

Summary
Macrophage scavenger receptor 1 (MSR1) plays an important role in host defense to bacterial infections, M2 macrophage polarization, and lipid homeostasis. However, its physiological function in viral pathogenesis remains poorly defined. Herein, we report that MSR1 facilitates vesicular stomatitis virus (VSV) infection in the central nervous system. Msr1-deficient (Msr1−/−) mice presented reduced morbidity, mortality, and viral loads in the spinal cord following lethal VSV infection, along with normal viremia and innate immune responses, compared to Msr1+/− littermates and wild-type mice. Msr1 expression was most significantly upregulated in the spinal cord, the predominant target of VSV. Mechanistically, through its extracellular domains, MSR1 interacted with VSV surface glycoprotein and facilitated its cellular entry in a low-density lipoprotein receptor-dependent manner. In conclusion, our results demonstrate that MSR1 serves as a cofactor for VSV cellular entry and facilitates its infection preferentially in the spinal cord.
Subject areas: Molecular physiology, Neuroscience, Virology, Cell biology
Graphical abstract
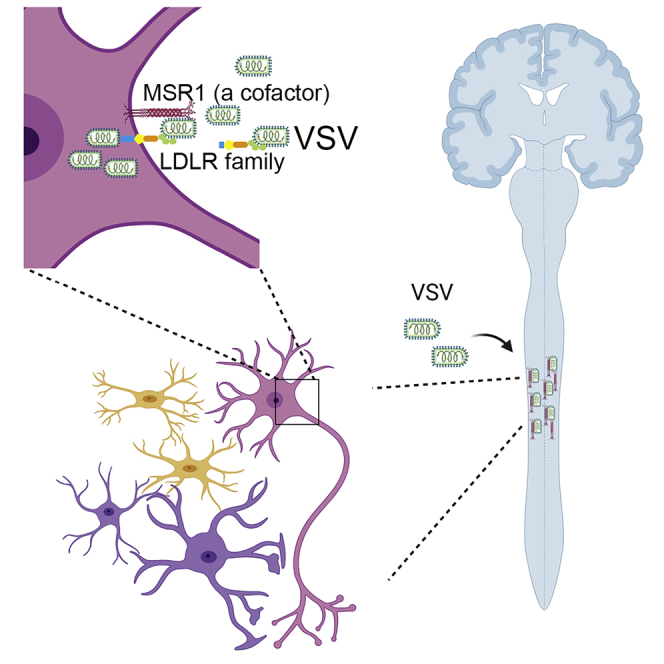
Highlights
-
•
MSR1 contributes to VSV pathogenesis in mice
-
•
MSR1 is highly upregulated and facilitates VSV infection in the central nervous system
-
•
MSR1 facilitates cellular entry of VSV in an LDLR family-dependent manner
-
•
MSR1 interacts with VSV glycoprotein G via its extracellular domains
Molecular physiology; Neuroscience; Virology; Cell biology
Introduction
Macrophage scavenger receptor 1 (MSR1; also known as CD204, SCARA1, and SR-A1), a member of class A scavenger receptors, performs many functions in homeostasis and immunity, including pathogen clearance, lipid metabolism, and macrophage polarization (Canton et al., 2013). MSR1 recognizes a wide range of ligands including oxidized low-density lipoprotein, endogenous proteins, beta-amyloid, lipoteichoic acid (LTA), and bacterial lipopolysaccharide (LPS) (Pluddemann et al., 2011; Pombinho et al., 2018). The ligand promiscuity of MSR1 is probably due to its function as a component of other pathogen pattern recognition receptor signaling complexes. For instance, MSR1 interacts with receptor tyrosine kinase MER to enable apoptotic cell clearance (Todt et al., 2008). MSR1 partners with TLR4 to sense LPS that leads to activation of nuclear factor-κB (NF-κB) signaling (Yu et al., 2012) and therefore contributes to either a pro- or anti-inflammatory responses in Alzheimer disease, sepsis, cancer, and atherosclerosis conditions (Kelley et al., 2014; Mukhopadhyay et al., 2011; Zhang et al., 2017). MSR1 recognizes and internalizes dead cells or debris through interaction with spectrin in macrophages (Cheng et al., 2019). MSR1 also promotes M2 macrophage polarization and the Th2 cell response to bacterial infection by suppressing nuclear translocation of interferon regulatory factor 5 (Xu et al., 2017). Moreover, the functions of MSR1 during viral infection may also vary with virus species and disease conditions. MSR1 signaling may protect mice against lethal herpes simplex virus infection (Suzuki et al., 1997), activate autophagy to control chikungunya virus infection (Yang et al., 2020), and limit replication-defective adenovirus type 5-elicited hepatic inflammation and fibrosis by promoting M2 macrophage polarization (Labonte et al., 2017). MSR1 appears to function as a carrier of extracellular dsRNA of hepatitis C virus and presents it to endosomal TLR3 and cytoplasmic RIG-I-like receptors, resulting in an antiviral type I interferon (IFN) response (Dansako et al., 2013; DeWitte-Orr et al., 2010). However, MSR1 signaling could aggravate the pathogenesis of murine hepatitis virus-induced fulminant hepatitis by enhancing neutrophil NETosis formation-induced complement activation (Tang et al., 2018).
Vesicular stomatitis virus (VSV) is a negative-sense, single-stranded RNA virus of the Rhabdoviridae family. This bullet-shaped virus infects a wide range of cells through its surface glycoprotein (VSV-G) (Ge et al., 2010; Nikolic et al., 2018). Although VSV infection in humans is rare and very mild, it is common in livestock (e.g. cattle, horses, and swine) and may contribute to significant economic losses, particularly in the southwestern United States (Rozo-Lopez et al., 2018). Because of its broad cell tropism, adaptability for genetic engineering, and lack of preexisting human immunity, VSV is widely used as a model virus for fundamental research and vaccine development (Bukreyev et al., 2006; Mátrai et al., 2010). Like rabies virus, VSV is a neurotropic virus that primarily infects animals and occasionally humans (Gagnidze et al., 2016; Ludlow et al., 2016; Quiroz et al., 1988). Although the VSV neurotropism has been well established, there are still areas of ambiguity regarding the host factors that impact VSV infection and pathogenesis. Herein, we report that MSR1 expression is most dramatically upregulated by VSV in the spinal cord and facilitates its infection. Mechanistically, through its extracellular domain, MSR1 interacts with VSV surface glycoprotein and facilitates its cellular entry in a low-density lipoprotein receptor-dependent manner.
Results
Msr1 contributes to VSV pathogenesis in mice
MSR1 plays an important role in host defense to microbial infections (Canton et al., 2013). However, its physiological function in viral pathogenesis may vary significantly with viral species. We investigate this using VSV, a model RNA virus for the study of innate antiviral immune responses. Msr1 mRNA was expressed in various tissues of specific pathogen-free mice (Figure 1A). We then infected Msr1−/− and wild-type (WT, C57BL/6) control mice with a lethal dose of VSV. Intriguingly, ∼90% of the WT mice (n = 12) succumbed to VSV infection, but only ∼30% Msr1−/− mice died (n = 12) (p = 0.001, log rank test) (Figure 1B). In addition, the disease severity of WT mice continuously progressed from day 4 after infection and to a much greater extent than Msr1−/− mice (Figure 1C). Almost all infected WT mice developed severe symptoms, such as hindlimb paralysis, as well as decreased mobility (Figure 1D). To validate the phenotypic results, we produced Msr1−/− and Msr1+/− littermates and repeated VSV infection. We confirmed that the MSR1 protein level in Msr1+/− was the same as that in WT bone-marrow-derived macrophages (BMDMs), which express abundant Msr1 (Figure 1E). Consistently, the survival rate and disease severity of Msr1+/− mice infected with VSV were similar with WT mice: ∼ 80% of infected Msr1+/− mice (n = 6) presented severe hindlimb paralysis and succumbed to lethal VSV infection; while none of Msr1−/− mice (n = 6) showed severe symptoms (Figures 1F and 1G). We next assessed VSV-elicited morbidity in WT and Msr1−/− mice by intranasal instillation of 1 × 106 plaque-forming units (PFUs) of VSV. Although no mice succumbed to infection at this dose, the disease severity was much reduced in Msr1−/− mice compared to WT mice (Figure S1). These data suggest that Msr1 contributes to the pathogenesis of VSV-induced disease.
Figure 1.

Msr1 contributes to VSV pathogenesis in mice
(A) Msr1 mRNA expression in various tissues of wild-type (WT, C57BL/6) and Msr1−/− mice. LOD, limit of detection; N = 3 mice per genotype.
(B and C) The survival curves (B) and disease scores (C) of WT and Msr1−/− mice challenged with 1×107 plaque-forming units (PFUs) per mouse of VSV by retro-orbital injection, N = 12 mice/genotype. p = 0.001 (log rank test), ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001, non-parametric Mann-Whitney U test. All the error bars: mean ± standard error of the mean (S.E.M.).
(D) The WT mice with paralyzed hindlimbs at day 6 after infection.
(E) Immunoblots of Msr1 protein expression in bone-marrow-derived macrophages (BMDMs) of WT, Msr1−/−, and Msr1+/− littermates. β-actin is a housekeeping control.
(F and G) The survival curves (F) and disease scores (G) of Msr1−/− and Msr1+/− littermates challenged with 1 × 107 PFU/mouse of VSV by retro-orbital injection, N = 6 mice/genotype. For percent survival, p < 0.01 (log rank test); for disease score, ∗∗p < 0.01, non-parametric Mann-Whitney U test. All the error bars: mean ± S.E.M.
Msr1 plays an important role in the maintenance of immune homeostasis and is reported to aggravate virus-induced fulminate hepatitis pathogenesis by mediating a C5a-induced proinflammatory response (Tang et al., 2018). We thus assessed VSV viremia and the serum levels of type I IFN and inflammatory cytokines in VSV-infected WT, Msr1+/−, and Msr1−/− littermates by multiplex enzyme-linked immunosorbent assay (ELISA) and quantitative real time-PCR. Interestingly, the viremia and cytokine levels (i.e. IFN-α, IFN-γ, TNF-α, IL-10, CXCL-10, IL-1β) in Msr1−/− mice were similar to those in WT and Msr1+/− littermates (Figures 2A, 2B, S2A, and S2B). These results demonstrate that Msr1 is dispensable for systemic VSV dissemination and innate immune responses.
Figure 2.

Msr1 is dispensable for systemic VSV dissemination and innate immune responses
(A) The RNA viremia in the whole blood of WT and Msr1−/− mice, Msr1+/−, and Msr1−/− littermates infected with VSV, assessed by quantitative RT-PCR, N = 6 mice/genotype in each experiment. The VSV RNA is expressed as a fold change over limit of detection (LOD)s.
(B) The serum levels of type I IFN and inflammatory cytokines (IFN-α, IFN-γ, TNF-α, IL-10, CXCL-10, IL-1β, IL-6) in Msr1+/− and Msr1−/− littermates after VSV infection, assessed by multiplex ELISA, N = 6 mice/genotype. All the data are presented as mean ± S.E.M., and statistical significance is analyzed by non-parametric Mann-Whitney U tests.
Msr1 is critical for VSV infection in the central nervous system
VSV shows a strong neurotropism and causes severe neurological disorders in a variety of rodent and primate animals, although the underlying mechanism remains elusive (Hastie et al., 2013). Indeed, we observed overt hindlimb paralysis and immobility in WT mice (Figures 1B–1G), indicative of severe infection in the central nervous system (CNS). We therefore evaluated viral loads in different tissues after VSV infection. As shown in Figure 3A, the VSV load in the spinal cord was much higher than that of other tissues in VSV-infected Msr1+/− mice. We next examined the CNS specimens from VSV-infected Msr1+/− and Msr1−/− littermates. We found that the viral load in the spinal cord of Msr1−/− mice was significantly lower than that of Msr1+/− mice, while the difference in the brain was moderate (Figures 3B–3D). This result is consistent with VSV-induced severe paraplegia, which is most often caused by spinal cord injury (Smith et al., 2013). Interestingly, the Msr1 expression was sharply induced by VSV in the spinal cord and modestly in the brain and liver, while decreased in the spleen after infection (Figure 3E). A strong positive correlation between the Msr1 expression level and VSV load in the spinal cord was noted; the association was moderate in the brain and spleen (Figure 3F), suggesting that Msr1 is critical for VSV infection in the spinal cord.
Figure 3.

Msr1 is critical for VSV infection in the central nervous system
(A–C) Quantitative RT-PCR analyses of VSV loads in (A) different tissues of Msr1+/− and Msr1−/− littermates on day 6 after infection (N = 3 mice/genotype), (B) the spinal cords, and (C) the brains of Msr1+/− and Msr1−/− littermates on day 6 after infection (N = 6 mice/genotype). The viral RNA load is expressed as fold changes over the limit of detection (LOD).
(D) The VSV titers in the spinal cord and brain of Msr1+/− and Msr1−/− littermates on day 6 after infection, assessed by a plaque-forming assay (N = 6 mice/genotype).
(E) Msr1 mRNA expression (expressed as a fold change over the mock spinal cord group) in different tissues of mock and VSV-infected Msr1+/− mice on day 6 after infection, N = 3 mice/group.
(F–H) (F) Correlation between Msr1 expression and VSV load in various tissues after VSV infection, N = 3 mice/group. The mRNA expression of class A and B scavenger receptors in (G) the spinal cord and (H) the brain of mock and VSV-infected Msr1+/− and Msr1−/− littermates on day 6 after virus inoculation, assessed by quantitative RT-PCR and expressed as a fold change over the Msr1+/--mock group, N = 3 mice/group.
(I) Ldlr mRNA expression in the brain and spinal cord of mock and VSV-infected Msr1+/− and Msr1−/− littermates, expressed as a fold change over the brain of Msr1+/--mock group, N = 3 mice/group, ∗p < 0.05 vs brain, analyzed by non-parametric Mann-Whitney U test. Mock: no virus infection control.
(J) Colocalization of MSR1 with microglia, astrocytes, and neurons in the mouse spinal cord by dual immunofluorescence staining. IBA1: a marker of microglia; GFAP: a marker of astrocyte; MAP2: a marker of neuron; DAPI: nuclei. The yellow regions in the overlay indicate colocalizations. Scale bar represents 10 μm. All the data are presented as mean ± S.E.M., and statistical significances are analyzed by non-parametric Mann-Whitney U test, ∗p < 0.05, ∗∗p < 0.01.
The scavenger receptor family consists of 10 classes, which are structurally heterogeneous with little or no homology (Canton et al., 2013). MSR1 belongs to the class A receptors, which all have an extracellular collagenous domain (Canton et al., 2013). The aforementioned results suggest that MSR1 is upregulated by VSV in the spinal cord. We then asked if other members of class A and B may be induced similarly by VSV. The results show that CD36 (Scarb3) was also markedly induced in the spinal cord of Msr1+/− mice after VSV infection (Figure 3G), but none of the tested class A/B scavenger receptor expression levels were significantly changed in the infected brain (Figure 3H). The LDL receptor (LDLR) family members serve as the cellular receptors for VSV (Finkelshtein et al., 2013) and are also expressed in the nervous system (Beffert et al., 2004); we thus examined the expression of Ldlr in the brain and spinal cord. Intriguingly, Ldlr expression was higher in the spinal cord than in the brain, which might be the reason of higher VSV infectivity in the spinal cord; however, it was not significantly changed after VSV infection in either tissue (Figure 3I). We next pinpointed the resident cell types that express Msr1 in the spinal cord by immunofluorescence microscopy. The microglia (IBA1 positive), astrocytes (glial fibrillary acidic protein [GFAP] as a specific marker), and neurons (MAP2 positive) were stained positive for Msr1 (Figure 3J). These findings suggest that Msr1 is critical for VSV infection, especially in the spinal cord.
MSR1 facilitates cellular entry of VSV
The above mentioned data suggest that MSR1 may facilitate VSV infection directly. Of note, MSR1 is known to recognize a wide range of self- and microbial ligands, in particular, oxidized low-density lipoproteins (Pluddemann et al., 2011; Pombinho et al., 2018). We thus hypothesized that MSR1 could aid in cellular entry of VSV. To this end, we employed primary neurons from neonatal Msr1+/− and Msr1−/− littermates and BMDMs which express abundant Msr1 (Figure 4C). Indeed, the VSV loads in Msr1−/− neurons (Figure 4A) and the virus titer in the culture medium (Figure 4B) were significantly decreased compared to Msr1+/− littermates. The intracellular VSV-G protein level and extracellular infectious virions in Msr1−/− BMDMs were also much lower than those in WT cells (Figures 4D–4F). We next determined if cellular entry of VSV was impaired in Msr1−/− cells. BMDMs were inoculated with VSV at 4°C for 2 hr, which allows attachment of VSV to the cell surface, and then washed 3 times with cold phosphate-buffered saline (PBS) to remove unbound virions. The cells were then divided into two groups: one was kept at 4°C for 30 min, and the other one was incubated at 37°C for 30 min, allowing VSV entry into cells, and lastly, these were washed extensively to remove extracellular virions. Indeed, there were fewer VSV virions attached to Msr1−/− than WT cells at 4°C and consequently fewer virions inside Msr1−/− cells at 37°C (Figure 4G). These data show that Msr1 aids in cellular entry of VSV.
Figure 4.

MSR1 mediates cellular entry of VSV in primary mouse cells and human cells
(A and B) VSV loads in primary neurons of Msr1+/− and Msr1−/− littermates at 36 h after inoculation (MOI = 1, multiplicity of infection = 3), as assessed by (A) quantitative RT-PCR (expressed as a fold change over the lowest viral load) and (B) plaque forming assay. Each dot represents an individual mouse.
(C) Immunoblots of MSR1 protein expression in WT and Msr1−/− bone-marrow-derived macrophages (BMDMs) and peritoneal macrophages (PMs).
(D–F) (D) VSV-G protein expression in BMDMs, (E) virus titer in culture medium, and (F) VSV-GFP fluorescence under microscopy in BMDMs at 48 h after VSV-GFP infection (MOI = 5), N = 3 biological replicates. Objective: 20×, scale bar: 100 μm.
(G) Quantitative RT-PCR analyses of VSV virions attached to BMDMs (4°C for 2 h) and entry into BMDMs (37°C for 30 min). The cells were washed 3 times with cold PBS before switching from 4°C to 37°C and after 37°C as well. MOI = 10, N = 3 biological replicates.
(H) GFP expression in WT and MSR1−/− trophoblasts, 24 h after transduction with GFP-encoding VSV-G-pseudotyped lentiviral vectors (VSV-G-LV-GFP) without polybrene. Scale bar: 50 μm. The statistic VSV-G-LV-GFP was acquired with a fluorescence microscope from 9 random fields of three biological replicates.
(I) GFP expression in WT and LDLR−/−+ RAP trophoblasts, 24 h after transduction with VSV-G-LV-GFP. The LDLR−/− trophoblasts were pre-treated with RAP (200 nM, 30 min, 37°C) followed by VSV-G-LV-GFP transduction.
(J and K) The GFP fluorescence (J) and immunoblots of protein level (K) in VSV-GFP-infected WT and LDLR−/−+RAP trophoblasts with or without MSR1 overexpression; the WT and LDLR−/− trophoblasts were transfected with human MSR1 plasmid for 24 h, followed by VSV-GFP infection for 18 h (MOI = 0.5). The LDLR−/− trophoblasts were pre-treated with RAP (200 nM, 30 min, 37°C) before inoculation of VSV-GFP. Scale bar: 50 μm.
(L) Immunofluorescence staining for different Myc-tagged MSR1 fragments in human trophoblasts, 24 h after transfection of plasmids. Myc proteins were stained by a mouse anti-Myc antibody, followed by an Alexa Fluor 488 (green)-conjugated secondary antibody. The cell nuclei were stained by DAPI (blue). Scale bar: 10 μm. β-actin is a housekeeping control. Mock: no virus infection control. All the data are presented as mean ± S.E.M., and statistical significances are analyzed by a standard two-tailed unpaired Student's t-test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Human MSR1 protein shares 70% identity and 81% similarity with mouse Msr1. We then asked if the function of MSR1 during VSV infection is evolutionarily conserved. To this end, we evaluated VSV infection in a human MSR1−/− trophoblast line that was generated recently in our hands (Yang et al., 2020). Indeed, the VSV-G protein level in MSR1−/− trophoblasts was much less than that in WT cells at 12 hr after VSV infection (Figure 5A). Consistently, the fluorescence intensity of VSV-green fluorescent protein (GFP) in MSR1−/− trophoblasts was also decreased compared to WT cells (Figure 5B), and plaque assay further confirmed a significant reduction in VSV virions in the culture medium of MSR1−/− compared to WT trophoblasts (Figure 5C). To exclude any role of MSR1 in VSV replication after entry, we employed a VSV-G-pseudotyped GFP lentivirus (VSV-G-LV-GFP) which is replication defective. After transduction, MSR1−/− trophoblasts expressed a lower level of GFP than WT trophoblasts (Figure 4H), indicating that MSR1 facilitates VSV lentiviral vector binding but not post-binding events.
Figure 5.

MSR1 facilitates cellular entry of VSV through its extracellular domains
(A–C) WT and MSR1−/− trophoblasts were examined at 12 h after VSV-GFP inoculation, (A) immunoblots of VSV-G protein level, (B) VSV-GFP fluorescence intensity, and (C) VSV titers in the cell culture medium, MOI = 0.5. N = 3 biological replicates.
(D–F) WT and MSR1-overexpressed trophoblasts were examined at 12 h after VSV-GFP inoculation, (D) MSR1 protein expression, VSV-G protein level, (E) VSV-GFP fluorescence intensity, and (F) VSV titers in the culture medium, MOI = 0.5. N = 3 biological replicates. The VSV-GFP fluorescence intensity in (B) and (E) was acquired with a fluorescence microscope from 9 random fields of three biological replicates and quantified by ImageJ. Objective: 4x (top) and 20x, scale bar: 100 μm.
(G) Immunoblots of VSV-G protein level in MSR1−/− trophoblasts with epichromosomal complementation of an empty vector or FLAG-MSR1 expression plasmid at 12 h after VSV infection, MOI=0.5.
(H) The protein expression of different FLAG-tagged MSR1 fragments at 24 h after transfection of plasmids in trophoblasts. IB: immunoblotting. Amino residues 1-50 (cytoplasmic N-tail), 1-80 (cytoplasmic N-tail plus transmembrane), 51-end (extracellular domains with transmembrane), and 1-end (full length).
(I) VSV-G protein level and (J) VSV-GFP fluorescence intensity in trophoblasts overexpressing FLAG-MSR1 fragments at 12 h after VSV-GFP infection at an MOI = 0.5. The GFP fluorescence intensity was acquired with a fluorescence microscope from random regions of three biological replicates (N = 3). β-actin is a housekeeping control. Mock: no virus infection control. Scale bar: 100 μm. All the data are presented as mean ± S.E.M., and statistical significances are analyzed by a standard two-tailed unpaired Student's t-test, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, compare with WT or vector.
The LDLRs are ubiquitously expressed and have been shown to serve as the cellular receptors for VSV (Finkelshtein et al., 2013). We then asked if MSR1 functions in the LDLR pathway. To this end, we generated and selected LDLR−/- single clones by CRISPR-Cas9 (Figure S4). We then added a receptor-associated protein (RAP), a complete blocker of other LDLR family members, to LDLR−/- cells. Indeed, the VSV-GFP expression was almost completely abolished in LDLR−/−+ RAP cells either with or without MSR1 overexpression, while it was significantly increased in WT cells with MSR1 overexpression compared to vectors (Figures 4J and 4K). Similarly, the VSV-G-LV-GFP expression was completely abolished in LDLR−/−+ RAP cells, and MSR1 overexpression increased its expression only in WT cells (Figure 4I). These data suggest that MSR1 facilitates VSV cellular entry in an LDLR family-dependent manner. Consistently, overexpression of MSR1 significantly increased the VSV-G protein level (Figure 5D), VSV-GFP expression (Figure 5E), and VSV titer in the cell culture medium (Figure 5F), compared to the vector control. Epichromosomal complementation of MSR1 gene in MSR1−/− trophoblasts restored VSV infection (Figure 5G).
MSR1 is a cell surface protein with a short intracellular N terminus, a transmembrane domain, and a long extracellular portion. We postulated that the extracellular portion aids in VSV entry. To identify if the extracellular portion mediates cellular entry of VSV, we dissected MSR1 into several fragments: amino residues 1-50 (cytoplasmic N-tail), 1-80 (cytoplasmic N-tail plus transmembrane), and 51-end (extracellular domains with transmembrane), expressed them in trophoblasts (Figure S3A, 4L, and 5H), and infected cells with VSV-GFP for 12 hr. Intriguingly, both the fragment (51-end) and full-length (1-end) MSR1 enhanced VSV-G protein level and VSV-GFP fluorescence intensity, while the fragments without the extracellular domains (1-50 and 1-80) failed to do so (Figures 5I and 5J). The immunofluorescence staining showed MSR1 localization to the plasma membrane and punctate structures, while the fragment 1-50 was completely cytoplasmic and nucleoplasmic (Figure 4L). These results demonstrate that MSR1 facilitates cellular entry of VSV through its extracellular domains.
MSR1 interacts with VSV glycoprotein G via its extracellular domains
Based on the aforementioned data, we hypothesized that MSR1 serves as a cofactor of VSV infection. We thus employed an immunoprecipitation assay to investigate if full-length MSR1 (1-end) and/or fragments (1-80 and 51-end) directly interact with intact VSV-GFP virions. In agreement with Figures 5H–5J, both the 51-end and full-length MSR1 pulled down infectious virions, while the MSR1 (1-80) did not (Figures 6A–6C). Of note, with considerably lower expression, the 51-end fragment pulled down the same amount of VSV-GFP as full-length MSR1 did (Figures 6A–6C), suggesting that the extracellular fragment mediates MSR1 binding to VSV virions. We further asked if MSR1 interacts with VSV surface glycoprotein G that mediates VSV entry into host cells. We performed co-transfection of VSV-G and FLAG-MSR1/fragment plasmids into HEK293 cells and immunoprecipitation using an anti-FLAG antibody. The results show that the fragment (51-end) and full-length (1-end) FLAG-MSR1 pulled down VSV-G, while the empty vector and the fragment 1-80 did not (Figure 6D).
Figure 6.

MSR1 interacts with VSV glycoprotein G via its extracellular domains
(A–C) Binding of MSR1 to VSV virions. FLAG-MSR1 (human, 1-end) and its fragments (aa1-80, 51-end) were expressed in HEK293T cells and immunoprecipitated (IP) using anti-FLAG antibody-coated magnetic beads, which were then incubated with intact VSV-GFP virions (4°C for 2 h). The bound virions were eluted for (A) immunoblotting (IB) with a mouse monoclonal anti-FLAG and rabbit anti-VSV-G antibody
(B and C) Re-infection in Vero cells for 12 h followed by detection of (B) VSV-GFP by fluorescence microscopy and (C) VSV-G protein level by immunoblotting. Mock: no virion control.
(D) co-IP of FLAG-MSR1 and its fragments with VSV-G in HEK293T cells transfected with plasmids. WCE: whole-cell extraction. Arrows point to the faint protein bands.
Discussion
Although MSR1 has proven to perform a variety of functions in tissue homeostasis and host immunity due to its ability to recognize a diverse range of ligands (Pluddemann et al., 2011; Pombinho et al., 2018), its role in viral pathogenesis remains poorly defined. MSR1 signaling may be protective against chikungunya virus and herpes simplex virus infection (Suzuki et al., 1997; Yang et al., 2020) but could also contribute to hepatitis virus-induced liver damage (Tang et al., 2018). In this study, our data collectively demonstrate that MSR1 serves as a cofactor facilitating VSV infection, and this conclusion is substantiated by several lines of experimental evidence. First, Msr1−/− mice were resistant to lethal VSV infection and hindlimb paralysis compared to their WT littermates. However, Msr1 was dispensable for innate immune responses. Second, the viral loads in the CNS were much lower in Msr1−/− than those in WT mice. Third, VSV attachment, entry, and consequently replication were reduced in both mouse primary Msr1−/− macrophages and human MSR1−/− trophoblasts. Overexpression of MSR1 enhanced cellular entry of VSV and subsequent replication; however, MSR1 failed to do so when the function of all LDLR proteins was inactivated. Fourth, MSR1, through its extracellular domains, interacted with VSV surface glycoprotein G, and overexpression of the extracellular domains alone was sufficient to promote VSV infection. These observations suggest that MSR1 is a cofactor facilitating VSV infection in a LDLR family-dependent manner (Ge et al., 2010; Nikolic et al., 2018). Additionally, MSR1 could contribute to neuronal inflammation and apoptosis by activating NF-κB signaling pathway in the spinal cord (Kong et al., 2020).
The LDLR and its family members were recently characterized as essential entry receptors for VSV in both human and mouse cells (Finkelshtein et al., 2013). The widespread expression pattern of the LDLR family may thus in part underlie VSV pantropism. However, tissue tropisms of VSV are determined by not only its receptor abundance but also immune barriers, route of infection, etc (Chauhan et al., 2010; Hastie et al., 2013; Ireland and Reiss, 2006; Trottier et al., 2005, 2007). The CNS has a weaker immune system than peripheral organs, rendering it much vulnerable to neurotropic virus infection. In natural infection, the Rhabdoviridae viruses (e.g. rabies virus, VSV) primarily invade and replicate in motor neurons that exist predominately in the spinal cord (Bradbury and McMahon, 2006), and subsequent retrograde or anterograde spread of progeny virions in the motor nerve system results in paraplegia (Beier et al., 2011; Koyuncu et al., 2013). In mouse models of VSV infection, intranasal and intravenous inoculation also resulted in primarily neurotropism (Hastie et al., 2013; Steinhoff et al., 1995; Stojdl et al., 2000). Consistent with these studies, our data showed that VSV primarily targeted the spinal cord and secondarily the brain (Gagnidze et al., 2016; Hastie et al., 2013; Ludlow et al., 2016; Preble et al., 1980). Of note, Ldlr mRNA expression was threefold higher and VSV-induced Msr1 was tenfold higher in the spinal cord than in the brain, partly underlying a VSV tropism skewed toward the spinal cord. However, the basal expression level of Msr1 was high in peripheral organs than in the CNS, and yet there was no difference in the VSV loads between the peripheral organs (liver, heart, lung, and spleen) of WT and Msr1−/− mice. One reason could be the timing of analysis, e.g. day 6 post infection. The peak of VSV loads in the peripheral organs could be much earlier but very transient; thus, VSV had been cleared from the periphery at the time of analysis (day 6 post infection) by the immune system. However, VSV replicated much more productively and longer in the CNS due to a weaker immune response. Thus, our data could not completely rule out a possible role of MSR1 in facilitating transient VSV infection of peripheral tissues at early time points.
Although low at the basal level, MSR1 expression was most dramatically upregulated in the spinal cord by VSV infection, likely a result of predominant VSV replication there. This could further enhance VSV reinfection and form a positive feedback loop. Of note, among all tested class A and B scavenger receptors, MSR1 was the most significantly upregulated member following VSV infection and was expressed by neurons, microglia (the CNS-resident macrophages), and astrocytes in the spinal cord. This induction of MSR1 may happen not only in infected cells but also in uninfected cells in response to immunological cues such as Th2-polarizing factors, IL-4, macrophage colony-stimulating factor (Józefowski et al., 2005), and type I interferons (Schoggins et al., 2014) produced by infected cells. These cytokine-primed cells may be more permissive to initial VSV infection.
In sum, we herein define MSR1 as a cofactor for LDLR-mediated cellular entry of VSV. Given that VSV and its pseudotyped viruses have been widely applied to vaccine development and oncolytic therapy (Felt and Grdzelishvili, 2017; Liang et al., 2013; Lin et al., 2020), it seems plausible to improve their efficacy by manipulating MSR1 expression.
Limitations of the study
-
1.
Our results did not address a putative role of MSR1 in neuronal inflammation and apoptosis by activating NF-κB signaling pathway.
-
2.
Our data could not completely rule out a possible role of MSR1 in facilitating transient VSV infection of peripheral tissues.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-β-Actin | Cell Signaling Technology | Cat # 4967s |
| mouse anti-FLAG | Origene | Cat# TA50011 |
| rabbit anti-human/mouse MSR1 | Origene | Cat# TA336699 |
| mouse anti-human MSR1 | R&D Systems | Cat# MAB2708 |
| rabbit anti-VSV-G | Sigma-Aldrich | Cat#V4888 |
| anti-FLAG M2 magnetic beads(Clone M2) | Sigma-Aldrich | Cat# M8823 |
| rabbit anti-LDLR | Proteintech Group, Inc | Cat#10785-1-AP |
| Bacterial and virus strains | ||
| vesicular stomatitis virus (VSV)-Indiana strain | American Type Culture Collection (ATCC) | Cat# VR-1238 |
| Chemicals, peptides, and recombinant proteins | ||
| RAP, Human, Recombinant, E. coli | Millipore Sigma | Cat #553506 |
| Critical commercial assays | ||
| BioLegend's LEGENDplex™ kit | BioLegend | https://www.biolegend.com/en-us/search-results?PageSize=25&Keywords=Virus+Response+Panel&k1=Virus&k2=Response&k3=Panel&Applications=Multiplex-Panel |
| Experimental models: Cell lines | ||
| Vero cells(monkey kidney epithelial cells) | American Type Culture Collection (ATCC) | Cat # CCL-81 |
| human trophoblasts HTR-8/SVneo | American Type Culture Collection (ATCC) | Cat #CRL-3271 |
| L929 (mouse fibroblast cells) | American Type Culture Collection (ATCC) | Cat # CCL-1 |
| Experimental models: Organisms/strains | ||
| C57BL/6J mice | Jackson Laboratory | Stock No: 000664 |
| Msr1-/- mice (B6.Cg-Msr1tm1Csk/J) | Jackson Laboratory | Stock No: 006096 |
| Oligonucleotides | ||
| Human MSR1 primer, Forward: CTTGCGGCCGCGGA GCAGTGGGATCACTTTCA, Reverse: TTGGA TCCCTATAAAGTGCAAGTGACTCCAGC |
In this paper | N/A |
| Human MSR1 fragment 1-50 Forward: CTTGCGGC CGCGGAGCAGTGGGATCACTTTCA, Reverse: TTGGATCCCTAAGCTTTGAAGGACTTCAGTT |
In this paper | N/A |
| Human MSR1 fragment 1-80 Forward: CTTGCGGC CGCGGAGCAGTGGGATCACTTTCA, Reverse: TTGGATCCCTACGTTTCCCACTTCAGGAG |
In this paper | N/A |
| Human MSR1 fragment 51-end Forward: CTTG CG CCCGCGGCACTGATTGCCCTTTAC, Reverse: TTGGATCCCTATAAAGTGCAAGTGACTCCAGC |
In this paper | N/A |
| VSV qPCR primer: F: TGATACAGTACAATTATTT TGGGAC, R: GAGACTTTCTGTTACGGGATCTGG |
In this paper | N/A |
| Ldlr qPCR primer: F: GAATCTACTGGTCCGAC CTGTC, R: CTGTCCAGTAGATGTTGCGGTG |
In this paper | N/A |
| Ms-Msr1 qPCR primer: F: AGTGCTGTCTTCTTTAC CAGC, R: GTGAGGAAGGGATGCTGTA |
In this paper | N/A |
| Ms-Scara2 qPCR primer: F: ATGGCACCAAGGG AGACAAAGG, R: GCCTGGTTTTCCAGCATCACCT |
In this paper | N/A |
| Ms-Scara3 qPCR primer: F: CCACGGAGAAATCC TTCGCAATG, R: TAGGTCCTCTGCTACCAACAGG |
In this paper | N/A |
| Ms-Scara4 qPCR primer: F: ACTCCAAGCACGG TCAGCTCAT, R: CTTGTTGCCAGTTGGACCAGGT |
In this paper | N/A |
| Ms-Scara5 qPCR primer: F: TTTGATGGCAGGAG CCTGTCCA, R: CCCACAAGAATCAGGAAGACCAG |
In this paper | N/A |
| Ms-Scarb1 qPCR primer: F: ACACCCGAATCCTC GCTGGAAT, R: CCGTTGGCAAACAGAGTATCGG |
In this paper | N/A |
| Ms-Scarb2 qPCR primer: F: TAGCCAACACCTCC GAAAACGC, R: CGAACTTCTCGTCGGCTTGGTA |
In this paper | N/A |
| Ms-Scarb3 qPCR primer: F: GGACATTGAGATTC TTTTCCTCTG, R: CAAAGGCATTGGCTGGAAGAAC |
In this paper | N/A |
| Ms-Msr2 qPCR primer: F: CCTGATCCAGAGT GCAATCGTG, R: CACATCTCCGATGAAGGGCAAG |
In this paper | N/A |
| Ms-Actin qPCR primer: F: AGAGGGAAATCGTG CGTGAC, R: CAATAGTGATGATGACCTGGCCGT |
In this paper | N/A |
| Ms-Ifna qPCR primer: F: CTTCCACAGGATCA CTGTGTACCT, R: TTCTGCTCTGACCACCTCCC |
In this paper | N/A |
| Ms-Tnfa qPCR primer: F: CTCCAGGCGGTGCC TATGT, R: GAAGAGCGTGGTGGCCC |
In this paper | N/A |
| Ms-Il6 qPCR primer: F: CCAGAAACCGCTATG AAGTTCC, R: TCACCAGCATCAGTCCCAAG |
In this paper | N/A |
| Ms-Cxcl10 qPCR primer: F: ATCATCCCTGCGAG CCTATCCT, R: GACCTTTTTTGGCTAAACGCTTTC |
In this paper | N/A |
| Recombinant DNA | ||
| MSR1 (NM_138715) Human Tagged ORF Clone | Origene | Cat# RC209609 |
| pLVX-EF1alpha-GFP-2xStrep-IRES-Puro | Gordon et al., 2020 | N/A |
| Software and algorithms | ||
| Prism8 | GraphPad | https://www.graphpad.com |
| The LEGENDplex™ Data Analysis Software Suite | BioLegend | https://www.biolegend.com/en-us/legendplex#software |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Penghua Wang, Ph.D (pewang@uchc.edu).
Material availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate/analyse any datasets/code.
Experimental model and subject details
Animals
All mice were purchased from the Jackson Laboratory and housed under the same conditions. C57BL/6J mice (Stock No: 000664) were employed as the wild-type control mice. Msr1-/- mice (B6.Cg-Msr1tm1Csk/J, stock No: 006096) were made from 129 embryonic stem (ES) cells and then backcrossed to C57BL/6J mice for 12 generations. Msr1+/- mice were made by backcrossing Msr1-/- mice to C57BL/6J mice. All of the gene mutant and wild-type mice were normal in body weight and general health. Sex- and age-matched (6-8 weeks) mice were used for all the experiments. The animal protocols were approved by the Institutional Animal Care & Use Committee at University of Connecticut adhering to the National institutes of Health recommendations for the care and use of laboratory animals.
Cell culture and viruses
Vero cells (monkey kidney epithelial cells, # CCL-81), human trophoblasts HTR-8/SVneo (#CRL-3271), L929 (mouse fibroblast cells, # CCL-1) and vesicular stomatitis virus (VSV) (Indiana strain, Cat# VR-1238) were purchased from American Type Culture Collection (ATCC) (Manassas, VA20110, USA). MSR1-/- trophoblasts were generated in our previous study (Yang et al., 2020). Vero cells were grown in Corning® DMEM (Dulbecco’s Modified Eagle’s Medium) supplemented with 10% FBS and 1% penicillin/streptomycin. Human trophoblasts were cultured in Corning® RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Bone-marrow-derived macrophages (BMDMs) were differentiated from femur bone-derived bone marrows in L929 conditioned medium (RPMI1640, 20%FBS, 30% L929 culture medium and 1% penicillin/streptomycin) in 10-cm Petri-dishes at 37°C, 5% CO2 for 7 days. The BMDMs were then seeded in culture plates and cultured in regular RPMI1640 medium overnight before further treatment. MycoZap (Lonza) was used regularly to prevent mycoplasma contamination. Green fluorescence protein (GFP) tagged VSV was made by inserting a VSV-G/GFP fusion sequence between the VSV G and L genes (Dalton and Rose, 2001). These viruses were propagated in Vero cells.
Unless specified, BMDMs were infected with VSV or VSV-GFP at a multiplication of infection (MOI) of 5 (MOI=10 for virions attachment and entry experiments), trophoblasts at a MOI of 0.5, neurons at a MOI of 1.
Method details
Antibodies and plasmids
The rabbit anti-LDLR (Cat#10785-1-AP) antibody was purchased from Proteintech Group, Inc (Rosemont, IL 60018, USA). The rabbit anti-Actin (Cat # 4967s) antibody was purchased from Cell Signaling Technology (Danvers, MA 01923, USA). The mouse anti-FLAG (Cat# TA50011) and rabbit anti-human/mouse MSR1 (Cat# TA336699) antibodies were from Origene (Rockville, MD 20850, USA). The mouse anti-human MSR1 (Cat# MAB2708) antibody was obtained from R&D Systems (Minneapolis, MN 55413, USA). Then rabbit anti-VSV-G (Cat#V4888) antibody and the anti-FLAG M2 magnetic beads (Clone M2, Cat# M8823) were from Sigma-Aldrich (St. Louis, MO, USA). The human MSR1 (Cat# RC209609) were obtained from Origene (Rockville, MD 20850, USA). Human MSR1 and its fragment 1-50, 1-80 and 51-end were amplified by PCR and inserted into pcDNA3.1 (Zeo)-FLAG vector. The GFP-encoding lentiviral vector (pLVX-EF1alpha-GFP-2xStrep-IRES-Puro) was a kind gift from Dr. Nevan J. Krogan lab (Gordon et al., 2020). The primers are listed in the Table S1. The receptor-associated protein (RAP, Human, Recombinant, E. coli) was purchased from Millipore Sigma #553506.
Assessment of VSV virions attachment and entry into cells
After 7-day differentiation, BMDM were seeded with regular RPMI1640 medium in 12-well plate overnight before adding virus. BMDMs were inoculated with VSV at 4°C for 2hrs to allow VSV attachment to the cell surface, and then washed 3 times by cold PBS to remove unbound virions. The cells were then divided into two groups, one was kept at 4°C for 30 min, and the other one was incubated at 37 °C for 30 min to allow for VSV entry into cells, and lastly were washed extensively to remove extracellular virions. The RNA samples were then collected and analyzed by qPCR.
Production of VSV-G pseudotyped lentivirus
Production of VSV-G pseudotyped lentiviral particles was described in our published studies (Yang et al., 2020) (Yang et al., 2018). Briefly, the lentiviral vector (pLVX-EF1alpha-GFP-2xStrep-IRES-Puro) was transfected with lipofectamine into HEK293T cells together with the packaging plasmids pVSV-G and psPAX2 at a molar ratio 2:1:1. The supernatant was collected between 48-96hrs after transfection.
Generation of LDLR gene knockout cells using the CRISPR-Cas9 system
The gene-specific single-guide RNA was biosynthesized by Integrated DNA Technologies, Inc. and cloned into lentiCRISPRv2 vector followed by being co-transfected into HEK293T cells with the packaging plasmids pVSVg and psPAX2 (Ran et al., 2013; Sanjana et al., 2014). Half change9 the medium at 24 hours after transfection, the lentiviral particles in the cell culture media were harvested at 60 hours after transfection and applied to trophoblasts for 48 h. The transduced cells were then selected with puromycin at 1 μg/ml for 5 days until the mock transduced cells were all killed. The knockout efficiency was confirmed by immunoblotting. One of the best knockout candidates was chosen for clonal isolation of cell lines by serial dilutions in 96-well plate. Successful knockout clones were then confirmed by immunoblotting. The wild-type control was lentiCRISPRv2 vector. The sequence of sgRNAs for human LDLR were list as below: Forward: CACCGGACGAGTTTCGCTGCCACGA, Reverse: AAACTCGTGGCAGCGAAACTCGTCC.
Primary neuron culture
Primary neuron culturing was performed according to a previous method (Beaudoin et al., 2012). The brain of a 3-day old neonate was dissected dorsally under a microscope; the vascular tunic was removed, and then the brain was washed in ice-cold DMEM medium. The brain was homogenized by pipetting up and down in 0.125% trypsin with DNAse I, and then digested for 30 min in a 37 °C water bath, vortexing every 10 min. Digestion of the brain was terminated by 10% FBS+DMEM, and the lysate was filtered through a 70μm cell strainer (ThemoFisher brand). The filtrate was centrifuged at 500 x g at 4°C for 5min to pellet down neurons. The neurons were then plated in 0.0125% poly-L-lysine-coated culture plates (overnight at 37°C), and cultured in neurobasal medium (Gibco, U.S) with 2% B27 supplement (Gibco, U.S) at 37°C, 5% CO2 for 5-6 days.
Mouse infection and disease monitoring
VSV stock [∼108 plaque forming units (PFU/ml)] was diluted in sterile phosphate-buffered saline (PBS), and ∼1x107 PFU was injected into mice retro-orbitally (Yardeni et al., 2011). The mice were monitored for disease progression over a period of 8 days after infection. The main symptoms were immobility and hind-limb paralysis. The severity of disease was measured using a scale from 0 (no symptom), 1 (no plantar stepping in one hind leg), 2 (no plantar stepping in two hind legs or slight ankle movement in one hind leg), 3 (slight ankle movement in two hind legs or no ankle movement in one hind leg), 4 (no ankle movement in one hind leg and slight in the other one) to 5 (no ankle movement in both hind legs, almost loss of all movement ability), as described previously (Smith et al., 2013). The mice scored for 5 were euthanized as a humane endpoint according to the animal protocol. For intranasal infection, the mice were inoculated intranasally with 1×106 PFU of VSV per mouse in a 25-μl volume.
Real-time reverse transcription PCR
Homogenized animal tissues or whole blood (30μl) were collected in 350μl of lysis buffer (Invitrogen RNAasy mini-prep kit). The heart, liver, lung, brain and spleen were minced with a pair of scissors and homogenized in RLT buffer using an electrical pellet pestle (Kimble Chase LLC, USA). RNA was extracted following the Invitrogen RNAasy mini-prep kit protocol, and reverse-transcribed into cDNA using the TAKARA PrimeScriptTM RT Reagent Kit with gDNA Eraser (Perfect Real Time) (#RR047A). qPCR was performed with gene-specific primers and Bio-Rad SYBR Premix. qPCR results were calculated using the –ΔΔCt method and beta actin gene as an internal control. The qPCR primers are summarized in Table S2.
Plaque-forming assay
The viral particles in tissue homogenates or cell culture medium were quantitated by plaque forming assays as previously described (Wang, 2016). Briefly, samples were serially diluted by 10-fold using DMEM without FBS, and then 500 μL of each diluted sample plus 500 μL DMEM without FBS were added onto a Vero cell monolayer in a 6-well plate. The 6-well plate was then incubated at 37 °C for 2 h. The inoculum was replaced with 1% SeaPlaque agarose (Cat# 50100, Lonza) in complete DMEM medium (2 ml). The plate was left at room temperature for 30 min to allow the agarose to solidify, and then moved to a cell culture incubator (37 °C, 5% CO2). Plaques were visualized by a Neutral Red exclusion assay after 3 days.
Western blotting assay
Western blotting analysis was performed using standard procedures. Briefly, protein samples were resolved by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and transferred to a nitrocellulose membrane. The membrane blot was incubated with a primary antibody over night at 4°C, washed briefly and incubated with an HRP-conjugated secondary antibody for 1 hour at room temperature. An ultra-sensitive ECL substrate was used for detection (ThermoFisher, Cat# 34095).
Immunofluorescence staining
For immunofluorescence staining, cells or spinal cord tissues were fixed with 4% PFA for 15 min or 12 h, respectively. The cells or frozen section of spinal cord were sequentially permeabilized with 0.3% Triton X-100 in PBS (PBST) for 10 min, blocked with 0.1% BSA plus 1% goat serum in PBST at RT for 1 h, and then incubated with primary antibodies (1:200 dilution in 0.1% BSA of PBST) at 4 °C overnight. Cells were washed 3 times with PBST, and incubated with an Alexa Fluor 488/594-conjugated donkey anti-rabbit/mouse IgG (1:200, ThermoFisher) for 1 h in the dark at RT. Nuclei were stained with DAPI. Images were acquired using a Zeiss 880 confocal microscope (objective 65x oil).
Co-immunoprecipitation
Trophoblasts or HEK293 cells were seeded at a density of 5x105 cells/ml and transfected with protein expression plasmids (pVSVG, 1-80, 51-end, 1-end of FLAG-MSR1, and vector) using TransIT-X2® Transfection Reagent. Twenty-four hours after transfection, whole-cell extracts were prepared from transfected trophoblasts in lysis buffer (150 mM NaCl, 50 mM Tris pH 7.5, 1 mM EDTA, 0.5% NP40, 10% Glycerol) and were incubated with 50μl of anti-FLAG magnetic beads for 2hrs at 4°C. The whole procedure of co-immunoprecipitation was performed according to manufacturer’s protocol (Anti-Flag Magnetic Beads, Sigma-Aldrich). For co-immunoprecipitation of live VSV particles, 5x105 cells/ml HEK293 cells were transfected with protein expression plasmids (1-80, 51-end, 1-end of FLAG-MSR1, and vector). Twenty-four hours later, cells were lysed in lysis buffer and incubated with 50μl of anti-FLAG magnetic beads for 2hrs at 4°C. The beads were washed three times with sterile PBS to completely remove detergent and unbound cellular proteins, and were then incubated with VSV-GFP virions (500μl, 1x107 PFU/ml) for 2hrs at 4°C. The beads were washed with sterile PBS three times to remove unbound virions and the bound virions were eluted with 3xFLAG peptide in sterile PBS. The VSV-GFP pulled down by FLAG-MSR1 and fragments were then used to infect Vero cells or immunoblotting (Figure S3B).
Multiplex ELISA (enzyme-linked immunosorbent assay)
BioLegend’s LEGENDplex™ bead-based immunoassays of serum samples were performed exactly according to the instruction of BioLegend's LEGENDplex™ kit. The flow cytometry data were analyzed using the LEGENDplex™ data analysis software.
Quantification and statistical analysis
The data from all experiments were analyzed using a Prism GraphPad Software and expressed as mean ± S.D. Survival data were analyzed using a Log-rank (Mantel-Cox) test. For in vitro cell culture results, a standard two-tailed unpaired Student’s t-test was used. For animal studies, an unpaired two-tailed nonparametric Mann-Whitney U test was applied to statistical analysis. P values ≤ 0.05 were considered significant. The statistic statements of experiments are detailed in each figure legend.
Acknowledgments
This work was in part supported by a National Institutes of Health grant R01AI132526 and UConn Health startup fund to P.W.
Author contributions
D.Y. designed and performed all the experimental procedures and data analyses. T.L., A.G.H., and T.G. performed and provided technical support and/or data analyses. C.L. contributed to mouse infection studies. P.W. conceived and oversaw the study. D.Y. and P.W. wrote the paper, and all the authors reviewed and/or modified the manuscript.
Declaration of interests
The authors declare no competing interest.
Published: June 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102678.
Supplemental information
References
- Beaudoin G.M., 3rd, Lee S.H., Singh D., Yuan Y., Ng Y.G., Reichardt L.F., Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- Beffert U., Stolt P.C., Herz J. Functions of lipoprotein receptors in neurons. J. Lipid Res. 2004;45:403–409. doi: 10.1194/jlr.R300017-JLR200. [DOI] [PubMed] [Google Scholar]
- Beier K.T., Saunders A., Oldenburg I.A., Miyamichi K., Akhtar N., Luo L., Whelan S.P., Sabatini B., Cepko C.L. Anterograde or retrograde transsynaptic labeling of CNS neurons with vesicular stomatitis virus vectors. Proc. Natl. Acad. Sci. U S A. 2011;108:15414–15419. doi: 10.1073/pnas.1110854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury E.J., McMahon S.B. Spinal cord repair strategies: why do they work? Nat. Rev. Neurosci. 2006;7:644–653. doi: 10.1038/nrn1964. [DOI] [PubMed] [Google Scholar]
- Bukreyev A., Skiadopoulos M.H., Murphy B.R., Collins P.L. Nonsegmented negative-strand viruses as vaccine vectors. J. Virol. 2006;80:10293–10306. doi: 10.1128/JVI.00919-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton J., Neculai D., Grinstein S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013;13:621–634. doi: 10.1038/nri3515. [DOI] [PubMed] [Google Scholar]
- Chauhan V.S., Furr S.R., Sterka D.G., Jr., Nelson D.A., Moerdyk-Schauwecker M., Marriott I., Grdzelishvili V.Z. Vesicular stomatitis virus infects resident cells of the central nervous system and induces replication-dependent inflammatory responses. Virology. 2010;400:187–196. doi: 10.1016/j.virol.2010.01.025. [DOI] [PubMed] [Google Scholar]
- Cheng C., Hu Z., Cao L., Peng C., He Y. The scavenger receptor SCARA1 (CD204) recognizes dead cells through spectrin. J. Biol. Chem. 2019;294:18881–18897. doi: 10.1074/jbc.RA119.010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton K.P., Rose J.K. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology. 2001;279:414–421. doi: 10.1006/viro.2000.0736. [DOI] [PubMed] [Google Scholar]
- Dansako H., Yamane D., Welsch C., McGivern D.R., Hu F., Kato N., Lemon S.M. Class A scavenger receptor 1 (MSR1) restricts hepatitis C virus replication by mediating toll-like receptor 3 recognition of viral RNAs produced in neighboring cells. PLoS Pathog. 2013;9:e1003345. doi: 10.1371/journal.ppat.1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitte-Orr S.J., Collins S.E., Bauer C.M., Bowdish D.M., Mossman K.L. An accessory to the 'Trinity': SR-As are essential pathogen sensors of extracellular dsRNA, mediating entry and leading to subsequent type I IFN responses. PLoS Pathog. 2010;6:e1000829. doi: 10.1371/journal.ppat.1000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felt S.A., Grdzelishvili V.Z. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: a 5-year update. J. Gen. Virol. 2017;98:2895–2911. doi: 10.1099/jgv.0.000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelshtein D., Werman A., Novick D., Barak S., Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. U S A. 2013;110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnidze K., Hajdarovic K.H., Moskalenko M., Karatsoreos I.N., McEwen B.S., Bulloch K. Nuclear receptor REV-ERBα mediates circadian sensitivity to mortality in murine vesicular stomatitis virus-induced encephalitis. Proc. Natl. Acad. Sci. U S A. 2016;113:5730–5735. doi: 10.1073/pnas.1520489113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge P., Tsao J., Schein S., Green T.J., Luo M., Zhou Z.H. Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science. 2010;327:689–693. doi: 10.1126/science.1181766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O'Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie E., Cataldi M., Marriott I., Grdzelishvili V.Z. Understanding and altering cell tropism of vesicular stomatitis virus. Virus Res. 2013;176:16–32. doi: 10.1016/j.virusres.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland D.D., Reiss C.S. Gene expression contributing to recruitment of circulating cells in response to vesicular stomatitis virus infection of the CNS. Viral Immunol. 2006;19:536–545. doi: 10.1089/vim.2006.19.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Józefowski S., Arredouani M., Sulahian T., Kobzik L. Disparate regulation and function of the Class A scavenger receptors SR-AI/II and MARCO. J. Immunol. 2005;175:8032. doi: 10.4049/jimmunol.175.12.8032. [DOI] [PubMed] [Google Scholar]
- Kelley J.L., Ozment T.R., Li C., Schweitzer J.B., Williams D.L. Scavenger receptor-A (CD204): a two-edged sword in health and disease. Crit. Rev. Immunol. 2014;34:241–261. doi: 10.1615/critrevimmunol.2014010267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F.Q., Zhao S.J., Sun P., Liu H., Jie J., Xu T., Xu A.D., Yang Y.Q., Zhu Y., Chen J. Macrophage MSR1 promotes the formation of foamy macrophage and neuronal apoptosis after spinal cord injury. J. Neuroinflammation. 2020;17:62. doi: 10.1186/s12974-020-01735-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyuncu O.O., Hogue I.B., Enquist L.W. Virus infections in the nervous system. Cell Host Microbe. 2013;13:379–393. doi: 10.1016/j.chom.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonte A.C., Sung S.-S.J., Jennelle L.T., Dandekar A.P., Hahn Y.S. Expression of scavenger receptor-AI promotes alternative activation of murine macrophages to limit hepatic inflammation and fibrosis. Hepatology. 2017;65:32–43. doi: 10.1002/hep.28873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M., Yan M., Lu Y., Chen I.S. Retargeting vesicular stomatitis virus glycoprotein pseudotyped lentiviral vectors with enhanced stability by in situ synthesized polymer shell. Hum. Gene Ther. Methods. 2013;24:11–18. doi: 10.1089/hgtb.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K., Zhong X., Ying M., Li L., Tao S., Zhu X., He X., Xu F. A mutant vesicular stomatitis virus with reduced cytotoxicity and enhanced anterograde trans-synaptic efficiency. Mol. Brain. 2020;13:45. doi: 10.1186/s13041-020-00588-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow M., Kortekaas J., Herden C., Hoffmann B., Tappe D., Trebst C., Griffin D.E., Brindle H.E., Solomon T., Brown A.S. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 2016;131:159–184. doi: 10.1007/s00401-015-1511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mátrai J., Chuah M.K.L., VandenDriessche T. Recent advances in lentiviral vector development and applications. Mol. Ther. 2010;18:477–490. doi: 10.1038/mt.2009.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S., Varin A., Chen Y., Liu B., Tryggvason K., Gordon S. SR-A/MARCO-mediated ligand delivery enhances intracellular TLR and NLR function, but ligand scavenging from cell surface limits TLR4 response to pathogens. Blood. 2011;117:1319–1328. doi: 10.1182/blood-2010-03-276733. [DOI] [PubMed] [Google Scholar]
- Nikolic J., Belot L., Raux H., Legrand P. Structural basis for the recognition of LDL-receptor family members by VSV glycoprotein. Nat. Commun. 2018;9:1029. doi: 10.1038/s41467-018-03432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluddemann A., Mukhopadhyay S., Gordon S. Innate immunity to intracellular pathogens: macrophage receptors and responses to microbial entry. Immunol. Rev. 2011;240:11–24. doi: 10.1111/j.1600-065X.2010.00989.x. [DOI] [PubMed] [Google Scholar]
- Pombinho R., Sousa S., Cabanes D. Scavenger receptors: promiscuous players during microbial pathogenesis. Crit. Rev. Microbiol. 2018;44:685–700. doi: 10.1080/1040841X.2018.1493716. [DOI] [PubMed] [Google Scholar]
- Preble O.T., Costello L.E., Huang D.D., Barmada M.A. Neurovirulence mutant of vesicular stomatitis virus with an altered target cell tropism in vivo. Infect. Immun. 1980;29:744–757. doi: 10.1128/iai.29.2.744-757.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz E., Moreno N., Peralta P.H., Tesh R.B. A human case of encephalitis associated with vesicular stomatitis virus (Indiana serotype) infection. Am. J. Trop. Med. Hyg. 1988;39:312–314. doi: 10.4269/ajtmh.1988.39.312. [DOI] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozo-Lopez P., Drolet B.S., Londoño-Renteria B. Vesicular stomatitis virus transmission: a comparison of incriminated vectors. Insects. 2018;9:190. doi: 10.3390/insects9040190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., MacDuff D.A., Imanaka N., Gainey M.D., Shrestha B., Eitson J.L., Mar K.B., Richardson R.B., Ratushny A.V., Litvak V. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P.D., Bell M.T., Puskas F., Meng X., Cleveland J.C., Jr., Weyant M.J., Fullerton D.A., Reece T.B. Preservation of motor function after spinal cord ischemia and reperfusion injury through microglial inhibition. Ann. Thorac. Surg. 2013;95:1647–1653. doi: 10.1016/j.athoracsur.2012.11.075. [DOI] [PubMed] [Google Scholar]
- Steinhoff U., Müller U., Schertler A., Hengartner H., Aguet M., Zinkernagel R.M. Antiviral protection by vesicular stomatitis virus-specific antibodies in alpha/beta interferon receptor-deficient mice. J. Virol. 1995;69:2153–2158. doi: 10.1128/jvi.69.4.2153-2158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl D.F., Abraham N., Knowles S., Marius R., Brasey A., Lichty B.D., Brown E.G., Sonenberg N., Bell J.C. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J. Virol. 2000;74:9580–9585. doi: 10.1128/jvi.74.20.9580-9585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., Kurihara Y., Takeya M., Kamada N., Kataoka M., Jishage K., Ueda O., Sakaguchi H., Higashi T., Suzuki T. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- Tang Y., Li H., Li J., Liu Y., Li Y., Zhou J., Zhou J., Lu X., Zhao W., Hou J. Macrophage scavenger receptor 1 contributes to pathogenesis of fulminant hepatitis via neutrophil-mediated complement activation. J. Hepatol. 2018;68:733–743. doi: 10.1016/j.jhep.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todt J.C., Hu B., Curtis J.L. The scavenger receptor SR-A I/II (CD204) signals via the receptor tyrosine kinase Mertk during apoptotic cell uptake by murine macrophages. J. Leukoc. Biol. 2008;84:510–518. doi: 10.1189/jlb.0307135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trottier M.D., Jr., Palian B.M., Reiss C.S. VSV replication in neurons is inhibited by type I IFN at multiple stages of infection. Virology. 2005;333:215–225. doi: 10.1016/j.virol.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Trottier M.D., Lyles D.S., Reiss C.S. Peripheral, but not central nervous system, type I interferon expression in mice in response to intranasal vesicular stomatitis virus infection. J. Neurovirol. 2007;13:433–445. doi: 10.1080/13550280701460565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. Exploration of west nile virus infection in mouse models. Methods Mol. Biol. 2016;1435:71–81. doi: 10.1007/978-1-4939-3670-0_7. [DOI] [PubMed] [Google Scholar]
- Xu Z., Xu L., Li W., Jin X., Song X., Chen X., Zhu J., Zhou S., Li Y., Zhang W. Innate scavenger receptor-A regulates adaptive T helper cell responses to pathogen infection. Nat. Commun. 2017;8:16035. doi: 10.1038/ncomms16035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Geng T., Yang G., Ma J., Wang L., Ketkar H., Yang D., Lin T., Hwang J., Zhu S. Macrophage scavenger receptor 1 controls Chikungunya virus infection through autophagy in mice. Commun. Biol. 2020;3:556. doi: 10.1038/s42003-020-01285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Wang L., Ketkar H., Ma J., Yang G., Cui S., Geng T., Mordue D.G., Fujimoto T., Cheng G. UBXN3B positively regulates STING-mediated antiviral immune responses. Nat. Commun. 2018;9:2329. doi: 10.1038/s41467-018-04759-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardeni T., Eckhaus M., Morris H.D., Huizing M., Hoogstraten-Miller S. Retro-orbital injections in mice. Lab. Anim. (Ny) 2011;40:155–160. doi: 10.1038/laban0511-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Ha T., Liu L., Wang X., Gao M., Kelley J., Kao R., Williams D., Li C. Scavenger receptor A (SR-A) is required for LPS-induced TLR4 mediated NF-kappaB activation in macrophages. Biochim. Biophys. Acta. 2012;1823:1192–1198. doi: 10.1016/j.bbamcr.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wei Y., Jiang B., Chen L., Bai H., Zhu X., Li X., Zhang H., Yang Q., Ma J. Scavenger receptor A1 prevents metastasis of non–small cell lung cancer via suppression of macrophage serum amyloid A1. Cancer Res. 2017;77:1586. doi: 10.1158/0008-5472.CAN-16-1569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyse any datasets/code.