

Abstract
Inflammatory bowel disease (IBD) affects 6.8 million people globally. A variety of factors have been implicated in IBD pathogenesis, including host genetics, immune dysregulation and gut microbiota alterations. Emerging evidence implicates intestinal epithelial glycosylation as an underappreciated process that interfaces with these three factors. IBD is associated with increased expression of truncated O-glycans as well as altered expression of terminal glycan structures. IBD genes, glycosyltransferase mislocalization, altered glycosyltransferase and glycosidase expression and dysbiosis drive changes in the glycome. These glycan changes disrupt the mucus layer, glycan–lectin interactions, host–microbe interactions and mucosal immunity, and ultimately contribute to IBD pathogenesis. Epithelial glycans are especially critical in regulating the gut microbiota through providing bacterial ligands and nutrients and ultimately determining the spatial organization of the gut microbiota. In this Review, we discuss the regulation of intestinal epithelial glycosylation, altered epithelial glycosylation in IBD, and mechanisms for how these alterations contribute to disease pathobiology. We hope that this Review provides a foundation for future studies on IBD glycosylation and the emergence of glycan-inspired therapies for IBD.
Intestinal epithelial glycosylation is influenced by host genetics, the environment and the gut microbiota. In this Review, Kudelka et al. describe the functions of epithelial glycans and discuss the role of epithelial glycosylation in Crohn’s disease and ulcerative colitis.
Introduction
Inflammatory bowel disease (IBD), including Crohn’s disease and ulcerative colitis, affects 6.8 million people globally and is thought to be the result of inappropriate immune activation in response to the gut microbiota in susceptible individuals1–5. Multiple factors contribute to disease pathogenesis, including genes, the environment, the gut microbiota and the immune system. Although early studies focused on identifying host factors that contribute to IBD pathogenesis, advances in next-generation sequencing have led to an emerging interest in the gut microbiome.
The human gut microbiota consists of 1011 bacteria, 108 archaea, 108 viruses and 106 fungi per gram of stool and has diverse roles in regulating metabolism, immunity and health 6–8. Although many bacterial genes are shared across individuals, gut microbiota composition (such as the relative levels of specific species) is highly diverse between people as well as in different parts of the gut9,10. The gut microbiota in human IBD demonstrates reduced diversity, a shift in the abundance of specific taxa, and altered functional capacity7. Patients with IBD have a reduction in levels of Bacteroides, Firmicutes, Clostridia, Ruminococcaceae, Bifidobacterium, Lactobacillus and Faecalibacterium prausnitzii and an increase in levels of Gammaproteobacteria, Escherichia coli and Fusobacterium species7. Functionally, IBD is associated with loss of protective factors, such as short-chain fatty acids (a major nutrient source for colonocytes), as well as with an increase in pro-inflammatory factors, such as pathways involved in auxotrophy (a feature of pathobionts), sulfate transport, oxidative stress and toxin secretion, in addition to changes in lipopolysaccharide (LPS) structure that are critical for establishing an inflammatory versus tolerogenic milieu7,11,12.
A major question is how genes, the environment and the gut microbiota interact to contribute to IBD pathogenesis. Epithelial glycans [G] are a major component of the intestinal mucosa, facilitate interactions between the gut microbiota and the intestinal epithelia, and are regulated by host genetics and the environment13–16. Thus, they are well-placed to integrate host, microbial, and environmental cues. Epithelial glycans provide ligands and nutrients and induce host signalling to regulate the gut microbiota, and are altered in IBD15,17–27. These alterations to glycans result in several changes that contribute to IBD pathogenesis, including compromised mucin 2 (MUC2) barrier function, disrupted glycan–lectin [G] interactions, pathological host-microorganism interactions and altered mucosal immunity15,16,19,22,23,28–32. We review the regulation of epithelial glycans, their contribution to disease pathogenesis, and opportunities to target host glycans in IBD. Specifically, we discuss mechanisms of protein glycosylation in general, the normal glycan structures in the gut and their regulation, as well as glycan alterations in IBD and how those alterations arise. Additionally, we compare glycome alterations in IBD and colorectal cancers and discuss downstream effects of altered glycosylation in IBD, including in mucin biology, glycan–lectin interactions, host–microorganism interactions, and immune regulation. Finally, we discuss translational opportunities of glycobiology in IBD. Although the gut contains a variety of types of microorganisms, we primarily focus on glycan–bacteria interactions.
Protein glycosylation
Glycans, or polysaccharides, are an abundant post-translational modification found on the cell surface, in intracellular membranes, and in the cytosol (Fig. 1, Box 1). In mammals, combinations of ten monosaccharides (galactose (Gal); glucose (Glc); mannose (Man); fucose (Fuc); xylose (Xyl); N-acetylgalactosamine (GalNAc); N-acetylglucosamine (GlcNAc); glucuronic acid (GlcA); iduronic acid (IdoA); and sialic acids (Sia; this is a family of monosaccharides, the most common of which is N-acetylneuraminic acid (Neu5Ac))) are attached in branched and linear patterns with α or β glycosidic linkages to form thousands of unique structures, sometimes called the glycocode33–35. In vertebrates, this complex system is regulated by >700 genes (called glycogenes), accounting for 1–2% of the genome, that include those encoding glycosyltransferases, sugar transporters and glycan-binding proteins36. In addition, many more genes indirectly regulate glycosylation37. Although glycans can form free structures, they are typically attached to other macromolecules to form glycoconjugates, including glycoproteins and glycolipids, which are either membrane-associated (glycolipids and glycoproteins) or cytosolic (glycoproteins)38 (Fig. 1).
Figure 1. Mammalian glycan classes and monosaccharides.
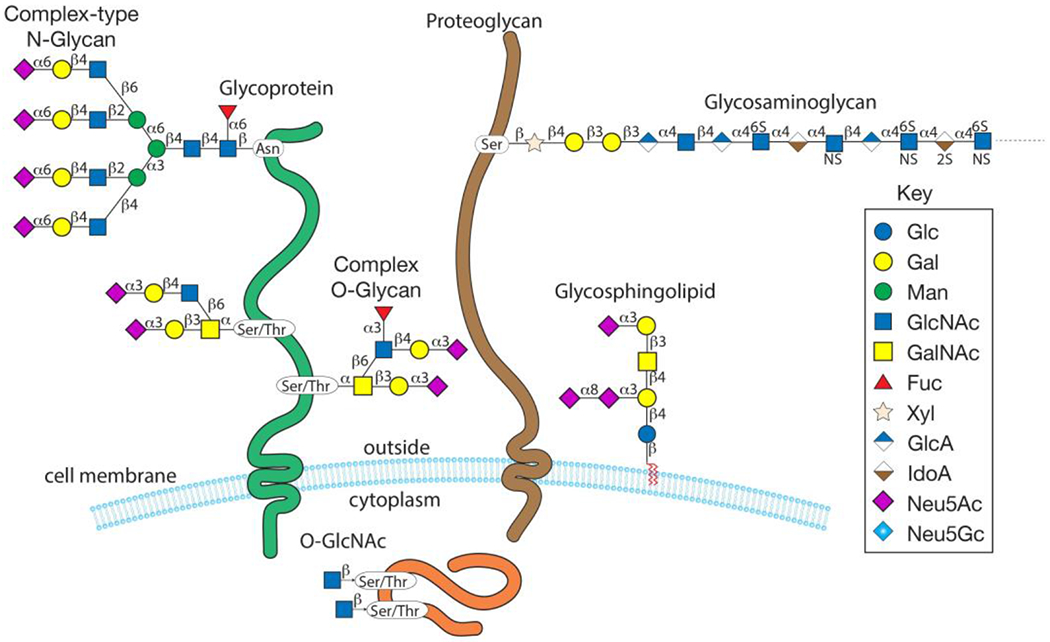
Cell-surface glycans, including N-glycans, O-glycans, glycosaminoglycans, glycosphingolipids and intracellular O-GlcNAc, are shown. NS, N-sulfated glucosamine (GlcNSO3); 6S, 6-O-sulfate.
Box 1 | Glycosylation nomenclature.
Glycan nomenclature
There are multiple ways to refer to glycans, including by glycan classes (for example, mucin-type O-glycans and N-glycans), subclasses based on common motifs or core structures within a glycan class (such as high-mannose, hybrid or complex N-glycans, and core 1-based, core-2 based and core-3 based mucin-type O-glycans), common epitopes (such as Lewisa, CHO-131, CAD and ABO), partial structural descriptions (for example, mucin-type core 1 O-glycan (Gal-GalNAc-Ser/Thr)), or complete structural descriptions that include linkage information (for example, mucin-type core 1 O-glycan (Galβ1,3GalNAcα1-Ser/Thr)). In the latter case, β refers to the anomeric linkage (which can be either α or β) of galactose to GalNAc. ‘1,3’ indicates that position 1 on the galactose ring is attached to position 3 on the GalNAc ring. The level of detail used to describe a glycan in general and in this Review often depends on the biological function of the glycan. In some cases, the presence or absence of a glycan class, the specific epitope (that, for example, interacts with a glycan-binding protein) or the core structure that determines the number and type of terminal epitopes (for example, high-mannose N-glycans terminating in mannose versus complex N-glycans that can be decorated with diverse sialylated and fucosylated termini) are most important in defining function. Further, depending on the type of structural analysis performed in general, linkage information is often unavailable, whereas in other cases it is empirically defined. Thus, linkage information is often omitted from the structural description (for example, in GalGalNAc versus Galβ1,3GalNAc), although the linkage information can sometimes be inferred. This is similar to how peptide mass fingerprinting can be used for protein identification in the absence of de novo sequencing.
Glycosyltransferase nomenclature
Glycosyltransferases are divided into families on the basis of amino-acid sequence and named on the basis of enzymatic activity. Sometimes a single glycosyltransferase can have multiple names, in part reflecting the history of its discovery. For example, the enzyme that extends GalNAc-Ser/Thr (also known as Tn antigen) by adding a galactose (at the β1,3 linkage) to form Galβ1,3GalNAc-Ser/Thr (also known as core 1 (or T antigen)) is called core 1 synthase (core 1 β1,3-galactosyltransferase 1 (C1GALT1)) or T-synthase, to reflect the two names used for the glycan (core 1 or T antigen).
Glycans that are attached to proteins regulate protein stability and oligomerization, and serve as ligands for glycan-binding proteins, including antibodies and lectins. Functionally, glycans have essential roles in a variety of biological processes, such as cell adhesion, cell growth, cell death, cell migration, embryonic development, homeostasis and immunity. Deletion of genes encoding major classes of glycans, such as N-glycans or O-glycans, is typically embryonically lethal, whereas disruption of monosaccharides within terminal epitopes of glycan chains, rather than monosaccharides directly attached to the protein backbone, results in diverse phenotypes39.
N-linked and O-linked glycans
N-linked and O-linked glycans, a major component of both the cell surface glycocalyx and the secreted glycoproteome38,40, constitute the two major classes of glycoproteins, and are defined by their glycan–protein linkage. N-linked glycans are linked by the nitrogen of asparagine to form a Man3GlcNAc2Asn core in the Asn-X-Ser/Thr sequon [G] and are added en bloc to proteins in the endoplasmic reticulum before being processed in the endoplasmic reticulum and Golgi (Supplementary Figure 1). O-linked glycans are linked to the oxygen atom of serine or threonine in glycoproteins. Mucin-type O-glycans, which are attached to mucins (a family of glycoproteins), are the most abundant membrane-associated glycans41,42. Unlike N-glycans, mucin-type O-glycans (hereafter referred to as O-glycans unless otherwise specified) do not attach to a conserved sequon; however, machine learning algorithms predict O-glycosylation sites reasonably well41. Unlike N-glycan biosynthesis, O-glycans are added progressively as glycoproteins traverse the Golgi (Supplementary Figure 2).
N-glycan biosynthesis occurs in the endoplasmic reticulum and the Golgi apparatus (Supplementary Figure 1). In brief, a 14-sugar precursor glycan, Glc3Man9GlcNAc2, is synthesized and transferred to the nascent polypeptide chain and deglucosylated to a monoglucosylated species. The protein folding and quality control machinery evaluates the protein; properly folded proteins are marked by removal of the last glucose. In one synthetic pathway, mannose residues are removed followed by addition of GlcNAc by GlcNAc-transferase 1 (GnT1) and then eventually by GnT2 to form a biantennary GlcNAc2Man3GlcNAc2 structure. This structure is then modified by the addition of a core fucose to the first GlcNAc. This structure can be further branched, modified by addition of a bisecting GlcNAc, and/or modified by extension with galactose, GlcNAc, fucose and/or sialic acid. N-glycans that terminate in mannose (high-mannose), galactose or sialic acid (complex) or some combination (hybrid) can be found on the cell surface.
Mucin-type O-glycan biosynthesis is described in detail in Supplementary Figure 2. Mucin-type O-glycans consist of hundreds to thousands of unique structures built out of nine core structures that are attached to >80% of the cell surface proteins and secreted proteins33,34,41,43–45 (Fig. 2). Cores 1 – 4 are the most common cores, with expression of core 1 (also known as T antigen and Thomsen–Friedenreich (TF) antigen; Galβ1,3GalNAc) and 2 [GlcNAcβ1,6(Galβ1,3)GalNAc] in all cell types and core 3 (GlcNAcβ1,3GalNAc) and 4 [GlcNAcβ1,6(GlcNAcβ1,3)GalNAc] exclusively in the intestinal epithelium (Supplementary Figure 2). All mucin-type O-glycans are built from a single GalNAc attached via α linkage to serine or threonine in a glycoprotein by a family of 20 polypeptide GalNAc-transferases to form GalNAc-α-Ser/Thr (also known as Tn antigen)46. These enzymes have been extensively studied and can be differentiated according to expression patterns (whether their expression is ubiquitous or tissue-specific) and by their ability to add GalNAc to either an unmodified or a Tn-antigen-containing glycopeptide46. Although Tn can be expressed in diseased cells, healthy tissues extend Tn beyond a single GalNAc to form the core O-glycans35,47,48. T-synthase (encoded by C1GALT1), which requires the molecular chaperone COSMC to form a properly folded active glycosyltransferase, extends a single Tn antigen by adding galactose to Tn to form core 1 (Supplementary Figure 2)49. Similarly, core 3 β1,3-N-acetylglucosaminyltransferase (C3GnT) adds GlcNAc to Tn to form core 350. Either core 1 or core 3 can be extended by the addition of a branching GlcNAc at the C6 hydroxyl group of GalNAc to form core 2 or core 4 structures, respectively35. These structures can then be further modified by addition of galactose, GlcNAc, fucose, sialic acid and sulfate to form glycans of various lengths that are decorated by a variety of terminal epitopes. Although some epitopes are unique to O-glycans (for example, CHO-131 (a core 2 structure terminating in sialyl-Lewis x, a ligand for P-selectin)), many terminal extensions are present on both O-glycans and N-glycans, including blood group antigens and Lewis antigens [G] (Supplementary figure 3).
Figure 2. O-glycan core structures.
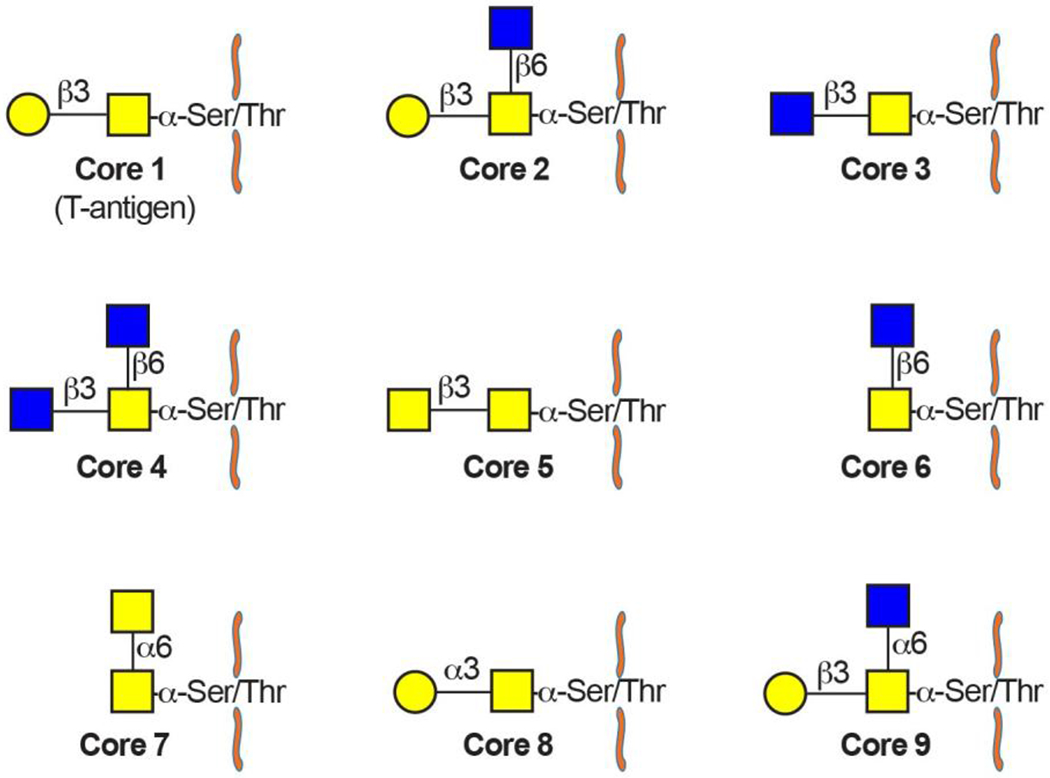
Cores 1–9 are shown. Core 1–4-based structures constitute the majority of O-glycans, whereas cores 5–9 are generally absent or a relatively minor component. Cores 1 and 2 are ubiquitous whereas cores 3 and 4 are restricted to the gastrointestinal tract.
ABO blood group antigens
ABO blood group antigens are formed by modification of terminal galactose residues by addition of fucose in an α1,2 linkage to form Fucα1,2Gal (also known as the H antigen) (Supplementary figure 3a)51. Whereas FUT2 (historically Se or secretor) encodes the H antigen in epithelial linings of the gastrointestinal tract, FUT1 encodes these structures on erythrocytes51,52. The A and B blood group antigens are formed by addition of GalNAc and Gal in an α1,3-linkage to the galactose of H antigen by the A (A3GALNT) and B (A3GALT1) transferases, respectively. By contrast, individuals with O blood type have a functional FUT2 but lack a functional A or B glycosyltransferase. In that case, H antigen is produced, but it is not modified to form A or B antigens.
Lewis antigens
Lewis antigens are present on glycoproteins and glycolipids on red blood cells and gastrointestinal epithelia or their secreted products. These structures are only made in the gastrointestinal tract and are not red blood cell precursors. Thus, gastrointestinal epithelia release glycolipids that are adsorbed on the red blood cell surface from plasma. Although many healthy individuals express Lewis antigens, these structures are often upregulated in inflammatory states53,54. Lewis antigens are formed by addition of one or two fucose residues to a terminal Gal-GlcNAc structure with either a β1,3 linkage (Galβ1,3GlcNAc, type 1 chain) or a β1,4 linkage (Galβ1,4GlcNAc, type 2 chain). All Lewis structures have a fucose residue attached to the subterminal GlcNAc. If this residue is the sole fucose, then the structure is called Lewisa (for type 1 chains) or Lewisx (for type 2 chains). If a second fucose is also present on the terminal galactose, forming a difucosylated structure, the Lewis structure is called Lewisb (for type 1 chains) or Lewisy (for type 2 chains), respectively (Supplementary figure 3b)51. If only one fucose is added, it is first attached to the GlcNAc. By contrast, if two fucoses are added, the first fucose is attached to the galactose, rather than to the sub-terminal GlcNAc, forming the H antigen51,55,56. Enzymatically, FUT2 (Se), which encodes the H antigen, first synthesizes H (for Lewisb and Lewisy). Then, FUT3 adds fucose to the subterminal GlcNAc. Alternative fucosyltransferases are sometimes involved51,55,56.
The healthy gut glycome
The intestinal epithelial glycome is developmentally and regionally regulated by host and environmental factors13,57,58. Mucin-type O-glycans are the major class of glycan in the gut, accounting for 80% of the mass of human MUC2, the most abundant intestinal mucin 59–61. In humans, the stomach and duodenum contain core 1 and core 2-based structures, the rest of the small intestine contains core 3-based structures, and the colon contains core 3 (primarily in the sigmoid colon; Figure 3a) and core 4-based structures62. These structures are further modified by Gal, GlcNAc, GalNAc, fucose, sialic acid and sulfate and regulated by genes implicated in IBD(Fig. 3)62,63. Terminal epitopes follow a rostral–caudal gradient, with increasing levels of sialic acid, sulfate and Cad antigen [G] and decreasing levels of fucose, blood group antigens and Lewis antigens from the stomach to the rectum57. Although this gradient is present across species, the direction appears to vary. In contrast to humans, mice exhibit an increase in fucose and a reduction in charged species from the stomach to the rectum64. As Lewis and ABO blood group antigens (which are expressed in the proximal gut) vary across the human population, the proximal gut glycome, including that of the caecum and small intestine, is highly variable across the population, in contrast to the relatively invariant distal gut glycome63. The variability in the proximal gut glycome mirrors the variability seen in other secretory materials, such as milk, saliva, lungs and cervix, leading to the hypothesis that the invariant distal gut glycome selects for commensal microorganisms whereas the variable proximal gut glycome and the glycomes of other secretory organs defend against pathogens by providing decoy glycan receptors63. Accordingly, pathogens that are able to subvert this system and bind to epithelial-associated rather than secretory glycans in the gut can cause inflammation19. This hypothesis is further supported by computational studies that suggest that mucus glycan–microorganism interactions support positive selection of commensal microorganisms during the slow transit through the colon and negative selection of pathogens during the fast transit through the small intestine65. Experimental studies involving engineered deletion of mucus glycans support these computational findings15. For example, glycan-deficient mice exhibit loss of colonic mucosa-associated commensal microorganisms without affecting the composition of microbial communities in the small intestine, resulting in spontaneous colitis but no inflammation of the small intestine15. Such mechanisms could be relevant for ulcerative colitis or Crohn’s disease, in both of which the balance between commensal microorganisms, pathobionts, and pathogens influence interactions with the immune system8,66.
Figure 3. Altered glycan structures and genes in inflammatory bowel disease.
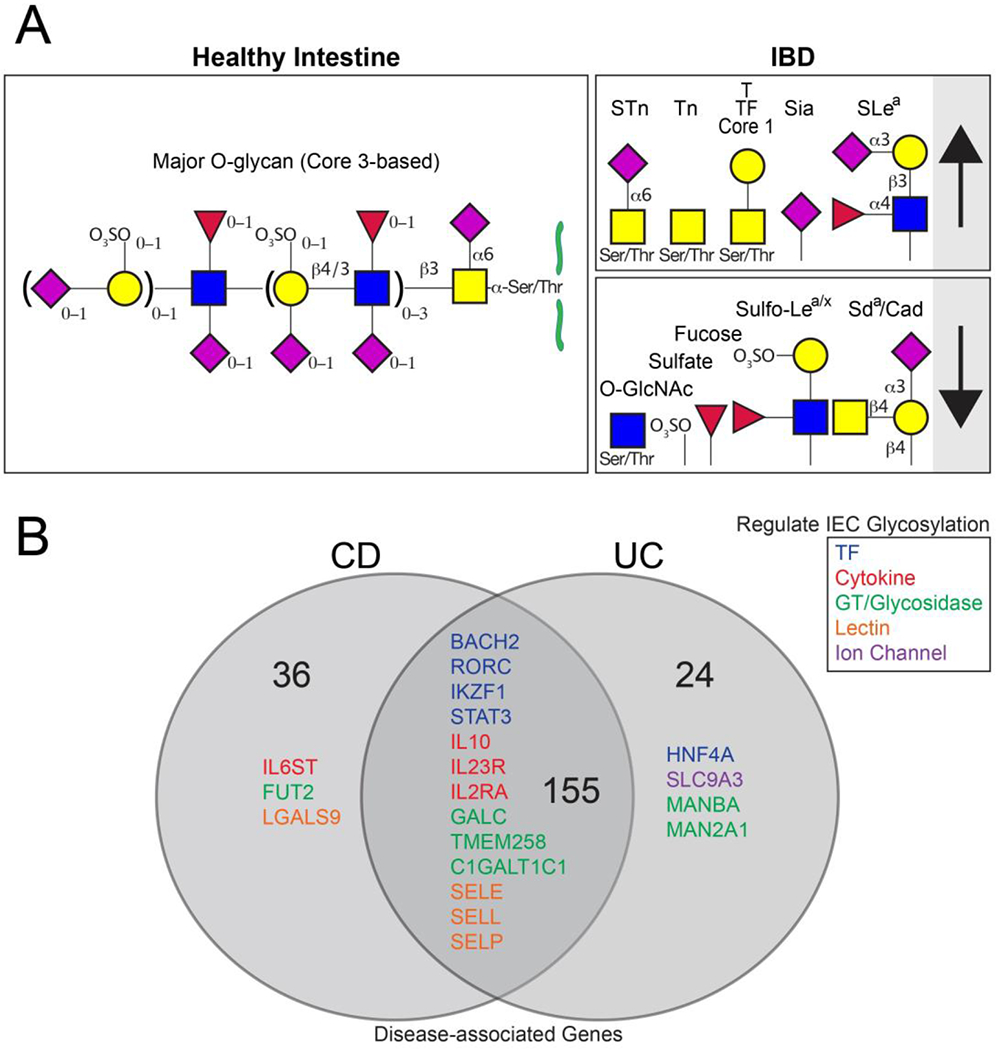
(A) Major O-glycans of the healthy sigmoid colon and alterations in inflammatory bowel disease (IBD) are depicted20,26,28,63,72,112,113,115. (B) All candidate genes for Crohn’s disease, ulcerative colitis or both (IBD) are enumerated, with those implicated in glycosylation listed80,83–89. Genes are divided by function as indicated.
Sulfated motifs on the termini of glycans are one of the structures that most reliably distinguish ileal from colonic glycoproteins by immunohistochemistry: there is increased sulfation in the human colon. Sulfation is likely induced by the gut microbiota: surgical construction of an ileal pouch, which results in a shift to a more colonic morphology and microbiota, results in elevated sulfate67–69. Sulfation is likely to be highly relevant in IBD, as patients have reduced levels of sulfate compared with healthy individuals, which is partly due to increased production of bacterial sulfatases70–72. Interestingly, the increased sulfatase activity is observed in patients with ulcerative colitis but not those with Crohn’s disease70.
Different glycans are expressed in the human fetal gut than in the adult gut, indicating that epithelial glycosylation is developmentally regulated58. In particular, the human fetal gut primarily expresses neutral core 2-based glycans, whereas the adult gut primarily expresses glycans with charged termini (for example, those modified by Neu5Ac and/or sulfate). In addition, Cad antigen is not expressed in the human fetus, and there is an absence of the characteristic gradient of sulfate, sialic acid and fucose seen in the adult gut. Also, some oncofetal antigens are expressed in fetuses but not in adults, such as T antigen (Galβ1,3GalNAc-Ser/Thr) and sialyl-Tn (STn) antigen (Neu5Acα2,6GalNAc-Ser/Thr).
Regulation of epithelial glycosylation
Studies investigating fetal versus adult intestinal glycosylation indicate that the glycome is developmentally regulated. However, whether this regulation depends solely on host cues or also on environmental signals has been a major unanswered question. In contrast to humans, the mouse intestinal glycome primarily consists of core 2-based glycans, and the regional distribution of some terminal epitopes varies compared with humans13,64. Nonetheless, both mice and humans express terminal Cad, sialic acid, sulfate and fucose. Thus, mice are an ideal model to study regulation of epithelial glycosylation.
Epithelial fucosylation has been a model epitope to understand regulation of intestinal glycosylation. Fucose can be added to proteins in α1,6, α1,3 and α1,2 linkages. The lectin Ulex europaeus agglutinin I (UEAI) recognizes α1,2-linked fucose, the expression of which depends on two enzymes: FUT1 and FUT2. α1,2-linked fucose is the major class of fucosylation in the intestinal epithelium in humans and mice 73. Importantly, in mouse models FUT2 regulates fucosylation in most of the intestinal epithelia, whereas FUT1-dependent fucosylation is restricted to Paneth cells and microfold cells (M cells) in the small intestine73,74. Interestingly, preliminary work shows that distinct populations of murine Paneth cells express either FUT1 or both FUT1 and FUT275.
One of the first observations that the gut microbiota can induce epithelial glycosylation arose in studies comparing UEAI staining in conventionally housed with germ-free mice27,76. These studies demonstrated that conventionally housed mice, but not germ-free mice, express α1,2 fucose in the ileum27,76 . Further, colonization of germ-free mice with a commensal gut microorganism, Bacteroides thetaiotaomicron, also induced α1,2 fucose76. Many other microorganisms can also probably induce FUT2 expression, as has been shown for the commensal segmented filamentous bacteria and the pathogen Salmonella enterica subsp. enterica serovar Typhimurium27. Bacteria are able to metabolize fucose, and, interestingly, deleting genes associated with fucose sensing or metabolism inhibits the ability of B. thetaiotaomicron to induce epithelial fucosylation24,76. Some bacteria, such as Bacteroides fragilis, can salvage free fucose and incorporate it into its own glycoconjugates77. This ability provides a competitive advantage in colonization experiments, perhaps by making the bacteria more closely resemble host cells and thus avoid immunological detection, in a process called molecular mimicry [G] .
Although initial experiments proposed that bacteria interact directly with the intestinal epithelia to induce fucosylation, later work suggested an indirect mechanism involving immune cells (Fig. 4). Using a variety of null mutant mouse models, Goto et al. and Pham et al. showed that bacteria interact with group 3 innate lymphoid cells (ILC3s) in the gut to induce expression of IL-22, which interacts with IL-22R on intestinal epithelial cells to induce FUT2 expression in the intestinal epithelia27,78. In addition to the bacteria-inducible IL-22 signal, ILC3s constitutively express lymphotoxin, which is also required for FUT2 expression. These observations suggest a system in which ILC3s secrete lymphotoxin in a microbiota-independent manner to induce baseline intestinal epithelial fucosylation, which is then supplemented by microbially induced IL-22 that achieves increased intestinal epithelial fucosylation. Other immunological signals might also regulate intestinal epithelial fucosylation. Goto et al. showed that T cell-deficient mice, but not B cell-deficient mice, had elevated intestinal epithelial fucosylation, and that IL-10-producing CD4+ T cells, but not soluble IL-10 alone, were responsible for suppressing epithelial fucosylation via transcriptional regulation of FUT279. Thus, components of the innate immune system induce fucosylation whereas components of the adaptive immune system repress it. This microorganism-induced fucosylation seems to be crucial in the small intestine but not in the colon. In contrast to the absence of α1,2 fucose in the ileum of germ-free mice, levels of colonic α1,2 fucose are similar in germ-free and conventionally housed mice76. Therefore, distinct mechanisms regulate epithelial glycosylation in different regions of the gut. Importantly, microbiota-induced glycosylation is not limited to fucosylation. Compared with germ-free mice, conventionally raised mice express a more-complex, extended O-glycome throughout the intestine due to increased levels of many, but not all, glycosyltransferases13.
Figure 4. Immune regulation of epithelial fucosylation.
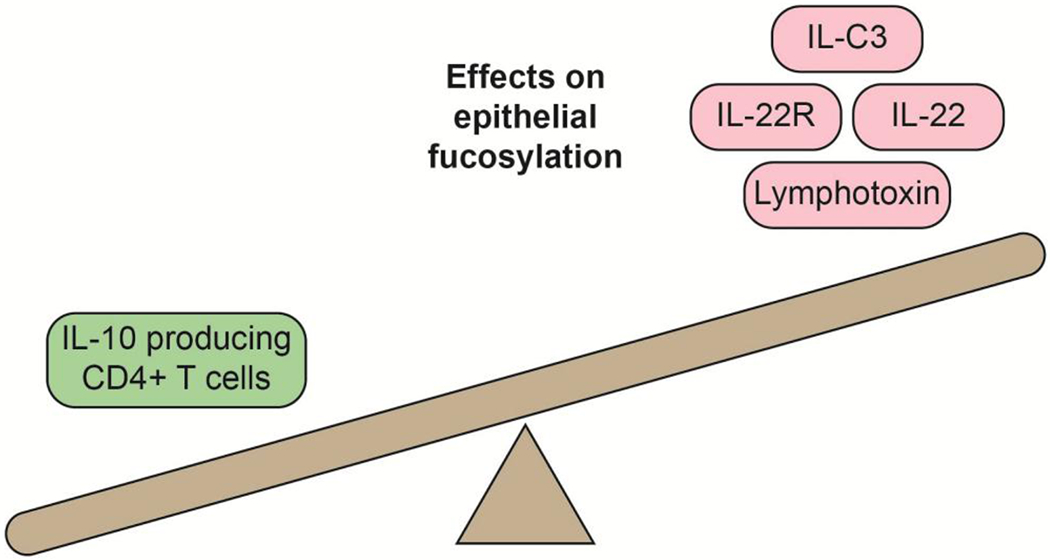
Cytokines, cytokine receptors and immune cells that reduce (left) or increase (right) epithelial fucosylation are depicted.
Epithelial glycosylation in IBD
Alterations in glycosylation in human IBD
Multiple lines of evidence implicate altered epithelial glycosylation in IBD pathogenesis. Such evidence includes the effects of disruption of mucin-type O-glycans, N-glycans and termini common to multiple classes of glycosylation 15,16,20,23,28,80,81. Genome-wide association studies (GWAS) have identified human genes that directly regulate epithelial glycosylation, such as FUT2 and COSMC, and those that indirectly regulate glycosylation via the microbiota–immune–intestinal epithelia axis, such as interleukin-23 (IL-23) and signal transducer and activator of transcription 3 (STAT3), which help regulate FUT2 expression in the intestinal epithelium (Fig. 3b, Table 1, Supplementary Table 1)16,73,80,82–89. In addition to genes associated with O-glycan synthesis, genes that are important for N-glycan synthesis, such as those that encode components of the oligosaccharyltransferase complex81, are also implicated in IBD. Although most glycan-associated genes are altered in both ulcerative colitis and Crohn’s disease (such as C1GALT1C1 and TMEM258), which is consistent with overall disease genetics, a few are specific to either ulcerative colitis (such as MAN2A1) or Crohn’s disease (such as FUT2)15,16,23,27,79–81,83–85,87–105 (Supplementary Table 1).
Table 1.
Genes regulating glycosylation that are implicated in inflammatory bowel disease
| Gene | Change in IBDa | Role in regulating glycosylationb | Refs |
|---|---|---|---|
| Cytokines | |||
| IL6ST | SNP/Increased | Increased N-glycan galactose | 89,90,93,95 |
| IL10 | Decreased | Decreased fucose | 79,90,92,93,101 |
| IL23R | SNP | Increased fucose | 27,85,90,93,99,104 |
| IL2RA | SNP/Decreased | Decreased N-glycan branching | 90,93,97 |
| Lectins | |||
| SELE | Increased | Glycan binding protein | 89,90,93 |
| SELL | Increased | Glycan binding protein | 89,90,93 |
| SELP | Increased | Glycan binding protein | 89,90,93 |
| LGALS9 | Increased | Glycan binding protein; immunosuppressive | 90,91,93 |
| Glycosyltransferases and glycosidases | |||
| FUT2 | Decreased | α1,2-fucosyltransferase | 16,23,27,85,90,93 |
| C1GALT1C1 | Decreased | Chaperone for T-synthase/core 1 O-glycan synthase | 15,80,90,93 |
| GALC | SNP | Lysosomal galactosidase | 90,93 |
| TMEM258 | SNP/Decreased | Oligosaccharyltransferase complex, N-glycan synthesis | 86,90,93 |
| MANBA | SNP | Lysosomal mannosidase | 90,93 |
| MAN2A1 | SNP/Increased | N-glycan processing and maturation | 90,93,105 |
| Transcription factors | |||
| BACH2 | Increased | Increased biantennary monogalactosylated N-glycan with core fucose | 89,90,93 |
| IKZF1 | SNP/Decreased | Decreased fucose, increased bisecting GlcNAc, binds glycosyltransferase promoters | 89,90,93,95 |
| RORC | Decreased | Increased fucose, transcription factor for ILC3 and subsequent FUT2 induction | 27,90,93 |
| HNF4A | SNP/Decreased | Increased fucose, increased GDP-fucose and fucosyltransferase | 89,90,93,98,100,103 |
| STAT3 | Increased | Increased FUT2 via immune signalling | 27,85,90,93,99,102 |
| Gut pH/ion channel | |||
| SLC9A3 | Decreased | Na/H exchanger; gene deletion increases gut lumen pH and bacteria-induced fucose | 85,90,93,94,96 |
Single nucleotide polymorphisms (SNPs) or gene/transcript increase or decrease; SNP/increased or SNP/decreased implies that both SNPs and a change in transcript levels are implicated in IBD.
Increased or decreased implies positive or negative correlation with transcript level; for example, ‘increased’ in this column corresponds to an increase in IBD if the transcript is increased in IBD (see Change in IBD column), but if the transcript is decreased in IBD ‘increased’ in this column would imply a decrease in IBD.
In addition to disease genetics, immunohistochemical and structural studies indicate that IBD, including Crohn’s disease and ulcerative colitis, is associated with a simplified O-glycome106, characterized by increased levels of the truncated O-glycans T (in Crohn’s disease and ulcerative colitis)107, Tn (in ulcerative colitis; not well studied in Crohn’s disease)28, and STn (in Crohn’s disease more than in ulcerative colitis) antigens26,108–110. In addition to class-restricted changes in O-glycans, IBD is associated with alterations in terminal structures that are present on multiple classes of glycosylation, including O-glycans, N-glycans and glycolipids14,20,26,28,107,110–113. In particular, IBD is associated with increased terminal sialylation (in ulcerative colitis; not well studied in Crohn’s disease)112,114, fucosylation (in Crohn’s disease and ulcerative colitis)16,107, reduced terminal sulfation (in ulcerative colitis more than in Crohn’s disease)70,113, reduced Sda and Cad antigens (in ulcerative colitis; not well studied in Crohn’s disease)64 and either increased or decreased Lewis antigens (depending on the specific structure as described below; in ulcerative colitis and Crohn’s disease)26,64,115,116 (Fig. 3a). Sialyl-Lewisa (also known as CA19-9) is increased in Crohn’s disease and ulcerative colitis and Sulfo-Lewisa/x is decreased in ulcerative colitis, which might simply reflect an increased sialylation-to-sulfation ratio rather than altered Lewis antigen synthesis26,115,116. Interestingly, IBD is also associated with distinct changes in serum N-glycans, including an increase in large glycans with a corresponding decrease in hybrid and high-mannose structures, a decrease in fucosylation and galactosylation, and an increase in sialylation (in both Crohn’s disease and ulcerative colitis but more so in Crohn’s disease)117. These identified serum N-glycans can be found on secreted intestinal glycoproteins, immunoglobulins or as acute phase reactants117. In addition to changes in cell surface glycosylation, patients with IBD also have a reduction in intracellular O-GlcNAcylation in the intestinal epithelia (in Crohn’s disease and ulcerative colitis)118. Intriguingly, STn expression in humans seems to predict progression from ulcerative colitis to colitis-associated colorectal cancer 119.
In some cases, changes in glycosylation precede inflammation. Unaffected monozygotic twins of patients with Crohn’s disease or ulcerative colitis exhibit elevated T antigen expression in the epithelial cells of the intestinal crypt surface with concomitant elevation of nuclear factor kappa-B (NF-κB) activation in the same cells, which suggests that genetic or possibly environmental factors induce T antigen expression before disease onset and that T antigen expression might lead to increased inflammatory tone111. As well as altered glycosylation preceding inflammation, glycan structural alterations have also been shown to occur in response to inflammation. For example, the glycans of patients with ulcerative colitis in remission (n=13) resembled those of control patients (n=25; those with histologically normal colons who underwent colonoscopy for anaemia, rectal bleeding or polyp surveillance), whereas inflammation-associated glycan truncation was observed in patients with ulcerative colitis undergoing a flare (n=15)26. Importantly, in patients with active disease the IBD-associated changes in glycosylation normalized within the same individual after resolution of the ulcerative colitis flare, indicating that components of the IBD-associated glycome are reversible within the same individual over time 26. Thus, it is likely that some alterations in intestinal epithelial glycosylation precede inflammation and others result from inflammation.
Mechanisms of altered glycosylation in IBD
Altered cell surface glycosylation can arise from disrupted glycan synthesis, degradation and substrate expression (that is, expression of proteins or lipids to which the glycans attach). Glycan synthesis is complex and relies on coordinated regulation of synthesis and processing genes, the proteins they encode, and the distribution of glycans in organelles, notably the endoplasmic reticulum and the Golgi apparatus. Hundreds of genes are involved in glycan synthesis and at least twenty of these have been implicated in IBD, including those that encode transcription factors, cytokines, glycosyltransferases and glycosidases (N-linked, O-linked and glycolipids), lectins and ion channels. Whereas a few of these are specific to ulcerative colitis or Crohn’s disease, most are shared between the two diseases (Fig. 3b, Supplementary Table 1)15,16,23,27,79,80,85,86,89–105.
In terms of synthesis, single-nucleotide polymorphisms identified by GWAS affect protein structure, transcriptional regulation and alternative splicing regulation of human genes including glycogenes120. Growth factors, such as pro-inflammatory cytokines, activate signal transduction pathways that alter glycosyltransferase expression, such as increasing sialyltransferase and sulfotransferase expression and, subsequently, sialyl-Lewisx expression 121. Mutations in genes involved in extension of mucin-type O-glycans have been observed in small cohorts of patients with IBD 28.
Although the precise mechanisms for altered epithelial glycosylation in IBD are unknown, clues can be gained by analysing mechanisms for altered glycosylation in cancer, which exhibits a similar simplification of the glycome35,122,123. In cancer cell lines (fibroblast, kidney and breast), growth factors have been shown to mislocalize glycosyltransferases, including from the Golgi back to the endoplasmic reticulum, resulting in increased expression of truncated mucin-type O-glycans124,125. Additionally, a wide array of genetic and epigenetic disruptions have been observed in glycogenes in cancer, especially for those in the mucin-type O-glycosylation pathway35,126–128, and altered Golgi pH alters Golgi structure, in turn disrupting glycosyltranferase localization and/or the flow of substrates through the secretory apparatus, resulting in expression of truncated glycans such as T antigen129–131.
Enzymatic and non-enzymatic mechanisms degrade cell surface glycans and might contribute to altered glycosylation in IBD. The gut microbiota produces glycosidases that digest host mucin and non-mucin glycans132, and intestinal inflammation results in oxidative stress, which in other settings has been shown to chemically release or degrade the glycans of glycoconjugates133,134. These mechanisms are likely to be important in IBD pathogenesis but will need to be tested further.
IBD results in altered substrate expression, including altered and ectopic glycoprotein expression as a result of disrupted gene expression and alternative splicing. For example, levels of MUC2 are reduced in both Crohn’s disease and ulcerative colitis, whereas levels of MUC1 are decreased in Crohn’s disease and increased in ulcerative colitis135. The level of substrate expression inversely correlates with glycosite [G] density as well as the abundance of specific glycan structures34,136. In IBD, ectopic expression of gastric mucins in the intestine, as well as alternative splicing of glycoproteins already present in the intestine such as CD44, introduces novel glycosylation substrates and peptide-driven glycan motifs89.
Another clue to altered regulation of intestinal epithelial glycosylation comes from examining the glycomes of germ-free mice, which express a simplified glycome that in part mirrors changes observed in IBD, for example increased T antigen expression 13. Although the mechanisms for this simplification are largely unexplored, a few examples, such as microorganism-induced fucosylation, indicate that specific microbial signals induce intestinal epithelial expression of glycosyltransferases involved in glycan extension through lymphocyte-dependent mechanisms27. Thus, disrupted intestinal epithelial–microorganism crosstalk, for example via genetic disruption of microorganism-sensing genes, might contribute to expression of an IBD-associated glycome.
Epithelial glycome: IBD versus CRC
Colorectal cancer results in a number of glycan alterations that are also observed in IBD. These alterations include truncation of mucin-type O-glycans, such as increased Tn, STn and T antigens; altered terminal epitopes across glycan classes, such as increased sialyl-Lewisa; increased sialic acid; and reduced sulfate 20,28,35,70,110,112–114,116. Such alterations differ from changes that only occur in cancer (and not IBD), such as loss of ABO blood groups and increased N-glycan branching137. In contrast to increased N-glycan branching in breast and colon cancers, IBD exhibits a reduction in human and mouse T cell N-glycan branching that is pro-inflammatory138,139. Similarly, in humans, O-GlcNAcylation [G] is increased in colorectal cancer (CRC) but decreased in IBD 118,140,141.
Although the mechanisms underlying these changes are still under investigation, some mechanisms might be shared between colorectal cancer and IBD. Genetic disruptions in human mucin-type O-glycan biosynthesis have been observed in colorectal cancer cells and IBD tissue28,142. Addition of a single galactose to the Tn antigen to form the T antigen, which serves as a platform for the synthesis of all extended core 1 and core 2-based O-glycans, requires the molecular chaperone COSMC49. COSMC, in effect, serves as a master regulator of O-glycosylation. Disruption of this chaperone has been observed in cancer and IBD. In human cancer tissues or cancer cells (cervical, leukaemia, colorectal, melanoma and pancreas), mutation, loss of heterozygosity, and epigenetic silencing lead to loss of COSMC activity35,47,48,142–145, whereas in IBD, inactivating mutations in COSMC have been identified in patient biopsy samples, and GWAS studies have identified single-nucleotide polymporphisms in COSMC associated with IBD28,80. Loss of Cosmc in mice and humans leads to O-glycan truncation and expression of Tn antigen or STn antigen, its sialylated counterpart142,146,147. STn expression can also arise owing to overexpression of the ST6GalNAc-I sialyl transferase, which modifies Tn148–150. However, whether these or other mechanisms are responsible for pathological Tn and STn expression in the gut is still unclear128,151. In addition to Tn antigen and STn antigen, T antigen is also overexpressed in IBD and CRC19,20. The mechanisms for T antigen overexpression are less clear, but expression likely depends on glycosyltransferase mislocalization, either due to growth factor signalling or to an altered Golgi structure, arising for example from Golgi alkalinization 124,125,131.
IBD increases the risk of CRC in patients with ulcerative colitis more than it does in patients with Crohn’s disease152,153. Notably, in Crohn’s disease the risk of intestinal cancer is mainly elevated in people with colonic disease and not ileal disease154. Thus, some of these shared glycan alterations in IBD and CRC might have functional consequences. Indeed, STn predicts an increased rate of malignant progression in patients with ulcerative colitis109, whereas engineered expression of Tn (and concomitant STn) in the mouse gut results in ulcerative colitis-like pathogenesis and subsequently CRC15,22,28, with corresponding activation of oncogenic pathways143.
Food substances and bacteria both interact with these IBD and CRC-associated intestinal epithelial glycans. Plant lectins, such as those contained in peanuts, bind to T antigen and induce mitogenic activity in human cell line models155–157, whereas the gut microbiota, constituents of which bind to and consume host glycans, exhibits similar alterations in IBD and cancer, which suggests a possible link between IBD, CRC, epithelial glycans and the gut microbiota. In particular, in humans, IBD and CRC both exhibit a reduction in bacterial diversity, a reduction in the genus Bacteroides, and an enrichment of specific bacterial species, such as Fusobacterium nucleatum, Campylobacter species, colibactin-producing polyketide synthase (pks)+ E. coli, enterotoxigenic B. fragilis and Enterococcus faecalis158–162.
Altered glycosylation drives some of these bacterial changes. Engineered expression of Tn and STn and subsequent loss of extended O-glycans in the gut leads to loss of bacterial diversity, reduction of Bacteroides species, which consume host carbohydrates, and the emergence of pro-inflammatory Campylobacter species in mice15. Additionally, in mice, expression of T antigen recruits F. nucleatum to the tumour via fatty-acid binding protein 2 (Fap2), a Gal-GalNAc bacterial lectin19. These bacteria activate inflammatory cascades and/or damage DNA160. Thus, IBD-associated changes to the epithelial glycome, which persist in CRC, result in recruitment of pro-inflammatory and pro-tumorigenic bacteria.
Effects of altered glycosylation in IBD
Evidence from a variety of mouse models with engineered expression of IBD-associated glycosylation supports a functional role for altered glycosylation in IBD. These models include mice that express truncated Tn and STn antigens through deletion of Cosmc (also known as C1galt1c1), C1galt1 (the gene encoding T-synthase), or B3gnt6 (also known as core 3 synthase, which synthesizes Core 3 O-glycan); mice with loss of core 2 and core 2-based extensions as a result of deletion of core 2 GlcNAc-transferase-2 (Gcnt3); mice with reduced sulfation as a result of deletion of N-acetylglucosamine 6-O-sulfotransferase 2 (GlcNAc6ST2, Chst4) or the Na+–sulfate cotransporter (Slc13a1); and mice with reduced O-GlcNAcylation as a result of deletion of the gene encoding O-GlcNAc transferase (Ogt)15,28–32,118,163. In conjunction with data from human studies, the mechanisms by which these alterations to glycans contribute to inflammation are detailed in this section, with a focus on mucin biology, glycan–lectin and host–microorganism interactions, and altered immunity (Box 2).
Box 2 | Effects of disrupted intestinal epithelial glycosylation in IBD.
Altered immunity
- Altered antigen uptake
- M cells
- Transepithelial dendrites
- Goblet-associated passages
MUC2-dependent tolerogenic imprinting
Transepithelial leukocyte recruitment to the crypts
Glycan–lectin interactions
- Host lectins
- Altered glycosyltransferase binding
- Impaired protein quality control
- Impaired protein sorting
- Impaired Siglec immune inhibition
- Foreign lectins
- Altered binding of plant lectins, viruses, bacteria, toxins and parasites
MUC2 synthesis and stability
Impaired MUC2 synthesis
Increased MUC2 degradation
Increased intestinal permeability
Host–microorganism interactions
- Altered bacteria–intestinal epithelial cell (IEC) binding:
- Helicobacter pylori
- Lactobacillus
- Bifidobacterium
- Polysaccharide utilization loci
- Altered IEC glycan induction
- Fucose and N-glycans
Spatial dysbiosis
- Microorganism nutrient dysregulation
- Bacteroidales
MUC2 synthesis and stability
Epithelial glycosylation contributes to barrier formation, host–microorganism symbiosis and immunity. Thus, it is no surprise that altered glycosylation has a putative role in IBD. Mouse models have demonstrated that glycans are critical for synthesis of the MUC2 mucus layer, which forms a single loose layer in the small intestine and an outer loose layer and inner attached layer in the colon164. The loose layers in the small intestine and colon serve as habitats and nutritional substrates for the gut microbiota and are penetrable to bacteria, whereas the inner mucus layer is impenetrable to bacteria and prevents bacterial–epithelial interactions in the distal gut, where bacterial loads can reach 1012 bacteria per gram of stool164 (Fig. 5a).
Figure 5. Mucus structure and synthesis.
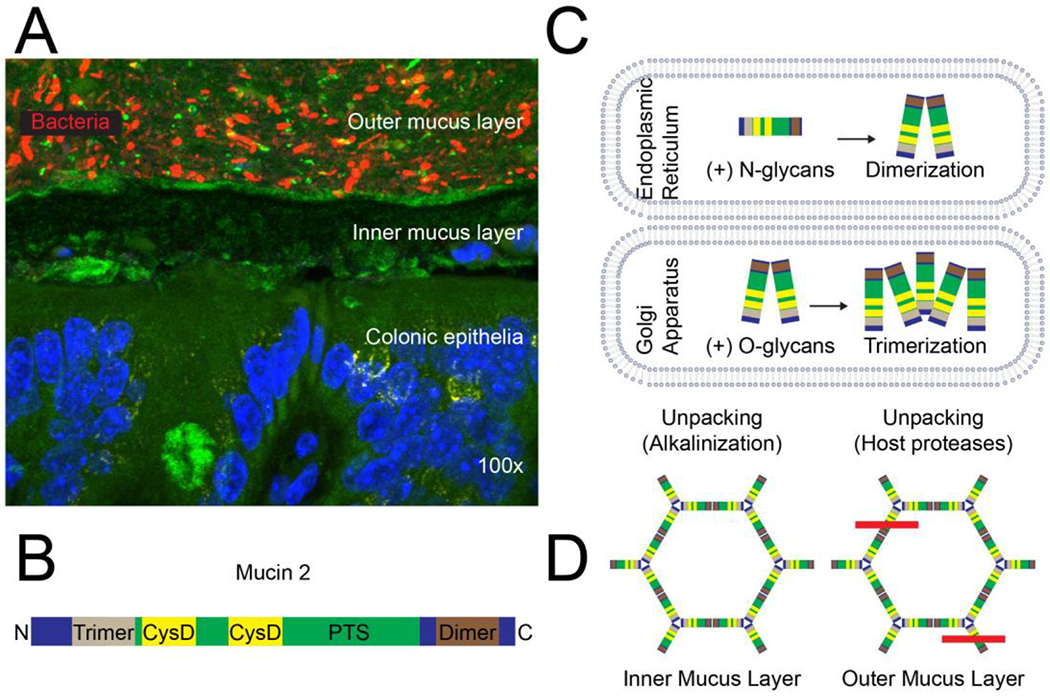
(A) Topography of the normal colonic mucus layer. Bacteria in the outer mucus layer overlay the inner mucus layer, which is normally impenetrable to bacteria; DAPI (blue), MUC2 (green; sc-15334, H-300), bacteria (16S FISH; red), magnification: 100x15. (B) The MUC2 glycoprotein has an N-terminal trimerization and a C-terminal dimerization domain with a PTS domain that is rich in proline, threonine, serine in between. The N-terminus contains three von Willebrand D domains and the C-terminus contains one (not shown). The PTS domain is highly O-glycosylated in the Golgi and interspersed by two CysD domains, contributing to intramolecular disulfide bonds and intermolecular non-covalent interactions. (C) MUC2 is synthesized in the endoplasmic reticulum, N-glycosylated and dimerized. It is then transported to the Golgi through an N-glycan-dependent mechanism, where it is O-glycosylated and forms trimers. (D) MUC2 is released and unpacked in the inner mucus layer through alkalinization and chelation of Ca2+ ions and then further unpacked in the outer mucus layer through proteolysis; the red line indicates cleavage by host proteases.
MUC2 contains hundreds of O-glycans, accounting for ~80% of its mass (Fig. 5b)165, and ~20 possible N-glycan attachment sites166. Nonetheless, both N- and O-glycosylation are important for MUC2 biology (Fig. 5c). MUC2 is translated and dimerized in the endoplasmic reticulum. Then, N-glycosylation in the endoplasmic reticulum is crucial for transfer of MUC2 to the Golgi167, where MUC2 is highly O-glycosylated and obtains many charged residues such as sialic acid and sulfate63,165 (Fig. 5c). Each MUC2 protein in the dimer then participates in additional interactions to form trimers, which, along with low pH and Ca2+ content in the Golgi, enables dense compaction of the MUC2 network168. MUC2 is stored in a specific structure within goblet cells called the theca and is released homeostatically or as a result of environmental triggers, for example by acetylcholine or Toll-like receptor ligands169,170. Nearby cystic fibrosis transmembrane conductance regulator (CFTR) channels release bicarbonate into the intestinal lumen, which neutralizes the pH and chelates Ca2+171, thereby facilitating unfolding of the MUC2 network into stratified sheets (Fig. 5d)172. In the small intestine, additional processing by the protease meprin-β releases the attached mucus layer, in a microbiota-dependent fashion, from the epithelium to form an unattached loose layer173. By contrast, expansion of the colonic mucus layer involves the host metalloprotease calcium-activated chloride channel regulator 1 (CLCA1), which cleaves MUC2 independent of the gut microbiota174. Accordingly, the colons of germ-free mice have an outer and inner mucus layer, but in explant cultures full mucus expansion does not occur in the presence of protease inhibitors; however, it can be induced by recombinant CLCA1165,174.
Although N-glycans are less abundant than O-glycans on MUC2, they are crucial for proper protein folding. Pharmacological or genetic disruption of N-glycosylation leads to the unfolded protein response, endoplasmic reticulum stress and subsequent inflammation in mice 81,175.
O-glycans likely have two major roles in MUC2 synthesis. First, O-glycans (as well as N-glycans) contain charged residues that interact with Ca2+ in the Golgi and goblet cell theca, thereby facilitating tight packing and storage of MUC2172. Although not yet experimentally tested, a loss of charged residues in MUC2 likely results in reduced packing, thereby reducing the amount of MUC2 that can be stored and released. Second, glycans on MUC2 might prevent mucus degradation by bacterial proteases, for example by blocking access to the polypeptide backbone. O-glycan extension blocks the degradation of defined peptides and murine gastrointestinal mucins by the model proteases RgpB (from Porphyromonas gingivalis) and pronase, respectively 176,177; however, whether proteases derived from commensal bacteria are able to access and degrade O-glycan-deficient MUC2 in the inner mucus layer has not been demonstrated. Nonetheless, T-synthase-knockout mice and Cosmc-knockout mice, which are deficient in O-glycans extended beyond a single GalNAc or their sialylated counterpart, both have loss of the outer and inner mucus layers and increased bacterial–epithelial contact in the distal colon15,28. Loss of the mucus layer results in increased gut permeability28. However, whether this increased permeability is due to an indirect effect of cytokine-induced remodelling of the tight junctions or due to a direct effect of O-glycans on an adhesion molecule (for example, via disrupted protein stability or disrupted glycan–protein interactions) is not known178. An additional mechanism for the loss of O-glycans and the resultant mucus degradation arises from the observation that administration of a Westernized diet (with fewer complex carbohydrates) to mice results in increased host glycan foraging by the gut microbiota and associated mucus thinning and increased permeability compared with standard chow179. Although this increased foraging can be driven by changes in diet, variations in the gut microbiota between animals alone (in the absence of dietary changes) can also result in enhanced mucus glycan degradation; when comparing colonies of mice housed in different rooms, differences in the microbiome between mice led to differences in bacterial inner mucus penetrability180. Thus, defects in N- and O-glycosylation could affect mucin synthesis and stability upon secretion into the intestinal lumen, leading to compromised barrier function.
Glycan–lectin interactions
Perturbed glycan–lectin interactions can contribute to inflammation. Host lectins from diverse species, including R, L, P, I and S-type lectins and many of the cluster of differentiation markers, interact with intestinal epithelial cell (IEC) glycans (Table 2)181,182. R-type lectins include members of the polypeptide GalNAc-transferases, which add GalNAc to serine or threonine in a peptide to form the Tn antigen and are largely conserved between mice and humans46. L-type lectins are involved in protein folding quality control (for example, calnexin and calreticulin) and vesicular sorting (for example, in mice and humans, endoplasmic reticulum–Golgi intermediate compartment 53 (ERGIC53), vesicular integral-membrane protein 36 (VIP36) and vesicular integral-membrane protein L (VIPL)) and interact with monoglucosylated or non-glucosylated high-mannose N-glycans, respectively183. The P-type lectin mannose-6-phosphate (M6P) receptor (MPR) binds to M6P on N-linked glycans to ensure proper shuttling of enzymes to the lysosome184,185. Mice and humans have two MPRs, a cation-independent mannose-6-phosphate receptor (CI-MPR) and a smaller cation-dependent mannose-6-phosphate receptor (CD-MPR), which are both critical for proper lysosomal hydrolase sorting184,185. C-type lectins, including dectin-2, the mannose receptor, and macrophage galactose type-lectin (MGL)), recognize high-mannose N-glycans, α-linked mannose and Tn antigen on mucins, respectively17,186–188. Interestingly, while human MGL (hMGL) has a single gene, mouse MGL (mMGL) includes two genes (mMGL1 and mMGL2)189–191. hMGL and mMGL2 both bind to terminal GalNAc, including Tn antigen, whereas mMGL1 binds to Lewis a/x structures189–191. Although selectins are probably the best-studied C-type lectin, they bind to endothelial or leukocyte glycoconjugates rather than to epithelia192. I-type lectins include Siglecs, which are sialic-acid-binding proteins that inhibit immune activation193. These lectins include human and mouse Siglec-15, human Siglec-5, and human Siglec-14, which bind to IBD and cancer-associated STn antigen193–196, and human Sigleg-7 and Siglec-9, which block natural killer cell activation197. Although sialic acid is increased in IBD, expression of specific Siglec ligands, such as the Siglec-9 ligands sulfo-Lewis antigens, is decreased in in IBD26,115,116,193. Thus, loss of Siglec inhibitory signals in human IBD might contribute to immune hyperactivity193,197–199. LacNAc (Galβ1,4GlcNAc) is recognized by galectins (previously known as S-type lectins), which regulate immune cells, bacteria and IECs (for example, they have roles in cell turnover, intestinal permeability and tissue repair)200–209. Furthermore, galectin expression levels in humans distinguish IBD from non-IBD control tissues (endoscopically examined for abdominal pain, constipation, irritable bowel syndrome or CRC screening) but not ulcerative colitis from Crohn’s disease 200–209.
Table 2.
Host and foreign lectins that bind to intestinal epithelial glycans
| Lectin type | Lectin | Ligand |
|---|---|---|
| Host lectins | ||
| R-type lectin | ppGalNAcT | GalNAα1-Ser/Thr |
| L-type lectin | Calnexin and calreticulin | Monoglucosylated high-mannose N-glycans |
| ERGIC-53, VIP36, and VIPL | High-mannose N-glycans | |
| P-type lectin | Mannose-6-phosphate receptor | Mannose-6-phosphate on N-glycans |
| C-type lectin | Dectin-1/2 | β-glucan, high-mannose N-glycan |
| Mannose receptor | α-linked mannose on microorganisms or mucins | |
| Macrophage galactose-type lectin (MGL) | GalNAc | |
| Siglec 5, Siglec 14 and Siglec 15 | SialylTn | |
| Siglec-7 | α2,8-linked disialic acid | |
| Siglec-9 | 6-sulfo sialyl Lewis x | |
| S-type lectin | Galectins | LacNAc (Galβ1,4GlcNAc) |
| Foreign lectins | ||
| Plant | Peanut agglutinin | GalGalNAα1-Ser/Thr |
| Viruses | Influenza, adenovirus, reovirus, rotavirus, murine respirovirus, polyomavirus | Sialic acid |
| VP8 (rotavirus) | Non-sialyl type 1 or 2 chains on O-glycans or N-glycans | |
| HSV, HIV, dengue, foot and mouth disease | Heparan sulfate | |
| Bacteria | Vibrio cholerae sialidase | Sialic acid |
| E. coli FimH | Mannose | |
| Enterotoxigenic E. coli F17-G adhesin | GlcNAc | |
| Bacterioides, Clostridium, E. coli, Lactobacillus | Galabiose (Galα1,4Gal) | |
| F. nucleatum Fap2 | GalGalNAα1-Ser/Thr | |
| Borrelia burgdorferi ErpG | Heparan sulfate | |
| Campylobacter jejuni flagella/LPS | H-antigen (Fucα1,2Galβ1,4GlcNAcβ), | |
| E. coli K99 fimbriae | GM3(Neu5Gc) (Neu5Gcα2,3Galβ1,4Glc-Cer) | |
| Helicobacter pylori BabA | Sialyl Lewis x | |
| Helicobacter pylori SabA | Lewis b | |
| Toxins | Pseudomonas aeruginosa PA1 | Galactose |
| Pseudomonas aeruginosa PA2 | Fucose or mannose | |
| Clostridioides difficile toxin A | GalNAcβ1,3Galβ1,4GlcNAcβ1,3Galβ1,4GlcβCer | |
| E. coli heat-labile toxin | GM1 | |
| Shigella dysenteriae Shiga toxin | Galα1,4GalβCer or Galα1,4Galβ1,4GlcβCer | |
| Vibrio cholerae cholera toxin | Galβ1,3GalNAc of GM1 | |
| Parasites | Entamoeba histolytica 260-kDa lectin | Gal/GalNAc |
| Trypanosoma cruzi surface ‘mucins’ | NeuAc, heparan sulfate | |
| Cryptosporidium parvum lectin p30 | Gal/GalNAc | |
| Giardia lamblia | Mannose | |
| Toxoplasma gondii TgMIC1 | NeuAcα2,3Galβ1,4GlcNAc | |
Intestinal epithelial glycans also interact with foreign lectins from plants, viruses, bacteria, toxins and parasites (Table 2). For example, peanuts produce peanut agglutinin (PNA), which binds to GalGalNAc on the intestinal epithelium and leads to human colorectal epithelial cell proliferation 156,157.
These interactions can have pathological consequences. For example, Fap2 on F. nucleatum binds to GalGalNAc, leading to inflammation and cancer in mouse models 19,210–212. Bacterial toxins that bind to IEC glycans include Pseudomonas aeruginosa 1 (PA1; binds to Gal; this binding is interferon-γ-inducible, and expression leads to intestinal permeabilization and sepsis) and PA2 (binds to fucose or mannose), Clostridioides difficile toxin A (binds to a GalNAc-terminating glycosphingolipid), E. coli heat-labile toxin (binds to GM1), Shigella dysenteriae Shiga toxin (binds to Galα1,4Galβ), and Vibrio cholerae cholera toxin (Galβ1,3GalNAc)3,213–217.
In addition to toxins, parasite lectins recognize IEC Gal/GalNAc (Entamoeba histolytica), sialic acid (Trypanosoma cruzi), heparan sulfate (T. cruzi), GalGalNAc (Cryptosporidium parum), mannose (Giardia lamblia) and Neu5Acα2,3Galβ1,4GlcNAc (Toxoplasma gondii)218–220. Collectively, the diverse interactions of host and foreign lectins with intestinal epithelial glycans influence inflammatory tone and gut homeostasis.
Host–microorganism interactions
Intestinal epithelial O-glycans can directly regulate host–microorganism interactions by providing ligands for bacterial adhesins and nutrients for bacterial metabolism. Indirectly, O-glycans have diverse roles, ranging from controlling epithelial gene and protein expression to regulating inflammatory tone by regulating the mucosal barrier as well as through antigen uptake and immune imprinting. Ultimately, these diverse mechanisms converge to select commensals for long-term colonization in the gut and to facilitate (or inhibit) pathogen binding to intestinal epithelia and subsequent damage.
Microbial host glycan-binding proteins were first studied in pathogens, including bacteria and viruses. Many of these proteins bind to fucose residues that are only present in secretor-positive individuals (that is, the 80% of the population who express FUT2 glycosyltransferase and are therefore able to synthesize blood group antigens on mucosal and salivary secretions in addition to on erythrocytes221). These glycan-binding proteins include BabA in H. pylori as well as adhesins in norovirus and rotavirus18,222–224. H. pylori is an interesting example because glycans both facilitate and inhibit binding. The deeper glands of the human stomach, including the fundic and pyloric glands, express terminal α1,4GlcNAc-containing O-glycans that block synthesis of a cholesterol-containing cell wall component by inhibiting cholesterol α-glucosyltransferase from H. pylori225–227. Thus, in contrast to BabA–glycan interactions, which promote H. pylori colonization, α1,4GlcNAc prevents gastric invasion, inflammation and oncogenesis228.
A number of glycan-binding adhesins of commensal bacteria have also been identified. These adhesins have been expressed by a number of Lactobacillus species: Lactobacillus plantarum-expressed glyceraldehyde 3-phosphate dehydrogenase (GAPDH) binds to blood group A and B-containing mucins on human colonic cells; Lactobacillus mucosae binds to human A and B blood groups through a newly identified adhesin (Lam29) that is expressed by Lactobacillus mucosae ME-340 and has homology with ATP-binding cassette (ABC) transporters; Lactobacillus rhamnosus GG binds to mucin through the subtilin biosynthesis protein SpaC in a glycan-dependent manner; and Lactobacillus reuteri binds to mucin through a family of mucus-binding proteins in a sialic acid-dependent manner229–234. In addition to glycans of lactobacilli, a collection of family 1 solute binding proteins (F1SBPs) from Bifidobacterium longum subsp. infantis bind to various host glycans235. In addition, polysaccharide utilization loci [G] require glycan-binding proteins to ultimately uptake and degrade extracellular glycans132. These glycan-binding proteins are homologous to starch utilization system C (SusC) and SusD molecules from the starch utilization locus and likely facilitate bacterial adhesion to host glycans in addition to aiding glycan metabolism236.
The small intestine degrades and absorbs proteins and simple sugars, but many complex plant polysaccharides and mucosal glycans are indigestible by the host, thereby providing a food source for the gut microbiota. The gut microbiota degrades these carbohydrates via a collection of glycosyl hydrolases, sugar transporters, sensors and regulatory proteins that bind to, import and degrade glycans62. The genes encoding these molecules are either organized as a single loci that contains all the necessary genes for glycan utilization, such as the polysaccharide utilization loci that are common in Bacteroides, or spread across the genome into individual operons dedicated to encoding specific monosaccharides, a strategy commonly used by members of the Firmicutes phylum, such as Ruminococcus gnavus and Ruminococcus torques, as well as in some Bifidobacterium species that lack an outer membrane and classic Bacteroidetes polysacchardide utilization loci components62,237. Bacteroides species contain a broad repertoire of polysaccharide utilization loci, with B. thetaiotamicron containing 88 different loci238. Studies of this model organism led to the idea that although some bacteria can degrade both dietary and host glycans, dietary polysaccharides are preferentially utilized when both are present239. However, recent work questions this idea15,240. Kudelka et al. found that host glycans were required for colonization of commensal bacteria in the mucosa of the distal intestine in a mouse model, even in the presence of dietary polysaccharides15. Additionally, further analysis revealed that although some bacteria, such as B. thetaiotomicron, are able to metabolize multiple classes of glycans, other bacterial species are more limited and preferentially consume either dietary or host glycans240. For example, Bifidobacterium species and Akkermansia muciniphilia contain a more limited repertoire of polysaccharide utilization loci that restricts foraging to host glycans241, which corresponds to A. muciniphilia’s enrichment in the human intestinal mucosa241.
Another emerging concept is that some microorganisms share resources. This concept was initially observed in pathogens that contain monosaccharide transporters but not the glycosidases needed to release them, and that therefore require commensals for colonization242,243. However, resource sharing has since been observed among members of the order Bacteroidales, which suggests that this might be a more common feature among commensals than has previously been believed244. Historically, theoretical models to describe selection of commensal bacteria in the intestine predicted that one type of bacteria would eventually dominate a mixed population given sufficient time. This prediction contrasts with the experimental evidence that the human gut microbiota is incredibly diverse and generally stable over time245. Resource sharing would explain mutualism and the persistence of two species as well as the idea that the gut contains many distinct ecological niches or biogeographies in both the mucosal–luminal and rostral–caudal axes10,246. Multiple studies have shown that bacterial inoculation into animals induces expression of polysaccharide utilization loci and that this expression is critical for colonization, transmission and maintenance of a long-term reservoir (for example, of Bacteroides within the crypt channel) following antibiotic or pathogen challenge25,238,247.
Additionally, microorganisms can reprogramme intestinal epithelial glycosylation. Salmonella directly modifies human glycans through bacterial glycosidases and induces host gene expression to alter host glycan biosynthesis248,249. These changes can have a major effect on host physiology. In addition to an increase in direct invasion of intestinal epithelial cells, Salmonella induced host neuraminidase activity, which led to enhanced intestinal alkaline phosphatase (IAP) turnover in the duodenum of mice 249,250. As IAP typically dephosphorylates and detoxifies LPS, these mice developed chronic inflammation even after clearing the initial infection. And as previously discussed, commensals also release host monosaccharides, such as sialic acid, which can facilitate pathogen expansion by providing a nutrient source242.
Intestinal epithelial glycosylation spatially regulates the gut microbiota and altered glycosylation contributes to the spatial pattern of dysbiosis in IBD15. Patients with IBD, including Crohn’s disease and ulcerative colitis, and healthy individuals both have spatial regulation of the gut microbiota, with patients with IBD exhibiting more pronounced alterations in the mucosal microbiome than in the luminal microbiome10,82,246,251. However, the mechanisms underlying spatial regulation of the microbiome in IBD were until the past few years poorly understood. But in 2014, COSMC, which controls the extension of O-glycans beyond a single GalNAc, was identified as an IBD risk gene in a GWAS80. As host glycans have a direct interface with the gut microbiota, Kudelka et al. tested the hypothesis that Cosmc spatially regulates the gut microbiome. Deletion of Cosmc in the mouse intestine led to loss of microbial diversity and emergence of a pathobiont in the mucosa of the distal colon but not in the overlying lumen or in the mucosa of the small intestine, indicating that Cosmc and the downstream O-glycans spatially regulate the gut microbiome (Fig. 6)15. Further, regional alterations in the microbial ecology led to regional pathology, with the most pronounced inflammation being in the distal colon. Thus, intestinal epithelial glycosylation regulates microbial biogeography in IBD.
Figure 6. COSMC and O-glycans spatially regulate the gut microbiota.
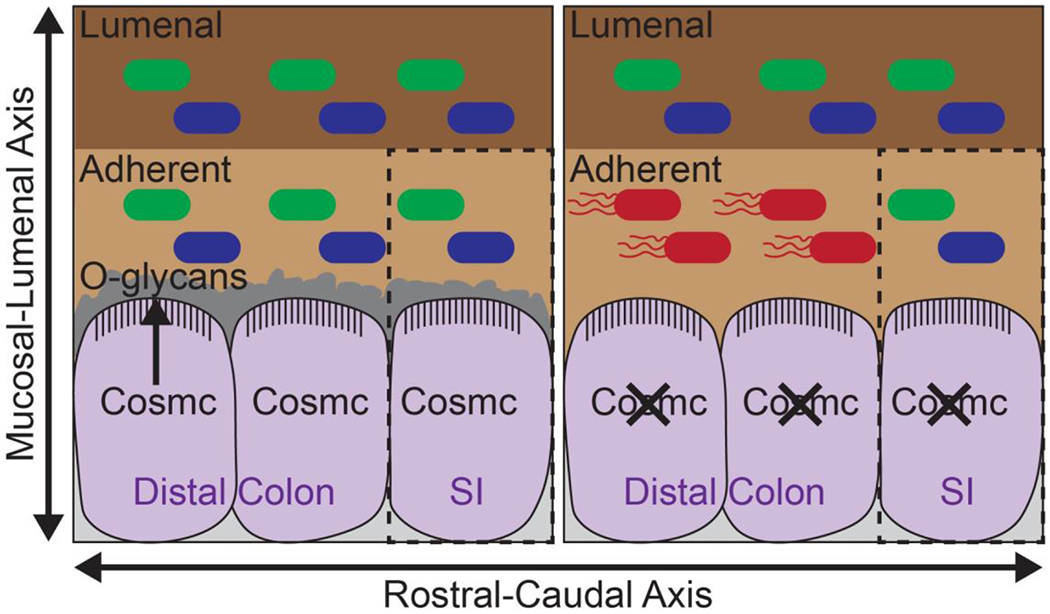
COSMC regulates extension of O-glycans in intestinal epithelia and formation of the colonic mucus layer, separating the gut microbiota from contact with the intestinal epithelia. In mice, loss of Cosmc leads to loss of extended O-glycans and increased bacterial–epithelial contact in the colon. This loss coincides with a loss of bacterial diversity and the emergence of a pro-inflammatory pathobiont in the colonic mucosa but not in the overlying lumen or ileal mucosa15. These changes correspond to the spatial dysbiosis observed in patients with inflammatory bowel disease (IBD)82.
Although it is difficult to determine which mechanisms are most important in vivo, especially in patients, these studies suggest that the simplified glycome observed in IBD contributes to an altered gut microbiota and reduced microbial diversity due to disrupted bacterial binding and reduced resource availability, leading to altered spatial geography and disease.
Altered immunity
Epithelial glycans and the mucins they decorate are involved in antigen uptake, immune imprinting and inflammatory cell recruitment. Although glycan-dependent disruption of intestinal immunity in IBD has not been rigorously evaluated, observations from basic immunology suggest that these mechanisms have an important role in disease pathogenesis. Antigen is taken up in the small intestine via multiple routes. First, in mice and humans, the small intestine can take up antigen in the Peyer’s patches [G] using specialized cells known as M cells252. M cells lack a mucin layer and have a relatively thin glycocalyx compared with enterocytes, thereby facilitating interaction of these cells with the luminal contents253. Expression of cell surface receptors (such as integrin β1, complement component 5a receptor (C5aR), glycans, glycoprotein 2 (GP2) and a poorly characterized immunoglobulin A receptor (IgAR)) facilitates uptake of microparticles via endocytosis, phagocytosis or micropinocytosis254,255. In addition, specialized dendritic cells called lysozyme-expressing dendritic cells can extend dendrites through M-cell pores to sample antigen in the gut lumen254. It has been demonstrated in a mouse model that antigen uptake by M cells is critical for the generation of faecal IgA against the commensal microbiota, thereby preventing aberrant immune activation against these resident bacteria256.
A second route of antigen uptake is CD11c+ dendritic cells in the lamina propria extending dendrites into the intestinal lumen and sampling antigens. During this process, dendritic cells maintain transepithelial resistance by expressing tight junction molecules that intercalate with a reorganized epithelial tight junction257. In contrast to the M cells of Peyer’s patches, this route occurs in villous epithelia, which therefore substantially expands the potential surface area for antigen sampling. A third route, identified in mice using in vivo imaging and confirmed in humans, uses goblet-associated passages (GAPs) that expand the surface area available for antigen sampling258. This route mechanistically differs from antigen sampling by transepithelial dendrites258. GAPs form a transepithelial passage, or tunnel, through the goblet cell by which soluble antigens can cross the intestinal epithelium and access CD103+ lamina propria dendritic cells258,259. Whereas antigen sampling by transepithelial dendrites likely has an important role during infection, GAPs are involved in steady-state sampling and result in tolerogenic responses260.
Although the three routes already described can sample antigens not coated with antibodies, a fourth route, involving the human neonatal Fc receptor (FcRn), is involved in uptake of immune complexes261,262. Human FcRn transports IgG in the basal to apical direction across the intestinal epithelium and into the faeces261. The faecal IgG then binds to antigen to form immune complexes that are then retrotranslocated by FcRn back across the intestinal epithelium to the lamina propria and its associated dendritic cells for processing and subsequent presentation to CD4+ T cells261. The translocated antigen is subsequently able to prime CD4+ T cell responses. FcRn mediates this uptake throughout the absorptive cells, or enterocytes, of the intestinal epithelium. The different routes of antigen uptake differ in the type and size of antigens as well as in the quality of immune response, whether it be inflammatory or regulatory. Thus, these routes differentially participate in the uptake of pathogens, commensals and food antigens in the human intestine254.
The first route, uptake via M cells, depends on intestinal epithelial glycosylation. There are at least five distinct classes of receptor–ligand interactions (integrin β1, C5aR, glycans, GP2 and putative IgAR are all M cell receptors) that mediate antigen uptake, and two of these clearly depend on glycosylation254. First, binding of α2,3-linked sialic acid as well as α1,2-linked fucose residues to M cell receptors facilitates antigen uptake263–265. Second, the bacterial lectin FimH interacts with GP2 in a carbohydrate-dependent manner266. The FimH of a subset of Gram-negative bacteria, such as E. coli and S. Typhimurium, recognize mannose residues on GP2 likely in the context of the GP2 peptide backbone, as free mannose or deletion of GP2 both abrogate binding and bacterial uptake 267. It is interesting that human serum antibodies against GP2 are highly expressed in Crohn’s disease and positively correlate with antibodies against mannose-rich phosphopeptidomannan (called anti-Saccharomyces cerevisiae antibodies (ASCA)), an established serological marker of Crohn’s disease268. Anti-GP2 human serum antibodies also exhibit a weaker positive correlation with the presence of anti-outer membrane porin C precursor (OmpC; an E. coli surface protein) antibodies269. Notably, although anti-GP2, ASCA and anti-OmpC antibodies are present in Crohn’s disease, they are minimally expressed in ulcerative colitis268,269. The co-occurrence of anti-GP2, ASCA and anti-OmpC antibodies in Crohn’s disease presents the interesting possibility that M cells with expression of GP2 engulf FimH+ E. Coli–S. cerevisiae clusters (via a FimH–mannan interaction), resulting in the generation of anti-E. coli (specifically, anti-OmpC) and anti-yeast (specifically, ASCA) allo-antibodies as well as of anti-GP2 autoantibodies. Although it is unclear whether these specific antibodies trigger dysbiosis or an autoinflammatory reaction in IBD, it is possible that the mannose–FimH axis might be a treatment target in addition to a biomarker in IBD. The increased abundance of ASCA and anti-GP2 antibodies in ileal Crohn’s disease compared with ulcerative colitis (or colonic-only Crohn’s disease) makes sense considering the location of M cells primarily in the small intestine268,269. In addition to these direct roles for glycans in mediating M-cell–antigen interactions, glycans also have an indirect role in antigen uptake via the regulation of glycocalyx bulk — the thin glycocalyx present on M cells facilitates interactions with luminal antigens253. Glycans, as well as the glycoconjugates to which they are attached, determine glycocalyx thickness. For example, overexpression of the polypeptide GalNAc transferase 7 (GALNT7), which adds GalNAc to Ser or Thr to initiate mucin-type O-glycosylation, increases glycocalyx bulk270.
Although not yet experimentally tested, it is likely that both paracellular dendritic cell sampling and GAP uptake depend on glycosylation. Glycans help to maintain important intercellular junctions in the intestinal epithelium, for example via increasing the stability of JAM-A and desmoglein-2 204,271. Similar glycan-dependent interactions are also likely important in regulating interactions of epithelial cells and dendritic cells during antigen sampling, as dendrites extend from the basal to the luminal side of the intestinal epithelial sheet257. Glycans are also crucial in multiple aspects of MUC2 synthesis and stability in goblet cells and in the mucus layer28. Thus, one would predict that disruption in epithelial glycosylation would perturb GAP-dependent uptake, although this will need to be tested.
Intestinal epithelial glycosylation also facilitates tolerogenic imprinting and leukocyte–epithelial interactions. Interactions between MUC2 and dendritic cells in the lamina propria and Peyer’s patches in the small intestine suppress inflammatory signals and induce tolerance21. This tolerogenic activity of MUC2 depends on its glycosylation. Mechanistically, MUC2 binds to soluble, cell surface galectin-3, which in turn interacts with dectin-1 and FcγRIIB, which are cell-surface receptors on dendritic cells. Deglycosylation of MUC2 or biochemical or genetic disruption of galectin-3, dectin-1 or FcγRIIB impairs the ability of MUC2 to bind to dendritic cells and induce downstream regulatory cytokines. Glycosylated MUC2 activates AKT and glycogen synthase kinase 3β (GSK3β) phosphorylation, thereby facilitating β-catenin nuclear translocation21, which is crucial for induction of tolerogenic signals in intestinal dendritic cells272. Given the role of MUC2 glycosylation in inducing anti-inflammatory signals in the gut, it is possible that the impaired MUC2 glycosylation observed in IBD results in a switch from tolerogenic to inflammatory dendritic cells, leading to immune activation against the commensal microbiota and host molecules273.
In addition to their crucial role in antigen sampling, intestinal epithelial glycans also directly regulate leukocyte recruitment via receptor–ligand interactions. The best-known example of glycan-mediated leukocyte recruitment is selectin-mediated leukocyte rolling. L-selectin on leukocytes and P-selectin and E-selectin on endothelia bind to cognate glycans to facilitate cell rolling. These interactions are a prerequisite for subsequent integrin-mediated tight binding and diapedesis [G]38,274. Glycans also mediate interactions between leukocytes and the epithelia. This role is especially important in IBD, as neutrophil transepithelial migration and accumulation in crypt bases of neutrophils to form crypt abscesses is a hallmark of disease275. Multiple steps are involved in the migration of neutrophils from the vasculature to the gut lumen. After extravasating from blood vessels, neutrophils undergo transepithelial migration in the basal to apical direction. Multiple receptors are involved in attaching neutrophils to the apical membrane, including intercellular adhesion molecule 1 (ICAM1), decay-accelerating factor (DAF; also known as CD55) and the CD44 isoform CD44v6275. Sialyl-Lewisa glycans expressed on epithelial-expressed, inflammation-induced CD44v6 are important regulators of neutrophil–epithelia interactions276. In particular, an antibody targeting Sialyl-Lewisa, GM35, blocks transepithelial migration by inhibiting apical detachment of neutrophils into the gut lumen276,277. Although the mechanism blocking neutrophil release is not fully clear, it seems to depend in part on GM35-mediated inhibition of ectodomain shedding of CD44275,276. Thus, both epithelial and non-epithelial glycans regulate multiple steps in leukocyte recruitment from the blood to the intestinal lumen.
Translational opportunities
Translational glycobiology is still in its infancy; however, clinical trials of glycan mimetics and antibodies that target protein–glycan interactions have generated great excitement, for example in inflammatory diseases such as sickle cell disease and cancer278–283. In IBD, a number of approaches targeting intestinal epithelial glycosylation could have a major effect on treatment and diagnosis. These approaches would broadly fall into therapeutics that remodel surface glycans or block protein–glycan interactions and diagnostics that evaluate epithelial glycans or anti-glycan antibodies.
Evidence from the past decade highlights a pathological role for aberrant intestinal epithelial glycosylation in IBD, which suggests that ‘normalizing’ the disease-associated glycome could benefit patients. A number of methods can be used to remodel the intestinal glycome, including direct methods, such as editing glycans with glyosyltransferases and/or glycosidases or their inhibitors or replacing glycans through the addition of antibodies or lipids linked to intestinal glycans284, and indirect methods, such as normalizing the gut microbiota or reprogramming the glycome using select bacterial products (that is, glycosylation inducers).
In terms of direct methods, one promising approach is developing inhibitors of glycosidases, including bacterial glyosidases involved in glycan simplification or host glycosidases such as O-GlcNAcase, that are involved in pathogenic signalling. Indeed, inhibiting O-GlcNAcase ameliorates gut inflammation in a pre-clinical mouse model118. In addition to loss of terminal glycosylation, IBD is also associated with a reduction in mucin sulfation, a post-glyosylational modification that contributes to disease pathogenesis. Reduced sulfation arises in part from increased mucin sulfatase activity. Although the nature (extracellular versus intracellular, host versus bacterial) and identify of this sulfatase is not clear, a number of sulfatase inhibitors have been developed for various applications and some have been tested in patients (although not in IBD)285–290. An alternative approach to increase sulfation could be to deliver sulfation precursors such as 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to patients either systemically or locally, although this would need to be tested291.
In terms of indirect methods, bacterial interactions with the intestinal epithelia result in host signalling that induces gut glycosylation in mice and probably also in humans76. Therefore, normalizing the gut microbiota through faecal microbiota transplantation, prebiotics, probiotics and engineered diets might correct components of glycan disruption in IBD. Similarly, in a feed-forward loop, inflammation itself drives pathological changes to the glycome, which in turn disrupts the gut microbiota and promotes recruitment of inflammatory cells in humans1–3,26. Given the role of immune cells in regulating the glycome in mice, anti-inflammatory drugs, such as biologics, immunomodulators, steroids and aminosalicylates, might in part act through reprogramming of the glycome; however, this will need to be tested27,75,79,85. Although gut microbiota normalization and use of immunomodulators are complex interventions that contribute to disease improvement via multiple mechanisms, it is possible that correcting the glycome is one such mechanism.
Protein–glycan interactions contribute to IBD pathogenesis by recruiting inflammatory cells or pathogenic bacteria and can be blocked through glycome reprogramming or by inhibitors such as antibodies that interrupt protein–glycan interaction or small molecules (for example, glycan mimetics) that target protein–glycan interactions or glycosyltransferases or glycosidases in experimental models235,276. Interestingly, dietary substances contain lectin–glycan inhibitors that might ameliorate gut inflammation. For example, ingestion of plant products high in galactose correlates with reduced incidence of CRC (512 CRC cases versus 512 controls; OR 0.67; CI 0.47–0.95)292, suggesting that galactose interferes with F. nucleatum FAP2–GalGalNAc interactions, which are pathogenic in IBD and cancer. This observation lays the groundwork for other dietary substances, such as selected or designed fibre oligosaccharides, to interfere with or promote pathogenic or beneficial host–microorganism interactions, respectively. High fibre intake is associated with reduced IBD incidence and disease severity, and specific dietary interventions such as exclusive enteral nutrition can induce disease remission in select patients with IBD293,294. However, such dietary interventions in general have not been designed or evaluated to date for specific glycan content, providing an opportunity for new investigation. Similarly, synthetic inhibitors, for example those that have been developed to block pathogenic E. coli FimH lectin activity, could also be useful and have entered clinical trials for Crohn’s disease (EB8018 FimH inhibitor)295,296.
Additionally, glycome simplification and loss of extended glycans unmasks the protein backbone, making mucins more accessible to host and bacterial proteases176,177. Loss of this protective mucin layer leads to enhanced bacterial–intestinal epithelia interactions, which in theory can be blocked with appropriate mucin protease inhibitors22,176,177.
Intestinal epithelial glycomics, serum glycomics, anti-glycan immunohistochemistry and glycan microarrays offer unique diagnostic opportunities. All of these approaches distinguish IBD from healthy or non-IBD controls26,115,297,298. However, perhaps the most exciting application of these technologies is in the field of precision medicine. Clinicians could use alterations in the epithelial glycome and the anti-glycome immune response to stratify patients and potentially influence therapeutic selection. In addition, some of these assays might augment current approaches for evaluating treatment response. The potential of the various approaches to target intestinal epithelial glycosylation to improve IBD is exciting.
Conclusions
IBD is the result of the dysregulation of immune cells, the gut microbiota and the gut epithelia. Current evidence suggests that intestinal epithelial glycosylation plays a central part in all of these processes. Further, studies of patients with IBD indicate that the presence of disease and worse disease activity correlate with the expression of truncated O-glycans, although the mechanisms underlying these alterations are currently unclear. In twin studies, altered glycosylation can in some cases precede disease initiation, whereas other glycan changes emerge during active disease. Glycosylation is crucial for biosynthesis and stability of MUC2, the major glycoprotein in the intestinal mucus layer, and loss of MUC2 results in spontaneous inflammation and CRC299,300. The mucus layer is often compromised in IBD, which suggests that altered glycosylation might be one explanation for this. Intestinal epithelial glycosylation and dietary glycans likely synergize to establish microbial networks in the intestine, and alterations in intestinal epithelial glycosylation might explain some aspects of dysbiosis in IBD. Additionally, epithelial glycosylation might be a key part of regulating the intestinal immune system. Glycans are involved in antigen uptake, tolerogenic imprinting and immune cell interactions with the intestinal epithelia. Alterations in the normal glycome might lead to disruptions in these processes that tip the gut from a regulatory to an inflammatory environment. Novel animal models and advances in disease glycomics and genetics will hopefully lead to new approaches to treat IBD (Box 3).
Box 3 | Unanswered questions in IBD glycosylation.
How does inflammatory bowel disease (IBD) genetics contribute to the structural changes in glycans that are observed in patient tissues?
How do host glycans interact with dietary glycans to regulate the gut microbiota?
What host factors are able to compensate for changes in glycosylation to maintain homeostasis?
How does the gut microbiota induce or degrade host glycosylation to regulate gut biology?
How does compromised intestinal epithelial glycosylation affect the intestinal immune system?
How do glycans select for or against specific gut bacteria?
How can we best engineer the epithelial glycome in vivo to repair the pro-inflammatory IBD-associated glycome?
What protein–glycan interactions would be best to target in IBD to ameliorate inflammation?
How can we develop better small molecule inhibitors for glycosyltransferases, glycosidases and sulfatases for IBD and other diseases?
How do current IBD therapies, including immunomodulators, dietary interventions and faecal microbiota transplantation, affect the epithelial glycome?
Can eating glycans, glycomimetics or lectins reduce inflammation in IBD?
How can we use next-generation glycomic technologies (for example, tissue or serum glycomics, glycan microarrays and glycan imaging) to improve diagnosis and treatment of IBD?
Supplementary Material
Key points.
A large set of transcriptional and enzymatic pathways spatially and developmentally regulate glycosylation in the gut.
Host genetics, environment and the gut microbiota influence intestinal epithelial glycosylation.
Epithelial glycans have many functions: they act as ligands and nutrient sources, and establish immunological tone, for the gut microbiota.
Genome-wide association studies and biochemical studies implicate altered intestinal epithelial glycosylation in Crohn’s disease and ulcerative colitis.
Disrupted glycosylation contributes to inflammation by perturbing intestinal barrier function, glycan–lectin interactions, the gut microbiota and mucosal immunity.
Targeting epithelial glycans in the intestine provides an opportunity to combat inflammatory bowel disease.
Acknowledgements
The authors were supported by NIH grants U01CA168930 and P41GM103694 (R.D.C.), the Burroughs Wellcome Trust Career Award for Medical Scientists and the NIH Early Independence Grants DP5OD019892 (S.R.S.) and DK89763 and AI64462 (A.S.N.).
Glossary
- Lectin
A non-antibody glycan binding protein isolated from animals or plants
- Cad antigen
A non-ABO blood group antigen present on red blood cells and secretions. Structurally, Cad antigen consists of a galactose modified by addition of sialic acid and GalNAc at two different positions on the galactose ring: GalNAcβ1,4(Neu5Acα2,3)Galβ-R
- Glycan
A carbohydrate consisting of at least two monosaccharides linked together
- Sequon
A defined series of amino acids that serve as an attachment site for a glycan
- Glycosite
The amino acid position within a protein where a specific glycan is attached
- Diapedesis
The process of blood cells travelling between endothelial cells as the blood cells exit the blood vessel and enter the tissue
- Polysaccharide utilization locus
A bacterial gene cluster that encodes proteins that bind, degrade and transport extracellular polysaccharides across the bacterial cell membrane and into the bacterial cell
- Peyer’s patches
An organized collection of immune cells with a specific microarchitecture that is found in the gut, most commonly in the mucosal and submucosal layers of the ileum
- Lewis antigens
Lewis (Le) antigens are fucosylated non-ABO blood group antigens. The basic Le structure consists of a terminal galactose attached via β-linkage to a subterminal N-acetylglucosamine (GlcNAc), which is modified by attachment of a fucose in α-linkage. The galactose can be attached to the 3 or 4 position of the GlcNAc and the fucose can be attached to the open 4 or 3 position, depending on which position is available. This basic Le structure (Lea or Lex, depending on the exact linkages) can be further modified by additionally adding a fucose to the galactose to form Ley or Leb depending on the exact linkages and/or by addition of sulfate or sialic acid to either galactose or GlcNAc. FUT3 attaches fucose to the GlcNAc, whereas FUT2 attaches fucose to the galactose
- Molecular mimicry
In glycobiology, this term refers to the process of foreign microorganisms, typically bacteria, expressing glycan structures that resemble host glycan structures. This prevents immune recognition of the bacteria as a foreign invader and is used by pathogens and commensals alike
- O-GlcNAcylation
This is a class of nucleocytoplasmic glycosylation defined by β-linkage of unmodified N-acetylglucosamine on serine or threonine on glycoproteins. Unlike the majority of cell surface glycans, which last for the lifespan of the protein, O-GlcNAc is added and removed rapidly throughout the lifespan of a protein. O-GlcNAcylation often occurs at the same site as phosphorylation and similarly regulates cell signalling
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 126, 1504–1517 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Kumar V, Abbas AK & Aster JC Robbins and Cotran pathologic basis of disease. Ninth edition. edn, (Elsevier/Saunders, 2015). [Google Scholar]
- 3.Strober W & Fuss IJ Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140, 1756–1767, doi: 10.1053/j.gastro.2011.02.016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahlhamer JM, Zammitti EP, Ward BW, Wheaton AG & Croft JB Prevalence of Inflammatory Bowel Disease Among Adults Aged >/=18 Years - United States, 2015. MMWR Morb Mortal Wkly Rep 65, 1166–1169, doi: 10.15585/mmwr.mm6542a3 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Collaborators, G. B. D. I. B. D. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol 5, 17–30, doi: 10.1016/S2468-1253(19)30333-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bojanova DP & Bordenstein SR Fecal Transplants: What Is Being Transferred? PLoS Biol 14, e1002503, doi: 10.1371/journal.pbio.1002503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kostic AD, Xavier RJ & Gevers D The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146, 1489–1499, doi: 10.1053/j.gastro.2014.02.009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sekirov I, Russell SL, Antunes LC & Finlay BB Gut microbiota in health and disease. Physiol Rev 90, 859–904, doi: 10.1152/physrev.00045.2009 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Turnbaugh PJ et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484, doi: 10.1038/nature07540 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donaldson GP, Lee SM & Mazmanian SK Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 14, 20–32, doi: 10.1038/nrmicro3552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan XC et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13, R79, doi: 10.1186/gb-2012-13-9-r79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vatanen T et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 165, 1551, doi: 10.1016/j.cell.2016.05.056 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Arike L, Holmen-Larsson J & Hansson GC Intestinal Muc2 mucin O-glycosylation is affected by microbiota and regulated by differential expression of glycosyltranferases. Glycobiology, doi: 10.1093/glycob/cww134 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell BJ, Yu LG & Rhodes JM Altered glycosylation in inflammatory bowel disease: a possible role in cancer development. Glycoconj J 18, 851–858 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Kudelka MR et al. Cosmc is an X-linked inflammatory bowel disease risk gene that spatially regulates gut microbiota and contributes to sex-specific risk. Proc Natl Acad Sci U S A 113, 14787–14792, doi: 10.1073/pnas.1612158114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study in mice describes how intestinal glycans regulate the spatial distribution of bacteria in the gut and how this regulation is disrupted in IBD.
- 16.McGovern DP et al. Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum Mol Genet 19, 3468–3476, doi: 10.1093/hmg/ddq248 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allavena P et al. Engagement of the mannose receptor by tumoral mucins activates an immune suppressive phenotype in human tumor-associated macrophages. Clin Dev Immunol 2010, 547179, doi: 10.1155/2010/547179 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao S et al. Structural basis for the recognition of blood group trisaccharides by norovirus. J Virol 81, 5949–5957, doi: 10.1128/JVI.00219-07 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abed J et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe 20, 215–225, doi: 10.1016/j.chom.2016.07.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study in mice shows that bacterial lectins that bind IBD-associated glycans can drive bacterial localization to regions expressing those glycans in vivo.
- 20.Campbell BJ, Finnie IA, Hounsell EF & Rhodes JM Direct demonstration of increased expression of Thomsen-Friedenreich (TF) antigen in colonic adenocarcinoma and ulcerative colitis mucin and its concealment in normal mucin. J Clin Invest 95, 571–576, doi: 10.1172/JCI117700 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shan M et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 342, 447–453, doi: 10.1126/science.1237910 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study in mice shows that glycans on intestinal MUC2 contribute to oral tolerance through interactions with dendritic cells and induction of immunoregulatory signals.
- 22.Bergstrom K et al. Defective Intestinal Mucin-Type O-Glycosylation Causes Spontaneous Colitis-Associated Cancer in Mice. Gastroenterology 151, 152–164 e111, doi: 10.1053/j.gastro.2016.03.039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rausch P et al. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc Natl Acad Sci U S A 108, 19030–19035, doi: 10.1073/pnas.1106408108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study in humans shows that a genetically determined IBD-associated glycome shapes the gut microbiota in Crohn’s disease.
- 24.Hooper LV, Xu J, Falk PG, Midtvedt T & Gordon JI A molecular sensor that allows a gut commensal to control its nutrient foundation in a competitive ecosystem. Proc Natl Acad Sci U S A 96, 9833–9838 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sonnenburg JL et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307, 1955–1959, doi: 10.1126/science.1109051 (2005). [DOI] [PubMed] [Google Scholar]; This study in mice shows that gut bacteria are programmed to consume epithelial carbohydrates.
- 26.Larsson JM et al. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm Bowel Dis 17, 2299–2307, doi: 10.1002/ibd.21625 (2011). [DOI] [PubMed] [Google Scholar]; This study in humans shows that gut inflammation in ulcerative colitis leads to reversible expression of truncated glycans.
- 27.Goto Y et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 345, 1254009, doi: 10.1126/science.1254009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study in mice provides mechanistic insight into how gut bacteria induce epithelial glycosylation: gut bacteria communicate with innate lymphoid cells, releasing IL-22, which induces epithelial glycosyltransferase transcription and subsequent fucosylation.
- 28.Fu J et al. Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J Clin Invest 121, 1657–1666, doi: 10.1172/JCI45538 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study in mice demonstrates a functional role for altered glycosylation, in particular loss of complex O-glycans, in IBD.
- 29.Tobisawa Y, Imai Y, Fukuda M & Kawashima H Sulfation of colonic mucins by N-acetylglucosamine 6-O-sulfotransferase-2 and its protective function in experimental colitis in mice. J Biol Chem 285, 6750–6760, doi: 10.1074/jbc.M109.067082 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stone EL et al. Glycosyltransferase function in core 2-type protein O glycosylation. Mol Cell Biol 29, 3770–3782, doi: 10.1128/MCB.00204-09 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dawson PA et al. Reduced mucin sulfonation and impaired intestinal barrier function in the hyposulfataemic NaS1 null mouse. Gut 58, 910–919, doi: 10.1136/gut.2007.147595 (2009). [DOI] [PubMed] [Google Scholar]
- 32.An G et al. Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J Exp Med 204, 1417–1429, doi: 10.1084/jem.20061929 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cummings RD The repertoire of glycan determinants in the human glycome. Mol Biosyst 5, 1087–1104, doi: 10.1039/b907931a (2009). [DOI] [PubMed] [Google Scholar]
- 34.Kudelka MR et al. Cellular O-Glycome Reporter/Amplification to explore O-glycans of living cells. Nat Methods 13, 81–86, doi: 10.1038/nmeth.3675 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kudelka MR, Ju T, Heimburg-Molinaro J & Cummings RD Simple sugars to complex disease--mucin-type O-glycans in cancer. Adv Cancer Res 126, 53–135, doi: 10.1016/bs.acr.2014.11.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nairn AV et al. Regulation of glycan structures in animal tissues: transcript profiling of glycan-related genes. J Biol Chem 283, 17298–17313, doi: 10.1074/jbc.M801964200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neelamegham S & Mahal LK Multi-level regulation of cellular glycosylation: from genes to transcript to enzyme to structure. Curr Opin Struct Biol 40, 145–152, doi: 10.1016/j.sbi.2016.09.013 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cummings RD “Stuck on sugars - how carbohydrates regulate cell adhesion, recognition, and signaling”. Glycoconj J 36, 241–257, doi: 10.1007/s10719-019-09876-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freeze HH, Baum L & Varki A in Essentials of Glycobiology (eds rd et al. ) 521–526 (2015). [Google Scholar]
- 40.Cummings RD & Pierce JM The challenge and promise of glycomics. Chem Biol 21, 1–15, doi: 10.1016/j.chembiol.2013.12.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steentoft C et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J 32, 1478–1488, doi: 10.1038/emboj.2013.79 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Apweiler R, Hermjakob H & Sharon N On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta 1473, 4–8, doi: 10.1016/s0304-4165(99)00165-8 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Liu J, Jin C, Cherian RM, Karlsson NG & Holgersson J O-glycan repertoires on a mucin-type reporter protein expressed in CHO cell pools transiently transfected with O-glycan core enzyme cDNAs. J Biotechnol 199, 77–89, doi: 10.1016/j.jbiotec.2015.02.017 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Xia B, Royall JA, Damera G, Sachdev GP & Cummings RD Altered O-glycosylation and sulfation of airway mucins associated with cystic fibrosis. Glycobiology 15, 747–775, doi: 10.1093/glycob/cwi061 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Kudelka MR et al. Isotopic labeling with cellular O-glycome reporter/amplification (ICORA) for comparative O-glycomics of cultured cells. Glycobiology 28, 214–222, doi: 10.1093/glycob/cwy005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bennett EP et al. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology 22, 736–756, doi: 10.1093/glycob/cwr182 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ju T, Aryal RP, Kudelka MR, Wang Y & Cummings RD The Cosmc connection to the Tn antigen in cancer. Cancer Biomark 14, 63–81, doi: 10.3233/CBM-130375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ju T et al. Tn and sialyl-Tn antigens, aberrant O-glycomics as human disease markers. Proteomics Clin Appl 7, 618–631, doi: 10.1002/prca.201300024 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ju T & Cummings RD A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci U S A 99, 16613–16618, doi: 10.1073/pnas.262438199 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwai T et al. Molecular cloning and characterization of a novel UDP-GlcNAc:GalNAc-peptide beta1,3-N-acetylglucosaminyltransferase (beta 3Gn-T6), an enzyme synthesizing the core 3 structure of O-glycans. J Biol Chem 277, 12802–12809, doi: 10.1074/jbc.M112457200 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Stanley P & Cummings RD in Essentials of Glycobiology (eds rd et al.) 161–178 (2015). [Google Scholar]
- 52.Uhlen M et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419, doi: 10.1126/science.1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 53.De Graaf TW, Van der Stelt ME, Anbergen MG & van Dijk W Inflammation-induced expression of sialyl Lewis X-containing glycan structures on alpha 1-acid glycoprotein (orosomucoid) in human sera. J Exp Med 177, 657–666, doi: 10.1084/jem.177.3.657 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steinberg W The clinical utility of the CA 19–9 tumor-associated antigen. Am J Gastroenterol 85, 350–355 (1990). [PubMed] [Google Scholar]
- 55.Barbe L et al. Histo-blood group antigen-binding specificities of human rotaviruses are associated with gastroenteritis but not with in vitro infection. Sci Rep 8, 12961, doi: 10.1038/s41598-018-31005-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trinchera M, Aronica A & Dall’Olio F Selectin Ligands Sialyl-Lewis a and Sialyl-Lewis x in Gastrointestinal Cancers. Biology (Basel) 6, doi: 10.3390/biology6010016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robbe C et al. Evidence of regio-specific glycosylation in human intestinal mucins: presence of an acidic gradient along the intestinal tract. J Biol Chem 278, 46337–46348, doi: 10.1074/jbc.M302529200 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Robbe-Masselot C, Maes E, Rousset M, Michalski JC & Capon C Glycosylation of human fetal mucins: a similar repertoire of O-glycans along the intestinal tract. Glycoconj J 26, 397–413, doi: 10.1007/s10719-008-9186-9 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Axelsson MA, Asker N & Hansson GC O-glycosylated MUC2 monomer and dimer from LS 174T cells are water-soluble, whereas larger MUC2 species formed early during biosynthesis are insoluble and contain nonreducible intermolecular bonds. J Biol Chem 273, 18864–18870, doi: 10.1074/jbc.273.30.18864 (1998). [DOI] [PubMed] [Google Scholar]
- 60.Carlstedt I et al. Characterization of two different glycosylated domains from the insoluble mucin complex of rat small intestine. J Biol Chem 268, 18771–18781 (1993). [PubMed] [Google Scholar]
- 61.Hollingsworth MA & Swanson BJ Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer 4, 45–60, doi: 10.1038/nrc1251 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Tailford LE, Crost EH, Kavanaugh D & Juge N Mucin glycan foraging in the human gut microbiome. Front Genet 6, 81, doi: 10.3389/fgene.2015.00081 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Larsson JM, Karlsson H, Sjovall H & Hansson GC A complex, but uniform O-glycosylation of the human MUC2 mucin from colonic biopsies analyzed by nanoLC/MSn. Glycobiology 19, 756–766, doi: 10.1093/glycob/cwp048 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Holmen Larsson JM, Thomsson KA, Rodriguez-Pineiro AM, Karlsson H & Hansson GC Studies of mucus in mouse stomach, small intestine, and colon. III. Gastrointestinal Muc5ac and Muc2 mucin O-glycan patterns reveal a regiospecific distribution. Am J Physiol Gastrointest Liver Physiol 305, G357–363, doi: 10.1152/ajpgi.00048.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McLoughlin K, Schluter J, Rakoff-Nahoum S, Smith AL & Foster KR Host Selection of Microbiota via Differential Adhesion. Cell Host Microbe 19, 550–559, doi: 10.1016/j.chom.2016.02.021 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Hooper LV, Littman DR & Macpherson AJ Interactions between the microbiota and the immune system. Science 336, 1268–1273, doi: 10.1126/science.1223490 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shepherd NA et al. Restorative proctocolectomy with ileal reservoir: pathological and histochemical study of mucosal biopsy specimens. J Clin Pathol 40, 601–607, doi: 10.1136/jcp.40.6.601 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Young VB et al. Multiphasic analysis of the temporal development of the distal gut microbiota in patients following ileal pouch anal anastomosis. Microbiome 1, 9, doi: 10.1186/2049-2618-1-9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Silva HJ et al. Mucosal characteristics of pelvic ileal pouches. Gut 32, 61–65, doi: 10.1136/gut.32.1.61 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsai HH, Dwarakanath AD, Hart CA, Milton JD & Rhodes JM Increased faecal mucin sulphatase activity in ulcerative colitis: a potential target for treatment. Gut 36, 570–576, doi: 10.1136/gut.36.4.570 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ulmer JE et al. Characterization of glycosaminoglycan (GAG) sulfatases from the human gut symbiont Bacteroides thetaiotaomicron reveals the first GAG-specific bacterial endosulfatase. J Biol Chem 289, 24289–24303, doi: 10.1074/jbc.M114.573303 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Corfield AP et al. Colonic mucins in ulcerative colitis: evidence for loss of sulfation. Glycoconj J 13, 809–822 (1996). [DOI] [PubMed] [Google Scholar]
- 73.Pickard JM & Chervonsky AV Intestinal fucose as a mediator of host-microbe symbiosis. J Immunol 194, 5588–5593, doi: 10.4049/jimmunol.1500395 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Terahara K et al. Distinct fucosylation of M cells and epithelial cells by Fut1 and Fut2, respectively, in response to intestinal environmental stress. Biochem Biophys Res Commun 404, 822–828, doi: 10.1016/j.bbrc.2010.12.067 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Mariko Kamioka YG, Jun Kunisawa, Aayam Lamichhane, Shintaro Sato, and Hiroshi Kiyono. in International Conference of Mucosal Immunology. [Google Scholar]
- 76.Bry L, Falk PG, Midtvedt T & Gordon JI A model of host-microbial interactions in an open mammalian ecosystem. Science 273, 1380–1383 (1996). [DOI] [PubMed] [Google Scholar]; This study in mice shows that gut bacteria induce fucosylation in the small intestine.
- 77.Coyne MJ, Reinap B, Lee MM & Comstock LE Human symbionts use a host-like pathway for surface fucosylation. Science 307, 1778–1781, doi: 10.1126/science.1106469 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Pham TA et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe 16, 504–516, doi: 10.1016/j.chom.2014.08.017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goto Y et al. IL-10-producing CD4(+) T cells negatively regulate fucosylation of epithelial cells in the gut. Sci Rep 5, 15918, doi: 10.1038/srep15918 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chang D et al. Accounting for eXentricities: analysis of the X chromosome in GWAS reveals X-linked genes implicated in autoimmune diseases. PLoS One 9, e113684, doi: 10.1371/journal.pone.0113684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Graham DB et al. TMEM258 Is a Component of the Oligosaccharyltransferase Complex Controlling ER Stress and Intestinal Inflammation. Cell Rep 17, 2955–2965, doi: 10.1016/j.celrep.2016.11.042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gevers D et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392, doi: 10.1016/j.chom.2014.02.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Afzali B et al. BACH2 immunodeficiency illustrates an association between super-enhancers and haploinsufficiency. Nat Immunol 18, 813–823, doi: 10.1038/ni.3753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Christodoulou K et al. Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut 62, 977–984, doi: 10.1136/gutjnl-2011-301833 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goto Y, Uematsu S & Kiyono H Epithelial glycosylation in gut homeostasis and inflammation. Nat Immunol 17, 1244–1251, doi: 10.1038/ni.3587 (2016). [DOI] [PubMed] [Google Scholar]
- 86.Graham DB et al. TMEM258 Is a Component of the Oligosaccharyltransferase Complex Controlling ER Stress and Intestinal Inflammation. Cell Rep 17, 2955–2965, doi: 10.1016/j.celrep.2016.11.042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jostins L et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124, doi: 10.1038/nature11582 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu JZ et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 47, 979–986, doi: 10.1038/ng.3359 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Theodoratou E et al. The role of glycosylation in IBD. Nat Rev Gastroenterol Hepatol 11, 588–600, doi: 10.1038/nrgastro.2014.78 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Momozawa Y et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat Commun 9, 2427, doi: 10.1038/s41467-018-04365-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Kivit S et al. Galectin-9 Produced by Intestinal Epithelial Cells Enhances Aldehyde Dehydrogenase Activity in Dendritic Cells in a PI3K- and p38-Dependent Manner. J Innate Immun 9, 609–620, doi: 10.1159/000479817 (2017). [DOI] [PubMed] [Google Scholar]
- 92.Zhu L et al. IL-10 and IL-10 Receptor Mutations in Very Early Onset Inflammatory Bowel Disease. Gastroenterology Res 10, 65–69, doi: 10.14740/gr740w (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McGovern DP, Kugathasan S & Cho JH Genetics of Inflammatory Bowel Diseases. Gastroenterology 149, 1163–1176 e1162, doi: 10.1053/j.gastro.2015.08.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Engevik MA et al. Loss of NHE3 alters gut microbiota composition and influences Bacteroides thetaiotaomicron growth. Am J Physiol Gastrointest Liver Physiol 305, G697–711, doi: 10.1152/ajpgi.00184.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lauc G et al. Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet 9, e1003225, doi: 10.1371/journal.pgen.1003225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fonseca-Camarillo G & Yamamoto-Furusho JK Gene expression of solute carrier family 9 (sodium/hydrogen exchanger) 3, (SLC9A3) is downregulated in patients with ulcerative colitis. Inflamm Bowel Dis 18, 1197–1198, doi: 10.1002/ibd.22968 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Mkhikian H et al. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat Commun 2, 334, doi: 10.1038/ncomms1333 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lauc G et al. Genomics meets glycomics-the first GWAS study of human N-Glycome identifies HNF1alpha as a master regulator of plasma protein fucosylation. PLoS Genet 6, e1001256, doi: 10.1371/journal.pgen.1001256 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bogaert S et al. Differential mucosal expression of Th17-related genes between the inflamed colon and ileum of patients with inflammatory bowel disease. BMC Immunol 11, 61, doi: 10.1186/1471-2172-11-61 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Consortium U. I. G. et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet 41, 1330–1334, doi: 10.1038/ng.483 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Glocker EO et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med 361, 2033–2045, doi: 10.1056/NEJMoa0907206 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sugimoto K Role of STAT3 in inflammatory bowel disease. World J Gastroenterol 14, 5110–5114, doi: 10.3748/wjg.14.5110 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ahn SH et al. Hepatocyte nuclear factor 4alpha in the intestinal epithelial cells protects against inflammatory bowel disease. Inflamm Bowel Dis 14, 908–920, doi: 10.1002/ibd.20413 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duerr RH et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463, doi: 10.1126/science.1135245 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Suzuki K et al. Intestinal Epithelial Cell-specific Deletion of alpha-Mannosidase II Ameliorates Experimental Colitis. Cell Struct Funct 43, 25–39, doi: 10.1247/csf.17022 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Clamp JR, Fraser G & Read AE Study of the carbohydrate content of mucus glycoproteins from normal and diseased colons. Clin Sci (Lond) 61, 229–234 (1981). [DOI] [PubMed] [Google Scholar]
- 107.Rhodes JM, Black RR & Savage A Altered lectin binding by colonic epithelial glycoconjugates in ulcerative colitis and Crohn’s disease. Dig Dis Sci 33, 1359–1363, doi: 10.1007/bf01536988 (1988). [DOI] [PubMed] [Google Scholar]
- 108.Ta A et al. Sialyl-tn antigen expression in Crohn’s colitis. Inflamm Bowel Dis 3, 254–259 (1997). [PubMed] [Google Scholar]
- 109.Itzkowitz SH et al. Sialosyl-Tn antigen: initial report of a new marker of malignant progression in long-standing ulcerative colitis. Gastroenterology 109, 490–497 (1995). [DOI] [PubMed] [Google Scholar]; This study in humans demonstrates that colonic expression of the truncated O-glycan sialyl-Tn predicts progression from ulcerative colitis to colitis-associated-carcinoma.
- 110.Itzkowitz SH et al. Sialosyl-Tn antigen is prevalent and precedes dysplasia in ulcerative colitis: a retrospective case-control study. Gastroenterology 110, 694–704 (1996). [DOI] [PubMed] [Google Scholar]
- 111.Bodger K et al. Altered colonic glycoprotein expression in unaffected monozygotic twins of inflammatory bowel disease patients. Gut 55, 973–977, doi: 10.1136/gut.2005.086413 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Parker N, Tsai HH, Ryder SD, Raouf AH & Rhodes JM Increased rate of sialylation of colonic mucin by cultured ulcerative colitis mucosal explants. Digestion 56, 52–56, doi: 10.1159/000201222 (1995). [DOI] [PubMed] [Google Scholar]
- 113.Raouf AH et al. Sulphation of colonic and rectal mucin in inflammatory bowel disease: reduced sulphation of rectal mucus in ulcerative colitis. Clin Sci (Lond) 83, 623–626 (1992). [DOI] [PubMed] [Google Scholar]
- 114.McMahon RFT, Jones CJP, Dutt S & Stoddart RW Altered oligosaccharide expression in ulcerative colitis with increasing grades of inflammation. Glycosylation & Disease 1, 235–245, doi: 10.1007/bf00919331 (1994). [DOI] [Google Scholar]
- 115.Cooper HS & Steplewski Z Immunohistologic study of ulcerative colitis with monoclonal antibodies against tumor-associated and/or differentiation antigens. Gastroenterology 95, 686–693 (1988). [DOI] [PubMed] [Google Scholar]
- 116.Frykholm G, Enblad P, Pahlman L & Busch C Expression of the carcinoma-associated antigens CA 19–9 and CA-50 in inflammatory bowel disease. Dis Colon Rectum 30, 545–548, doi: 10.1007/bf02554787 (1987). [DOI] [PubMed] [Google Scholar]
- 117.Clerc F et al. Plasma N-Glycan Signatures Are Associated With Features of Inflammatory Bowel Diseases. Gastroenterology 155, 829–843, doi: 10.1053/j.gastro.2018.05.030 (2018). [DOI] [PubMed] [Google Scholar]
- 118.Zhao M et al. Deficiency in intestinal epithelial O-GlcNAcylation predisposes to gut inflammation. EMBO Mol Med 10, doi: 10.15252/emmm.201708736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study in mice and humans identifies loss of intracellular glycosylation, in particular O-GlcNAcylation, as a driver of gut inflammation in IBD.
- 119.Karlen P et al. Sialyl-Tn antigen as a marker of colon cancer risk in ulcerative colitis: relation to dysplasia and DNA aneuploidy. Gastroenterology 115, 1395–1404 (1998). [DOI] [PubMed] [Google Scholar]
- 120.Yuan HY et al. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res 34, W635–641, doi: 10.1093/nar/gkl236 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dewald JH, Colomb F, Bobowski-Gerard M, Groux-Degroote S & Delannoy P Role of Cytokine-Induced Glycosylation Changes in Regulating Cell Interactions and Cell Signaling in Inflammatory Diseases and Cancer. Cells 5, doi: 10.3390/cells5040043 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stowell SR, Ju T & Cummings RD Protein glycosylation in cancer. Annu Rev Pathol 10, 473–510, doi: 10.1146/annurev-pathol-012414-040438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pinho SS & Reis CA Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 15, 540–555, doi: 10.1038/nrc3982 (2015). [DOI] [PubMed] [Google Scholar]
- 124.Gill DJ, Chia J, Senewiratne J & Bard F Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J Cell Biol 189, 843–858, doi: 10.1083/jcb.201003055 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gill DJ et al. Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc Natl Acad Sci U S A 110, E3152–3161, doi: 10.1073/pnas.1305269110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Noda M et al. Glycosyltransferase Gene Expression Identifies a Poor Prognostic Colorectal Cancer Subtype Associated with Mismatch Repair Deficiency and Incomplete Glycan Synthesis. Clin Cancer Res 24, 4468–4481, doi: 10.1158/1078-0432.CCR-17-3533 (2018). [DOI] [PubMed] [Google Scholar]
- 127.Dall’Olio F & Trinchera M Epigenetic Bases of Aberrant Glycosylation in Cancer. Int J Mol Sci 18, doi: 10.3390/ijms18050998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sun X, Ju T & Cummings RD Differential expression of Cosmc, T-synthase and mucins in Tn-positive colorectal cancers. BMC Cancer 18, 827, doi: 10.1186/s12885-018-4708-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Maeda Y & Kinoshita T The acidic environment of the Golgi is critical for glycosylation and transport. Methods Enzymol 480, 495–510, doi: 10.1016/S0076-6879(10)80022-9 (2010). [DOI] [PubMed] [Google Scholar]
- 130.Maeda Y, Ide T, Koike M, Uchiyama Y & Kinoshita T GPHR is a novel anion channel critical for acidification and functions of the Golgi apparatus. Nat Cell Biol 10, 1135–1145, doi: 10.1038/ncb1773 (2008). [DOI] [PubMed] [Google Scholar]
- 131.Rivinoja A, Pujol FM, Hassinen A & Kellokumpu S Golgi pH, its regulation and roles in human disease. Ann Med 44, 542–554, doi: 10.3109/07853890.2011.579150 (2012). [DOI] [PubMed] [Google Scholar]
- 132.Koropatkin NM, Cameron EA & Martens EC How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol 10, 323–335, doi: 10.1038/nrmicro2746 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Song X et al. Oxidative release of natural glycans for functional glycomics. Nat Methods 13, 528–534, doi: 10.1038/nmeth.3861 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Duan J, Avci FY & Kasper DL Microbial carbohydrate depolymerization by antigen-presenting cells: deamination prior to presentation by the MHCII pathway. Proc Natl Acad Sci U S A 105, 5183–5188, doi: 10.1073/pnas.0800974105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Boltin D, Perets TT, Vilkin A & Niv Y Mucin function in inflammatory bowel disease: an update. J Clin Gastroenterol 47, 106–111, doi: 10.1097/MCG.0b013e3182688e73 (2013). [DOI] [PubMed] [Google Scholar]
- 136.Graziani F et al. Ruminococcus gnavus E1 modulates mucin expression and intestinal glycosylation. J Appl Microbiol 120, 1403–1417, doi: 10.1111/jam.13095 (2016). [DOI] [PubMed] [Google Scholar]
- 137.Khalili H et al. ABO blood group and risk of colorectal cancer. Cancer Epidemiol Biomarkers Prev 20, 1017–1020, doi: 10.1158/1055-9965.EPI-10-1250 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dias AM et al. Metabolic control of T cell immune response through glycans in inflammatory bowel disease. Proc Natl Acad Sci U S A 115, E4651–E4660, doi: 10.1073/pnas.1720409115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fernandes B, Sagman U, Auger M, Demetrio M & Dennis JW Beta 1–6 branched oligosaccharides as a marker of tumor progression in human breast and colon neoplasia. Cancer Res 51, 718–723 (1991). [PubMed] [Google Scholar]
- 140.Slawson C & Hart GW O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer 11, 678–684, doi: 10.1038/nrc3114 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mi W et al. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim Biophys Acta 1812, 514–519, doi: 10.1016/j.bbadis.2011.01.009 (2011). [DOI] [PubMed] [Google Scholar]
- 142.Ju T et al. Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res 68, 1636–1646, doi: 10.1158/0008-5472.CAN-07-2345 (2008). [DOI] [PubMed] [Google Scholar]
- 143.Radhakrishnan P et al. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc Natl Acad Sci U S A 111, E4066–4075, doi: 10.1073/pnas.1406619111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ju T, Otto VI & Cummings RD The Tn antigen-structural simplicity and biological complexity. Angew Chem Int Ed Engl 50, 1770–1791, doi: 10.1002/anie.201002313 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mi R et al. Epigenetic silencing of the chaperone Cosmc in human leukocytes expressing tn antigen. J Biol Chem 287, 41523–41533, doi: 10.1074/jbc.M112.371989 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ju T & Cummings RD Protein glycosylation: chaperone mutation in Tn syndrome. Nature 437, 1252, doi: 10.1038/4371252a (2005). [DOI] [PubMed] [Google Scholar]
- 147.Wang Y et al. Cosmc is an essential chaperone for correct protein O-glycosylation. Proc Natl Acad Sci U S A 107, 9228–9233, doi: 10.1073/pnas.0914004107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Marcos NT et al. ST6GalNAc-I controls expression of sialyl-Tn antigen in gastrointestinal tissues. Front Biosci (Elite Ed) 3, 1443–1455 (2011). [DOI] [PubMed] [Google Scholar]
- 149.Sewell R et al. The ST6GalNAc-I sialyltransferase localizes throughout the Golgi and is responsible for the synthesis of the tumor-associated sialyl-Tn O-glycan in human breast cancer. J Biol Chem 281, 3586–3594, doi: 10.1074/jbc.M511826200 (2006). [DOI] [PubMed] [Google Scholar]
- 150.Ikehara Y et al. Cloning and expression of a human gene encoding an N-acetylgalactosamine-alpha2,6-sialyltransferase (ST6GalNAc I): a candidate for synthesis of cancer-associated sialyl-Tn antigens. Glycobiology 9, 1213–1224, doi: 10.1093/glycob/9.11.1213 (1999). [DOI] [PubMed] [Google Scholar]
- 151.Vazquez-Martin C, Cuevas E, Gil-Martin E & Fernandez-Briera A Correlation analysis between tumor-associated antigen sialyl-Tn expression and ST6GalNAc I activity in human colon adenocarcinoma. Oncology 67, 159–165, doi: 10.1159/000081003 (2004). [DOI] [PubMed] [Google Scholar]
- 152.Feagins LA, Souza RF & Spechler SJ Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol 6, 297–305, doi: 10.1038/nrgastro.2009.44 (2009). [DOI] [PubMed] [Google Scholar]
- 153.Keller DS, Windsor A, Cohen R & Chand M Colorectal cancer in inflammatory bowel disease: review of the evidence. Tech Coloproctol 23, 3–13, doi: 10.1007/s10151-019-1926-2 (2019). [DOI] [PubMed] [Google Scholar]
- 154.Canavan C, Abrams KR & Mayberry J Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment Pharmacol Ther 23, 1097–1104, doi: 10.1111/j.1365-2036.2006.02854.x (2006). [DOI] [PubMed] [Google Scholar]
- 155.Yu LG The oncofetal Thomsen-Friedenreich carbohydrate antigen in cancer progression. Glycoconj J 24, 411–420, doi: 10.1007/s10719-007-9034-3 (2007). [DOI] [PubMed] [Google Scholar]
- 156.Singh R, Subramanian S, Rhodes JM & Campbell BJ Peanut lectin stimulates proliferation of colon cancer cells by interaction with glycosylated CD44v6 isoforms and consequential activation of c-Met and MAPK: functional implications for disease-associated glycosylation changes. Glycobiology 16, 594–601, doi: 10.1093/glycob/cwj108 (2006). [DOI] [PubMed] [Google Scholar]
- 157.Ryder SD, Smith JA & Rhodes JM Peanut lectin: a mitogen for normal human colonic epithelium and human HT29 colorectal cancer cells. J Natl Cancer Inst 84, 1410–1416, doi: 10.1093/jnci/84.18.1410 (1992). [DOI] [PubMed] [Google Scholar]
- 158.Yang Y & Jobin C Novel insights into microbiome in colitis and colorectal cancer. Curr Opin Gastroenterol 33, 422–427, doi: 10.1097/MOG.0000000000000399 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kang M & Martin A Microbiome and colorectal cancer: Unraveling host-microbiota interactions in colitis-associated colorectal cancer development. Semin Immunol 32, 3–13, doi: 10.1016/j.smim.2017.04.003 (2017). [DOI] [PubMed] [Google Scholar]
- 160.Brennan CA & Garrett WS Gut Microbiota, Inflammation, and Colorectal Cancer. Annu Rev Microbiol 70, 395–411, doi: 10.1146/annurev-micro-102215-095513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gagniere J et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol 22, 501–518, doi: 10.3748/wjg.v22.i2.501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dejea CM et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592–597, doi: 10.1126/science.aah3648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bergstrom KS & Xia L Mucin-type O-glycans and their roles in intestinal homeostasis. Glycobiology 23, 1026–1037, doi: 10.1093/glycob/cwt045 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Johansson ME et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A 105, 15064–15069, doi: 10.1073/pnas.0803124105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Johansson ME, Larsson JM & Hansson GC The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A 108 Suppl 1, 4659–4665, doi: 10.1073/pnas.1006451107 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.van der Post S, Thomsson KA & Hansson GC Multiple enzyme approach for the characterization of glycan modifications on the C-terminus of the intestinal MUC2mucin. J Proteome Res 13, 6013–6023, doi: 10.1021/pr500874f (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Asker N, Axelsson MA, Olofsson SO & Hansson GC Dimerization of the human MUC2 mucin in the endoplasmic reticulum is followed by a N-glycosylation-dependent transfer of the mono- and dimers to the Golgi apparatus. J Biol Chem 273, 18857–18863 (1998). [DOI] [PubMed] [Google Scholar]
- 168.Godl K et al. The N terminus of the MUC2 mucin forms trimers that are held together within a trypsin-resistant core fragment. J Biol Chem 277, 47248–47256, doi: 10.1074/jbc.M208483200 (2002). [DOI] [PubMed] [Google Scholar]
- 169.Specian RD & Neutra MR Mechanism of rapid mucus secretion in goblet cells stimulated by acetylcholine. J Cell Biol 85, 626–640 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Birchenough GM, Nystrom EE, Johansson ME & Hansson GC A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 352, 1535–1542, doi: 10.1126/science.aaf7419 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gustafsson JK et al. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J Exp Med 209, 1263–1272, doi: 10.1084/jem.20120562 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ambort D et al. Calcium and pH-dependent packing and release of the gel-forming MUC2 mucin. Proc Natl Acad Sci U S A 109, 5645–5650, doi: 10.1073/pnas.1120269109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Schutte A et al. Microbial-induced meprin beta cleavage in MUC2 mucin and a functional CFTR channel are required to release anchored small intestinal mucus. Proc Natl Acad Sci U S A 111, 12396–12401, doi: 10.1073/pnas.1407597111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nystrom EEL et al. Calcium-activated Chloride Channel Regulator 1 (CLCA1) Controls Mucus Expansion in Colon by Proteolytic Activity. EBioMedicine 33, 134–143, doi: 10.1016/j.ebiom.2018.05.031 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Das I et al. Glucocorticoids alleviate intestinal ER stress by enhancing protein folding and degradation of misfolded proteins. J Exp Med 210, 1201–1216, doi: 10.1084/jem.20121268 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.van der Post S et al. Site-specific O-glycosylation on the MUC2 mucin protein inhibits cleavage by the Porphyromonas gingivalis secreted cysteine protease (RgpB). J Biol Chem 288, 14636–14646, doi: 10.1074/jbc.M113.459479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This in vitro study finds that glycans maintain the epithelial barrier by preventing mucin degradation via bacterial proteases.
- 177.Bergstrom K et al. Core 1- and 3-derived O-glycans collectively maintain the colonic mucus barrier and protect against spontaneous colitis in mice. Mucosal Immunol 10, 91–103, doi: 10.1038/mi.2016.45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Capaldo CT et al. Proinflammatory cytokine-induced tight junction remodeling through dynamic self-assembly of claudins. Mol Biol Cell 25, 2710–2719, doi: 10.1091/mbc.E14-02-0773 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Schroeder BO et al. Bifidobacteria or Fiber Protects against Diet-Induced Microbiota-Mediated Colonic Mucus Deterioration. Cell Host Microbe 23, 27–40 e27, doi: 10.1016/j.chom.2017.11.004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jakobsson HE et al. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep 16, 164–177, doi: 10.15252/embr.201439263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kumar S, Lutteke T & Schwartz-Albiez R GlycoCD: a repository for carbohydrate-related CD antigens. Bioinformatics 28, 2553–2555, doi: 10.1093/bioinformatics/bts481 (2012). [DOI] [PubMed] [Google Scholar]
- 182.Taylor ME, Drickamer K, Schnaar RL, Etzler ME & Varki A in Essentials of Glycobiology (eds rd et al.) 361–372 (2015). [Google Scholar]
- 183.Cummings RD, Etzler ME & Surolia A in Essentials of Glycobiology (eds rd et al.) 413–422 (2015). [Google Scholar]
- 184.Varki A & Kornfeld S in Essentials of Glycobiology (eds rd et al.) 423–433 (2015). [Google Scholar]
- 185.Kornfeld S Trafficking of lysosomal enzymes in normal and disease states. J Clin Invest 77, 1–6, doi: 10.1172/JCI112262 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Leclaire C et al. Molecular basis for intestinal mucin recognition by galectin-3 and C-type lectins. FASEB J 32, 3301–3320, doi: 10.1096/fj.201700619R (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.van Vliet SJ et al. Carbohydrate profiling reveals a distinctive role for the C-type lectin MGL in the recognition of helminth parasites and tumor antigens by dendritic cells. Int Immunol 17, 661–669, doi: 10.1093/intimm/dxh246 (2005). [DOI] [PubMed] [Google Scholar]
- 188.Taylor PR et al. The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J Immunol 169, 3876–3882, doi: 10.4049/jimmunol.169.7.3876 (2002). [DOI] [PubMed] [Google Scholar]
- 189.Singh SK et al. Characterization of murine MGL1 and MGL2 C-type lectins: distinct glycan specificities and tumor binding properties. Mol Immunol 46, 1240–1249, doi: 10.1016/j.molimm.2008.11.021 (2009). [DOI] [PubMed] [Google Scholar]
- 190.Tsuiji M et al. Molecular cloning and characterization of a novel mouse macrophage C-type lectin, mMGL2, which has a distinct carbohydrate specificity from mMGL1. J Biol Chem 277, 28892–28901, doi: 10.1074/jbc.M203774200 (2002). [DOI] [PubMed] [Google Scholar]
- 191.Suzuki N, Yamamoto K, Toyoshima S, Osawa T & Irimura T Molecular cloning and expression of cDNA encoding human macrophage C-type lectin. Its unique carbohydrate binding specificity for Tn antigen. J Immunol 156, 128–135 (1996). [PubMed] [Google Scholar]
- 192.McEver RP Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res 107, 331–339, doi: 10.1093/cvr/cvv154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Macauley MS, Crocker PR & Paulson JC Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol 14, 653–666, doi: 10.1038/nri3737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Angata T, Tabuchi Y, Nakamura K & Nakamura M Siglec-15: an immune system Siglec conserved throughout vertebrate evolution. Glycobiology 17, 838–846, doi: 10.1093/glycob/cwm049 (2007). [DOI] [PubMed] [Google Scholar]
- 195.Angata T, Hayakawa T, Yamanaka M, Varki A & Nakamura M Discovery of Siglec-14, a novel sialic acid receptor undergoing concerted evolution with Siglec-5 in primates. FASEB J 20, 1964–1973, doi: 10.1096/fj.06-5800com (2006). [DOI] [PubMed] [Google Scholar]
- 196.Varki A, Schnaar RL & Crocker PR in Essentials of Glycobiology (eds rd et al.) 453–467 (2015). [Google Scholar]
- 197.Adams OJ, Stanczak MA, von Gunten S & Laubli H Targeting sialic acid-Siglec interactions to reverse immune suppression in cancer. Glycobiology 28, 640–647, doi: 10.1093/glycob/cwx108 (2018). [DOI] [PubMed] [Google Scholar]
- 198.Xiao H, Woods EC, Vukojicic P & Bertozzi CR Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc Natl Acad Sci U S A 113, 10304–10309, doi: 10.1073/pnas.1608069113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hudak JE, Canham SM & Bertozzi CR Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat Chem Biol 10, 69–75, doi: 10.1038/nchembio.1388 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Stowell SR et al. Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens. J Biol Chem 283, 10109–10123, doi: 10.1074/jbc.M709545200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sundblad V et al. Galectins in Intestinal Inflammation: Galectin-1 Expression Delineates Response to Treatment in Celiac Disease Patients. Front Immunol 9, 379, doi: 10.3389/fimmu.2018.00379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Cummings RD, Liu FT & Vasta GR in Essentials of Glycobiology (eds rd et al.) 469–480 (2015). [Google Scholar]
- 203.Papa Gobbi R et al. A galectin-specific signature in the gut delineates Crohn’s disease and ulcerative colitis from other human inflammatory intestinal disorders. Biofactors 42, 93–105, doi: 10.1002/biof.1252 (2016). [DOI] [PubMed] [Google Scholar]
- 204.Jiang K et al. Galectin-3 regulates desmoglein-2 and intestinal epithelial intercellular adhesion. J Biol Chem 289, 10510–10517, doi: 10.1074/jbc.M113.538538 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Muglia C et al. The glycan-binding protein galectin-1 controls survival of epithelial cells along the crypt-villus axis of small intestine. Cell Death Dis 2, e163, doi: 10.1038/cddis.2011.44 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Stowell SR et al. Innate immune lectins kill bacteria expressing blood group antigen. Nat Med 16, 295–301, doi: 10.1038/nm.2103 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Santucci L et al. Galectin-1 suppresses experimental colitis in mice. Gastroenterology 124, 1381–1394, doi: 10.1016/s0016-5085(03)00267-1 (2003). [DOI] [PubMed] [Google Scholar]
- 208.Kavanaugh D, Kane M, Joshi L & Hickey RM Detection of galectin-3 interaction with commensal bacteria. Appl Environ Microbiol 79, 3507–3510, doi: 10.1128/AEM.03694-12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Nio J, Kon Y & Iwanaga T Differential cellular expression of galectin family mRNAs in the epithelial cells of the mouse digestive tract. J Histochem Cytochem 53, 1323–1334, doi: 10.1369/jhc.5A6685.2005 (2005). [DOI] [PubMed] [Google Scholar]
- 210.Gur C et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42, 344–355, doi: 10.1016/j.immuni.2015.01.010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kostic AD et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207–215, doi: 10.1016/j.chom.2013.07.007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Rubinstein MR et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14, 195–206, doi: 10.1016/j.chom.2013.07.012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Imberty A, wimmerova M, Mitchell EP & Gilboa-Garber N Structures of the lectins from Pseudomonas aeruginosa: insight into the molecular basis for host glycan recognition. Microbes Infect 6, 221–228 (2004). [DOI] [PubMed] [Google Scholar]
- 214.Nizet V, Varki A & Aebi M in Essentials of Glycobiology (eds rd et al.) 481–491 (2015). [Google Scholar]
- 215.Okuda J et al. Translocation of Pseudomonas aeruginosa from the intestinal tract is mediated by the binding of ExoS to an Na,K-ATPase regulator, FXYD3. Infect Immun 78, 4511–4522, doi: 10.1128/IAI.00428-10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wu L et al. Recognition of host immune activation by Pseudomonas aeruginosa. Science 309, 774–777, doi: 10.1126/science.1112422 (2005). [DOI] [PubMed] [Google Scholar]
- 217.Chichlowski M & Hale LP Bacterial-mucosal interactions in inflammatory bowel disease: an alliance gone bad. Am J Physiol Gastrointest Liver Physiol 295, G1139–1149, doi: 10.1152/ajpgi.90516.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Mohammadi R, Hosseini-Safa A, Ehsani Ardakani MJ & Rostami-Nejad M The relationship between intestinal parasites and some immune-mediated intestinal conditions. Gastroenterol Hepatol Bed Bench 8, 123–131 (2015). [PMC free article] [PubMed] [Google Scholar]
- 219.Cavalcanti MG et al. MIF participates in Toxoplasma gondii-induced pathology following oral infection. PLoS One 6, e25259, doi: 10.1371/journal.pone.0025259 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nishihira J & Mitsuyama K Overview of the role of macrophage migration inhibitory factor (MIF) in inflammatory bowel disease. Curr Pharm Des 15, 2104–2109 (2009). [DOI] [PubMed] [Google Scholar]
- 221.Jaff MS Higher frequency of secretor phenotype in O blood group - its benefits in prevention and/or treatment of some diseases. Int J Nanomedicine 5, 901–905, doi: 10.2147/IJN.S13980 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Aspholm-Hurtig M et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science 305, 519–522, doi: 10.1126/science.1098801 (2004). [DOI] [PubMed] [Google Scholar]
- 223.Yolken RH et al. Human milk mucin inhibits rotavirus replication and prevents experimental gastroenteritis. J Clin Invest 90, 1984–1991, doi: 10.1172/JCI116078 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Hu L et al. Cell attachment protein VP8* of a human rotavirus specifically interacts with A-type histo-blood group antigen. Nature 485, 256–259, doi: 10.1038/nature10996 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kawakubo M et al. Natural antibiotic function of a human gastric mucin against Helicobacter pylori infection. Science 305, 1003–1006, doi: 10.1126/science.1099250 (2004). [DOI] [PubMed] [Google Scholar]; This in vitro study of human mucins shows that epithelial glycans can directly inhibit the growth of harmful bacteria.
- 226.Lee H et al. Alpha1,4GlcNAc-capped mucin-type O-glycan inhibits cholesterol alpha-glucosyltransferase from Helicobacter pylori and suppresses H. pylori growth. Glycobiology 18, 549–558, doi: 10.1093/glycob/cwn037 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Nakayama J et al. Expression cloning of a human alpha1, 4-N-acetylglucosaminyltransferase that forms GlcNAcalpha1-->4Galbeta-->R, a glycan specifically expressed in the gastric gland mucous cell-type mucin. Proc Natl Acad Sci U S A 96, 8991–8996, doi: 10.1073/pnas.96.16.8991 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Karasawa F et al. Essential role of gastric gland mucin in preventing gastric cancer in mice. J Clin Invest 122, 923–934, doi: 10.1172/JCI59087 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kinoshita H et al. Cell surface glyceraldehyde-3-phosphate dehydrogenase (GAPDH) of Lactobacillus plantarum LA 318 recognizes human A and B blood group antigens. Res Microbiol 159, 685–691, doi: 10.1016/j.resmic.2008.07.005 (2008). [DOI] [PubMed] [Google Scholar]
- 230.Nishiyama K, Ueno S, Sugiyama M, Yamamoto Y & Mukai T Lactobacillus rhamnosus GG SpaC pilin subunit binds to the carbohydrate moieties of intestinal glycoconjugates. Anim Sci J 87, 809–815, doi: 10.1111/asj.12491 (2016). [DOI] [PubMed] [Google Scholar]
- 231.Watanabe M et al. Identification of a new adhesin-like protein from Lactobacillus mucosae ME-340 with specific affinity to the human blood group A and B antigens. J Appl Microbiol 109, 927–935, doi: 10.1111/j.1365-2672.2010.04719.x (2010). [DOI] [PubMed] [Google Scholar]
- 232.Bene KP et al. Lactobacillus reuteri Surface Mucus Adhesins Upregulate Inflammatory Responses Through Interactions With Innate C-Type Lectin Receptors. Front Microbiol 8, 321, doi: 10.3389/fmicb.2017.00321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Gunning AP et al. Use of Atomic Force Microscopy to Study the Multi-Modular Interaction of Bacterial Adhesins to Mucins. Int J Mol Sci 17, doi: 10.3390/ijms17111854 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Etzold S et al. Structural basis for adaptation of lactobacilli to gastrointestinal mucus. Environ Microbiol 16, 888–903, doi: 10.1111/1462-2920.12377 (2014). [DOI] [PubMed] [Google Scholar]
- 235.Garrido D, Kim JH, German JB, Raybould HE & Mills DA Oligosaccharide binding proteins from Bifidobacterium longum subsp. infantis reveal a preference for host glycans. PLoS One 6, e17315, doi: 10.1371/journal.pone.0017315 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sonnenburg JL, Angenent LT & Gordon JI Getting a grip on things: how do communities of bacterial symbionts become established in our intestine? Nat Immunol 5, 569–573, doi: 10.1038/ni1079 (2004). [DOI] [PubMed] [Google Scholar]
- 237.Martens EC, Koropatkin NM, Smith TJ & Gordon JI Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem 284, 24673–24677, doi: 10.1074/jbc.R109.022848 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Martens EC, Chiang HC & Gordon JI Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4, 447–457, doi: 10.1016/j.chom.2008.09.007 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study in mice shows that for some bacteria, the ability to consume epithelial carbohydrates provides a Darwinian advantage.
- 239.Lynch JB & Sonnenburg JL Prioritization of a plant polysaccharide over a mucus carbohydrate is enforced by a Bacteroides hybrid two-component system. Mol Microbiol 85, 478–491, doi: 10.1111/j.1365-2958.2012.08123.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Pudlo NA et al. Symbiotic Human Gut Bacteria with Variable Metabolic Priorities for Host Mucosal Glycans. MBio 6, e01282–01215, doi: 10.1128/mBio.01282-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Marcobal A, Southwick AM, Earle KA & Sonnenburg JL A refined palate: bacterial consumption of host glycans in the gut. Glycobiology 23, 1038–1046, doi: 10.1093/glycob/cwt040 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Ng KM et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99, doi: 10.1038/nature12503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kamada N et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 336, 1325–1329, doi: 10.1126/science.1222195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Rakoff-Nahoum S, Foster KR & Comstock LE The evolution of cooperation within the gut microbiota. Nature 533, 255–259, doi: 10.1038/nature17626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Faith JJ et al. The long-term stability of the human gut microbiota. Science 341, 1237439, doi: 10.1126/science.1237439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Tropini C, Earle KA, Huang KC & Sonnenburg JL The Gut Microbiome: Connecting Spatial Organization to Function. Cell Host Microbe 21, 433–442, doi: 10.1016/j.chom.2017.03.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lee SM et al. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 501, 426–429, doi: 10.1038/nature12447 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Arabyan N et al. Salmonella Degrades the Host Glycocalyx Leading to Altered Infection and Glycan Remodeling. Sci Rep 6, 29525, doi: 10.1038/srep29525 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Park D et al. Salmonella Typhimurium Enzymatically Landscapes the Host Intestinal Epithelial Cell (IEC) Surface Glycome to Increase Invasion. Mol Cell Proteomics 15, 3653–3664, doi: 10.1074/mcp.M116.063206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Yang WH et al. Recurrent infection progressively disables host protection against intestinal inflammation. Science 358, doi: 10.1126/science.aao5610 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Lo Presti A et al. Fecal and Mucosal Microbiota Profiling in Irritable Bowel Syndrome and Inflammatory Bowel Disease. Front Microbiol 10, 1655, doi: 10.3389/fmicb.2019.01655 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Owen RL Sequential uptake of horseradish peroxidase by lymphoid follicle epithelium of Peyer’s patches in the normal unobstructed mouse intestine: an ultrastructural study. Gastroenterology 72, 440–451 (1977). [PubMed] [Google Scholar]
- 253.Frey A et al. Role of the glycocalyx in regulating access of microparticles to apical plasma membranes of intestinal epithelial cells: implications for microbial attachment and oral vaccine targeting. J Exp Med 184, 1045–1059, doi: 10.1084/jem.184.3.1045 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Schulz O & Pabst O Antigen sampling in the small intestine. Trends Immunol 34, 155–161, doi: 10.1016/j.it.2012.09.006 (2013). [DOI] [PubMed] [Google Scholar]
- 255.Mantis NJ et al. Selective adherence of IgA to murine Peyer’s patch M cells: evidence for a novel IgA receptor. J Immunol 169, 1844–1851, doi: 10.4049/jimmunol.169.4.1844 (2002). [DOI] [PubMed] [Google Scholar]
- 256.Rios D et al. Antigen sampling by intestinal M cells is the principal pathway initiating mucosal IgA production to commensal enteric bacteria. Mucosal Immunol 9, 907–916, doi: 10.1038/mi.2015.121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Rescigno M et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol 2, 361–367, doi: 10.1038/86373 (2001). [DOI] [PubMed] [Google Scholar]
- 258.McDole JR et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 483, 345–349, doi: 10.1038/nature10863 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Miller MJ, Knoop KA & Newberry RD Mind the GAPs: insights into intestinal epithelial barrier maintenance and luminal antigen delivery. Mucosal Immunol 7, 452–454, doi: 10.1038/mi.2014.4 (2014). [DOI] [PubMed] [Google Scholar]
- 260.Knoop KA, Miller MJ & Newberry RD Transepithelial antigen delivery in the small intestine: different paths, different outcomes. Curr Opin Gastroenterol 29, 112–118, doi: 10.1097/MOG.0b013e32835cf1cd (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Yoshida M et al. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity 20, 769–783, doi: 10.1016/j.immuni.2004.05.007 (2004). [DOI] [PubMed] [Google Scholar]
- 262.Yoshida M et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J Clin Invest 116, 2142–2151, doi: 10.1172/JCI27821 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Chionh YT, Wee JL, Every AL, Ng GZ & Sutton P M-cell targeting of whole killed bacteria induces protective immunity against gastrointestinal pathogens. Infect Immun 77, 2962–2970, doi: 10.1128/IAI.01522-08 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Nochi T et al. A novel M cell-specific carbohydrate-targeted mucosal vaccine effectively induces antigen-specific immune responses. J Exp Med 204, 2789–2796, doi: 10.1084/jem.20070607 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Helander A et al. The viral sigma1 protein and glycoconjugates containing alpha2–3-linked sialic acid are involved in type 1 reovirus adherence to M cell apical surfaces. J Virol 77, 7964–7977, doi: 10.1128/jvi.77.14.7964-7977.2003 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Hase K et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature 462, 226–230, doi: 10.1038/nature08529 (2009). [DOI] [PubMed] [Google Scholar]; This study in mice shows that epithelial glycans can interact with foreign lectins to help uptake antigens and generate a productive immune response.
- 267.Ohno H & Hase K Glycoprotein 2 (GP2): grabbing the FimH bacteria into M cells for mucosal immunity. Gut Microbes 1, 407–410, doi: 10.4161/gmic.1.6.14078 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Bogdanos DP et al. Pancreatic-specific autoantibodies to glycoprotein 2 mirror disease location and behaviour in younger patients with Crohn’s disease. BMC Gastroenterol 12, 102, doi: 10.1186/1471-230X-12-102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zholudev A, Zurakowski D, Young W, Leichtner A & Bousvaros A Serologic testing with ANCA, ASCA, and anti-OmpC in children and young adults with Crohn’s disease and ulcerative colitis: diagnostic value and correlation with disease phenotype. Am J Gastroenterol 99, 2235–2241, doi: 10.1111/j.1572-0241.2004.40369.x (2004). [DOI] [PubMed] [Google Scholar]
- 270.Mockl L et al. Quantitative Super-Resolution Microscopy of the Mammalian Glycocalyx. Dev Cell 50, 57–72 e56, doi: 10.1016/j.devcel.2019.04.035 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Scott DW et al. N-glycosylation controls the function of junctional adhesion molecule-A. Mol Biol Cell 26, 3205–3214, doi: 10.1091/mbc.E14-12-1604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Manicassamy S et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 329, 849–853, doi: 10.1126/science.1188510 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Matricon J, Barnich N & Ardid D Immunopathogenesis of inflammatory bowel disease. Self/Nonself 1, 299–309, doi: 10.4161/self.1.4.13560 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Cummings RD & McEver RP in Essentials of Glycobiology (eds rd et al.) 435–452 (2015). [Google Scholar]
- 275.Brazil JC & Parkos CA Pathobiology of neutrophil-epithelial interactions. Immunol Rev 273, 94–111, doi: 10.1111/imr.12446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Brazil JC et al. alpha3/4 Fucosyltransferase 3-dependent synthesis of Sialyl Lewis A on CD44 variant containing exon 6 mediates polymorphonuclear leukocyte detachment from intestinal epithelium during transepithelial migration. J Immunol 191, 4804–4817, doi: 10.4049/jimmunol.1301307 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Brazil JC et al. Neutrophil migration across intestinal epithelium: evidence for a role of CD44 in regulating detachment of migrating cells from the luminal surface. J Immunol 185, 7026–7036, doi: 10.4049/jimmunol.1001293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ataga KI et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med 376, 429–439, doi: 10.1056/NEJMoa1611770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Sackstein R Translational glycobiology: Patient-oriented glycoscience research. Glycobiology 26, 544–545, doi: 10.1093/glycob/cww035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Telen MJ et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood 125, 2656–2664, doi: 10.1182/blood-2014-06-583351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Steentoft C et al. Glycan-directed CAR-T cells. Glycobiology 28, 656–669, doi: 10.1093/glycob/cwy008 (2018). [DOI] [PubMed] [Google Scholar]
- 282.Goonetilleke KS & Siriwardena AK Systematic review of carbohydrate antigen (CA 19–9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur J Surg Oncol 33, 266–270, doi: 10.1016/j.ejso.2006.10.004 (2007). [DOI] [PubMed] [Google Scholar]
- 283.Muz B et al. Inhibition of E-Selectin (GMI-1271) or E-selectin together with CXCR4 (GMI-1359) re-sensitizes multiple myeloma to therapy. Blood Cancer J 9, 68, doi: 10.1038/s41408-019-0227-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Woods EC, Yee NA, Shen J & Bertozzi CR Glycocalyx Engineering with a Recycling Glycopolymer that Increases Cell Survival In Vivo. Angew Chem Int Ed Engl 54, 15782–15788, doi: 10.1002/anie.201508783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Schelwies M et al. Glucosamine-6-sulfamate analogues of heparan sulfate as inhibitors of endosulfatases. Chembiochem 11, 2393–2397, doi: 10.1002/cbic.201000401 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Parker RB & Kohler JJ Regulation of intracellular signaling by extracellular glycan remodeling. ACS Chem Biol 5, 35–46, doi: 10.1021/cb9002514 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Stanway SJ et al. Phase I study of STX 64 (667 Coumate) in breast cancer patients: the first study of a steroid sulfatase inhibitor. Clin Cancer Res 12, 1585–1592, doi: 10.1158/1078-0432.CCR-05-1996 (2006). [DOI] [PubMed] [Google Scholar]
- 288.Hanson SR, Whalen LJ & Wong CH Synthesis and evaluation of general mechanism-based inhibitors of sulfatases based on (difluoro)methyl phenyl sulfate and cyclic phenyl sulfamate motifs. Bioorg Med Chem 14, 8386–8395, doi: 10.1016/j.bmc.2006.09.002 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Rho JH et al. A novel mechanism for desulfation of mucin: identification and cloning of a mucin-desulfating glycosidase (sulfoglycosidase) from Prevotella strain RS2. J Bacteriol 187, 1543–1551, doi: 10.1128/JB.187.5.1543-1551.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Nussbaumer P & Billich A Steroid sulfatase inhibitors. Med Res Rev 24, 529–576, doi: 10.1002/med.20008 (2004). [DOI] [PubMed] [Google Scholar]
- 291.Klaassen CD & Boles JW Sulfation and sulfotransferases 5: the importance of 3’-phosphoadenosine 5’-phosphosulfate (PAPS) in the regulation of sulfation. FASEB J 11, 404–418, doi: 10.1096/fasebj.11.6.9194521 (1997). [DOI] [PubMed] [Google Scholar]
- 292.Evans RC et al. Diet and colorectal cancer: an investigation of the lectin/galactose hypothesis. Gastroenterology 122, 1784–1792 (2002). [DOI] [PubMed] [Google Scholar]; This study in humans provides evidence that dietary lectins interact with intestinal glycans to influence gut biology and disease.
- 293.Ananthakrishnan AN Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol 12, 205–217, doi: 10.1038/nrgastro.2015.34 (2015). [DOI] [PubMed] [Google Scholar]
- 294.Grover Z, Muir R & Lewindon P Exclusive enteral nutrition induces early clinical, mucosal and transmural remission in paediatric Crohn’s disease. J Gastroenterol 49, 638–645, doi: 10.1007/s00535-013-0815-0 (2014). [DOI] [PubMed] [Google Scholar]
- 295.Hudak JE & Bertozzi CR Glycotherapy: new advances inspire a reemergence of glycans in medicine. Chem Biol 21, 16–37, doi: 10.1016/j.chembiol.2013.09.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.US National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03709628 (2020). [DOI] [PubMed] [Google Scholar]
- 297.Malickova K et al. Anticarbohydrate antibodies as markers of inflammatory bowel disease in a Central European cohort. Eur J Gastroenterol Hepatol 22, 144–150, doi: 10.1097/MEG.0b013e32832f5c7e (2010). [DOI] [PubMed] [Google Scholar]
- 298.Trbojevic Akmacic I et al. Inflammatory bowel disease associates with proinflammatory potential of the immunoglobulin G glycome. Inflamm Bowel Dis 21, 1237–1247, doi: 10.1097/MIB.0000000000000372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study in humans identifies differences in serum IgG glycosylation in ulcerative colitis and Crohn’s disease versus healthy controls.
- 299.Velcich A et al. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 295, 1726–1729, doi: 10.1126/science.1069094 (2002). [DOI] [PubMed] [Google Scholar]
- 300.Van der Sluis M et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129, doi: 10.1053/j.gastro.2006.04.020 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.