

Abstract
Autophagy is an ancient catabolic process used by cells to clear excess or dysfunctional organelles and large subcellular structures and thus performs an important housekeeping role for the cell. Autophagy is acutely sensitive to nutrient availability and is upregulated at a transcriptional and posttranslational level in response to nutrient deprivation. This serves to promote turnover of cellular content and recycling of nutrients for continued growth and survival. While important for most normal tissues, tumor cells appear to be particularly dependent on autophagy for survival under ischemic or therapeutic stress, and in response to loss of matrix attachment; autophagy is upregulated markedly in cancers as they progress to malignancy. Ras-driven tumors appear to be particularly dependent on autophagy and thus inhibition of autophagy is being pursued as a productive clinical approach for such cancers. However, this enthusiasm needs to be offset against possible negative effects of autophagy inhibition on normal tissue function and on limiting antitumor immune responses. In addressing all of these topics, we focus in on understanding how autophagy is induced by nutrient stress, its role in recycling metabolites for growing tumors, how selective forms of autophagy, such as mitophagy and ribophagy contribute specifically to tumorigenesis, how autophagy in the tumor microenvironment and throughout the animal affects access of the tumor to nutrients, and finally how different oncogenic pathways may determine which tumors respond to autophagy inhibition and which ones will not.
Macroautophagy is a catabolic process in which cellular constituents are engulfed by autophagic vesicles and targeted for degradation at the auto-phagolysosome (Dikic and Elazar, 2018; Galluzzi et al., 2017a; Simon et al., 2017). Autophagy assures cellular survival by eliminating dysfunctional organelles, trimming organelle mass as an adaptation to stress, recycling nutrients and other homeostatic functions in the cell. Autophagy also promotes tumor cell migration and invasion during metastasis (Mowers et al., 2017), modulates antitumor immunity (Zhong et al., 2016a) and is required for stemness in both normal tissues and in cancer (Smith and Macleod, 2019). Autophagy has been viewed as challenging to target in cancer therapy due to the pleotropic nature of its function and evidence suggesting it has both pro- and antitumorigenic properties (Galluzzi et al., 2015; Levy et al., 2017). However, recent in-depth “omics” analyses of human cancers and mouse models of cancer indicate that autophagy inhibition may be particularly effective for cancer therapy in combination with drugs that target the RAS-RAF-MEK-ERK pathway (Bryant et al., 2019; Kinsey et al., 2019; Lee et al., 2019). This sensitivity of RAS-driven cancers to autophagy inhibition is determined in part by the unique rewiring of tumor metabolism that makes these cancers highly dependent on autophagy for metabolite recycling (Kimmelman and White, 2017). Here, we review the role of autophagy in cancer cell metabolism including the mechanistic basis of how autophagy is induced by nutrient stress, how autophagy specifically promotes recycling of nucleotides, amino acids and lipids and finally how autophagy modulates cancer growth via effects on systemic metabolism and in the tumor microenvironment.
1. Autophagy, the process
Autophagosomes are double-membraned vesicles that form as nascent phagophore membranes at the endoplasmic reticulum (ER), the outer mitochondrial membrane (OMM) and parts of the trans-Golgi network (TGN) with membrane contribution also coming from recycling endosomes and other cellular membrane compartments (Carlsson and Simonsen, 2015; Lamb et al., 2013). There is particular interest in mitochondrial associated membrane (MAM) sites on the ER as specific sites of phagophore initiation due to the concentration of key autophagy protein complexes there (Hamasaki et al., 2013). Autophagy is orchestrated by autophagy-related genes/proteins (ATG) that are highly conserved from yeast to human and which make up the serine kinase preinitiation complex, the lipid kinase initiation complex and two major conjugation complexes (Fig. 1) (Dikic and Elazar, 2018; Galluzzi et al., 2017a; Kroemer et al., 2010; Weidberg et al., 2011).
Fig. 1.
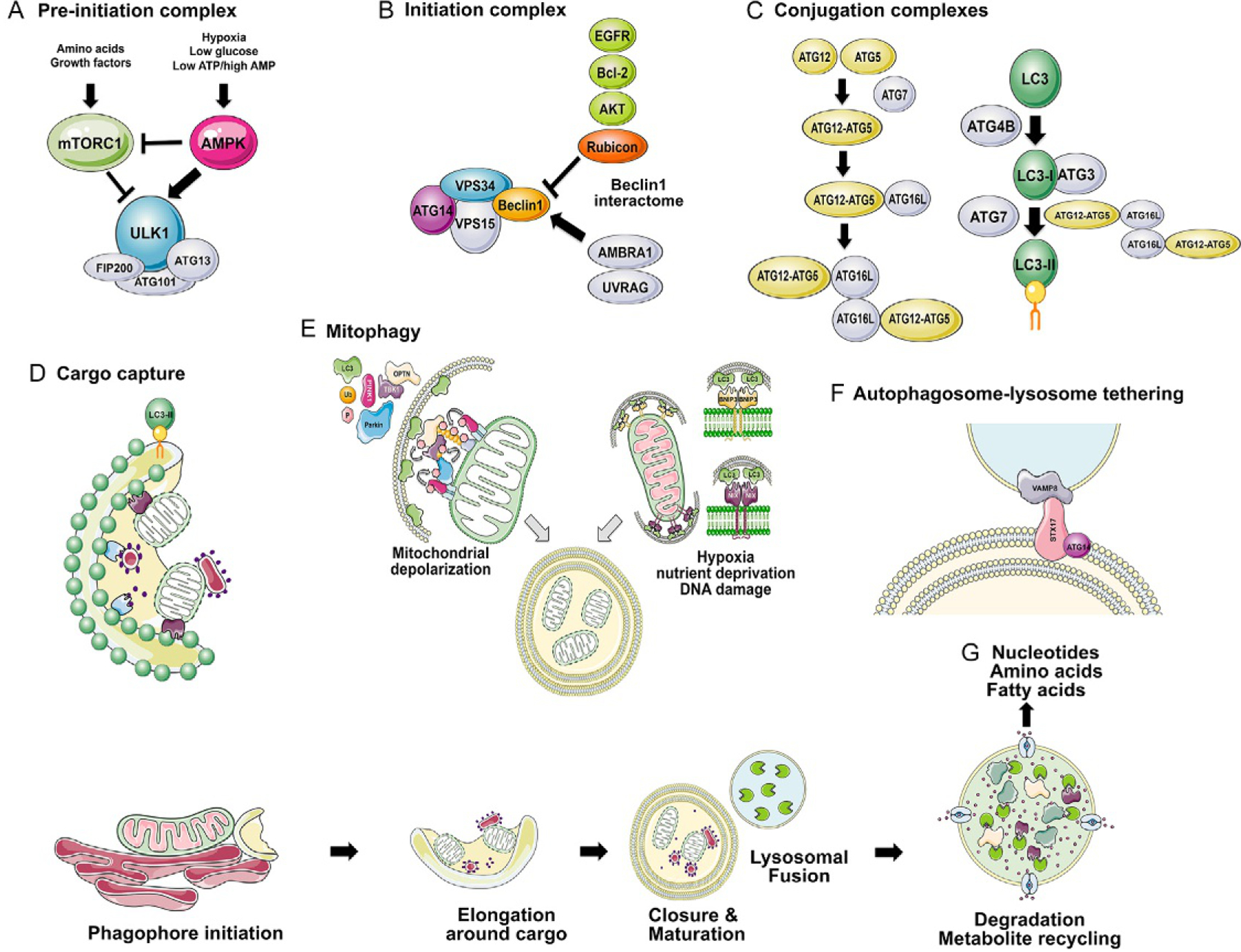
Overview of the molecular basis of autophagy initiation and completion. The process of autophagy is summarized above with (A) nutrient control of the preinitiation complex (containing the critical ULK1/ULK2, FIP200, ATG13 and ATG101) via activation of AMPK and repression of mTOR; (B) initiation complex made up of VPS34, VPS15, ATG14 and Beclin1 promotes phagophore formation and recruitment of the conjugation complexes, ATG12-ATG5/ATG16L and LC3-II-PE; (C) conjugation complexes ATG12-ATG5/ATG16L and LC3-II-PE are formed under the control of ATG7; (D) autophagic cargo is recruited to the growing phagophore through interaction of cognate cargo receptors, such as NIX (purple) with mitochondria and NUPIF1 (pale blue) with ribosomes via LIR motifs with LC3-II; (E) mitophagy is a selective form of autophagy in which mitochondria are targeted for degradation as a result of depolarization in a ubiquitin-dependent manner (PINK1/Parkin), or in response to nutrient stress in a ubiquitin-independent manner (BNIP3/NIX); (F) fusion of the autophagosome with the lysosome is promoted by the interaction of STX17 together with ATG14 at the autophagosome, with Vamp8 at the lysosome; (G) autophagic cargo is degraded at the lysosome via lysosomal hydrolases.
The preinitiation complex consists of the Unc51-like kinases (ULK1 or ULK2) bound to ATG13, ATG101 and FAK-interacting protein 200kDa (FIP200). This complex senses cellular nutrient stress via the ULK1/ULK2 kinases, which are substrates of AMP-activated Kinase (AMPK) activated by low ATP levels that arise from hypoxia, glucose deprivation and other metabolic stresses (Fig. 1A) (Egan et al., 2011). Conversely, mammalian Target of Rapamycin (mTOR) negatively regulates ULK1/ULK2, causing the preinitiation complex and autophagy to be inhibited in nutrient-replete conditions, but activated acutely when mTORC1 is inhibited, such as occurs following amino acid depletion. Following activation, ULK1/ULK2 phosphorylates Beclin1 (encoded by BECN1/ATG6) promoting its association with and activation of Vacuolar Protein Sorting 34 (VPS34), a class III PI3K that is part of the initiation complex with VPS15 and ATG14L (Russell et al., 2013). The ULK1 preinitiation complex also plays a role in delivering additional membrane to growing phagophores by controlling the trafficking of transmembrane protein ATG9 through the Golgi and endosomes. Beclin1 activity as part of the initiation complex is also controlled through its interaction with B Cell Lymphoma-2 (Bcl-2) in response to stress, such that phosphorylation of Bcl-2 by stress kinase cJun-kinase 1 (JNK1) induces dissociation of Bcl-2 from Beclin1 and activation of autophagy (Pattingre et al., 2005; Wei et al., 2008). The Beclin1 interactome also includes Autophagy and Beclin1 regulator-1 (Ambra1), UV radiation resistance associated gene (UVRAG), Rubicon and other proteins and represents a second major node for signal integration into autophagy activation or repression, particularly in response to Protein kinase B (AKT) and Epidermal Growth Factor Receptor (EGFR) signaling (Fig. 1B) (Kroemer et al., 2010; Wei et al., 2013).
Activation of the VPS34 lipid kinase increases phospho-inositol-3-phosphate (PIP3) production that results in recruitment of PIP3-binding proteins Double-FYVE-containing protein 1 (DFCP1) and WD repeat domain, phosphoinositide interacting protein 2 (WIPI2) that in turn recruit the ATG5-ATG12/ATG16L conjugation complex (Fig. 1C) (Carlsson and Simonsen, 2015; Mizushima and Komatsu, 2011). ATG7 and ATG10 promote covalent conjugation of ATG5 to ATG12 and this ATG12-ATG5 conjugate then interacts with ATG16L, which promotes multimerization of ATG12-ATG5/ATG16L via homodimerization of ATG16L. The ATG5-ATG12/ATG16L complex is required for the conjugation of ubiquitin-like microtubule-associated protein 1 light chain 3 (encoded by MAP1LC3/ATG3 and commonly known as LC3) and related GABA type A receptor-associated protein (GABARAP/L1/L2) molecules to phenylethanolamine (PE), a major membrane phospholipid in cells, to form LC3-II (Fig. 1C) (Carlsson and Simonsen, 2015, Mizushima and Komatsu, 2011). First, LC3 is cleaved by the redox-sensitive cysteine protease ATG4B to generate LC3-I, which is then activated by ATG7 and passed to ATG3 in the second major conjugation reaction. The ATG12-ATG5/ATG16L complex promotes the transfer of LC3-I from ATG3 to PE in the final step of the conjugation process. Levels of LC3-II at the growing phagophore determines the size of autophagosomes, promotes tethering of lipid bilayers, introduces membrane curvature, regulates closure of the phagophore and interacts with selective autophagy receptors.
Cellular content destined for degradation at the autophagosome, called “cargo” is targeted for degradation via cognate cargo receptors containing specific motifs known as LC3-interacting regions (LIR) which promote their interaction with LC3/GABARAP at nascent phagophores (Fig. 1D) (Rogov et al., 2014; Weidberg et al., 2011). Notable cargo receptors include SQSTM1 (p62/Sqstm1), Optineurin (OPTN), Near BRCA1 (NBR1), Nuclear Dot Protein 52 (NDP52) and Tax1 Binding protein (TAX1BP1), which mediate bivalent interaction with LC3-II and with phosphorylated ubiquitin moieties on the cargo, including protein aggregates and organelles (Harper et al., 2018; Moscat et al., 2016).
Other cargo receptors are more restricted in the cargo they deliver to the autophagosome and are involved in selective autophagy, including mitophagy wherein mitochondria are selectively targeted for degradation (Drake et al., 2017), or ribophagy in which ribosomes are selectively turned over (An and Harper, 2018). Bcl-2 interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L, or NIX as it is more commonly called to distinguish it clearly from BNIP3) are selective mitophagy receptors (Drake et al., 2017). Both BNIP3 and NIX integrate into the OMM via their carboxy-terminal transmembrane domains and interact with LC3-II via conserved LIR motifs at their amino-termini (Hanna et al., 2012; Macleod, in press; Schwarten et al., 2009) (Fig. 1E). Similarly, nuclear fragile X mental retardation-interacting protein 1 (NUFIP1) is a selective ribophagy receptor that also contains a LIR motif to interact with LC3B-II and translocates from the nucleus to the autophagosome under nutrient stress to promote ribosome turnover (Wyant et al., 2018).
Closure of the phagophore to form a mature autophagosome requires both the ATG12-ATG5-ATG16L and LC3-II conjugates that promote elongation of the phagophore around the cargo (Tsuboyama et al., 2016). Depletion of either conjugate causes incomplete autophagosomes to accumulate and failure to degrade the inner autophagosomal membrane (Tsuboyama et al., 2016). There is evidence that GABARAP-II plays a more significant role in closure of the autophagosome than does LC3-II that is more important for elongation around the cargo, although both classes of LC3 were essential for autophagy in cells (Weidberg et al., 2010). Neither conjugate is required for autophagosome-lysosome fusion (Tsuboyama et al., 2016).
Fusion of the autophagosome with the lysosome requires autophagosome tethering to the lysosome and membrane fusion (Nakamura and Yoshimori, 2017; Yu et al., 2018). A key component of the tethering mechanism is the homotypic fusion and protein sorting (HOPS) complex that in conjunction with RAB7 and other small GTPases promotes tethering and fusion of the autophagosome with the lysosome (Yu et al., 2018). HOPS interacts with Syntaxin-17 (STX17), an autophagosomal soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein that is essential for autophagosome-lysosomal fusion, such that expression of dominant negative STX17 caused mature autophagosomes to accumulate (Uematsu et al., 2017). STX17 translocates to the ER and mitochondria when phagophores are initiating, where it recruits ATG14L (Hamasaki et al., 2013). STX17 also concentrates at closed autophagosomes where ATG14L promotes the interaction of STX17 with vesicle-associated membrane protein 8 (VAMP8), a v-SNARE protein at the lysosome (Diao et al., 2015) (Fig. 1F). Thus, STX17 and ATG14L play a role in both autophagosome biogenesis at early stages of the process but also in completion of autophagy (Nakamura and Yoshimori, 2017). Additional autophagosomal SNARE proteins, YKT6 for example, can also promote autophagosome-lysosome fusion independent of STX17 (Matsui et al., 2018). Fusion of the autophagosome with the lysosome results in the degradation of autophagosomal cargo mediated by lysosomal hydrolases resident in the lysosome, including cathepsins, sphingomyelinase, DNase, RNase, acid phosphatases and other enzymes active at acid pH (Settembre et al., 2013b). This in turn promotes recovery and recycling of amino acids, nucleotides and fatty acids derived from the degraded cargo, back to the cytosol for use in biosynthetic processes and metabolism that promote cancer cell growth (Davidson and Vander Heiden, 2017; Kimmelman and White, 2017) (Fig. 1G).
Various ATG proteins also function in a noncanonical fashion in the secretion of key cytokines and danger-associated molecular patterns (DAMPs) from cells, in what is known as secretory autophagy (Ponpuak et al., 2015). In addition, critical ATG proteins (ATG5, ATG7, VPS34 and others, but not the preinitiation complex) function in LC3-associated phagocytosis (LAP) to eliminate extra-cellular material, including dead cells and pathogens preventing autoimmunity and promoting antitumor T cell responses (Cunha et al., 2018; Heckmann and Green, 2019). A putative role in degrading the content of vesicles captured by the cell via macropinocytosis, entosis, endocytic trafficking and/or exosome production represent other likely noncanonical contributions of ATG proteins to cellular homeostasis (Florey and Overholtzer, 2019). As a result, care needs to be taken when interpreting data, involving deletion of specific ATG genes, that resulting phenotypes are not mistakenly attributed to macroautophagy per se without additional validation. Understanding the biochemistry of ATG protein interaction and function and its relevance for cellular functions is an area of ongoing intense research interest.
2. Activation of autophagy by nutrient stress
Autophagy is active at low levels in most tissues where it performs a housekeeping role, particularly in the nervous system, liver, muscle and immune system, as evidenced by the tissue defects in mice inactivated for autophagy in those tissues (Hara et al., 2006; Karsli-Uzunbas et al., 2014; Komatsu et al., 2005, 2006). Acute systemic inhibition of autophagy in the whole mouse caused neurodegeneration and susceptibility to infection, as well as fatal hypoglycemia, liver and muscle damage and reduced long term survival of otherwise healthy mice (Karsli-Uzunbas et al., 2014). Autophagy is particularly critical for cell survival in most tissues exposed to nutrient stressors, including deprivation of oxygen (hypoxia), amino acids and glucose, such as occurs in ischemic tumors (Galluzzi et al., 2014; Kimmelman and White, 2017; Perera et al., 2015). Autophagy also is important in the response to proteotoxic and ER stress (Wilkinson, 2019) and in response to mitochondrial depolarization and dysfunction (D’Amico et al., 2017; Drake et al., 2017). In the context of human cancer, a marked increase in autophagy is detected as tumors progress to becoming invasive and malignant (Mowers et al., 2017) and increased LC3B-II staining was associated with increased metastasis and poor prognosis in breast cancer and melanoma (Lazova et al., 2010, 2012; Zhao et al., 2013). This role in promoting metastasis may be explained in part by the role of autophagy in promoting survival following matrix detachment that occurs during progression to invasive cancer (Avivar-Valderas et al., 2013; Fung et al., 2008). Autophagy is increased in these various settings promoting adaptation to the imposed stress and cellular survival. The mechanisms by which autophagy is regulated in cancer are discussed in more detail below.
2.1. Transcriptional control of autophagy in cancer
ATG genes are transcriptionally induced by the Activating transcription factor-4 (ATF4) transcription factor in response to amino acid deprivation, hypoxia and ER stress (Rzymski et al., 2009, 2010), by MiTF/TFE (Microphthalmia-associated transcription factor/Transcription Factor E-box Binding) factors in response to mTOR inhibition (Perera et al., 2015; Settembre et al., 2012) and by Forkhead Box (FoxO) transcription factors in stem cells, muscle atrophy and hepatic nutrient stress (Liu et al., 2009; Mammucari et al., 2007; Warr et al., 2013). Other stress-activated transcription factors induce specific cargo receptors or autophagy modulators. For example, hypoxia-inducible factor-1 (HIF-1) induces the mitophagy receptors BNIP3 and NIX in response to hypoxia (Bruick, 2000; Kasper et al., 2005), while p53 activates expression of damage-regulated autophagy modulator (DRAM) in response to DNA damage (Crighton et al., 2006). Similarly, nuclear factor kappa B (NF-kB) induces expression of the p62/Sqstm1 cargo receptor during inflammatory responses (Zhong et al., 2016b) while signal transducer and activator of transcription 3 (STAT3) can repress autophagy by recruiting histone deacetylase-3 (HDAC3) to the Beclin1 promoter to silence it (You et al., 2015). However, here we focus on discussion of transcription factors that play a central generic role in regulating autophagy, namely, ATF4, MiTF/TFE and FoxO transcription factors (Fig. 2).
Fig. 2.
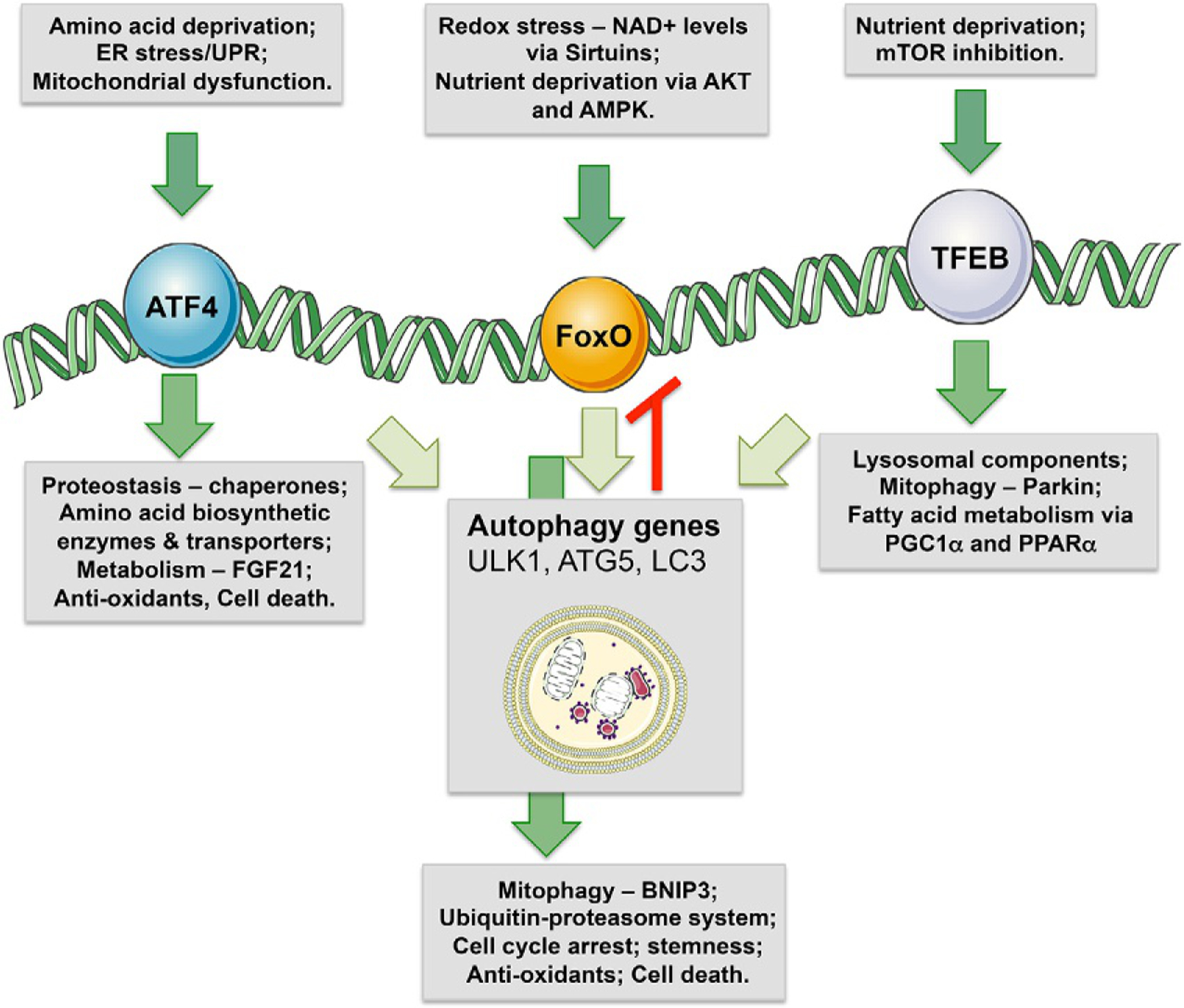
Transcriptional control of autophagy. Transcriptional control of autophagy is mediated by multiple transcription factors, including ATF4, FoxOs and MiTF/TFE factors. Other factors that also induce autophagy genes are PPARα, p53, HIF1, NFkB and others but ATF4, FoxOs and MiTF/TFE factors do so in a more general manner. ATF4 is induced by amino acid deprivation, ER stress and mitochondrial dysfunction. In addition to inducing autophagy, ATF4 also induces genes involved in amino acid metabolism, systemic metabolism, protein refolding, antioxidant responses, and cell death genes that are activated if stress is not resolved. FoxOs are transcription factors that promote stemness and reduce aging and are activated by nutrient deprivation and redox stress. In addition to inducing general autophagy via ATG genes, FoxOs potently induce mitophagy via BNIP3 in atrophying muscle, cell cycle arrest in high redox states, and like ATF4 promotes cell death via Bim and Puma if cell stress is not removed. TFEB is the key factor of the MiTF/TFE transcription factor family that induces numerous autophagy genes (ATG) and lysosomal genes, and is subject to nutrient control via mTOR.
2.1.1. ATF4 induces autophagy during the integrated stress response
ATF4 is a basic leucine zipper (bZIP) stress-responsive transcription factor that belongs to the CREB/ATF family of transcription factors that are able to homodimerize or heterodimerize to regulate gene expression (Rzymski et al., 2009; Wortel et al., 2017). ATF4 expression is stimulated in response to amino acid deprivation via General Control Nonderepressible-2 (GCN2) and during ER stress and unfolded protein response (UPR) via protein kinase R-like ER kinase (PERK). GCN2 and PERK kinases both phosphorylate translation initiation factor eIF2α, resulting in general translational inhibition but selective translation of ATF4 protein, which is derepressed due to suppression of upstream open reading frames (ORFs) (Harding et al., 2003). There is growing evidence that ATF4 is also activated in response to mitochondrial stress, including respiratory chain inhibition by metformin (Bao et al., 2016; Lee, 2015; Quiros et al., 2017) although the mechanistic basis of this activation is less clear. Once activated as part of this integrated stress response (IRS), ATF4 induces transcription of protein chaperones such as GRP78/BiP that promote protein refolding (Han et al., 2013; Luo et al., 2003); enzymes involved in amino acid biosynthesis and transport, including asparagine synthetase (ASNS), phosphoserine amino transferase-1 (PSAT1) and SLC6A9 (Zhang et al., 2018); antioxidants, such as cystathionase (CTH) and heme oxygenase (HO1) (Dey et al., 2015); metabolic modulators, including fibroblast growth factor-21 (FGF21) and Sestrin-2 (De Sousa-Coelho et al., 2012; Gomez-Samano et al., 2017; Ye et al., 2015); and finally, in response to hypoxia, amino acid deprivation or matrix detachment, ATF4 induces expression of autophagy genes including LC3B, ULK1 and ATG5 (Pike et al., 2013; Rouschop et al., 2010; Rzymski et al., 2010). By activating expression of ATG genes, ATF4 provides an important feedback mechanism wherein amino acid deprivation leads to increased protein catabolism and amino acid recycling, and tumor cell survival. ATF4-mediated induction of autophagy genes in response to matrix detachment also protected tumor cells from anoikis and promoted lung metastasis in mouse models (Dey et al., 2015; Mowers et al., 2017). ATF4 also specifically induces Parkin suggesting that ATF4 may respond to mitochondrial stress and/or ER stress by inducing mitophagy (Bouman et al., 2011).
Significantly, ATF4 protein levels are also regulated by proteasomal turnover through interaction with the Skp-Cullin-F box (SCF) ubiquitin ligase complex containing the beta-transducin repeat containing (β-TrCP) E3Ub ligase that targets ATF4 for degradation in a phosphorylation dependent manner (Lassot et al., 2001). Consequently, treatment of tumor cells with the proteasomal inhibitor Bortezomib promotes ATF4 expression and autophagy, events which are reported to contribute to Bortezomib resistance in cancers (Milani et al., 2009; Rzymski et al., 2009).
Curiously, ATF4 binding sites are over-represented in mTOR regulated genes and the ability of mTORC1 to promote purine biosynthesis for cell growth was shown to be dependent on ATF4 regulation of methylenetetrahydrofolate dehydrogenase-2 (MTHFD2), an enzyme in the mitochondrial tetrahydrofolate cycle (Ben-Sahra et al., 2016). However, this study did not address how mTORC1 induced ATF4 activity and other work has conversely suggested that ATF4 is phosphorylated by mTORC1 resulting in increased ATF4 turnover at the proteasome (Csibi et al., 2013). ATF4 is also induced downstream of KRas in nonsmall cell lung cancer (NSCLC) via nuclear factor erythroid 2 like 2 (NRF2) and Protein Kinase B (AKT) to promote amino acid replenishment via induction of amino acid biosynthetic enzymes, including ASNS (Gwinn et al., 2018); the authors did not address a role for ATF4 in promoting autophagy in this study.
2.1.2. TFEB and the CLEAR gene network promotes autophagy
MiTF/TFE are a related group of transcription factors (MiTF, TFEB, TFE3, TFEC) that regulate expression of genes involved in lysosomal biogenesis and function (Raben and Puertollano, 2016). They bind to a motif in the promoters of their target genes called a CLEAR (Coordinated Lysosomal Expression and Regulation) element. Interestingly, the CLEAR consensus binding motif (GTCACGTGAC) contains an E-box (CANNTG) raising the possibility that other E-box binding factors such as MYC and HIF may compete with TFE factors for binding to target gene promoters. TFEB was the first factor in the family to be identified and is considered the master regulator of the CLEAR gene network (Settembre et al., 2013b). TFEB promotes expression of numerous genes involved in autophagy (including ATG4B, ULK2, ATG14, LC3A and p62/Sqstm1), such that over-expression of TFEB promotes autophagosome numbers and rates of autophagosome fusion with the lysosome (Settembre et al., 2013b). TFEB is phosphorylated by mTOR and colocalizes with mTORC1 at the lysosome under nutrient-replete conditions (Roczniak-Ferguson et al., 2012; Settembre et al., 2012). Inactivation of mTORC1 or nutrient deprivation induces nuclear translocation of TFEB and expression of its target genes, thereby linking nuclear control of autophagy-lysosomal gene expression to changes in nutrient availability (Roczniak-Ferguson et al., 2012, Settembre et al., 2012).
Deregulation of MiT/TFE transcription factors has been determined in many tumor types, including human pancreatic ductal adenocarcinoma (PDAC), renal cell carcinoma (where TFE3 and TFEB are over-expressed due to translocation) and in melanoma where MiTF is upregulated (Garraway et al., 2005). There is tight nutrient control of MiT/TFE factors in untransformed cells but in PDAC, the nuclear translocation of MiT/TFE factors is no longer subject to mTORC1 inhibition due to upregulated expression of Importin-8 (Perera et al., 2015). TFEB feeds back to control of the expression of RagD, a core component of the Ragulator that promotes recruitment of mTORC1 to the lysosome, such that tumor-associated increases in expression of MiTF/TFE factors stimulates mTORC activity and tumor cell growth (Di Malta et al., 2017). The uncoupling of MiTF/TFE factors from control by mTOR in cancers, partly explains the conundrum of how tumors can maintain high rates of autophagy despite high mTOR activity that phosphorylates ULK1 to limit autophagy.
2.1.3. FoxO transcription factors promote tissue homeostasis and limit aging via autophagy
FoxO transcription factors are activated by metabolic and stress signaling and promote cellular homeostasis through control of genes involved in cell cycle, apoptosis, antioxidant responses, the ubiquitin-proteasome system and importantly, autophagy (Salih and Brunet, 2008; Webb and Brunet, 2014). As a result, FoxOs have been linked to longevity and reduced aging (Lapierre et al., 2015). There are four isoforms of FoxOs in mammals, FoxO1, FoxO3, FoxO4 and FoxO6 that are phosphorylated by AKT in response to insulin and growth factor signaling resulting in their nuclear export and repression of their transcriptional activity. Conversely, when cells are deprived of growth factors, FoxOs translocate to the nucleus and activate expression of genes involved in promoting cellular homeostasis. In addition to their regulation by AKT, FoxOs are regulated by JNK, AMPK and other stress-induced kinases (Greer et al., 2007; Webb and Brunet, 2014). FoxOs are also stimulated by NAD+-dependent deacetylation by nuclear Sirtuins, rendering FoxOs sensitive to NAD+/NADH balance and the redox state of the cell (Salih and Brunet, 2008, Webb and Brunet, 2014).
FoxO3 was first shown to induce the autophagy genes LC3B, GABARAPL1, ULK2, ATG12, Beclin/ATG6 and ATG4B, as well as the mitophagy receptor BNIP3, in skeletal muscle undergoing atrophy in response to starvation or denervation (Mammucari et al., 2007). FoxOs are also implicated in promoting lipophagy in the liver via induction of ATG14 (Xiong et al., 2012) and in neurons following JNK inactivation (Xu et al., 2011). They also drive autophagy in neural stems cells in response to oxidative stress responses to promote neurogenesis (Vazquez et al., 2012; Wang et al., 2013) and FoxO3A-induced autophagy survival programs are required for hematopoietic stem cell maintenance (Warr et al., 2013).
Multiple feedback pathways coordinate to control FoxO activity and autophagy. For example, FoxO3 inhibits autophagy by repressing transcription of FoxO1 (Zhu et al., 2014). Also, AKT both inhibits FoxO-dependent autophagy at a transcriptional level and inhibits autophagy via phosphorylation of Beclin1 which then inhibits the initiation complex (Wang et al., 2012). Under oxidative stress, FoxO1 dissociates from Sirtuins causing acetylated FoxO1 to accumulate in the cytoplasm where it binds to ATG7 to promote ATG12-ATG5 conjugation and autophagic flux (Zhao et al., 2010). As a result of being sequestered in the cytosol, FoxO1 is not active in promoting autophagy at a transcriptional level resulting in a self-limiting induction of autophagy. More recently, studies have shown FoxO3A turnover by autophagy in the cytoplasm (Fitzwalter et al., 2018). When autophagy was inhibited using chloroquine, FoxO3a accumulation in tumor cells lead to increased expression of the proapoptotic FoxO target gene, Puma which caused programmed cell death (Fitzwalter et al., 2018). FoxO3A-dependent induction of Puma was required to promote synergistic tumor cell killing by genotoxic agents, including doxorubicin and etoposide, in combination with chloroquine (Fitzwalter et al., 2018).
2.2. Energy sensing and posttranslational control of autophagy
AMPK and mTOR are two major growth regulatory kinases in the cell that oppose each others’ activities with respect to autophagy induction (Galluzzi et al., 2014). Their activity is frequently deregulated in cancer, for example, loss of the Liver Kinase B1 (LKB1) tumor suppressor results in reduced AMPK activity (Herzig and Shaw, 2018), and loss of the phosphatase and tensin homolog (PTEN) tumor suppressor results in elevated mTOR activity (Mossmann et al., 2018). Other regulators influence rates of autophagy, such as eIF2α, Sirtuins and acetyl transferases. But in most instances, their effect is indirect and mediated via either the transcriptional regulators detailed above, or via AMPK or mTOR (Fig. 3) (Galluzzi et al., 2014).
Fig. 3.
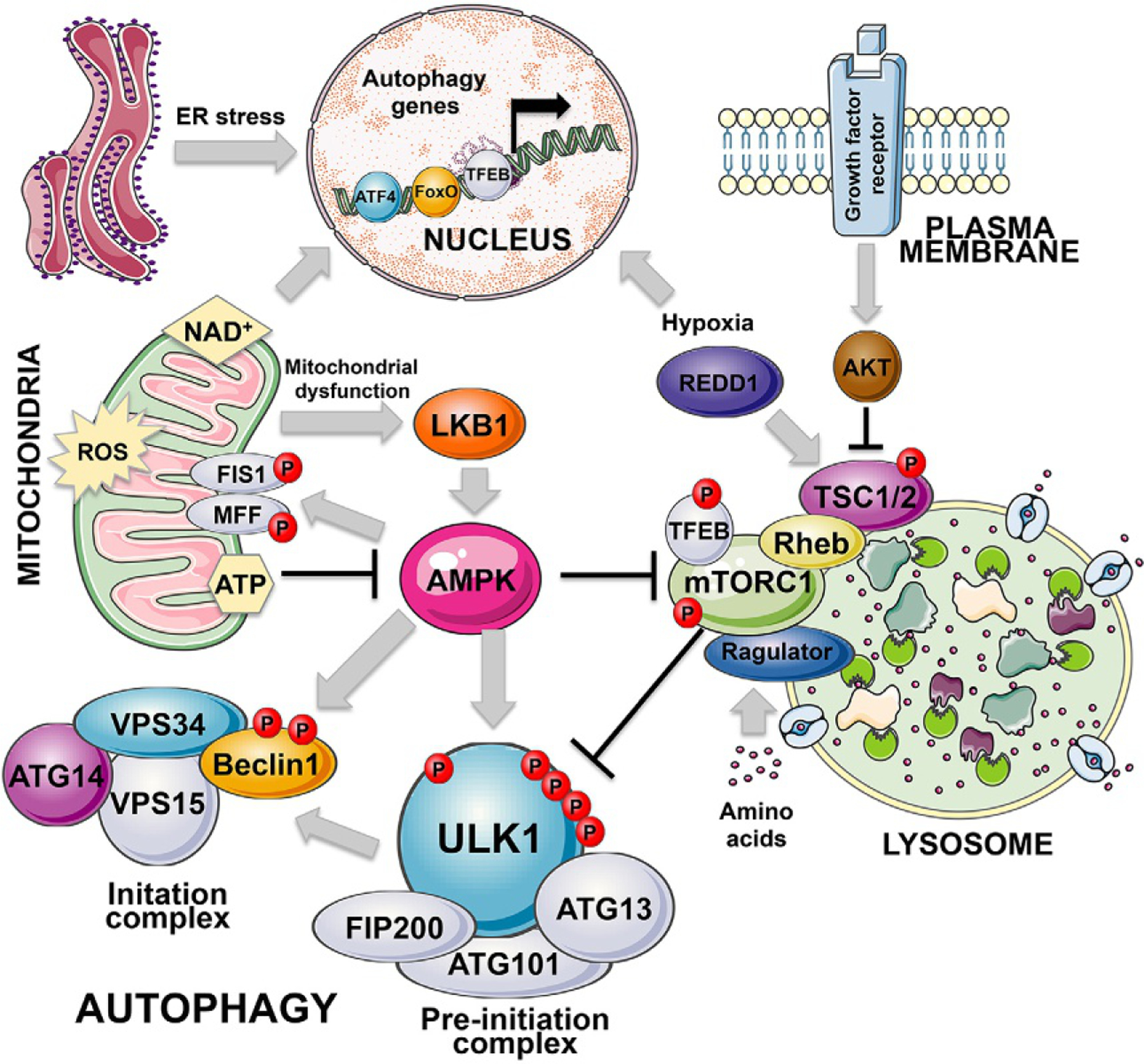
Posttranslational control of autophagy. The key posttranslational regulators of autophagy are AMPK and mTOR, kinases that sense the nutrient status of the cells to either inhibit or promote cell growth and conversely to either activate or inhibit autophagy as an adaptive response to nutrient stress. AMPK is activated by high AMP levels that arise when ATP levels are low, either due to the low energy state of the cell in general or due to mitochondrial dysfunction that can cause low ATP levels. LKB1 is a critical upstream regulator of AMPK, that is itself activated by mitochondrial dysfunction, including inhibition of the electron transport chain. AMPK in addition to promoting general autophagy by phosphorylating ULK1/2 and also Beclin1, promotes mitophagy via phosphorylation and activation of FIS1 and MFF, both receptors for DRP1 in mitochondrial fission, but emerging data suggests FIS1 plays a role in integrating mitochondrial remodeling with the autophagy machinery. mTOR by contrast with AMPK inhibits autophagy downstream of major growth stimulatory signals including growth factor receptor stimulation and AKT activation. Nutrient deprivation, including hypoxia, promotes autophagy via mTOR inhibition, mediated by specific regulators including REDD1 that is a HIF1 target. The mitochondria is a major signaling organelle that can induce mitophagy via production of reactive oxygen species (ROS), or suppress autophagy via sustained ATP production that prevents AMPK activation. Mitochondria also contribute to cellular redox balance by maintaining levels of NAD+ that keep Sirtuins in an active conformation ensuring activity of FoxOs and low PARP activity.
2.2.1. AMPK promotes autophagy
AMPK is a trimeric complex with a catalytic α-subunit and two regulatory subunits, β and γ that are modulated by glycogen and AMP, respectively, to activate the α-subunit (Herzig and Shaw, 2018). There is a critical threonine residue at T172 within the catalytic loop of AMPK’s α-subunit that is phosphorylated by upstream kinases, including the LKB1 tumor suppressor.Binding of the γ-subunit to AMP promotes phosphorylation of the α-subunit at T172 as a result of increased binding by LKB-1 and/or reduced dephosphorylation of T172 (Herzig and Shaw, 2018). Regulation of AMPK activity by AMP renders the kinase sensitive to cellular ATP and energy levels such that it becomes activated when ATP is hydrolyzed down to ADP and then AMP. In response to sensing low ATP levels, AMPK functions to limit anabolic activities in the cell that consume ATP, such as protein synthesis, lipid synthesis, glycogen synthesis and storage. Conversely, it promotes catabolic processes that generate ATP, including fatty acid oxidation, glycolysis and autophagy (Herzig and Shaw, 2018).
The initial determination that AMPK promotes autophagy came from data showing direct phosphorylation of ULK1 on four separate sites by AMPK resulting in increased ULK1-containing preinitiation complex activity and increased autophagy (Egan et al., 2011, Kim et al., 2011). In addition, AMPK has been proposed to control the assembly and activity of the initiation complex by phosphorylating Beclin1 on S91/S94 to promote its interaction with VPS34 in a manner stimulated by ATG14L (Kim et al., 2013; Russell et al., 2014).
Another key AMPK substrate that contributes to autophagy induction is Raptor, a critical subunit of mTORC1. Phosphorylation of Raptor on S722 and S792 by AMPK has been shown to inhibit mTORC1 activity (Gwinn et al., 2008). The Tuberous schlerosis-2 (TSC2) tumor suppressor is a key upstream negative regulator of mTOR. TSC2 is an AMPK substrate that when phosphorylated on S1227 and S1345 by AMPK causes mTOR inhibition (Inoki et al., 2003). AMPK also promotes autophagy by activating FoxO3 (Greer et al., 2007; Webb and Brunet, 2014) and by indirectly stimulating TFEB (via mTOR inhibition) (Herzig and Shaw, 2018) with both FoxO3 and TFEB inducing autophagy gene expression, as discussed above. Recently, AMPK regulation of TFE3-induced lysosomal gene expression was shown to be critical for growth of KRas activated/p53-mutant lung cancers in mice (Eichner et al., 2019).
In addition to its role in promoting general autophagy, AMPK has been implicated in promoting mitophagy via phosphorylation of fission, mitochondrial 1 (FIS1) and mitochondrial fission factor (MFF) (Pei et al., 2018; Toyama et al., 2016). FIS1 and MFF are both mitochondrial receptors for the mitochondrial fission GTPase, dynamin-related protein-1 (DRP1) and mitochondrial fission is required for mitophagy under certain circumstances (Gomes et al., 2011; Rambold et al., 2011). Interestingly, FIS1 is also required for PINK1 recruitment to mitochondria in leukemia stem cells (LSCs) to activate mitophagy in response to leukemia therapy (Pei et al., 2018). In addition, FIS1 interacts directly with STX17 to regulate STX17 and ATG14L recruitment to mitochondria during mitophagy, perhaps suggesting a role for FIS1 in mitophagy that is separate from its role in fission (Xian et al., 2019). Finally, AMPK was activated during mitophagy induced following extended mitotic arrest where AMPK phosphorylated the glycolysis regulator PFKFB3 to promote glycolysis and survival (Domenech et al., 2015). These more recent findings suggest that AMPK has additional functions in selective autophagy, particularly mitophagy, in addition to its role in general autophagy.
2.2.2. mTOR inhibits autophagy
Like AMPK, mTOR is also a master regulator of cell growth and metabolism in response to nutrient availability but where AMPK is the “brake” under nutrient deprivation, mTOR is the “accelerator/gas” under nutrient-replete conditions (Fig. 3) (Mossmann et al., 2018). Thus, in opposition to AMPK, mTOR promotes anabolic processes such as ribosome biogenesis and synthesis of protein, nucleotide and lipid (Saxton and Sabatini, 2017). Conversely, mTOR inhibits catabolic processes, including autophagy (Mossmann et al., 2018). mTOR is regulated in response to growth factor and nutrient availability and is particularly sensitive to levels of key amino acids, such as leucine and arginine (Saxton and Sabatini, 2017). Amino acid deprivation prevents mTOR activation at the lysosome by the Ragulator, a heteromeric complex of Rag GTPases that recruit mTOR to the lysosome to be activated by Rheb GTPase (Wolfson et al., 2016; Zoncu et al., 2011). Rheb is negatively regulated by the TSC1–TSC2 complex which acts as a GTPase-activating protein (GAP) for Rheb (Mossmann et al., 2018). TSC2 is a major signal integration point in the mTOR signaling pathway with growth factor receptor activity inhibiting TSC1–TSC2 complex formation via phosphorylation of TSC2 by AKT, and conversely, hypoxia inhibiting mTOR via induction of REDD1 which promotes TSC1–TSC2 complex formation and activity (Mossmann et al., 2018; Saxton and Sabatini, 2017).
mTOR represses autophagy in various ways, including via inhibition of TFEB and FoxOs that promote transcription of autophagy genes (Mossmann et al., 2018; Saxton and Sabatini, 2017; Webb and Brunet, 2014), as discussed above. Interestingly, ATF4 which is activated by amino acid deprivation, induces autophagy genes but also feeds back to suppress mTOR via induction of Sestrin-2 that represses mTOR in the absence of leucine (Saxton and Sabatini, 2017; Ye et al., 2015). In addition to these effects of mTOR on transcriptional control of autophagy, mTOR directly inhibits autophagy via phosphorylation of key autophagy proteins, including ULK1 and ATG13L of the preinitiation complex, and ATG14L in VPS34-containing initiation complexes (Yuan et al., 2013). mTOR directly phosphorylates ULK1 on S637 and S757 to inhibit its activity while mTOR inhibition activates ULK1 that undergoes autophosphorylation to become active (Kim et al., 2011; Shang et al., 2011). Recently, amino acid deprivation and mTOR inhibition has been shown to lead to enhanced ribophagy—the selective turnover of ribosomes that serves to supply the cell with both amino acids from ribosomal proteins and ribonucleotides from ribosomal RNA (Wyant et al., 2018).
Autophagy is required for tumor progression and metastasis and is upregulated as tumors progress to becoming invasive (Lazova et al., 2012; Mowers et al., 2017; Zhao et al., 2013). Malignant tumors also rely on high mTOR activity, which presents a conundrum in understanding how tumors maintain high rates of autophagy despite high mTOR activity. One explanation is that protein phosphatase 2A (PP2A), that is hyper-activated in tumor cells compared to normal cells, dephosphorylates phospho-S637 to maintain ULK1 activity in tumor cells despite high mTORC1 activity (Wong et al., 2015). Additionally, both TFEB and MiTF escape inhibition by mTOR in cancers to upregulate autophagy gene expression (Perera et al., 2015), as discussed above.
3. Functions of autophagy in cancer metabolism—An overview
Tumors evolve in increasingly difficult nutrient conditions as they grow and become ischemic (Gatenby and Gillies, 2008). They adapt and survive these conditions by rewiring their metabolism (Deberardinis et al., 2008), upregulating nutrient transporters (Zhang et al., 2018), finding new ways to capture food from the microenvironment, such as via macropinocytosis (Commisso et al., 2013) and by activating autophagy (Galluzzi et al., 2014; Kimmelman and White, 2017). What follows is a synthesis of how autophagy promotes recycling and cellular balance of three key classes of metabolites: nucleotides, amino acids and lipids, and how this equilibrium may be disrupted in cancer.
3.1. Autophagy and nucleotide homeostasis
Early work to understand the role of autophagy in cancer concluded that autophagy was tumor suppressive by inhibiting DNA damage downstream of a nucleotide deficit (Karantza-Wadsworth et al., 2007; Mathew et al., 2007). By recycling nucleotides through targeted degradation of mitochondria, pathogens and nuclei (that all have a degradable DNA genome), autophagy prevented replication fork stalling and DNA base-pairing mismatch, and indeed, inhibition of autophagy causes DNA damage and genome instability (Karantza-Wadsworth et al., 2007). Recent work has suggested that autophagy prevents genome instability by eliminating cells via autophagic cell death as they go through replicative crisis and that inhibition of autophagy resulted in accumulation of cells with end-to-end joined chromosomes arising from telomere attrition (Nassour et al., 2019). Nevertheless, rapidly dividing tumor cells do have a high nucleotide demand and autophagy plays a critical role in promoting tumor growth by satisfying this demand (Guo et al., 2016). Autophagy dependent ribosomal RNA degradation is essential for maintaining nucleotide homeostasis during worm development (Liu et al., 2018) and recent work in mammalian systems has identified a role for ribophagy in cellular recovery of ribonucleotides from rRNA following mTOR suppression (Wyant et al., 2018). Another major source of nucleotides is mitochondrial DNA (mtDNA) that is turned over by mitophagy (Drake et al., 2017). Mitophagy limits activation of the inflammasome and the GAS-STING pathway, both of which are activated by the release of oxidized mtDNA from damaged mitochondria to the cytosol (Chen et al., 2016; Vanpouille-Box et al., 2018; Zhou et al., 2011). By turning over mitochondria with multiple copy genomes, mitophagy likely contributes significantly to autophagic nucleotide recovery. Interestingly, autophagy plays an important role in maintaining a balance between the synthetic and degradative roles of PolG (the mitochondrial DNA polymerase/nuclease) (Medeiros et al., 2018). Following prolonged starvation, the 3′–5′ exonuclease activity of PolG dominates over its polymerase activity as a result of nucleotide insufficiency in the absence of functional autophagy, causing irreversible defects in respiration and emphasizing the importance of autophagy in maintaining mitochondrial function.
3.2. Amino acid recycling via autophagy
Amino acid deprivation potently induces autophagy and in response, autophagy acts to recycle amino acids through lysosomal degradation of cellular proteins thereby restoring amino acid levels (Perera et al., 2015; Wyant et al., 2017). Induction of autophagy by TFEB was required to maintain amino acid pools and tumorigenicity in pancreatic cancer (Perera et al., 2015). The recycling of leucine produced at the lysosome via autophagy relies on the SLC38A9 lysosomal amino acid transporter to replenish cytosolic leucine levels and to activate mTOR (Wyant et al., 2017). Thus amino acids produced by autophagy activate mTOR which in turn represses autophagy consistent with a negative feedback loop between autophagy and mTOR activity (Nicklin et al., 2009). When autophagy is defective, cells increase uptake of amino acids from their environment in part by upregulating expression of key amino acid transporters (AATs), including SLC6A9, SLC7A1, SLC7A5 and others (Chen et al., 2014; Zhang et al., 2018). Elevated AAT expression also correlated with low autophagy levels in vivo in primary colorectal cancers (Zhang et al., 2018). Many of these AATs were shown to be ATF4 transcriptional targets and upregulated in response to fasting signals particularly when autophagy was inhibited (Zhang et al., 2018) and inhibition of amino acid uptake caused death of autophagy-deficient tumor cells and limited tumor growth in response to glutamine deprivation (Zhang et al., 2018).
Apart from containing approximately 80% of cellular ribonucleotides, ribosomes also contain about half of all cellular amino acids (Wyant et al., 2018), and are particularly rich in the basic amino acids, arginine, lysine and histidine. In response to starvation, ribophagy is activated and ribosomes are turned over to provide amino acids and nucleosides (An and Harper, 2018; Wyant et al., 2018). Specifically, the nuclear protein NUFIP1 translocates to autophagosomes when cells are starved, where it interacts with LC3 and with small nucleolar ribonucleoproteins at the ribosome (Wyant et al., 2018). NUFIP1 has a LIR motif that promotes its interaction with LC3 and is required for delivering ribosomes to the lysosome for degradation (Wyant et al., 2018). NUFIP1 and ribophagy are required for cell survival under starvation conditions (Wyant et al., 2018). Interestingly, arginine is a key regulator of mTOR and thus ribophagy likely plays a important role in maintaining mTOR activity. Conversely, mTOR appears to inhibit ribophagy since inhibition of mTOR with Torin promoted translocation of NUFIP1 out of the nucleus to the autophagosome-lysosome (Wyant et al., 2018).
Another source of amino acids in the cell is the ubiquitin proteasome system (UPS) and there is clearly cross-talk between the UPS and autophagy. This is supported by evidence that UPS inhibition can activate autophagy via the ISR and UPR, while inhibition of autophagy causes p62/Sqstm1 to accumulate which can act as a sink for ubiquitin resulting in UPS inhibition (Korolchuk et al., 2010). Interestingly, FoxO transcription factors induce expression of UPS components, including various E3 Ub ligases, in addition to its activation of autophagy gene expression (Webb and Brunet, 2014). What is not clear is to what extent the UPS can compensate for autophagy deficiency. The UPS plays an important role in cancer with key cell cycle regulators, including cyclins, CDK inhibitors, turned over by the UPS and expression of various UPS components, such as Cul1 and Cks2, induced by the MYC oncogene (Bassermann et al., 2014). The use of the proteasome inhibitor, Bortezomib has been effective in the treatment of multiple myeloma and various types of lymphoma (Bassermann et al., 2014). If the proteasome can compensate for autophagy or vice versa, does this suggest that the combined use of autophagy inhibitors, such as chloroquine with Bortezomib might be more effective? Are MYC-driven tumors less reliant on autophagy than say RAS-driven tumors due to higher proteasome activity? These are all avenues for future investigation.
Finally, recent work has highlighted the role of autophagy in the tumor microenvironment in providing amino acids for growing tumor cells (Katheder et al., 2017; Sousa et al., 2016) and autophagy active in distant tissues, such as the liver appears to play important roles in maintaining circulating levels of key amino acids that sustain tumor growth (Poillet-Perez et al., 2018). This is discussed in more detail below but highlights the general importance of autophagy as a source of amino acids needed for cancer growth.
3.3. Autophagy in control of lipid metabolism
Lipophagy is a selective form of autophagy in which lipid droplets are turned over to generate free fatty acids used to generate ATP via β-oxidation (Singh and Cuervo, 2011; Singh et al., 2009). Lipophagy is induced in the liver by fasting or by high lipid loading of hepatocytes and LC3 localizes to lipid droplets during lipophagy (Singh et al., 2009). Inhibition of autophagy in the liver caused lipid droplets to accumulate and mice lacking ATG5, ATG7, ATG14 or VPS34 exhibit hepatic steatosis (Jaber et al., 2012; Singh et al., 2009; Xiong et al., 2012; Yang et al., 2010). Interestingly, liver-specific knockout of the transcriptional regulator Tfeb also caused lipid droplets to accumulate (Settembre et al., 2013a) and rescue of this defect by over-expressing Tfeb relied on functional autophagy (Settembre et al., 2013a). Tfeb likely plays multiple roles in lipid metabolism since loss of Tfeb also limited expression of PGC-1α and PPARα that regulate expression of genes involved in β-oxidation of fatty acids (Settembre et al., 2013a). FoxOs similarly coregulate lipophagy and fatty acid oxidation via direct interaction with PGC-1α and PPARα (Barthel et al., 2005).
In addition to its role inhibiting autophagy, mTOR promotes lipid synthesis (Thelen and Zoncu, 2017) and liver-specific inactivation of mTORC1 inhibits hepatic steatosis in mice on a high fat diet (Peterson et al., 2011). This ability of mTOR to promote lipogenesis is dependent on its activation of the SREBP1 transcription factor which induces expression of numerous lipid synthesis genes, including fatty acid synthase (Shao and Espenshade, 2012). It has been proposed that mTOR stimulates SREBP1 by phosphorylating and inhibiting Lipin-1, a phosphatidic acid phosphatase that regulates triglyceride synthesis (Peterson et al., 2011) but further work is required to fully appreciate the significance of this mechanism in vivo. Lipin-1 mutation causes dysfunctional mitochondria to accumulate in muscle where Lipin-1 is required for BNIP3-dependent mitophagy (Alshudukhi et al., 2018). Loss of BNIP3 also causes hepatic steatosis (Glick et al., 2012) but it is not clear whether the role of Lipin-1 in BNIP3-dependent mitophagy contributes to control of lipogenesis by mTOR in the liver.
In addition to direct turnover of lipid droplets, recent work has identified a role for autophagy in the turnover of NCoR1, nuclear receptor corepressor that inhibits PPARα, a key transcriptional regulator of fatty acid oxidation genes (Iershov et al., 2019; Saito et al., 2019), as well as autophagy genes, including LC3B, ATG7, ATG12, GABARAP and BNIP3 (Lee et al., 2014). NCoR1 interacts directly with GABARAP and is turned over by autophagy under fasted conditions. As a result, autophagy inhibition as a result of Atg5 or Atg7 knockout causes NCoR1 to accumulate and PPARα to be inhibited, resulting in impaired lipid oxidation (Saito et al., 2019). Similar findings were reported when Vps15 was knocked out in the liver (Iershov et al., 2019). Here, autophagy inhibition caused defective mitochondrial function and reduced fatty acid oxidation. This was associated with accumulation of both NCoR1 and HDAC3, also an inhibitor of PPARα (Iershov et al., 2019). PPARα activation or HDAC3 inhibition rescued both mitochondrial mass and fatty acid oxidation (Iershov et al., 2019).
In Ras-driven lung cancers, autophagy is required to maintain mitochondrial function and fatty acid oxidation but lipid accumulation in this model appeared to be dependent on loss of p53 (Guo et al., 2013) suggesting that p53 induction somehow compensates for the fatty acid oxidation defect. Autophagy was particularly critical for lung tumor growth driven by KRas activation combined with Lkb1 loss, where autophagy was required to maintain levels of free fatty acids and autophagy deficient tumor cells relied more on fatty acid oxidation than wild-type tumors (Bhatt et al., 2019). These findings suggest that Lkb1 deleted lung tumors may be effectively treated with inhibitors of fatty acid oxidation and/or autophagy inhibitors.
4. Autophagy versus mitophagy in cancer
Many of the genetically engineered mouse models used to study the role of autophagy in cancer have highlighted the role of autophagy in maintaining proper mitochondrial function (Guo et al., 2011, 2013, 2016; Rosenfeldt et al., 2013; Strohecker et al., 2013). Indeed, autophagy defects caused lung tumors to arrest in progression to become benign oncocytomas which are characterized by large cytoplasmic volume and high mitochondrial mass (Guo et al., 2013; Joshi et al., 2015). However, general autophagy contributes to tumorigenesis as a result of its role in amino acid recycling (Perera et al., 2015), and cell-cell interactions in the tumor microenvironment (Katheder et al., 2017, Sousa et al., 2016), among other pleiotropic effects of general autophagy on cellular homeostasis. To what extent mitophagy contributes to tumorigenesis more specifically has been partly addressed through analysis of the phenotypes in mice deleted for Parkin or PINK1 or for the BNIP3 and NIX mitophagy receptors (Drake et al., 2017).
Parkin and PINK1 are encoded by the genetic loci PARK2 and PARK6 respectively that are mutated in human Parkinson’s disease (PD) (Pickrell and Youle, 2015). Parkin and PINK1 function synergistically to eliminate depolarized mitochondria from the cell in a mechanism that relies on stabilization of PINK1 at the OMM when mitochondrial membrane potential is lost (Sekine and Youle, 2018). PINK1 is a ubiquitin kinase that phosphorylates key ubiquitinated substrates, including the Parkin E3 ubiquitin ligase (Harper et al., 2018). As a result of phosphorylation by PINK1, Parkin accumulates at the OMM where it ubiquitinates other substrates that are further phosphorylated by PINK1, including VDAC1, MFN2 and others (Harper et al., 2018). Ubiquitinated substrates at the OMM interact with cargo receptors, including OPTN, p62/Sqstm1, NDP52 and others, that bind to LC3B-II thereby targeting depolarized mitochondria for degradation (Harper et al., 2018; Lazarou et al., 2015). Parkin-null mice are susceptible to spontaneous hepatocellular carcinoma and sensitized to irradiation-induced lymphomagenesis (Fujiwara et al., 2008; Zhang et al., 2011) and loss of either Parkin or Pink1 promotes KRas-driven pancreatic ductal adenocarcinoma (PDAC) (Li et al., 2018). In human cancers, PARK2 maps to a common fragile site at chromosome 6q25–q26 that is frequently deleted in bladder, breast, lung and ovarian cancers (Cesari et al., 2003) while PARK2 mutations are linked to glioblastoma, colon cancer and lung cancer (Veeriah et al., 2010). PARK6 (PINK1) expression is downregulated in ovarian cancer and glioblastoma, and occasionally mutated in neuroblastoma (Agnihotri et al., 2016). Thus, in contrast to inhibition of general autophagy, mitophagy inhibition via Parkin or PINK1 disruption appears to be promote tumorigenesis. This supports a tumor suppressor function for mitophagy that is distinct from the tumor-promoting activities of general autophagy.
Similarly, epigenetic silencing of the BNIP3 mitophagy receptor in human cancers and knockout of BNIP3 in mouse models of cancer, indicates that BNIP3-dependent mitophagy is also tumor suppressive (Chourasia et al., 2015a,b). BNIP3 is a transcriptional target of HIF-1, FoxO3A and PPARα and thus is induced in response to hypoxia, nutrient deprivation and oxidative stress (Bruick, 2000; Lee et al., 2014; Mammucari et al., 2007). NIX (or BNIP3L) is also a HIF1 target and both BNIP3 and NIX appear to function analogously to promote mitophagy through interaction with LC3B-II/GABARAP via a conserved LIR motif in their amino termini (Hanna et al., 2012; Novak et al., 2010; Schwarten et al., 2009). By promoting mitophagy, it has been suggested that NIX promotes a switch to glycolytic metabolism (Esteban-Martinez et al., 2017) and this has recently been linked to a protumorigenic role for NIX in PDAC (Humpton et al., 2019) although previous studies also indicated a tumor suppressive function for NIX (Fei et al., 2005). Of note, BNIP3 and NIX are both regulated by p53 but while NIX is induced by p53 (Fei et al., 2005), BNIP3 is repressed by p53 (Feng et al., 2011). During development, BNIP3 and NIX are expressed in different tissues to differing extents with BNIP3 playing an important role in mitophagy in liver and muscle (Glick et al., 2012; Mammucari et al., 2007), while NIX is critical for mitophagy during erythroid differentiation (Sandoval et al., 2008; Schweers et al., 2007). It is possible that BNIP3 and NIX function distinctly in tumorigenesis due to differences in the timing and/or tissue-specificity of their expression, or it is also possible that BNIP3 or NIX possess additional nonmitophagy functions that cause them to contribute differentially to tumorigenesis. These are areas of ongoing research that will hopefully clarify whether mitophagy is a relevant target for cancer treatment, or not.
5. Autophagy in the tumor microenvironment
Cancer cells coevolve with their microenvironment and the role of autophagy in determining how tumor cells interact with each other and with other cell types in the tumor microenvironment (TME) is an area of intense interest (Levy et al., 2017). These interactions have come strongly into focus with the realization that autophagy in the TME generates metabolites for neighboring tumor cells (Karsli-Uzunbas et al., 2014; Katheder et al., 2017; Sousa et al., 2016; Yang et al., 2018), and indeed that autophagy at distant normal tissues, such as the liver, can also influence cancer cell metabolism remotely (Poillet-Perez et al., 2018).
5.1. Autophagy and antitumor immunity
Autophagy is required for the proper function of cells of the immune system (Jia et al., 2011; Pua et al., 2009; Puleston et al., 2014; Xu et al., 2014). Autophagy inhibition results in loss of T cell function due to, for example, reduced survival of memory T cells (Jia et al., 2011; Pua et al., 2009). Autophagy also modulates how immune cells interact with tumor cells to either promote or inhibit tumorigenesis (Ma et al., 2013; Wei et al., 2016; Zhong et al., 2016a) and since inhibiting autophagy is being promoted as an antitumor therapeutic strategy, it is important to know how autophagy inhibition affects tumor immune surveillance (Galluzzi et al., 2017b). Release of damage-associated molecular patterns (DAMPs) and ATP from dying tumor cells is dependent on autophagy and required to recruit CD8+ cytotoxic T lymphocytes (CTLs) to the tumor (Ladoire et al., 2016; Michaud et al., 2011). Autophagy also stimulates tumor-antigen production and trafficking through the lysosome for cross-presentation on dendritic cells that promotes tumor immune surveillance (Ladoire et al., 2016). Similarly, functional autophagy in the tumor suppressed infiltration of tumor-promoting FoxP3+ T-regulatory cells in a mouse model of lung cancer (Rao et al., 2014). These studies would argue that autophagy inhibition could have negative outcomes in cancer treatment (Galluzzi et al., 2017b).
However, a recent study suggested that autophagy inhibition with chloroquine does not adversely affect antitumor T cell function significantly in immunocompetent orthotopic models of breast cancer and melanoma (Starobinets et al., 2016). Indeed some studies suggest that in contrast to its role in the tumor, that autophagy may limit antitumor responses in the immune system (Noman et al., 2011; Wei et al., 2011), making it an ideal clinical target. For example, autophagy deficiency in FoxP3+ Tregs caused loss of viability and reduced the ability of Tregs to suppress antitumor immune responses (Wei et al., 2016). Furthermore, loss of autophagy in CD8+ T cells was recently reported to impair the growth of syngeneic mammary tumors as a result of increased reliance of T cells on glucose metabolism and associated changes in gene expression arising from wholesale genomic re-programming (DeVorkin et al., 2019). The context-dependent roles of autophagy in the immune response to tumors may be due to secretory autophagy or LAP in tumorigenesis, as opposed to macroautophagy itself. As discussed above, LAP has potent antitumor effects and it is not clear at this time to what extent effects of ATG5 or ATG7 deletion (or deletion of other autophagy genes common to both LAP and macroautophagy) are being mediated by LAP versus autophagy proper.
5.2. Autophagy in tumor-associated macrophages
Autophagy is required for macrophage polarization, including via NIX-dependent mitophagy promoting a glycolytic switch (Esteban-Martinez et al., 2017). In the tumor microenvironment, macrophages polarized to a M2 phenotype are recognized to play a protumorigenic role through secretion of promigratory cytokines, growth factors like EGF and vascular endothelial growth factor (VEGF), production of matrix metalloproteinases (MMPs) and inhibition of antitumor T cells responses (Qian and Pollard, 2010). A recent study identified a novel role for autophagy in suppressing the antitumor activity of macrophages in a mouse model of PDAC (Yang et al., 2018). Inhibiting autophagy through inducible expression of a dominant negative Atg4B allele (Atg4BCA) decreased tumor growth and induced tumor regression of already formed tumors in an autochthonous model of PDAC (Yang et al., 2018). This was associated with macrophage accumulation in tumors, and depletion of macrophages blocked tumor regression induced by autophagy inhibition, without altering tumor cell growth. When autophagy was similarly inhibited in an orthotopic transplant model using primary tumor cells from the same autochthonous model, tumor growth was effectively slowed but macrophage accumulation in the tumor and tumor regression was not observed (Yang et al., 2018). These findings suggest that autophagy plays a cell-intrinsic role in tumor cells promoting tumor growth and a noncell autonomous role in macrophages suppressing tumor growth (Yang et al., 2018).
5.3. Autophagy in cancer-associated fibroblasts
Carcinoma-associated fibroblasts (CAFs) are a major component of the tumor microenvironment that promote both tumor initiation and progression to carcinoma (Bhowmick et al., 2004; Kalluri, 2006). Pancreatic stellate cells (PSCs) are a specialized type of CAF that constitute a significant component of the tumor microenvironment in PDAC. Autophagy is upregulated in PSCs cocultured with pancreatic tumor cells but not when they are grown with normal ductal epithelia (Sousa et al., 2016). Autophagy-dependent protein catabolism in PSCs fueled metabolism in adjacent PDAC tumor cells as alanine generated in PSCs fed into the TCA cycle, oxygen consumption and lipid synthesis in associated tumor cells (Sousa et al., 2016). Increased alanine uptake by tumor cells redirected glucose-derived carbon to serine and glycine biosynthesis and also promoted nucleotide levels. Inhibiting autophagy in PSCs or knocking down GPT1 (alanine transaminase) in tumor cells negated the growth-stimulating effect of PDAC/PSC coculture. Autophagy is also required to activate PSCs to produce promigratory and invasive cytokines such as IL-6 in addition to extra-cellular matrix proteins (Endo et al., 2017). Thus, autophagy inhibition in PSCs attenuated IL-6 and ECM secretion and limited metastasis of associated PDAC tumor cells to the liver (Endo et al., 2017). PSC activation and associated desmoplasia in PDAC is a barrier to therapy that physically prevents access of the vasculature and drugs to the tumor (Gore and Korc, 2014). Approaches that degrade the tumor microenvironment to improve delivery of drugs to PDAC tumors have largely failed as a result of increased escape of aggressive tumor cells to secondary sites and outgrowth of metastatic disease (Ozdemir et al., 2014; Rhim et al., 2014). However, autophagy inhibition opens up a new avenue for therapy since it would both cut off the supply of amino acids from PSCs to the tumor and prevent secretion of promigratory cytokines such as IL-6 (Endo et al., 2017; Sousa et al., 2016) (Fig. 4A). These studies focused on PDAC but CAFs are known to play important tumor-promoting activities in other cancers also, including ovarian, breast and prostate cancers (Kalluri, 2006). The role of autophagy in CAFs in other types of cancer remains to be fully examined.
Fig. 4.
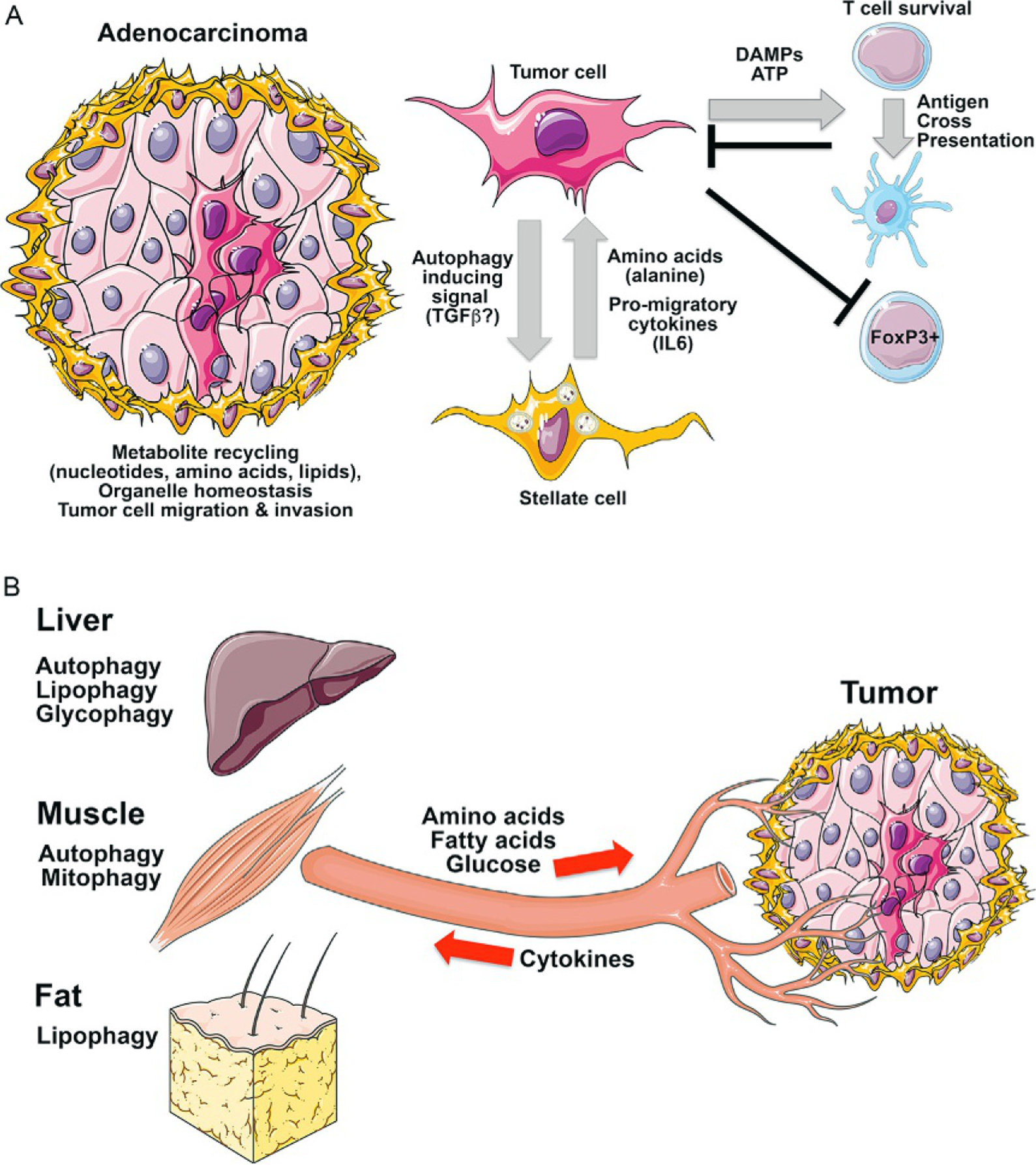
Non-cell autonomous roles of autophagy in tumorigenesis. (A) Beyond work showing a central role for autophagy in promoting tumor cell survival via metabolite recycling, organelle homeostasis and tumor cell migration, data from mouse models have identified a non-cell autonomous role for autophagy in pancreatic stellate cells supporting PDAC tumor cells by providing amino acids (specifically alanine in published studies) while other work shows associated PSCs stimulate tumor cell migration by secreting IL-6 and other pro-migratory cytokines. In Drosophila, tumor cells released signals that promoted autophagy both in the tumor microenvironment and systemically. Autophagy also plays a critical role in anti-tumor immune responses. (B) Inhibition of autophagy systemically has a more marked inhibitory effect on repression of tumor growth than inhibition of autophagy in the tumor itself. This has been attributed to the role for autophagy in distant tissues, such as liver, muscle and fat, in providing amino acids, fatty acids and glucose to the tumor via the circulation.
5.4. Systemic effects of autophagy on cancer growth
Several studies now attest to the importance of systemic autophagy in the animal for tumor growth in vivo (Fig. 4B). When whole body deletion of Atg7 to inhibit autophagy was systemically carried out, more dramatic tumor regression of KRas-driven lung cancers was observed than when autophagy was inhibited only in the tumor (Karsli-Uzunbas et al., 2014). However, systemic autophagy inhibition induced widespread metabolic defects in numerous tissues as nutrient stores were depleted urging some caution in applying autophagy inhibitors in a clinical setting (Karsli-Uzunbas et al., 2014). In related studies, systemic expression of dominant negative Atg4B to inhibit whole-body autophagy provoked marked regression of KRas driven PDAC and to a greater extent than autophagy inhibition in the tumor alone (Yang et al., 2018). Interestingly, autophagy is upregulated systemically in Drosophila with malignant RasV12;scrib−/−tumors but not with benign RasV12 tumors (Katheder et al., 2017). Again, autophagy inhibition in the tumor had only a modest effect on tumor growth whereas inhibition of autophagy in the tumor microenvironment or in the whole animal had a much more marked repressive effect on tumorigenesis (Katheder et al., 2017). Transplant of quiescent RasV12;scrib−/−; Atg13−/−autophagy deficient tumors into autophagy competent host flies rescued tumor growth while transplant into autophagy deficient hosts did not (Katheder et al., 2017). Induction of autophagy in the tumor microenvironment was referred to as noncell-autonomous autophagy (NAA) and occurred independent of tumor growth but was dependent on tumor-specific activation of Yorkie transcriptional targets upd1 and upd3 (Drosophila IL6-like cytokines) to induce NAA (Katheder et al., 2017).
To understand which metabolites and which tissues play the critical autophagy-dependent function in promoting tumor growth at a distance, metabolite profiling of serum from control mice or autophagy deficient mice was performed (Poillet-Perez et al., 2018). These studies revealed a deficit in serum arginine levels when autophagy was systemically inhibited and more specifically when Atg7 was deleted and autophagy was inhibited in the liver (Poillet-Perez et al., 2018). Significantly, tumor growth in autophagy-deficient hosts was rescued by dietary supplementation with arginine. This effect was observed for growth of syngeneic melanoma, urothelial tumors and lung tumors that were arginine auxotrophs dependent on exogenous sources of arginine (Poillet-Perez et al., 2018). Serum deficit for arginine was associated with elevated expression of arginase in autophagy-deficient liver that secretes arginase into the circulation under conditions that cause liver damage (Poillet-Perez et al., 2018). These studies provide new perspectives on the role of autophagy in tumorigenesis and suggest that metabolic stresses that induce autophagy in distant organs, such as liver or muscle, could influence tumor growth. Conversely, the Drosophila studies suggest that tumor growth may influence systemic metabolism by promoting autophagy in adjacent and remote tissues. This may be particularly relevant for cancer cachexia in which loss of muscle and fat mass in cancer patients reduces body weight and the ability of the patient to tolerate treatment regimens and survive (Fearon et al., 2012). Also, there is the possibility that other metabolites in addition to alanine or arginine, generated by autophagy in nontumor tissues may play a signaling or metabolic role in promoting tumorigenesis. These are all avenues of future investigation.
6. Autophagy in RAS driven cancers and therapeutic vulnerabilities exposed
Extensive studies on the role of autophagy in GEM models of cancer have largely focused on Ras driven cancer models, including KRas-driven NSCLC (Bhatt et al., 2019; Guo et al., 2011, 2013, 2016; Karsli-Uzunbas et al., 2014; Rao et al., 2014) and KRas-driven PDAC (Perera et al., 2015; Rosenfeldt et al., 2013; Santana-Codina et al., 2018; Sousa et al., 2016; Yang et al., 2011, 2018). These studies showed collectively that autophagy is required for progression of Ras-driven tumors to malignancy. Inhibition of autophagy in these tumors disrupted mitochondrial metabolism, including lipid oxidation, amino acid recycling, metabolite support for the tumor by the tumor microenvironment, nucleotide homeostasis, survival in response to nutrient deprivation and other key aspects of cancer metabolism (Kimmelman and White, 2017). This led to the suggestion that Ras-driven tumors are “autophagy addicted” (Guo et al., 2011) and that inhibiting autophagy would be particularly effective as a therapeutic effect in Ras-driven cancer (Kimmelman and White, 2017).
These extensive observations made it all the more surprising when recent work showed a nearly 10-fold increase in autophagic flux when KRas was acutely inhibited in PDAC lines, or equally if extracellular regulated MAP kinase (ERK) or MAP-kinase-ERK kinase (MEK) signaling was acutely inhibited (Bryant et al., 2019; Kinsey et al., 2019). The increased rate of autophagic flux detected upon acute inhibition of Ras, MEK or ERK was associated with increased AMPK activity, reduced mTOR activity, increased expression of key autophagy and lysosomal genes including ATG5, but decreased expression of glycolysis and mitochondrial biogenesis genes (Bryant et al., 2019). Nucleotide metabolism was also disrupted possibly contributing to the observed AMPK activation following Ras/MEK/ERK inhibition (Bryant et al., 2019, Kinsey et al., 2019). Oncogenic KRas promotes nucleotide biosynthesis by driving glycolysis intermediates along the nonoxidative pentose phosphate pathway (PPP) and via upregulation of MYC-driven nonoxidative PPP gene ribose 5-phosphate isomerase-A (RPIA) (Santana-Codina et al., 2018; Ying et al., 2012). Of relevance here are the observations that MYC is downregulated via proteasomal degradation when KRas or ERK is inhibited (Hayes et al., 2016; Vaseva et al., 2018). Does this make Ras-driven tumors more dependent on autophagy for nucleotide recycling when oncogenic signaling is acutely inhibited? Also, can the induction of autophagy-lysosomal genes observed with Ras/MEK/ERK inhibition be explained by downregulation of MYC? Does this result in increased TFEB activation of these genes, given that both TFEB and MYC are E-box binding factors that may compete with each other for promoter element occupancy? These are all areas for future investigation.
The significance of these findings for clinical management of Ras-driven tumors is potentially immense with a related study showing that combining Trametinib therapy (to inhibit MEK) with hydroxychloroquine (HCQ) (to inhibit autophagy) had a synergistic killing effect on PDAC cells in culture and on xenografted tumors in vivo (Kinsey et al., 2019). Importantly, this approach of combining Trametinib with HCQ was highly effective in reducing tumor burden in a PDAC patient (Kinsey et al., 2019) and these studies are now being expanded in clinical trials (NCT03825289). This therapeutic concept could also be extended to NRas/BRaf-driven melanomas, BRaf-driven pediatric brain tumors, PTEN-driven prostate cancers and KRas-driven NSCLCs that are all Ras/Raf driven malignancies that rely on autophagy for survival (Lee et al., 2019; Mulcahy Levy et al., 2017; Santanam et al., 2016; Strohecker et al., 2013; Xie et al., 2015).
In addition to these new therapeutic combinations, clinical interventions to block autophagy are now more promising due to the development of more potent dimeric analogs of CQ that are active at lower doses and have fewer side-effects, including Lys05 and DQ661 (McAfee et al., 2012; Rebecca et al., 2017). DQ661 in particular has improved lysosomal targeting capability and in addition to inhibiting autophagy, impairs the activity of palmitoyl-protein thioesterase (PPT1) that is required for mTOR interaction with Rheb at the lysosome (Rebecca et al., 2018). Furthermore, development of catalytic inhibitors of ULK1 and VPS34 will likely offer additional and more specific autophagy inhibitors to the clinic in the future. As mentioned above, some caution in the clinical use of autophagy inhibitors is advisable due to uncertainty over effects of systemic autophagy inhibition on normal tissue homeostasis, most notably on the liver and brain (Karsli-Uzunbas et al., 2014). There are also unanswered questions about whether autophagy inhibition would have negative consequences for cancer treatment outcomes due to the requirement for autophagy in tumor immune surveillance (Galluzzi et al., 2017b). Finally, it is not clear that all types of cancers will respond positively to autophagy inhibition, even if Ras-driven tumors are responsive. However, these recent findings augur well for the future of autophagy inhibition in cancer treatment and ongoing research will likely identify the specific contexts where its use in the clinic is scientifically justified.
7. Future directions
Understanding the role of autophagy in cancer has moved rapidly in the past decade to the point where the use of autophagy inhibitors in the clinic holds significant promise. These advances have primarily leveraged the role of autophagy in recycling metabolites to promote survival, although the role of autophagy in other aspects of tumorigenesis, and particularly in cancer metastasis, is also very relevant. Of note, most of the work addressing the role of autophagy in cancer has focused on the primary tumor and assessment of the role of autophagy in the metabolism of metastatic cancers and how this may differ from primary disease is an area that deserves further attention. Autophagy also underpins tumor dormancy and cancer stemness properties but the underlying mechanisms explaining how autophagy is activated during dormancy or how it promotes stemness warrant further investigation. Its role in cancer stem cells may be related to its ability to promote glycolysis and maintain tumor cells in a quiescent state. Much of the current focus on autophagy in cancer focuses on Ras-driven cancers which represent an important group of currently intractable tumors but the role of autophagy in other cancers, for example, cancers driven by MYC over-expression, remains an under-examined line of research where avenues to assess whether MYC, or indeed HIF-1, interferes with TFEB-induced autophagy gene expression via E-box competition is an intriguing line of research. Understanding the extent to which other noncanonical forms of autophagy, including LAP, contribute to cancer will need to be further examined. Finally, the development of new drugs to target autophagy, including ULK1 and VPS34 inhibitors, and thereby prevent cancers from adapting to altered nutrient availability seems to have arrived just at the right time as the scientific justification for the use of autophagy inhibitors in the clinic is gaining momentum.
References
- Agnihotri S, Golbourn B, Huang X, Remke M, Younger S, Cairns RA, Chalil A, Smith CA, Krumholtz SL, Mackenzie D, Rakopoulos P, Ramaswamy V, Taccone MS, Mischel PS, Fuller GN, Hawkins C, Stanford WL, Taylor MD, Zadeh G, Rutka JT, 2016. PINK1 is a negative regulator of growth and the Warburg effect in glioblastoma. Cancer Res. 76, 4708–4719. [DOI] [PubMed] [Google Scholar]
- Alshudukhi AA, Zhu J, Huang D, Jama A, Smith JD, Wang QJ, Esser KA,Ren H, 2018. Lipin-1 regulates Bnip3-mediated mitophagy in glycolytic muscle. FASEB J. fj201800374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Harper JW, 2018. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol 20, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avivar-Valderas A, Bobrovnikova-Marjon E, Alan Diehl J, Bardeesy N, Debnath J, Aguirre-Ghiso JA, 2013. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 32, 4932–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, Vafai SB, Cox AG, Marutani E, Ichinose F, Goessling W, Regev A, Carr SA, Clish CB, Mootha VK, 2016. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 5, e10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel A, Schmoll D, Unterman TG, 2005. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab 16, 183–189. [DOI] [PubMed] [Google Scholar]
- Bassermann F, Eichner R, Pagano M, 2014. The ubiquitin proteasome system—implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 1843, 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD, 2016. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt V, Khayati K, Hu ZS, Lee A, Kamran W, Su X, Guo JY, 2019. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev. 33, 150–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Neilson EG, Moses HL, 2004. Stromal fibroblasts in cancer initiation and progression. Nature 432, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D, Gasser T, Augustin R, Trumbach D, Irrcher I, Park DS, Wurst W, Kilberg MS, Tatzelt J, Winklhofer KF, 2011. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 18, 769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick RK, 2000. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. U. S. A 97, 9082–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, Tomar G, Papke B, Hobbs GA, Yan L, Hayes TK, Diehl JN, Goode GD, Chaika NV, Wang Y, Zhang GF, Witkiewicz AK, Knudsen ES, Petricoin EF 3rd, Singh PK, Macdonald JM, Tran NL, Lyssiotis CA, Ying H, Kimmelman AC, Cox AD, Der CJ, 2019. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med 25, 628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson SR, Simonsen A, 2015. Membrane dynamics in autophagosome biogenesis. J. Cell Sci 128, 193–205. [DOI] [PubMed] [Google Scholar]
- Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, Trapasso F, Drusco A, Shimizu M, Masciullo V, D’andrilli G, Scambia G, Picchio MC, Alder H, Godwin AK, Croce CM, 2003. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc. Natl. Acad. Sci. U. S. A 100, 5956–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zou Y, Mao D, Sun D, Gao G, Shi J, Liu X, Zhu C, Yang M, Ye W, Hao Q, Li R, Yu L, 2014. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J. Cell Biol 206, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun L, Chen ZJ, 2016. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol 17, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Chourasia AH, Boland ML, Macleod KF, 2015a. Mitophagy and cancer. Cancer Metab. 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS, Macleod KF, 2015b. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 16, 1145–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG, Bar-Sagi D, 2013. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O’prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM, 2006. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134. [DOI] [PubMed] [Google Scholar]
- Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, Henske EP, Haigis MC, Cantley LC, Stephanopoulos G, Yu J, Blenis J, 2013. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153, 840–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, Quarato G, Boada-Romero E, Kalkavan H, Johnson MDL, Natarajan S, Turnis ME, Finkelstein D, Opferman JT, Gawad C, Green DR, 2018. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell 175, 429–441.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’amico D, Sorrentino V, Auwerx J, 2017. Cytosolic proteostasis networks of the mitochondrial stress response. Trends Biochem. Sci 42, 712–725. [DOI] [PubMed] [Google Scholar]
- Davidson SM, Vander Heiden MG, 2017. Critical functions of the lysosome in cancer biology. Annu. Rev. Pharmacol. Toxicol 57, 481–507. [DOI] [PubMed] [Google Scholar]
- De Sousa-Coelho AL, Marrero PF, Haro D, 2012. Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem. J 443, 165–171. [DOI] [PubMed] [Google Scholar]
- Deberardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB, 2008. The biology of cancer: metabolic programming fuels cell growth and proliferation. Cell Metab. 7, 11–20. [DOI] [PubMed] [Google Scholar]
- DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, McNamara E, Huang H, Kim P, Zacharias LG, Mizushima N, Saitoh T, Akira S, Beckham W, Lorzadeh A, Moksa M, Cao Q, Murthy A, Hirst M, Deberardinis RJ, Lum JJ, 2019. Autophagy regulation of metabolism is required for CD8(+) T cell anti-tumor immunity. Cell Rep. 27, 502–513.e5. [DOI] [PubMed] [Google Scholar]
- Dey S, Sayers CM, Verginadis I, Lehman SL, Cheng Y, Cerniglia GJ, Tuttle SW, Feldman MD, Zhang PJ, Fuchs SY, Diehl JA, Koumenis C, 2015. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Invest 125, 2592–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, Eastes AN, Davis O, De Cegli R, Zampelli A, Di Giovannantonio LG, Nusco E, Platt N, Guida A, Ogmundsdottir MH, Lanfrancone L, Perera RM, Zoncu R, Pelicci PG, Settembre C, Ballabio A, 2017. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 356, 1188–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, Pfuetzner RA, Brunger AT, Zhong Q, 2015. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 520, 563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, Elazar Z, 2018. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol 19, 349–364. [DOI] [PubMed] [Google Scholar]
- Domenech E, Maestre C, Esteban-Martinez L, Partida D, Pascual R, Fernandez-Miranda G, Seco E, Campos-Olivas R, Perez M, Megias D, Allen K, Lopez M, Saha AK, Velasco G, Rial E, Mendez R, Boya P, Salazar-Roa M, Malumbres M, 2015. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell Biol 17, 1304–1316. [DOI] [PubMed] [Google Scholar]
- Drake LE, Springer MZ, Poole LP, Kim CJ, Macleod KF, 2017. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol 47, 110–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor RC, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ, 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner LJ, Brun SN, Herzig S, Young NP, Curtis SD, Shackelford DB, Shokhirev MN, Leblanc M, Vera LI, Hutchins A, Ross DS, Shaw RJ, Svensson RU, 2019. Genetic analysis reveals AMPK is required to support tumor growth in murine kras-dependent lung cancer models. Cell Metab. 29, 285–302.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T, Koikawa K, Okumura T, Sada M, Horioka K, Zheng B, Mizuuchi Y, Iwamoto C, Murata M, Moriyama T, Miyasaka Y, Ohtsuka T, Mizumoto K, Oda Y, Hashizume M, Nakamura M, 2017. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology 152, 1492–1506.e24. [DOI] [PubMed] [Google Scholar]
- Esteban-Martinez L, Sierra-Filardi E, Mcgreal RS, Salazar-Roa M, Marino G, Seco E, Durand S, Enot D, Grana O, Malumbres M, Cvekl A, Cuervo AM, Kroemer G, Boya P, 2017. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 36, 1688–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon KC, Glass DJ, Guttridge DC, 2012. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 16, 153–166. [DOI] [PubMed] [Google Scholar]
- Fei P, Wang W, Kim SH, Wang S, Burns TF, Sax JK, Buzzai M, Dicker DT, McKenna WG, Bernhard EJ, El-Deiry WS, 2005. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell 6, 597–609. [DOI] [PubMed] [Google Scholar]
- Feng X, Liu X, Zhang W, Xiao W, 2011. p53 directly suppresses BNIP3 expression to protect against hypoxia-induced cell death. EMBO J. 30, 3397–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzwalter BE, Towers CG, Sullivan KD, Andrysik Z, Hoh M, Ludwig M, O’prey J, Ryan KM, Espinosa JM, Morgan MJ, Thorburn A, 2018. Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev. Cell 44, 555–565.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Overholtzer M, 2019. Macropinocytosis and autophagy crosstalk in nutrient scavenging. Philos. Trans. R. Soc. Lond. B Biol. Sci 374, 20180154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara M, Marusawa H, Wang HQ, Iwai A, Ikeuchi K, Imai Y, Kataoka A, Nukina N, Takahashi R, Chiba T, 2008. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 27, 6002–6011. [DOI] [PubMed] [Google Scholar]
- Fung C, Lock R, Gao S, Salas E, Debnath J, 2008. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 19, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Levine B, Kroemer G, 2014. Metabolic control of autophagy. Cell 159, 1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, Levine B, Maiuri MC, Martin SJ, Penninger J, Piacentini M, Rubinsztein DC, Simon HU, Simonsen A, Thorburn AM, Velasco G, Ryan KM, Kroemer G, 2015. Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, Cuervo AM, Debnath J, Deretic V, Dikic I, Eskelinen EL, Fimia GM, Fulda S, Gewirtz DA, Green DR, Hansen M, Harper JW, Jaattela M, Johansen T, Juhasz G, Kimmelman AC, Kraft C, Ktistakis NT, Kumar S, Levine B, Lopez-Otin C, Madeo F, Martens S, Martinez J, Melendez A, Mizushima N, Munz C, Murphy LO, Penninger JM, Piacentini M, Reggiori F, Rubinsztein DC, Ryan KM, Santambrogio L, Scorrano L, Simon AK, Simon HU, Simonsen A, Tavernarakis N, Tooze SA, Yoshimori T, Yuan J, Yue Z, Zhong Q, Kroemer G, 2017a. Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G, 2017b. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov 16, 487–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, Lee C, Wagner SN, Li C, Golub TR, Rimm DL, Meyerson ML, Fisher DE, Sellers WR, 2005. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ, 2008. A microenvironmental model of carcinogenesis. Nat. Rev. Cancer 8, 56–61. [DOI] [PubMed] [Google Scholar]
- Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, Hart J, Dorn GW, Brady MJ, Macleod KF, 2012. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol. Cell. Biol 32, 2570–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L, 2011. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol 13, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Samano MA, Grajales-Gomez M, Zuarth-Vazquez JM, Navarro-Flores MF, Martinez-Saavedra M, Juarez-Leon OA, Morales-Garcia MG, Enriquez-Estrada VM, Gomez-Perez FJ, Cuevas-Ramos D, 2017. Fibroblast growth factor 21 and its novel association with oxidative stress. Redox Biol. 11, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore J, Korc M, 2014. Pancreatic cancer stroma: friend or foe? Cancer Cell 25, 711–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A, 2007. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem 282, 30107–30119. [DOI] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzumbas G, Kamphorst JJ, Chen G, Lemmons JMS, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E, 2011. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, Snyder E, Santanam U, Dipaola RS, Jacks T, Rabinowitz JD, White E, 2013. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 27, 1447–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, Khor S, Chan CS, Rabinowitz JD, White E, 2016. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 30, 1704–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shaclekford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ, 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D, Sweet-Cordero EA, 2018. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell 33, 91–107.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T, 2013. Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. [DOI] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ, 2013. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB, 2012. Microtubule-associated protein 1 light chain 3 (LC3) interacts with BNip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem 287, 19094–19104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N, 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D, 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Harper JW, Ordureau A, Heo JM, 2018. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol 19, 93–108. [DOI] [PubMed] [Google Scholar]
- Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, Marayati R, Kher S, George SD, Xu M, Wang-Gillam A, Samatar AA, Maitra A, Wennerberg K, Petricoin EF 3rd, Yin HH, Nelkin B, Cox AD, Yeh JJ, Der CJ, 2016. Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell 29, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann BL, Green DR, 2019. LC3-associated phagocytosis at a glance. J. Cell Sci 132, jcs231472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Shaw RJ, 2018. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol 19, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humpton TJ, Alagesan B, Denicola G, Lu D, Yordanov GN, Leonhardt CS, Yao MA, Alagesan P, Zaatari MN, Park Y, Skepper JN, Macleod KF, Perez-Mancera PA, Murphy MP, Evan GI, Vousden KH, Tuveson DA, 2019. NIX-mediated mitophagy promotes pancreatic cancer. Cancer Discov (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iershov A, Nemazanyy I, Alkhoury C, Girard M, Barth E, Cagnard N, Montagner A, Chretien D, Rugarli EI, Guillou H, Pende M, Panasyuk G, 2019. The class 3 PI3K coordinates autophagy and mitochondrial lipid catabolism by controlling nuclear receptor PPARalpha. Nat. Commun 10, 1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL, 2003. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. [DOI] [PubMed] [Google Scholar]
- Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, Zong WX, 2012. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. U. S. A 109, 2003–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W, Pua HH, Li QJ, He YW, 2011. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J. Immunol 186, 1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Tolkunov D, Aviv H, Hakimi AA, Yao M, Hsieh JJ, Ganesan S, Chan CS, White E, 2015. The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep. 13, 1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, 2006. Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401. [DOI] [PubMed] [Google Scholar]
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E, 2007. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 21, 1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, White E, 2014. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4, 914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, Baudino T, Cleveland JL, Brindle PK, 2005. Two transcriptional mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 24, 3846–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katheder NS, Khezri R, O’farrell F, Schultz SW, Jain A, Rahman MM, Schink KO, Theodossiou TA, Johansen T, Juhasz G, Bilder D, Brech A, Stenmark H, Rusten TE, 2017. Microenvironmental autophagy promotes tumour growth. Nature 541, 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL, 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL, 2013. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC, White E, 2017. Autophagy and tumor metabolism. Cell Metab. 25, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, Burrell LD, Lum DH, Whisenant JR, Gilcrease GW 3rd, Cavalieri CC, Rehbein KM, Cutler SL, Affolter KE, Welm AL, Welm BE, Scaife CL, Snyder EL, McMahon M, 2019. Protective autophagy elicited by RAF–>MEK–>ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med 25, 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueono T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T, 2005. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol 169, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata S, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K, 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Menzies FM, Rubinsztein DC, 2010. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 584, 1393–1398. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B, 2010. Autophagy and the integrated stress response. Mol. Cell 40, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladoire S, Enot D, Senovilla L, Ghiringhelli F, Poirier-Colame V, Chaba K, Semeraro M, Chaix M, Penault-Llorca F, Arnould L, Poillot ML, Arveux P, Delaloge S, Andre F, Zitvogel L, Kroemer G, 2016. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy 12, 864–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb CA, Yoshimori T, Tooze SA, 2013. The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol 14, 759–774. [DOI] [PubMed] [Google Scholar]
- Lapierre LR, Kumsta C, Sandri M, Ballabio A, Hansen M, 2015. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 11, 867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassot I, Segeral E, Berlioz-Torrent C, Durand H, Groussin L, Hai T, Benarous R, Margottin-Goguet F, 2001. ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(betaTrCP) ubiquitin ligase. Mol. Cell Biol 21, 2192–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ, 2015. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazova R, Klump V, Pawelek J, 2010. Autophagy in cutaneous malignant melanoma. J. Cutan. Pathol 37, 256–268. [DOI] [PubMed] [Google Scholar]
- Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM, 2012. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res 18, 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, 2015. Effect of mitochondrial stress on systemic metabolism. Ann. N. Y. Acad. Sci 1350, 61–65. [DOI] [PubMed] [Google Scholar]
- Lee JM, Wagner M, Xiao R, Kim KH, Lazar MA, Moore DD, 2014. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 516, 112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, Caplen NJ, McCormick F, Luo J, 2019. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. U. S. A 116, 4508–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JMM, Towers CG, Thorburn A, 2017. Targeting autophagy in cancer. Nat. Rev.Cancer 17, 528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Zhang Y, Cheng X, Yuan H, Zhu S, Liu J, Wen Q, Xie Y, Liu J, Kroemer G, Klionsky DJ, Lotze MT, Zeh HJ, Kang R, Tang D, 2018. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev. Cell 46, 441–455.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, Liu Z, Cao W, 2009. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem 284, 31484–31492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zou W, Yang P, Wang L, Ma Y, Zhang H, Wang X, 2018. Autophagy-dependent ribosomal RNA degradation is essential for maintaining nucleotide homeostasis during C. elegans development. Elife 7, e36588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS, 2003. Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem 278, 37375–37385. [DOI] [PubMed] [Google Scholar]
- Ma Y, Galluzzi L, Zitvogel L, Kroemer G, 2013. Autophagy and cellular immune responses. Immunity 39, 211–227. [DOI] [PubMed] [Google Scholar]
- Macleod KF, Mitophagy & mitochondrial dysfunction in cancer, Ann. Rev. Cancer Biol in press, AOP. [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M, 2007. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471. [DOI] [PubMed] [Google Scholar]
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E, 2007. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 21, 1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Jiang P, Nakano S, Sakamaki Y, Yamamoto H, Mizushima N, 2018. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J. Cell Biol 217, 2633–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, Winkler JD, Amaravadi RK, 2012. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. U. S. A 109, 8253–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros TC, Thomas RL, Ghillebert R, Graef M, 2018. Autophagy balances mtDNA synthesis and degradation by DNA polymerase POLG during starvation. J. Cell Biol 217, 1601–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, Vacchelli E, Galluzzi L, Ghiringhelli F, Di Virgilio F, Zitvogel L, Kroemer G, 2011. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577. [DOI] [PubMed] [Google Scholar]
- Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL, 2009. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 69, 4415–4423. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M, 2011. Autophagy: renovation of cells and tissues. Cell 147, 728–741. [DOI] [PubMed] [Google Scholar]
- Moscat J, Karin M, Diaz-Meco MT, 2016. p62 in cancer: signaling adaptor beyond autophagy. Cell 167, 606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossmann D, Park S, Hall MN, 2018. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757. [DOI] [PubMed] [Google Scholar]
- Mowers EE, Sharifi MN, Macleod KF, 2017. Autophagy in cancer metastasis. Oncogene 36, 1619–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy Levy JM, Zahedi S, Griesinger AM, Morin A, Davies KD, Aisner DL, Kleinschmidt-Demasters BK, Fitzwalter BE, Goodall ML, Thorburn J, Amani V, Donson AM, Birks DK, Mirsky DM, Hankinson TC, Handler MH, Green AL, Vibhakar R, Foreman NK, Thorburn A, 2017. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 6, e19671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Yoshimori T, 2017. New insights into autophagosome-lysosome fusion. J. Cell Sci 130, 1209–1216. [DOI] [PubMed] [Google Scholar]
- Nassour J, Radford R, Correia A, Fuste JM, Schoell B, Jauch A, Shaw RJ, Karlseder J, 2019. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565, 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO, 2009. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, Buart S, Berchem G, Romero P, Mami-Chouaib F, Chouaib S, 2011. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 71, 5976–5986. [DOI] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, LÖhr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, DÖtsch V, Ney PA, Dikic I, 2010. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses HL, Weaver VM, Maitra A, Allison JP, Lebleu VS, Kalluri R, 2014. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Linag XH, Mizushima N, Packer M, Schneider MD, Levine B, 2005. Bcl-2 anti-apoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. [DOI] [PubMed] [Google Scholar]
- Pei S, Minhajuddin M, Adane B, Khan N, Stevens BM, Mack SC, Lai S, Rich JN, Inguva A, Shannon KM, Kim H, Tan AC, Myers JR, Ashton JM, Neff T, Pollyea DA, Smith CA, Jordan CT, 2018. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell 23, 86–100.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, Settleman J, Stephanopoulos G, Dyson NJ, Zoncu R, Ramaswamy S, Haas W, Bardeesy N, 2015. Transcriptional control of the autophagy-lysosome system in pancreatic cancer. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM, 2011. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ, 2015. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LR, Singleton DC, Buffa F, Abramczyk O, Phadwal K, Li JL, Simon AK, Murray JT, Harris AL, 2013. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem. J 449, 389–400. [DOI] [PubMed] [Google Scholar]
- Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, Su X, Maganti A, Jiang C, Lu W, Zheng H, Bosenberg MW, Mehnert JM, Guo JY, Lattime E, Rabinowitz JD, White E, 2018. Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V, 2015. Secretory autophagy. Curr. Opin. Cell Biol 35, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pua HH, Guo J, Komatsu M, He YW, 2009. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol 182, 4046–4055. [DOI] [PubMed] [Google Scholar]
- Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, Watson AS, Cerundolo V, Townsend AR, Klenerman P, Simon AK, 2014. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife 3, e03706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW, 2010. Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Prado MA, Zamboni N, D’amico D, Williams RW, Finley D, Gygi SP, Auwerx J, 2017. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol 216, 2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Puertollano R, 2016. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell Dev. Biol 32, 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J, 2011. Tubular network formation protects mitochondrial from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. U. S. A 108, 10190–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, Sykacek P, Frank L, Schramek D, Komnenovic V, Sigl V, Aumayr K, Schmauss G, Fellner N, Handschuh S, GlÖsmann M, Pasierbek P, Schlederer M, Resch GP, Ma Y, Yang H, Popper H, Kenner L, Kroemer G, Penninger JM, 2014. A dual role for autophagy in a murine model of lung cancer. Nat. Commun 5, 3056–3070. [DOI] [PubMed] [Google Scholar]
- Rebecca VW, Nicastri MC, McLaughlin N, Fennelly C, McAfee Q, Ronghe A, Nofal M, Lim CY, Witze E, Chude CI, Zhang G, Alicea GM, Piao S, Murugan S, Ojha R, Levi SM, Wei Z, Barber-Rotenberg JS, Murphy ME, Mills GB, Lu Y, Rabinowitz J, Marmorstein R, Liu Q, Liu S, Xu X, Herlyn M, Zoncu R, Brady DC, Speicher DW, Winkler JD, Amaravadi RK, 2017. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 7, 1266–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecca VW, Nicastri MC, Fennelly C, Chude CI, Barber-Rotenberg JS, Ronghe A, McAfee Q, McLaughlin NP, Zhang G, Goldman AR, Ojha R, Piao S, Noguera-Ortega E, Martorella A, Alicea GM, Lee JJ, Schuchter LM, Xu X, Herlyn M, Marmorstein R, Gimotty PA, Speicher DW, Winkler JD, Amaravadi RK, 2018. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov. 9 (2), 220–229. AOP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, Stanger BZ, 2014. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM, 2012. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal 5, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov V, Dotsch V, Johansen T, Kirkin V, 2014. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 53, 167–178. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt MT, O’prey J, Morton JP, Nixon C, Mackay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, Sansom OJ, Ryan KM, 2013. p53 status determines the role of autophagy in pancreatic tumour development. Nature 504, 296–300. [DOI] [PubMed] [Google Scholar]
- Rouschop KM, Van Den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P, Van Der Kogel AJ, Koritzinsky M, Wouters BG, 2010. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Invest 120, 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL, 2013. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol 15, 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Yuan HX, Guan KL, 2014. Autophagy regulation by nutrient signaling. Cell Res. 24, 42–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzymski T, Milani M, Singleton DC, Harris AL, 2009. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 8, 3838–3847. [DOI] [PubMed] [Google Scholar]
- Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL, 2010. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 29, 4424–4435. [DOI] [PubMed] [Google Scholar]
- Saito T, Kuma A, Sugiura Y, Ichimura Y, Obata M, Kitamura H, Okuda S, Lee HC, Ikeda K, Kanegae Y, Saito I, Auwerx J, Motohashi H, Suematsu M, Soga T, Yokomizo T, Waguri S, Mizushima N, Komatsu M, 2019. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun 10, 1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih DA, Brunet A, 2008. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol 20, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Scumacker A, Prchal JT, Chen M, Wang J, 2008. Essential role for Nix in autophagic maturation of red cells. Nature 454, 232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana-Codina N, Roeth AA, Zhang Y, Yang A, Mashadova O, Asara JM, Wang X, Bronson RT, Lyssiotis CA, Ying H, Kimmelman AC, 2018. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun 9, 4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E, Dipaola RS, 2016. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 30, 399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM, 2017. mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarten M, Mohrluder J, Ma P, Stoldt M, Thielman Y, Stangler T, Hersch N, Hoffman B, Merkel R, Wilbold D, 2009. Nix binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 5, 690–698. [DOI] [PubMed] [Google Scholar]
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA, 2007. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. U. S. A 104, 19500–19505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S, Youle RJ, 2018. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 16, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A, 2012. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and Tfeb. EMBO J. 31, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, Irazoqui JE, Ballabio A, 2013a. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol 15, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A, 2013b. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol 14, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X, 2011. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U. S. A 108, 4788–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Espenshade PJ, 2012. Expanding roles for SREBP in metabolism. Cell Metab. 16, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon HU, Friis R, Tait SW, Ryan KM, 2017. Retrograde signaling from autophagy modulates stress responses. Sci. Signal 10, eaag2791. [DOI] [PubMed] [Google Scholar]
- Singh R, Cuervo AM, 2011. Autophagy in the cellular energetic balance. Cell Metab. 13, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka AJ, Cuervo AM, Czaja MJ, 2009. Autophagy regulates lipid metabolism. Nature 458, 1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG, Macleod KF, 2019. Autophagy, cancer stem cells and drug resistance. J. Pathol 247, 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, Asara JM, Evans RM, Cantley LC, Lyssiotis CA, Kimmelman AC, 2016. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starobinets H, Ye J, Broz M, Barry K, Goldsmith J, Marsh T, Rostker F, Krummel M, Debnath J, 2016. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J. Clin. Invest 126, 4417–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, White E, 2013. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 3, 1272–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelen AM, Zoncu R, 2017. Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol. 27, 833–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ, 2016. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N, 2016. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354, 1036–1041. [DOI] [PubMed] [Google Scholar]
- Uematsu M, Nishimura T, Sakamaki Y, Yamamoto H, Mizushima N, 2017. Accumulation of undegraded autophagosomes by expression of dominant-negative STX17 (syntaxin 17) mutants. Autophagy 13, 1452–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille-Box C, Demaria S, Formenti SC, Galluzzi L, 2018. Cytosolic DNA sensing in organismal tumor control. Cancer Cell 34, 361–378. [DOI] [PubMed] [Google Scholar]
- Vaseva AV, Blake DR, Gilbert TSK, Ng S, Hostetter G, Azam SH, Ozkan-Dagliyan I, Gautam P, Bryant KL, Pearce KH, Herring LE, Han H, Graves LM, Witkiewicz AK, Knudsen ES, Pecot CV, Rashid N, Houghton PJ, Wennerberg K, Cox AD, Der CJ, 2018. KRAS suppression-induced degradation of MYC is antagonized by a MEK5-ERK5 compensatory mechanism. Cancer Cell 34, 807–822.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez P, Arroba AI, Cecconi F, De La Rosa EJ, Boya P, De Pablo F, 2012. Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 8, 187–199. [DOI] [PubMed] [Google Scholar]
- Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, Holland EC, Paty PB, Mischel PS, Liau L, Cloughesy TF, Mellinghoff IK, Solit DB, Chan TA, 2010. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet 42, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White MF, Reichelt J, Levine B, 2012. Akt-mediated regulation of autophagy and tumorigenesis through Beclin1 phosphorylation. Science 338, 956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liang CC, Bian ZC, Zhu Y, Guan JL, 2013. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat. Neurosci 16, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegue E, 2013. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494, 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AE, Brunet A, 2014. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem. Sci 39, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G, Pattingre S, Sinha S, Bassik MC, Levine B, 2008. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL, 2011. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 25, 1510–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, Grishin NV, Peyton M, Minna J, Bhagat G, Levine B, 2013. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 154, 1269–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, Wu C, Vogel P, Neale G, Green DR, Chi H, 2016. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol 17, 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z, 2010. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 29, 1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Elazar Z, 2011. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem 80, 125–156. [DOI] [PubMed] [Google Scholar]
- Wilkinson S, 2019. ER-phagy: shaping up and de-stressing the endoplasmic reticulum. FEBS J. in press, AOP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, Sabatini DM, 2016. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PM, Feng Y, Wang J, Shi R, Jiang X, 2015. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun 6, 8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortel IMN, Van Der Meer LT, Kilberg MS, Van Leeuwen FN, 2017. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab 28, 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, Vander Heiden MG, Sabatini DM, 2017. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171, 642–654.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyant GA, Abu-Remaileh M, Frenkel EM, Laqtom NN, Dharamdasani V, Lewis CA, Chan SH, Heinze I, Ori A, Sabatini DM, 2018. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian H, Yang Q, Xiao L, Shen HM, Liou YC, 2019. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun 10, 2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Koh JY, Price S, White E, Mehnert JM, 2015. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. 5, 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Tao R, Depinho RA, Dong XC, 2012. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J. Biol. Chem 287, 39107–39114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Das M, Reilly J, Davis RJ, 2011. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 25, 310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, Green DR, Jones DP, Virgin HW, Ahmed R, 2014. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat. Immunol 15, 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, Hotamisligil GS, 2010. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’Antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC, 2011. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong KK, Kimmelman AC, 2018. Autophagy sustains pancreatic cancer growth through both cell autonomous and non-autonomous mechanisms. Cancer Discov. 8, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Palm W, Peng M, King B, Lindsten T, Li MO, Koumenis C, Thompson CB, 2015. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev. 29, 2331–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, Depinho RA, 2012. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You L, Wang Z, Li H, Shou J, Jing Z, Xie J, Sui X, Pan H, Han W, 2015. The role of STAT3 in autophagy. Autophagy 11, 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Chen Y, Tooze SA, 2018. Autophagy pathway: cellular and molecular mechanisms. Autophagy 14, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan HX, Russell RC, Guan KL, 2013. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 9, 1983–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lin M, Wu R, Wang X, Yang BG, Levine AJ, Hu W, Feng Z, 2011. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. U. S. A 108, 16259–16264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Yang X, Yuan F, Zhang L, Wang Y, Wang L, Mao Z, Luo J, Zhang H, Zhu WG, Zhao Y, 2018. Increased amino acid uptake supports autophagy-deficient cell survival upon glutamine deprivation. Cell Rep. 23, 3006–3020. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG, 2010. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol 12, 665–675. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yang M, Zhao J, Wang J, Zhang Y, Zhang Q, 2013. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med. Oncol 30, 475. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Sanchez-Lopez E, Karin M, 2016a. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell 166, 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, Mcgeough MD, Ellisman MH, Seki E, Gustafsson AB, Hoffman HM, Diaz-Meco MT, Moscat J, Karin M, 2016b. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J, 2011. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. [DOI] [PubMed] [Google Scholar]
- Zhu WL, Tong H, Teh JT, Wang M, 2014. Forkhead box protein O3 transcription factor negatively regulates autophagy in human cancer cells by inhibiting forkhead box protein O1 expression and cytosolic accumulation. PLoS One 9, e115087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM, 2011. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]