

Abstract
(R)-ND-336—designated as compound (R)-5—is a highly selective inhibitor of matrix metalloproteinase (MMP)-9 with efficacy in accelerating diabetic wound healing in murine models. (R)-ND-336 belongs to the class of thiirane inhibitors of MMPs and it is currently undergoing Investigation New Drug (IND)-enabling studies. We investigated the in vitro metabolism of (R)-ND-336 using S9 fractions obtained from mice, rats, dogs, minipigs, monkeys, and humans in order to select the rodent and nonrodent species for toxicology studies. Three metabolites were observed. One metabolite, M3, was observed across all species. Metabolite M2 was found in rats, monkeys, and humans. Metabolite M1 was observed only in rats. The identities of the metabolites were suggested by liquid chromatography/tandem mass spectroscopy (LC/MS-MS) analyses, which were authenticated by comparison to synthetic samples. Metabolites M2 and M3 arise from oxidative deamination of (R)-ND-336 by monoamine oxidase to give the arylaldehyde as a transient (and unobserved) intermediate. Reductive metabolism of this aldehyde gives the alcohol metabolite M2, while further oxidative metabolism of the aldehyde produces the carboxylate metabolite M3. A minor route of metabolism, seen only in rats, is N-acetylation of (R)-ND-336 to give the acetamide M1. The metabolism of (R)-ND-336 is distinctly different from that of the prototype member of this thiirane class ((±)-1, lacking the 4-aminomethyl aryl substituent) which is metabolized primarily by oxidation α to the sulfone to lead to a benzenesulfinate metabolite. All three metabolites are poorer MMP-9 inhibitors, compared to (R)-ND-336 (MMP-9, Ki = 19 nM): M3, MMP-9 IC50 > 100 μM; M2, Ki = 390 nM; and M1, IC50 > 100 μM). The rat and the minipig were selected as the rodent and nonrodent species, respectively, for toxicology studies.
Keywords: MMP-9, metabolism, (R)-ND-336
Thiirane
(±)-1 (known in the literature as SB-3CT) is a selective
inhibitor of
two zinc-dependent gelatinases: matrix metalloproteinase-2 (MMP-2)
and MMP-9.1,2 Compound 1 is the prototype
of the thiirane class of MMP inhibitors. Both enantiomers of (±)-1 are effective inhibitors of MMP-2 and MMP-9. Thiirane (±)-1 shows efficacy in animal models for the metastases of lymphoma,3 breast cancer,4 and
prostate cancer.5 Metabolism of (±)-1, both in vitro and in vivo, follows two primary routes.6,7 The first route is hydroxylation
of the terminal aromatic ring to give compound 2. The
second route is hydroxylation α to the sulfone. Fragmentation
of the resulting unstable α-hydroxysulfone gives the sulfinic
acid 4. While thiirane (±)-2 is a more
potent inhibitor of MMP-9 than is (±)-1, sulfinic
acid 4 is inactive.6
The hydrochloric acid salt of the p-aminomethyl
analogue (R)-5 (known in the literature
as (R)-ND-336) is a 20-fold more potent inhibitor
of MMP-9 than compound 1 and has in vivo activity in accelerating wound healing in murine models of diabetic
ulcers.8,9 Diabetic wound pathology is exacerbated
by the presence of MMP-9, hence its inhibition is desirable.8,10−12 While the p-aminomethyl moiety of
(R)-5 blocks hydroxylation of the terminal
phenoxy ring (as occurs for (±)-1), the question
remained if α-hydroxylation to its sulfone would give the sulfinic
acid 6 as a metabolite or whether (R)-5 would experience an entirely distinct metabolism.
These questions are addressed in the present report.
Results and Discussion
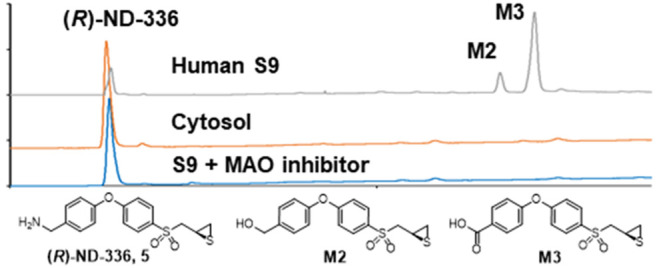
The in vitro metabolism of (R)-ND-336 was investigated in mouse, rat, dog, minipig, monkey, and human S9 fractions that perform phase 1 and phase 2 metabolism. Phase 1 metabolism reactions typically convert a parent drug to more polar metabolites by oxidation, reduction, and/or hydrolysis. S9 fractions include cytochrome P450s (CYPs), flavin monooxygenases, aldehyde oxidase, xanthine oxidase, and monoamine oxidases (MAOs). Phase 2 involves conjugation reactions such as glucuronidation, acetylation, and sulfation. In these experiments we observed three metabolites: M1, M2, and M3. The M3 metabolite (eluting at 6.2 min, Figure 1) was seen for all species.
Figure 1.

Species comparison of the metabolism of (R)-ND-336. (A) Synthetic standards for sulfinic acid 6, sulfonic acid 12, (R)-ND-336 (5), N-acetyl derivative 9, alcohol derivative 8, and carboxylate 7. (B) Incubation of (R)-ND-336 for 60 min in S9 fractions of human, monkey, minipig, dog, rat, and mouse in the presence of NADPH. HPLC conditions: Atlantis T3 column (3.0 μm, 2.1 × 100 mm, 0.5 mL/min elution with 85% A/15% B for 1.5 min, followed by a 7.8 min linear gradient to 15% A/85% B, where A = 0.1% formic acid in water, B = 0.1% formic acid in acetonitrile, UV detection at 255 nm.
The tandem negative ion ESI MS spectrum of M3 showed an [M–H]− ion at m/z 349, which is 15 atomic mass units (amu) higher than the parent compound. A major daughter ion of the ESI– m/z 349 ion is seen at m/z 305 (Figure 2A). This mass difference is consistent with the loss of CO2 from a carboxylic acid. The further loss of mass from this daughter ion to give an m/z 232 ion indicated the loss of methylthiirane. These data were consistent with M3 as the carboxylic acid metabolite of 5. This identity for M3 was confirmed by comparison to the synthetic standard (described below). The MS/MS spectrum of synthetic compound (R)-7 matched that of M3 (Figure 2B).
Figure 2.

Structure characterization of M3 by tandem mass spectrometry in ESI negative mode. MS/MS spectra of (A) metabolite M3 and (B) synthetic compound (±)-7.
Metabolite M2 was observed in human, monkey, and rat S9 incubations (Figure 1). M2 showed an [M–H]− ion at m/z 335, one amu higher than the parent compound. This difference was consistent with an oxygen atom replacing a nitrogen atom. High-resolution ESI–MS gave an m/z 335.0433, consistent with the empirical formula [C16H15O4S2]– (calcd 335.0417). The daughter ions at m/z 263, 199, and 123 (Figure 3A) further substantiated replacement of the aminomethyl moiety in (R)-ND-336 by the hydroxylmethyl group. The identify of M2 was confirmed by comparison to synthetic standard (±)-8 (synthesized according to the literature, the MS spectrum is given in Figure 3B).13
Figure 3.

Structure characterization of M2 by tandem mass spectrometry in ESI negative mode. MS/MS spectra of (A) M2 and (B) synthetic compound (±)-8.
Metabolite M1 was observed only in rat S9, which increased in concentration on incubation with acetyl-CoA as the source of the acetyl group. The observation indicated this metabolite to be an N-acetyl conjugate. The ESI+ tandem mass spectrum of M1 showed ions at m/z 319, 246, 230, 213, and 183, indicating that the biphenyl sulfonyl thiirane core was intact (Figure 4A). Confirmation of the identify of M1 as the N-acetyl metabolite was achieved by comparison to the retention time and product-ion mass spectrum of synthetic standard (R)-9 (Figure 4B). Acetamide (R)-9 was prepared in 61% yield, using a modified synthesis.14
Figure 4.

Structure characterization of M1 by tandem mass spectrometry in ESI positive mode. MS/MS spectra of (A) M1 and (B) synthetic compound (R)-9.
We also investigated the metabolism of (R)-ND-336 in human liver microsomes in the presence of glutathione (Figure 5A) as a surrogate for potential covalent modification of biomolecules. We observed only M2 and M3 (Figure 5B) via ultraviolet (UV) detection in this experiment. Further screening for the glutathione adduct by mass spectrometry at m/z 643 revealed that the adduct was not present (Figure 5C).
Figure 5.

Metabolism of (R)-ND-336 (5) in human liver microsomes in the presence of added glutathione. (R)-ND-336 (20 μM) was incubated for 40 min in the presence of NADPH and glutathione. Chromatogram (A) at time 0 and (B) at 40 min, showing the presence of M2 and M3 using UV detection at 255 nm. (C) Screening for the ion at m/z 643 corresponding to the glutathione adduct of (R)-ND-336 indicated that it was not observed (the trace is of background noise). HPLC conditions are the same as those given in Figure 1.
The metabolism of (R)-ND-336 was NADPH-independent (Figure 6A), indicating that CYPs or flavin monooxygenases were not involved, as the latter require NADPH.15 This was confirmed by incubation of (R)-ND-336 individually with CYP1A2, CYP2C8, CYP2C9, CYP2D6, CYP3A4, and CYP3A5, which are responsible for the metabolism of ∼80% of the clinically used drugs.16 (R)-ND-336 was not metabolized by these P450 isoforms. We next incubated (R)-ND-336 in human liver cytosol to distinguish among the reactions of aldehyde oxidase, xanthine oxidase, and MAO. Aldehyde oxidase and xanthine oxidase are cytosolic enzymes, whereas MAO is present in mitochondria.15 (R)-ND-336 was not metabolized in human liver cytosol (Figure 6B), indicating that aldehyde oxidase or xanthine oxidase were not involved. We used deprenyl, which is a compound that selectively inhibits MAO-B with a Ki value of 0.091 μM; it poorly inhibits MAO-A with a Ki value of 9.06 μM.17 The metabolism of (R)-ND-336 was not affected in the presence of 5 μM concentration of deprenyl (Figure 6C), but was suppressed at 500 μM of deprenyl (Figure 6D), indicating that MAO-A is likely involved in the metabolism of (R)-ND-336 to M2 and M3.
Figure 6.

Metabolism of (R)-ND-336 (5). (R)-ND-336 (20 μM) was incubated for 60 min in the absence of NADPH. (A) Human S9 fraction in the absence of NADPH generated M2 and M3. (B) No metabolites were generated in human cytosol containing aldehyde oxidase and xanthine oxidase. (C) Human S9 in the presence of the MAO inhibitor deprenyl at 5 μM did not suppress metabolism of (R)-ND-336, whereas (D) M2 and M3 were not generated at 500 μM. The HPLC conditions were the same as those given in Figure 1.
The potential sulfinic acid metabolite of (R)-ND-336,
compound 6 (corresponding to compound 4 for
SB-3CT) was not observed. Nonetheless, we synthesized compound 6. The authentic sample eluted at the solvent front as ascertained
by UV and ESI–MS analysis. The ion at m/z 262 corresponding to [M–H]− for 6 was not observed in the reconstructed ion chromatograms
for the S9 fractions, indicating that (R)-ND-336
was not metabolized to this sulfinic acid. As sulfinic acids disproportionate
(albeit slowly) to sulfonic acids,18 we
also synthesized sulfonic acid 12. Compound 12 also was not observed as a metabolite.
The MMP kinetics for the synthetic sulfinic acid 6, N-acetyl derivative (R)-9, carboxylic acid derivative (±)-7, and hydroxymethyl derivative (±)-8 were investigated with representative MMPs (Table 1). Where inhibition was poor, we evaluated the level of activity of the enzyme at a single high concentration (100 μM). At 100 μM compounds 6 and (±)-7 were very poor inhibitors of MMP-2, MMP-9, and MMP-14. Compound (R)-9 (seen only in rats) was a slow-binding inhibitor of MMP-2 (Ki = 164 ± 11 nM), but was a poor inhibitor of MMP-9 and MMP-14. The difference is explained by the S1′ subsite of MMP-2 accommodating the bulkier p-acetamido group, in contrast to an inability to perform this accommodation for MMP-9 and MMP-14.14 Compound (R)-9 inhibits MMP-8 as a linear competitive inhibitor with a Ki value of 440 ± 26 nM. Compound (±)-7 was essentially inactive across the board. We had reported previously on the kinetics of the hydroxymethyl derivative (±)-8 for most of the enzymes in Table 1, except for MMP-8, ADAM9, and ADAM10. All values are compiled for (±)-8 in Table 1. Compound (±)-8 is a noncompetitive inhibitor of MMP-8 with a Ki value of 1330 ± 50 nM, it and inhibits ADAM9 and ADAM10 even more poorly.
Table 1. Inhibition Profiles of the Synthetic Samples for the Metabolites of (R)-ND-336.
| compound (R)-5a | compound 6 | compound (±)-7 | compound (±)-8 | compound (R)-9 | |
|---|---|---|---|---|---|
| MMP-1cat | 22% @ 100 μM | 31% @ 100 μM | 21% @ 100 μM | 205 000 ± 8000 nMd,e | 27% @ 100 μM |
| MMP-2 | 127 ± 1 nMb | 36% @ 100 μM | 9% @ 100 μM | 78 ± 13 nMb,e | 164 ± 11 nMb |
| MMP-3cat | 37% @ 100 μM | 20% @ 100 μM | 24% @ 100 μM | 4100 ± 500 nMd,e | 8% @ 100 μM |
| MMP-7 | 20% @ 100 μM | 27% @ 100 μM | 43% @ 100 μM | 48 500 ± 5000 nMd,e | 15% @ 100 μM |
| MMP-8cat | 8590 ± 230 nMc | 18% @ 100 μM | 16% @ 100 μM | 1330 ± 50 nMc | 440 ± 26 nMd |
| MMP-9cat | 19 ± 3 nMb | 27% @ 100 μM | 10% @ 100 μM | 390 ± 85 nMb,e | 73% @ 50 μM |
| MMP-14cat | 119 ± 3 nMb | 36% @ 100 μM | 36% @ 100 μM | 215 ± 12 nMb,e | 45% @ 50 μM |
| ADAM9 | 41% @ 100 μM | 19% @ 100 μM | 39% @ 100 μM | 40% @ 100 μM | 18% @ 100 μM |
| ADAM10 | 13% @ 100 μM | 8% @ 100 μM | 24% @ 100 μM | 29% @ 100 μM | 16% @ 100 μM |
The preparation of each of the synthetic samples for the purpose of authentication is described. As shown in Scheme 1, allylation of thiophenol 13 (allylbromide, K2CO3) afforded 14 in 82% yield.19 The S-allyl diphenyl ether 15 was synthesized from 14 using p-boronic-acid-substituted benzoic acid ester as the SNAr acceptor (Cu(OAc)2, TEA) in 35% yield. The synthesis of 17 was completed using functional group transformations as reported previously, as follows.8 Epoxidation of the S-allyl moiety (m-CPBA) gave oxirane 16 in 45% yield, which was converted to thiirane 17 (thiourea) in 67% yield. Compound 7 (M3) was obtained by Brønsted acid-catalyzed (HCO2H) deprotection of the t-butyl ester 17 to afford 7 in 55% yield.
Scheme 1.

The synthesis of the sulfinic acid 6 presented challenges (see Schemes 2 and 3). Sulfinic acids are sulfur oxyacids with reactivity as Brønsted acids, as radical precursors, and (as the conjugate base) as nucleophiles. The greater acidity of arylsulfinic acids, compared to arylcarboxylic acids (the pKa of the benzenesulfinic acid functional group is 2),20,21 enhances the reactivity of the sulfinate anion as a soft nucleophile.22 Although the synthetic value of the sulfinic acid is well-recognized in industrial process chemistry,23 for some time, their relative reactivity limited their application in small-molecule synthesis. However, benzenesulfinates are stable and easy to handle. Literature methods for the synthesis of aryl-substituted arylsulfinic acids are limited.24−27 Our objectives also included the arylsulfonic acid 12 as a control compound. Since both 6 and 12 combine acidic and basic functional groups, the synthetic routes for each used sequential, final-stage unmasking of these two functional groups. The viable synthetic routes to sulfinic acid 6 incorporated the functional group equivalence of the sulfone (obtained easily by oxidation of the thioether) and the sulfinic acid. In particular, the masking of the sulfinic acid as a β-sulfonylester was seen as a perfect match to the reagents required to assemble, and then also to ultimately deprotect, the amine. Under mild basic conditions, β-sulfonyl esters fragment to give the desired sulfinic acid, by elimination from what becomes the acrylate ester.28 The exploratory routes, and the successful route wherein this fragmentation is the penultimate synthetic transformation, are summarized in Schemes 2 and 3.
Scheme 2.

Scheme 3.

Our synthesis of sulfinic acid 6 used the inexpensive 4-halophenols (X = Br, I) as starting materials. Diphenylethers 18a and 18b were synthesized in good yields (75%–80%) from the 4-halophenols by SNAr reaction (K2CO3 in DMF, 100 °C) with 4-fluorobenzonitrile. Reduction of the nitrile of 18a and 18b (BH3·SMe2 in THF at reflux) was followed by N-protection of the amine using standard procedures, to yield 19a–19d in good yields (65%–72%). Compounds 20a and 20b were synthesized from the N-Boc bromophenol 19a and the N-Cbz bromophenol 19b (methyl 3-mercaptopropanoate, Pd2(dba)3 catalyst), both in 80% yields. In order to expand the deprotection methodology with N-acetyl (Ac) and N-trifluoroacetyl (TfAc), N-Boc removal from 20a was followed by N-acetylation to give 21a (reaction with Ac2O, R = Ac, 70%) and 21b (reaction with [(F3CC(O)]2O, R = TfAc, 82%). Sulfones 22a–22d were synthesized in 70%–80% isolated yields from thioethers 20a, 20b, 21a, and 21b, respectively, using 30% H2O2/AcOH as the oxidizing agent.
The suitability of each sulfone 22a–22d with respect to N-deprotection was assessed (Scheme 3). Basic deprotection (NaOMe/MeOH) of sulfones 22a to sulfinic acid 26 and of 22b to sulfinic acid 24 was successful. In contrast, base solvolysis of the N-acetyl-protected 22c and the N-trifluoroacetyl-protected 22d, although giving the respective product 23, did so with substantially lower purity, so this route was abandoned. Attempted deprotection of the N-Cbz-protected sulfinate 24 by catalytic hydrogenolysis (10 mol % Pd/C, H2, MeOH) failed, so this route was not pursued. The N-Boc sulfone ester 22a was the optimal precursor to the target sulfinic acid 6. Reaction of 26 with HCl in MeOH gave the desired product 6 as the hydrochloride salt. Final purification by reverse phase chromatography gave the desired sulfinic acid 6 in 65% yield.
Our initial effort toward the synthesis of sulfonic acid 12 used the iodo-substituted 19c as the starting material (Scheme 4). Copper-catalyzed (CuI, TEA, toluene at 110 °C) coupling of thiobenzoic acid gave 27. While solvolysis of its thioester (K2CO3, MeOH) produced thiol 28 in an acceptable yield of 75%, efforts toward the sequential oxidation and deprotection of compound 28 failed to give the desired product 12. Hence, a different but equally direct approach using 4-hydroxybenzenesulfonic acid as the starting material was used. The two-step sequence (SNAr with 4-fluorobenzonitrile to give 29, followed by BH3·SMe2 reduction) gave the desired benzenesulfonic acid product 12 in 10% overall yield. The yield was not optimized as this compound was merely being prepared as a reference compound.
Scheme 4.

The overall proposed metabolism of (R)-ND-336 is summarized in Figure 7. (R)-ND-336 is metabolized to the aryl aldehyde 11 by MAO. Reduction of 11 produces the hydroxymethyl derivative M2, whereas oxidation of this aldehyde results in the carboxylic acid M3. A minor metabolite that is seen only in rats is N-acetylated (R)-ND-336, metabolite M1. These results led us to select the rat and the minipig as the rodent and nonrodent species for repeated toxicology studies.
Figure 7.
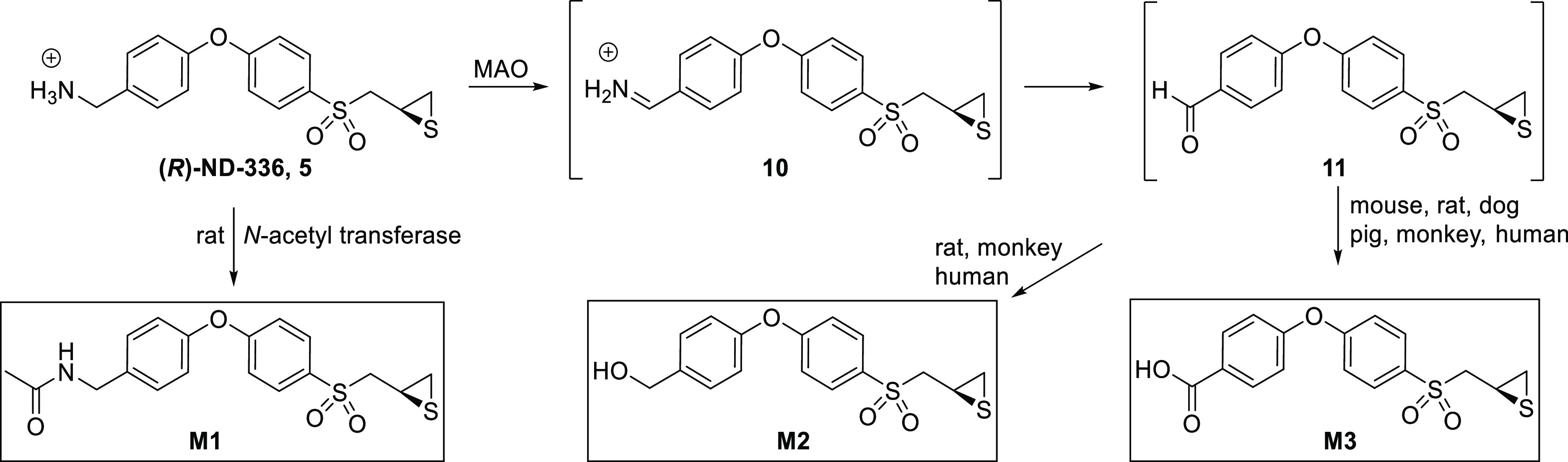
Metabolism of (R)-ND-336.
Methods
Synthesis of M2 (8)
The hydroxymethyl 8 (M2) was synthesized as reported earlier.13
Incubation with S9 Fractions, Cytosol, and CYP450s
The metabolism of (R)-ND-336 was investigated in pooled human, monkey, pig, dog, rat, and mouse S9 fractions and in human cytosol (XenoTech, Lenexa, KS, USA). Incubations were performed with 20 μM of (R)-ND-336 (final concentration) in a volume of 1 mL with 1 mM NADPH (Cayman Chemical, Ann Arbor, MI, USA) in 100 mM potassium phosphate buffer (pH 7.4) containing 10 mM MgCl2, 1 mM acetyl-CoA (Sigma-Aldrich, St. Louis, MO, USA), and 1 mg/mL of S9 fraction at 37 °C. For human incubations, aliquots were drawn at 0, 5, 15, 30, and 60 min and added to one volume of cold acetonitrile in order to precipitate protein. For other species, a fixed time point of 60 min was analyzed for S9 fractions. To investigate the metabolism of (R)-ND-336 in the absence of NADPH, similar incubations were conducted as described above with human cytosol, pooled human S9, and pooled human S9 in the presence of deprenyl (Cayman Chemical, Ann Arbor, MI, USA) at 5 μM and 500 μM final concentration. The metabolism of (R)-ND-336 (20 μM final concentration) in recombinant human CYP1A2, CYP2C8, CYP2C9, CYP2D6, CYP3A4, and CYP3A5 enzymes (200 pmol protein/ml, XenoTech) was investigated in 50 mM potassium phosphate buffer (pH 7.4) containing 3 mM MgCl2. The reaction was started by addition of 1 mM NADPH and was terminated after 60 min with one volume of cold acetonitrile. The mixture was centrifuged at 15 000 rpm for 20 min. The supernatant was analyzed by ultraperformance chromatography (UPLC) using UV and mass spectrometry detection.
Incubation with Human Liver Microsomes in the Presence of Glutathione
(R)-ND-336 (20 μM final concentration) was incubated with human liver microsomes (Xenotech, 1 mg/mL) in 1 mL volume of 100 mM potassium phosphate buffer (pH 7.4) containing 1 mM NADPH, 5 mM l-glutathione (Sigma–Aldrich), and 10 mM MgCl2 at 37 °C. Aliquots were drawn at several time points and protein was precipitated with one volume of acetonitrile. Samples were analyzed by UPLC with UV and mass spectrometry detection. Samples were screened for the glutathione adduct at m/z 643 in positive ESI mode.
UPLC Chromatography and Mass Spectrometry
A Waters Acquity UPLC System (Waters Corporation, Milford, MA, USA) equipped with a binary solvent manager, an autosampler, a column heater, and a photodiode array detector was used. The chromatographic conditions included an Atlantis T3 column (3.0 μm, 2.1 × 100 mm; Waters Corporation), elution at 0.5 mL/min with 85% A/15% B for 1.5 min, followed by a 7.8 min linear gradient to 15% A/85% B, then held at 15% A/85% B for 4.7 min, immediately followed by 1 min at 85% A/15% B (A = 0.1% formic acid in water; B = 0.1% formic acid in acetonitrile). (R)-ND-336 and metabolites were monitored by UV detection at 255 nM.
Mass spectrometry was performed with a Waters TQD tandem quadrupole detector (Milford, MA, USA) monitored with MassLynx MS software. The acquisition was performed in the positive or negative ESI mode. The capillary voltage, cone voltage, extractor voltage, and RF lens voltage were set at 1.50 kV, 50, 3, and 2 V, respectively. Desolvation gas flow rate was 650 L/h (nitrogen), and the cone gas flow rate was 100 L/h (nitrogen). The source temperature and desolvation temperature were held at 150 and 200 °C, respectively. Using these parameters, the metabolite masses 335 and 349 were identified in negative ESI mode, whereas the mass 378 was identified in positive ESI mode. Product ion mass spectra of the metabolites and synthetic standards used the same conditions as described above and a collision energy of 25 V.
(R)-N-(4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)benzyl)acetamide (M1, 9, C18H19NO4S2)
To an ice-cold solution of 5 (200 mg, 0.6 mmol) in THF (1 mL) was added slowly neat acetyl chloride (150 mg, 1.91 mmol) and Et3N (193 mg, 1.91 mmol). The reaction mixture was stirred for 3 h at ice–water temperature. The ice bath was removed as the reaction mixture was stirred overnight. The reaction was quenched with saturated NaHCO3. The product was taken up into CH2Cl2 (2 × 20 mL). The combined organic layer were dried (Na2SO4), the suspension was filtered, and the filtrate was evaporated to dryness. The residue was purified by preparative TLC (8:2 hexanes/EtOAc) to give 9, M1 (124 mg, 0.33 mmol, 61%) as an off-white-colored solid: 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 8.9 Hz, 2H), 7.28 (d, J = 8.5 Hz, 2H), 7.01 (d, J = 8.9 Hz, 2H), 6.99 (d, J = 8.5 Hz, 2H), 5.77 (s, 1H), 4.39 (d, J = 5.9 Hz, 2H), 3.44 (dd, J = 14.2, 5.6 Hz, 1H), 3.12 (dd, J = 14.2, 7.8 Hz, 1H), 3.00 (dddd, J = 11.4, 7.6, 5.7, 1.9 Hz, ω = 26.6 Hz, 1H), 2.47 (dd, J = 6.1, 1.4 Hz, 1H), 2.10 (dd, J = 5.1, 1.8 Hz, 1H), 1.99 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.00, 162.86, 154.13, 135.47, 132.02, 130.77, 129.74, 120.70, 117.73, 62.66, 43.07, 26.09, 24.23, 23.35; HRMS (ESI+) m/z calcd for C18H20NO4S2 (M+H)+ 378.0828, found 378.0825. These NMR and MS data are identical with the reported values.14
4-(Allylthio)phenol [5656–44–0] (14, C9H10OS)
To a stirred solution of 4-hydroxythiophenol (13, 2 g, 15.9 mmol) in DMF (25 mL) were added K2CO3 (2.19 g, 15.9 mmol) and allyl bromide (1.92 g, 15.9 mmol) at ice-water temperature. The mixture was stirred for 15 min. The ice bath was removed. The reaction mixture was stirred overnight at room temperature. A solution of 1 M aqueous HCl (20 mL) was added, and the resulting solution was washed with ethyl ether (3 × 25 mL). The combined organic layer was washed with water and brine. The solution was dried (MgSO4), the suspension was filtered and the filtrate was concentrated. The oily residue was purified by silica chromatography (8:2 hexanes/EtOAc) to give 14 (2.16 g, 13 mmol, 82%) as a liquid: 1H NMR (400 MHz, CDCl3) δ 7.23 (d, J = 8.7 Hz, 2H), 6.70 (d, J = 8.7 Hz, 2H), 5.76 (ddt, J = 17.1, 10.1, 7.1 Hz, 1H), 4.93 (dd, J = 9.96, 1.3 Hz, 1H), 4.91 (dd, J = 16.8, 1.4 Hz, 1H), 3.35 (d, J = 7.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 155.07, 134.21, 133.97, 125.98, 117.35, 115.92, 39.39. The 1H and 13C NMR spectra and mass spectrum were identical with reported values.19
tert-Butyl 4-(4-(allylthio)phenoxy)benzoate (15, C20H22O3S)
A 250 mL round-bottomed flask was charged with 14 (2.5 g, 15.0 mmol), 4-tert-butoxycarbonylphenylboronic acid (6.68 g, 30 mmol), Cu(OAc)2 (3.71 g, 15 mmol), Et3N (7.61 g, 75 mmol), 2.5 g of activated 4 Å molecular-sieve powder, and CH2Cl2 (0.1 L). The mixture was stirred at room temperature for 24 h. The reaction mixture was filtered through Celite. The filtrate was concentrated to dryness under reduced pressure. The residue was purified by column chromatography on silica gel (8:2 hexanes/EtOAc) to provide 15 as a colorless oil (1.8 g, 5.25 mmol, 35%): 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 8.9 Hz, 2H), 7.30 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 1.7 Hz, 2H), 6.88 (d, J = 1.8 Hz, 2H), 5.85–5.75 (m, 1H), 4.99–5.04 (m, 2H), 3.44 (d, J = 7.0 Hz, 2H), 1.51 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 165.26, 160.99, 154.95, 133.61, 132.70, 131.52, 130.82, 126.74, 120.20, 117.73, 117.46, 80.92, 38.30, 28.24; HRMS (ESI+) m/z calcd for C20H23O3S (M+H) 343.1362, found 343.1356.
tert-Butyl 4-(4-((oxiran-2-ylmethyl)sulfonyl)phenoxy)benzoate (16, C20H22O6S)
meta-Chloroperbenzoic acid (5.04 g, 29.2 mmol) was added portionwise to an ice-cold solution of 15 (2 g, 5.84 mmol) in CH2Cl2 (20 mL) over 10 min. After completion of the addition, the ice-water bath was removed. The reaction mixture was stirred at room temperature for 4 days. A second portion of mCPBA (5.04 g, 29.2 mmol) was added and the mixture was stirred at room temperature for an additional 4 days. The suspension was filtered. The filtrate was diluted with CH2Cl2. The resulting solution was washed sequentially with 10% aqueous sodium thiosulfate, saturated NaHCO3, and brine. The organic solution was dried (Na2SO4), the suspension was filtered and the filtrate was concentrated. The residue was purified using silica chromatography (7:3 hexanes/EtOAc) to give 16 as an oil (1.02 g, 2.61 mmol, 45%): 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.8 Hz, 1H), 7.77 (d, J = 8.8 Hz, 1H), 7.04 (d, J = 8.8 Hz, 1H), 7.00 (d, J = 8.8 Hz, 1H), 5.74 (ddt, J = 17.4, 10.1, 7.4 Hz, 1H), 5.29 (d, J = 10.1 Hz, 1H), 5.12 (dd, J = 17.1, 1.0 Hz, 1H), 3.75 (d, J = 7.4 Hz, 1H), 1.53 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 164.94, 161.37, 158.71, 132.74, 131.85, 130.97, 128.55, 124.83, 124.74, 119.30, 118.35, 81.33, 61.05, 28.21; HRMS (ESI+) m/z calcd for C20H22O6S (M+H)+ 391.1210, found 391.1215.
tert-Butyl 4-(4-((thiiran-2-ylmethyl)sulfonyl)phenoxy)benzoate (17, C20H22O5S2)
To a solution of 16 (800 mg, 2.0 mmol) in 1:1 MeOH/CH2Cl2 (10 mL) was added thiourea (343 mg, 4.5 mmol). The resulting mixture was stirred at room temperature for 24 h. The solvent was removed under reduced pressure, and the residue was partitioned between CH2Cl2 and water. The organic solution was washed with a second portion of water and with brine and dried over Na2SO4. The suspension was filtered and filtrate was evaporated to dryness. The residue was purified by silica-gel chromatography (7:3 hexanes/EtOAc) to give 17 as an oil (0.56 g, 1.38 mmol, 67%): 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.8 Hz, 2H), 7.84 (d, J = 8.9 Hz, 2H), 7.08 (d, J = 8.9 Hz, 2H), 7.02 (d, J = 8.8 Hz, 2H), 3.45 (dd, J = 14.2, 5.8 Hz, 1H), 3.15 (dd, J = 14.2, 7.6 Hz, 1H), 2.95 (dddd, J = 7.2, 6.2, 6.2, 5.2 Hz, ω = 24.8 Hz, 1H), 2.49 (dd, J = 6.1, 1.4 Hz, 1H), 2.11 (dd, J = 5.1, 1.8 Hz, 1H), 1.54 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 164.89, 161.81, 158.59, 132.94, 131.89, 130.92, 128.70, 119.37, 118.67, 81.35, 62.65, 28.22, 26.06, 24.19; HRMS (ESI+) m/z calcd for C20H23O5S2 (M+H)+ 407.0981, found 407.0971.
4-(4-((Thiiran-2-ylmethyl)sulfonyl)phenoxy)benzoic acid (M3, 7, C16H14O5S2)
To a solution of dry CH2Cl2:HCO2H (1:1, 5 mL) in a round-bottom flask was added 17 (500 mg, 1.23 mmol) under an argon atmosphere. The solution was stirred overnight at room temperature and then evaporated. The residue was redissolved in DCM/MeOH and filtered through silica gel to give M3, 7 (237 mg, 0.68 mmol, 55%) as an off-white-colored solid: 1H NMR (400 MHz, acetone-d6) δ 8.00 (d, J = 8.8 Hz, 2H), 7.88 (d, J = 8.8 Hz, 2H), 7.20 (d, J = 8.8 Hz, 2H), 7.12 (d, J = 8.8 Hz, 2H), 3.49 (dd, J = 14.4, 6.2 Hz, 1H), 3.32 (dd, J = 14.4, 7.2 Hz, 1H), 2.95 (dddd, J = 7.0, 6.0, 6.0, 5.0 Hz, ω = 25 Hz, 1H), 2.43 (dd, J = 6.2, 1.0 Hz, 1H), 2.07 (dd, J = 5.2, 1.4 Hz, 1H); 13C NMR (100 MHz, acetone-d6) δ 165.95, 161.15, 159.62, 134.24, 132.10, 131.17, 126.75, 119.18, 119.12, 100.00, 61.68, 26.29, 23.49; HRMS (ESI+) m/z calcd for C16H15O5S2 (M+H)+ 351.0355, found 351.0357.
4-(4-Bromophenoxy)benzonitrile) [330792-93-3] (18a, C13H8BrNO)
To a round-bottom flask containing DMF (20 mL) under an argon atmosphere were added 4-fluorobenzonitrile (1.00 g, 8.26 mmol), 4-bromophenol (1.43 g, 8.26 mmol) and K2CO3 (1.37 g, 9.9 mmol). The mixture was heated to 110 °C for 15 h (reaction was complete by TLC). The mixture was diluted with water (10 mL) and washed with Et2O (2 × 20 mL). The combined Et2O extracts were washed with brine and dried (Na2SO4). The suspension was filtered and the filtrate was evaporated to dryness. The residue was purified by silica chromatography (9:1 hexanes:EtOAc) to give product 18a (1.7 g, 6.2 mmol, 75%) as an off-white-colored solid: 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 8.9 Hz, 2H), 7.44 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 6.88 (d, J = 8.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 161.18, 154.13, 134.35, 133.37, 122.16, 118.76, 118.18, 118.00, 106.48; HRMS (ESI+) m/z calcd for C13H9BrNO (M+H)+ 273.9868, found 273.9852.
tert-Butyl 4-(4-bromophenoxy)benzylcarbamate (19a, C18H20BrNO3)
To a round-bottom flask containing dry THF (20 mL) under an argon atmosphere was added 18a (1.7 g, 6.2 mmol). A solution of BH3·SMe2 (1 M in THF, 7.45 mL, 1.2 equiv) was added. The mixture was refluxed for 12 h (reaction was complete by TLC). It was cooled to room temperature and methanolic HCl (5 mL, 3 M HCl in MeOH) was added slowly. The solvent was evaporated in vacuo. The resulting solid was triturated with Et2O (10 mL) to give the product as the HCl salt. The crude products were carried forward to protection without further purification. The entire solid HCl salt was suspended in dry THF (20 mL). To this stirred mixture were added sequentially 2 M aqueous NaOH (5 mL), and Boc2O (1.6 g, 7.43 mmol). The mixture was stirred at room temperature for 15 h. The reaction mixture was extracted with Et2O (2 × 20 mL). The combined Et2O extracts were washed with brine and dried (Na2SO4). The suspension was filtered. The filtrate was concentrated to dryness in vacuo. The resulting crude was purified by silica chromatography (2:8 hexanes/EtOAc) to afford the product 19a (1.7 g, 4.49 mmol, 72%): 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 8.9 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 6.79 (d, J = 8.9 Hz, 2H), 4.81 (s, 1H), 4.22 (d, J = 5.0 Hz, 2H), 1.39 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 156.58, 155.92, 155.90, 134.44, 132.69, 129.05, 120.34, 119.19, 115.64, 79.61, 44.08, 28.43; HRMS (ESI+) m/z calcd for C18H21BrNO3 (M+H)+ 378.0699, found 378.0694.
Synthesis of Methyl 3-((4-(4-(((tert-butoxycarbonyl)amino)methyl)phenoxy)phenyl)thio)propanoate (20a, C22H27NO5S)
Pd(dba)3 (0.1 mmol) and Xantphos (0.2 mmol) were suspended in a round-bottom flask with toluene (20 mL) under an argon atmosphere. The starting material 19a (1.5 g, 4.0 mmol) was added, followed by methyl 3-mercaptopropanoate (662 mg, 4 mmol) and DIPEA (1.179 g, 9.1 mmol). The reaction mixture was refluxed overnight (reaction was complete by TLC). The mixture was cooled to room temperature and filtered through Celite. The Celite plug was washed with toluene. The combined toluene solutions were evaporated. The residue was purified by silica chromatography (8:2 hexanes/EtOAc) to give the product 20a as a pale yellow colored solid (1.32 g, 3.16 mmol, 80%): 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.7 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 4.82 (s, 1H), 4.22 (d, J = 5.6 Hz, 2H), 3.61 (s, 3H), 3.03 (t, J = 7.3 Hz, 3H), 2.54 (t, J = 7.3 Hz, 3H), 1.39 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 172.22, 156.92, 155.94, 134.35, 133.32, 129.03, 128.50, 119.30, 119.17, 51.85, 44.09, 34.33, 30.45, 28.43; HRMS (ESI+) m/z calcd for C22H28NO5S (M+H)+ 440.1502, found 440.1493.
Methyl 3-((4-(4-(((tert-butoxycarbonyl)amino)methyl)phenoxy)phenyl)sulfonyl)propanoate (22a, C22H27NO7S)
The thioether 20a (1 g, 2.39 mmol) was dissolved in a solution of acetic acid (10 mL) under an argon atmosphere. To this solution was added 30% H2O2 (2 mL). The mixture was stirred at room temperature overnight (reaction was complete by TLC). The solvent was removed by evaporation. The residue was taken up into EtOAc (2 × 20 mL). The combined EtOAc solution was washed sequentially with saturated NaHCO3 and brine, and dried over anhydrous Na2SO4. The mixture was filtered and the filtrate was concentrated to dryness. The residue was purified by silica chromatography (6:4 hexanes/EtOAc) to give the product 22a as off-white-colored solid (807 mg, 1.79 mmol, 75%): 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 8.9 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H), 7.00 (d, J = 8.9 Hz, 2H), 6.97 (d, J = 8.6 Hz, 2H), 4.86 (s, 1H), 4.27 (d, J = 4.6 Hz, 2H), 3.60 (s, 3H), 3.35 (t, J = 7.9 Hz, 2H), 2.69 (t, J = 7.9 Hz, 2H), 1.40 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 170.52, 162.84, 155.91, 153.93, 136.17, 131.81, 130.52, 129.32, 120.63, 117.65, 52.37, 51.72, 28.42, 27.73; HRMS (ESI+) m/z calcd for C22H27NNaO7S (M+H)+ 472.1400, found 472.1406.
Synthesis of the Sodium Salts of Sulfinic Acid 26
To round-bottom flasks containing the respective sulfone intermediate 22a (R = Boc: 500 mg, 1.11 mmol) in methanol (5 mL) under an argon atmosphere was added NaOMe (61 mg, 1.11 mmol). The reaction mixture was stirred overnight at room temperature (reaction was complete by TLC). The solvent was evaporated to dryness. The solid was triturated with two portions of Et2O (20 mL, discarded) and collected by filtration. Formation of the sulfinate salt was confirmed by LC-MS analysis. The sulfinate salt 26 was subjected to the acidification step without further purification.
4-(4-(Aminomethyl)phenoxy)benzenesulfinic Acid Hydrochloride (6, C13H13NO3S·HCl)
A solution of the entire portion of the above sulfinic acid (26, 200 mg, 0.519 mmol) in MeOH (10 mL) was cooled to ice-water temperature. Methanolic HCl (2 mL) was added. The mixture was stirred at room temperature for 1 h. The solvent was removed by evaporation. The resulting solid was triturated with Et2O (5 mL, discarded). The crude product was purified by reverse-phase chromatography (acetonitrile/water) to give 6 (101 mg, 0.34 mmol, 65%) as an off-white solid: 1H NMR (400 MHz, DMSO) δ 8.45 (s, 1H), 7.62 (d, J = 8.6 Hz, 2H), 7.53 (d, J = 8.6 Hz, 2H), 7.06 (d, J = 8.6 Hz, 2H), 6.93 (d, J = 8.6 Hz, 2H), 3.99 (d, J = 5.6 Hz, 2H); 13C NMR (100 MHz, DMSO) δ 161.91, 156.99, 144.15, 131.56, 129.83, 128.07, 119.18, 118.04, 41.96; The sulfinic acid compound 6 oxidizes to corresponding sulfonic acid due to its poor stability.
4-(4-Cyanophenoxy)benzenesulfonic Acid (29, C13H9NO4S)
To a suspension of 4-hydroxybenzenesulfonic acid (2 g, 11.5 mmol), K2CO3 (3.2 g, 23 mmol), and molecular sieves (1 g) in DMF (10 mL) was added 4-fluorobenzonitrile (1.4 g, 11.5 mmol). The mixture was stirred at 100 °C for 12 h. The mixture was cooled to room temperature and filtered through Celite. The Celite was washed with EtOAc (10 mL). The combined extracts were evaporated to dryness. The crude mass was partitioned between water (10 mL) and EtOAc (1 × 20 mL). The aqueous layer was back-extracted with EtOAc (20 mL). The combined EtOAc fractions were washed with brine and dried (Na2SO4). The mixture was filtered and the filtrate was evaporated to dryness. The residue was purified by silica chromatography (DCM/EtOAc) to give 29 as an off-white-colored solid (1000 mg, 3.63 mmol, 35%): 1H NMR (400 MHz, DMSO) δ 7.84 (d, J = 8.8 Hz, 2H), 7.68 (d, J = 8.5 Hz, 2H), 7.12 (d, J = 8.8 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H); 13C NMR (100 MHz, DMSO) δ 161.39, 154.89, 145.74, 135.15, 128.32, 119.72, 119.17, 118.73, 105.69; HRMS (ESI+) m/z calcd for C13H10NO4S (M+H)+ 276.0325, found 276.0334.
4-(4-(Aminomethyl)phenoxy)benzenesulfonic Acid Hydrochloride (12, C13H13NO4S·HCl)
To a solution of 29 (200 mg, 0.73 mmol) in dry THF (5 mL) in a flask under an argon atmosphere was added a solution of BH3·SMe2 (0.20 mL of 1 M in THF, 1 equiv). The reaction mixture was refluxed for 24 h (reaction was complete by TLC). The mixture was cooled to room temperature. To this mixture was added methanolic HCl (2 mL). The solvent was removed by evaporation. The resulting solid was triturated with Et2O, and the Et2O was discarded. The product as the HCl salt was purified by reverse-phase chromatography (MeOH/H2O) to give 12 (64 mg, 0.21 mmol, 28%) as an off-white-colored solid: 1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H), 7.62 (d, J = 8.6 Hz, 1H), 7.47 (d, J = 7.9 Hz, 2H), 7.08 (d, J = 7.8 Hz, 2H), 6.92 (d, J = 8.6 Hz, 2H), 4.03 (dd, J = 7.1, 14.2 Hz, 2H); 13C NMR (100 MHz, DMSO) δ 157.28, 156.83, 144.55, 131.36, 128.08, 119.23, 118.13; HRMS (ESI+) m/z calcd for C13H14NO4S (M+H) 280.0638, found 280.0644.
MMP Kinetics
The experiments were performed with the same reported methodology for enzyme-inhibition studies.29 The following enzymes were purchased from EMD Chemicals, Inc. (Burlington, MA): human recombinant active MMP-2 and MMP-7, and the catalytic domains of MMP-3 and MMP-14. The following enzymes were purchased from Enzo Life Sciences, Inc. (Farmingdale, NY): human recombinant catalytic domains of MMP-1, MMP-8, and MMP-9. The following enzymes were purchased from R&D Systems (Minneapolis, MN): human recombinant active ADAM9 and ADAM10. The fluorogenic substrates MOCAc-Pro-Leu-Gly-Leu-A2pr(Dnp)-Ala-Arg-NH2 (for MMP-2, MMP-7, MMP-9, and MMP-14) and MOCAc-Arg-Pro-Lys-Pro-Val-Glu-Nva-Trp-Arg-Lys(Dnp)-NH2 (for MMP-3) were purchased from Peptides International (Louisville, KY); Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 (for MMP-1, MMP-8, and ADAM10) and Mca-Pro-Leu-Ala-Gln-Ala-Val-Dpa-Arg-Ser-Ser-Ser-Arg-NH2 (for ADAM9) from R&D Systems. The Km values used for MMP-2, MMP-9, and MMP-14 were determined to be similar to the published values.30 Inhibitor stock solutions of 10 mM for each compound were prepared freshly in DMSO before the enzyme-inhibition studies were performed on a Cary Eclipse fluorescence spectrophotometer (Varian, Agilent Technologies, Santa Clara, CA).
Acknowledgments
This work was supported by the Department of Defense Therapeutic Development Award No. W81XWH19-1-0493.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00063.
Experimental details; 1H and 13C NMR spectra of compounds (PDF)
Author Contributions
C.E.R.G. synthesized compounds under the direction of J.F. and S.M. T.T.N. and E.M.G. conducted metabolism experiments under the direction of M.C. M.C. and S.M. wrote the manuscript with contributions from all authors.
The authors declare the following competing financial interest(s): U.S. Patent 9,604,957, U.S. Patent 9,951,035, U.S. Patent 10,253,103, Eur. Patent 3107905, Jpn. Patent 6,5,21,995, and Chin. Patent ZL201580023723.6 have been issued for (R)-ND-336. T.T.N., M.C., and S.M. report involvement in an organization with a financial interest in the subject matter of this manuscript.
Supplementary Material
References
- Brown S.; Bernardo M. M.; Li Z. H.; Kotra L. P.; Tanaka Y.; Fridman R.; Mobashery S. (2000) Potent and Selective Mechanism-Based Inhibition of Gelatinases. J. Am. Chem. Soc. 122, 6799–6800. 10.1021/ja001461n. [DOI] [Google Scholar]
- Nguyen T. T.; Ding D.; Wolter W. R.; Champion M. M.; Hesek D.; Lee M.; Perez R. L.; Schroeder V. A.; Suckow M. A.; Mobashery S.; Chang M. (2018) Expression of active matrix metalloproteinase-9 as a likely contributor to the clinical failure of aclerastide in treatment of diabetic foot ulcers. Eur. J. Pharmacol. 834, 77–83. 10.1016/j.ejphar.2018.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger A.; Arlt M. J.; Gerg M.; Kopitz C.; Bernardo M. M.; Chang M.; Mobashery S.; Fridman R. (2005) Antimetastatic activity of a novel mechanism-based gelatinase inhibitor. Cancer Res. 65, 3523–3526. 10.1158/0008-5472.CAN-04-3570. [DOI] [PubMed] [Google Scholar]
- Martin M. D.; Carter K. J.; Jean-Philippe S. R.; Chang M.; Mobashery S.; Thiolloy S.; Lynch C. C.; Matrisian L. M.; Fingleton B. (2008) Effect of ablation or inhibition of stromal matrix metalloproteinase-9 on lung metastasis in a breast cancer model is dependent on genetic background. Cancer Res. 68, 6251–6259. 10.1158/0008-5472.CAN-08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfil R. D.; Dong Z.; Trindade Filho J. C.; Sabbota A.; Osenkowski P.; Nabha S.; Yamamoto H.; Chinni S. R.; Zhao H.; Mobashery S.; Vessella R. L.; Fridman R.; Cher M. L. (2007) Prostate cancer-associated membrane type 1-matrix metalloproteinase: a pivotal role in bone response and intraosseous tumor growth. Am. J. Pathol. 170, 2100–2111. 10.2353/ajpath.2007.060720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Villegas-Estrada A.; Celenza G.; Boggess B.; Toth M.; Kreitinger G.; Forbes C.; Fridman R.; Mobashery S.; Chang M. (2007) Metabolism of a Highly Selective Gelatinase Inhibitor Generates Active Metabolite. Chem. Biol. Drug Des. 70, 371–382. 10.1111/j.1747-0285.2007.00577.x. [DOI] [PubMed] [Google Scholar]
- Celenza G.; Villegas-Estrada A.; Lee M.; Boggess B.; Forbes C.; Wolter W. R.; Suckow M. A.; Mobashery S.; Chang M. (2008) Metabolism of (4-Phenoxyphenylsulfonyl)methylthiirane, a Selective Gelatinase Inhibitor. Chem. Biol. Drug Des. 71, 187–196. 10.1111/j.1747-0285.2008.00632.x. [DOI] [PubMed] [Google Scholar]
- Nguyen T. T.; Ding D.; Wolter W. R.; Perez R. L.; Champion M. M.; Mahasenan K. V.; Hesek D.; Lee M.; Schroeder V. A.; Jones J. I.; Lastochkin E.; Rose M. K.; Peterson C. E.; Suckow M. A.; Mobashery S.; Chang M. (2018) Validation of Matrix Metalloproteinase-9 (MMP-9) as a Novel Target for Treatment of Diabetic Foot Ulcers in Humans and Discovery of a Potent and Selective Small-Molecule MMP-9 Inhibitor That Accelerates Healing. J. Med. Chem. 61, 8825–8837. 10.1021/acs.jmedchem.8b01005. [DOI] [PubMed] [Google Scholar]
- Peng Z.; Nguyen T. T.; Song W.; Anderson B.; Wolter W. R.; Schroeder V. A.; Hesek D.; Lee M.; Mobashery S.; Chang M. (2021) Selective MMP-9 Inhibitor (R)-ND-336 Alone or in Combination with Linezolid Accelerates Wound Healing in Infected Diabetic Mice. ACS Pharmacol. Transl. Sci. 4, 107–117. 10.1021/acsptsci.0c00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M. (2016) Restructuring of the extracellular matrix in diabetic wounds and healing: A perspective. Pharmacol. Res. 107, 243–248. 10.1016/j.phrs.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Gao M.; Nguyen T. T.; Suckow M. A.; Wolter W. R.; Gooyit M.; Mobashery S.; Chang M. (2015) Acceleration of diabetic wound healing using a novel protease-anti-protease combination therapy. Proc. Natl. Acad. Sci. U. S. A. 112, 15226–15231. 10.1073/pnas.1517847112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. I.; Nguyen T. T.; Peng Z.; Chang M. (2019) Targeting MMP-9 in Diabetic Foot Ulcers. Pharmaceuticals 12, 79. 10.3390/ph12020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Ikejiri M.; Klimpel D.; Toth M.; Espahbodi M.; Hesek D.; Forbes C.; Kumarasiri M.; Noll B. C.; Chang M.; Mobashery S. (2012) Structure-Activity Relationship for Thiirane-Based Gelatinase Inhibitors. ACS Med. Chem. Lett. 3, 490–495. 10.1021/ml300050b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.; Zhang H.; Trivedi A.; Mahasenan K. V.; Schroeder V. A.; Wolter W. R.; Suckow M. A.; Mobashery S.; Noble-Haeusslein L. J.; Chang M. (2016) Selective Inhibition of MMP-2 Does Not Alter Neurological Recovery after Spinal Cord Injury. ACS Chem. Neurosci. 7, 1482–1487. 10.1021/acschemneuro.6b00217. [DOI] [PubMed] [Google Scholar]
- Bohnert T.; Patel A.; Templeton I.; Chen Y.; Lu C.; Lai G.; Leung L.; Tse S.; Einolf H. J.; Wang Y. H.; Sinz M.; Stearns R.; Walsky R.; Geng W.; Sudsakorn S.; Moore D.; He L.; Wahlstrom J.; Keirns J.; Narayanan R.; Lang D.; Yang X. (2016) Evaluation of a New Molecular Entity as a Victim of Metabolic Drug-Drug Interactions-an Industry Perspective. Drug Metab. Dispos. 44, 1399–1423. 10.1124/dmd.115.069096. [DOI] [PubMed] [Google Scholar]
- Zanger U. M.; Schwab M. (2013) Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 138, 103–141. 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Jagrat M.; Behera J.; Yabanoglu S.; Ercan A.; Ucar G.; Sinha B. N.; Sankaran V.; Basu A.; Jayaprakash V. (2011) Pyrazoline based MAO inhibitors: synthesis, biological evaluation and SAR studies. Bioorg. Med. Chem. Lett. 21, 4296–4300. 10.1016/j.bmcl.2011.05.057. [DOI] [PubMed] [Google Scholar]
- Aziz J.; Messaoudi S.; Alami M.; Hamze A. (2014) Sulfinate derivatives: dual and versatile partners in organic synthesis. Org. Biomol. Chem. 12, 9743–9759. 10.1039/C4OB01727G. [DOI] [PubMed] [Google Scholar]
- Goux C.; Lhoste P.; Sinou D. (1994) Palladium(O)-catalyzed alkylation of thiols. Tetrahedron 50, 10321–10330. 10.1016/S0040-4020(01)81764-6. [DOI] [Google Scholar]
- Glass R. S. (2018) Sulfur Radicals and Their Application. Top Curr. Chem. (Z) 376, 22. 10.1007/s41061-018-0197-0. [DOI] [PubMed] [Google Scholar]
- Alfassi Z. B. (1999) S-Centered Radicals, pp 311–354, Wiley. [Google Scholar]
- Denisov E. T., and Afanas’ev I. B. (2005) Oxidation and Antioxidants in Organic Chemistry and Biology; CRC Press. [Google Scholar]
- Nagy P.; Winterbourn C. C. (2010) Redox Chemistry of Biological Thiols. Adv. Mol. Toxicol. 4, 183–222. 10.1016/S1872-0854(10)04006-3. [DOI] [Google Scholar]
- Fier P. S.; Maloney K. M. (2019) NHC-Catalyzed Deamination of Primary Sulfonamides: A Platform for Late-Stage Functionalization. J. Am. Chem. Soc. 141, 1441–1445. 10.1021/jacs.8b11800. [DOI] [PubMed] [Google Scholar]
- Day J. J.; Neill D. L.; Xu S.; Xian M. (2017) Benzothiazole Sulfinate: A Sulfinic Acid Transfer Reagent under Oxidation-Free Conditions. Org. Lett. 19, 3819–3822. 10.1021/acs.orglett.7b01693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier D. R. Jr.; Yoshikawa N. (2016) A General, One-Pot Method for the Synthesis of Sulfinic Acids from Methyl Sulfones. Org. Lett. 18, 5994–5997. 10.1021/acs.orglett.6b02723. [DOI] [PubMed] [Google Scholar]
- Truce W. E.; Murphy A. M. (1951) The Preparation of Sulfinic Acids. Chem. Rev. 48, 69–124. 10.1021/cr60149a004. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Bauer P.; Baumeister T.; Buckmelter A. J.; Caligiuri M.; Clodfelter K. H.; Han B.; Ho Y. C.; Kley N.; Lin J.; Reynolds D. J.; Sharma G.; Smith C. C.; Wang Z.; Dragovich P. S.; Gunzner-Toste J.; Liederer B. M.; Ly J.; O’Brien T.; Oh A.; Wang L.; Wang W.; Xiao Y.; Zak M.; Zhao G.; Yuen P. W.; Bair K. W. (2013) Structure-based discovery of novel amide-containing nicotinamide phosphoribosyltransferase (nampt) inhibitors. J. Med. Chem. 56, 6413–6433. 10.1021/jm4008664. [DOI] [PubMed] [Google Scholar]
- Ikejiri M.; Bernardo M. M.; Bonfil R. D.; Toth M.; Chang M.; Fridman R.; Mobashery S. (2005) Potent mechanism-based inhibitors for matrix metalloproteinases. J. Biol. Chem. 280, 33992–34002. 10.1074/jbc.M504303200. [DOI] [PubMed] [Google Scholar]
- Gooyit M.; Song W.; Mahasenan K. V.; Lichtenwalter K.; Suckow M. A.; Schroeder V. A.; Wolter W. R.; Mobashery S.; Chang M. (2013) O-Phenyl Carbamate and Phenyl Urea Thiiranes as Selective Matrix Metalloproteinase-2 Inhibitors that Cross the Blood-Brain Barrier. J. Med. Chem. 56, 8139–8150. 10.1021/jm401217d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.