

SUMMARY
Enzyme-mediated damage repair or mitigation, whilst common for nucleic acids, is rare for proteins. Examples of protein damage are elimination of phosphorylated Ser/Thr to dehydroalanine/dehydrobutyrine (Dha/Dhb) in pathogenesis and aging. Bacterial LanC enzymes use Dha/Dhb to form carbon-sulfur linkages in antimicrobial peptides, but the functions of eukaryotic LanC-like (LanCL) counterparts are unknown. We show that LanCLs catalyze addition of glutathione to Dha/Dhb in proteins, driving irreversible C-glutathionylation. Chemoenzymatic methods were developed to site-selectively incorporate Dha/Dhb at phospho-regulated sites in kinases. In human MAPK-MEK1 such ‘elimination damage’ generated aberrantly activated kinases, which were deactivated by LanCL-mediated C-glutathionylation. Surveys of endogenous proteins bearing damage from elimination (the eliminylome) also suggest it is a source of electrophilic reactivity. LanCLs thus remove these reactive electrophiles and their potentially dysregulatory effects from the proteome. As knockout of LanC2 in mice can result in premature death, repair of this kind of protein damage appears important physiologically.
In brief
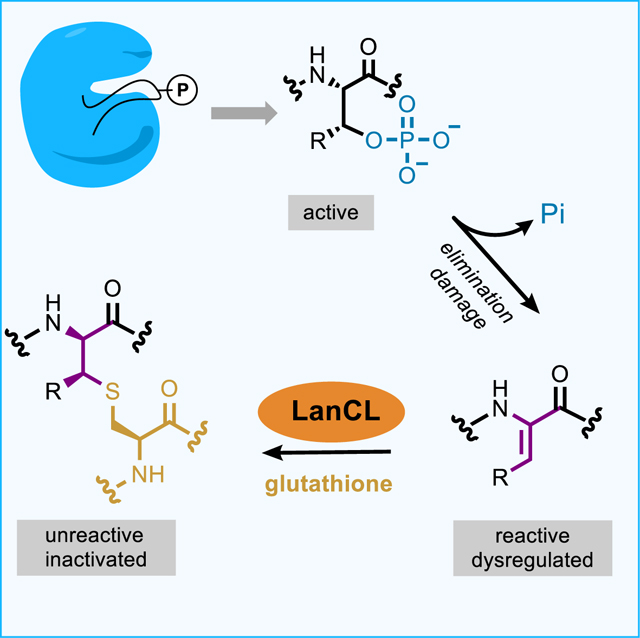
LanCL enzymes respond to sites of protein damage arising from conversion of phosphorylated Ser and Thr residues into highly reactive non-canonical amino acids, catalyzing irreversible capping of the residues with glutathione.
Graphical Abstract
INTRODUCTION
Lanthionine-containing peptides or lanthipeptides are ribosomally synthesized and post-translationally modified peptides produced by bacteria (Repka et al., 2017). Lanthionines are installed through dehydratase-mediated conversion of Ser/Thr residues in precursor peptides to dehydroamino acids. Subsequently, intramolecular addition of thiols of cysteines onto the dehydroamino acid residues is mediated by LanC enzymes or LanC-type domains in bifunctional LanM proteins (Repka et al., 2017). Mammalian genomes encode multiple LanC-like proteins (LanCLs) but lack genes encoding an obvious dehydratase ortholog. The structure of human LanCL1 resembles NisC (a bacterial LanC); both structures contain two-layered α-helix barrels that bind Zn2+ (Figure 1A) (Li et al., 2006; Zhang et al., 2009). LanCL1 and 2 are ubiquitously expressed in various tissues (Bauer et al., 2000; Eley et al., 2002; Mayer et al., 2001b; Mayer et al., 1998), with the highest expression levels in brain, heart and testes, whereas the expression pattern of LanCL3 remains to be determined due to the lack of a specific antibody. Human LanCL1 and LanCL2 bind to GSH and LanCL1 possesses weak glutathione S-transferase activity in assays with a xenobiotic substrate (Chung et al., 2007; Huang et al., 2014; Mladkova et al., 2010; Zhang et al., 2009). However, the endogenous function of LanCL proteins remains unknown.
Figure 1. Structural and pulldown analyses of LanC-type domains and LanCL proteins reveal common architectures and affinity for kinases.

(A) Crystal structures of human LanCLs, bacterial NisC (PDB ID 2G0D), and a LanC-domain in a LanM (CylM; PDB ID 5DZT). (B) Partial sequence alignment of NisC, the cyclase domain of CylM, and human LanCLs. The residues proposed to protonate the enolate intermediate during Michael-type addition to dehydroamino acids are in red. Zinc ion binding residues are in yellow. (C) GSH binding pocket is conserved in LanCL1 and LanCL2 but not LanC/LanMs. The apo structure of Δ1–18-LanCL2 (green sticks) was superimposed with the LanCL1-GSH co-crystal structure (blue sticks; PDB: 3E73). Arg22 is not observed in the electron density of LANCL2. Pink sticks show GSH. (D) Crystal structure of CylM showing interaction of a LanC-type cyclase domain (red) with a kinase-like dehydratase domain (gold). (E) His-pulldown assay with LanCL proteins. Recombinant His-tagged human LanCL1 and LanCL2 show affinity for several cellular kinases from HEK293 cell extracts. For pulldown of endogenous LanCL2 with a subset of these kinases, see Figure S1.
Bacterial LanC catalyze the addition of thiols of Cys to dehydroamino acids. In mammals, dehydroamino acids are thought to be rare, but under certain conditions they are formed in proteins as a result of enzymatic or background non-specific chemical elimination of the phosphate group of phosphoserine/phosphothreonine (pSer/pThr) or of oxidized Cys residues (Cooper et al., 2011). For instance, pathogens like Salmonella or Shigella contain effector proteins as virulence factors that eliminate the phosphate from pSer/pThr in the activation loop of kinases to generate Dha/Dhb (Li et al., 2007; Zhu et al., 2007). In addition to this enzymatic process during infections, non-enzymatic elimination of phosphate is thought to lead to irreversible damage to proteins during aging since Dha/Dhb residues cannot be rephosphorylated (Wang et al., 2014). Because Dha and Dhb are reactive electrophilic residues, they can also lead to protein crosslinking during aging (Linetsky et al., 2004). Collectively, we will call Dha/Dhb-containing proteins in the proteome the ‘eliminylome’.
Scattered clues to the enigmatic function of LanCLs exist. The crystal structure of a bacterial LanM unexpectedly showed structural homology of its dehydratase domain with mammalian kinases. In particular, the adjoining LanC-type cyclase domain in the LanM structure makes intramolecular interactions with the dehydratase domain (Dong et al., 2015). LanCL proteins have previously been shown to interact with kinases such as Akt and mammalian target of rapamycin (mTOR) (Zeng et al., 2014). Based on such inferred domain-domain interaction of kinase-type domains with LanC-type domains, we explored its fuller possible significance in mammalian systems.
Here we report that LanCL proteins catalyze conjugate addition in an intermolecular manner. They activate the Cys thiol of GSH and catalyze C–S bond-forming C-glutathionylation of dehydroamino acid-containing peptides and kinases in their activation loops. Unexpected partial activation of MEK1 by dehydroamino acids in the activation loop and then deactivation by such LanCL ‘scavenging’ identifies a putative role for this reaction in the removal of potentially dysregulatory protein function within the eliminylome. Mice with all three LanCL knocked out (triple knockout – TKO) mostly show no phenotype, but in response to a currently unknown stimulus ~25–30% of them display a striking phenotype of premature death by six months of age.
RESULTS
Structure of LanCL2
The NisC and LanCL1 crystal structures were previously reported (Li et al., 2006; Zhang et al., 2009). In this work we solved the structure of Δ1–18-LanCL2 (PDB ID 6WQ1), which proved more amenable to crystallization than full length protein. Its overall structure resembles that of NisC and LanCL1 and is made up of two layers of 14 α-helices with a zinc ion binding site located on one side (Figure 1A). The Zn2+-binding residues in LanCL2 are Cys321, Cys367, and His368 and corresponding residues are both conserved in NisC and essential for NisC activity (Figure 1B) (Li and van der Donk, 2007; Zhang et al., 2009). Bovine LanCL1 binds GSH (Chung et al., 2007) using five residues: Arg4, Lys317, Arg364, Asp367, Glu374. The corresponding residues in LanCL2 are Arg22, Lys362, Arg409, Asp412, and Glu419. Importantly, these residues are not conserved in bacterial LanC proteins or the LanC domains of LanM proteins but highly conserved in eukaryotic LanCLs suggestive of a common yet distinct and undiscovered function for the eukaryotic LanCLs compared to bacterial LanC/LanMs.
LanCL proteins interact with a range of kinases
Beyond its binding of GSH, LanCL2 interacts with Akt and mTOR (Zeng et al., 2014). This finding was intriguing since in bacterial LanM proteins, the LanC-like domain is part of the same polypeptide as a kinase domain (Figure 1D). We investigated here whether LanCLs also interact with other cellular kinases. We expressed C-terminal His-tagged recombinant human LanCL2 in bacteria and incubated the purified protein with HEK293 cell lysate followed by His-pulldown. As expected, mTOR was detected in the pulldown by Western blotting (Figure 1E). The pulldown samples also contained several other protein and lipid kinases, including ATR (a PIK-related kinase like mTOR), p110α (catalytic subunit of a class I phosphatidylinositol 3-kinase), PIP4K2A (a phosphatidylinositol-5-phosphate 4-kinase), Vps34 (class III phosphatidylinositol 3-kinase), and the Ser/Thr kinases p70-S6K1, ERK1/2 and MEK1/2 (Figure 1E and Figure S1A). On the other hand, glyceraldehyde-3-phosphate dehydrogenase and leucyl-tRNA synthetase were not present in the pulldown samples (Figure 1E). These data suggested that LanCL2 might have a broad binding affinity towards kinases.
We also tested whether LanCL1 demonstrates binding affinity toward kinases. Using His6-LanCL1 pulldown assays, LanCL1 was shown to interact with mTOR, ATR, Akt, p110α PIP4K2A and MEK1/2 but not p70-S6K1 and ERK1/2 (Figure 1E and Figure S1B). The seemingly general interaction of LanCLs with a range of kinases was surprising and somewhat difficult to explain. We also analyzed the pulldown samples by mass spectrometry. Several of the aforementioned kinases were again observed compared to control experiments, but also many non-kinases suggesting LanCLs interact directly or indirectly with a wide range of proteins (Supplemental Table 1).
LanCLs add GSH to dehydroamino acids in peptides corresponding to kinase activation loops
The structural and interaction data suggested that LanCLs bind both GSH and broad protein motifs. Bacterial LanC enzymes catalyze the intramolecular addition of Cys thiols to Dha/Dhb (Li et al., 2006). By extension of mechanistic logic, we investigated whether LanCLs could catalyze intermolecular GSH addition to eliminated (Dha- or Dhb-containing) peptide sequences corresponding to functional motifs (e.g. the activation loops of kinases). Indeed, the possibility that LanCL proteins might catalyze Michael-type additions to dehydroamino acids has been previously, presciently postulated but not experimentally tested (Brennan and Barford, 2009).
The bacterial pathogen pThr lyases SpvC and OspF catalyze phosphate elimination from mitogen activated protein kinase (MAPK) activation loop peptides containing pThr to form the corresponding Dhb-containing peptides (Chambers et al., 2018; Li et al., 2007; Zhu et al., 2007). We therefore used these lyases to mimic such pathogen-induced damage to prepare model, Dhb-containing peptides corresponding to the activation loops of ERK and Akt (Figure 2A; Figure S1C). Following this generation of eliminative damage, we tested LanCL activity. The Dhb-containing ERK peptides (containing either Tyr or pTyr, Figure S1D–F) were reacted with GSH and recombinant LanCLs and analyzed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). Strikingly, a glutathione adduct was observed when the Dhb-ERK peptides were treated with wild-type (wt) LanCL1/2, but not with mutant LanCL1/2 in which residues corresponding in sequence and structure to amino acids that are critical for NisC activity (see below) were mutated (Figure 2B; Figure S1G) (Li and van der Donk, 2007). This observation constitutes not only an experimentally-demonstrated catalytic function for LanCLs but also implicates some of the key catalytic residues responsible (see following section).
Figure 2. Eliminative damage in peptides catalyzed by the pathogen pThr lyases SpvC or OspF and addition of GSH to these eliminated peptides by wt LanCL1 and LanCL2 but not mutants.

(A) MALDI-TOF mass spectra of phospho-ERK peptide treated with His6-SpvC and phospho-Akt peptide treated with His6-OspF resulting in a loss of 98 Da. See Supplemental Table 2 for calculated and observed masses and Figure S1C for fragmentation data. (B) MALDI-TOF mass spectra of Dhb-containing ERK peptide treated with wt LanCL1- and 2 or mutants in the presence of GSH. (C) MALDI-TOF mass spectra of Dhb-containing Akt peptide treated with wt LanCL1 and LanCL2 or mutants. (D) Active sites of LanCL1 bound to GSH (PDB ID 3E73) and the intra-molecular C-S bond forming catalyst NisC (PDB ID 2G0D). (E,F) Loss of catalytic activity upon mutation reveals the importance of the His proposed to protonate the enolate during intramolecular nisin cyclase C–S-bond-forming activity as well as intermolecular LanCL-catalyzed C-glutathionylation. The zinc ion binding Cys residues are also essential for LanCL activity.
Next, we evaluated the substrate tolerance of LanCL1/2-mediated catalysis. The Dhb-containing Akt peptide was incubated with wt LanCL1/2 and again a 307 Da shift corresponding to GSH was observed when compared to negative controls using mutant LanCL1/2 (Figure 2C). Together these data suggested LanCLs are capable of potentially broad ‘trapping’ of eliminated peptide motifs. We also evaluated alternative nucleophiles. The dipeptide γ-glutamyl-Cys and free Cys were both substrates but less efficient than GSH with Dhb-Erk peptide, whereas the dipeptide GlyCys or the reactive Cys-containing protein thioredoxin were not accepted (Figure S1H).
LanCLs use residues conserved in LanC cyclases to catalyze intermolecular C–S-bond-forming C-glutathionylation
His212 in NisC has been proposed to protonate the enolate intermediate generated upon Cys addition to Dha/Dhb (Li and van der Donk, 2007; Yang and van der Donk, 2015) and this residue is conserved in LanCLs (His219 in LanCL1; His264 in LanCL2). Furthermore, the zinc binding residues (Cys284, Cys330, His331) in NisC are conserved in LanCL1 (Cys276, Cys322, His323) and LanCL2 (Cys321, Cys367, His368) (Figure 2D) (Li and van der Donk, 2007; Zhang et al., 2009). The H212N and C330A mutants of NisC cannot produce correctly cyclized nisin (Li and van der Donk, 2007). The corresponding variants LanCL1-H219A and C322A and LanCL2-H264A and C321A/C367A were expressed in E. coli Rosetta 2 (DE3) with N-terminal or C-terminal hexahistidine-tags and purified by affinity chromatography. None of the variants was able to catalyze GSH addition to Dhb-ERK (Figure 2C,E,F). These data suggest that the active site residues have similarly important functions in NisC and LanCL.
Kinases with dehydro amino acids in the activation loop show unexpected activation
Activation by phosphorylation in kinases is attributed to conformational changes in activation loops (Alessi et al., 1994; Gopalbhai et al., 2003; Zheng and Guan, 1994). Given the greater planarity (flattening) at Cα caused by elimination we wondered if this too might act as a source of conformational alteration in kinases. Whilst access to cleanly eliminated peptide motifs from the activation loop of representative kinases could be accomplished enzymatically (see above) we chose to also explore a scaleable chemical method for site-selectively and cleanly installing dehydroamino acids in the activation loops of full-length MAPKKs.
We have previously developed a three-step, one-pot method (involving bis-alkylation/elimination) that chemically converts free Cys residues to Dha using the reagent DBHDA (Figure 3A) (Chalker et al., 2011). We considered applying this strategy to a human kinase identified above as interacting with LanCLs, MEK1. However, MEK1 contains several existing, native Cys residues and, in preliminary experiments, removal of these native Cys created functional alterations and/or expression problems (Wagle et al., 2011; Zhao et al., 2014); these observations therefore prevented direct application of prior methods that relied upon complete removal of native Cys residues.
Figure 3. A strategy for chemical generation of eliminated MEK1 proteins.

(A) Overall sequence for site-selective incorporation of Dha at regulatory Ser sites in kinases. Reaction of the most reactive Cys over less reactive Cys was used to allow chemical, regioselective incorporation of Dha at different sites. SDM, site directed mutagenesis. (B) MEK1, its six native free Cys (black, grey, red) and two activating Ser sites 218 (cyan) and 222 (blue); Table: Predicted side-chain accessibility of sites with/without ‘masking’ nucleotide and Mg(II); (C) Schematic of predicted structural and accessibility effects of ‘masking’ and enhancing ligands; Cys207 is blocked by ATPγS; 218 & 222 are more accessible with Mg(II). See also Figure S2.
We therefore considered a more challenging approach based on regio-selective chemical conversion of target Cys residues to Dha in the presence of other non-target native Cys residues. Current chemistries for targeting one free Cys residue in proteins amongst other Cys are rare; they require engineering (Dai et al., 2016) or exploitation of privileged arrangements of residues that were not applicable in MEK1 (Willwacher et al., 2016). In principle, however, the different protein environments around each Cys residue could allow direct, chemical differentiation. In the three-step conversion of Cys to Dha (Figure 3A, inset), the first irreversible alkylation step is rate- and hence regio-selectivity determining. We reasoned therefore that reagent tuning (without losing reactivity in steps 2–3) could allow the site-to-site control required for this chemical process.
Wild-type MEK1 contains six free, native Cys residues at positions 121, 142, 207, 277, 341, and 376. To discriminate potential reactivities of these residues, relative side-chain accessibility was estimated using a predictive computational approach (Hubbard and Thornton, 1993) based on accessible surfaces determined from x-ray crystal structures (Figure 3B) (Lee and Richards, 1971). A similar approach has proven successful previously for predicting chemical regioselectivity in other protein modifications (van Kasteren et al., 2007). This analysis, when applied to the apo-MEK1 structure (Figure 3B), predicted that a MEK1 mutant in which Cys was introduced at activation loop phosphorylation sites 218 and/or 222 (and removed from non-functional sites 277 and 376) would allow regioselective elimination. Importantly, when expressed, the MEK1-C277S/C376S mutant kinase displayed essentially identical enzymatic properties to wt MEK1 (Supplemental Table 2; STAR Methods). The same predictive analysis of reactivity (based on analyses of binary (MEK1•ATPγS [PDB 3W8Q]) or ternary (MEK1•ATPγS•Mg [PDB 3EQD] (Fischmann et al., 2009) and MEK1•ADP•Mg [PDB 3EQI]) complexes) as well as prior studies (Lamoureaux and Lee, 2011) also suggested the additional consideration of nucleotides (ATPγS, ATP, ADP) and metals (Mg(II)) as masking or enhancing ligands to usefully enhance the selectivity of this chemistry (Figure 3C and STAR methods).
In this way, we designed and generated three mutant variants of MEK1 for testing with this regioselective elimination chemistry: MEK1-C218, MEK1-C222, and MEK1-C218C222 (bearing five, five or six Cys residues, respectively – see STAR methods for full mutational patterns used to remove and introduce Cys). Next, we tuned a panel of systematically-varied 1,4-bis-alkylating reagents derived from parent reagent DBHDA (Figure S2A). These varied in their reactivity, hydrophobicity, sterics and charge whilst all retaining the ability to create 5-membered ring sulfonium intermediates that are critical to elimination of the side chain of the Cys residues introduced (Chalker et al., 2011). Finally, consistent with our design predictions, use of these reagents in combination with masking ligands and metals (see STAR methods) allowed clean access (avoiding competing pathways, Figure 4) to different MEK1 kinases containing complete elimination at different sites in their activation loops: MEK1-Dha222, MEK1-Dha218 and MEK1-Dha218Dha222 (Figure 4A–C, respectively; Figures S2–S5).
Figure 4. Reagent-controlled, regioselective, chemical elimination reactions of MEK1 allow single site and double site elimination.

Switching of reagents and conditions selectively avoids unwanted, competing chemical pathways (dotted) and yields clean chemical elimination (bold) to desired proteins: (A) MEK1-Dha222 (B) MEK1-Dha218, or (C) MEK1-Dha218Dha222. Intact protein LC-ESI-MS shown; see Supplemental Table 2 for calculated and observed masses, Figure S2 for structures of reagents, and Figures S3–S5 for all data.
The enzymatic activities of these ‘eliminated’ MEK1 variants on the native substrate protein ERK (both mono- (kf1) and di-phosphorylation (kf2)) were assessed directly by real time monitoring using total protein ESI-MS (Figure S6). The ERK1-K71R mutant that cannot autophosphorylate (Robbins et al., 1993) was utilized to allow cleaner analysis of ERK by MS. Strikingly, all Dha-containing MEK1 variants showed activated phosphorylation kinetics over wt MEK1. Moreover, His6-MEK1-Dha218 and His6-MEK1-Dha218Dha222 displayed similar or even higher activity than ‘constitutively active’ MEK1-Ser→Glu mutants (Supplemental Table 2; STAR Methods). Notably, despite different behaviour as a kinase, elimination in an alternative construct His6-Δ1–60-MEK1-Dha218 (which displays altered selectivity due to deletion of the ERK-docking domain (Xu et al., 1999)) also caused significant activation, suggesting a general function of manipulation of the activation loop of MEK1 and not peculiar artefacts of one kinase structure. Notably, His6-MEK1-S218A and His6-MEK1-S222A variants of MEK1 are inactive (Alessi et al., 1994) although they differ only from the activated His6-MEK1-Dha218 and His6-MEK1-Dha222 in unsaturation. This precisely implicates the presence of the sp2 centre at the α-carbon of Dha218 and Dha222 in this activation, thereby highlighting the eliminylome as a putative source of unwanted dysregulation through activation. This reasoning is also consistent with the greater effect seen at the α-helical site 218 (c.f. site 222, which is in a flexible loop). Any such conformational effects were too localized to be detected by global measures, such as circular dichroism (Figure S5F).
LanCLs add GSH to dehydroamino acids in representative kinases and deactivate dysregulated MEK1
Next, we tested LanCL activity against full-length MEK1 proteins. First, we investigated the interaction between LanCL proteins and dehydroamino acid containing MEK1. MBP-tagged LanCL2 was expressed in bacterial cells and purified. The MBP-tagged LanCL2 was incubated with His6-MEK1-Dha218 and pulled down by cobalt resin. The pulldown assay shows that LanCL2 has a direct physical interaction with His6-MEK1-Dha218 (Figure 5A).
Figure 5. LanCL proteins catalyze non-canonical C-glutathionylation of dehydroamino acid containing proteins.

(A) In vitro binding analysis of MBP-LanCL2 with His6-MEK1-Dha218 by His-pulldown assay. (B) LanCL1 and LanCL2 (2 μM) C-glutathionylate His6-MEK1-Dha218. The conversions of Dha-MEK1 (2 μM) to GS-MEK1 after 40-min incubation with 1.0 mM GSH at 25 °C (Dha-MEK1: LanCL = 1:1) are shown as deconvoluted LC mass spectra. (C) Time course of GSH (2.5 mM) addition to MEK1-Dha218 (6 μM) catalyzed by LanCL1/2 (1.2 μM). The formation of GS-MEK1 was monitored according to the intensities of each product from deconvoluted spectra on LCMS. (D) Pseudo-single substrate Michaelis-Menten plot in the presence of LanCL1 or LanCL2 (Dha-MEK1, 6 μM; LanCL1/2, 1.2 μM). All kinetic assays were performed in triplicate. Initial reaction velocity at different GSH concentrations was calculated by linear regression fit to the Michaelis-Menten equation of the formation of GS-MEK1 over time using OriginPro9.7 for apparent kinetic parameters KM and kcat. For spectra and data with Dhb-Erk2 see Fig. S7; for kinase activity of Dha- and GS-MEK1, see Fig. S6.
Second, we investigated C-glutathionylation activity. The Dha218-containing MEK1 was treated with wt LanCL1/2 and GSH, and the reactions were monitored by MS. A GSH adduct was observed, whereas no reactions occurred with LanCL mutants (Figure 5B). Kinetic analyses using intact protein MS were performed in the presence of increasing amounts of GSH, and initial rates of forming glutathione conjugate were plotted against substrate concentration. The data demonstrated that C-glutathionylation of His6-MEK1-Dha218 was catalyzed by LanCL1 and LanCL2, with LanCL1 exhibiting a higher catalytic activity than LanCL2 (Figure 5C and 5D; Figure S6). Pseudo-single substrate plots of initial reaction velocity versus GSH concentration and non-linear Michaelis-Menten regression analyses (Figure 5D) provided apparent KM and kcat values (Table 1).
Table 1.
Kinetic parameters of addition of GSH to various substrates catalyzed by LanCL1/2.
| Substrate | Enzyme | KM (μM) | kcat (min−1) | Vmax (μM/min) |
|---|---|---|---|---|
| MEK-Dha218 | LanCL1 | 28.1 ± 6.1a | 0.15 ± 0.01a | 0.18 ± 0.01a |
| MEK-Dha218 | LanCL2 | 63.4 ± 10.0a | 0.021 ± 0.002a | 0.025 ± 0.002a |
| Dhb-ERK peptide | LanCL1 | 115 ± 21b | 7.7 ± 0.5 | 3.9 ± 0.3 |
| Dhb-ERK peptide | LanCL2 | 210 ± 75b | 10.1 ± 1.9 | 5.0 ± 1.0 |
| Dhb-CylLs” | LanCL2 | 250 ± 51b | 11.8 ± 1.2 | 5.9 ± 0.6 |
Apparent Km,GSH and kcat values at fixed [MEK-Dha218] of 6.0 μM.
Km,peptides at 1 mM GSH. The error bars are derived from fits of the data from triplicate experiments by global least-square regression to solutions of the Michaelis Menten equation using OriginPro9.7.
Third, to test other targets, we also investigated the addition of GSH to Dha-containing ERK (chemically-generated in a similar manner to that used for eliminated MEK1 – see STAR Methods). GSH addition experiments were conducted using Erk1-Dha202. Compared with controls (background and mutant LanCL proteins), higher percentages of ERK1-GS202 adducts were formed in samples treated with wt LanCL1 or LanCL2 (Figure S7). Some background reactivity of Erk1-Dha202 was observed, and therefore we also generated the Dhb version of ERK2 (ERK2-Dhb185) by phosphorylating ERK2 using MEK-Dha218Dha222 and then using the pThr lyase OspF to eliminate the pThr (see STAR Methods). Dhb-containing peptides and proteins showed negligible background reaction with GSH, but LanCL2 again catalyzed addition of GSH to ERK2-Dhb185 as demonstrated by MS and Western blot analysis (Figure S1J,S7C). Interestingly, a doubly eliminated variant of ERK1 (ERK1-Dha82Dha202, created in essentially the same manner as for ERK1-Dha202) was C-glutathionylated once by LanCL2 with essentially the same kinetics as ERK1-Dha202, suggesting that LanCL selectively addresses elimination at the natural phosphosite 202 but not when found at non-phosphorylated site 82. Taken together, our data demonstrate that LanCL proteins not only catalyze GSH addition to Dhb-containing peptides but also catalyze glutathionylation of Dha/Dhb-containing MEK1 and ERK1/2.
The observed LanCL activity generates a carbon-sulfur thioether bond between GSH and the protein (C-glutathionylation) as opposed to the well-known (reversible) S-glutathionylation of Cys residues via disulfide linkages (Cooper et al., 2011; Dalle-Donne et al., 2009; Grek et al., 2013; Yang et al., 2016). As such, the non-canonical glutathionylation activity demonstrated here produces an irreversible modification of dehydroamino acids in proteins (Cooper et al., 2011).
Notably, the GSH-adducts of MEK1 showed reduced kinase activity compared to their Dha-containing counterparts (Supplemental Table 2) suggesting the possibility not only of an effective scavenging/trapping pathway to remove reactive electrophiles, but also one that directly reduces kinase activity dysregulation caused by elimination (see above).
LanCL proteins have plastic but selective substrate specificity
Based on the data with the ERK and Akt peptides and the MEK and ERK proteins, LanCLs appear to have considerable substrate tolerance with respect to the sequences in which the dehydroamino acids can be embedded. To further investigate the substrate scope, we assessed the reactivity of several bacterial peptides with LanCL2. We first focused on a prochlorosin peptide (Li et al., 2010) (ProcA 2.8-mut, Figure 6A) that contains a single Dha and reacted it with wt or mutant LanCL2 proteins. MALDI-TOF MS analysis demonstrated glutathionylation with wt LanCL2 protein but not with mutant LanCL2 (Figure 6A). Next, we used the cytolysin peptides (CylLL” and CylLS”) that are virulence factors in Enterococcus faecalis and that contain several dehydroamino acids and lanthionine rings (Figure 6B) (Duan et al., 2019; Tang and van der Donk, 2013). CylLS” contains two lanthionine rings and two Dhb residues. LanCL2 added one GSH whereas no adduct was formed by a LanCL2 mutant (Figure 6B). MS/MS analysis showed that the glutathionylation site on CylLS” was Dhb7 that is outside of the lanthionine rings (Figure S1K). CylLL” contains a Dha and Dhb located within rings and two Dhb residues that are outside of rings (Figure 6C). LanCL2 catalyzed glutathionylation of the two Dhb residues located outside of lanthionine rings (Dhb10 and Dhb30; Figure 6C; Figure S1K). Lanthionine rings might prevent LanCL2 from adding GSH to dehydroamino acid residues within rings through steric hindrance.
Figure 6. LanCL-catalyzed C-glutathionylation shows flexible substrate selectivity, high stereoselectivity, is the major C-glutathionylation activity in cells, and under certain circumstances the absence of LanCL proteins leads to lethality.

(A, B, C) LanCL2, but not an active site variant, adds GSH to (A) ProcA 2.8 mut, (B) CylLS”, and (C) CylLL”. Eliminated (Dha or Dhb) residues are highlighted in red; Cys residues involved in thioether linkages are in green. (D) Comparative activity of LanCL2-His6 and His6-GSTA4 with Dhb-containing ERK peptide. (E) Non-canonical glutathionylation was observed with His6-MEK1-Dha222 when incubated with wt MEF cell extracts. No glutathionylation was observed when His6-MEK1-Dha222 was treated with cell extract of LanCL1–3 triple knockout (TKO) MEF cells. (F) Structure of DL- and LL-MeLan. (G) GC-MS analysis of the methyllanthionine in glutathionylated Dhb-ERK peptide. Deep blue: DL-MeLan synthetic standard; Red: LL-MeLan synthetic standard; Cyan: derivatized residue from GS-ERK peptide; Co-injection of derivatized MeLan from the GS-ERK peptide with DL standard (orange) or LL standard (pink). (H) Kaplan-Meier survival curve of WT (n = 58) and LanCL TKO (n = 47) FVB mice. Censored individual mice are shown as a tick mark (see STAR methods). Gehan-Breslow-Wilcoxon test showed a P value of 0.0004. Statistical significance was accepted at P < 0.05.
We also investigated other dehydroamino acid-containing proteins. Glutathione peroxidase (GPX) contains a selenocysteine in its active site that is important for catalysis but is prone to oxidative elimination generating Dha (Cho et al., 2010; Ma et al., 2003; Wang et al., 2011). LanCL1/2 were unable to add GSH to the Dha-containing GPX (Fig. S1I), presumably because the Dha is in a protected active site pocket.
Mammalian systems contain multiple glutathione S-transferases (Wu and Dong, 2012). The glutathione transferase A4 (GSTA4) has been demonstrated to catalyze GSH addition to another α,β-unsaturated compound, 4-hydroxynonenal (Bruns et al., 1999; Hubatsch et al., 1998). Because GSTA4 catalyzes Michael-type addition, we tested its activity with Dhb-containing ERK peptide in parallel with LanCL2 and the reaction mixtures were monitored by MALDI-TOF MS. GSH addition was observed with LanCL2-His6 treatment, however, no glutathionylation was observed with His6-GSTA4 (Figure 6D). These data suggest that LanCL proteins might be specialized in catalyzing GSH addition to eliminated (Dha- and Dhb-containing) peptides or proteins.
To test if LanCLs have substrate preference, we compared the kinetics of LanCL2 with Dhb-ERK peptide and CylLS”. Two-substrate steady-state kinetic analysis was performed with saturating GSH at a concentration similar to that found in cells (1 mM) (Meister and Anderson, 1983). The production of glutathionylated product was monitored and quantified by LC/MS. The KM and kcat values observed for the Dhb-ERK and CylLS” substrates are similar (Table 1) suggesting that LanCL2 does not have a clear sequence preference and Dha/Dhb might serve as the recognition feature. LanCL1 displayed similar kinetic parameters as LanCL2 with the Dhb-ERK peptide (Table 1).
Other cellular proteins do not add glutathione to dehydroamino acids
To test if endogenous LanCL adds GSH to MEK1-Dha222, the latter protein was incubated with cell lysates from wt or LanCL1–3 TKO mouse embryonic fibroblast (MEF) cells (He et al., 2017). The primary MEF cells were isolated and cultured as described in the STAR Methods, and expression of LanCL1 and LanCL2 in wt MEF cells but not TKO MEF cells was observed by Western blotting (Figure 6E). Equal numbers of wt and TKO MEF cells were collected, lysed, and incubated with His6-MEK1-Dha222. Cobalt-bound resin was used to enrich the His-tagged MEK1 protein, and the protein was glutathionylated in the presence of wt MEF cell extract but not in the presence of TKO MEF cell extract as detected by Western blot with an anti-GSH antibody (Figure 6E and Figures S1J, S1L). These data suggest that endogenous LanCL proteins are the major, if not only, enzymes that can catalyze glutathione addition to His6-MEK1-Dha222 in cells.
Glutathione addition by LanCL is stereoselective
Methyllanthione (MeLan) formed by LanC enzymes typically have the DL-configuration (Figure 6F) (Chatterjee et al., 2005). The α−carbon from the former Thr residue possesses the D-configuration whereas the α−carbon on the Cys retains the L-configuration. In order to determine the stereochemistry of the GSH adduct generated by LanCL2, the glutathionylated ERK peptide was hydrolyzed and derivatized. The derivatized sample was analyzed by gas chromatography-mass spectrometry (GC/MS) with a chiral stationary phase. The MeLan residue in the glutathionylated peptide has the (2S, 3S, 6R) configuration as shown by coinjections with synthetic MeLan standards (Liu et al., 2011; Tang and van der Donk, 2012) (Figure 6G). The data indicate that LanCL2 catalyzes stereoselective GSH addition to Dhb forming a D-stereocenter, which may contribute to the aforementioned deactivation of MEK1-Dha218 upon glutathionylation by creating non-natural stereochemistry.
Survey of the eliminylome from proteomics databases and MEF cells
To examine the extent of the kinase-associated eliminylome we conducted analyses of published proteomics data on human cell kinomes (Médard et al., 2015) (Supplemental Table 3). These analyses confirmed that Dha and Dhb are detected in many proteins. Amongst these are at least 30 protein kinases (MAPK1, MAPK3, MAPK8, MAPK9, MAPK10, MAPK14, MAP2K3/6, MAP3K19, MAP4K5, PRKAA1, CAMK2B, CAMK2D, CAMK2G, PRKX/Y, CDK5, PTK2, PTK2B, GSK3B, Src, Akt1/2, Ribosomal protein S6 kinase, Stk3, MRCKβ, N1, Nek9, TBK1, CSK, Fer, Fes, Lck, Lyn). About ~50% of these Dha/Dhb sites map to corresponding Ser/Thr sites that have been observed to be phosphorylated (including MAPK1, MAPK14, MAPK3, MAP3K19, MAP2K3/6, CAMK2D, PTK2, Ribosomal protein S6 kinase, MRCKβ, N1, Nek9, TBK1, Lck, Lyn). Another smaller number of Dha and Dha-adducts detected in the proteomic databases are at Cys sites suggesting that elimination also takes place at these residues (Supplemental Table 3).
We attempted to detect dehydroamino acid-containing proteins by incubating lysates of TKO and wt MEF cells with GSH and LanCL1/2, and conducting Western blot analysis with anti-GSH antibodies. No clear differences were observed. We attribute the absence of an abundance of dehydroamino acid containing proteins in the TKO MEF cells to the expected low rate and hence low occurrence of elimination as well as the relatively short timepoints (days) of these cell-based assays. Therefore, the eliminylome is not anticipated to be highly abundant and catalytic effects due to misregulation of enzymatic signalling activity such as shown for MEK1 may be more detrimental than stoichiometric effects through the formation of electrophiles in the proteome.
LanCL1/2/3 TKO mice are at risk of premature death
We previously reported that TKO mice did not display any obvious phenotype (He et al., 2017). However, at two different time periods separated by several years (see STAR methods), we observed a strikingly similar and prominent phenotype for LanCL KO mice. In both cases, roughly 25–30% of KO mice died prematurely at ages between 2 and 6 months whereas age-matched wt mice that were co-housed with the KO mice did not display such a phenotype (Figures 6H and S7D&E). The KO mice that did not die by 6 months did not display any further phenotype. The TKO mice that died were submitted for histopathology analysis but no common underlying abnormalities were detected that could explain the premature death (see STAR methods). In addition, heart and brain tissue of mice that died prematurely as well as age- and gender matched wt and surviving TKO mice were incubated with LanCL1/2 and GSH and analyzed by Western blot for glutathionylated proteins. No differences were observed for the three populations. Thus, the cause of death is not known and neither is the stimulus that causes the highly similar phenotype at two different times (2011 and 2020), but not in the intervening period. However, the premature death of a large number of LanCL KO mice (Figures 6H and S7D,E) clearly demonstrates that under certain circumstances the absence of these proteins causes lethality.
DISCUSSION
LanCL proteins were first identified two decades ago (Bauer et al., 2000; Mayer et al., 2001b; Mayer et al., 1998), but their activity remained unknown. The possibility that these proteins might be responsible for generation of lanthionine in the brain was recently ruled out (He et al., 2017). In this study, we uncovered a non-canonical glutathionylation activity of LanCL proteins. Pathogen effector pThr lyases were utilized to generate dehydroamino acid containing peptides that mimic the activation loop of ERK and Akt kinases. With these peptides, LanCL proteins added GSH to the Dhb residues, which are much less electrophilic than Dha residues and do not show appreciable non-enzymatic background reactivity with GSH. Importantly, the glutathionylation activity results in a thioether-linked structure as opposed to the well known disulfide linked S-glutathionylation generated in response to oxidative stress (Cooper et al., 2011; Dalle-Donne et al., 2009; Grek et al., 2013; Yang et al., 2016).
Additionally, Dha-containing MEK and ERK were generated using a chemical mutagenesis strategy and glutathionylation activity was observed when treating Dha-MEK1 and Dha-ERK1 with LanCL1/2. These data potentially explain why LanCL proteins have affinities toward many different cellular kinases in mammalian systems (Figure 1E), but unbiased proteomic analysis suggests LanCL1/2 interact with many other proteins that are not kinases. Consistent with a potentially wide array of substrates, LanCL proteins appear tolerant of the protein sequence in which the dehydroamino acids occur as GSH was added to both mammalian and bacterial substrates. Collectively, these findings potentially indicate that LanCL proteins might act on a variety of dehydroamino acid-containing proteins, which may be formed by damage to phosphorylated proteins as well as proteins that undergo eliminative damage at Cys. Our data also show that His6-MEK1-Dha222 is glutathionylated by wt MEF cell extracts but not by triple LanCL knockout MEF cell extracts. These data imply that no other GSH activating proteins can catalyze non-canonical C-glutathionylation of Dha-MEK1 in these cells. Importantly, the glutathione-conjugated MEK1 proteins lost most of the activity present in Dha-containing MEK1.
Overall, our study provides insights into the longstanding puzzle of LanCL protein activities. Glutathione adducts have been observed previously in cells with long-lived proteins in tissues such as the human lens, cardiac myocytes, and neurons, but were believed to be formed non-enzymatically (Cooper et al., 2011; Friedman, 1999; Linetsky et al., 2004; Linetsky and LeGrand, 2005; Wang et al., 2014). Importantly, these adducts are found at sites that are phosphorylated during normal physiology and the dehydroamino acids are thought to be formed by non-enzymatic phosphate elimination (Wang et al., 2014). Intriguingly, LanCL levels in eyes, brain, cardiac myocytes and neurons are elevated at the transcriptomic and proteomic level (Forrest et al., 2014; Geiger et al., 2013; Huttlin et al., 2010; Kawaji et al., 2017; Kim et al., 2014; Mayer et al., 2001a; Mayer et al., 2001b), providing a potential link between tissue distribution of the enzyme and substrate presence. Whereas non-enzymatic GSH addition to Dha has a detectable background rate (Figure 5), non-enzymatic addition to Dhb is not detected under physiological conditions. Therefore, the GSH adducts observed in proteomic studies at positions that are known Thr phosphorylation sites (Wang et al., 2014) would very likely require enzymatic catalysis. The data with the cell extracts of wt and TKO mouse MEF cells demonstrate that endogenous LanCL, but not other proteins, can catalyze addition of GSH to Dha-MEK1. The high sequence tolerance with respect to dehydroamino acid containing peptides/proteins accepted as substrates by LanCL1/2 would ensure that electrophilic sites in the form of dehydroamino acids present in diverse proteins could be removed. It is possible that LanCL1–3 will have substrate selectivity in cells and organisms and indeed such selectivity is observed in vitro (e.g. Figure 5C).
In addition to the glutathione adducts that have been detected previously, protein cross-links such as lanthionine (Lan), histidinoalanine (HAL) and lysinoalanine (LAL) have been identified in various settings including the human lens and the abundance of these cross-links are increased in aged human cataractous lens (Friedman, 1999; Linetsky et al., 2004). These crosslinks are thought to be formed by nucleophilic addition of the side-chains of Cys, His, and Lys to Dha and Dhb generated in turn by phosphate elimination of pSer and pThr (Linetsky et al., 2004; Wang et al., 2014). It has been predicted that dehydroamino acids are likely generated in other tissues as well by β-elimination of activated Ser/Thr/Cys residues (Cooper et al., 2011), leading to the suggestion that an eliminylome may be present in cells (Brennan and Barford, 2009). Indeed, our initial survey of proteomics data suggests that elimination at known Cys and phosphorylated Ser sites to Dha and phosphorylated Thr sites to Dhb may be quite common. It would be surprising if mammals would not have evolved a protection mechanism to prevent protein cross-linking and irreversible kinase (in)activation involving dehydroamino acids. Non-canonical glutathionylation catalyzed by LanCL proteins might serve this function. Glutathionylation does not repair eliminated proteins, unlike for instance the repair methyltransferases that convert isoAsp back to Asp (McFadden and Clarke, 1987). But LanCL proteins would provide containment of the eliminylome. The importance of such containment is implied by the phenotype of LanCL KO mice. At present we do not know why the striking premature death phenotype is not observed continuously and suspect that a currently unknown environmental factor likely induces the phenotype when in a LanCL(−) background. The absence of a clear pathological cause of death makes it challenging to identify the molecular mechanism especially since a potential misregulated enzyme such as that shown for MEK1 could be formed in very small amounts, and yet, due to its catalytic signalling potential, have major and/or amplified effects. Future studies will need to investigate the many lines of research that may be able to provide more insights for such transient and/or amplified events.
Notably, the existence of an eliminylome was previously suggested to be part of a regulated suppressing mechanism (Brennan and Barford, 2009). Here we reveal that it can be part of an activating mechanism. Given the natural occurrence of elimination and the existence of pathogens that utilize virulence-determining enzymes that drive phosphate elimination at kinase activation sites, we speculate that such elimination processes might have a further, dysregulatory effect in addition to primary, dynamic phosphorylation-dephosphoryation. Conflicting observations have been made on the effect of the pathogen effector protein OspF upon host kinase activity: whilst some suggest decreased activity (Arbibe et al., 2007), other studies suggest that OspF instead drives higher kinase activities (Zurawski et al., 2006). Similarly, in the phosphoproteomes of Shigella-infected human epithelial cells both decreases and increases of global phosphorylation levels have been reported (Schmutz et al., 2013). The basis of the previously unexplained activation of the MEK/ERK pathway in cellulo following Shigella infection (Zurawski et al., 2009) might be explained by effects similar to the elimination-activation effects that we demonstrate in vitro for MEK1. Kinases that would be activated by phosphate elimination would no longer be sensitive to dephosphorylation by phosphatases and therefore a subsequent glutathionylation event would be essential for preventing dysregulation.
Limitations of the Study
The current study demonstrates that LanCL1 and −2 add GSH to dehydro amino acids in peptides and proteins. We cannot rule out that these enzymes also have activity with other electrophilic substrates. Our data show that elimination of phosphate in the activation loop of MEK1 leads to activation of its kinase activity but the generality of this effect in other enzyme contexts needs to be investigated. The premature death phenotype of TKO mice appears triggered by one or more currently unknown factor(s) that need to be identified. We propose that the phenotype is caused by dysregulation caused by eliminative damage to one or more unknown proteins, but this needs to be confirmed in future studies.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wilfred van der Donk (vddonk@illinois.edu).
Materials Availability
Plasmids generated in this study for preparation of LanCL1/2, their variants, and their protein substrates were deposited to Addgene.
Data and Code Availability
The published article includes all datasets analyzed during this study. Structural data has been deposited in the PDB under ID 6WQ1. Mass spectral data and Western blot data have been deposited at Mendeley Data, http://dx.doi.org/10.17632/sgjzwcxgsr.1 and at https://ora.ox.ac.uk/objects/uuid:d8729aad-ca98–4f06-b14a-0a75ea306584.
EXPERIMENTAL MODEL DETAILS
Cell culture and primary cell isolation
Human embryonic kidney (HEK) 293 cells and primary mouse embryonic fibroblasts (MEFs) were cultured in Dulbecco’s Modified Eagle Media (DMEM) with 10% (v/v) fetal bovine serum (FBS) (Gibco) and 1X penicillin/streptomycin (final concentration for penicillin: 100 unit/mL, streptomycin: 100 μg/mL) (DMEM complete media). MEFs were isolated from 13-day mouse embryos. In brief, the mouse embryos were harvested and rinsed with 70% ethanol. The embryos were transferred into PBS buffer and organs were removed for each embryo. After all embryos were dissected, the tissues were minced with a scalpel and then incubated with 0.25% trypsin-EDTA (Thermo Fisher) at 37 °C for 15 min. Trypsin was inactivated by adding DMEM complete media. The isolated cells were re-suspended in DMEM complete media and cultured in T-75 flasks. LanCL1/2/3 triple knockout (TKO) MEF cells were obtained from TKO mice (He et al., 2017).
Animal husbandry
All animal strains were generated in a FVB background (He et al., 2017). All animal experiments in this study followed protocols approved by the Animal Care and Use Committee at the University of Illinois at Urbana-Champaign. FVB/NJ mice were purchased from the Jackson Laboratory (Stock Number #001800). LanCL1–3 TKO FVB mice were generated at the University of Illinois at Urbana Champaign. Neither wt nor TKO FVB mice were involved in previous procedures and were drug and test naïve. All mice were housed and maintained in the Institute for Genomic Biology on a 12 h/12 h light/dark cycle with access to clean water and food (2018 Teklad global 18% protein rodent diet). No special treatment was given to experimental animals. Sick animals were given moist food by mixing their diet with clean water.
Bacterial growth conditions for protein expression
His6-SpvC, His6-OspF, His6-LanCL1, His6-LanCL1-H219, His6-LanCL1-C322A, LanCL2-His6, LanCL2-His6-H264A, LanCL2-His6-C321A/C367A and MBP-LanCL2 were expressed in E. coli Rosetta 2 (DE3) cells as either His6- or maltose binding protein (MBP) fusion proteins. Cells were grown in LB media with appropriate antibiotics (final concentrations: ampicillin, 100 μg/mL; chloramphenicol, 25 μg/mL) until OD600 reached 0.7. Cells were then induced with 0.2 mM IPTG after cooling on ice and incubated at 18 °C for 18 h unless otherwise noted. Cells were isolated by centrifugation and stored at −80 °C until protein purification.
The His6-ERK2-MEK1-R4F plasmid was expressed in E. coli T7 lysY cells (New England Biolabs). Cells were grown in LB media with appropriate antibiotics (final concentrations: ampicillin, 100 μg/mL; chloramphenicol, 25 μg/mL) until the OD600 reached 0.7. Cells were then induced with 0.5 mM IPTG after cooling on ice and incubated at 18 °C for 20 h. Cells were isolated by centrifugation and stored at −80 °C until protein purification.
The mutant plasmids MEK-C218 and MEK-C222 were used to transform E. coli BL21 (DE3) competent cells and expressed as N-terminal His6-fusion proteins. Cells were grown in LB media with 50 μg/mL kanamycin and induced with 1 mM IPTG when the OD600 reached 0.6. The incubation was continued for another 12–16 h at 25 °C. Cells were isolated by centrifugation and stored at −80 °C until protein purification.
METHOD DETAILS
Plasmids
The gene encoding LanCL1 was cloned into pET15b using XhoI and NdeI cloning sites (pET-15b-LanCL1; Addgene 154189). For bacterial expression of C-terminal His-tagged LanCL2, the human LanCL2 cDNA was subcloned into pETDuet-1 using NcoI and NotI cloning sites in multiple cloning site I (MCSI; pETDuet1-WT-LanCL2-C-Ter-His; Addgene 154186). The gene encoding LanCL2 was cloned into pMAL-p2x vector using BamHI and SalI sites (pMAL-p2x-LanCL2; Addgene 154192). All LanCL1 and LanCL2 mutants were generated by site-directed mutagenesis (Addgene 154187, 154188, 154190, 154191). The cDNA encoding the phosphothreonine lyase OspF was synthesized by Integrated DNA technologies and amplified by polymerase chain reaction (PCR). The SpvC encoding gene was directly amplified by PCR using genomic DNA as template from S. typhimurium strain 14028 (gift from Dr. James M. Slauch at the University of Illinois at Urbana-Champaign). The cDNA encoding SpvC or OspF was cloned into the pET15b vector using NdeI and BamHI cloning sites. The human glutathione transferase A4 (GSTA4) cDNA was subcloned into the pET15b vector using NdeI and BamHI sites. All lanthipeptides were co-expressed using the pRSFDuet vector system with the corresponding modification enzyme as previously reported (Dong et al., 2015; Yang et al., 2018). The ProcA2.8 mutant (C3A/C19A/S13A) was generated by site directed mutagenesis. A list of primers for generating all constructs is provided in the KEY RESOURCES TABLE and Supplemental Table 4.
pET28a-MEK1-S222C/C277S/C376S plasmid was prepared by Genscript (1 μL, 200 ng/μL). This plasmid encodes for an N-terminal His6 tag. In order to overcome gluconoylation, the plasmid was mutated at G(−19)F to afford MEK1 G(−19)F/S222C/C277S/C376S referred to as MEK1C222 herein. All desired mutations and deletions were performed from this original plasmid. MEK1-C218 (S218C/C277S/C376S; Addgene 164639), MEK1-C222 (S222C/C277S/C376S; Addgene 164638), MEK1-C218C222 (S218C/S222C/C277S/C376S: Addgene 164640), ERK1-C202 (K71R/C82S/C144S/C178S/C183S/T202C/ C271S; Addgene 164647) and ERK1-C82C202 (K71R/C144S/C178S/C183S/T202C/C271S; Addgene 164650) mutants were obtained by site-directed mutagenesis using standard protocols and were deposited at Addgene. A list of primers for generating all constructs is provided in the KEY RESOURCES TABLE and Supplemental Table 4. The N-terminal truncated (residues 1–60) MEK1 C277SC377S, referred as Δ1–60MEK1 mutant with a C-terminal His6 tag (residues 323–329), was constructed and cloned by SGC (Dr. Apirat Chaikuad) from the MEK1 plasmids described above. The gene encoding MEK1 was cloned into pET28a between NcoI and BamHI restriction sites (C222: Addgene 164641; C218 Addgene 164643; C218C222 Addgene 164645).
The following plasmids were obtained from Addgene: human ERK1 gene inserted between EcoRI and BamHI sites of NpT7–5 vector (gift from Melanie Cobb; RRID: Addgene_39229).pGEX-4T-1–3XFlag-ERK2 (gift from Kevin Janes; RRID:Addgene_47573) (Bose and Janes, 2013); pET-His6-ERK2-MEK1_R4F_coexpression (gift from Melanie Cobb; RRID:Addgene_39212) (Khokhlatchev et al., 1997).
Solid-phase peptide synthesis
Peptide synthesis was performed on a CEM Liberty microwave peptide synthesizer. Standard Fmoc protected amino acids were used unless otherwise noted. 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) was used as activator, 2 M N,N-diisopropylethylamine (DIPEA) in N-methyl-2-pyrrolidone (NMP) as activator base and 20% piperidine in dimethylformamide (DMF) as deprotection agent. Synthetic peptides were cleaved from the resin by adding cleavage cocktail (90/7.5/2.5 TFA/triisopropylsilane/H2O) for 2 h and crude peptides were precipitated in ice-cold diethyl ether. Precipitants were then air-dried to remove residual diethyl ether. The dried precipitants were dissolved in water and purified by RP-HPLC (Macherey-Nagel, VP 250/10 Nucleodur C18 HTec, 5 μm) at 1 mL/min flow rate with the following gradient of solvent A (0.1% trifluoroacetic acid in H2O) and B (0.1% trifluoroacetic acid in MeCN): linear gradient from 2% to 85% B in 40 min, followed by a gradient from 85% to 98% B in 2 min.
Protein purification
For His-tagged proteins, cell pellets from 1 L of cell culture were resuspended and lysed in 25 mL of lysis buffer (20 mM Tris, 500 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 5 mM imidazole, 10% glycerol, pH 8.0). Lysates were centrifuged at 4 °C for 30 min at 75,000 ×g and supernatants were filtered with centrifugal filters (Fisher Scientific). NiNTA purification was performed using a 5 mL HisTrap coumn (GE Healthcare). After loading the clarified lysate, the column was applied to an Äkta Pure FPLC system (GE Healthcare) using solvent A (20 mM Tris, 500 mM NaCl, 1 mM TCEP, 5 mM imidazole, 10% glycerol, pH 8.0) and solvent B (20 mM Tris, 500 mM NaCl, 1 mM TCEP, 500 mM imidazole, 10% glycerol, pH 8.0). The loaded column was washed at 1.5 mL/min with a linear gradient from 0 to 20% solvent B over 23 column volumes (CV) and eluted with 100% solvent B for 10 CV. The purest fraction was identified by SDS-PAGE and concentrated by 10 kDa cutoff Amicon ultracentrifugal filters (Millipore). For MBP-tagged protein purification, cell pellets from 1 L of cell culture were resuspended and lysed in 10 mL of MBP purification buffer (20 mM Tris, 150 mM NaCl, pH 8.0). Lysates were centrifuged at 4 °C for 30 min at 75,000 × g and supernatants were filtered with centrifugal filters (Fisher Scientific). The filtered lysate was applied to 3 mL of amylose resin (New England Biolabs) and eluted with MBP elution buffer (20 mM Tris, 150 mM NaCl, 200 mM maltose, pH 8.0). Fractions were identified by SDS-PAGE and concentrated using 10 kDa cutoff Amicon ultracentrifugal filters (Millipore).
For purification of the phosphorylated His6-ERK2 protein, cell pellets from 1 L of cell culture were resuspended and lysed in 25 mL of lysis buffer (20 mM Tris, 500 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 25 mM NaF, 1 mM NaVO3, 25 mM β-glycerophosphate, 5 mM imidazole, 10% glycerol, pH 8.0). Lysates were centrifuged at 4 °C for 30 min at 75,000 ×g and supernatants were filtered with centrifugal filters (Fisher Scientific). The His6-ERK2 was purified using the same procedure as described above, and its phosphorylation was confirmed by intact protein MS.
Pellets of cells expressing MEK-C218 and MEK-C222 were resuspended in 30 mL of lysis buffer (50 mM HEPES, 500 mM NaCl, 20 mM imidazole, 5% glycerol, 0.5 mM TCEP, 1% Triton X-100, pH 7.5) for 1 h and sonicated using a microtip. The supernatant was separated by centrifugation (20,000 rpm, 30 min at 4 °C) and loaded onto a 5 mL HisTrap column. The column was washed using HEPES buffer with increased amounts of imidazole (20–60 mM). The target protein was eluted with 80 to 500 mM imidazole in HEPES buffer, identified with SDSPAGE and mass spectrometry, and concentrated by the use of Vivaspin 6 10kDa-cutoff concentrator.
Expression of ERK1-C202 and ERK1-C82C202 mutants
For in vitro MEK1 activity assay and the generation of dehydroalanine-containing ERK1, a series of plasmids encoding wt ERK1 or mutants (ERK1-C202 and ERK1-C82C202) was constructed. ERK1 wt or mutant gene with a His6-tag at its N-terminus was inserted into the expression vector NpT7–5 between EcoRI and BamHI sites, and the plasmid was used to transform E. coli BL21(DE3) competent cells. °C Then 1 mM IPTG was added to induce protein expression, and after further 16-h incubation at 25 °C, cell pellets were harvested. ERK1 protein was purified by the use of Ni-NTA column, and its purity verified through SDS-PAGE and LC-MS.
His-pulldown assays for Western blot analysis
HEK293 cells were rinsed with ice-cold PBS buffer and lysed in His-pulldown buffer (20 mM Tris, 150 mM NaCl, 25 mM NaF, 25 mM β-glycerolphosphate, 0.1 mM NaVO3, 20 mM imidazole, 0.3% Triton X-100, pH 8.0 and 1X Halt protease inhibitor cocktail [Thermo Fisher]). HEK293 cell extracts were micro-centrifuged at 14,100 ×g for 20 min at 4 °C and supernatants were transferred into clean vials. Recombinant His-tagged LanCL1 or LanCL2 (4 μM) were added into HEK293 cell extracts and incubated at 4 °C. After 2 h, cell extracts were treated with 30 μL of HisPur Cobalt resin (Thermo Fisher) for His-pulldown at 4 °C for 1 h. The cobalt resin was washed with 500 μL of His-pulldown buffer three times and boiled with 30 μL of 2X Laemmli buffer (Bio-Rad). All samples were analyzed by Western blotting.
His-pulldown assays for unbiased MS-based proteomics
HEK293 cells were rinsed with ice-cold PBS buffer and lysed in His-pulldown buffer (20 mM Tris, 150 mM NaCl, 25 mM NaF, 25 mM β-glycerolphosphate, 0.1 mM NaVO3, 20 mM imidazole, 0.3% Triton X-100, pH 8.0 and 1X Halt protease inhibitor cocktail [Thermo Fisher]). HEK293 cell extracts were micro-centrifuged at 14,100 ×g for 20 min at 4 °C and supernatants were pre-cleaned with 20 μL of HisPur Cobalt resin (Thermo Fisher) for 30 min at 4 °C. The pre-cleaned cell extracts were transferred into clean vials. Recombinant His-tagged LanCL1 or LanCL2 (4 μM) were added to the HEK293 cell extracts and incubated at 4 °C. Equivalent amounts of LanCL storage buffer were added to the negative controls. After 2 h, cell extracts were treated with 30 μL of HisPur Cobalt resin for His-pulldown at 4 °C for 1 h. The cobalt resin was washed with 500 μL of His-pulldown buffer three times and boiled with 30 μL of 2X Laemmli buffer (Bio-Rad). A small fraction was used for Western blot analysis as described above and most of the samples were resolved on a 4–20% TGX gel (Bio-Rad). The gel was cut into slices that were submitted to the Harvard Taplin Mass Spectrometry Facility for proteomics analysis where they were treated with trypsin and the resulting digest analyzed by MS.
Proteomics Sequence Analysis
Excised gel pieces were washed and dehydrated with acetonitrile and completely dried with a speed-vac. Acetonitrile-free gel pieces were rehydrated with 50 mM ammonium bicarbonate solution containing 12.5 ng/μL of modified sequencing-grade trypsin (Promega, Madison, WI) at 4 ºC for 45 min. Trypsin solution was replaced with 50 mM ammonium bicarbonate solution and samples were left at 37 ºC overnight. Peptides were extracted by removing the ammonium bicarbonate solution followed by one wash with 50% acetonitrile and 1% formic acid. The extracts were dried in a speed-vac for approximately 1 h and stored at 4 ºC until analysis. Samples were reconstituted in 5 – 10 μL of 5% acetonitrile and 0.1% formic acid solution. A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter x ~30 cm length) with a flame-drawn tip (Peng and Gygi, 2001). Samples were loaded on to the column via a Famos auto sampler (LC Packings, San Francisco CA). A gradient was formed, and peptides were eluted with increasing concentrations of 97.5% acetonitrile, 0.1% formic acid solution.
Eluted peptides were subjected to electrospray ionization and then entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences were determined by matching protein databases with the acquired fragmentation pattern by the software program Sequest (Thermo Fisher Scientific, Waltham, MA). All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate. The intensity of ions corresponding to individual proteins were summed and compared to the negative control. If the protein was also observed in the negative control the data are expressed as fold-change compared to the negative control with a 10-fold change used as cut-off for inclusion. If the protein was not observed in the negative control, the protein is indicated as unique to the LanCL pulldown sample. The data are presented in Supplemental Table 1.
Purification of ProcA 2.8 mut, CylLs” and CylLL”
E. coli BL21(DE3) cells were transformed with the corresponding plasmid and protein expression was induced with 0.3 mM IPTG in TB media at 18 °C for 18 h (Dong et al., 2015; Yang et al., 2018). Cell pellets were resuspended at 0.2 mg/mL in LanA buffer B1 (4 M guanidine-HCl, 20 mM NaH2PO4, 500 mM NaCl, 0.5 mM imidazole, pH 7.5) and lysed by sonication. Insoluble debris was removed by centrifugation at 16,500 ×g for 30 min at 4 °C. The supernatant was purified by immobilized metal affinity chromatography using 2–4 mL of His60 Ni Superflow Resin (Takara). Following 30 min incubation at room temperature, the resin was washed with LanA B2 buffer (4 M guanidine-HCl, 20 mM NaH2PO4, 300 mM NaCl, 30 mM imidazole, pH 7.5). For the ProcA 2.8 mutant (ProcA 2.8 mut: C3A/S13A/C19A), the peptide was eluted with LanA elution buffer (4 M guanidine hydrochloride, 20 mM NaH2PO4, 100 mM NaCl, 0.5 M imidazole, pH 7.5). The elution fractions were desalted by C4 SPE column and purified on a Machery-Nagel NUCLEODUR® C18 HTec C18 column, (250 mm × 7 μm; part no. 762566.100). For CylLS” and CylLL”, resins were washed again with LanA co-expression buffer (20 mM NaH2PO4, 300 mM NaCl, 30 mM imidazole, pH 7.5) and eluted with LanA co-expression elution buffer (20 mM NaH2PO4, 300 mM NaCl, 0.5 M imidazole, pH 7.5). To the eluted fractions, the protease CylA was added to a final concentration of 10 μg/mL to remove the leader peptides (Tang et al., 2019) and produce CylLL” and CylLS”. The core peptide was purified by preparatory HPLC (Agilent) equipped with a Phenomenex Jupiter® 4 μm Proteo 90 Å LC column (250 × 10 mm; Part No. 00G-4396-N0) using the gradient described in the section on peptide synthesis.
Generation of Dhb-containing ERK peptide by phosphothreonine lyase and glutathionylation by LanCL2
Phosphate elimination was carried out using 5 μM phosphothreonine lyase and 25 μΜ peptide in reaction buffer (25 mM Tris, 150 mM NaCl, pH 8.0). For phospho-peptides that mimic the activation loop of ERK, peptides were treated with His6-SpvC or His6-OspF at 30 °C for 90 min. For the phospho-Akt peptide, peptide was treated with His6-OspF at 30 °C for 18 h. The phosphate elimination process was monitored by matrix-assisted laser-desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) and dehydroamino acid containing peptides were purified by RP-HPLC (Macherey-Nagel, VP 250/10 Nucleodur C18 HTec, 5 μm). For glutathionylation of the ERK peptide, 25 μM Dhb-containing peptide was dissolved in reaction buffer (25 mM Tris, 150 mM NaCl, 1 mM glutathione (GSH), 1 μM TCEP, pH 8.0) and incubated with 5 μM LanCL2-His6 at 25 °C for 18 h. The glutathionylation process was monitored by MALDI-TOF MS and the resulting peptide was purified by RP-HPLC (Macherey-Nagel, VP 250/10 Nucleodur C18 HTec, 5 μm). All peptides described in this section were purified by HPLC as described in the peptide synthesis section.
Hydrolysis of glutathionylated ERK peptide (GS-ERK), derivatization of the amino acids and GC-MS analysis of GS-ERK peptide
GS-ERK peptide was enzymatically synthesized by LanCL2 from Dhb-ERK and purified by RP-HPLC (Macherey-Nagel, VP 250/10 Nucleodur C18 HTec, 5 μm). GS-ERK peptide (1.5 mg) was dissolved in 6 M DCl in D2O (3 mL). The reaction mixture was heated in an oil bath with stirring at 110 °C for 24 h. After cooling down the reaction mixture to RT, the solvent was removed by rotary evaporator. Acetyl chloride (1.5 mL) was added dropwise to pre-chilled MeOH (5 mL). The mixed solution (3 mL) was added to the hydrolyzed peptide and heated to 110 °C for 1 h. The reaction mixture was cooled to RT and the solvent was removed by rotary evaporation. The residue was dissolved in CH2Cl2 (3 mL) and pentafluoropropionic anhydride (1 mL) was added into the reaction vessel. The reaction vessel was heated to 110 °C for 1 h with reflux. After cooling down the reaction vessel to RT, the sample was dried under a stream of argon. The dried residue was dissolved in MeOH and transferred to a clean vial. The samples were analyzed by GC-MS on an Agilent HP 6890N mass spectrometer with an Agilent CP-Chirasil-L-Val column (25 m × 0.25 mm × 0.12 μm). The derivatized sample was diluted with MeOH and injected into the instrument with helium gas at a flow rate of 2.0 mL/min. Samples were analyzed by the following gradient: the injection was held at 160 °C for 5 min and increased to 190 °C at 3 °C /min. The selected ion mode (SIM) was used to monitor 379 Da for derivatized methyllanthionine (MeLan) (Tang and van der Donk, 2012). The derivatized sample was co-injected with characterized synthetic standards of DL- and LL-methyllanthionine to identify the stereochemistry of GS-ERK peptide (Tang and van der Donk, 2012, 2013).
LanCL1/2 kinetic assays with dehydroamino acid containing peptides
Kinetic assays were performed with His6-LanCL1 or LanCL2-His6 and Dhb-ERK/CylLs” purified as described previously. Assay mixtures consisted of 0.5 mL containing 1 mM GSH, different concentrations of Dhb-containing peptides (50/100/150/200/250/300/350 μM), 0.5 mM TCEP, 0.1 mM ZnCl2, 150 mM NaCl and 100 mM Tris, pH 7.5. The assay was performed at room temperature for 30 min. After addition of His6-LanCL2 at a final concentration of 0.5 μM, reaction aliquots of 45 μL were collected and quenched with 5 μL of LanCL quench buffer (1.1 M citric acid and 5.5 mM EDTA) at 0, 1, 2, and 3 min timepoints. Quenched reaction aliquots (5 μL) were injected onto a Macherey-Nagel NUCLEODUR® 100–5 C18 ec column (125 mm × 2 mm; catalog no. 7600001.20) attached to a quadrupole/time-of-flight (Q/TOF) Synapt-G1 mass spectrometer (Waters). All kinetic and control experiments were performed in triplicate. The column was eluted with solvent A (100% H2O, 0.1% formic acid) and solvent B (100% MeCN, 0.1% formic acid) with a gradient of 2–100% solvent B over 15 min at a flow rate of 0.18 mL/min. The Synapt instrument settings were as follows: positive ion mode, V optics, capillary voltage = 2.0 V, cone gas = 40 L/h, desolvation gas = 600 L/h, souce temperature = 120 °C, desolvation temperature = 200 °C. The instrument was externally calibrated with a 0.1% phosphoric acid standard. Data were collected over a m/z window of 50–2000 Da in continuous mode with a 0.2 s scan rate using a leucine-enkephalin lockspray solution for internal calibration. Spectra were smoothed and baselines were subtracted using the MassLynx software package (Waters). Extracted ion chromatograms (EICs) were generated by applying a mass window of 2.5 Da around the center of the most intense peak of each product. The EICs were integrated using MassLynx software to generate peak areas. The EIC peak areas were used to calculate the concentration of glutathionylated product at each time point by comparison with standard curves. Initial rates at different substrate concentrations were calculated by linear regression fit of product concentration over time and fit to the Michaelis-Menten model using Prism 6 (GraphPad) for kinetic parameters KM,peptide and kcat.
In vitro binding assays
To determine the direct physical interaction of MEK1-Dha218 and LanCL2, 500 nM of MBP or MBP-LanCL2 and 700 nM of His6-MEK1-Dha218 were mixed at 4 °C for 30 min and subsequently incubated with 30 μL HisPur cobalt resin (Thermo Fisher) for 7 min. The resin was washed with 500 μL of His-pulldown buffer three times and boiled with 30 μL of 2X Laemmli buffer (Bio-Rad). The samples were analyzed by Western blotting. MBP or MBP-LanCL2 was detected by anti-MBP tag antibody and His6-MEK1-Dha218 was detected by an anti-His tag antibody.
Preparation of MEF cell extracts for enzymatic analysis
MEF cells (2×106) were seeded in 100 mm cell culture dishes and cultured with DMEM complete media for two days. The MEFs were then washed with ice-cold PBS buffer and cell pellets resuspended with enzymatic buffer (25 mM Tris, 150 mM NaCl, 1 mM GSH, 1 μM TCEP, pH 8.0). Cells were lysed by sonication followed by microcentrifugation at 14,100 ×g for 30 min at 4 °C. The supernatant was transferred into fresh tubes and the protein concentration was determined by bicinchoninic acid (BCA) assay (Thermo Fisher). Equal amounts of total protein (500 μg) from wt and triple LanCL knockout MEFs were incubated with His6-MEK1-Dha222 (final concentration 1.5 μM) at 25 °C for 4 h. His6-MEK1-Dha222 was enriched by incubating with 30 μL of HisPur Cobalt resin (Thermo Fisher) for 10 min followed by washing the resin with 500 μL of His-pulldown buffer three times and boiled with 30 μL of 2X Laemmli buffer (Bio-Rad). All samples were analyzed by Western blotting.
Site-selective formation of MEK-Dha218, MEK-Dha222, and MEK-Dha218Dha222
MEK1C proteins in reaction buffer at pH 8.0 were treated with bisalkylating/elimination agents (added from a stock solution in DMSO, <2% v/v). The mixtures were shaken at 600 rpm at 37 °C or at room temperature. The reactions were monitored using LCMS-ESI-TOF. For more details see Figures (S2–S6 and Supplemental Table 2). For example, specifically for use of 2,5-methyldibromopentonate (MDBP), the purified MEK-C218 or MEK-C222 protein was desalted into reaction buffer (20 mM Tris, 100 mM NaCl, 25 mM MgCl2, and 10 mM ADP, pH 8.0) via PD SpinTrap G25 column. The concentration of desalted protein was measured by BCA protein assay. In order to obtain the dehydroalanine (Dha) form of MEK1 proteins through in vitro site-directed modification, MEK-C218 or MEK-C222 protein was incubated with 0.5 or 0.2 mM TCEP, and 400 (or 200) molar equivalents of MDBP. The mixtures were kept at 25 °C for 1 h, followed by increase of the reaction temperature to 37 °C. After 2 h, MEK-C218 or MEK-C222 reached 100% conversion to its Dha form (MEK-Dha218 or MEK-Dha222), as monitored by LC-MS using a XEVO mass spectrometer (Waters). The excess MDBP and TCEP were removed by the use of a SpinTrap G25 column. MEK-Dha proteins were stored in reaction buffer and kept at −80 °C.
Generation of Full Length MEK-Dha/MEK-GSH ERK Variants for Phosphorylation Assay
Eliminated MEK1 variants, 30 μM MEK-Dha222 or MEK-Dha218Dha222 were treated with 50 mM glutathione (GSH) in Tris buffer (20 mM Tris, 100 mM NaCl, pH 8.0). After 2-hour incubation at 25 °C, the complete conversion to glutathione adduct (MEKGS222 or MEKGS218GS222) was achieved and verified by LC-ESI-MS analysis (Xevo). The excess of glutathione in each MEK glutathione adduct was removed by a SpinTrap G25 column. The target product was obtained and confirmed by LC-ESI-MS analysis (Xevo).
For eliminated MEKGS218 variant, 30 μM MEK-Dha218 was incubated with 20 mM glutathione and 3.5 μM LanCL1 protein. After 1-hour incubation at 25 °C, the complete conversion to glutathione adduct was achieved and verified by LC-ESI-MS analysis (Xevo). The excess of glutathione in each MEK glutathione adduct was removed by a SpinTrap G25 column. The target product was obtained and confirmed by LC-ESI-MS analysis (Xevo).
MEK variants – ERK Phosphorylation Assay
Purified ERK1 (0.5, 1.0 or 1.8 μM) was incubated with MEK1 variants in kinase buffer (50 mM Tris pH 7.5, 30 mM NaCl, 10 mM MgCl2, 0.2 mM DTT) at 30 °C. 1 mM ATP was added to the reaction mixture. At the indicated time, an aliquot was removed and analyzed by LC-ESI-MS after the addition of 50 mM EDTA to quench the reaction. The amounts of phosphorylated ERK were quantified using the intensities of ERK deconvoluted peaks from mass spectra and fitted by global least-square regression to solutions of kinetic models (see Figure S6) using OriginPro9.7. To control for the presence of residual potential LanCL1 protein (in any MEK-GS adducts that were synthesized using LanCL1) in phosphorylation assays, a control experiment of ERK phosphorylation also in the presence of 3.5 μM LanCL1 protein was performed under the same conditions; the additional presence of LanCL1 had no significant effect on activity.
Formation of ERK1-Dha202 and ERK1-Dha82Dha202
In order to generate ERK1-Dha202, the mutant protein ERK1-C202 was desalted into the reaction buffer consisting of 20 mM Tris, 100 mM NaCl, 0.1 mM TCEP pH 8.0. After 1 h incubation, TCEP was removed with a PD SpinTrap G25 column. ERK1-C202 protein was treated with 100 molar equivalents of MDBP immediately and the reaction was monitored by MS using a XEVO mass spectrometer. ERK1-C202 protein reached fully convertion to ERKDha202 within 24 h at 25 °C and excess MDBP was removed with a SpinTrap G25 desalting column. ERK-1-Dha82Dha202 was generated using the same method as ERK1-Dha202.
MS/MS of Eliminated MEK and ERK variants
‘In-solution’ tryptic digestions were performed following an MS-compatible protocol. Specifically, for 10 μg ERK1-Dha202 or ERK1-Dha82Dha202 protein sample was diluted into 100 μl with 8 M urea, 100 mM Triethylammonium bicarbonate (TEAB) buffer and treated with 10 mM TCEP at room temperature for 30 mins. The mixture was incubated with 50 mM freshly prepared iodoacetamide at room temperature for 30 mins in dark. Then a ten-time dilution was performed with a buffer containing 8 M urea and 50 mM TEAB. The mixture was treated with trypsin (1:20 w/w trypsin:protein) at 37 °C overnight. The salts within the digested samples were extracted by an Oasis HLB 1cc Vac cartridge (Waters) and the eluted peptide fragments were concentrated overnight on SpeedVac. The dried sample was dissolved in 10 μl MilliQ water containing 0.1% formic acid and 2% acetonitrile for LC-MS/MS analysis on Orbitrap Elite mass spectrometer (Thermo Fisher).
For MEK-Dha218, MEK-Dha222 and MEK-Dha218Dha222 samples (25 μL, around 1 mg/mL) were diluted with 8 M urea solution (75 μL). 200 mM aqueous DTT (2 μL) was then added and the mixture was heated at 56 °C for 15 min. The mixtures were cooled down to room temperature and treated with a 36 mg/mL iodoacetamide in 50 mM ammonium carbonate solution (2 μL). The mixture was left standing in the dark for 30 min. 200 mM aqueous DTT (2 μL) was then added to neutralize the iodoacetamide. The mixture was diluted four times with 50 mM ammonium bicarbonate solution and treated with trypsin (1:50 w/w ratio enzyme:protein). The mixture was incubated at 37 °C overnight. Some samples were also treated with AspN (1:50) and incubated at 37 °C for 4 h. The sample was diluted down to 200 fmol/μL with, either milliQ water or 1% formic acid before analysis LC-MS/MS analysis on an Orbitrap Elite mass spectrometer (Thermo Fisher). Data was processed and analysed using PEAKS software v7.0.
Kinetics of glutathionylation of MEK-Dha by LanCLs
LanCL1/2 were incubated with 0.1 mM zinc chloride before adding to the reaction due to its Zn2+-binding property. The reaction mixtures, consisting of 6 μM MEK-Dha218 protein and 1.2 μM LanCL1/2, were incubated at 25 °C with up to 120 μM glutathione (GSH). At each time point, an aliquot (1 μL) was diluted with 49 μL of 100 mM ammonium acetate solution (pH 8.0), and the conversion of MEK-Dha218 to MEK-GS218 was monitored and quantified by using a XEVO mass spectrometer. Initial rates of forming MEK-GS218 conjugate were plotted against the concentrations of substrate GSH, the apparent KM,GSH and kcat values were obtained using a fit of the data to the Michaelis-Menten equation using Origin9.7.
The following MS set-up was used: Xevo G2-S and G2-XS mass spectrometer systems (Waters) were coupled to an Acquity UPLC using a Thermo ProSwiftTM RP-2H column (4.6 mm x 50 mm; catalog no. 064296). All kinetic experiments were carried out in triplicate. Water (as solvent A) and acetonitrile (as solvent B) both containing 0.1% formic acid were used as the mobile phase with a gradient of 5–95-5% solvent B over 10 min at a flow rate of 0.3 mL/min. The electrospray source was operated with a capillary voltage of 3 kV and a cone voltage of 20 V. Spectra were calibrated by use of an internal lock-spray. Desolvation temperature was 400 °C and desolvation gas at a total flow of 700 L/h or 650 L/h for Xevo G2-S or G2-XS mass spectrometer, respectively. Total mass spectra were deconvoluted from multiply charged ion series using the MaxEnt algorithm preinstalled on MassLynx Software (V4.1).
Glutathionylation of ERK1-Dha202 and ERK1-Dha82Dha202 Comparative Time Course
LanCL1/2 wt or mutant proteins (LanCL1-C322A or LanCL2-C321A/C367A) were pre-treated with 0.1 mM ZnCl2 for 30 mins. 6 μM ERK1-Dha202 or ERK1-Dha82Dha202 was incubated with 1.0 mM glutathione (GSH) and 1.2 μM LanCL proteins at 25 °C up to 3 h. A negative control experiment without the addition of LanCL proteins was performed under the same conditions. At each time interval, an aliquot from the above mixture was diluted with 100 mM ammonia acetate (pH 8.0) buffer and analysed by Xevo mass spectrometer. The deconvoluted spectra of LC-MS were used to quantify the conversion to target ERK1-GS202 or ERK1-Dha82GS202.
Generation of dehydrobutyrine-containing ERK2 (ERK2-Dhb185pY187)
GST-Flag-ERK2 purification
The plasmid pGEX-4T-1-3xFlag-ERK2 with a GST-tag and triple Flag-tag at the N-terminus was obtained from Addgene (plasmid no. 47573; gift from Kevin Janes). The 3xFlag tag was inserted at BamHI site, and a new BamHI site was regenerated at its downstream. erk2 gene was inserted between BamHI and SalI sites of this pGEX-4T1 vector. GST-Flag-ERK2 protein was expressed in E. coli Rosetta 2 (DE3) cells. The overnight cultures were inoculated in a 1L fresh LB media with 100 μg/ml ampicillin at 37 °C. Cells were induced with 0.5 mM IPTG until OD600 reaches 0.7 and incubated at 18 °C for further18 h. Cell pellets were harvested and then lysed in a 25 ml ice-cold PBS buffer with 1X protease inhibitor. The supernatants were centrifugated at 76,000 x g for 30 mins at 4 °C, filtered by centrifugal filters and then applied onto a GSTrap column (GE healthcare) at a flow rate of 1.5 mL/min. The loaded column was washed with 10 column volumes of PBS to remove non-bound components and the purified GST-Flag-ERK2 protein was recovered from the column with a elution buffer (1xPBS buffer containing 10 mM reduced glutathione and 1X protease inhibitor). The fractions with desired protein were analysed by SDS-PAGE and verified by Xevo mass spectrometer.
Phosphorylation of ERK2 using MEK-Dha218Dha222
The mutant protein His6-MEK-C218/C222 was expressed in E. coli BL21(DE3) cells and purified by Ni2+-NTA column according to the same methods as shown for MEK-C218 and MEK-C222 proteins. The purified MEK-C218C222 protein in reaction buffer (20 mM Tris, 100 mM NaCl, 25 mM MgCl2, 10 mM ADP, pH 8.0) was incubated with 0.2 mM TCEP for 1 h at 25 °C and then treated with 600 molar equivalents of bis-alkylating reagent MDBP gradually. The mixture was kept at 25 °C for 1 h, followed by incubating at 37 °C for 2 h. The fully converted MEK-Dha218Dha222 protein was verified by LC-ESI-MS using Xevo mass spectrometer (Waters) and desalted by a SpinTrap column to remove excess of MDBP and TCEP.
Double phosphorylation of GST-Flag-ERK2 at Thr185 and Tyr187 residues was achieved by incubating 200 μM GST-Flag-ERK2 protein with 4 μM His6-MEK-Dha218Dha222 in the reaction buffer (20 mM Tris, 100 mM NaCl, 25 mM MgCl2, 25 mM ATP, pH 8.0) at 25 °C for 4 h and then at 4 °C for 16 h. The fully phosphorylated GST-Flag-ERK2-pT185pY187 was confirmed by Xevo mass spectrometer. To remove His6-MEK-Dha218Dha222 protein from double phosphorylated ERK, the reaction mixture was applied to a 1 ml pre-equilibrated HisTrap column and incubated at 4 °C for 1 h. The column was washed with 3 column volumes of Tris buffer (20 mM Tris, 100 mM NaCl, pH 8.0) and the proteins were eluted with increased amounts of imidazole (20 to 500 mM). Each fraction was analysed by SDS-PAGE. The purified GST-Flag-ERK2-pT185pY187 was in the fractions containing 20 mM imidazole and concentrated by a Vivaspin6 10 kDa-cutoff concentrator.
Elimination of ERK2-pThr185-using Phosphothreonine Lyase
Phosphate elimination was achieved with 80 μM GST-Flag-ERK2-pT185pY187 and 16 μM His6-OspF at 25 °C for 1 h. The reaction process was monitored by Xevo mass spectrometer. The dehydrobutyrine-containing ERK protein (GST-Flag-ERK2-Dhb185pY187) was separated from His6-OspF by a 1 mL HisTrap column using the similar procedures as mentioned above. The fractions of purified GST-Flag-ERK2-Dhb185pY187 (containing 20 mM imidazole in Tris buffer) were combined and desalted to remove imidazole.
Test of glutathionylation of Dhb-containing ERK2 (ERK2-Dhb185pY187) by LanCL1/2
For the glutathionylation of ERK2-Dhb185pY187 protein, 10 μM Dhb-containing ERK2 was incubated with 1 mM GSH and 10 μM LanCL protein (LanCL1 or LanCL2) for up to 7 h at 25 °C. At each time interval, an aliquot (2 μL) of reaction mixture was diluted into 48 μL 100 mM ammonia acetate (pH 8.0) buffer (with 1 mM DTT) and monitored by Xevo mass spectrometer. The conversion of ERK2-Dhb185pY187 protein to ERK2-GS185pY187 was monitored and quantified by the deconvoluted spectra of LC-MS.
Western blot analysis of phosphate elimination and glutathionylation of His6-ERK2
Phosphate elimination was carried out using 5 μM bisphosphorylated His6-ERK2 (obtained by co-expression with MEK1-R4F (Khokhlatchev et al., 1997)) and 1 μM His6-OspF and incubated at 30 °C for 1 h. A total of 1 μM MBP-LanCL2 was added into the reaction mixture and incubated at 25 °C for 3 h. Phosphorylation of ERK2 and Dhb formation were first verified by MS. His6-ERK2-pThr185pTyr187 was analyzed with a Thermo Q Exactive Ultra High Mass Range (UHMR) Mass Spectrometer. First buffer exchange was performed for phospho-ERK2 (50 μM) using a Zeba spin column with 25 mM ammonium acetate. The resulting sample was analyzed. Calculated mass: 42329.53 Da Observed mass: 42333.72 Da. For the Dhb-containing ERK2 in solution tryptic digestion was performed and the resulting peptides were analyzed by using a Thermo Fusion Orbitrap Tribrid Mass Spectrometer. Calculated mass for the relevant peptide phosphorylated on Tyr and containing Dhb at the position of the former pThr: 2204.9508 Da Observed: 2204.9528 Da. All reaction mixtures were also boiled with 2X Laemmli buffer (Bio-Rad) and analyzed by Western blotting. Phosphate elimination was detected using anti-phospho-ERK1/2 antibody (Cell Signaling Technology). Glutathionylation was monitored using anti-GSH antibody (Virogen). The amount of total ERK2 protein was detected using anti-ERK1/2 antibody (Cell Signaling Technology).
Accessibility analysis for Cys residues
Accessibility analysis applied to unbound apo-MEK1 structure [generated from the binary MEK1-ATPγS structure (PDB 3W8Q) minus ligand ATPγS] predicted (Figure 3B) that Cys might be added through SDM at sites 218 and/or 222 to give residues with highest predicted reactivity, as long as higher accessibility residues C277, C376 (93.5, 21.9% accessibility, respectively, Figure 3B) were removed. Thus, mutation to near-isosteric, unreactive Ser yielded MEK1-C277S/C376S with essentially identical enzymatic properties to MEK1wt (Supplemental Table 2). This protein was used as the base mutant sequence for all subsequent alterations.
Critically, in the mutated construct used in this study, C207, C121, C142 and C341 were retained. C207 lies within the ATP-binding pocket; C121, 207, 341 mutants variously (Wagle et al., 2011; Zhao et al., 2014) alter activity and C207 is a key target site for covalent inhibitors (Liu et al., 2013).
Importantly, the same predictive analysis of reactivity suggested that in binary (MEK1•ATPγS [PDB 3W8Q]) or ternary (MEK1•ATPγS•Mg [PDB 3EQD] & MEK1•ADP•Mg [PDB 3EQI]) complexes accessibility to C207 is dramatically decreased (accessibility 22.4→~0.1%, Figure 3B,C), consistent with C207’s location in the nucleotide binding site of MEK1. In these complexes, predicted accessibilities of proposed reaction sites C222 and C218 were also importantly either unaffected or enhanced (Fig. 3C). Notably, Mg(II) induces a conformational change that usefully enhances accessibility of both sites (C222: 38.4→93.4%, C218: 8.9→15.2%, respectively, Figure 3C). These analyses prompted the additional consideration of nucleotides (ATPγS, ATP, ADP) (Lamoureaux and Lee, 2011) and metals (Mg(II)) as masking or enhancing ligands to usefully modulate regioselectivity (Figure 3C). In this way, we designed and generated three key mutant variants of MEK1 for testing as substrates in regioselective elimination chemistry: MEK1-C218, MEK1-C222, MEK1-C218C222 (bearing five, five or six Cys residues, respectively).
Prior to reactivity testing, interactions with masking agents were confirmed: differential scanning fluorimetry (Niesen et al., 2007) showed strong binding of putative masking/enhancing ligands to form MEK1•ATP(±Mg) and MEK1•ATPγS(±Mg) (Figure S2). Consistent with predictions, all of the MEK1 mutant proteins, in the absence of masking nucleotide/metal gave products of unwanted reaction at Cys207 with parent reagent DBHDA (Figure 4A, Figure S2, Supplemental Table 2). For elimination of MEK1-C222, use of alternative alkylating agents did not prevent unwanted C207 reaction (Figure S2, Supplemental Table 2). However, use of nucleotide additive proved successful: thus, whilst ATP only partially masked C207 in MEK1-C222 (Figure S2), ATP•Mg with DBHDA masked it fully, successfully allowing elimination reaction at site 222 (Figure 4A). Concomitant observation of ~25% phosphorylated MEK1-Dha222 (plus ADP release, Figure 4A) also revealed competitive autophosphorylation during reaction. To solve this problem, the same reaction was performed ‘masked’ instead by ADP•Mg and this led to clean, selective formation of desired MEK1-Dha222 after 5 h (Figure 4A, Figure S3). Use of other reagents from the panel similarly allowed clean formation of MEK1-Dha222 at tuneable rates (Figure S3, Supplemental Table 2). Reaction of MEK1-C218 with less accessible site 218 was slower: DBHDA + ADP•Mg gave fully converted MEK1-Dha218 in 15 h (Figure 4B) along with unwanted side-products of overalkylation without elimination that increased with longer reaction (Figure S4, likely pyrrolidine-Lys). Other conditions were therefore sought: screening of alternative reagents (Supplemental Table 2) revealed clean, site-selective Dha formation (Figure 4B, Figure S4) using DBDGla (20 h, 37 °C) or MDBP (Morrison et al., 2015) (4 h, 37 °C).
Following successful regio-selective formation of both MEK1-Dha218 and MEK1-Dha222 through single site elimination, we tested double elimination in MEK1-C218C222. DBHDA alone showed no Dha formation and only partial alkylation (Fig. 4C, Figure S2). Promisingly, DBHDA + ATP•Mg gave partial conversion (~80%) to MEK1 bearing Dha, albeit with concomitant autophosphorylation (Figure S5). Screening of reagents and masking agents (Supplemental Table 2) revealed that DBDGla + ADP•Mg gave only low conversion to MEK1-Dha218Dha222 (23%, 14 h, Figure S5). Pleasingly, however, MDBP + ADP•Mg cleanly gave MEK1-Dha218Dha222 (Fig. 4C, Figure S5).
Synthetic chemistry procedures
General consideration
Reagents
Chemicals and solvents were purchased from Sigma-Aldrich UK, Acros UK, Alfa Aesar UK, or Fischer UK and were used as delivered unless stated otherwise. This includes 2,5-methyldibromopentanoate (MDBP).
Small molecule characterisation
Proton nuclear magnetic resonance (δH) spectra were recorded on a Bruker AV400 (400 MHz), or on a Bruker AVII500 (500 MHz) spectrometer. Carbon nuclear magnetic resonance (δC) spectra were recorded on a Bruker AV400 (100.7 MHz) spectrometer or on a Bruker AVII500 (125.8 MHz) spectrometer. All chemical shifts are quoted on the δ scale in ppm using residual solvent as the internal standard (1H NMR: CDCl3 = 7.26, CD3OD = 4.87; 13C NMR: CDCl3 = 77.0; CD3OD = 49.0; D2O = 4.80). The following splitting abbreviations were used: s = singlet, d = doublet, t = triplet, q = quartet, a = apparent. All NMR spectra are found in Supplemental Methods S1. Infrared spectra were recorded on a Bruker Tensor 27 Fourier Transform spectrophotometer by attenuated total reflectance (ATR) attachment –oil or crystals of the compound were placed directly against the stage. A background measurement was subtracted from each spectrum and a new background measurement was recorded before each spectrum was recorded.. Absorption maxima (υmax) are reported in wavenumbers (cm−1) and classified as strong (s) or broad (br). Only signals representing functional groups are reported; CH absorptions as well as the fingerprint region are not listed. Low resolution mass spectra were recorded on a Micromass Platform 1 spectrometer using electrospray ionization (ESI). High resolution mass spectra were recorded on a Walters 2790-Micromass LCT electrospray ionization mass spectrometer. m/z values are reported in Daltons. Thin layer chromatography (TLC) was carried out using Merck aluminium backed sheets coated with 60F254 silica gel. Visualization of the silica plates was achieved using a UV lamp (λmax = 254 nm), and/or ammonium molybdate (5% in 2M H2SO4), or potassium permanganate (5% in 1M NaOH). Flash column chromatography was carried out using BDH PROLAB® 40–63 mm silica gel (VWR).
Synthesis of bisalkylating/elimination agents
2,5-Dibromohexanediamide, DBHDA
Adipic acid (25 g, 171.1 mmol) was suspended in thionylchloride (75 mL, 1034 mmol). The reaction was heated to reflux (open to air, bath temp 80 ºC). After 30 min at reflux, the solid had dissolved. The reaction was stirred for an additional 60 min at reflux and then cooled to room temperature. Carbontetrachloride (100 mL) was added to the reaction followed by N-bromosuccinimide (73 g, 411 mmol). The reaction was stirred vigorously and 10 drops of HBr (48% aq.) was added by pipette. The reaction was heated to reflux, again open to air. The reaction gradually turns from red to black over the course of an hour. After 2 h at reflux, the reaction was cooled to room temperature and then to 0 ºC. The mixture was stirred at 0 ºC to ensure all succinimide had precipitated. The solid was removed by filtration. Diethylether (50 mL) was used to rinse and complete the filtration. The filtrate was concentrated under reduced pressure to give a thick, dark red liquid. In a 500 mL round bottom flask, 200 mL of NH4OH (25% aq.) was cooled to 0 ºC. The crude acid chloride was added dropwise over 20 min to the ammonia solution with rapid stirring. After the addition was complete, the reaction was stirred vigorously at 0 ºC for 1 h. The bis-amide product precipitated from the reaction mixture. The dark solid was isolated by filtration and partially dried. The product was purified by triturating in MeOH/H2O: The solid was suspended in H2O (100 mL) and MeOH (100 mL) and heated to 60 ºC. The mixture was stirred rapidly at 60 ºC for 30 min. After this time, the mixture was cooled to room temperature. The resulting white solid (a mixture of meso and D/L diastereomers) was isolated by filtration and washed with MeOH (200 mL). The title compound was dried under high vacuum to afford 28.4 g (55 %) as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 1.75–2.08 (4H, m, H3, H4), 4.28–4.36 (2H, m, H2, H5), 7.30 (2H, s, H7, H8), 7.69 (2H, s, H7, H8). 13C NMR (100 MHz, DMSO-d6): (both diastereomers reported), δ ppm 32.5 (s, C3, C4), 32.6 (s, C3, C4), 48.2 (s, C2, C5), 48.5 (s, C2, C5), 169.87 (s, C1), 169.92 (s, C5) (Supplemental Methods S1). IR (υmax, KBr): 3302, 2946, 2801, 1684, 1418, 1319, 1277, 1250, 1220, 1194, 978, 876. HRMS m/z (ESI+): Found 300.9178 [M+H]+; C8H11O2N279Br2 requires 300.9182.
2,5-Dibromohexanedioic acid, DBHDAc
Adipic acid (2.0 g, 13.7 mmol) and thionyl chloride (2.98 mL, 41.1 mmol) were stirred at reflux for 1 h. The mixture was cooled down to room temperature and treated with N-bromosuccinimide (7.3 g, 41.1 mmol). The mixture was stirred at 75 °C for 3 h. The mixture was cooled down to 0 °C before addition of a 1:1 mixture of water/tetrahydrofuran (20 mL). The mixture was stirred vigorously for 2 h. The mixture was filtered. The residue was stirred in dichloromethane for 1 h. Filtration afforded 1.3 g (32 %) of the title compound (mixture of racemate and meso diastereoisomer) as an off-white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 1.83 – 2.22 (m, 4H, H3, H4), 4.45 – 4.60 (m, 2H, H2, H5), 13.32 (br. s., 2H, H7, H8). 13C NMR (101 MHz, DMSO-d6) δ ppm 32.05 (s, C3, C4), 32.17 (s, C3, C4), 46.74 (s, C2, C5), 46.88 (s, C2, C5), 170.44 (s, C1, C6). (Supplemental Methods S1).HRMS m/z (ES-): Found 300.8718 [M-H]-; C6H7O479Br2 requires 300.8717.
The compound (50 mg, 0.016 mmol) was stirred in water (1 mL) for 16 h. The solid was filtered to afford meso-2,5-dibromohexanedioic acid.
2,5-Dibromo-N,N’-dimethylhexanediamide, DBDMet
2,5-Dibromohexanedioic acid (100 mg, 0.33 mmol) and thionyl chloride (239 μL, 3.3 mmol) was stirred at reflux for 2 h. The solution was cooled down to room temperature before evaporation under reduced pressure. The residue was then added to a stirred 40% wt aqueous solution of methyl amine (1 mL, 13.6 mmol) at 0 °C. The mixture was stirred vigorously for 2 h. The precipitate was filtered and stirred in a minimum of dichloromethane. Filtration afforded 35 mg (32 %) of title compound as a white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 1.67 – 2.13 (m, 4H, H3, H4), 2.61 (d, J = 4.58 Hz, 6H, H10, H12), 4.23 – 4.45 (m, 2H, H2, H5), 8.13 – 8.36 (m, 2H, H9, H11). 13C NMR (101 MHz, DMSO-d6) δ ppm 25.87 (s, C10, C12), 32.67 (s, C3, C4), 48.24 (s, C2, C5), 168.31 (s, C1, C6) (Supplemental Methods S1). IR (υmax, KBr): 3279, 1650, 1571. HRMS m/z (ESI+): Found 328.9493 [M+H]+; C8H15O2N279Br2 requires 328.9495.
meso-N,N’-bis(2-amino-2-oxoethyl)-2,5-dibromohexanediamide, DBDGla
2,5-Dibromohexanedioic acid (100 mg, 0.33 mmol) and thionyl chloride (100 μL, 1.38 mmol) was stirred at reflux for 1.5 h. The solution was cooled down to room temperature before evaporation under reduced pressure. The residue was then diluted in diethyl ether (1 mL). The solution was added dropwise to a stirred solution of glycinamide hydrochloride (72 mg, 0.66 mmol) in a mixture of diethyl ether (500 μL) and 2 M aqueous sodium hydroxide solution (660 μL) at 0 °C. The mixture was stirred for 1 h at room temperature. The organic layer was separated. The aqueous was acidified to pH~2 with addition of a 1 N hydrochloric acid solution. The mixture was washed with ethyl acetate (2 mL). The aqueous layer (suspension) was separated, evaporated under reduced pressure and azeotroped twice with toluene. The residue was stirred in acetonitrile for 1 h at room temperature. The precipitate was filtered to afford 130 mg of crude material. 40 mg of this material were stirred in water for 16 h. Filtration afforded 7 mg (17 %) of the title compound as a white solid.
1H NMR (400 MHz, DMSO-d6) δ ppm 1.73 – 1.92 (m, 2H, H3, H4), 2.02 – 2.19 (m, 2 H, H3, H4), 3.55 – 3.83 (m, 4 H, H10, H12), 4.43 – 4.63 (m, 2H, H2, H5), 7.09 (br. s., 2 H, H14, H16), 7.39 (br. s., 2 H, H14, H16), 8.44 (t, J = 5.62 Hz, 2H, H9, H11). 13C NMR (101 MHz, DMSO-d6) δ ppm 32.65 (s, C3, C4), 42.04 (s, C10, C12), 48.01 (s, C2, C3), 168.27 (s, C13, C14), 170.24 (s, C1, C6) (Supplemental Methods S1). IR (υmax, KBr): 3292, 1679, 1651, 1600, 1556. HRMS m/z (ESI-): Found 412.9467 [M-H]-; C10H15O4N479Br2 requires 412.9466.
({(meso)-2,5-dibromo-6-[(carboxymethyl)amino]-6-oxohexanoyl}amino)acetic acid, DBDGly
2,5-Dibromohexanedioic acid (100 mg, 0.33 mmol) and thionyl chloride (100 μL, 1.38 mmol) was stirred at reflux for 1.5 h. The solution was cooled down to room temperature before evaporation under reduced pressure. The residue was then diluted in diethyl ether (1 mL). The solution was added dropwise to a stirred solution of glycine (50 mg, 0.66 mmol) in a mixture of diethyl ether (500 μL) and 2 M aqueous sodium hydroxide solution (660 μL) at 0 °C. The mixture was stirred for 1 h at room temperature. The organic layer was separated. The aqueous was acidified to pH~2 with addition of a 1 N hydrochloric acid solution. The mixture was washed with ethyl acetate (2 mL). The aqueous layer (suspension) was separated, evaporated under reduced pressure and azeotroped twice with toluene. The residue was stirred in acetonitrile for 1 h at room temperature. The precipitate was filtered to afford 100 mg of crude material. 40 mg of this material were stirred in water for 16 h. Filtration afforded 4 mg (9 %) of the title compound as a white solid. The solid was highly soluble in water.
1H NMR (400 MHz, DMSO-d6) δ ppm 1.77 – 1.93 (m, 2H, H3, H4), 2.01 – 2.15 (m, 2H, H3, H4), 3.80 (dd, J = 5.87, 3.42 Hz, 4H, H10, H12), 4.38 – 4.59 (m, 2H, H2, H5), 8.60 (t, J = 5.75 Hz, 2H, H9, H11), 12.66 (br. s, 2H, H14, H16). 13C NMR (101 MHz, DMSO-d6) δ ppm 32.69 (s, C3, C4), 40.95 (s, C10, C12), 47.56 (s, C2, C5), 168.44 (s, C13, C15), 170.78 (s, C1, C6) (Supplemental Methods S1). IR (υmax, KBr): 3279, 1650, 1571. HRMS m/z (ESI+): Found 416.9292 [M+H]+; C10H15O6N279Br2 requires 416.9291.
Eliminylome Analysis
Previously deposited MS raw data by Medard et al. (Médard et al., 2015) were processed using MaxQuant software integrated with the Andromeda search engine (Cox and Mann, 2008; Cox et al., 2011).
For identification of putative eliminylome components of serine and threonine residues, the peak lists were searched using MaxQuant software against the human protein database with carbamidomethyl as fixed modification for cysteine residues. Elimination of serine and threonine residues to form Dha and Dhb, respectively, as well as their Michael adducts with DTT, beta-mercaptoethanol (bME), and glutathione were used along with acetylation (at N-terminus), oxidation (at M), and phosphorylation (at S, T, or Y) as variable modifications. Trypsin was selected as the proteolytic enzyme, and up to two missed cleavages were allowed. The mass tolerance of the precursor ion was set to 4.5 ppm and for fragmentation ion to 0.5 Da. Protein and PSM false discovery rate were set at 0.01. The threshold score for modified protein selected for inclusion was set at 40. A parallel search was performed for identification of putative eliminylome components of cysteine residues. Elimination of cysteine residues to form Dha as well as their Michael adducts with DTT, beta-mercaptoethanol (bME), and glutathione were used along with carbamidomethyl (at C), acetylation (at N-terminus), oxidation (at M), and phosphorylation (at S,T, or Y) as variable modifications. All other processing parameters were kept same as previously mentioned.
The resulting data were then manually curated for correlation of elimination to Dha or Dhb at S or T phosphorylation sites in protein kinases, respectively, and Cys sites in kinase domains using at least one occurrence as noted under the post-translational modification function in UniProt (The Uniprot Consortium 2019; Veuthey et al., 2013) and PhosphoSitePlus (Hornbeck et al., 2015).
Generation of Dha-GPX by H2O2 oxidation
Glutathione peroxidase (GPX) from bovine erythrocytes was purchased from Sigma-Aldrich in the form of lyophilized powder and contained DTT. To remove DTT, the initial powder was dissolved in Tris buffer (25 mM Tris, 150 mM NaCl, pH 7.5) and purified by Zeba spin column. The concentration of the resulting GPX solution was quantified by nanodrop. Following a reported protocol (Cho et al., 2010), Dha-GPX was generated by incubating the GPX solution with 1 mM H2O2 for 1 h at 37 ºC. The oxidation product was purified by Zeba spin column to remove excess H2O2 and quantified by nanodrop.
Synthesis of biotinylated cysteamine to detect dehydroalanine (Dha) in GPX
Nonenzymatic thiol Michael addition was used to detect whether Dha was successfully generated in GPX upon H2O2 oxidation (Rhee and Cho, 2010). For this purpose, biotinylated cysteamine was synthesized. Cysteamine hydrochloride (11.4 mg, 0.1 mmol) was treated with 0.5 mL of PBS solution of NaHCO3 (1 M) and 0.1 mL of a DMF solution of EZ-linked N-hydroxysuccinimide biotin (0.5 M) for 3 h at room temperature. To the above reaction mixture was added TCEP hydrochloride (45 mg, 0.15 mmol). The resulting solution was incubated at room temp for another 2 h. After lyophilizing the reaction mixture, the sample was dissolved in 0.1% TFA, filtered through a 0.2 μM filter, and purified by reversed-phase HPLC on a C18 column. The mass of biotinylated cysteamine was confirmed by MS (ESI): [M + H]+ calculated, 304.1075; found, 304.1145. The detection of Dha in GPX was performed by following an established protocol (Rhee and Cho, 2010). Briefly, Dha-containing GPX (~3 μg) was treated with a PBS solution containing 0.1 M NaHCO3, 0.5 mM biotinylated cysteamine, 1 mM TCEP, and 1% SDS for 20 h at 37 ºC. The reaction mixture was then boiled with 2X Laemmli buffer (Bio-Rad) and separated by SDS-PAGE. The separated proteins were transferred to a nitrocellulose membrane for blot analysis with HRP-conjugated streptavidin (Thermo Scientific). GPX was used as the control in the Western blot analysis (Figure S1H).
Investigating the possibility of LanCL-catalyzed GSH addition to Dha-GPX
Dha-containing GPX (10 μM) was dissolved in reaction buffer (25 mM Tris, 150 mM NaCl, 1 mM GSH, 1 mM TCEP, pH 7.5) and incubated with 2 μM His6-LanCL1 or LanCL2-His6 (preactivated with 0.1 mM ZnCl2 at room temperature for 0.5 h) at room temperature for 2 h. The reaction mixtures were boiled with 2X Laemmli buffer and analyzed by Western blotting. No protein bands were detected upon staining with anti-GSH antibody (Virogen) compared to positive control, which suggests that LanCL does not catalyze GSH addition to Dha-GPX.
Investigating the possibility of LanCL2-catalyzed crosslinking of thioredoxin and Dhb-ERK peptide
To investigate whether LanCL may catalyze protein crosslinking, we performed a preliminary exploration based on a model system in which thioredoxin was chosen as the thiol nucleophile given its reactive and accessible Cys and Dhb-ERK was chosen as the electrophile. Dhb-ERK (25 μM) and thioredoxin (Trx, 100 μM) in reaction buffer (25 mM Tris, 150 mM NaCl, 1 mM TCEP, pH 7.5) were incubated with 5 μM LanCL2-His6 (preactivated with 0.1 mM ZnCl2) at room temperature. The reaction mixture was monitored by MALDI-TOF MS over 5 h. No crosslinking products were detected. To test whether LanCL2-His6 was still active, 1 mM GSH was introduced to the reaction at the end of the above incubation. The GSH-containing reaction mixture was further incubated at room temperature for an additional 1 h and then analyzed by MALDI-TOF MS. The glutathionylation of Dhb-ERK was clearly detected in the presence of LanCL2-His6. Thus, LanCL2 was active but did not add Cys from Trx to Dhb-ERK.
Knock-out mice phenotype
In 2011 we first generated LanCL2−/− KO mice. The first generation of these mice displayed the phenotype shown in Figure S7E where about 30% of the KO mice died between ~2 and 6 months. Histopathology analysis did not identify any common cause of death and for most mice no obvious cause of death was found. The surviving KO mice did not display any phenotype and subsequent generations did not display the premature death phenotype. Hence, we assumed that a subset of mice for unknown reasons possibly associated with other genetic differences died and that we had lost the phenotype in the surviving mice. In the subsequent years we generated LanCL1−/− and LanCL3−/− mice and through breeding generated TKO mice in 2016. These mice did not display any obvious phenotype when we used them to test the hypothesis that LanCL proteins are responsible for lanthionine formation in the brain (He et al., 2017). However, a colony of TKO mice that were born in early 2020 displayed near-identical premature death phenotype (Fig. 6H & S7D) as observed in 2011.
Multiple seizures were observed before death in several of the TKO mice that died prematurely. Necropsy specimen from nine mice were examined, including one TKO male, six TKO females, one sacrificed wt male, and one wt female. Four out of six TKO female mice examined had necrotic livers containing coagula of hypereosinophilic, hepatocellular debris with degenerative neutrophils. Two TKO female mice had necrotic endometrium with bacterial infection. Steiner stain of necrotizing mouse livers was negative. Gram stain on endometrium and placentae showed intralesional colonies of gram-positive cocci. No significant histopathological abnormality was seen in wt mice or TKO males. Overall no common cause of death was evident in TKO mice that died prematurely.
Survival analysis
A total of 105 mice (58 WT and 47 LanCL TKO) were studied and used to generate Kaplan-Meier (KM) survival plots (Fig. 6H and S7D). Survival times were monitored for up to 210 days. The following data were collected, i) age (if mice were alive then, age = age at end of study, if mice died then, age = age at death), ii) sex; differences between wt and TKO survival were determined by the Gehan-Breslow-Wilcoxon test. Statistical significance in all tests was accepted at P < 0.05. The KM plots were generated using GraphPad Prism 8.0.2. The older LanCL2 data involved 50 LanCL2 −/− mice and 24 wt mice and provided similar overall numbers (Figure S7E). Censored subjects are healthy mice that were sacrificed to compare tissues containing high LanCL levels (based on previous studies) to monitor for potential differences in GSH-adduct in wt and TKO mice (see below). In addition, all surviving mice at the end of the time interval are considered censored as their long term survival was not known at the end of the study.
Protein extraction from mouse brain and heart tissue
Mouse brain and heart from deceased TKO mice, sacrificed TKO, or wt mice were dissected and snap frozen immediately in liquid nitrogen. Frozen tissue was homogenized and lysed in tissue extraction buffer (200 nM Tris HCl pH 7.4, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 2.5 mM sodium pyrophosphate, 1 mM β-glycerol phosphate, 1 mM sodium orthovanadate, 2% Triton X-100 and a protease inhibitor) using a tissue homogenizer. Homogenized tissue was rotated for 30 min and centrifuged at 14,100 ×g for 10 min at 4 °C. Supernatant was transferred to a clean vial and protein concentration was determined using Pierce™ BCA Protein Assay Kit. LanCL1/2 and GSH, or GSH only were added to the protein, and the samples were analyzed by Western blot after SDS-PAGE under reducing conditions using anti-glutathione antibody (Virogen). No difference was observed between TKO and wt samples.
Immunoprecipitation of ERK1/2 from total mouse brain protein and reaction with LanCL2
In an effort to potentially concentrate a dehydroamino acid containing protein, mouse brain total protein was pre-cleaned with Protein A Agarose Beads (Cell Signaling Technology) to remove endogenous IgG and incubated with ERK1/2 antibody (Cell Signaling Technology) at 4 °C overnight. The solution was then incubated with Protein A Agarose Beads at 4 °C for 4 h and micro-centrifuged at 250 ×g for 1 min. Supernatant was removed and beads were washed with tissue extraction buffer 3 times.
Beads with ERK1/2 immunoprecipitated from 200 μg of WT or TKO mouse brain total protein were treated with 4 μM LanCL2, 500 μg glutathione (GSH) (Sigma-Aldrich), and 0.5 mM tris(2-carboxyethyl) phosphine (TCEP), and incubated at 37 °C for 1 h. Samples were washed three times with tissue extraction buffer and boiled with 20 μL of 2X Laemmli buffer (Bio-Rad). All samples were analyzed by Western blotting to detect C-glutathionylated Erk using anti-glutathione antibody (Virogen). No difference was observed between TKO and wt brain.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all non-quantitative experiments such as mass spectrometric analysis, experiments were performed in triplicate with a typical outcome shown that was representative of all three experiments. Kinetic experiments with dehydroamino acid containing peptides were performed in triplicate. The amounts of products formed were quantified by LC-MS as previously reported (Thibodeaux et al., 2014). The EICs were integrated using MassLynx software to generate peak areas. The EIC peak areas were used to calculate the concentration of glutathionylated product at each time point by comparison with standard curves. Initial rates at different substrate concentrations were calculated by linear regression fit of product concentration over time and these intitial rates were fit to the Michaelis-Menten model using Prism 6 (GraphPad) for kinetic parameters KM,peptide and kcat. For kinetic experiments to determine kinase activity of MEK variants, the amounts of phosphorylated ERK were quantified using the intensities of ERK observed in deconvoluted peaks from mass spectra obtained by LC-MS. The results, shown as mean ± standard deviation of triplicate experiments, were fitted by global least-square regression to solutions of kinetic models (see Figure S6) using OriginPro9.7.
Supplementary Material
(A) His-pulldown assay for LanCL2-His6 and His6-LanCL1. Only a small fraction of each kinase was pulled down by His6-LanCL1 and LanCL2-His6. (B) Endogenous LanCL1/2 were co-immunoprecipitated by anti-p110, anti-mTOR and anti-Vps34 antibodies in HEK293 cells. (C) MSMS for Dhb-ERK peptide and Dhb-Akt peptide. (D-F) Phosphate elimination by His6-SpvC and glutathionylation of Dhb-ERK (with pTyr) by LanCL2-His6. (D) Phosphate elimination by His6-SpvC using phospho-ERK peptide containing a pTyr residue as substrate. (E) MSMS for Dhb-ERK (with pTyr residue). (F) ESI-LC-MS analysis of LanCL2 activity using Dhb-ERK (with pTyr residue) as substrate. EIC, extracted ion chromatogram; ESI, electrospray ionization. (G) Generation of Dha in GSH peroxidase (GPX) by H2O2 treatment, which was verified by addition of biotinylated cysteamine. No GSH adduct was observed upon incubation of the Dha-GPX with LanCL1/2 and GSH. (H) Left, partial conversion with γ-Glu-Cys (γ-EC) (1 mM) and Dhb-Erk by LanCL2 but not its double mutant after 12 h. Right, near complete conversion with free Cys (1 mM) and Dhb-Erk by LanCL2 but not its double mutant after 12 h. (I) Anti-GSH antibody can recognize non-canonical glutathionylation. (J) His6-phospho-ERK2 was eliminated by His6-OspF and the resulting His6-ERK2-Dhb185pTyr187 was glutathionylated by MBP-LanCL2 as demonstrated by Western blot. (K) Tandem MS analysis of native and glutathionylated CylLS” and CylLL”. (L) Non-canonical glutathionylation is catalyzed by endogenous LanCLs. Glutathionylation of His6-MEK1-Dha222 was observed by Western blotting.
(a) Panel of DBHDA derivatives used in this study. 1,4-Bis-alkylating/elimination agents ‘tuned’ by indicated properties (magenta). (b) Reagent Synthesis: i. SOCl2, reflux, 1.5h then NBS,HBr/CCl4, reflux, 3 h; ii. NH3OH, 0 °C, 1.5h; iii. NaI, acetone, reflux, 4 h; iv. H2O/THF (1:1), 0 °C, 2 h; v. SOCl2, reflux, 1.5h then RNH2 then trituration in H2O. (c) Differential scanning fluorimetry (Niesen et al., 2007) showed strong binding of putative masking/enhancing ligands to form MEK1•ATP(±Mg) and MEK1•ATPγS(±Mg2+) for model mutant substrate MEK1-C222. (d) MEK1 mutant proteins, in the absence of masking nucleotide / metal gave products of unwanted reaction at Cys207 with parent reagent DBHDA. MEK1-C218 (22 μM) in 20 mM Tris, 100 mM NaCl, pH 8 buffer treated with DBHDA (4.4 mM) at 37 °C afforded overalkylated MEK1Dha218 (Mcalc.=45862, Mobs.=45866) after 4 h; MEK1-C222 (22 μM) in the same buffer treated with DBHDA (4.4 mM) at 37 °C afforded overalkylated MEK1-Dha222 (Mcalc.=45862, Mobs.=45867) after 4 h, MSMS of the tryptic peptide 206–227 confirmed the overalkylation at C207; MEK1-C218C222 (22 μM) in the same buffer treated with DBHDA (4.4 mM) at 37 °C afforded overalkylated stapled MEK1-C218C222 (Mcalc.=46052, Mobs.=46057) after 4 h. (e) Use of alternative alkylating agents did not prevent unwanted C207 reaction in MEK1-C222. MEK1-C222 (33 μM) in 20 mM Tris, 100 mM NaCl, pH 8 buffer treated with DBDMet (6.6 mM) at 37 °C afforded a mixture 85% MEK1-Dha222 (Mcalc.=45642, Mobs.=45646) and 15% overalkylated MEK1-Dha222 (Mcalc.=45890, Mobs.=45891) after 3 h; MEK1-C222 (22 μM) in the same buffer treated with DBDGla (4.4 mM) at 37 °C afforded overalkylated MEK1Dha222 (Mcalc.=45982, Mobs.=45981) after 3 h; MEK1-C222 (33 μM) in the same buffer treated with DBHDAc (6.6 mM) at room temperature afforded substituted MEK1 (Mcalc.=45979, Mobs.=45983) after 24 h. (f) Treatment of MEK1-C222 with DBHDA in the presence of ATP. MEK1-C222 (22 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ATP, pH 8 buffer treated with DBHDA (4.4 mM) at 37 °C afforded a mixture of 85% MEK1-Dha222 (Mcalc.=45642, Mobs.=45645) and 15% overalkylated MEK1-Dha222 (Mcalc.=45862, Mobs.=45866) after 6 h.
(a) Treatment of MEK1-C222 with DBHDA in the presence of ADP-Mg. MEK1-C222 (33 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBHDA (6.6 mM) at 37 °C afforded pure MEK1-Dha222 (Mcalc.=45642, Mobs.=45646); (b) LCMSMS of the tryptic/AspN peptides containing cysteine residues confirmed the formation of Dha exclusively at position 222. (c) MEK1-C222 (44 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBDGla (8.8 mM) at 37 °C afforded MEK1-Dha222 (Mcalc.=45642, Mobs.=45641) after 4 h; Dha position was confirmed by LC-MSMS of peptide 206–227; (d) MEK1-C222 (44 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DIB (2000 equiv) at 37 °C afforded MEK1-Dha222 (Mcalc.=45642, Mobs.=45645) after 20 h; Dha position was confirmed by LC-MSMS of peptide 207–227; (e) MEK1-C222 (44 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with MDBP (4.4 mM) at 37 °C afforded MEK1-Dha222 (Mcalc.=45642, Mobs.=45642) after 5 h, Dha position was confirmed by LC-MSMS of peptide 217–227.
(a) Treatment of MEK1C-218 with DBHDA in the presence of ADP-Mg. MEK1-C218 (22 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBHDA (4.4 mM) at 37 °C afforded a mixture of MEK1Dha218 (Mcalc.=45642, Mobs.=45645) and 25% overalkylated MEK1-Dha222 (Mcalc.=45785, Mobs.=45785) after 15 h; (b) LC-MSMS of the tryptic peptide 206–227 confirmed the formation of Dha at position 218; (c) Longer reaction time lead to the formation of more adducts. (d) MEK1-C218 (44 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBDGla (4.4 mM) at 37 °C afforded exclusively MEK1-Dha218 (Mcalc.=45642, Mobs.=45647) after 20 h, the position was confirmed by LCMSMS analysis of peptide 206–227; (e) MEK1-C218 (44 μM) in the same buffer treated with MDBP (4.4 mM) at 37 °C afforded exclusively MEK1-Dha218 (Mcalc.=45642, Mobs.=45647) after 4 h, the position was confirmed by LC-MSMS analysis of peptide 206–227. (f) Treatment of Δ160MEK1-C218 with MDBP in the presence of ADP-Mg. Δ1–60MEK1-C218 (55 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with MDBP (5.5 mM) at room temperature only afforded Δ160MEK1-Dha218 (Mcalc.=36611, Mobs.=36616) after 14 h.
(a) Treatment of MEK1-C218C222 with DBHDA analogues in the presence of nucleotide-Mg. MEK1-C218C222 (22 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ATP, 25 mM MgCl2, pH 8 buffer treated with DBHDA (4.4 mM) at 37 °C afforded a mixture 60% pMEK1-Dha222 (Mcalc.=45738, Mobs.=45741), 20% MEK1-Dha222 (Mcalc.=45658, Mobs.=45661) and 20% phosphorylated stapled MEK1-C218C222 (Mcalc.=45912, Mobs.=45914) after 5 h; (b) MEK1-C218C222 (22 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBHDA (4.4 mM) at 37 °C afforded a mixture 50% MEK1-Dha222 (Mcalc.=45658, Mobs.=45658) and 50% stapled MEK1C218C222 (Mcalc.=45832, Mobs.=45831) after 5 h; (c) MEK1-C218C222 (44 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBHDA (8.8 mM) at 37 °C afforded a mixture 23% MEK1-Dha218Dha222 (Mcalc.=45624, Mobs.=45627), 18% MEK1-Dha222 (Mcalc.=45658, Mobs.=45668) and 59% stapled MEK1-C218C222 (Mcalc.=45946, Mobs.=45951) after 14 h. (d) Treatment of MEK1-C218C222 with MDBP in the presence of ADP-Mg. MEK1-C218C222 (22 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with MDBP (4.4 mM) at room temperature afforded exclusively MEK1-Dha218Dha222 (Mcalc.=45624, Mobs.=45628) after 15 h; (e) LCMSMS of the tryptic peptide 206–227 confirmed the formation of Dha at position 218 and 222. (f) Comparative circular dichroism measurement of MEK1-Cs and MEK1-Dhas.
(a) Sequential and distributive proposed mechanism of ERK1 phosphorylation by MEK1. (b) The phosphorylation of ERK1K71R by MEK1 construct was monitored by LCMS. The intensities of the deconvoluted peaks corresponding to ERK1 (Mcalc. = 44058), pERK1 (Mcalc. = 44138), and ppERK1 (Mcalc. = 44218) were recorded over time (Figure S18). The conversions were calculated from these intensities and plotted. (c) Differential equation defined the phosphorylation constants kf1 and kf2. These constants were calculated by fitting the analytical equations. (d) ESI-MS progress curves allowing time course analysis of ERK1 by MEK1 variants. ERK1-K71R was incubated with MEK1 variant in 50 mM Tris, 30 mM NaCl, 1 mM ATP, 10 mM MgCl2, 0.2 mM DTT, pH 7.5 at 30 °C.
(A) End-point assays of LanCL1 and LanCL2 (6.0 μM) with His6- MEK1-Dha218 as the substrate for glutathionylation. The conversions of MEK1-Dha218 (6 μM) to MEK1-GS218 after 2-h incubation with 1.0 mM GSH at 25 °C were shown by deconvoluted spectra of LC-MS. (B) Wild-type or mutant LanCL catalyze C-S glutathionylation of ERK1-Dha202. The Michael addition of 0.5 mM GSH to Dha-containing ERK protein (6 μM) was monitored by LC-MS after 3-h incubation with LanCL proteins (6.0 μM) at 25 °C. Top right inset: Time-dependent GSH addition to ERK1-Dha202 catalyzed by wild-type (LanCL1 or LanCL2) or mutant LanCL proteins (LanCL1-C322A or LanCL2-C321A/C367A). The conversion to ERK1-GS202 was quantified using intact mass LC-ESI-MS and is plotted here as the value over background chemical GSH addition (~0.13 M−1 s−1 under these conditions) in the absence of enzyme. (C) LanCL2 adds GSH to GST-3XFlag-ERK2-Dhb185pY187 generated using the pThr lyase OspF. (D) Kaplan-Meier survival curve of female WT (n = 33) and LanCL TKO (n = 27) FVB mice (P = 0.0052) and male WT (n = 25) and LanCL TKO (n = 20) FVB mice (P = 0.0332). Gehan-Breslow-Wilcoxon P test. (E) Survival curve of age-matched FVB WT mice (n = 24) and LanCL2 −/− mice (n = 50).
Supplemental Table S1. Unbiased protein pulldown and proteomics analysis. Related to Figure 1E.
Supplemental Table S2. Chemical generation of Dha-containing MEK1 variants and their kinase activity. Related to Figures 3 and 4, and Supplemental Figures S2–S6.
Supplemental Table S3. Analysis of the proteomic literature for the presence of Dha and Dhb proteins and their conjugates with GSH, β-mercaptoethanol, Cys, and dithiothreitol. Related to Figures 5, S7.
Supplemental Table S4. Oligonucleotide sequences used in this study. Related to the STAR Methods.
Highlights.
pSer/pThr and Cys are converted to reactive dehydroamino acids during protein damage
MEK1 with dehydroalanine in its activation loop is an active, deregulated kinase
LanCL adds GSH to dehydroamino acids, removing electrophiles from the proteome
C-glutathionylation of MEK1-dehydroalanine inactivates its kinase activity
Acknowledgements
This study was supported by the Howard Hughes Medical Institute (to W.A.V.), NIH GM089771 (to J.C.), the BBSRC with AstraZeneca (S.R.G.G.), the EPSRC UK Catalysis Hub (EP/R026815/1, YZ, G.J.H, B.G.D), the EU Horizon 2020 Programme under the Marie Sklodowska-Curie (700124, J.R.), the Felix Foundation (R.R.) and the EPSRC with Pfizer (K.P.C.). A MALDI TOF mass spectrometer was purchased with a grant from the National Institutes of Health (S10 RR027109 A). We thank Dr. Ian R. Bothwell and Dr. Alex V. Ulanov (Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign) for assistance with GC-MS, Elisabete Pires for assistance with MS/MS analysis, Dr. Shih-Hsuan Hsiao (Veterinary Medicine, UIUC) for histopathology of deceased TKO mice, and Drs J. Read and G. Pairaudeau (AstraZeneca) for useful discussions.
Footnotes
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alessi DR, Saito Y, Campbell DG, Cohen P, Sithanandam G, Rapp U, Ashworth A, Marshall CJ, and Cowley S. (1994). Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J 13, 1610–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C, Parsot C, and Sansonetti PJ (2007). An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat Immunol 8, 47–56. [DOI] [PubMed] [Google Scholar]
- Bauer H, Mayer H, Marchler-Bauer A, Salzer U, and Prohaska R. (2000). Characterization of p40/GPR69A as a peripheral membrane protein related to the lantibiotic synthetase component C. Biochem Biophys Res Commun 275, 69–74. [DOI] [PubMed] [Google Scholar]
- Bose AK, and Janes KA (2013). A high-throughput assay for phosphoprotein-specific phosphatase activity in cellular extracts. Mol Cell Proteomics 12, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan DF, and Barford D. (2009). Eliminylation: a post-translational modification catalyzed by phosphothreonine lyases. Trends Biochem Sci 34, 108–114. [DOI] [PubMed] [Google Scholar]
- Bruns CM, Hubatsch I, Ridderstrom M, Mannervik B, and Tainer JA (1999). Human glutathione transferase A4–4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J Mol Biol 288, 427–439. [DOI] [PubMed] [Google Scholar]
- Chalker JM, Gunnoo SB, Boutureira O, Gerstberger SC, Fernndez-Gonzlez M, Bernardes GJL, Griffin L, Hailu H, Schofield CJ, and Davis BG (2011). Chem Sci 2, 1666–1676. [Google Scholar]
- Chambers KA, Abularrage NS, and Scheck RA (2018). Selectivity within a family of bacterial phosphothreonine lyases. Biochemistry 57, 3790–3796. [DOI] [PubMed] [Google Scholar]
- Chatterjee C, Paul M, Xie L, and van der Donk WA (2005). Biosynthesis and mode of action of lantibiotics. Chem Rev 105, 633–684. [DOI] [PubMed] [Google Scholar]
- Cho CS, Lee S, Lee GT, Woo HA, Choi EJ, and Rhee SG (2010). Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid Redox Signal 12, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CH, Kurien BT, Mehta P, Mhatre M, Mou S, Pye QN, Stewart C, West M, Williamson KS, Post J, et al. (2007). Identification of lanthionine synthase C-like protein-1 as a prominent glutathione binding protein expressed in the mammalian central nervous system. Biochemistry 46, 3262–3269. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Pinto JT, and Callery PS (2011). Reversible and irreversible protein glutathionylation: biological and clinical aspects. Expert Opin Drug Metab Toxicol 7, 891–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, and Mann M. (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- Dai P, Zhang C, Welborn M, Shepherd JJ, Zhu T, Van Voorhis T, and Pentelute BL (2016). Salt effect accelerates site-selective cysteine bioconjugation. ACS Cent Sci 2, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Colombo G, Giustarini D, and Milzani A. (2009). Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci 34, 85–96. [DOI] [PubMed] [Google Scholar]
- Dong SH, Tang W, Lukk T, Yu Y, Nair SK, and van der Donk WA (2015). The enterococcal cytolysin synthetase has an unanticipated lipid kinase fold. eLife 4, e07607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, White RC, Clarke TH, Nguyen K, Torralba M, et al. (2019). Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 575, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eley GD, Reiter JL, Pandita A, Park S, Jenkins RB, Maihle NJ, and James CD (2002). A chromosomal region 7p11.2 transcript map: its development and application to the study of EGFR amplicons in glioblastoma. Neuro Oncol 4, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischmann TO, Smith CK, Mayhood TW, Myers JE, Reichert P, Mannarino A, Carr D, Zhu H, Wong J, Yang RS, et al. (2009). Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry 48, 2661–2674. [DOI] [PubMed] [Google Scholar]
- Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, et al. (2014). A promoter-level mammalian expression atlas. Nature 507, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman M. (1999). Chemistry, biochemistry, nutrition, and microbiology of lysinoalanine, lanthionine, and histidinoalanine in food and other proteins. J Agric Food Chem 47, 1295–1319. [DOI] [PubMed] [Google Scholar]
- Geiger T, Velic A, Macek B, Lundberg E, Kampf C, Nagaraj N, Uhlen M, Cox J, and Mann M. (2013). Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol Cell Proteomics 12, 1709–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalbhai K, Jansen G, Beauregard G, Whiteway M, Dumas F, Wu C, and Meloche S. (2003). Negative regulation of MAPKK by phosphorylation of a conserved serine residue equivalent to Ser212 of MEK1. J Biol Chem 278, 8118–8125. [DOI] [PubMed] [Google Scholar]
- Grek CL, Zhang J, Manevich Y, Townsend DM, and Tew KD (2013). Causes and consequences of cysteine S-glutathionylation. J Biol Chem 288, 26497–26504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Zeng M, Dutta D, Koh TH, Chen J, and van der Donk WA (2017). LanCL proteins are not involved in lanthionine synthesis in mammals. Sci Rep 7, 40980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E. (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Chen M, Pang D, Bi D, Zou Y, Xia X, Yang W, Luo L, Deng R, Tan H, et al. (2014). Developmental and activity-dependent expression of LanCL1 confers antioxidant activity required for neuronal survival. Dev Cell 30, 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubatsch I, Ridderstrom M, and Mannervik B. (1998). Human glutathione transferase A4–4: an alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem J 330 ( Pt 1), 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard S, and Thornton J. (1993). NACCESS (University College London; ). [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaji H, Kasukawa T, Forrest A, Carninci P, and Hayashizaki Y. (2017). The FANTOM5 collection, a data series underpinning mammalian transcriptome atlases in diverse cell types. Sci Data 4, 170113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokhlatchev A, Xu S, English J, Wu P, Schaefer E, and Cobb MH (1997). Reconstitution of mitogen-activated protein kinase phosphorylation cascades in bacteria. Efficient synthesis of active protein kinases. J Biol Chem 272, 11057–11062. [DOI] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. (2014). A draft map of the human proteome. Nature 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoureaux TL, and Lee DH (2011). Chemical activation of MEK1--a redox trigger for evaluating the effects of phosphorylation. Chem Commun 47, 8623–8625. [DOI] [PubMed] [Google Scholar]
- Lee B, and Richards FM (1971). J Mol Biol 55, 379–400. [DOI] [PubMed] [Google Scholar]
- Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW, and van der Donk WA (2010). Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria. Proc Natl Acad Sci USA 107, 10430–10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and van der Donk WA (2007). Identification of essential catalytic residues of the cyclase NisC involved in the biosynthesis of nisin. J Biol Chem 282, 21169–21175. [DOI] [PubMed] [Google Scholar]
- Li B, Yu JP, Brunzelle JS, Moll GN, van der Donk WA, and Nair SK (2006). Structure and mechanism of the lantibiotic cyclase involved in nisin biosynthesis. Science 311, 1464–1467. [DOI] [PubMed] [Google Scholar]
- Li H, Xu H, Zhou Y, Zhang J, Long C, Li S, Chen S, Zhou JM, and Shao F. (2007). The phosphothreonine lyase activity of a bacterial type III effector family. Science 315, 1000–1003. [DOI] [PubMed] [Google Scholar]
- Linetsky M, Hill JM, LeGrand RD, and Hu F. (2004). Dehydroalanine crosslinks in human lens. Exp Eye Res 79, 499–512. [DOI] [PubMed] [Google Scholar]
- Linetsky M, and LeGrand RD (2005). Glutathionylation of lens proteins through the formation of thioether bond. Mol Cell Biochem 272, 133–144. [DOI] [PubMed] [Google Scholar]
- Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, and Gray NS (2013). Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chan ASH, Liu H, Cochrane SA, and Vederas JC (2011). Solid supported chemical syntheses of both components of the lantibiotic lacticin 3147. J Am Chem Soc 133, 14216–14219. [DOI] [PubMed] [Google Scholar]
- Ma S, Caprioli RM, Hill KE, and Burk RF (2003). Loss of selenium from selenoproteins: conversion of selenocysteine to dehydroalanine in vitro. J Am Soc Mass Spectrom 14, 593–600. [DOI] [PubMed] [Google Scholar]
- Mayer H, Bauer H, Breuss J, Ziegler S, and Prohaska R. (2001a). Characterization of rat LANCL1, a novel member of the lanthionine synthetase C-like protein family, highly expressed in testis and brain. Gene 269, 73–80. [DOI] [PubMed] [Google Scholar]
- Mayer H, Pongratz M, and Prohaska R. (2001b). Molecular cloning, characterization, and tissue-specific expression of human LANCL2, a novel member of the LanC-like protein family. DNA Seq 12, 161–166. [DOI] [PubMed] [Google Scholar]
- Mayer H, Salzer U, Breuss J, Ziegler S, Marchler-Bauer A, and Prohaska R. (1998). Isolation, molecular characterization, and tissue-specific expression of a novel putative G protein-coupled receptor. Biochim Biophys Acta 1395, 301–308. [DOI] [PubMed] [Google Scholar]
- McFadden PN, and Clarke S. (1987). Conversion of isoaspartyl peptides to normal peptides: implications for the cellular repair of damaged proteins. Proc Natl Acad Sci U S A 84, 2595–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Médard G, Pachl F, Ruprecht B, Klaeger S, Heinzlmeir S, Helm D, Qiao H, Ku X, Wilhelm M, Kuehne T, et al. (2015). Optimized chemical proteomics assay for kinase inhibitor profiling. J Proteome Res 14, 1574–1586. [DOI] [PubMed] [Google Scholar]
- Meister A, and Anderson ME (1983). Glutathione. Annu Rev Biochem 52, 711–760. [DOI] [PubMed] [Google Scholar]
- Mladkova J, Sanda M, Matouskova E, and Selicharova I. (2010). Phenotyping breast cancer cell lines EM-G3, HCC1937, MCF7 and MDA-MB-231 using 2-D electrophoresis and affinity chromatography for glutathione-binding proteins. BMC Cancer 10, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison PM, Foley PJ, Warriner SL, and Webb ME (2015). Chemical generation and modification of peptides containing multiple dehydroalanines. Chem Commun 51, 13470––13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen FH, Berglund H, and Vedadi M. (2007). The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protocols 2, 2212––2221. [DOI] [PubMed] [Google Scholar]
- Peng J, and Gygi SP (2001). Proteomics: the move to mixtures. J Mass Spectrom 36, 1083–1091. [DOI] [PubMed] [Google Scholar]
- Repka LM, Chekan JR, Nair SK, and van der Donk WA (2017). Mechanistic understanding of lanthipeptide biosynthetic enzymes. Chem Rev 117, 5457–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG, and Cho CS (2010). Blot-based detection of dehydroalanine-containing glutathione peroxidase with the use of biotin-conjugated cysteamine. Methods Enzymol 474, 23–34. [DOI] [PubMed] [Google Scholar]
- Robbins DJ, Zhen E, Owaki H, Vanderbilt CA, Ebert D, Geppert TD, and Cobb MH (1993). Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J Biol Chem 268, 5097–5106. [PubMed] [Google Scholar]
- Schmutz C, Ahrne E, Kasper CA, Tschon T, Sorg I, Dreier RF, Schmidt A, and Arrieumerlou C. (2013). Systems-level overview of host protein phosphorylation during Shigella flexneri infection revealed by phosphoproteomics. Mol Cell Proteomics 12, 2952–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Bobeica SC, Wang L, and van der Donk WA (2019). CylA is a sequence-specific protease involved in toxin biosynthesis. J Ind Microbiol Biotechnol 46, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, and van der Donk WA (2012). Structural characterization of four prochlorosins: a novel class of lantipeptides produced by planktonic marine cyanobacteria. Biochemistry 51, 4271–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, and van der Donk WA (2013). The sequence of the enterococcal cytolysin imparts unusual lanthionine stereochemistry. Nat Chem Biol 9, 157–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The_Uniprot_consortium (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–d515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeaux CJ, Ha T, and van der Donk WA (2014). A price to pay for relaxed substrate specificity: a comparative kinetic analysis of the class II lanthipeptide synthetases ProcM and HalM2. J Am Chem Soc 136, 17513–17529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kasteren SI, Kramer HB, Jensen HH, Campbell SJ, Kirkpatrick J, Oldham NJ, Anthony DC, and Davis BG (2007). Expanding the diversity of chemical protein modification allows post-translational mimicry. Nature 446, 1105–1109. [DOI] [PubMed] [Google Scholar]
- Veuthey AL, Bridge A, Gobeill J, Ruch P, McEntyre JR, Bougueleret L, and Xenarios I. (2013). Application of text-mining for updating protein post-translational modification annotation in UniProtKB. BMC Bioinformatics 14, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, et al. (2011). Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol 29, 3085–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SK, Weaver JD, Zhang S, and Lei XG (2011). Knockout of SOD1 promotes conversion of selenocysteine to dehydroalanine in murine hepatic GPX1 protein. Free Radic Biol Med 51, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Lyons B, Truscott RJ, and Schey KL (2014). Human protein aging: modification and crosslinking through dehydroalanine and dehydrobutyrine intermediates. Aging Cell 13, 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willwacher J, Raj R, Mohammed S, and Davis BG (2016). Selective metal-site-guided arylation of proteins. J Am Chem Soc 138, 8678–8681. [DOI] [PubMed] [Google Scholar]
- Wu B, and Dong D. (2012). Human cytosolic glutathione transferases: structure, function, and drug discovery. Trends Pharmacol Sci 33, 656–668. [DOI] [PubMed] [Google Scholar]
- Xu B, Wilsbacher JL, Collisson T, and Cobb MH (1999). The N-terminal ERK-binding site of MEK1 is required for efficient feedback phosphorylation by ERK2 in vitro and ERK activation in vivo. J Biol Chem 274, 34029–34035. [DOI] [PubMed] [Google Scholar]
- Yang J, Carroll KS, and Liebler DC (2016). The expanding landscape of the thiol redox proteome. Mol Cell Proteomics 15, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lennard KR, He C, Walker MC, Ball AT, Doigneaux C, Tavassoli A, and van der Donk WA (2018). A lanthipeptide library used to identify a protein-protein interaction inhibitor. Nat Chem Biol 14, 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, and van der Donk WA (2015). Michael-type cyclizations in lantibiotic biosynthesis are reversible. ACS Chem Biol 10, 1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, van der Donk WA, and Chen J. (2014). Lanthionine synthetase C-like protein 2 (LanCL2) is a novel regulator of Akt. Mol Biol Cell 25, 3954–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang L, Liu Y, Xu J, Zhu G, Cang H, Li X, Bartlam M, Hensley K, Li G, et al. (2009). Structure of human lanthionine synthetase C-like protein 1 and its interaction with Eps8 and glutathione. Genes Dev 23, 1387–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Ju Y, Li S, Altaany Z, Wang R, and Yang G. (2014). S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair. EMBO Rep 15, 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng CF, and Guan KL (1994). Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. Embo J 13, 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Li H, Long C, Hu L, Xu H, Liu L, Chen S, Wang DC, and Shao F. (2007). Structural insights into the enzymatic mechanism of the pathogenic MAPK phosphothreonine lyase. Mol Cell 28, 899–913. [DOI] [PubMed] [Google Scholar]
- Zurawski DV, Mitsuhata C, Mumy KL, McCormick BA, and Maurelli AT (2006). OspF and OspC1 are Shigella flexneri type III secretion system effectors that are required for postinvasion aspects of virulence. Infect Immun 74, 5964–5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurawski DV, Mumy KL, Faherty CS, McCormick BA, and Maurelli AT (2009). Shigella flexneri type III secretion system effectors OspB and OspF target the nucleus to downregulate the host inflammatory response via interactions with retinoblastoma protein. Mol Microbiol 71, 350–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) His-pulldown assay for LanCL2-His6 and His6-LanCL1. Only a small fraction of each kinase was pulled down by His6-LanCL1 and LanCL2-His6. (B) Endogenous LanCL1/2 were co-immunoprecipitated by anti-p110, anti-mTOR and anti-Vps34 antibodies in HEK293 cells. (C) MSMS for Dhb-ERK peptide and Dhb-Akt peptide. (D-F) Phosphate elimination by His6-SpvC and glutathionylation of Dhb-ERK (with pTyr) by LanCL2-His6. (D) Phosphate elimination by His6-SpvC using phospho-ERK peptide containing a pTyr residue as substrate. (E) MSMS for Dhb-ERK (with pTyr residue). (F) ESI-LC-MS analysis of LanCL2 activity using Dhb-ERK (with pTyr residue) as substrate. EIC, extracted ion chromatogram; ESI, electrospray ionization. (G) Generation of Dha in GSH peroxidase (GPX) by H2O2 treatment, which was verified by addition of biotinylated cysteamine. No GSH adduct was observed upon incubation of the Dha-GPX with LanCL1/2 and GSH. (H) Left, partial conversion with γ-Glu-Cys (γ-EC) (1 mM) and Dhb-Erk by LanCL2 but not its double mutant after 12 h. Right, near complete conversion with free Cys (1 mM) and Dhb-Erk by LanCL2 but not its double mutant after 12 h. (I) Anti-GSH antibody can recognize non-canonical glutathionylation. (J) His6-phospho-ERK2 was eliminated by His6-OspF and the resulting His6-ERK2-Dhb185pTyr187 was glutathionylated by MBP-LanCL2 as demonstrated by Western blot. (K) Tandem MS analysis of native and glutathionylated CylLS” and CylLL”. (L) Non-canonical glutathionylation is catalyzed by endogenous LanCLs. Glutathionylation of His6-MEK1-Dha222 was observed by Western blotting.
(a) Panel of DBHDA derivatives used in this study. 1,4-Bis-alkylating/elimination agents ‘tuned’ by indicated properties (magenta). (b) Reagent Synthesis: i. SOCl2, reflux, 1.5h then NBS,HBr/CCl4, reflux, 3 h; ii. NH3OH, 0 °C, 1.5h; iii. NaI, acetone, reflux, 4 h; iv. H2O/THF (1:1), 0 °C, 2 h; v. SOCl2, reflux, 1.5h then RNH2 then trituration in H2O. (c) Differential scanning fluorimetry (Niesen et al., 2007) showed strong binding of putative masking/enhancing ligands to form MEK1•ATP(±Mg) and MEK1•ATPγS(±Mg2+) for model mutant substrate MEK1-C222. (d) MEK1 mutant proteins, in the absence of masking nucleotide / metal gave products of unwanted reaction at Cys207 with parent reagent DBHDA. MEK1-C218 (22 μM) in 20 mM Tris, 100 mM NaCl, pH 8 buffer treated with DBHDA (4.4 mM) at 37 °C afforded overalkylated MEK1Dha218 (Mcalc.=45862, Mobs.=45866) after 4 h; MEK1-C222 (22 μM) in the same buffer treated with DBHDA (4.4 mM) at 37 °C afforded overalkylated MEK1-Dha222 (Mcalc.=45862, Mobs.=45867) after 4 h, MSMS of the tryptic peptide 206–227 confirmed the overalkylation at C207; MEK1-C218C222 (22 μM) in the same buffer treated with DBHDA (4.4 mM) at 37 °C afforded overalkylated stapled MEK1-C218C222 (Mcalc.=46052, Mobs.=46057) after 4 h. (e) Use of alternative alkylating agents did not prevent unwanted C207 reaction in MEK1-C222. MEK1-C222 (33 μM) in 20 mM Tris, 100 mM NaCl, pH 8 buffer treated with DBDMet (6.6 mM) at 37 °C afforded a mixture 85% MEK1-Dha222 (Mcalc.=45642, Mobs.=45646) and 15% overalkylated MEK1-Dha222 (Mcalc.=45890, Mobs.=45891) after 3 h; MEK1-C222 (22 μM) in the same buffer treated with DBDGla (4.4 mM) at 37 °C afforded overalkylated MEK1Dha222 (Mcalc.=45982, Mobs.=45981) after 3 h; MEK1-C222 (33 μM) in the same buffer treated with DBHDAc (6.6 mM) at room temperature afforded substituted MEK1 (Mcalc.=45979, Mobs.=45983) after 24 h. (f) Treatment of MEK1-C222 with DBHDA in the presence of ATP. MEK1-C222 (22 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ATP, pH 8 buffer treated with DBHDA (4.4 mM) at 37 °C afforded a mixture of 85% MEK1-Dha222 (Mcalc.=45642, Mobs.=45645) and 15% overalkylated MEK1-Dha222 (Mcalc.=45862, Mobs.=45866) after 6 h.
(a) Treatment of MEK1-C222 with DBHDA in the presence of ADP-Mg. MEK1-C222 (33 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBHDA (6.6 mM) at 37 °C afforded pure MEK1-Dha222 (Mcalc.=45642, Mobs.=45646); (b) LCMSMS of the tryptic/AspN peptides containing cysteine residues confirmed the formation of Dha exclusively at position 222. (c) MEK1-C222 (44 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBDGla (8.8 mM) at 37 °C afforded MEK1-Dha222 (Mcalc.=45642, Mobs.=45641) after 4 h; Dha position was confirmed by LC-MSMS of peptide 206–227; (d) MEK1-C222 (44 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DIB (2000 equiv) at 37 °C afforded MEK1-Dha222 (Mcalc.=45642, Mobs.=45645) after 20 h; Dha position was confirmed by LC-MSMS of peptide 207–227; (e) MEK1-C222 (44 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with MDBP (4.4 mM) at 37 °C afforded MEK1-Dha222 (Mcalc.=45642, Mobs.=45642) after 5 h, Dha position was confirmed by LC-MSMS of peptide 217–227.
(a) Treatment of MEK1C-218 with DBHDA in the presence of ADP-Mg. MEK1-C218 (22 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBHDA (4.4 mM) at 37 °C afforded a mixture of MEK1Dha218 (Mcalc.=45642, Mobs.=45645) and 25% overalkylated MEK1-Dha222 (Mcalc.=45785, Mobs.=45785) after 15 h; (b) LC-MSMS of the tryptic peptide 206–227 confirmed the formation of Dha at position 218; (c) Longer reaction time lead to the formation of more adducts. (d) MEK1-C218 (44 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBDGla (4.4 mM) at 37 °C afforded exclusively MEK1-Dha218 (Mcalc.=45642, Mobs.=45647) after 20 h, the position was confirmed by LCMSMS analysis of peptide 206–227; (e) MEK1-C218 (44 μM) in the same buffer treated with MDBP (4.4 mM) at 37 °C afforded exclusively MEK1-Dha218 (Mcalc.=45642, Mobs.=45647) after 4 h, the position was confirmed by LC-MSMS analysis of peptide 206–227. (f) Treatment of Δ160MEK1-C218 with MDBP in the presence of ADP-Mg. Δ1–60MEK1-C218 (55 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with MDBP (5.5 mM) at room temperature only afforded Δ160MEK1-Dha218 (Mcalc.=36611, Mobs.=36616) after 14 h.
(a) Treatment of MEK1-C218C222 with DBHDA analogues in the presence of nucleotide-Mg. MEK1-C218C222 (22 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ATP, 25 mM MgCl2, pH 8 buffer treated with DBHDA (4.4 mM) at 37 °C afforded a mixture 60% pMEK1-Dha222 (Mcalc.=45738, Mobs.=45741), 20% MEK1-Dha222 (Mcalc.=45658, Mobs.=45661) and 20% phosphorylated stapled MEK1-C218C222 (Mcalc.=45912, Mobs.=45914) after 5 h; (b) MEK1-C218C222 (22 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBHDA (4.4 mM) at 37 °C afforded a mixture 50% MEK1-Dha222 (Mcalc.=45658, Mobs.=45658) and 50% stapled MEK1C218C222 (Mcalc.=45832, Mobs.=45831) after 5 h; (c) MEK1-C218C222 (44 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with DBHDA (8.8 mM) at 37 °C afforded a mixture 23% MEK1-Dha218Dha222 (Mcalc.=45624, Mobs.=45627), 18% MEK1-Dha222 (Mcalc.=45658, Mobs.=45668) and 59% stapled MEK1-C218C222 (Mcalc.=45946, Mobs.=45951) after 14 h. (d) Treatment of MEK1-C218C222 with MDBP in the presence of ADP-Mg. MEK1-C218C222 (22 μM) in 20 mM Tris, 100 mM NaCl, 10 mM ADP, 25 mM MgCl2, pH 8 buffer treated with MDBP (4.4 mM) at room temperature afforded exclusively MEK1-Dha218Dha222 (Mcalc.=45624, Mobs.=45628) after 15 h; (e) LCMSMS of the tryptic peptide 206–227 confirmed the formation of Dha at position 218 and 222. (f) Comparative circular dichroism measurement of MEK1-Cs and MEK1-Dhas.
(a) Sequential and distributive proposed mechanism of ERK1 phosphorylation by MEK1. (b) The phosphorylation of ERK1K71R by MEK1 construct was monitored by LCMS. The intensities of the deconvoluted peaks corresponding to ERK1 (Mcalc. = 44058), pERK1 (Mcalc. = 44138), and ppERK1 (Mcalc. = 44218) were recorded over time (Figure S18). The conversions were calculated from these intensities and plotted. (c) Differential equation defined the phosphorylation constants kf1 and kf2. These constants were calculated by fitting the analytical equations. (d) ESI-MS progress curves allowing time course analysis of ERK1 by MEK1 variants. ERK1-K71R was incubated with MEK1 variant in 50 mM Tris, 30 mM NaCl, 1 mM ATP, 10 mM MgCl2, 0.2 mM DTT, pH 7.5 at 30 °C.
(A) End-point assays of LanCL1 and LanCL2 (6.0 μM) with His6- MEK1-Dha218 as the substrate for glutathionylation. The conversions of MEK1-Dha218 (6 μM) to MEK1-GS218 after 2-h incubation with 1.0 mM GSH at 25 °C were shown by deconvoluted spectra of LC-MS. (B) Wild-type or mutant LanCL catalyze C-S glutathionylation of ERK1-Dha202. The Michael addition of 0.5 mM GSH to Dha-containing ERK protein (6 μM) was monitored by LC-MS after 3-h incubation with LanCL proteins (6.0 μM) at 25 °C. Top right inset: Time-dependent GSH addition to ERK1-Dha202 catalyzed by wild-type (LanCL1 or LanCL2) or mutant LanCL proteins (LanCL1-C322A or LanCL2-C321A/C367A). The conversion to ERK1-GS202 was quantified using intact mass LC-ESI-MS and is plotted here as the value over background chemical GSH addition (~0.13 M−1 s−1 under these conditions) in the absence of enzyme. (C) LanCL2 adds GSH to GST-3XFlag-ERK2-Dhb185pY187 generated using the pThr lyase OspF. (D) Kaplan-Meier survival curve of female WT (n = 33) and LanCL TKO (n = 27) FVB mice (P = 0.0052) and male WT (n = 25) and LanCL TKO (n = 20) FVB mice (P = 0.0332). Gehan-Breslow-Wilcoxon P test. (E) Survival curve of age-matched FVB WT mice (n = 24) and LanCL2 −/− mice (n = 50).
Supplemental Table S1. Unbiased protein pulldown and proteomics analysis. Related to Figure 1E.
Supplemental Table S2. Chemical generation of Dha-containing MEK1 variants and their kinase activity. Related to Figures 3 and 4, and Supplemental Figures S2–S6.
Supplemental Table S3. Analysis of the proteomic literature for the presence of Dha and Dhb proteins and their conjugates with GSH, β-mercaptoethanol, Cys, and dithiothreitol. Related to Figures 5, S7.
Supplemental Table S4. Oligonucleotide sequences used in this study. Related to the STAR Methods.
Data Availability Statement
The published article includes all datasets analyzed during this study. Structural data has been deposited in the PDB under ID 6WQ1. Mass spectral data and Western blot data have been deposited at Mendeley Data, http://dx.doi.org/10.17632/sgjzwcxgsr.1 and at https://ora.ox.ac.uk/objects/uuid:d8729aad-ca98–4f06-b14a-0a75ea306584.