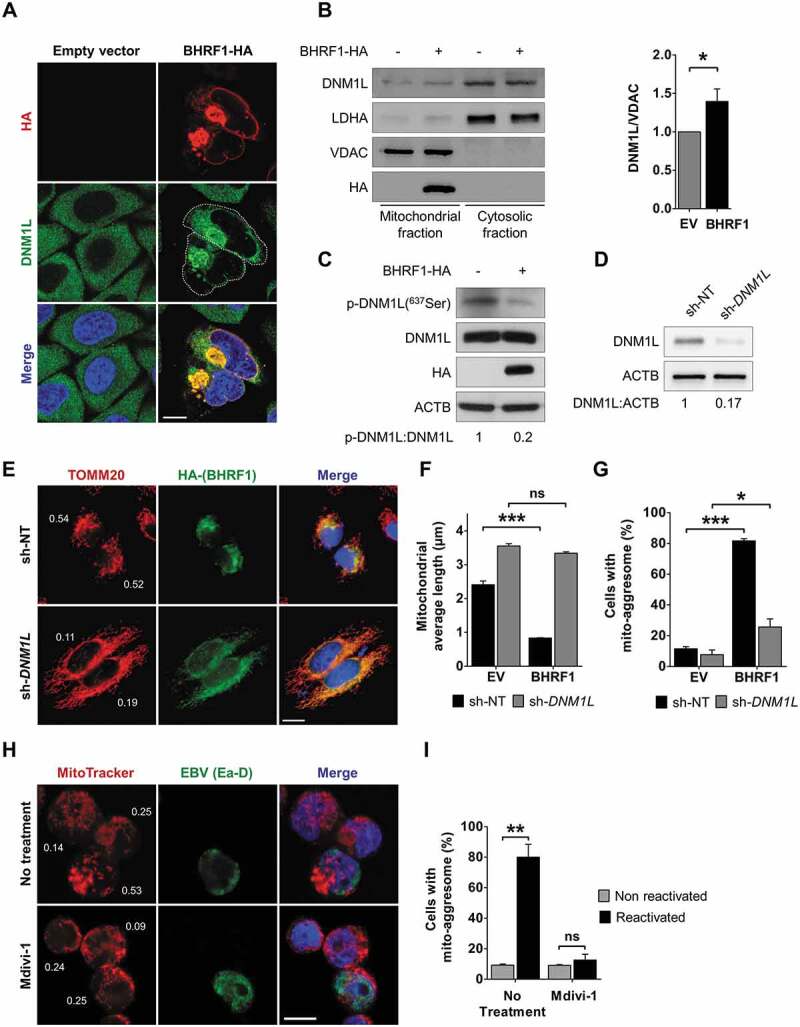
BHRF1 induces DNM1L-dependent mitochondrial fission. (A) Confocal images of HeLa cells transfected with EV or BHRF1-HA plasmids for 24 h and immunostained for DNM1L and HA. Nuclei were stained with DAPI. Cells outline was drawn to better visualize the recruitment of DNM1L into mito-aggresome structures. Scale bar: 10 µm. (B) Subcellular fractionation of HeLa cells transfected for 48 h with EV or BHRF1-HA plasmids. VDAC and LDHA were used as markers for mitochondrial and cytosolic fractions, respectively. Left: immunoblot analysis of DNM1L, LDHA, and VDAC. Right: quantification of DNM1L expression in the mitochondrial fraction. (C) Immunoblot analysis of phosphorylated (Serine 637) and total DNM1L upon BHRF1-HA expression. ACTB was used as a loading control. (D) Silencing level of DNM1L in HeLa knockdown cell line (sh-DNM1L), compared to the control cell line (sh-NT). ACTB was used as a loading control. See Figure S2 for deficient cell line characterization. (E-G) sh-NT and sh-DNM1L HeLa cells were transfected with EV or BHRF1-HA plasmids for 24 h and immunostained for TOMM20 and HA. Nuclei were stained with DAPI. (E) Representative images of BHRF1-HA-expressing cells. Scale bar: 10 µm. Values of mitochondrial CI are indicated on representative cells. (F) Mitochondrial average length. (G) Percentage of cells presenting a mito-aggresome. (H) Confocal images of EBV-reactivated Akata cells treated for 1 h with DNM1L-inhibitor Mdivi-1 (50 µM) and labeled with MitoTracker and an antibody against EBV (Ea-D). Nuclei were stained with Hoechst. Scale bar: 5 µm. Values of mitochondrial CI are indicated on representative cells. (I) Percentage of Akata cells presenting a mito-aggresome. Data represent the mean ± SEM of three independent experiments. ns = non-significant; * P < 0.05; ** P < 0.01; *** P < 0.001 (Student’s t-test)
BHRF1 triggers autophagy
Cellular BCL2 is known to inhibit autophagy. Since BHRF1 shares notable homology with cellular BCL2, we wondered whether it might modulate autophagy as well [13]. To study the impact of BHRF1 on autophagy, we first used HeLa cells that stably express GFP-LC3, a classical marker of autophagosomes, and added chloroquine (CQ) 4 h before harvesting the cells [28]. CQ neutralizes the lysosomal pH and causes the accumulation of GFP-LC3-positive vesicles by inhibiting endogenous protein degradation [29]. As shown in Figure 3A and Figure 3B, BHRF1-expressing cells displayed increased numbers of GFP-LC3 dots compared to control cells, with and without CQ. Because an accumulation of autophagosomes in cells can result either from an increase in the rate of their formation or a decrease in their fusion with lysosomes, we next explored the autophagic flux, which reflects the autophagic degradation activity. We, therefore, analyzed the accumulation of the lipidated LC3 (LC3-II) by immunoblotting in cells that were or were not treated with CQ. An increased level of LC3-II was observed in BHRF1-expressing cells (Figure 3B and Figure 3C), which was even higher in the presence of CQ. This result led us to conclude that BHRF1 stimulates the autophagic flux. We also monitored the abundance of the autophagy substrate SQSTM1 by immunoblot and concordantly observed a decrease in SQSTM1 when BHRF1 was expressed (Figure 3C). To confirm our data, the autophagic flux was investigated using the mRFP-GFP-LC3 HeLa cells (Figures 3D and S3) [30]. Because GFP fluorescence is quenched in acidic compartments, such as autolysosomes, this probe makes it possible to differentiate between autophagosomes (GFP+ RFP+ or yellow dots) and autolysosomes (GFP− RFP+ or red dots) [31]. As a control, cells were treated with CQ to block autophagic flux or maintained in Earle’s balanced salt solution (EBSS; starvation) to induce autophagy. We observed an increase in the total amount of autophagic vacuoles (autophagosomes and autolysosomes) in BHRF1-expressing cells (Figure S3B). Furthermore, BHRF1, as well as starvation, led to a significant percentage of autolysosomes over autophagosomes, whereas CQ-induced vacuoles were mainly autophagosomes (>95%) (Figures 3D and S3B). Altogether, these findings clearly indicated that BHRF1 stimulates the biosynthesis of autophagosomes and autophagic flux in HeLa cells. BECN1 is a critical component of highly regulated complexes that control the formation and maturation of autophagosomes [32,33]. Since BCL2 and several viral proteins have been described to interact with BECN1, we investigated the putative interaction between BECN1 and BHRF1 [34–36]. First, we analyzed the distribution of BHRF1 and BECN1 in HeLa cells, and we observed a partial colocalization of the two proteins in the cytoplasm (Figure 3E), which suggests an interaction. To confirm this interaction, co-immunoprecipitation experiments were performed using HeLa cells that are transiently transfected by plasmids encoding for HA-tagged BHRF1 and BECN1 (Figure 3F). Since herpes simplex virus type 1 (HSV-1) ICP34.5 has been previously reported to interact with BECN1, we used a Flag-tagged ICP34.5 plasmid as a positive control [36,37]. Immunoprecipitation of BECN1 pulled down both BHRF1 and ICP34.5. Reversely, endogenous BECN1 was co-immunoprecipitated with HA-tagged BHRF1 using an anti-HA antibody (Figure 3G). Therefore, we can conclude that BHRF1 interacted with BECN1. Altogether, these findings suggested that BHRF1 stimulated autophagy through its interaction with BECN1.
Figure 3.
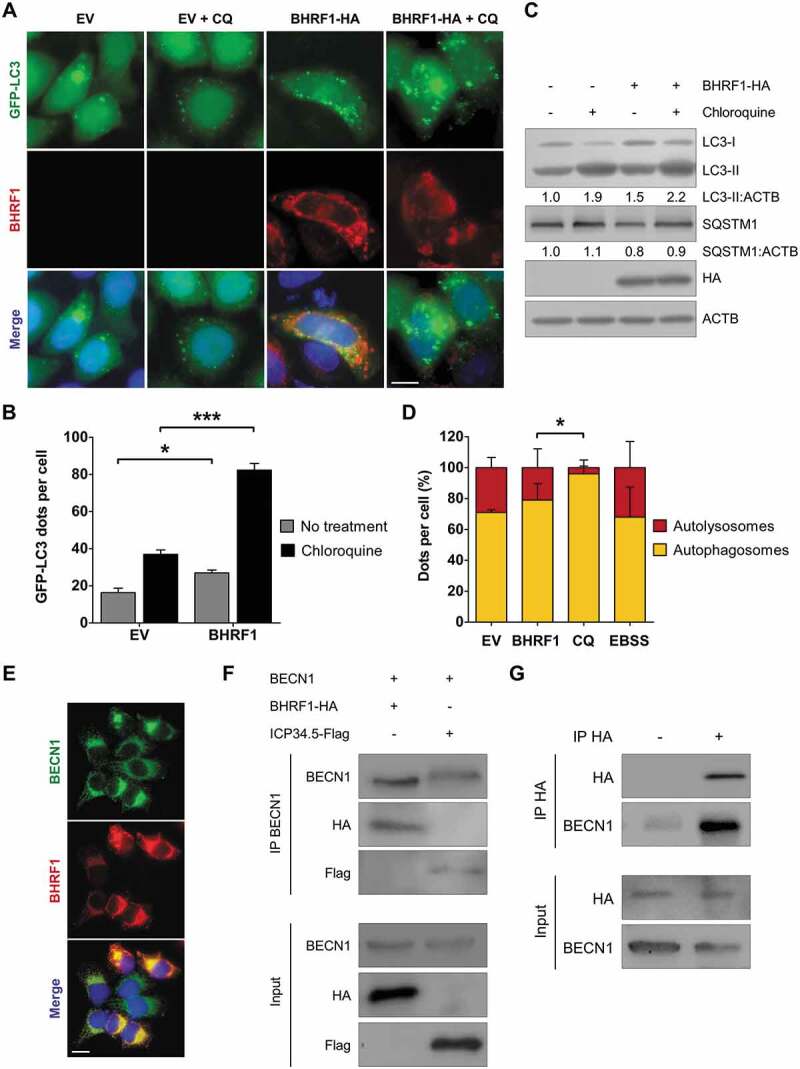
BHRF1 stimulates autophagy and interacts with BECN1. (A and B) GFP-LC3 HeLa cells were transfected with EV or BHRF1-HA plasmids for 48 h and fixed after CQ treatment when indicated. Nuclei were stained with DAPI. (A) Representative images. Scale bar: 20 µm. (B) Number of GFP-LC3 dots. (C) Immunoblot analysis of LC3 and SQSTM1 expression in HeLa cells treated with CQ when indicated. ACTB was used as a loading control. (D) To study autophagic flux, mRFP-GFP-LC3 HeLa cells were transfected with BHRF1-HA plasmids, or treated with CQ to block the autophagic flux, or maintained in EBSS (starvation) to induce the autophagic flux. Representative images are presented in Figure S3. Results are shown as the percentage of total autophagic vesicles. (E) Representative images of BHRF1-HA-transfected HeLa cells immunostained for BECN1 and BHRF1. Scale bar: 20 µm. (F) HeLa cells were transiently transfected to express BECN1 and either BHRF1-HA or ICP34.5-Flag vector. After immunoprecipitation with a goat polyclonal anti-BECN1 antibody, proteins were detected by immunoblotting with anti-BECN1, anti-Flag, or anti-HA antibodies. (G) In HeLa cells, BHRF1-HA was transiently transfected in both lanes. BHRF1-HA was immunoprecipitated in the absence of antibody (-) or with an anti-HA antibody (+). Endogenous BECN1 and BHRF1-HA proteins were detected by immunoblotting. Data represent the mean ± SEM of three independent experiments. * P < 0.05; *** P < 0.001 (Student’s t-test)
Interplay between BHRF1-induced autophagy and mitochondrial morphology alterations
Since BHRF1 induced both mitochondrial fission and autophagy in an apparently concomitant manner, we wondered whether fission was required for the induction of autophagy. To answer this question, we monitored the level of BHRF1-induced autophagy in DNM1L-deficient cells by staining endogenous LC3 (Figure 4A). We first demonstrated that DNM1L deficiency did not impair starvation-induced autophagy, since EBSS treatment of sh-DNM1L cells induced LC3 dot accumulation, as well as SQSTM1 degradation (Figure S4). Then, we confirmed that BHRF1 induced the accumulation of endogenous LC3 dots in sh-NT control cells, and even more after CQ treatment (Figure 4A and Figure 4B). However, BHRF1 did not promote the accumulation of LC3 dots in sh-DNM1L-treated cells, even in the presence of CQ (Figure 4B). These results demonstrated that DNM1L, and by extension, DNM1L-mediated mitochondrial fission, is required for BHRF1-induced autophagy. In order to seek whether autophagy is required to form mito-aggresomes, HeLa cells expressing BHRF1 were treated with two different autophagy inhibitors, i.e., Spautin-1 and 3-methyladenine (3-MA), while immunostaining the mitochondrial network and BHRF1 (Figure 4C–E). When autophagy was blocked, mitochondria were homogeneously distributed in the cytoplasm and were not organized into mito-aggresomes even in the presence of BHRF1, as visualized by the CI values (Figure 4C). However, autophagy was not required for BHRF1 to induce mitochondrial fission since a significant reduction in the length of the mitochondria was observed in BHRF1-expressing cells both in the presence and absence of autophagy inhibitors. (Figure 4E). Altogether, these findings demonstrated that mitochondrial fission induced by BHRF1 leads to autophagy activation, which in turn promotes mito-aggresomes formation.
Figure 4.
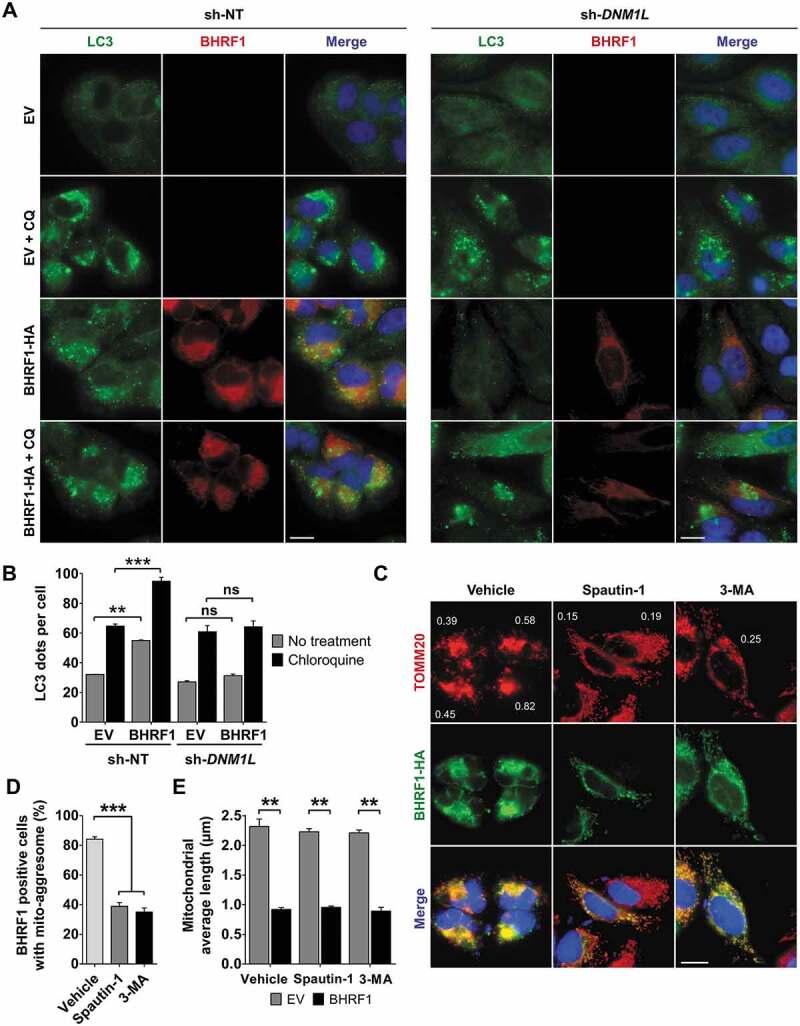
Interplay between BHRF1-induced autophagy and mito-aggresome formation. (A and B) Mitochondrial fission is required for BHRF1-induced autophagy. sh-NT and sh-DNM1L HeLa cells were transfected with EV or BHRF1-HA plasmids for 24 h and treated with CQ when indicated. LC3-positive vesicles and BHRF1-HA-transfected cells were visualized, respectively, using anti-LC3 and anti-BHRF1 antibodies. Nuclei were stained with DAPI. See Figure S4 for deficient cell line characterization. (A) Representative images. Scale bar: 20 µm. (B) Quantification of LC3 dots. (C-E) Autophagy is necessary to BHRF1-induced mito-aggresome formation. HeLa cells were transfected with EV or BHRF1-HA plasmids for 24 h. When indicated, cells were treated with Spautin-1 or 3-MA to inhibit autophagy. After fixation, cells were immunostained for TOMM20 and HA. (C) Representative images of BHRF1-HA-expressing cells. Values of mitochondrial CI are indicated on representative cells. Scale bar: 20 µm. (D) Percentage of BHRF1-positive cells presenting a mito-aggresome. (E) Mitochondrial average length. Data represent the mean ± SEM of three independent experiments. (B and E) ns = non-significant; ** P < 0.01; *** P < 0.001 (Student’s t-test). (D) *** P < 0.001 (One-way ANOVA test)
BHRF1 expression induces mitophagy
Considering that BHRF1 induces mitochondrial fission and stimulates autophagy, we wondered whether BHRF1 could induce mitophagy. Indeed, the fragmentation of mitochondria facilitates their envelopment into autophagosomes, which eventually leads to their degradation into lysosomes via mitophagy [38]. Moreover, the appearance of mito-aggresomes was previously described following the depolarization of mitochondria and preceded by mitophagy in a Parkinson disease model [20]. Finally, during mitophagy, it has been observed that dysfunctional mitochondria undergo retrograde transport to the perinuclear region, where they cluster to be degraded [39]. A sensitive dual fluorescence reporter expressing mito-mRFP-EGFP, fused in-frame with a mitochondrial targeting sequence, was used to monitor the completion of the mitophagic process [40]. In the presence of BHRF1, numerous mitochondria were detected in acidic compartments (mitochondria labeled in red), suggesting that BHRF1 triggered mitophagy (Figure 5A). Confocal microscopy analysis confirmed that mitochondria (TOMM20 labeling) and autophagosomes (GFP-LC3 labeling) significantly colocalized in the presence of BHRF1 (Figure 5B). CQ treatment increased the colocalization intensity, measured by Manders’ coefficient, possibly because it inhibited the autophagic degradation of sequestered mitochondria (Figure 5C). Because autophagosomes are expected to fuse with the lysosomes, we also monitored the impact of BHRF1 on the colocalization between the mitochondria and lysosomes using LAMP1 labeling as a lysosomal marker (Figure S5). In BHRF1-expressing cells, the presence of LAMP1-positive vesicles containing mitochondria could be detected both in the presence and absence of CQ (Figure S5A and B), together with increased LAMP1 expression (Figure S5C).
Figure 5.
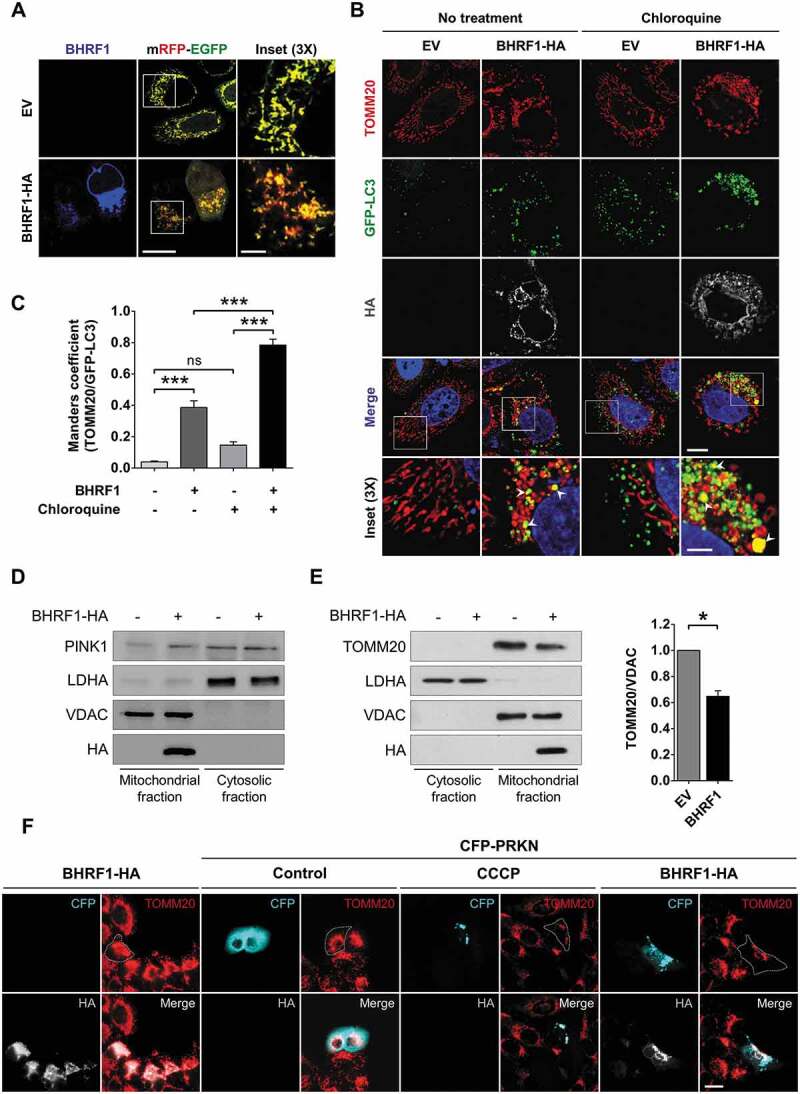
BHRF1 induces mitophagy and recruits PRKN to the mitochondria. (A) Representative images of HeLa cells co-transfected for 24 h with the mRFP-EGFP probe targeted to the mitochondria and EV or BHRF1-HA plasmids. Cells were immunostained for BHRF1. Insets show part of the cytoplasm at a higher magnification. Red staining corresponds to the delivery of mitochondria to the acidic compartments. Scale bars: 20 µm and 5 µm for insets. (B and C) GFP-LC3 HeLa cells were transfected with EV or BHRF1-HA plasmids for 24 h and treated with CQ when indicated. Cells were immunostained for TOMM20 and HA. Nuclei were stained with DAPI. (B) Representative images of cells. Insets show part of the cytoplasm at a higher magnification to better visualize colocalization (see arrows) between mitochondria (TOMM20) and autophagosomes (GFP-LC3), as assessed by confocal microscopy. Scale bars: 10 µm and 4 µm for insets. (C) Colocalization level (Manders split coefficient) between mitochondria and autophagosomes. (D and E) Subcellular fractionation of BHRF1-HA- and EV-transfected HeLa cells. VDAC and LDHA were used as markers for mitochondrial and cytosolic fractions, respectively. (D) Immunoblot analysis of PINK1 showing its accumulation at the outer mitochondrial membrane. Same subcellular fractionation experiment as Figure 2B (E). Left: immunoblot analysis of TOMM20. Right: quantification of TOMM20 expression in the mitochondrial fraction showing that BHRF1 decreases the TOMM20 protein level. (F) BHRF1 recruits CFP-PRKN to the mitochondria. Representative images of HeLa cells co-transfected with BHRF1-HA plasmid and either EV or CFP-PRKN plasmids for 48 h and immunostained for TOMM20 and HA. Treatment with CCCP was used as a positive control of mitophagy. Cells outline was drawn to better visualize the degradation of mitochondria under BHRF1 and CFP-PRKN expression. Scale bar: 20 µm. See Figure S5 for visualization of mitochondria inside LAMP1-positive vesicles in BHRF1-HA-expressing cells. Data represent the mean ± SEM of two independent experiments. (C) ns = non-significant; *** P < 0.001 (one-way ANOVA test). (E) * P < 0.05 (Student’s t-test)
Mitochondria need to recruit autophagy receptor proteins for mitophagy to occur. One of the best-studied mitophagy mechanisms in mammalian cells is the PINK1 (PTEN induced kinase 1)-PRKN (parkin RBR E3 ubiquitin protein ligase)-mediated mitophagy pathway [41]. PINK1 is constitutively imported into healthy mitochondria through the TOMM and TIMM membrane translocation complexes, where it is degraded by the mitochondrial inner membrane protease PARL. When mitochondria are dysfunctional, PINK1 is not imported anymore and starts to accumulate on the MOM, triggering the recruitment of PRKN/Parkin [41]. Once activated, PRKN ubiquitinates proteins on the MOM surface, and this ubiquitination initiates the engulfment of the mitochondria into autophagosomes to complete mitophagy. We thus wondered whether BHRF1 induced mitophagy through the PINK1-PRKN pathway. First, we monitored the expression of PINK1 in the cytosolic and mitochondrial fractions of BHRF1-expressing cells. As shown on Figure 5D, PINK1 was significantly translocated to the mitochondrial fraction following BHRF1 expression. Then, HeLa cells, which lack a detectable endogenous PRKN, were co-transfected with CFP-PRKN and BHRF1 plasmids for 48 h. Since treatment with the protonophore carbonyl cyanide 3-chlorophenylhydrazone (CCCP) for 24 h leads to mitochondrial depolarization, it was herein used as a positive control for mitophagy induction. PRKN distribution was diffuse in the cytoplasm of control cells, whereas PRKN was clearly recruited to mitochondrial clusters in cells expressing CFP-PRKN following mitophagy induction (CCCP treatment) (Figure 5F). Similarly, in cells expressing both CFP-PRKN and BHRF1, CFP-PRKN was recruited to BHRF1-positive structures and mito-aggresomes (Figure 5F). Moreover, in cells expressing CFP-PRKN and BHRF1, the mitochondrial population seemed to decrease compared to the population found in the absence of CFP-PRKN (Figure 5F). These results demonstrated that CFP-PRKN expression potentiated mitochondrial degradation induced by BHRF1 in HeLa cells. However, our results suggested that BHRF1-induced mitophagy could occur independently of PRKN since we observed it in HeLa cells, which have little or no endogenous PRKN expression. Finally, we observed a clear decrease in TOMM20 accumulation in the mitochondrial fraction upon BHRF1 expression, which confirmed a partial degradation of mitochondria (Figure 5E). Altogether, these results confirmed that BHRF1 induces mitophagy.
BHRF1 expression blocks type I IFN induction via the MAVS-STING1 signaling pathway
BHRF1-induced mitochondrial alterations and mitophagy suggested that BHRF1 might impact mitochondria-dependent signaling pathways, such as antiviral signaling and IFN production. Indeed, MAVS, an innate immunity adaptor, is localized at the surface of mitochondria and its interaction with STING1 (stimulator of interferon response cGAMP interactor 1), an ER-resident adaptor, plays a crucial role in the induction of type I IFN production [42]. MAVS and STING1 recruit and activate TBK1 (TANK-binding kinase 1), which phosphorylates the transcription factor IRF3 (interferon regulatory factor 3), leading to its nuclear translocation and subsequent activation of type I IFN promoter [43]. It has been previously reported that fragmentation of the mitochondrial network disrupted the interaction between MAVS and STING1, thus leading to reduced signaling by IRF3 [42]. Moreover, Ding et al. demonstrated that the sequestration of mitochondria in autophagosomes during infection with the human parainfluenza virus type 3 (HPIV3) blocked the IFN response [44].
To investigate the potential significance of BHRF1-mediated mitochondrial network alterations on innate immunity, we performed an IFNB/IFN-β promoter-driven luciferase reporter assay (Figure 6) [45]. We explored the effect of BHRF1 expression on IFNB promoter activation in HEK293T or HeLa cells. To activate the IFN-signaling pathway in a MAVS-dependent manner, we used a dominant-positive mutant form of DDX58/RIG-I (∆DDX58) (Figure 6A) that contains two CARD (caspase recruitment domains), which mimic its oligomerization but lacks the helicase domain [46]. As MAVS contains one CARD [47], it becomes constitutively activated by CARD-CARD interaction in the presence of ΔDDX58. Transfection of the double-stranded DNA (dsDNA) was used to activate CGAS, a DNA sensor, to stimulate the STING1 signaling cascade [48] (Figure 6B). BHRF1 expression dramatically blocked the IFNB promoter activation by either pathway in a dose-dependent manner (Figure 6A and Figure 6B). We next examined whether BHRF1 modulated the activation of the transcription factor IRF3. IRF3 staining was diffused in the cytoplasm of non-stimulated cells, but it became mainly nuclear in response to ∆DDX58 or dsDNA transfection in cells expressing an empty control vector (Figure 6C). Conversely, IRF3 nuclear translocation was clearly abrogated in the BHRF1-expressing cells following MAVS or STING1 activation. We measured the percentage of cells that exhibited nuclear IRF3 in EV or BHRF1-expressing cells and observed a ten-fold decrease when BHRF1 was expressed (Figure 6D). Together, these observations showed that BHRF1 prevented MAVS and STING1-mediated IFNB pathway activation.
Figure 6.
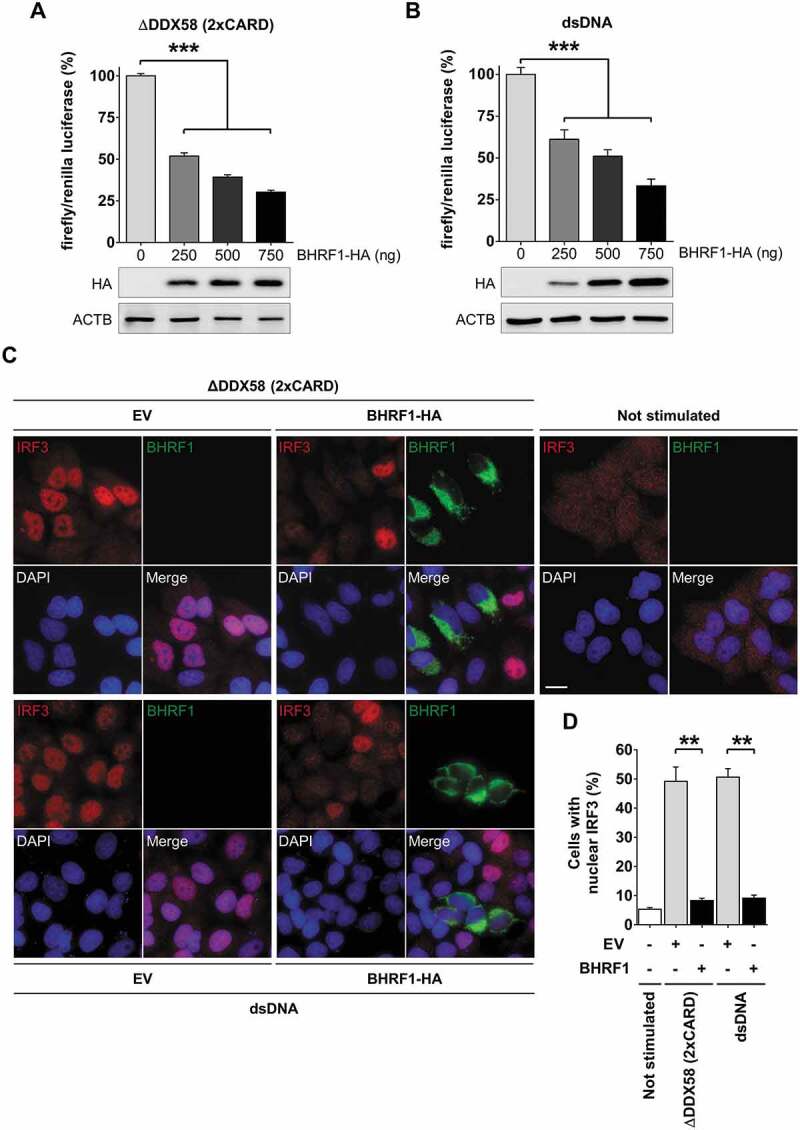
BHRF1 inhibits type I IFN induction via MAVS or STING1 signaling pathways. (A and B) Luciferase reporter assay on HEK293T or HeLa cells co-transfected with plasmids expressing BHRF1-HA, the reporter plasmid expressing firefly luciferase under the control of the IFNB promoter (IFNB-Luc) and renilla luciferase (RL-TK; to standardize the transfection efficiency). The activation of the IFNB promoter was analyzed 24 h after transfection. Below the histograms, immunoblot analysis of BHRF1-HA from HEK293T or HeLa cells transfected with increasing amounts of BHRF1-HA plasmid are shown. ACTB was used as a loading control. (A) To study the MAVS pathway, HEK293T cells were transfected with the Flag-∆DDX58 (2xCARD) plasmid to express a dominant-positive form of DDX58. (B) To study the STING1 pathway, HeLa cells were stimulated with dsDNA for the last 8 h to activate the CGAS-STING1 axis. Firefly/renilla luciferase ratios were calculated and normalized to control condition (EV) to assess the activation level of the IFNB promoter. (C and D) HeLa cells were transfected with EV or BHRF1-HA for 24 h. Cells were also transfected with Flag-∆DDX58 (2xCARD) plasmid or stimulated with dsDNA. After fixation, cells were immunostained for IRF3 and BHRF1. Nuclei were stained with DAPI. (C) Representative images. Scale bar: 20 µm. (D) Percentage of cells with IRF3 nuclear localization. Data represent the mean ± SEM of three independent experiments. (A and B) *** P < 0.001 (one-way ANOVA test). (D) ** P < 0.01 (Student’s t-test)
Mitochondrial fission and autophagy induction are both necessary to block IFNB induction
We speculated that mitochondrial fission might be required for BHRF1 to downregulate the IFNB response. To test this hypothesis, we used sh-DNM1L HEK293T or sh-DNM1L HeLa cells and measured the IFNB promoter activity, as before (Figure 6A and Figure 6B). We observed that DNM1L knockdown abolished the inhibitory effect of BHRF1 on the IFNB promoter activation triggered by ∆DDX58 or dsDNA (Figure 7A). Similarly, nuclear translocation of IRF3 was abrogated by BHRF1 in sh-NT cells, whereas BHRF1 lost this ability in sh-DNM1L cells (Figure 7B and Figure 7C). Altogether, these results showed that BHRF1 required DNM1L-dependent mitochondrial fission to block the IFNB induction. Since we previously demonstrated that BHRF1 activated the autophagic flux, we speculated that autophagy might be required for BHRF1 to block the IFNB induction as well. To test this hypothesis, cells were treated with two different autophagy inhibitors, Spautin-1 and 3-MA. As shown in Figure 7D, both treatments totally abrogated the ability of BHRF1 to inhibit the IFNB induction in response to ∆DDX58 transfection, suggesting that autophagy induction is necessary for BHRF1 to inhibit the IFNB system. This result was further confirmed by the inability of BHRF1 to block nuclear IRF3 translocation in response to ∆DDX58 or dsDNA activation when autophagy was inhibited by Spautin-1 or 3-MA (Figure 7E and Figure 7F).
Figure 7.
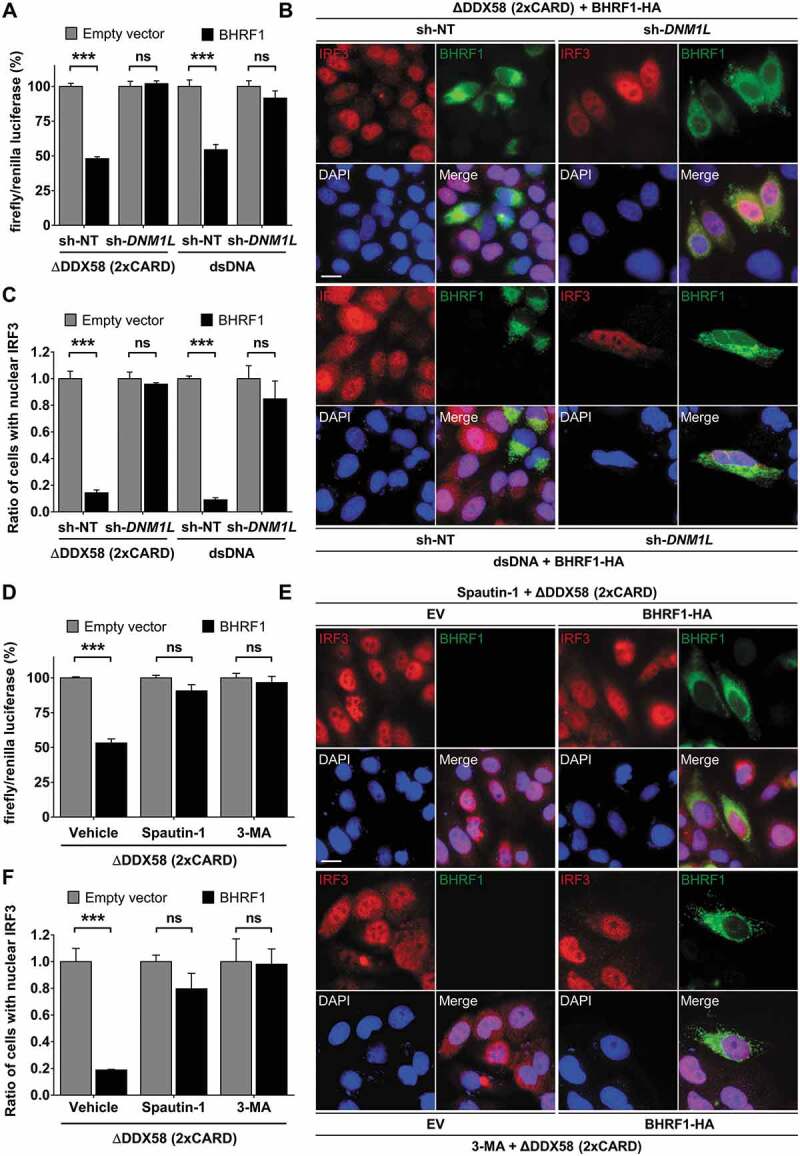
Mitochondrial fission and autophagy are required for BHRF1-inhibitor effect on type I IFN induction. (A) Luciferase reporter assay on sh-DNM1L HEK293T or sh-DNM1L HeLa cells and their corresponding control cell lines (sh-NT). Cells were co-transfected with EV or BHRF1-HA, IFNB-Luc, and RL-TK plasmids for 24 h. HEK293T cells were also transfected with Flag-∆DDX58 (2xCARD) plasmid and HeLa cells were stimulated with dsDNA. Firefly/renilla luciferase ratios were calculated and normalized to control conditions (EV). (B and C) sh-NT and sh-DNM1L HeLa cells were transfected with EV or BHRF1-HA plasmids. Cells were also transfected with Flag-∆DDX58 (2xCARD) plasmid or stimulated with dsDNA and immunostained for IRF3 and BHRF1. Nuclei were stained with DAPI. (B) Representative images of BHRF1-HA-expressing cells. Scale bar: 20 µm. (C) Quantification of IRF3 nuclear localization, normalized to EV condition. (D) Luciferase reporter assay, as described in Figure 6A. HEK293T cells were transfected with Flag-∆DDX58 (2xCARD) plasmid to stimulate IFN production. To inhibit autophagy, cells were treated by Spautin-1 or 3-MA. Firefly/renilla luciferase ratios were calculated and normalized to control conditions (EV). (E and F) HeLa cells were co-transfected with Flag-∆DDX58 (2xCARD) and EV or BHRF1-HA plasmids. Cells were treated with Spautin-1 or 3-MA, and immunostained for IRF3 and BHRF1. Nuclei were stained with DAPI. (E) Representative images. Scale bar: 20 µm. (F) Quantification of IRF3 nuclear localization, normalized to EV condition. Data represent the mean ± SEM of three independent experiments. ns = non-significant; ** P < 0.01; *** P < 0.001 (Student’s t-test)
Discussion
Whereas it has been reported that BHRF1, an EBV-encoded protein, acts on the mitochondria to control apoptosis, only one study previously noticed that BHRF1 might modify the distribution of the mitochondria [19]. However, the mechanisms leading to BHRF1-induced mitochondrial reorganization, as well as their functional consequences, have not been explored so far. In the present study, we shed new light on the role of BHRF1 and provide evidence that contributes to innate immunity evasion. It is shown here that BHRF1 dampens the type I IFN activation pathway by promoting the fragmentation and subsequent sequestration of mitochondria in autophagosomes, which eventually leads to their degradation by mitophagy (Figure 8). This notion could be explained by the fact that MAVS, which is localized on the mitochondrial surface, might become unavailable for functional interaction with STING1.
Figure 8.
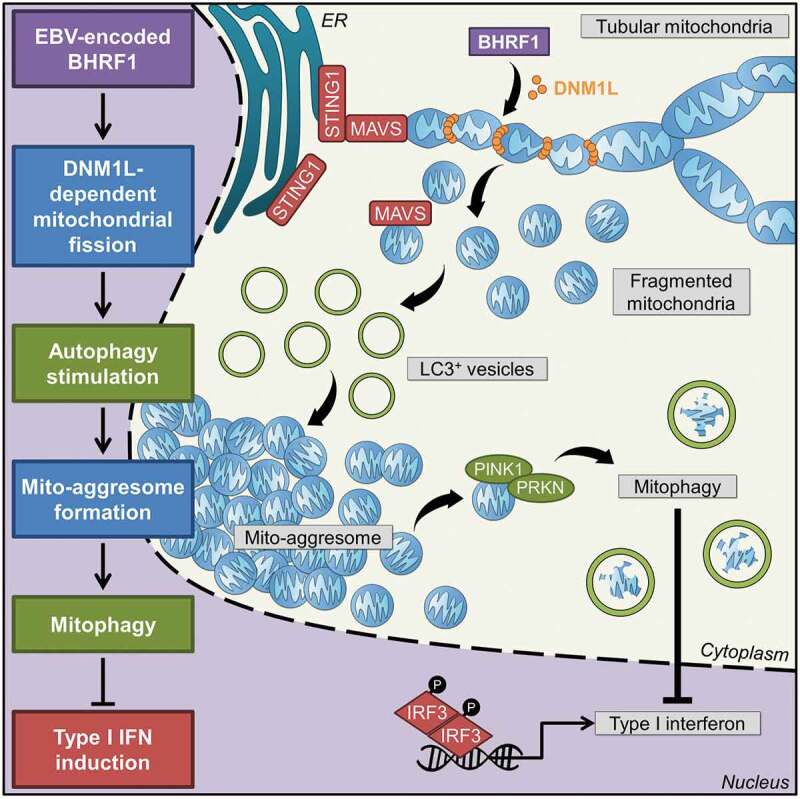
Working model. Expression of BHRF1, a BCL2 viral homolog encoded by the Epstein-Barr virus, is able to modify mitochondrial dynamics. Recruitment of a DNM1L active form upon BHRF1 expression induces mitochondrial fission. This fission is required to stimulate autophagy by BHRF1, leading to the formation of mito-aggresomes. Then, numerous mitochondria are sequestrated inside autophagosomes in order to be degraded by BHRF1-induced mitophagy. The innate immune signaling adaptors MAVS and STING1 are located at the mitochondria and at the ER, respectively. They can interact together and play a pivotal role in the antiviral response by inducing type I interferons. BHRF1 modulates the mitochondrial fate by the sequestration of mitochondria in autophagic vacuoles and mitophagy and finally leads to inhibition of type I IFN induction via both MAVS and STING1 pathways
Throughout evolution, herpesviruses, such as EBV, HSV-1, or human cytomegalovirus (HCMV), have developed numerous strategies to escape the innate immune response [49], and several EBV-encoded proteins have been described to block type I IFN production by different mechanisms. Hahn et al. initially reported that the trans-activator protein BZLF1/ZEBRA/Zta interfers with type I IFN production. During EBV reactivation, BZLF1 counteracts IFNA4 and IFNB production by interacting with IRF7, although BZLF1 does not affect the nuclear localization of IRF7 [50]. The other immediate early activator BRLF1/Rta can also suppress IFNB production by decreasing both IRF3 and IRF7 expression in reactivated Akata cells [51]. EBV-encoded BGLF4 kinase can interact with IRF3 and inhibits its phosphorylation [52]. Additionally, the tegument protein LF2 blocks IRF7 dimerization directly inside the nucleus [53]. Finally, it has been recently demonstrated that EBV reactivation is associated with the induction of cellular TRIM29, which leads to the proteasomal degradation of STING1 and, therefore, inhibits the downstream signaling cascade [54]. In this report, we discovered that BHRF1 inhibits the type I IFN activation pathway through an original mechanism, which involves mitochondrial dynamics and autophagy.
Besides its role in the maintenance of cellular homeostasis, autophagy is a process with multiple effects on immunity. Indeed, during viral infections, autophagy plays a crucial role by promoting the clearance of viral components but also by activating innate immunity in order to produce antiviral cytokines. However, some viruses have developed different strategies to impair the immune response by subverting autophagy [55]. One mechanism relies on the suppression of the signal transduction related to the autophagic degradation of signaling proteins, such as MAVS or DDX58. Following viral infection, BST2/tetherin recruits MARCHF8, an E3 ubiquitin ligase, in order to mediate the K27-ubiquitination of MAVS for CALCOCO2/NDP52-dependent degradation [56]. LRRC25, another key negative regulator of type I IFN, stimulates the interaction between DDX58 and the autophagic cargo receptor SQSTM1, therefore, promoting DDX58 degradation by selective autophagy [57]. Autophagic proteins can also interfere with innate immunity by directly inhibiting signaling molecules. To that respect, ATG12–ATG5 conjugate downregulates the DDX58 signaling by directly binding to DDX58 and MAVS CARD domains [58]. J. Cui and collaborators have demonstrated that BECN1 can also block the interaction between DDX58 and MAVS [59]. They have shown that the USP19 (ubiquitin-specific peptidase 19), which stabilizes BECN1 by deubiquitination, allows its interaction with the MAVS CARD domain and, therefore, inhibits the DDX58-mediated type I IFN signaling. Members of the IRF family are also targeted by autophagic proteins to downregulate type I IFN production. Kim et al. have reported that RUBCN/Rubicon, an autophagic inhibitor, can suppress IRF3 dimerization by interacting with the IRF association domain [60]. Mitochondrial sequestration into autophagosomes may be an additional mechanism to downregulate type I IFN signaling [44]. Indeed, it has been recently reported that the HPIV3-encoded matrix protein M triggers mitochondrial sequestration and inhibits the type I IFN response. However, the final degradation of mitochondria does not occur in infected cells, thanks to the expression of the phosphoprotein P that inhibits the maturation of autophagosomes [44]. Here, we show that BHRF1 protein induces the sequestration of fragmented mitochondria in autophagosomes, which leads to their degradation by mitophagy, thereby suppressing type I IFN production (Figure 8). Three EBV-encoded proteins (BRLF1, BZLF1, and BALF0/1) have been described to induce autophagy when ectopically expressed, but their involvement regarding the IFN signaling pathway was not investigated [17,61,62].
Modifications of mitochondrial distribution, mitochondrial fission, and/or mitophagy have been reported to occur during several viral infections. Indeed, other herpesviruses than EBV, such as HSV-1, pseudo rabies virus (PRV), and HCMV, have been reported to disrupt the mitochondrial network and promote aberrant mitochondrial dynamics [63,64]. Interestingly, the hepatitis B virus (HBV) and hepatitis C virus (HCV) also induce the formation of mito-aggresomes in a juxtanuclear region, mitochondrial fission, and subsequent mitophagy [40,65]. The activation of mitophagy during HBV or HCV infection seems essential for the clearance of damaged mitochondria, contributes to cell survival and attenuates the virus-induced mitochondrial apoptotic cell death. The expression of proteins encoded by different viral families can also reproduce the effects on mitochondria observed during viral infection. For example, the expression of HBx, encoded by HBV, promotes mitochondrial fragmentation and PRKN-dependent mitophagy [40]. The anti-apoptotic protein vMIA (viral mitochondrion-localized inhibitor of apoptosis), encoded by HCMV, induces mitochondrial fragmentation either alone or in the context of viral infection [63]. Nevertheless, vMIA induces only minor modifications of the membrane potential and seems unable to initiate mitophagy [63,66]. Until now, the LMP2A (latent membrane protein 2A), is the only EBV protein that has been reported to modify the mitochondrial network and increase DNM1L-dependent mitochondrial fission [67]. BHRF1 is, therefore, the first EBV protein described to trigger mitophagy. It is tempting to speculate that BHRF1 participates to cell survival in EBV-infected cells by the attenuation of mitochondrial apoptosis, similar to HBV and HCV, by inducing mitophagy and autophagy. Moreover, due to its expression during some latency programs and by inhibiting type I IFN activation pathways, BHRF1 may favor the establishment of latent EBV infection. Indeed, BHRF1, currently described as the tenth latent protein, is constitutively expressed during Wp-restricted latency and from Wp-initiated transcripts in Latency III [1]. Since type I and II IFN were described for their antiproliferative properties, BHRF1 could also participate in EBV-related cancer development by impairing type I IFN production [68]. However, further studies are required to confirm the significance of our findings in the context of latency.
Materials and methods
Antibodies, plasmids, and reagents
References of materials used in this study are summarized in Table 1. The BHRF1-HA expression vector was constructed by inserting the BHRF1 open reading frame in the pcDNA3.1 expression vector (Invitrogen, V79020). Briefly, BHRF1 ORF was amplified by PCR with AmpliTaq Gold® Master Mix (Applied Biosystems™, 10460085) using genomic DNA extracted from Burkitt lymphoma Akata cell line and BHRF1-specific primers (Table 1). The Reverse primer has been designed so that an HA-tag (YPYDVPDYA) is fused in frame at the C terminus of BHRF1. The PCR product was inserted in the EcoRI site of pcDNA3.1 by recombination using the Cold Fusion Cloning kit (Ozyme, MC100A-1) according to the manufacturer’s instructions.
Table 1.
Materials used in this study
| Antibodies | Source | Identifier |
|---|---|---|
| Mouse Anti-Human SQSTM1 Monoclonal Antibody, Unconjugated, Clone 2C11 | Abnova Corporation | H00008878-M01 |
| IgG antibody | Agilent | A0423 |
| BECN1/Beclin 1 antibody | BD Biosciences | 612,112 |
| DNM1L/Drp1 antibody | BD Biosciences | 611,738 |
| TIMM23/Tim23 antibody | BD Biosciences | 611,223 |
| TOMM20/Tom20 antibody | BD Biosciences | 612,278 |
| DNM1L/DRP1 (D6C7) rabbit mAb antibody | Cell Signaling Technology | 8570 |
| LAMP1 (D2D11) XP rabbit Antibody | Cell Signaling Technology | 9091 |
| Phospho-DNM1L/DRP1 (Ser637) (D3A4) rabbit mAb antibody | Cell Signaling Technology | 6319 |
| Alexa Fluor 488-AffiniPure goat anti-mouse IgG (H + L) antibody | Jackson ImmunoResearch | 115-545-003 |
| Alexa Fluor 488-AffiniPure goat anti-rabbit IgG (H + L) antibody | Jackson ImmunoResearch | 111-545-003 |
| Peroxidase-AffiniPure goat anti-mouse IgG (H + L) antibody | Jackson ImmunoResearch | 115-035-003 |
| Peroxidase-AffiniPure goat anti-rabbit IgG (H + L) antibody | Jackson ImmunoResearch | 111-035-003 |
| Anti-human LC3 polyclonal antibody | MBL International | PM036 |
| Anti-ACTB/β-actin, clone C4 antibody | Millipore | MAB1501 |
| Anti-EBV EA-R-p17, clone 5B11 antibody | Millipore | MAB8188 |
| BECN1/Beclin 1 human, mouse | Novus | NB500-249 |
| PINK1 antibody | Novus | BC100-494 |
| BECN1 (D-18) antibody | Santa Cruz Biotechnology | 10,086 |
| EBV Ea-D (0261) antibody | Santa Cruz Biotechnology | 58,121 |
| EBV BZLF1/ZEBRA (BZ1) antibody | Santa Cruz Biotechnology | 53,904 |
| HA-probe (Y-11) antibody | Santa Cruz Biotechnology | 805 |
| IRF3/IRF-3 (SL-12) antibody | Santa Cruz Biotechnology | 33,641 |
| IRF3/IRF-3 (FL-425) antibody | Santa Cruz Biotechnology | 9082 |
| Anti-LC3B antibody produced in rabbit | Sigma-Aldrich | L7543 |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | F3165 |
| Donkey anti-mouse IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor 647 | Thermo Fisher Scientific | A-31,571 |
| Goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor 350 | Thermo Fisher Scientific | A-11,045 |
| Goat anti-mouse IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor 555 | Thermo Fisher Scientific | A-21,424 |
| Goat anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody, Alexa Fluor 555 | Thermo Fisher Scientific | A-21,429 |
| LDHA polyclonal antibody | Thermo Fisher Scientific | PA5-27,406 |
| VDAC antibody | Provided by C Brenner (Inserm UMR-S 1180) | N/A |
| Dye and markers | Source | Identifier |
| DAPI (4ʹ,6-diamidino-2-phenylindole, dihydrochloride) | Thermo Fisher Scientific | D1306 |
| Hoechst 33,342 | Thermo Fisher Scientific | H3570 |
| MitoTracker Red CMXRos | Life technologies | M7512 |
| Protein ladder prestained | Euromedex | 06P-0111 |
| Oligonucleotides and DNA | Source | Identifier |
| BHRF1 ORF forward primer: AGTCCAGTGTGGAAGATGGCCTATTCAACA | This paper | N/A |
| BHRF1 ORF reverse primer: GATATCTGCAGAATTTTAAGCGTAATCTGGAACATCGTATGGGTAGTGTCTTCCTCTGGAGAT | This paper | N/A |
| Deoxyribonucleic acid, low molecular weight from salmon sperm | Sigma-Aldrich | 31,149-10 G-F |
| Plasmids | Source | Identifier |
| BECN1 expression vector | [33] | N/A |
| BHRF1-HA expression vector | This paper | N/A |
| CFP-Parkin | Addgene | 47,560 |
| pFlag-ICP34.5 | [37] | N/A |
| pFlag-DDX58 (2xCARD) | [46] | N/A |
| pIFN-β-Luc | [45] | N/A |
| pMito mRFP-EGFP | Provided by Andreas Till [40] | N/A |
| pcDNA3.1 empty vector (EV) | Invitrogen | N/A |
| pRL Renilla Luciferase Control Reporter Vectors | Promega Corporation | E2241 |
| BZLF1 expression vector | [23] | N/A |
| Reagents and kit | Source | Identifier |
| Bovine serum albumin (BSA) | Euromedex | 04-100-812-C |
| Deoxycholic acid | Sigma | D-6750 |
| Dulbecco’s modified eagle medium (DMEM) | Gibco | 41,965-039 |
| Dual-luciferase reporter assay system | Promega Corporation | E1910 |
| Fetal calf serum (FCS) | Dominique Dutscher | S181B-500 |
| Fugene HD transfection reagent | Promega Corporation | E2311 |
| Glycergel Mounting Medium | Dako | C0563 |
| Hepes, Sodium | Sigma-Aldrich | H-3662 |
| Hexadimethrine bromide (Polybrene) | Sigma-Aldrich | H9268 |
| Immobilon western | Merck Millipore | MA01821 |
| Oligofectamine | Thermo Fisher Scientific | 12,252,011 |
| Opti-MEM | Gibco | 31,985-047 |
| Phosphate buffered saline (PBS) | Gibco | 14,200-067 |
| Protease inhibitor cocktail | Sigma-Aldrich | P8340 |
| RPMI medium 1640 | Gibco | A10491-01 |
| Triton X-100 | Sigma-Aldrich | T-8787 |
| Software and algorithms | Source | Identifier |
| Prism (GraphPad 5.00) | GraphPad Software | https://www.graphpad.com/ |
| ImageJ (Fiji) | ImageJ software | https://fiji.sc/ |
| LAS AF Lite | Leica Microsystems CMS GmbH | https://www.leica-microsystems.com/fr/ |
| Viruses | Source | Identifier |
| MISSION pLKO.1-puro non-target shRNA control transduction particles | Sigma-Aldrich | SHC016V |
| MISSION lentiviral transduction particles (Drp1 sh-RNA) | Sigma-Aldrich | SHCLNV |
Cell culture and treatments
HeLa and HEK293T cells (ATCC, CRL11268) were cultured at 37°C under 5% CO2 in DMEM supplemented with 10% fetal calf serum (FCS) as previously described [69,70]. DNM1L knockdown (sh-DNM1L) and corresponding control (sh-NT) cell lines were generated from HeLa or HEK293T cells and were cultured at 37°C under 5% CO2 in DMEM supplemented with 10% FCS and puromycin (2.5 µg/mL). HEK293/EBV+ cells harboring WT and ΔBHRF1 bacmids were kindly provided by Wolfgang Hammerschmidt (German Research Center for Environmental Health and German Center for Infection Research, Munich, Germany) [7] and were grown at 37°C under 5% CO2 in DMEM supplemented with 10% FCS and hygromycin B (100 µg/mL). GFP-LC3 stably transfected HeLa cells were kindly provided by Aviva Tolkovsky (Cambridge Center for Brain Repair, Cambridge, UK) [28] and mRFP-GFP-LC3 stably transfected HeLa cells were kindly provided by David Rubinsztein (University of Cambridge School of Clinical Medicine, Cambridge, UK) [30]. They were grown in DMEM supplemented with 10% FCS and G418 (500 µg/mL). The EBV-positive Burkitt lymphoma cell line Akata was cultured at 37°C under 5% CO2 in RPMI-1640 supplemented with 10% FCS as previously described [62].
For stress induction, culture media was replaced by freshly prepared media containing the indicated drugs. Mitochondrial fission was induced by culturing cells in media supplemented with NaCl (125 mM) for 30 min and mitophagy was promoted by CCCP treatment (10 µM) for 4 h. Starvation-induced autophagy was carried out by placing the cells in EBSS for 4 h before cell lysis. Autophagic flux was monitored by the addition of CQ (50 µM) 4 h before cell lysis. Autophagy inhibition was performed in two different ways: addition of spautin-1 (20 µM) or 3-MA (5 mM) in culture media for 6 h.
Cell transfection
DNA plasmid transfections were performed using Fugene HD transfection reagent (Promega Corporation, E2311) according to the manufacturer’s protocol. One-day prior transfection, the cells were seeded in 24-well plates and incubated at 37°C. On the day of transfection, DNA plasmids were diluted in opti-MEM and Fugene HD was added to the mix (ratio DNA:Fugene equals 1:3). After incubation for 10 min at room temperature (RT), the transfection mix was added to the cells. The cells were incubated at 37°C and 6 h post-transfection DMEM supplemented with 10% FCS was added. Depending on the expression plasmids, cells were fixed 24 h or 48 h post-transfection.
Cells transfection with linear dsDNA was performed using oligofectamine transfection reagent (Thermo Fisher Scientific, 12,252,011) according to the manufacturer’s protocol. One-day prior transfection, the cells were seeded in 24-well plates and incubated at 37°C. On the day of transfection, linear dsDNA (final concentration of 5 µg/mL) and oligofectamine were diluted in opti-MEM and separately incubated for 5 min at RT. Both mixes were then pooled and incubated for 20 min at RT. After one phosphate-buffered saline (PBS) wash, the culture medium was changed to opti-MEM, and the transfection mix was added. The cells were incubated at 37°C and 4 h post-transfection, DMEM supplemented with 30% FCS was added to the cells. Finally, cells were fixed 8 h post-transfection.
Co-immunoprecipitation assays
To study the interaction between BHRF1 and BECN1, HeLa cells cultured in 6-well plates were transfected to co-express BECN1, and different molecular partners tested (EV, BHRF1-HA or Flag-ICP34.5). Two days (48 h) after transfection, cells were washed with cold, sterile PBS and then lysed at 4°C for 2 h in lysis buffer (50 mM Tris HCl, pH 7.5, 50 mM NaCl, 0.5% Triton X-100 [Sigma-Aldrich, T-8787], 0.5% deoxycholic acid [Sigma-Aldrich, D-6750], 0.2% bovine serum albumin [BSA], 25 mM NaPPi [Sigma-Aldrich, P8010], 50 mM NAF [Merck Millipore, 7681-49-4], 1 mM Na3VO4) followed by centrifugation at 20,000 g at 4°C for 30 min to remove cell debris. BECN1 was immunoprecipitated with a goat anti-BECN1 antibody (20 µg/mL). The interaction between BHRF1-HA and endogenous BECN1 was also explored by immunoprecipitation with a rabbit anti-HA antibody (20 µg/mL). Immunoprecipitations were performed overnight at 4°C. Protein G-Sepharose beads (Ademtech, 04332) were added for 2 h at 4°C and were washed three times with lysis buffer and twice with wash buffer (20 mM Tris HCl, pH 7.5, 50 mM NaCl, 0.2% BSA). The immune complexes were finally boiled for 5 min in loading buffer (62.5 mM Tris, pH 6.8, 10% glycerol [Euromedex, EU3550], 1.5% SDS [Euromedex, EU0660], 0.025% bromophenol blue [Sigma-Aldrich, B8026], 8% beta-mercaptoethanol [Sigma-Aldrich, M3148]) before being analyzed by SDS-PAGE.
Cytosol extraction and purification of mitochondrial fractions
Mitochondrial fractions were prepared using a digitonin-based subcellular fractionation technique, as described previously [26]. Briefly, HeLa cells transfected with an EV or BHRF1-HA plasmids were plated 48 h before the experiment in 6-well plates. After being washed twice with PBS, cells were permeabilized with lysis buffer (250 mM sucrose, 70 mM KCl, 137 mM NaCl, 4.3 mM Na2 HPO4, 1.4 mM KH2PO4, pH 7.2, supplemented with 200 µg/mL digitonin [Sigma-Aldrich, D141], and a cocktail of protease/phosphatase inhibitors) for 5 min on ice and then collected before being centrifugated at 1,000 g for 10 min. The cytosolic fraction consisted of the supernatant whereas the pellet, containing mitochondria, was resuspended in mitochondrial lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA [Euromedex, EU0084-A], 2 mM EGTA [Merck Millipore, 324,626], 0.2% Triton X-100, 0.3% NP-40 [Sigma-Aldrich, 74,385] and a cocktail of protease and phosphatase inhibitors [Sigma-Aldrich, PPC1010]) and incubated for 5 min on ice. After centrifugation at 10,000 g for 10 min, the pellet was discarded and the supernatant corresponded to the mitochondrial fraction. Purity of the mitochondrial fraction was confirmed by the presence of the mitochondrial marker VDAC (voltage dependent anion channel) and quasi-absence of a cytosolic marker, LDHA (lactate dehydrogenase A).
EBV-positive cells and EBV reactivation
Akata cells correspond to EBV-positive Burkitt lymphoma B cells, where EBV gene expression is limited to type I latency. To induce the EBV lytic cycle, cells were treated with polyclonal rabbit anti-human IgG at 7.5 µg/mL for 8 h. Then, the cells were washed with PBS and incubated at 37°C with MitoTracker Red CMXRos (250 nM) for 30 min. Following incubation, cells were washed with PBS, fixed with 4% paraformaldehyde in PBS for 10 min, and permeabilized in PBS, Triton X-100 (0.2%) for 5 min. The cells were pelleted and resuspended with PBS. The cell suspension was then fixed to microscope slides via cytocentrifugation using a cytospin (A78300003; Thermo Fisher Scientific, USA) at 1,200 g under medium acceleration mode for 1 min. Cells were blocked and immunostained as described below. We also used HEK293/EBV WT and ΔBHRF1 mutant cells. These cells harbor the genome of the EBV strain B95.8 [7]. To induce EBV reactivation, cells were transfected with a BZLF1 (also called ZEBRA, Zta or EB1) expression plasmid for 48 h, or an EV as a control [23]. Then, the cells were washed with PBS and incubated at 37°C with MitoTracker Red CMXRos (250 nM) for 30 min. Following incubation, cells were washed with PBS, fixed with 4% paraformaldehyde in PBS for 10 min and permeabilized in PBS, Triton X-100 (0.2%) for 5 min. Cells were blocked and immunostained as described below.
Establishment of stable DNM1L knockdown cell lines
Stable HeLa and HEK293T cells knockdown for DNM1L expression were established using lentiviral transduction. DNM1L and non-target shRNA (sh-NT) were purchased from Sigma (MISSION lentiviral transduction particles). One-day before transduction, the cells were seeded in 96-well plates. On the day of transduction, the cells were placed in transduction media (DMEM supplemented with 10% FCS, Hepes 10 mM and polybrene [Sigma-Aldrich, H9268] 8 µg/mL). Then, the cells were infected with lentiviruses at a multiplicity of infection of 0.5, 1 or 5. One-day post-transduction, the culture medium was replaced by DMEM supplemented with 10% FCS. Two days post-transduction, sh-NT and sh-DNM1L-expressing cells were selected by the addition of puromycin (5 µg/mL) in the culture medium. To assess the knockdown, proteins level in cells stably expressing the sh-DNM1L were compared to cells expressing a sh-NT by immunoblot.
Immunoblot analysis
Cells were lysed in lysis buffer (65 mM Tris, pH 6.8, 4% SDS, 1.5% beta-mercaptoethanol and a cocktail of protease/phosphatase inhibitors) and held at 100°C for 10 min. Protein extracts were resolved on SDS-PAGE (12.5%) and electro-transferred onto a polyvinylidene difluoride membrane (Amersham, 10,600,002). After 1 h of incubation in blocking buffer (PBS, 0.1% Tween 20 [Euromedex, 2001] and 5% BSA [Euromedex, 04-100-812] or nonfat dry milk), the membranes were probed overnight at 4°C with primary antibodies. Horseradish peroxidase-labeled antibodies were used as secondary antibody, and revelation was performed using the ECL detection system according to the manufacturer’s instructions (Merck Millipore, WBKLS0500). Scanning for quantification of protein levels was monitored using ImageJ software. An anti-ACTB (actin beta) antibody was used to ensure equal loadings and normalize quantification.
Immunofluorescence microscopy
For indirect immunofluorescence, cells were cultured on glass coverslips in 24-well plates. Cell monolayers were washed with PBS and cells were fixed with paraformaldehyde (4%) in PBS or methanol or acetone/water, depending on the antibodies. The cells were treated with PBS, NH4 Cl (50 mM) for 10 min, permeabilized using PBS, Triton X-100 (0.2%) for 4 min, washed twice with PBS and then incubated for 1 h in PBS, gelatin (0.2%; Sigma-Aldrich, G7765) supplemented with FCS (5%) for blocking. Then the cells were incubated for 1 h with appropriate primary antibody diluted in PBS, gelatin (0.2%) at RT. Cells were washed three times with PBS and then incubated with appropriate secondary antibody diluted in PBS, gelatin (0.2%) for 1 h at RT. After washing, the nuclei were counterstained with DAPI or Hoechst 33,342. Coverslips were mounted in glycergel (Agilent Dako, C056330-2) and observed by microscopy as described below.
For the labeling of autophagic vesicles, cells were fixed with paraformaldehyde (4%) in PBS. Permeabilization and blocking were performed by treating the cells with PBS, NH4 Cl (50 mM) for 10 min followed by an incubation of 30 min with PBS, saponin (0.075%; Sigma-Aldrich, 84,510), BSA (1%) and FCS (5%). Then the appropriate primary antibody diluted in PBS, saponin (0.075%) and BSA (1%) was incubated for 1 h at RT. The cells were then washed twice in PBS, saponin (0.075%), BSA (1%), once with PBS, saponin (0.037%), BSA (0.5%) and twice in PBS. Then, cells were incubated with appropriate secondary antibody diluted in PBS, saponin (0.075%), BSA (1%). After washing, nuclei were counterstained with DAPI. Coverslips were mounted in glycergel and observed using a Nikon Eclipse 80i epifluorescence microscope (Nikon Instruments) or a Leica TCS SP8 X inverted confocal microscope (Leica, USA). Photographic images were resized, organized and labeled using ImageJ software or LAS AF Lite. For colocalization analysis, line profiles were performed using LAS AF Lite software and Manders split coefficients were determined using ImageJ software.
Luciferase reporter assays
HEK293T or HeLa cells were cultured in 24-well plates and transfected for 24 h using Fugene HD, with plasmids encoding the IFNB luciferase reporter (firefly luciferase; 100 ng), pRL-TK (Renilla luciferase; 25 ng) and increasing concentrations (0, 250, 500, or 750 ng) of plasmids expressing BHRF1-HA. EV was used to maintain equal amounts of DNA among wells. To study the IFNB promoter activation dependent on the MAVS pathway, a transfection with 150 ng of a plasmid encoding a positive dominant form of DDX58/RIG-I (Flag-∆DDX58 [2xCARD]) was performed. In order to study the IFNB promoter activation dependent on STING1 pathway, a stimulation by salmon sperm dsDNA (5 µg/mL) was performed for 8 h using oligofectamine as previously described [48]. 24 h post-transfection, cells were lysed and measurement of firefly and renilla luciferase activities was performed using the dual-luciferase reporter assay system (Promega Corporation, E1910) according to the manufacturer’s protocol. Relative expression levels were calculated by dividing the firefly luciferase values by those of renilla luciferase and normalized to the control condition (without BHRF1-HA expression).
Results quantification
Mito-aggresomes are scored by measuring the CI of mitochondrial staining, as previously described [21]. Briefly, the CI corresponds to the fact that compacted mitochondria occupied a limited area of the cell compared to mitochondria that are dispersed throughout the cell. The CI was calculated from the perimeter and the area occupied by mitochondria using the formula developed by Narendra et al [21]. A CI close to 1 corresponds to mitochondria being maximally compact, whereas a CI close to 0 corresponds to mitochondria being minimally compact. We arbitrary determined that a cell presents a mito-aggresome when the CI is above 0.4. The percentage of cells presenting a mito-aggresome was determined by counting at least 20 random cells in each condition from three independent experiments. Mitochondria average length was determined using ImageJ software. Images were converted into 8-bit, and brightness/contrast was adjusted in order to better visualize isolated mitochondria. For each condition, the length of 20 isolated mitochondria per cell was measured on 10 random cells from three independent experiments. The number of endogenous LC3-dots (or GFP-LC3 dots) per cell was determined using ImageJ software. All images were taken with the same microscope settings to be compared to each other. Images were converted into 8-bit. Cells outline was drawn and nuclei cropped to conserve only cytoplasm part. Thresholding of images was performed to detect only LC3 vesicles. Note that the value of the threshold was conserved and used for each analyzed cell. Finally, the “analyze particles” plug-in was used in order to count the total number of LC3-dots per cell. For each condition, at least 25 cells were analyzed in three independent experiments. To quantify colocalization degree between red channel and green channel, the measurement of Manders split coefficient was performed using the “colocalization threshold” plug-in. For each condition, 10 random cells were analyzed. Proportion of cells presenting an IRF3 nuclear localization was determined by counting at least 50 random cells in each condition from three independent experiments. Scanning for quantification of protein levels was monitored using ImageJ software. An anti-ACTB antibody was used to ensure equal loadings and normalize quantification.
Statistical analysis
Data are expressed as mean ± SEM (standard error of the mean) and were analyzed with Prism software (GraphPad) by using Student’s t-test or one-way ANOVA (analysis of variance) test comparisons. P values less than 0.05 were considered statistically significant. Three independent experiments were performed.
Supplementary Material
Acknowledgments
We would like to thank D. Rubinsztein (University of Cambridge School of Clinical Medicine, Cambridge, UK) for providing us the mRFP-GFP-LC3 HeLa cells. We are also very grateful to A. Till (University of California, San Diego, USA) for providing us the mito-mRFP-EGFP plasmid. We would like to thank W. Hammerschmidt (German Research Center for Environmental Health and German Center for Infection Research, Munich, Germany) for providing us the WT and ∆BHRF1 EBV bacmids. We wish to thank V. Nicolas (University of Paris-Saclay, Châtenay-Malabry, France) for her technical assistance from the cellular imaging MIPSIT facility. Also, the present work has benefited from the core facilities of Imagerie‐Gif, (http://www.i2bc.paris‐saclay.fr), member of IBiSA (http://www.ibisa.net), supported by “France‐BioImaging” (ANR‐ 10‐INBS‐04‐01), and the Labex “Saclay Plant Science” (ANR‐11‐ IDEX‐0003‐02). Finally, special thanks to B. Trimbach (University of Paris-Saclay, Châtenay-Malabry, France) for her critical reading of the manuscript.
Funding Statement
This work was supported by institutional funding from CNRS and from Univ. Paris-Sud, DIM MALINF Région Ile de France to GV and GS and grants from the Agence Nationale de la Recherche (ANR-14-CE14-0022) to AE; Agence Nationale de la Recherche [ANR-14-CE14-0022]; Centre National de la Recherche Scientifique [2019]; Conseil Régional, Île-de-France [DIM MALINF]; Conseil Régional, Île-de-France [DIM MALINF]; Université Paris-Sud [2019].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Rowe M, Kelly GL, Bell AI, et al. Burkitt’s lymphoma: the rosetta stone deciphering Epstein-Barr virus biology. Semin Cancer Biol. 2009;19:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kang MS, Kieff E.. Epstein-Barr virus latent genes. Exp Mol Med. 2015;47:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jha HC, Pei Y, Robertson ES. Epstein-Barr virus: diseases linked to infection and transformation. Front Microbiol. 2016;7:1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Longnecker RM, Kieff E, Cohen JI. Epstein-Barr virus. In: editors, Fields BNKD, Howley PM. Fields virology. 6th ed. Philadelphia: Lippincott-Raven; 2013. p. 1898–1959. [Google Scholar]
- [5].Fitzsimmons L, Kelly GL. EBV and apoptosis: the viral master regulator of cell fate? Viruses. 2017;9. DOI: 10.3390/v9110339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Austin PJ, Flemington E, Yandava CN, et al. Complex transcription of the Epstein-Barr virus BamHI fragment H rightward open reading frame 1 (BHRF1) in latently and lytically infected B lymphocytes. Proc Natl Acad Sci U S A. 1988;85:3678–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Altmann M, Hammerschmidt W. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005;3:e404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Khanim F, Dawson C, Meseda CA, et al. BHRF1, a viral homologue of the Bcl-2 oncogene, is conserved at both the sequence and functional level in different Epstein-Barr virus isolates. J Gen Virol. 1997;78(Pt 11):2987–2999. [DOI] [PubMed] [Google Scholar]
- [9].Kvansakul M, Hinds MG. Structural biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis. 2013;4:e909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kvansakul M, Wei AH, Fletcher JI, et al. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 2010;6:e1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Desbien AL, Kappler JW, Marrack P. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc Natl Acad Sci U S A. 2009;106:5663–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Henderson S, Huen D, Rowe M, et al. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc Natl Acad Sci U S A. 1993;90:8479–8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. [DOI] [PubMed] [Google Scholar]
- [14].Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nowag H, Guhl B, Thriene K, et al. Macroautophagy proteins assist Epstein Barr virus production and get incorporated into the virus particles. EBioMedicine. 2014;1:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Granato M, Santarelli R, Farina A, et al. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J Virol. 2014;88:12715–12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cirone M. EBV and KSHV infection dysregulates autophagy to optimize viral replication, prevent immune recognition and promote tumorigenesis. Viruses. 2018;10. DOI: 10.3390/v10110599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bellows DS, Howell M, Pearson C, et al. Epstein-Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. J Virol. 2002;76:2469–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lee JY, Nagano Y, Taylor JP, et al. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol. 2010;189:671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Narendra D, Kane LA, Hauser DN, et al. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Delecluse HJ, Hilsendegen T, Pich D, et al. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc Natl Acad Sci U S A. 1998;95:8245–8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gruffat H, Manet E, Sergeant A. MEF2-mediated recruitment of class II HDAC at the EBV immediate early gene BZLF1 links latency and chromatin remodeling. EMBO Rep. 2002;3:141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Palmer CS, Osellame LD, Stojanovski D, et al. The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell Signal. 2011;23:1534–1545. [DOI] [PubMed] [Google Scholar]
- [25].Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann N Y Acad Sci. 2010;1201:34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Perdiz D, Lorin S, Leroy-Gori I, et al. Stress-induced hyperacetylation of microtubule enhances mitochondrial fission and modulates the phosphorylation of Drp1 at (616)Ser. Cell Signal. 2017;39:32–43. [DOI] [PubMed] [Google Scholar]
- [27].Cereghetti GM, Stangherlin A, Martins de Brito O, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A. 2008;105:15803–15808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bampton ET, Goemans CG, Niranjan D, et al. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. [DOI] [PubMed] [Google Scholar]
- [29].Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sarkar S, Korolchuk V, Renna M, et al. Methodological considerations for assessing autophagy modulators: a study with calcium phosphate precipitates. Autophagy. 2009;5:307–313. [DOI] [PubMed] [Google Scholar]
- [31].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. [DOI] [PubMed] [Google Scholar]
- [32].Levine B, Liu R, Dong X, et al. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 2015. DOI: 10.1016/j.tcb.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mouna L, Hernandez E, Bonte D, et al. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy. 2016;12:327–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lussignol M, Esclatine A. Herpesvirus and autophagy: “All right, everybody be cool, this is a robbery!”. Viruses. 2017;9(12):372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Orvedahl A, Alexander D, Talloczy Z, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. [DOI] [PubMed] [Google Scholar]
- [37].Verpooten D, Ma Y, Hou S, et al. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J Biol Chem. 2009;284:1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Burman JL, Pickles S, Wang C, et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol. 2017;216:3231–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe. 2014;15:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim SJ, Khan M, Quan J, et al. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013;9:e1003722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Castanier C, Garcin D, Vazquez A, et al. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010;11:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ding B, Zhang L, Li Z, et al. The matrix protein of human parainfluenza virus Type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe. 2017;21:538–47 e4. [DOI] [PubMed] [Google Scholar]
- [45].Yoneyama M, Suhara W, Fukuhara Y, et al. Autocrine amplification of type I interferon gene expression mediated by interferon stimulated gene factor 3 (ISGF3). J Biochem. 1996;120:160–169. [DOI] [PubMed] [Google Scholar]
- [46].Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. [DOI] [PubMed] [Google Scholar]
- [47].Vazquez C, Horner SM. MAVS coordination of antiviral innate immunity. J Virol. 2015;89:6974–6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Choi HJ, Park A, Kang S, et al. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat Commun. 2018;9:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Garcia-Sastre A. Ten strategies of interferon evasion by viruses. Cell Host Microbe. 2017;22:176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hahn AM, Huye LE, Ning S, et al. Interferon regulatory factor 7 is negatively regulated by the Epstein-Barr virus immediate-early gene, BZLF-1. J Virol. 2005;79:10040–10052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bentz GL, Liu R, Hahn AM, et al. Epstein-Barr virus BRLF1 inhibits transcription of IRF3 and IRF7 and suppresses induction of interferon-beta. Virology. 2010;402:121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang JT, Doong SL, Teng SC, et al. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J Virol. 2009;83:1856–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wu L, Fossum E, Joo CH, et al. Epstein-Barr virus LF2: an antagonist to type I interferon. J Virol. 2009;83:1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xing J, Zhang A, Zhang H, et al. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. 2017;8:945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jin S, Tian S, Luo M, et al. Tetherin suppresses Type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol Cell. 2017;68:308–22 e4. [DOI] [PubMed] [Google Scholar]
- [57].Du Y, Duan T, Feng Y, et al. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. Embo J. 2018;37:351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jounai N, Takeshita F, Kobiyama K, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104:14050–14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jin S, Tian S, Chen Y, et al. USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. Embo J. 2016;35:866–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kim JH, Kim TH, Lee HC, et al. Rubicon modulates antiviral Type I interferon (IFN) signaling by targeting IFN regulatory factor 3 dimerization. J Virol. 2017;91(14):e00248-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hung CH, Chen LW, Wang WH, et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J Virol. 2014;88:12133–12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shao Z, Borde C, Quignon F, et al. Epstein-Barr virus BALF0 and BALF1 modulate autophagy. Viruses. 2019;11(12):1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].McCormick AL, Smith VL, Chow D, et al. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J Virol. 2003;77:631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kramer T, Enquist LW. Alphaherpesvirus infection disrupts mitochondrial transport in neurons. Cell Host Microbe. 2012;11:504–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kim SJ, Syed GH, Khan M, et al. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci U S A. 2014;111:6413–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pal AD, Basak NP, Banerjee AS, et al. Epstein-Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis. 2014;35:1592–1601. [DOI] [PubMed] [Google Scholar]
- [68].Bekisz J, Baron S, Balinsky C, et al. Antiproliferative properties of Type I and Type II interferon. Pharmaceuticals. 2010;3:994–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lussignol M, Queval C, Bernet-Camard MF, et al. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol. 2013;87:859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Pourcelot M, Zemirli N, Silva Da Costa L, et al. The Golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC Biol. 2016;14:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.