

Abstract
Relative free energy calculations are fast becoming a critical part of early stage pharmaceutical design, making it important to know how to obtain the best performance with these calculations in applications which could span hundreds of calculations and molecules. In this work, we compared two different treatments of long-range electrostatics, Particle Mesh Ewald (PME) and Reaction Field (RF), in relative binding free energy calculations using a non-equilibrium switching protocol. We found simulations using RF achieve comparable results as those using PME but gain more efficiency when using CPU and similar performance using GPU. The results from this work encourage more use of RF in molecular simulations.
Graphical Abstract
INTRODUCTION
The lead optimization stage is an important part of pharmaceutical drug discovery, involving optimization of several key chemical and biophysical properties in order to ensure candidate compounds have adequate selective binding to their target while also having appropriate other properties to potentially become a new pharmaceutical. Lead optimization efforts always require adequate ligand binding affinity for the target, making this a critical design criterion, and one which is a target for predictive methods. Alchemical relative binding free energy (RBFE) calculations based on molecular dynamics (MD) simulations and statistical mechanics have shown promise in providing reliable predictions to guide experimental work in the context of real drug discovery projects.1
RBFE calculations compare the potency of two structurally related ligands by transforming one ligand into another via an unphysical or alchemical pathway. This transformation is performed in both the protein-ligand complex and in the solution state to form a closed thermodynamic cycle. The RBFE of the ligands simulated can be calculated through two opposite legs in this cycle.2
To calculate free energies, these alchemical transformations can be performed via either an equilibrium or non-equilibrium protocol. In general, a number of intermediate simulations are conducted along an alchemical path between the two physical end states, with these simulations being either equilibrium or nonequilibrium depending on the approach chosen. While the free energy different of interest depends only on the physical end states in the limit of adequate sampling, these intermediate states serve to help obtain converged results and provide sampling which is hopefully adequate. Equilibrium free energy calculations run an equilibrium simulation at each intermediate state as well as the end states which are physically meaningful. In contrast, the non-equilibrium protocol simulates the end states at equilibrium, potentially spending considerably more time there, and only runs short simulations to switch between end states. However, running a large number of switching trials is critical in this case. It is still under debate which of the two protocols is more efficient, with different studies drawing different conclusions,3,4 and the choice of protocol is beyond the scope of this paper. This work follows the protocol deployed in a previous work5 in which a non-equilibrium approach was used, though our results may generalize to equilibrium approaches as well.
Generally, modern MD engines (e.g., AMBER, GROMACS, CHARMM, etc) support different approaches for simulations, including RBFE calculations. Among these software packages, GROMACS is an open source package which is widely used for molecular simulations and reproduces calculated free energies within about 0.2 kcal/mol.6 Moreover, tools (e.g., pmx7) allow easy high-throughput applications of GROMACS RBFE simulations, providing a workflow spanning from initial coordinate files to final free energy estimates. This leads to relatively user-friendly RBFE calculations using GROMACS.
Long-range electrostatics interactions are critical in modeling molecular motions in simulations. Due to the computational complexity of such long-range interactions, it becomes challenging to design accurate and efficient methods to describe such interactions. An excellent review of methods for computing the long-range electrostatics interactions in biomolecular simulations can be found here.8
Among a number of existing methods, Particle Mesh Ewald (PME)9,10 is perhaps the most broadly used. PME and other Particle-Mesh methods (e.g., particle-particle particle-mesh method (P3M)11,12) are based on the Ewald approach which is a classic method to exactly calculate the electrostatic potential13 and is chosen as a starting point for further adjustments for better efficiency. Historically, the PME methods were inspired by the P3M method and the similarities and differences of the two methods are reviewed by previous work.14,15 It is also somewhat controversial in the literature as to whether P3M and PME should be considered completely equivalent (and share the same name, e.g. P3M); here, however, we do not enter this debate and simply refer to the approach as PME, as this is how the implementation we use is described in the GROMACS documentation.
Another popular option to compute electrostatics interactions is Reaction Field (RF).16,17 The RF method only computes the interactions up to a cutoff distance and implicitly treats any interactions beyond the cutoff in a mean-field manner using an appropriate dielectric constant. RF sees considerable use in the field and can be appealing for suitable system geometries.18,19
Both Ewald based approaches (e.g., PME) and RF methods may introduce artifacts and lead to incorrect modeling of the system. Such artifacts have been extensively explored and summarized in previous work.20
In this work, we compare the performance of PME and RF methods in RBFE calculations. There is extensive literature comparing RF and Particle-Mesh methods (e.g., PME, P3M) in plain MD simulations on different systems (e.g., liquids, proteins, etc) which is well summarized in the work of Reif and Oostenbrink.21 Our focus in this work is specifically on whether these methods yield equivalent relative binding free energies, given the tremendous industrial interest in such calculations. While both RF and PME methods are widely used in free energy calculations, 5,18,19,22 they have not yet been compared head-to-head to see if they yield equivalent results in RBFE calculations, to our knowledge.
METHODS
Selected targets.
We selected the targets TYK2 and CDK2 which are part of several RBFE benchmark studies23–25 including the set commonly referred to as Schrödinger’s “JACS set”23 from a key paper in JACS on large-scale free energy calculations. Among all 8 target systems in the JACS set, TYK2 has a moderate system size (∼ 60000 atoms including water and ions) which yields representative results in performance. CDK2 is the largest system (∼ 110000 atoms including water and ions) among these targets and we selected it to verify the trends observed in TYK2 simulations. For TYK2, we used all 24 edges in our calculations and randomly selected 6 edges of CDK2. The ligands simulated in this work are neutral so no net charge change is involved in each perturbation. A table of successfully simulated perturbations can be found in Table S1, S2.
Molecular Dynamics Simulations.
The simulation protocol follows a previous work.5 Simulation details can be found in the Supporting Information.
Both CPU and GPU simulations were performed using PME or RF electrostatics. In the rest of the paper, we denote CPU-PME, CPU-RF, GPU-PME and GPU-RF to represent the hardware and methods for long-range electrostatic interactions treatment.
RESULTS AND DISCUSSION
Predicted relative binding free energies from simulations using RF show good agreement with those from PME.
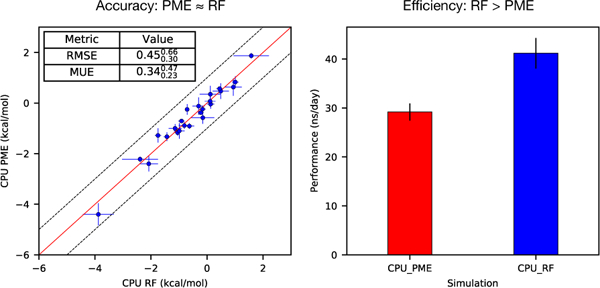
The relative binding free energies (∆∆G) were calculated for a set of modification of TYK2 (24 ∆∆G values in total). Figure 1 summarizes the computed values using RF/PME on CPU/GPU. The uncertainty estimates were performed by 1000 bootstrapping trial and reported in Figure 1 as where x is the mean value, xhigh and xlow indicate 95% confidence intervals. The averaged root-mean-square error (RMSE) and mean unsigned error (MUE) of ∆∆G for CPU-PME versus CPU-RF are 0.45 and 0.34 kcal/mol and for GPU-PME versus GPU-RF are 0.52, 0.40 kcal/mol, respectively (Figure 1a–b). A cross-platform (CPU vs GPU) comparison also shows essentially the same level of agreement between results using RF and PME (Figure 1c–d) suggesting the same trend holds true on both CPU and GPU.
Figure 1:
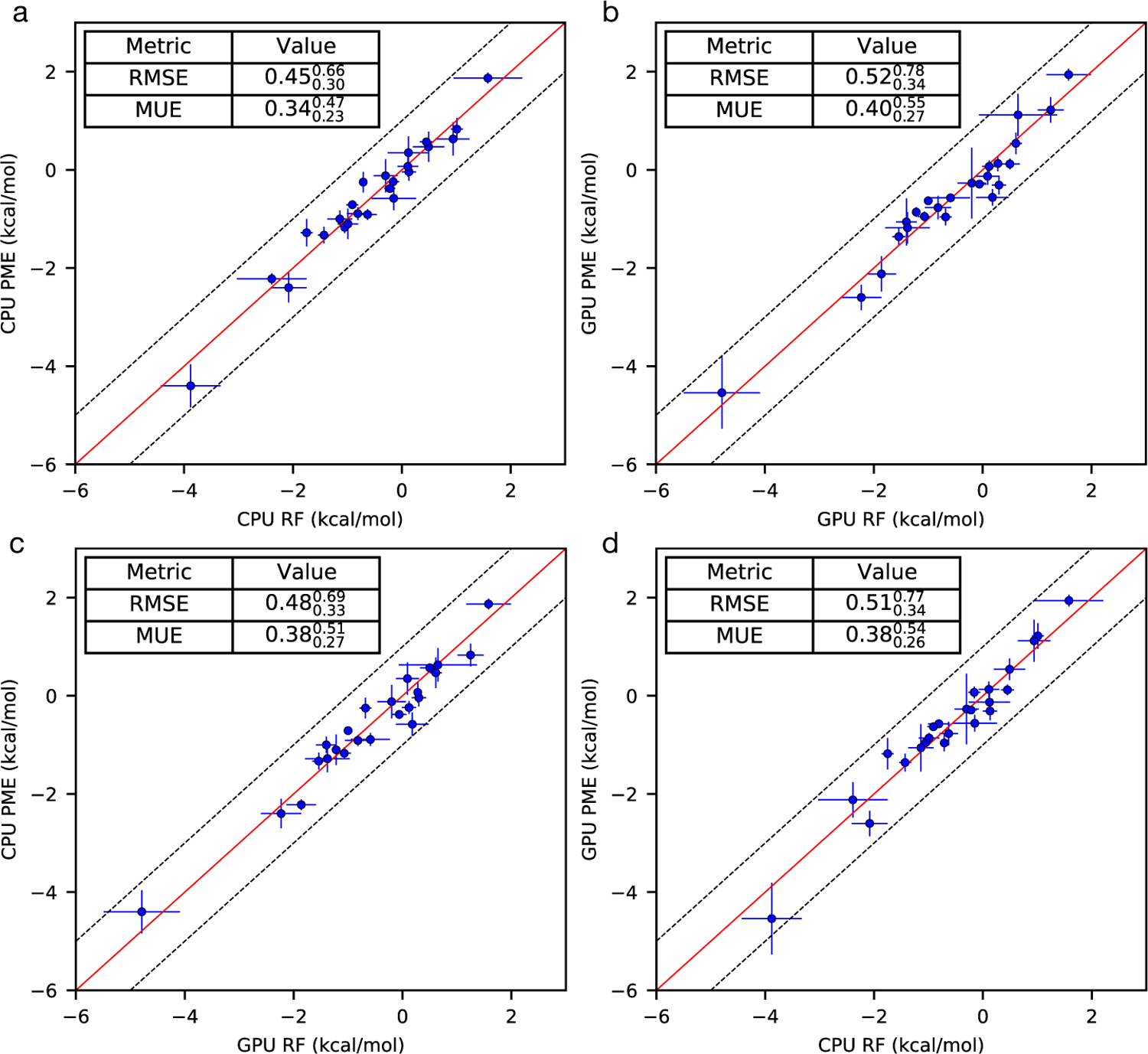
Calculated ∆∆G values of TYK2 ligands using RF/PME on CPU/GPU. Overall, a good agreement is achieved between using PME and RF on CPU/GPU. The uncertainty estimates are calculated by bootstrapping, using 100 bootstrap samples.
In this work, we are focused on the comparison between RF and PME methods on relative binding free energy calculations. While comparing calculated ∆∆G with experimental measured values is more important to benchmarking force field/methodologies, it is not the top interest in this work. However, we summarize our calculated values and the corresponding experimental results in Table S3. In conclusion, the calculated values of both methods are similarly accurate when compared to the experimental values.
Simulations using RF are more efficient than using PME.
Given that PME and RF achieve a good level of agreement of calculated ∆∆G values, our focus shifts to computational efficiency, where RF is generally less computationally demanding than using PME26,27 when the same cut-off is used since more computational time is needed for PME in reciprocal space and tabulated interactions. The simulation performance (in the unit of ns/day) was analyzed from the 6-ns equilibrium NPT simulation and is summarized in Figure 2 where different colors represent using RF/PME on CPU/GPU. The uncertainties were estimated using the standard deviations across different edges. For the (less costly) in-solution ligand simulations, using RF on CPU is ∼30% faster than using PME on average (Figure 2a) and is similar to PME when simulated on GPU considering the uncertainties (Figure 2b). For the more costly bound state simulation, a similar trend is observed in CPU simulations (Figure 2c). However, using RF on GPU is ∼10% slower than using PME on average (Figure 2d). A summary of simulation wallclock time can be found in Table S4, S5.
Figure 2:
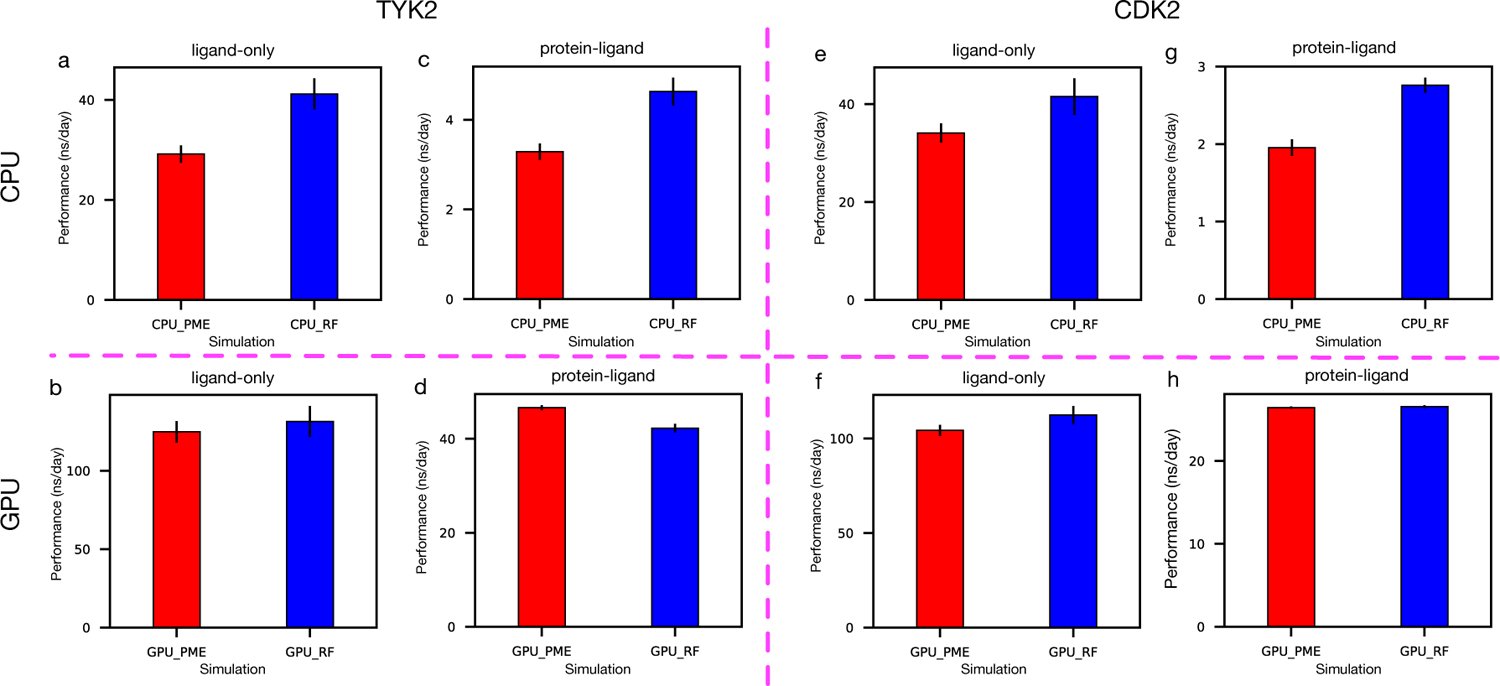
Performance different between using RF/PME on CPU/GPU in TYK2 simulations of (a–b) ligand-only in-solution, (c–d) protein-ligand complex and in CDK2 simulations of (e–f) ligand-only in-solution, (g-h) protein-ligand complex.
The results from Figure 2 show that using RF is faster than PME in most cases for the different system sizes and hardware tested here. It is notable that the GROMACS version used in these simulations was specifically optimized for PME performance on GPU. Thus, it is not surprising that PME outperformed RF on GPUs (slightly) in our tests here. However, the different between PME and RF on GPU performance is only minor (∼10%). Possibly a similar optimization of GROMACS for RF on GPUs could yield substantial performance gains.
To verify the observed trends in TYK2 simulations, 6 selected edges from CDK2 were simulated using the same protocol. Similar to the results of TYK2, simulations using RF are also faster (∼20–40%) than using PME on CPU (Figure 2e,g). Notably, GPU simulations using RF achieves a similar efficiency to PME (Figure 2f,h) which means even with optimizations, PME still cannot surpass RF in efficiency (Figure 2d).
We expect the results observed in this work will be generalizable to other simulation packages and force field in that results will not be highly dependent on PME vs RF, but (a) that is outside of scope for this work, and (b) details will depend on the exact implementation in those software packages. Previous work has shown that different simulation engines are able to take in the same force field and same systems and give identical free energies (with PME) within a reasonable tolerance, which would strongly suggest the implementations are equivalent or nearly so,28,29 at least for PME. Meanwhile, the results observed here are with present-day force field and may not always be the case in future studies (i.e, optimized force field based on the choice of electrostatics treatment).
The conclusions presented in this work are true only for neutral perturbations and are not guaranteed in other perturbations (e.g., charge-changing mutations). When charged species are involved, different correction schemes must be applied to correct the bias induced by electrostatics finite-size effects depending on electrostatic treatment methods used (e.g., RF, PME).30–33
CONCLUSION
The treatment of long-range electrostatic interactions is critical for a correct modeling of (bio)molecular systems in molecular dynamics simulations. Due to the long-range nature and N2-scaling of electrostatic interactions, they are computationally the most demanding terms in the force field evaluation. Given the results presented in this work, we suggest that RF may be a promising option for relative free energy simulations because, at least in GROMACS, it is less computationally demanding while retaining comparable accuracy to PME. This advantage may be particularly helpful in cases where a large number of simulations are needed (e.g., in the lead optimization stage of the drug discovery process). Thus we recommend free energy calculations with RF be considered as a viable option, at least for homogeneous systems.
Supplementary Material
ACKNOWLEDGEMENTS
DLM appreciates financial support from the National Institutes of Health (R01GM108889 and R01GM132386). We appreciate the Open Force Field Consortium for its support of the Open Force Field Initiative, which provided software infrastructure used in this work.
Footnotes
ASSOCIATED CONTENT
Supporting Information Available
Supporting information (simulated system size, calculated/experimental relative binding free energies, simulation wallclock time) is available free of charge via the Internet at http://pubs.acs.org.
Data and Software Availability
Datasets. All input files for simulations, data and scripts for analysis are freely available at https://github.com/MobleyLab/PME-RF-benchmark.
Software. Simulations were performed using the open-source molecular dynamics package GROMACS with a specific patch optimizing PME performance on GPUs. Analysis was performed using pmx and PLBenchmarks. The versions of both packages used in this work are freely distributed at https://github.com/MobleyLab/PME-RF-benchmark/tree/main/SI/analysis with installation instructions.
D.L.M is a member of the Scientific Advisory Board of OpenEye Scientific Software and an Open Science Fellow with Silicon Therapeutics.
References
- (1).Cournia Z; Allen B; Sherman W Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model 2017, 57, 2911–2937. [DOI] [PubMed] [Google Scholar]
- (2).Tembre BL; Mc Cammon J Ligand-receptor interactions. Comput. Chem. (Oxford, U. K.) 1984, 8, 281–283. [Google Scholar]
- (3).Ytreberg FM; Swendsen RH; Zuckerman DM Comparison of Free Energy Methods for Molecular Systems. J. Chem. Phys 2006, 125, 184114. [DOI] [PubMed] [Google Scholar]
- (4).Goette M; Grubmüller H Accuracy and Convergence of Free Energy Differences Calculated from Nonequilibrium Switching Processes. J. Comput. Chem 2009, 30, 447–456. [DOI] [PubMed] [Google Scholar]
- (5).Gapsys V; Pérez-Benito L; Aldeghi M; Seeliger D; van Vlijmen H; Tresadern G; de Groot BL Large Scale Relative Protein Ligand Binding Affinities Using Non-Equilibrium Alchemy. Chem. Sci 2020, 11, 1140–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Loeffler HH; Bosisio S; Duarte Ramos Matos G; Suh D; Roux B; Mobley DL; Michel J Reproducibility of Free Energy Calculations across Different Molecular Simulation Software Packages. J. Chem. Theory Comput 2018, 14, 5567–5582. [DOI] [PubMed] [Google Scholar]
- (7).Gapsys V; Michielssens S; Seeliger D; de Groot BL Pmx: Automated Protein Structure and Topology Generation for Alchemical Perturbations. J. Comput. Chem 2015, 36, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cisneros GA; Karttunen M; Ren P; Sagui C Classical Electrostatics for Biomolecular Simulations. Chem. Rev 2014, 114, 779–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Darden T; York D; Pedersen L Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (10).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- (11).Pollock EL; Glosli J Comments on P3M, FMM, and the Ewald method for large periodic Coulombic systems. Comput. Phys. Commun 1996, 95, 93–110. [Google Scholar]
- (12).Hockney RW; Eastwood JW Computer Simulation Using Particles; McGraw-Hill: New York, 1981. [Google Scholar]
- (13).Ewald PP Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys 1921, 369, 253–287. [Google Scholar]
- (14).Ballenegger V; Cerdà JJ; Holm C How to Convert SPME to P3M: Influence Functions and Error Estimates. J. Chem. Theory Comput 2012, 8, 936–947. [DOI] [PubMed] [Google Scholar]
- (15).Sagui C; Darden TA P3M and PME: A comparison of the two methods. AIP Conf. Proc 1999, 492, 104–113. [Google Scholar]
- (16).Barker JA; Watts RO Monte Carlo studies of the dielectric properties of water-like models. Mol. Phys 1973, 26, 789–792. [Google Scholar]
- (17).Barker JA Reaction field screening, and long-range interactions in simulations of ionic and dipolar systems. Mol. Phys 1994, 83, 1057–1064. [Google Scholar]
- (18).Stjernschantz E; Oostenbrink C Improved Ligand-Protein Binding Affinity Predictions Using Multiple Binding Modes. Biophys. J 2010, 98, 2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lai B; Oostenbrink C Binding free energy, energy and entropy calculations using simple model systems. Theor. Chem. Acc 2012, 131, 23–13. [Google Scholar]
- (20).Kastenholz MA; Hünenberger PH Influence of Artificial Periodicity and Ionic Strength in Molecular Dynamics Simulations of Charged Biomolecules Employing Lattice-Sum Methods. J. Phys. Chem. B 2004, 108, 774–788. [Google Scholar]
- (21).Reif MM; Kräutler V; Kastenholz MA; Daura X; Hünenberger PH Molecular Dynamics Simulations of a Reversibly Folding β-Heptapeptide in Methanol: Influence of the Treatment of Long-Range Electrostatic Interactions. J. Phys. Chem. B 2009, 113, 3112–3128. [DOI] [PubMed] [Google Scholar]
- (22).Loeffler HH; Bosisio S; Matos GDR; Suh D; Roux B; Mobley DL; Michel J Reproducibility of Free Energy Calculations across Different Molecular Simulation Software Packages. J. Chem. Theory Comput 2018, 14, 5567–5582. [DOI] [PubMed] [Google Scholar]
- (23).Wang L; Wu Y; Deng Y; Kim B; Pierce L; Krilov G; Lupyan D; Robinson S; Dahlgren MK; Greenwood J; Romero DL; Masse C; Knight JL; Steinbrecher T; Beuming T; Damm W; Harder E; Sherman W; Brewer M; Wester R; Murcko M; Frye L; Farid R; Lin T; Mobley DL; Jorgensen WL; Berne BJ; Friesner RA; Abel R Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc 2015, 137, 2695–2703. [DOI] [PubMed] [Google Scholar]
- (24).Kuhn M; Firth-Clark S; Tosco P; Mey ASJS; Mackey M; Michel J Assessment of Binding Affinity via Alchemical Free-Energy Calculations. J. Chem. Inf. Model 2020, 60, 3120–3130. [DOI] [PubMed] [Google Scholar]
- (25).Song LF; Lee T-S; Zhu C; York DM; Merz KM Using AMBER18 for Relative Free Energy Calculations. J. Chem. Inf. Model 2019, 59, 3128–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Monticelli L; Simöes C; Belvisi L; Colombo G Assessing the influence of electrostatic schemes on molecular dynamics simulations of secondary structure forming peptides. J. Phys.: Condens. Matter 2006, 18, S329–S345. [Google Scholar]
- (27).Garrido NM; Jorge M; Queimada AJ; Economou IG; Macedo EA Molecular simulation of the hydration Gibbs energy of barbiturates. Fluid Phase Equilib 2010, 289, 148–155. [Google Scholar]
- (28).Shirts MR; Klein C; Swails JM; Yin J; Gilson MK; Mobley DL; Case D a.; Zhong, E. D. Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J. Comput.-Aided Mol. Des 2016, 31, 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Rizzi A; Jensen T; Slochower DR; Aldeghi M; Gapsys V; Ntekoumes D; Bosisio S; Papadourakis M; Henriksen NM; de Groot BL; Cournia Z; Dickson A; Michel J; Gilson MK; Shirts MR; Mobley DL; Chodera JD The SAMPL6 SAMPLing challenge: assessing the reliability and efficiency of binding free energy calculations. J. Comput.-Aided Mol. Des 2020, 34, 601–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rocklin GJ; Mobley DL; Dill KA; Hünenberger PH Calculating the binding free energies of charged species based on explicit-solvent simulations employing lattice-sum methods: An accurate correction scheme for electrostatic finite-size effects. J. Chem. Phys 2013, 139, 184103–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kastenholz MA; Hünenberger PH Computation of methodology-independent ionic solvation free energies from molecular simulations. I. The electrostatic potential in molecular liquids. J. Chem. Phys 2006, 124, 124106–28. [DOI] [PubMed] [Google Scholar]
- (32).Kastenholz MA; Hünenberger PH Computation of methodology-independent ionic solvation free energies from molecular simulations. II. The hydration free energy of the sodium cation. J. Chem. Phys 2006, 124, 224501–21. [DOI] [PubMed] [Google Scholar]
- (33).Reif MM; Oostenbrink C Net charge changes in the calculation of relative ligand-binding free energies via classical atomistic molecular dynamics simulation. J. Comput. Chem 2013, 35, 227–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.