

Abstract
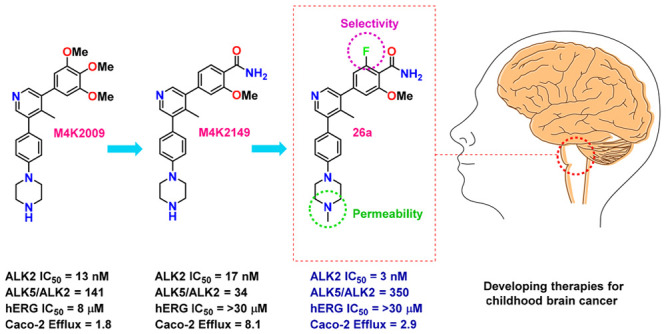
Diffuse intrinsic pontine glioma is an aggressive pediatric cancer for which no effective chemotherapeutic drugs exist. Analysis of the genomic landscape of this disease has led to the identification of the serine/threonine kinase ALK2 as a potential target for therapeutic intervention. In this work, we adopted an open science approach to develop a series of potent type I inhibitors of ALK2 which are orally bio-available and brain-penetrant. Initial efforts resulted in the discovery of M4K2009, an analogue of the previously reported ALK2 inhibitor LDN-214117. Although highly selective for ALK2 over the TGF-βR1 receptor ALK5, M4K2009 is also moderately active against the hERG potassium channel. Varying the substituents of the trimethoxyphenyl moiety gave rise to an equipotent benzamide analogue M4K2149 with reduced off-target affinity for the ion channel. Additional modifications yielded 2-fluoro-6-methoxybenzamide derivatives (26a–c), which possess high inhibitory activity against ALK2, excellent selectivity, and superior pharmacokinetic profiles.
Introduction
The design and development of brain-penetrant kinase inhibitors as a therapy for the treatment of primary central nervous system (CNS) tumors entail numerous challenges. This is in part due to the remarkably different structural properties that CNS drugs and kinase inhibitors have. Approved CNS drugs, for instance, have fewer hydrogen bond donors (HBDs), lower molecular weights, and half the topological polar surface area (tPSA) of kinase inhibitors on average.1 Elevated expression levels of efflux transporters at the blood–brain barrier (BBB) constitute an additional obstacle that drugs must overcome in order to reach therapeutically relevant concentrations at sites of lesion.1 CNS drug exposure is further limited by the endothelial tight junctions of the BBB, which impede paracellular transport.2 Despite these difficulties, the recent approval of Lorlatinib by the FDA for the treatment of metastatic anaplastic lymphoma kinase-positive nonsmall cell lung cancer demonstrates that the development of BBB penetrant kinase inhibitors is possible. There are multiple kinases in addition to the anaplastic lymphoma kinase that play pivotal roles in oncogenesis. Of interest to us are proteins involved in the bone morphogenetic protein (BMP) signaling pathway.
BMPs are a group of cytokines that modulate a plethora of physiological processes, including musculoskeletal development and neural differentiation.3 The signal elicited by BMP binding to type II BMP receptors is transduced by type I BMP receptors, which promote the translocation of downstream effector proteins (SMADs) to the nucleus where they can regulate the transcription of target genes via chromatin remodeling.4,5 Aberrant BMP signaling is implicated in a number of diseases,5 such as fibrodysplasia ossificans progressiva (FOP). Germline mutations (c.617G>A; p.R206H) in the juxtamembrane glycine–serine (GS)-rich domain of activin receptor-like kinase-2 (ALK2) confer gain-of-function activity to the type I BMP receptor and contribute to the abnormal skeletal phenotype observed in individuals affected by FOP.3,6 Somatic missense mutations in the ACVR1 gene encoding ALK2 have also been reported in approximately 24% of children with the rare pediatric disease diffuse intrinsic pontine glioma (DIPG),6 with a higher prevalence of mutation occurring in the serine/threonine kinase domain of the receptor.7
DIPG is a grade IV tumor originating in the glial tissue of the pons.3 Children affected by the disease have a 5-year relative survival rate of less than 1%.8 Treatment options are limited to focal radiation therapy because of the sensitive area in which the tumor resides and the failure of currently available chemotherapeutic drugs to prolong survival.8,9 The mechanism by which ALK2 contributes to DIPG pathogenesis has not yet been elucidated.3,7,10 However, a recent study by Carvalho and coworkers demonstrated that shRNA knockdown of ACVR1 elicits apoptosis in HSJD-DIPG-007 cells, harboring ACVR1 R206H mutations in conjunction with histone H3.3 K27M mutations.11 Their work suggests that DIPG cells are dependent on enhanced BMP signaling. This was further recapitulated in their orthotopic patient-derived xenograft model in which administration of two ALK2 inhibitors extended survival compared to controls.11 Although targeting the serine/threonine kinase may constitute a viable treatment, monotherapies are seldom efficacious for DIPG.12 Targeting proteins with the potential to restore normal epigenetic signatures, such as histone deacetylase (HDAC), has gained momentum in recent years.12 It is likely that the most beneficial treatment option for patients will consist of combinatorial therapies.
Several inhibitors of ALK2 have emerged in the past decade,13 including the pyrazolo[1,5-α]pyrimidine compound LDN-193189,14,15 as well as a relatively new class of 3,5-diarylpyridine analogues: K02288, LDN-213844, and LDN-214117.16−18 Triazolamine CP466722 represents another novel chemotype with moderate binding affinity for ALK2 and unparalleled selectivity over other proteins in the serine/threonine kinase receptor (STKR) family.19 Structure–activity relationship (SAR) studies surrounding this new scaffold should also be explored. As LDN-214117 has been reported to have low cytotoxic activity and excellent kinome-wide selectivity,17 we sought to explore whether additional modifications to the hinge-binding pyridyl core could improve ALK2 potency and selectivity over the closely related TGF-βRI receptor ALK5. Cardiotoxicity and gastrointestinal inflammation are adverse effects of ALK5 inhibition.20 Therefore, a major focus of our SAR studies was to synthesize analogues with reduced off-target affinity for this receptor. Shifting the methyl substituent from the C-2 position of the pyridyl core of LDN-214117 to the C-4 position (M4K2009) maintained potency and selectivity as determined by the in vitro and cell-based assays employed throughout our study (Figure 1).21
Figure 1.
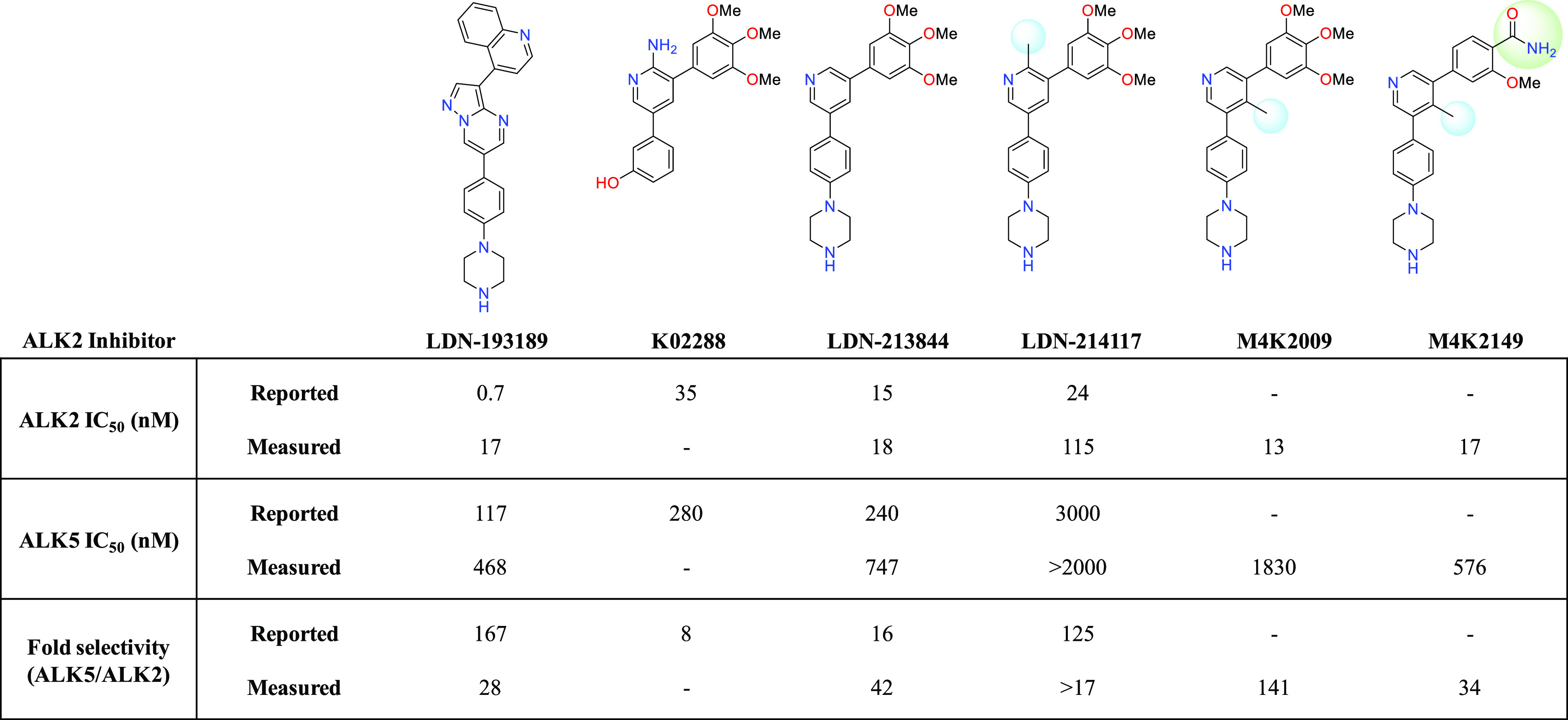
Inhibitory and off-target activity of previously reported ALK2 inhibitors and novel analogues. Reported values were obtained from the corresponding references.15,17,18 Measured values were determined utilizing a radioactive biochemical kinase assay.
Although M4K2009 has good structural and physicochemical properties, it poses the risk of eliciting torsades de pointes arrythmia in vivo because of the moderate affinity it has for the protein product encoded by the human ether-a-go-go related gene (hERG) (IC50 = 8 μM).21 Additional modifications made to the trimethoxyphenyl moiety led to the identification of the benzamide analogue M4K2149, which has a hERG IC50 of >50 μM and comparable inhibitory activity against ALK2 (Figure 1). In our pursuit of a potent, selective, orally bioavailable, and brain-penetrant type I inhibitor of ALK2, M4K2149 was an excellent starting point from which to expand our SAR studies.
Our initial work with M4K2009 revealed that the inhibitor was equipotent against both wild-type (WT) and mutant ALK2 (G328V, R206H, and R258G) in the biochemical kinase assay.21 These results are in alignment with the data generated by Mohedas and coworkers.17 Utilizing a thermal shift kinase assay, they were able to demonstrate that FOP-causing mutations in both the GS and serine/threonine kinase domain of ALK2 had negligible effects on the kinase’s affinity for type I inhibitors.17 As FOP and DIPG patients harbor very similar mutations in the ACVR1 gene,3 the inhibitory activity of the compounds in our series was determined primarily against WT ALK2.
The potency and selectivity of our analogues was assessed using a radioactive in vitro kinase assay, employing LDN-193189 as a control. To test the activity of the compounds in cells, a HEK293 cell-based NanoBRET assay from Promega was used. In this assay, the competitive displacement of a fluorescent tracer (PBI-6908) from the binding pocket of ALK2 by test compounds elicits reductions in BRET ratios, which are used to generate IC50 values. Cell-based potency against ALK5 was subsequently determined using a dual luciferase assay (DLA) in HEK293 cells.
The biological evaluation of these compounds was made possible by the pro bono contributions of Reaction Biology Corporation. This work, which was initiated by the open science pharmaceutical company M4K Pharma Inc., was performed in collaboration with the not-for-profit organizations, the Ontario Institute for Cancer Research (OICR) and the Structural Genomics Consortium (SGC). Adopting an open science approach enabled us to freely share and discuss results with experts in the field, forging collaborations that advanced the science without the delays associated with confidentiality agreements and intellectual property ownership.22
Results and Discussion
Synthesis of Analogues
The synthetic route employed to prepare M4K2149 and related analogues ultimately depended on the commercial availability of starting materials, ease of synthesis, and reaction efficiency. The compounds were initially accessed as depicted in Scheme 1. Suzuki–Miyaura coupling of 3,5-dibromo-4-methylpyridine (2) with 1 generated intermediate 3, which subsequently underwent Miyaura borylation to yield the boronate ester 4a. Aromatic methyl esters 5a–b were coupled with 4a to afford intermediates 6a–b, which were transformed to the corresponding primary amides 7a–b by refluxing in methanolic ammonia, which was then followed by the removal of the carbamate protecting groups with trifluoroacetic acid (TFA). The preparation of analogues 8a–c followed a similar synthetic route in which 4a was coupled with the aromatic amides 5c–e and then deprotected with TFA. Approximately 50% of intermediate 3 was converted to the undesired dehalogenated side product 4b in step b, which contributed to significantly low yields for the final products.
Scheme 1. Synthesis of Compounds 7a–b and 8a–c.
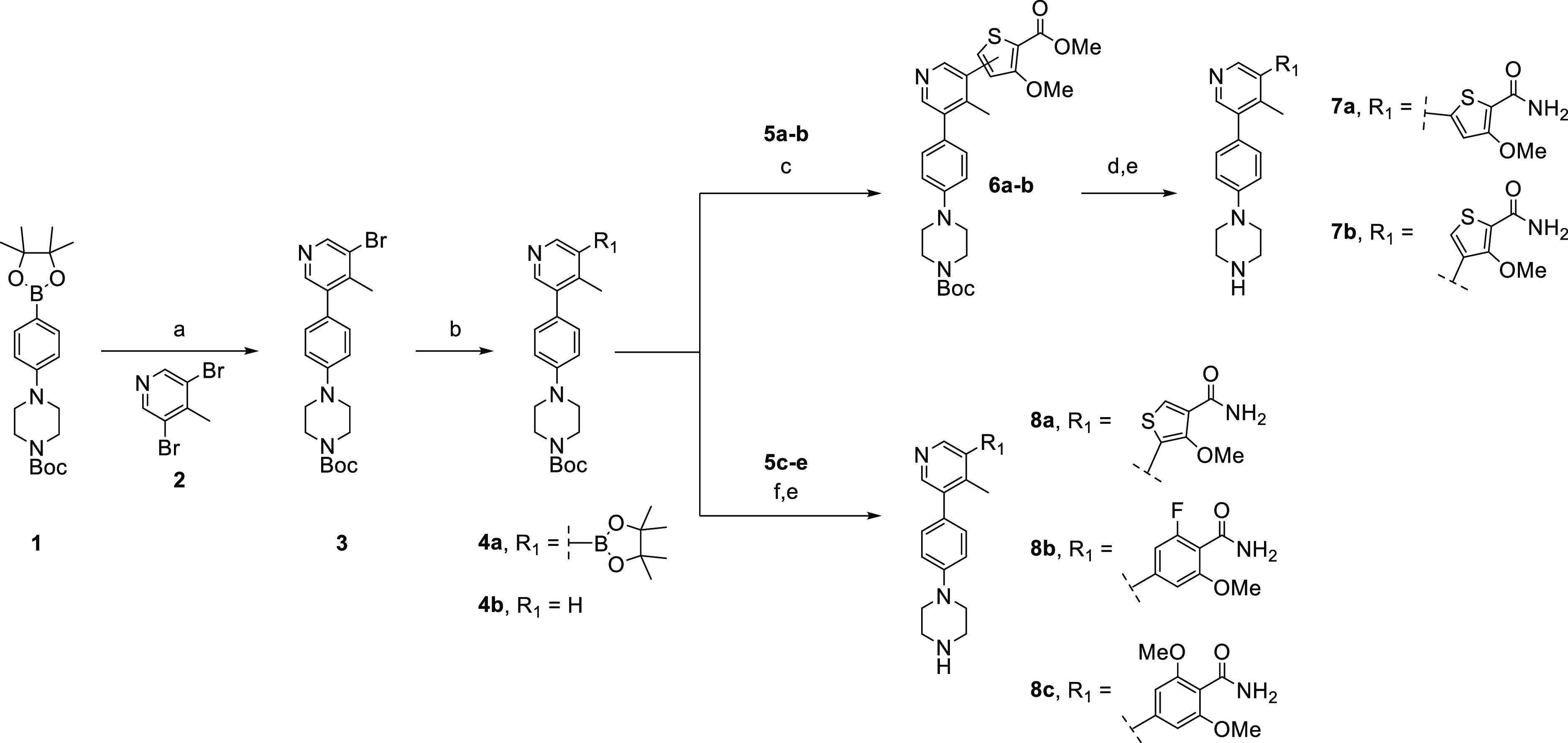
Reagents and conditions: (a) Pd(dppf)Cl2·DCM, Na2CO3, dioxane/H2O, 85 °C, overnight; (b) B2pin2, Pd(dppf)Cl2·DCM, KOAc, dioxane, 110 °C, 4 h; (c) aryl halide (5a–b), Pd(dppf)Cl2·DCM, Na2CO3, dioxane/H2O, 100 °C, 2 h; (d) 7 N NH3 in MeOH, 90 °C, 3 d; (e) TFA, DCM, rt, overnight; (f) aryl halide (5c–e), XPhos Pd G2, K3PO4, dioxane/H2O, 100 °C, 3 h.
To overcome yield constraints, a second synthetic scheme was devised in which a wide variety of boronate esters were coupled with the pyridyl derivatives 3, 10, or 15 (Scheme 2). The synthesis of the carboxylic acid intermediates 13a–b was accomplished via a two-step, one-pot Suzuki–Miyaura coupling sequence. HATU-mediated coupling with ammonium chloride followed by deprotection furnished the final amide regioisomers 14a–b.
Scheme 2. Synthesis of Compounds 14a–b, M4K2149, 18a–b, and 20a–e.
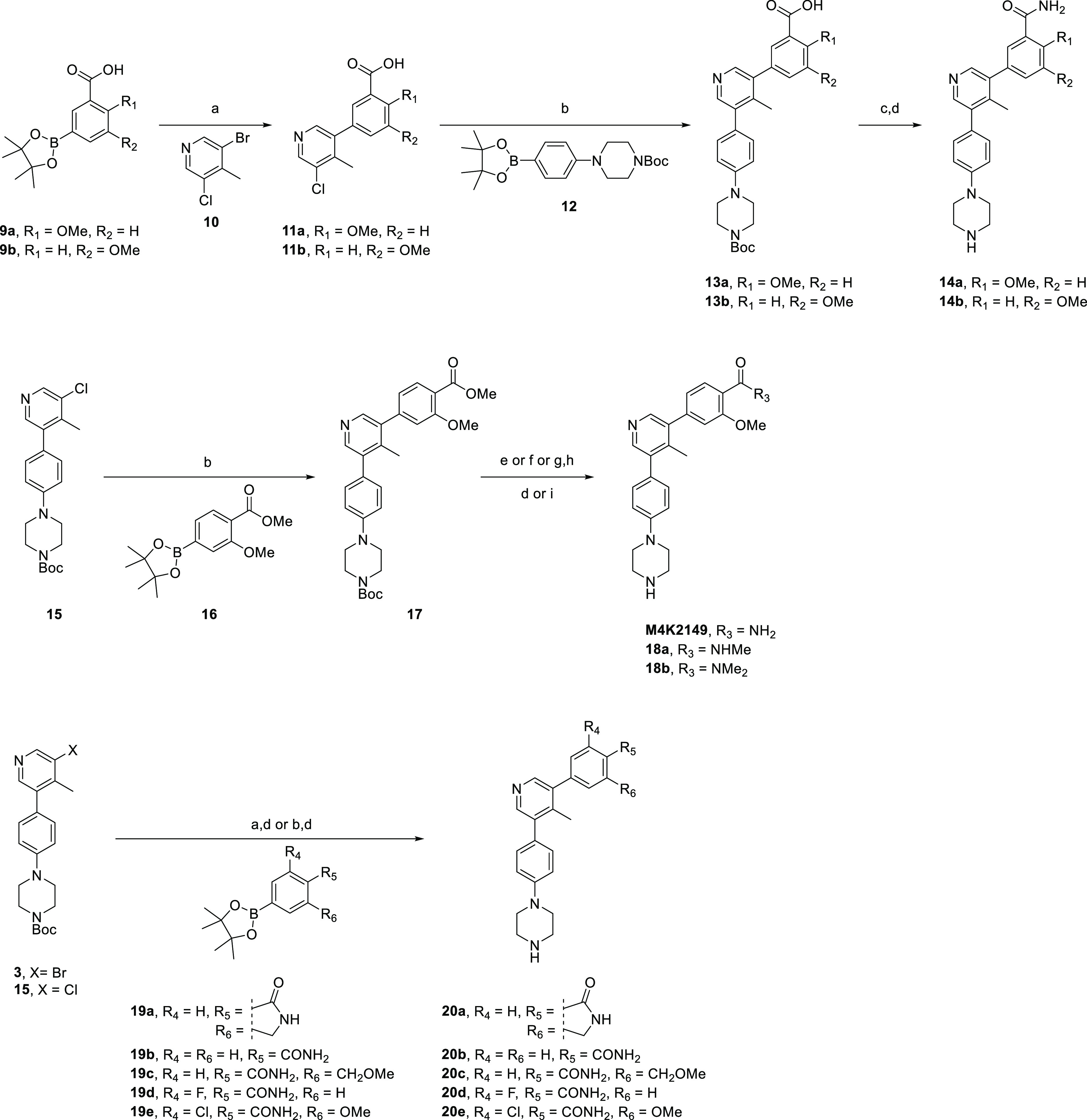
Reagents and conditions: (a) Pd(dppf)Cl2·DCM, Na2CO3, dioxane/H2O, 100 °C, 3 h; (b) XPhos Pd G2, K3PO4, dioxane/H2O, 100 °C, 3 h; (c) NH4Cl, HATU, DIPEA, DCM, rt, 3 h; (d) TFA, DCM, rt, 1 h; (e) 7 N NH3 in MeOH, 75 °C, 3 d; (f) methylamine, MeOH, 85 °C, 5 h; (g) KOH, THF/H2O, rt, 2 h; (h) dimethylamine, HOBt, EDC, DIPEA, DCM/DMF, 50 °C, overnight; (i) 4 M HCl in dioxane, MeOH, rt, 30 min.
Suzuki–Miyaura coupling of 15 with the boronate ester 16 afforded the methyl ester intermediate 17, which was converted to the corresponding primary, secondary, or tertiary amide via aminolysis or base-catalyzed hydrolysis followed by EDC-mediated coupling with the desired amine. Deprotection using TFA or HCl afforded the compounds M4K2149 and 18a–b. A similar synthetic route was used to access analogues 20a–e, although additional transformations beyond the standard Suzuki–Miyaura coupling and deprotection were not required, as the boronate esters 19a–e already had the desired amide substituents installed.
The synthesis of analogues 26a–f (Scheme 3) was initiated by the nucleophilic aromatic substitution of 4-bromo-2,6-difluorobenzonitrile (21) with sodium methoxide to yield both 4-bromo-2-fluoro-6-methoxybenzonitrile (22a) and 2,6-dimethoxybenzonitrile (22b). Both intermediates were hydrolyzed to the corresponding amides 5d–e using hydrogen peroxide and an aqueous solution of sodium hydroxide. Miyaura borylation of 5d–e followed by Suzuki–Miyaura coupling with 3-bromo-5-chloro-4-methylpyridine (10) afforded 24a–b, which were subjected to a second coupling reaction with a variety of (4-(piperazin-1-yl)phenyl)boronate ester derivatives (25a–c) to furnish the final compounds 26a–f in excellent yields.
Scheme 3. Synthesis of Compounds 26a–f.
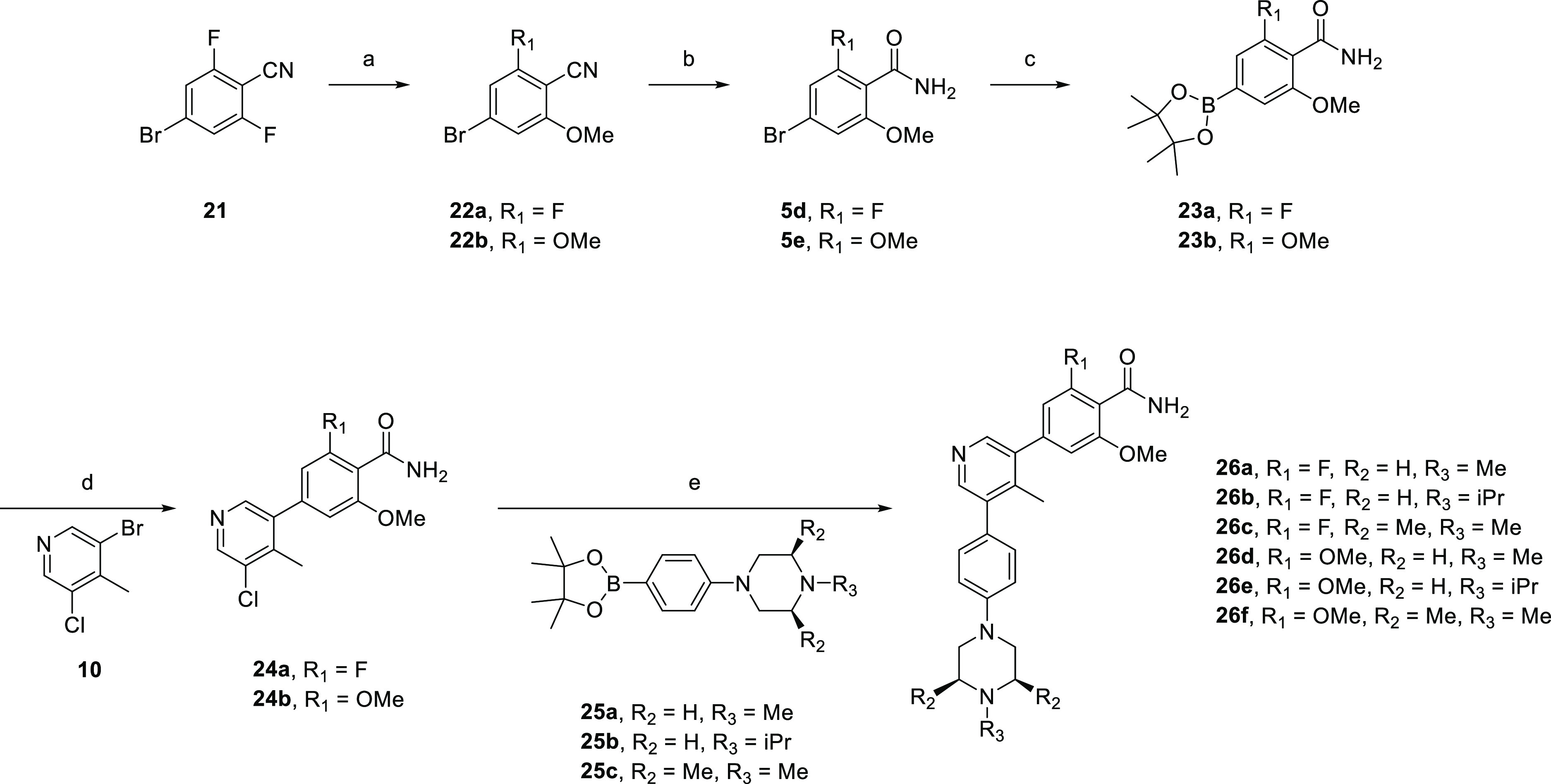
Reagents and conditions: (a) NaH, MeOH, dioxane, rt, overnight; (b) H2O2, NaOH, EtOH/H2O, overnight; (c) B2pin2, Pd(dppf)Cl2·DCM, KOAc, dioxane, 110 °C, 4 h; (d) Pd(dppf)Cl2·DCM, Na2CO3, dioxane/H2O, 100 °C, 3 h; (e) XPhos Pd G2, K3PO4, dioxane/H2O, 100 °C, 3 h.
Structure Activity Relationship Studies
It has been disclosed by Mohedas and coworkers that the pyridyl nitrogen of LDN-213844 participates in a key hydrogen bond interaction with the backbone amide of H286 in the hinge region of ALK2 (Figure 2B).17 Our own crystallographic efforts led to the generation of a co-crystal structure of M4K2149 with the kinase in high resolution, which revealed that the same interaction had been preserved (PDB code 6T6D). Furthermore, the trimethoxyphenyl motif of LDN-213844 was reported to occupy a hydrophobic pocket of ALK2, where the meta-methoxy group participates in a water-mediated hydrogen bond with K235 (PDB code 4BGG).17 Substitution of the para-methoxy group of LDN-213844 with a primary amide results in the establishment of a direct hydrogen bond between the carbonyl O of the amide and the NH3+ group of K235 (Figure 2A). Additionally, the phenyl ring of M4K2149 stacks between G289 and V214,23 while the protonated piperazine NH2+ is in close proximity to D293. This is suggestive of an electrostatic interaction. An intramolecular hydrogen bond between the amide NH2 and O of the ortho-methoxy substituent can also be observed in the co-crystal structure.
Figure 2.

(A) Cocrystal structure of M4K2149 (light yellow) with ALK2 (PDB code 6T6D). Hydrogen bonds are established with H286 and K235. The benzamide moiety of M4K2149 occupies a hydrophobic pocket (green) of ALK2 and is flanked by several hydrogen bond-donating (blue; K235) and hydrogen bond-accepting residues (red; D354 and E248). The protonated piperazine motif is in close proximity to D293, indicative of an electrostatic interaction. (B) Cocrystal structure of LDN-213844 (light yellow) with ALK2 (PDB code 4BGG). M4K2149 and LDN-213844 have similar modes of binding.
Our initial SAR studies focused on varying the substitution pattern of the amide in order to determine if the group could interact with other residues in the pocket, such as D354 of the DLG motif or E248.23 Inverting the amide and methoxy substituents (14a) resulted in a complete loss of activity against ALK2 in the biochemical kinase assay. This correlated well with the results obtained by NanoBRET and DLA (Table 1). Moving the methoxy group from an ortho- to a meta-position with respect to the amide (14b) improved the biochemical potency compared to 14a; however, this did not translate into a significant improvement in cell-based potency. Replacement of the phenyl ring with five-membered heterocycles gave rise to the thiophene analogues 7a–b and 8a, which were profiled to further investigate the effect that the amide geometry had on potency and selectivity. A greater than 40-fold decrease in inhibitory activity against ALK2 was measured for 7a and 7b, while 8a was found to be completely inactive against the kinase in both assays. It became evident that positioning the primary amide on a six-membered aromatic ring para to the hinge-binding pyridyl core was critical for maintaining key binding interactions with ALK2.
Table 1. Inhibitory and Off-Target Activities of 14a–b, 7a–b, and 8a.

Average of duplicate measurements.
Average of triplicate measurements.
The consideration of several physicochemical parameters, such as lipophilicity (cLogP), tPSA, and number of HBD, is pertinent in the design of small molecule inhibitors that must penetrate the BBB to exert their pharmacological effects. The ideal values that brain-penetrant drugs should have vary between reviews.24 In the case of lipophilicity, the consensus is that immoderate increases in cLogP should be avoided. Although increasing the lipophilicity of a drug typically enhances potency and permeability, concomitant increases in nonspecific tissue binding also occur, which would ultimately decrease the concentration of the free drug at its intended site of action within the brain.24 The number of HBD that a molecule possesses, in addition to its tPSA, can also influence its ability to permeate the BBB. Increasing the value of either physicochemical parameter also risks recognition by efflux transporters, such as P-glycoprotein (P-gp).1,2
With these guidelines in mind, we sought to determine if mono- and di-N-methylation of the primary amide, as well as its cyclization to form the corresponding isoindolinone analogue, could be tolerated. The effects of these modifications were two-fold. In addition to decreasing the number of HBD, the tPSA was reduced to below 70 Å2 for 18a–b and 20a,25 which is in an optimal range for CNS-penetrant drugs.24 These structural changes had only moderate effects on the lipophilicity of the three analogues. Unfortunately, these compounds were discovered to be completely inactive against ALK2 (Table 2), indicating that the binding affinity of the benzamides may be sensitive to steric effects. Consequently, we decided to incorporate only the primary amide motif in the rest of our analogues.
Table 2. Inhibitory and Off-Target Activities of 8b–c, 18a–b, 20a–e.

Average of duplicate measurements.
Average of triplicate measurements.
To determine if the methoxy group of M4K2149 was critical for maintaining potency, compound 20b was profiled. Removal of the methoxy substituent reduced inhibitory activity against ALK2 by 28-fold. This result led us to suspect that the methoxy group oriented the amide into a conformation that was ideal for ALK2 binding. The incorporation of intramolecular hydrogen bonds and electrostatic interactions to mask HBD is a technique commonly employed to enhance brain penetration.1,24,26−28 In an attempt to exploit these interactions and probe for additional ones in the vicinity of the benzamide ring, we profiled compounds 20c and 20d, which featured a one-carbon homologation of the methoxy group and a bioisosteric replacement of the methoxy for a fluorine atom, respectively. Both modifications, however, failed to improve biochemical potency against ALK2.
Incorporation of both a fluorine and methoxy substituent ortho to the amide gave rise to compound 8b, which not only had a biochemical ALK2 potency comparable to that of M4K2149 (ALK2 IC50 = 24 nM) but also an improved selectivity profile over ALK5. These results correlated well with those obtained in the NanoBRET and DLA assays (Table 2). Exactly how the fluorine substituent contributes to this enhancement in selectivity is yet to be elucidated, although two possible explanations exist. We surmised that the electron-withdrawing nature of the fluorine atom decreases the ability of the carbonyl O to act as a hydrogen bond acceptor,28 thereby reducing the strength of intermolecular interactions that may be more critical for ligand binding to ALK5 than ALK2. The second possible explanation focuses on how the halogen substituent affects the conformation of the amide with respect to the phenyl ring. For anisole and benzamide motifs, which typically adopt planar topologies, ortho substituents can force the methoxy or amide groups out of the plane of the benzene ring in order to reduce allylic strain.28,29 We postulated that a similar phenomenon could be occurring in the case of 8b and that this change in conformation is better tolerated by ALK2 than ALK5.
We decided to introduce a larger substituent ortho to the amide in order to confirm whether the latter hypothesis was correct. Replacing the fluorine atom of 8b with a chlorine atom (20e) increased the biochemical selectivity over ALK5 to greater than 200-fold, suggesting that our proposed explanation is a reasonable one. However, NanoBRET ALK2 IC50 values were similar for both 8b and 20e, indicating that the modification has little impact on ALK2 potency. To determine if electron-donating groups could be tolerated at this position as well, 8c was prepared. This analogue had the greatest structural similarity to our lead compound M4K2009. Although 8c was the most potent analogue in our series, it suffered from poor selectivity (biochemical selectivity of 9-fold over ALK5). Additionally, there was a substantial difference in its biochemical and cell-based potencies (biochemical ALK2 IC50 = 5 nM vs NanoBRET ALK2 IC50 = 178 nM).
Permeability, Selectivity, and Pharmacokinetic Studies
Having identified several potent analogues, we decided to focus our efforts on improving the pharmacokinetic (PK) profiles of two: 8b and 8c. In order to assess the permeability of these compounds, they were tested in a Caco-2 assay, which revealed that both analogues were poorly permeable and being recognized by efflux transporters (efflux ratios for both 8b and 8c were >30) (see Tables 4 and 2 in the Supporting Information). In an attempt to reduce efflux, the terminal piperazine nitrogens of 8b and 8c were capped with various alkyl groups to generate 1-methyl-, 1-isopropyl-, and 1,2,6-trimethylpiperazine analogues (26a–f).30 In addition to reducing the number of HBD, methylation of the terminal piperazine nitrogen offered the additional advantage of attenuating pKa,25,31 which is often associated with a decrease in P-gp-mediated efflux.24 The rationale behind incorporating methyl groups at positions 1, 2, and 6 of the piperazine groups was to increase the molecular rigidity of analogues 26c and 26f. This is a strategy that is commonly employed to enhance brain penetration and oral bioavailability.24 As it has been reported that the piperazine motif of LDN-193189 is a metabolic liability,32 increasing the steric bulk around this group was also done to improve the inhibitors’ ADME profile in vivo.
As anticipated, the permeability (Papp_AB) of the 2-fluoro-6-methoxybenzamide analogues was increased from 0.3 × 10–6 cm/s (8b) to around 5.0 × 10–6 cm/s for 26a–c. This was accompanied by a concomitant reduction in the efflux ratio (from >30 for 8b to less than 3.0 for 26a and 26b) (Table 4). An enhancement in selectivity over ALK5 was also observed for these compounds. This was more pronounced in the biochemical kinase assay (Table 3). Unfortunately, similar results were not obtained for the 2,6-dimethoxybenzamide analogues (26d–f). Piperazine alkylation appeared to have little effect on reducing efflux (see the Supporting Information, Table 2). However, 26d–f demonstrated excellent inhibitory activity against ALK2 in the NanoBRET assay. Although M4K2149 and 8c differ by only one methoxy group, the latter analogue had a significantly higher efflux ratio (8.1 vs >30).21 We suspected that the extra electron-donating group was increasing the hydrogen bond acceptor potential of the amide carbonyl, which was being recognized by one of the efflux transporters expressed by the Caco-2 cells. Consequently, these analogues were excluded from further profiling.
Table 4. In Vitro Permeability and Oral In Vivo PK Studies of 2-Fluoro-6-methoxybenzamide Analogues.

Table 3. Inhibitory and Off-Target Activity of 2-Fluoro-6-methoxybenzamide and 2,6-Dimethoxybenzamide Analogues, 26a–f.

Average of duplicate measurements.
Average of triplicate measurements.
Average of quadruplicate measurements.
Average of quintuplicate measurements.
Prior to assessing the PK profiles of the 2-fluoro-6-methoxybenzamide analogues in vivo, the metabolic stabilities of 8b and 26a–c were evaluated in mouse and human liver microsomal (MLM and HLM) stability assays. All four analogues exhibited moderate to high stability in both in vitro assays, with compounds 8b and 26b demonstrating the highest degree of stability after a 60 min incubation period at 37 °C (>85% remaining) (see the Supporting Information, Table 3). Oral administration of a 10 mg/kg dose of 8b in female CB17 SCID mice (n = 3) gave rise to suboptimal values for Cmax (97 ng/mL), t1/2 (1.31 h), and AUCinf (296 ng·h/mL). Given the poor intrinsic permeability of 8b, these results were not surprising. A significant improvement in PK properties was observed for analogues 26a and 26b, both of which yielded a greater than 17-fold increase in Cmax, 13-fold increase in AUCinf, and a doubling of t1/2 (Table 4). The two analogues were also assessed for their ability to penetrate the BBB in the same strain of mice. Oral administration of these compounds at a 100 mg/kg dose gave rise to average total brain concentrations of 777 and 1595 ng/g and total brain-to-plasma ratios (B/P) of 0.178 and 0.132 for 26a and 26b, respectively. Although these values are moderate, the use of B/P ratios to assess brain permeability is generally not encouraged. The extent of brain penetration is typically evaluated based on the ratio of the unbound brain concentration to the unbound plasma concentration (Kp,uu).2 Whether these analogues require additional modifications to enhance BBB permeability will ultimately depend on the value of this parameter.
To ensure that a favorable hERG profile had been maintained for the 2-fluoro-6-methoxybenzamide analogues, their potencies against the hERG potassium channel were assessed using a HEK293 cell-based patch-clamp assay. 26a and 26b had optimal hERG IC50 values of >30 μM, while 26c was slightly more potent against the ion channel (IC50 = 19 μM) (see the Supporting Information, Table 4). To further investigate the off-target activity of these analogues, we profiled them in a 375-member kinase panel. At a concentration of 1 μM for each of the three compounds, fewer than 5% of the kinases showed a greater than 50% reduction in enzymatic activity. Excluding ALK1, 2, 3, and 6, the kinases ARAF, MAP4K4, MINK, and TNK1 were the most sensitive to inhibition by 26a–c (see the Supporting Information, Table 5). We were also encouraged by the results obtained in an in vitro CYP inhibition assay, which showed that these analogues had negligible inhibitory activity (IC50 > 50 μM) against 7 CYP isoforms (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4) (see the Supporting Information, Table 4). Altogether, these results demonstrate that we were able to meet our objective of designing selective and orally bioavailable inhibitors of ALK2. Furthermore, profiling 7 analogues against 3 ALK2 mutants (R206H, G328V, and R258G) in a radioactive in vitro kinase assay revealed that, similar to M4K2009, the analogues had comparable potencies against both WT and mutant ALK2 (Table 5). These findings confirm that the benzamide inhibitors developed in this series have the potential to regulate aberrant BMP signaling in patients harboring these mutations.
Table 5. Inhibitory Activity of Benzamide Analogues against WT and DIPG-Linked Mutant Forms of ALK2.

Conclusions
Advances in the development of effective chemotherapeutic agents for the treatment of DIPG have been limited. This is in part due to the convoluted genomic signatures of DIPG, which has made our understanding of its pathogenesis difficult. Recent identification of ALK2 as a target for therapeutic intervention has prompted the emergence of several classes of type I kinase inhibitors. In this work, we expanded the SAR of the 3,5-diarylpyridine inhibitor LDN-214117, which led to the discovery of a potent benzamide analogue M4K2149 with an attenuated affinity for the hERG potassium channel. We determined that we could tailor the selectivity of our analogues over ALK5 by incorporating halogen substituents at a position ortho to the amide group of M4K2149. We were also able to address issues of permeability by capping the NH of the solvent-exposed piperazine group. The resulting 2-fluoro-6-methoxybenzamide derivatives 26a–c demonstrated excellent kinome-wide selectivity and had improved PK properties compared to their parent compound 8b. Furthermore, the co-crystal structure of M4K2149 with ALK2 helped us rationalize potency differences between analogues in the series and highlighted structural motifs that were crucial for maintaining key interactions with the protein. Despite these optimizations, total brain-to-plasma ratios are inadequate for accurately assessing the pharmacological activity of 26a–b in the brain. Measuring the unbound brain concentrations (Cb,u) of these analogues in vivo is therefore warranted. These data would ultimately determine whether additional modifications should be made to reduce efflux/enhance permeability. Nonetheless, these benzamides represent a new chemotype possessing high inhibitory activity against both WT and mutant ALK2. Implementation of an open science model accelerated the development of these analogues by promoting communication between the chemists involved in their design and establishing a pipeline for rapidly generating biological data. Future work will continue to use open science to develop novel classes of ALK2 inhibitors. The analogues presented in this study have the potential to deepen our understanding of the biology of DIPG and will hopefully pave the way for future chemotherapies.
Experimental Section
Chemistry
All reagents were purchased from commercial vendors and used without further purification. Volatiles were removed under reduced pressure by rotary evaporation or using the V-10 solvent evaporator system by Biotage. Very high boiling point (6000 rpm, 0 mbar, 56 °C), mixed volatile (7000 rpm, 30 mbar, 36 °C), and volatile (6000 rpm, 30 mbar, 36 °C) methods were used to evaporate solvents. The yields given refer to chromatographically purified and spectroscopically pure compounds. Compounds were purified using a Biotage Isolera One system by normal phase chromatography using Biotage SNAP KP-Sil or Sfär Silica D columns (part no.: FSKO-1107/FSRD-0445) or by reverse-phase chromatography using Biotage SNAP KP-C18-HS or Sfär C18 D columns (part no.: FSLO-1118/FSUD-040). If additional purification was required, compounds were purified by solid phase extraction (SPE) using Biotage Isolute Flash SCX-2 cation exchange cartridges (part nos.: 532-0050-C and 456-0200-D). Products were washed with two cartridge volumes of MeOH and eluted with a solution of MeOH and NH4OH (9:1 v/v). Preparative chromatography was carried out using a Waters 2767 injector with the collector attached to PDA UV/Vis and SQD mass detectors. An XSelect CSH Prep C18 5 μm OBD 19 mm × 100 mm (part no.: 186005421) or Xselect CSH Prep C18 5 μm 10 mm × 100 mm (part no.: 186005415) column was used for purification. Final compounds were dried using the Labconco Benchtop FreeZone Freeze-Dry System (4.5 L Model). 1H and proton-decoupled 19F NMRs were recorded on a Bruker AVANCE-III 500 MHz spectrometer at ambient temperature. Residual protons of CDCl3, DMSO-d6, and CD3OD solvents were used as internal references. Spectral data are reported as follows: chemical shift (δ in ppm), multiplicity (br = broad, s = singlet, d = doublet, dd = doublet of doublets, m = multiplet), coupling constants (J in Hz), and proton integration. Compound purity was determined by UV absorbance at 254 nm during tandem liquid chromatography/mass spectrometry (LCMS) using a Waters Acquity separation module. All final compounds had a purity of ≥95% as determined using this method. Low-resolution mass spectrometry was conducted in the positive ion mode using a Waters Acquity SQD mass spectrometer (electrospray ionization source) fitted with a PDA detector. Mobile phase A consisted of 0.1% formic acid in water, while mobile phase B consisted of 0.1% formic acid in acetonitrile. One of three types of columns were used: column 1: Acquity UPLC CSH C18 (2.1 × 50 mm, 130 Å, 1.7 μm. part no. 186005296), column 2: Acquity UPLC BEH C8 (2.1 × 50 mm, 130 Å, 1.7 μm. part no. 186002877), or column 3: Acquity UPLC HSS T3 (2.1 × 50 mm, 100 Å, 1.8 μm. part no. 186003538). For columns 1 and 2, the gradient went from 90 to 5% mobile phase A over 1.8 min, maintained at 5% for 0.5 min, then increased to 90% over 0.2 min for a total run time of 3 min. For column 3, the gradient went from 98 to 5% mobile phase A over 1.8 min, maintained at 5% for 0.5 min, then increased to 98% over 0.2 min for a total run time of 3 min, as well. The flow rate was 0.4 mL/min throughout both runs. All columns were used with the temperature maintained at 25 °C. High-resolution mass spectrometry was conducted using a Waters Synapt G2-S quadrupole-time-of-flight (QTOF) hybrid mass spectrometer system coupled with an Acquity ultra-performance liquid chromatography (UPLC) system. Chromatographic separations were carried out on an Acquity UPLC CSH C18 (2.1 × 50 mm, 130 Å, 1.7 μm. part no. 186005296), Acquity UPLC BEH C8 (2.1 × 50 mm, 130 Å, 1.7 μm. part no. 186002877), or Acquity UPLC HSS T3 (2.1 × 50 mm, 100 Å, 1.8 μm. part no. 186003538). The mobile phases were 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). Leucine Enkephalin was used as the lock mass. MassLynx 4.1 was used for data analysis.
tert-Butyl 4-(4-(5-Bromo-4-methylpyridin-3-yl)phenyl)piperazine-1-carboxylate (3)
A solution of tert-butyl 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)piperazine-1-carboxylate (1) (1.549 g, 3.99 mmol), 3,5-dibromo-4-methylpyridine (2) (0.953 g, 3.80 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II)·DCM complex (0.310 g, 0.38 mmol), and sodium carbonate monohydrate (1.414 g, 11.40 mmol) in 1,4-dioxane (16.3 mL) and water (2.7 mL) was heated to 85 °C and stirred overnight. The reaction mixture was concentrated under reduced pressure prior to dilution with water (30 mL) and extraction with EtOAc (3 × 30 mL). The combined extracts were dried over MgSO4 and concentrated to yield brown oil. The crude material was purified by silica gel chromatography (0–50% EtOAc in hexanes) to afford a white solid (0.770 g, 47% yield). 1H NMR (500 MHz, CDCl3): δ 8.61 (s, 1H), 8.31 (s, 1H), 7.20 (d, J = 8.6 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 3.65–3.56 (m, 4H), 3.26–3.18 (m, 4H), 2.36 (s, 3H), 1.49 (s, 9H). MS (ESI) m/z: 432.31 [M + H]+, 434.38 [M + H]+ + 2.
(5-(4-(4-(tert-Butoxycarbonyl)piperazin-1-yl)phenyl)-4-methylpyridin-3-yl)boronic Acid (4a)
A solution of 3 (171 mg, 0.396 mmol), bis(pinacolato)diboron (201 mg, 0.792 mmol), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II)·DCM complex (32 mg, 0.040 mmol), and potassium acetate (78 mg, 0.792 mmol) in 1,4-dioxane (4 mL) was microwaved at 110 °C for 4 h. The mixture was transferred to a 15 mL Falcon tube and centrifuged for 1 min at 4000 rpm. The dark brown supernatant was used without further purification in subsequent reactions (190 mg, 57% yield). MS (ESI) m/z: 397.90 [M + H]+.
5-Chloro-4-methoxythiophene-3-carboxamide (5c)
The title compound was prepared using a modified literature procedure.33 To a solution of 5-chloro-4-methoxythiophene-3-carboxylic acid (100.0 mg, 0.519 mmol) in DCM (1.5 mL) was added ammonium chloride (33.3 mg, 0.623 mmol), HATU (237.0 mg, 0.623 mmol), and DIPEA (271 μL, 1.558 mmol). The reaction mixture was stirred at room temperature for 3 h. Volatiles were removed under reduced pressure, and the crude product was purified by silica gel chromatography (0–100% EtOAc in hexanes) to afford an off-white solid (71.1 mg, 72% yield). 1H NMR (500 MHz, CDCl3): δ 7.91 (s, 1H), 7.21 (br s, 1H), 5.72 (br s, 1H), 4.06 (s, 3H). MS (ESI) m/z: 192.27 [M + H]+, 194.28 [M + H]+ + 2.
4-Bromo-2-fluoro-6-methoxybenzamide (5d)
The title compound was prepared using modified literature procedures.34,35 A solution of 4-bromo-2-fluoro-6-methoxybenzonitrile (22a) (5.00 g, 21.74 mmol) in EtOH (100.0 mL) was cooled in an ice bath prior to the addition of an aqueous solution of sodium hydroxide (0.43 M, 65.0 mL). This was followed by the addition of hydrogen peroxide (30 wt % solution in water) (26.6 mL). The solution was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure prior to dilution with water (250 mL) and extraction with EtOAc (3 × 250 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a white crystalline solid (4.63 g, 80% yield). 1H NMR (500 MHz, DMSO): δ 7.84 (br s, 1H), 7.58 (br s, 1H), 7.17 (d, J = 8.5 Hz, 1H), 7.13 (s, 1H), 3.82 (s, 3H). 19F NMR (471 MHz, DMSO): δ −115.38. MS (ESI) m/z: 248.20 [M + H]+, 250.27 [M + H]+ + 2.
4-Bromo-2,6-dimethoxybenzamide (5e)
The title compound was synthesized according to the procedure described for 5c from 4-bromo-2,6-dimethoxybenzoic acid (157 mg, 0.600 mmol). The final product was a white solid (100 mg, 64% yield). 1H NMR (500 MHz, DMSO): δ 7.51 (br s, 1H), 7.23 (br s, 1H), 6.87 (s, 2H), 3.75 (s, 6H). MS (ESI) m/z: 260.35 [M + H]+, 262.29 [M + H]+ + 2.
The title compound was alternatively synthesized according to the procedure described for 5d from 4-bromo-2,6-dimethoxybenzonitrile (22b) (968 mg, 4.00 mmol), hydrogen peroxide (30 wt % solution in water) (9.8 mL), and an aqueous solution of sodium hydroxide (2 M, 25.0 mL). The reaction mixture was heated to 110 °C for 8 h. The solvents were evaporated and the crude material was suspended in water, filtered, and dried under high vacuum to afford a white crystalline solid (903 mg, 87% yield).
tert-Butyl 4-(4-(5-(4-Methoxy-5-(methoxycarbonyl)thiophen-2-yl)-4-methylpyridin-3-yl)phenyl)piperazine-1-carboxylate (6a)
A solution of 4a (80 mg, 0.167 mmol), methyl 5-bromo-3-methoxythiophene-2-carboxylate (42 mg, 0.167 mmol) (5a), [1,12-bis(diphenylphosphino)ferrocene]dichloropalladium(II)·DCM complex (14 mg, 0.017 mmol), and sodium carbonate monohydrate (62 mg, 0.501 mmol) in 1,4-dioxane (2.9 mL) and water (477 μL) was heated to 100 °C for 2 h. The reaction mixture was adsorbed onto Celite, and the volatiles were removed under reduced pressure. The crude product was purified by silica gel chromatography (0–100% EtOAc in hexanes) to afford an off-white powder (45 mg, 50% yield). MS (ESI) m/z: 524.70 [M + H]+.
tert-Butyl 4-(4-(5-(4-Methoxy-5-(methoxycarbonyl)thiophen-3-yl)-4-methylpyridin-3-yl)phenyl)piperazine-1-carboxylate (6b)
The title compound was synthesized according to the procedure described for 6a from 4a (80 mg, 0.167 mmol) and methyl 4-bromo-3-methoxythiophene-2-carboxylate (5b) (42 mg, 0.167 mmol). The final product was a light yellow powder (47 mg, 52% yield). MS (ESI) m/z: 524.70 [M + H]+.
3-Methoxy-5-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)thiophene-2-carboxamide (7a)
A 5 mL MW vial was charged with 6a (20.0 mg, 0.038 mmol). The material was dissolved in a solution of ammonia in methanol (7 N) (4 mL). The vial was sealed, and the solution was stirred at 90 °C for 3 days. Volatiles were removed under reduced pressure, and the crude material was purified by silica gel chromatography (0–100% EtOAc in hexanes). The purified product was dissolved in DCM (1 mL) and treated with trifluoroacetic acid (88 μL, 1.146 mmol). The solution was stirred overnight. The product was purified by SPE. Drying under high vacuum overnight afforded an off-white powder (7.8 mg, 47% yield). 1H NMR (500 MHz, DMSO): δ 8.37 (s, 1H), 8.36 (s, 1H), 7.76 (s, 1H), 7.66 (br s, 1H), 7.31–7.25 (m, 3H), 7.05 (d, J = 8.7 Hz, 2H), 3.51 (s, 3H), 3.22–3.20 (m, 4H), 2.98–2.94 (m, 4H), 2.13 (s, 3H). HRMS (ESI) for C22H24N4O2S [M + H]+m/z: calcd, 409.1693; found, 409.1691.
3-Methoxy-4-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)thiophene-2-carboxamide (7b)
The title compound was synthesized according to the procedure described for 7a from 6b (20.0 mg, 0.038 mmol). The final product was an off-white powder (8.9 mg, 53% yield). 1H NMR (500 MHz, DMSO): δ 8.38 (s, 1H), 8.36 (s, 1H), 7.76 (s, 1H), 7.66 (br s, 1H), 7.30–7.26 (m, 3H), 7.04 (d, J = 8.7 Hz, 2H), 3.52 (s, 3H), 3.19–3.17 (m, 4H), 2.95–2.91 (m, 4H), 2.13 (s, 3H). HRMS (ESI) for C22H24N4O2S [M + H]+m/z: calcd, 409.1693; found, 409.1689.
4-Methoxy-5-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)thiophene-3-carboxamide (8a)
A solution of 4a (60.0 mg, 0.125 mmol), 5c (20.0 mg, 0.104 mmol), XPhos Pd G2 (8.2 mg, 0.010 mmol), and potassium phosphate tribasic (44.3 mg, 0.209 mmol) in 1,4-dioxane (1.8 mL) and water (298 μL) was heated to 100 °C and stirred for 3 h. The reaction mixture was adsorbed onto Celite, and volatiles were removed under reduced pressure. The crude product was purified by silica gel chromatography (0–100% EtOAc in hexanes). Further purification was carried out by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)]. The product was dissolved in DCM (1 mL) and treated with trifluoroacetic acid (479 μL, 6.26 mmol). The solution was stirred for 1 h. The product was purified by SPE. Freeze-drying for 3 days afforded an off-white powder (6.5 mg, 15% yield). 1H NMR (500 MHz, DMSO): δ 8.44 (s, 1H), 8.39 (s, 1H), 8.11 (s, 1H), 7.47 (br s, 1H), 7.44 (br s, 1H), 7.30 (d, J = 8.5 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 3.57 (s, 3H), 3.24–3.21 (m, 4H), 3.01–2.96 (m, 4H), 2.17 (s, 3H). HRMS (ESI) for C22H24N4O2S [M + H]+m/z: calcd, 409.1693; found, 409.1694.
2-Fluoro-6-methoxy-4-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)benzamide (8b)
The title compound was synthesized according to the procedure described for 8a from 4a (77 mg, 0.161 mmol) and 5d (40 mg, 0.161 mmol). The final product was an off-white powder (20 mg, 29% yield). 1H NMR (500 MHz, DMSO): δ 8.38 (s, 1H), 8.34 (s, 1H), 7.89 (br s, 1H), 7.58 (br s, 1H), 7.30 (d, J = 8.6 Hz, 2H), 7.04 (d, J = 8.6 Hz, 2H), 6.98–6.95 (m, 2H), 3.85 (s, 3H), 3.19–3.15 (m, 4H), 2.94–2.89 (m, 4H), 2.19 (s, 3H). 19F NMR (471 MHz, DMSO): δ −116.73. HRMS (ESI) for C24H25FN4O2 [M + H]+m/z: calcd, 421.2034; found, 421.2040.
2,6-Dimethoxy-4-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)benzamide (8c)
The title compound was synthesized according to the procedure described for 8a from 4a (190 mg, 0.396 mmol) and 5e (82 mg, 0.317 mmol). The final product was an off-white powder (45 mg, 32% yield). 1H NMR (500 MHz, DMSO): δ 8.36 (s, 1H), 8.35 (s, 1H), 7.57 (br s, 1H), 7.31 (d, J = 8.6 Hz, 2H), 7.23 (br s, 1H), 7.04 (d, J = 8.7 Hz, 2H), 6.72 (s, 2H), 3.78 (s, 6H), 3.19–3.15 (m, 4H), 2.93–2.89 (m, 4H), 2.20 (s, 3H). HRMS (ESI) for C25H28N4O3 [M + H]+m/z: calcd, 433.2234; found, 433.2228.
5-(5-Chloro-4-methylpyridin-3-yl)-2-methoxybenzoic Acid (11a)
The title compound was synthesized according to the procedure described for 6a from 3-bromo-5-chloro-4-methylpyridine (10) (41 mg, 0.200 mmol) and 5-borono-2-methoxybenzoic acid (9a) (39 mg, 0.200 mmol). DMF (1.6 mL) and water (428 μL) were used as the solvents.
The crude material was used without purification in the subsequent cross-coupling reaction (56 mg, 87% yield). MS (ESI) m/z: 278.30 [M + H]+, 280.30 [M + H]+ + 2.
3-(5-Chloro-4-methylpyridin-3-yl)-5-methoxybenzoic Acid (11b)
The title compound was synthesized according to the procedure described for 6a from 3-bromo-5-chloro-4-methylpyridine (10) (41 mg, 0.200 mmol) and 3-carboxy-5-methoxyphenylboronic acid (9b) (39 mg, 0.200 mmol). DMF (1.6 mL) and water (428 μL) were used as the solvents.
The crude material was used without purification in the subsequent cross-coupling reaction (56 mg, 90% yield). MS (ESI) m/z: 278.23 [M + H]+, 280.30 [M + H]+ + 2.
5-(5-(4-(4-(tert-Butoxycarbonyl)piperazin-1-yl)phenyl)-4-methylpyridin-3-yl)-2-methoxybenzoic Acid (13a)
The title compound was synthesized according to the procedure described for 8a from 11a (56 mg, 0.200 mmol) and 1 (140 mg, 0.360 mmol). The crude product was purified by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)]. The purified intermediate was used immediately in the subsequent reaction.
3-(5-(4-(4-(tert-Butoxycarbonyl)piperazin-1-yl)phenyl)-4-methylpyridin-3-yl)-5-methoxybenzoic Acid (13b)
The title compound was synthesized according to the procedure described for 8a from 11b (56 mg, 0.200 mmol) and 1 (140 mg, 0.360 mmol). The crude product was purified by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)]. The purified intermediate was used immediately in the subsequent reaction.
2-Methoxy-5-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)benzamide (14a)
The title compound was synthesized according to the procedure described for 5c from 13a. Deprotection with trifluoroacetic acid (459 μL, 6.00 mmol), purification by SPE, and freeze-drying for 2 days afforded the final product as a yellow powder (5.96 mg, 7% yield over 4 steps). 1H NMR (500 MHz, DMSO): δ 8.32 (s, 1H), 8.29 (s, 1H), 7.81 (d, J = 2.1 Hz, 1H), 7.71 (br s, 1H), 7.59 (br s, 1H), 7.56 (dd, J = 8.5, 2.2 Hz, 1H), 7.31 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.6 Hz, 1H), 7.04 (d, J = 8.6 Hz, 2H), 3.95 (s, 3H), 3.21–3.18 (m, 4H), 2.97–2.93 (m, 4H), 2.14 (s, 3H). HRMS (ESI) for C24H26N4O2 [M + H]+m/z: calcd, 403.2129; found, 403.2126.
3-Methoxy-5-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)benzamide (14b)
The title compound was synthesized according to the procedure described for 5c from 13b. Deprotection with trifluoroacetic acid (0.459 mL, 6.00 mmol), purification by reverse-phase chromatography and SPE and freeze drying for 2 days afforded the final product as an off-white powder (4.52 mg, 6% yield over 4 steps). 1H NMR (500 MHz, DMSO): δ 8.36 (s, 1H), 8.35 (s, 1H), 8.02 (br s, 1H), 7.52–7.49 (m, 1H), 7.49–7.47 (m, 1H), 7.44 (br s, 1H), 7.31 (d, J = 8.6 Hz, 2H), 7.17–7.15 (m, 1H), 7.05 (d, J = 8.7 Hz, 2H), 3.85 (s, 3H), 3.23–3.20 (m, 4H), 2.99–2.95 (m, 4H), 2.15 (s, 3H). HRMS (ESI) for C24H26N4O2 [M + H]+m/z: calcd, 403.2129; found, 403.2137.
tert-Butyl 4-(4-(5-Chloro-4-methylpyridin-3-yl)phenyl)piperazine-1-carboxylate (15)
The title compound was synthesized according to the procedure described for 6a from 10 (320 mg, 1.550 mmol) and 1 (722 mg, 1.860 mmol). The final product was an off-white crystalline solid (510 mg, 85% yield). 1H NMR (500 MHz, CDCl3): δ 8.48 (s, 1H), 8.30 (s, 1H), 7.21 (d, J = 8.7 Hz, 2H), 6.99 (d, J = 8.7 Hz, 2H), 3.63–3.59 (m, 4H), 3.24–3.19 (m, 4H), 2.33 (s, 3H), 1.49 (s, 9H). MS (ESI) m/z: 388.56 [M + H]+, 390.57 [M + H]+ + 2.
tert-Butyl 4-(4-(5-(3-Methoxy-4-(methoxycarbonyl)phenyl)-4-methylpyridin-3-yl)phenyl)piperazine-1-carboxylate (17)
The title compound was synthesized according to the procedure described for 8a from 15 (120 mg, 0.309 mmol) and 3-methoxy-4-methoxycarbonylphenylboronic acid, pinacol ester (16) (90 mg, 0.309 mmol). The solvents used were butan-1-ol (2 mL) and water (476 μL). The reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic fractions were dried over Na2SO4, filtered, and concentrated under reduced pressure to afford a light beige solid (160 mg, 99% yield), which was used without further purification in the subsequent reaction. MS (ESI) m/z: 518.57 [M + H]+.
tert-Butyl 4-(4-(5-(4-Carbamoyl-3-methoxyphenyl)-4-methylpyridin-3-yl)phenyl)piperazine-1-carboxylate (M4K2149)
To a solution of 17 (0.160 g, 0.309 mmol) in MeOH (3.0 mL) at room temperature was added a solution of ammonia in MeOH (7 N) (4.4 mL). The resulting mixture was heated to 75 °C for 3 days prior to cooling back down to room temperature, removing all solvents under reduced pressure, and triturating the residue from EtOAc with hexanes. The beige precipitate was collected by filtration and washed with hexanes. The product was subsequently dissolved in MeOH (5.0 mL) and treated with HCl (4.0 M in dioxane, 1.0 mL). The solution was stirred for 30 min prior to the removal of solvents under reduced pressure. The product was purified by SPE. The final compound was dried under vacuum overnight to give an off-white solid (75 mg, 60% yield). 1H NMR (500 MHz, MeOD): δ 8.35 (s, 1H), 8.34 (s, 1H), 8.10 (d, J = 7.9 Hz, 1H), 7.33 (d, J = 8.6 Hz, 2H), 7.20 (s, 1H), 7.12 (d, J = 8.3 Hz, 3H), 4.04 (s, 3H), 3.32–3.26 (m, 4H), 3.13–3.07 (m, 4H), 2.24 (s, 3H). HRMS (ESI) for C24H26N4O2 [M + H]+m/z: calcd, 403.2129; found, 403.2128.
tert-Butyl 4-(4-(5-(3-Methoxy-4-(methylcarbamoyl)phenyl)-4-methylpyridin-3-yl)phenyl)piperazine-1-carboxylate (18a)
To a solution of 17 (42 mg, 0.081 mmol) in MeOH (811 μL) was added methylamine, 33 wt % in EtOH (1.0 mL). The solution was stirred at 85 °C for 5h. The solvents were removed under reduced pressure prior to the crude material being triturated from a minimum amount of EtOAc and hexanes. The product was filtered and dried under air, then dissolved in DCM (213 μL), treated with trifluoroacetic acid (100 μL, 1.310 mmol), and stirred for 1 h. The solution was concentrated under reduced pressure prior to purification by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)]. The product was purified by SPE. Freeze-drying for 2 days afforded a white powder (7.68 mg, 20% yield). 1H NMR (500 MHz, MeOD): δ 8.32 (s, 1H), 8.30 (s, 1H), 8.01 (d, J = 7.9 Hz, 1H), 7.30 (d, J = 8.6 Hz, 2H), 7.15 (s, 1H), 7.12–7.06 (m, 3H), 4.00 (s, 3H), 3.30–3.27 (m, 4H), 3.12–3.08 (m, 4H), 2.98 (s, 3H), 2.21 (s, 3H). HRMS (ESI+) for C25H28N4O2 [M + H]+m/z: calcd, 417.2285; found, 417.2288.
4-(5-(4-(4-(tert-Butoxycarbonyl)piperazin-1-yl)phenyl)-4-methylpyridin-3-yl)-2-methoxybenzoic Acid
To a suspension of 17 (54 mg, 0.104 mmol) in THF (695 μL) and water (695 μL) was added potassium hydroxide pellets (12 mg, 0.209 mmol). The suspension was stirred at room temperature for 2 h. The reaction mixture was diluted with water (35 mL) and extracted with Et2O (1 × 20 mL). The aqueous layer was carefully acidified to a pH of 5 and extracted with DCM (3 × 20 mL). The Et2O and DCM layers were combined, dried over MgSO4, filtered, and concentrated under reduced pressure to afford an off-white solid (50 mg, 93% yield). MS (ESI) m/z: 504.60 [M + H]+.
2-Methoxy-N,N-dimethyl-4-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)benzamide (18b)
To a solution of 4-(5-(4-(4-(tert-butoxycarbonyl)piperazin-1-yl)phenyl)-4-methylpyridin-3-yl)-2-methoxybenzoic acid (50 mg, 0.099 mmol), HOBt (16 mg, 0.119 mmol), and EDC (18 mg, 0.119 mmol) in DCM (894 μL) and DMF (99 μL) was added DIPEA (43 μL, 0.248 mmol) and dimethylamine, 2.0 M in THF (50 μL, 0.099 mmol). The solution was stirred at 50 °C overnight. The reaction mixture was diluted with water (5 mL) and DCM (5 mL). The organic layer was separated, dried over MgSO4, filtered, and concentrated under reduced pressure to afford a sticky yellow solid. The solid was dissolved in DCM (886 μL) and treated with trifluoroacetic acid (339 μL, 4.430 mmol). The solution was stirred for 45 min prior to purification by reverse-phase chromatography [2–95% ACN (0.1% formic acid) in water (0.1% formic acid)]. The product was purified by SPE. Freeze-drying for 2 days afforded a white powder (13 mg, 23% yield). 1H NMR (500 MHz, MeOD): δ 8.33–8.30 (m, 2H), 7.35–7.28 (m, 3H), 7.12–7.04 (m, 4H), 3.90 (s, 3H), 3.28–3.24 (m, 4H), 3.12 (s, 3H), 3.08–3.04 (m, 4H), 2.94 (s, 3H), 2.22 (s, 3H). HRMS (ESI) for C25H28N4O2 [M + H]+m/z: calcd, 431.2442; found, 431.2439.
4-Bromo-2-(hydroxymethyl)benzonitrile
To a solution of 4-bromo-2-formylbenzonitrile (630 mg, 3.00 mmol) in MeOH (7.5 mL) cooled in an ice bath was added sodium borohydride (125 mg, 3.30 mmol). The reaction mixture was stirred for an hour at 0 °C prior to quenching with water (20 mL). Volatiles were removed under reduced pressure, and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic fractions were washed with brine, dried over NaSO4, filtered, and concentrated under reduced pressure to afford a yellow-brown solid, which was used without further purification in the subsequent reaction (637 mg, 85% yield). MS (ESI) m/z: 212.28 [M + H]+, 214.22 [M + H]+ + 2.
4-Bromo-2-(methoxymethyl)benzonitrile
To a solution of 4-bromo-2-(hydroxymethyl)benzonitrile (400 mg, 1.89 mmol) in THF (6.3 mL) cooled in an ice bath was added sodium hydride, 60% in mineral oil (181 mg, 7.54 mmol). The solution was stirred for 30 min prior to the addition of iodomethane (1.4 mL, 22.63 mmol). The reaction mixture was stirred for an additional 2 h, then quenched with water (50 mL) and extracted with EtOAc (3 × 50 mL). The combined organic fractions were dried over Na2SO4, filtered, and concentrated under reduced pressure prior to purification by silica gel chromatography (0–80% EtOAc in hexanes) to afford the final product (89 mg, 20% yield).
4-Bromo-2-(methoxymethyl)benzamide
The title compound was synthesized according to the procedure described for 5d from 4-bromo-2-(methoxymethyl)benzonitrile (80 mg, 0.354 mmol). The reaction mixture was stirred at 90 °C for 2 h and then at room temperature overnight. The crude mixture was diluted with water (10 mL) and extracted with EtOAc (3 × 10 mL). The organic layers were combined and dried over Mg2SO4 to afford an off-white solid (71 mg, 81% yield). 1H NMR (500 MHz, DMSO): δ 7.84 (br s, 1H), 7.64 (d, J = 1.9 Hz, 1H), 7.55 (dd, J = 8.2, 2.0 Hz, 1H), 7.46 (br s, 1H), 7.42 (d, J = 8.2 Hz, 1H), 4.58 (s, 2H).
2-(Methoxymethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzamide (19c)
The title compound was synthesized according to the procedure described for 4a from 4-bromo-2-(methoxymethyl)benzamide (50 mg, 0.205 mmol). The dark brown supernatant was used without purification in the subsequent reaction (60 mg, 78% yield). MS (ESI) m/z: 292.53 [M + H]+.
4-Bromo-2-chloro-6-fluorobenzonitrile
The title compound was prepared using a modified literature procedure.36 To a solution of 4-bromo-2-chloro-6-fluoroaniline (2.00 g, 8.91 mmol) in DCM (17.8 mL) was added nitrosonium tetrafluoroborate (1.14 g, 9.80 mmol). The solution was stirred for 1 h at room temperature and then cooled in an ice bath. Potassium cyanide (1.16 g, 17.82 mmol) was added. A solution of copper (II) sulfate pentahydrate (4.45 g, 17.82 mmol) in water (35.0 mL) was then added gradually. The suspension was stirred for 1 h on ice and then at room temperature for an additional hour. The reaction mixture was diluted with DCM and a saturated sodium bicarbonate solution, and then it filtered through Celite. The organic layer was washed with brine, separated, dried over Na2SO4, filtered, and concentrated under reduced pressure prior to purification by silica gel chromatography (0–50% EtOAc in hexanes) to give the final product (0.398 g, 10% yield).
4-Bromo-2-chloro-6-methoxybenzonitrile
The title compound was prepared using a modified literature procedure.37 To a solution of 4-bromo-2-chloro-6-fluorobenzonitrile (0.398 g, 1.697 mmol) in 1,4-dioxane (4.6 mL) was added MeOH (178 μL, 4.412 mmol). Sodium hydride, 60% in mineral oil (106 mg, 4.412 mmol) was added gradually over 1 h. The reaction mixture was stirred for 1 h at room temperature. The solvents were removed under reduced pressure, and the crude material was suspended in water and filtered. The filter cake was dissolved in DCM, concentrated, and purified by silica gel chromatography (0–100% DCM in hexanes) to give the final product (223 mg, 49% yield). MS (ESI) m/z: 246.26 [M + H]+, 248.26 [M + H]+ + 2, 250.21 [M + H]+ + 4. 1H NMR (500 MHz, CDCl3): δ 7.27 (d, J = 1.5 Hz, 1H), 7.04 (d, J = 1.4 Hz, 1H), 3.95 (s, 3H).
4-Bromo-2-chloro-6-methoxybenzamide
4-bromo-2-chloro-6-methoxybenzamide was synthesized according to the procedure described for 5d from 4-bromo-2-chloro-6-methoxybenzonitrile (219 mg, 0.889 mmol). The solution was stirred at 90 °C for 6 h. The crude was diluted with water (50 mL) and extracted with EtOAc (3 × 50 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to give the final product (219 mg, 77% yield). MS (ESI) m/z: 264.25 [M + H]+, 266.26 [M + H]+ + 2, 268.20 [M + H]+ + 4. 1H NMR (500 MHz, CDCl3): δ 7.20 (d, J = 1.4 Hz, 1H), 6.99 (d, J = 1.3 Hz, 1H), 5.93 (br s, 1H), 5.72 (br s, 1H), 3.85 (s, 3H).
2-Chloro-6-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzamide (19e)
The title compound was synthesized according to the procedure described for 4a from 4-bromo-2-chloro-6-methoxybenzamide (100 mg, 0.378 mmol). The dark supernatant was used without purification in the subsequent reaction (113 mg, 36% yield). MS (ESI) m/z: 312.46 [M + H]+, 314.41 [M + H]+ + 2.
5-(4-Methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)isoindolin-1-one (20a)
The title compound was synthesized according to the procedure described for 8a from 15 (75 mg, 0.193 mmol) and 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isoindolin-1-one (19a) (50 mg, 0.193 mmol). The final product was a white powder (33 mg, 44% yield). 1H NMR (500 MHz, DMSO): δ 8.62 (br s, 1H), 8.37 (s, 1H), 8.34 (s, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.65 (s, 1H), 7.54 (d, J = 7.7 Hz, 1H), 7.31 (d, J = 8.7 Hz, 2H), 7.04 (d, J = 8.8 Hz, 2H), 4.44 (s, 2H), 3.19–3.16 (m, 4H), 2.94–2.90 (m, 4H), 2.15 (s, 3H). HRMS (ESI) for C24H24N4O [M + H]+m/z: calcd, 385.2023; found, 385.2018.
4-(4-Methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)benzamide (20b)
The title compound was synthesized according to the procedure described for 8a from 15 (19 mg, 0.048 mmol) and 4-aminocarbonylphenylboronic acid (19b) (8 mg, 0.048 mmol). The final product was a light pink-orange powder (8 mg, 45% yield). 1H NMR (500 MHz, DMSO): δ 8.36 (s, 1H), 8.33 (s, 1H), 8.06 (br s, 1H), 7.98 (d, J = 8.3 Hz, 2H), 7.53 (d, J = 8.2 Hz, 2H), 7.42 (br s, 1H), 7.31 (d, J = 8.5 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 3.23–3.19 (m, 4H), 3.02–2.92 (m, 4H), 2.15 (s, 3H). HRMS (ESI) for C23H24N4O [M + H]+m/z: calcd, 373.2023; found, 373.2014.
2-(Methoxymethyl)-4-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)benzamide (20c)
The title compound was synthesized according to the procedure described for 8a from 15 (73 mg, 0.188 mmol) and 19c (60 mg, 0.205 mmol). The final compound was an off-white powder (23 mg, 29% yield). 1H NMR (500 MHz, CDCl3): δ 8.45 (s, 1H), 8.37 (s, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.44 (dd, J = 7.9, 1.7 Hz, 1H), 7.40 (d, J = 1.4 Hz, 1H), 7.28–7.26 (m, 2H), 7.01 (d, J = 8.7 Hz, 2H), 5.74 (br s, 1H), 4.65 (s, 2H), 3.46 (s, 3H), 3.26–3.22 (m, 4H), 3.10–3.06 (m, 4H), 2.17 (s, 3H). HRMS (ESI) for C25H28N4O2 [M + H]+m/z: calcd, 417.2285; found, 417.2283.
2-Fluoro-4-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)benzamide (20d)
The title compound was synthesized according to the procedure described for 8a from 15 (100 mg, 0.258 mmol) and 4-carbamoyl-3-fluorophenylboronic acid (19d) (47 mg, 0.258 mmol). The final compound was a white powder (50 mg, 50% yield). 1H NMR (500 MHz, MeOD): δ 8.37 (s, 1H), 8.32 (s, 1H), 7.96 (t, J = 8.0 Hz, 1H), 7.38–7.30 (m, 4H), 7.11 (d, J = 8.7 Hz, 2H), 3.29–3.23 (m, 4H), 3.08–3.02 (m, 4H), 2.24 (s, 3H). 19F NMR (471 MHz, MeOD): δ −114.60. HRMS (ESI) for C23H23FN4O [M + H]+m/z: calcd, 391.1929; found, 391.1926.
2-Chloro-6-methoxy-4-(4-methyl-5-(4-(piperazin-1-yl)phenyl)pyridin-3-yl)benzamide (20e)
The title compound was synthesized according to the procedure described for 6a from 3 (66 mg, 0.153 mmol) and 19e (59 mg, 0.189 mmol). The material was deprotected with trifluoroacetic acid (350 μL, 4.579 mmol) and purified by SPE. Freeze-drying for a day and a half afforded an off-white powder (24 mg, 36% yield). 1H NMR (500 MHz, CDCl3): δ 8.45 (s, 1H), 8.31 (s, 1H), 7.26–7.24 (m, 2H), 7.03 (d, J = 1.1 Hz, 1H), 7.01 (d, J = 8.7 Hz, 2H), 6.81 (d, J = 1.0 Hz, 1H), 5.94–5.87 (br m, 2H), 3.89 (s, 3H), 3.26–3.22 (m, 4H), 3.10–3.06 (m, 4H), 2.18 (s, 3H). HRMS (ESI) for C24H25ClN4O2 [M + H]+m/z: calcd, 437.1739; found, 437.1740.
4-Bromo-2-fluoro-6-methoxybenzonitrile (22a) and 4-Bromo-2,6-dimethoxybenzonitrile (22b)
The title compounds were prepared according to the procedure described for 4-bromo-2-chloro-6-methoxybenzonitrile from 4-bromo-2,6-difluorobenzonitrile (21) (2.00 g, 9.17 mmol), MeOH (744 μL, 18.34 mmol) and sodium hydride, 60% in mineral oil (0.733 g, 18.34 mmol). 4-Bromo-2-fluoro-6-methoxybenzonitrile was a white crystalline solid (965 mg, 46% yield). 4-Bromo-2,6-dimethoxybenzonitrile was also a white crystalline solid (754 mg, 34% yield). 4-Bromo-2-fluoro-6-methoxybenzonitrile: 1H NMR (500 MHz, CDCl3): δ 7.00 (d, J = 8.0 Hz, 1H), 6.94 (s, 1H), 3.95 (s, 3H). 19F NMR (471 MHz, CDCl3): δ −103.78 (s). MS (ESI) m/z: 230.21 [M + H]+, 232.21 [M + H]+ + 2. 4-Bromo-2,6-dimethoxybenzonitrile: 1H NMR (500 MHz, CDCl3): δ 6.73 (s, 2H), 3.91 (s, 6H). MS (ESI) m/z: 242.25 [M + H]+, 244.19 [M + H]+ + 2.
(4-Carbamoyl-3-fluoro-5-methoxyphenyl)boronic Acid (23a)
The title compound was synthesized according to the procedure described for 4a from 5d (2.48 g, 10.0 mmol). The dark brown supernatant was used without further purification in subsequent reactions (2.46 g, 84% yield). MS (ESI) m/z: 214.34 [M + H]+.
2,6-Dimethoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzamide (23b)
The title compound was synthesized according to the procedure described for 4a from 5e (800 mg, 3.08 mmol). The dark brown supernatant was used without further purification in subsequent reactions (945 mg, 70% yield). MS (ESI) m/z: 308.26 [M + H]+.
4-(5-Chloro-4-methylpyridin-3-yl)-2-fluoro-6-methoxybenzamide (24a)
The title compound was synthesized according to the procedure described for 6a from 10 (2.06 g, 10.0 mmol) and 23a (2.95 g, 10.0 mmol). The final compound was an off-white crystalline solid (1.67 g, 56% yield). 1H NMR (500 MHz, DMSO): δ 8.63 (s, 1H), 8.38 (s, 1H), 7.90 (br s, 1H), 7.60 (br s, 1H), 6.97–6.94 (m, 2H), 3.83 (s, 3H), 2.33 (s, 3H). 19F NMR (471 MHz, DMSO): δ −116.46. MS (ESI) m/z: 294.97 [M + H]+, 297.10 [M + H]+ + 2.
4-(5-Chloro-4-methylpyridin-3-yl)-2,6-dimethoxybenzamide (24b)
The title compound was synthesized according to the procedure described for 6a from 10 (954 mg, 4.62 mmol) and 23b (946 mg, 3.08 mmol). The final compound was an off-white crystalline solid (747 mg, 79% yield). 1H NMR (500 MHz, DMSO): δ 8.61 (s, 1H), 8.38 (s, 1H), 7.58 (br s, 1H), 7.26 (br s, 1H), 6.70 (s, 2H), 3.77 (s, 6H), 2.34 (s, 3H). MS (ESI) m/z: 307.26 [M + H]+.
(2R,6S)-4-(4-Bromophenyl)-1,2,6-trimethylpiperazine
The title compound was prepared using modified literature procedures.38,39 A solution of (2R,6S)-1,2,6-trimethylpiperazine (100 mg, 0.780 mmol), 1-bromo-4-iodobenzene (221 mg, 0.780 mmol), bis(dibenzylideneacetone)palladium(0) (22 mg, 0.039 mmol), xantphos (68 mg, 0.117 mmol), and lithium tert-butoxide 1.0 M in THF (2.34 mL, 2.340 mmol) in 1,4-dioxane (3.12 mL) was heated to 110 °C and stirred overnight. Volatiles were removed under reduced pressure, and the crude material was purified by silica gel chromatography (0–10% MeOH in EtOAc). 1H NMR (500 MHz, MeOD): δ 7.32 (d, J = 9.0 Hz, 2H), 6.87 (d, J = 9.0 Hz, 2H), 3.55–3.49 (m, J = 11.5 Hz, 2H), 2.52–2.46 (m, J = 11.4 Hz, 2H), 2.44–2.38 (m, 2H), 2.33 (s, 3H), 1.19 (d, J = 6.1 Hz, 6H). MS (ESI) m/z: 283.37 [M + H]+, 285.32 [M + H]+ + 2.
(2R,6S)-1,2,6-Trimethyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)piperazine (25c)
The title compound was prepared according to the procedure described for 4a from (2R,6S)-4-(4-bromophenyl)-1,2,6-trimethylpiperazine (100 mg, 0.353 mmol). The dark brown reaction mixture was used without purification in subsequent reactions (117 mg, 91% yield). MS (ESI) m/z: 331.46 [M + H]+.
2-Fluoro-6-methoxy-4-(4-methyl-5-(4-(4-methylpiperazin-1-yl)phenyl)pyridin-3-yl)benzamide (26a)
The title compound was synthesized according to the procedure described for 8a from 24a (150 mg, 0.509 mmol) and 4-(4-methylpiperazin-1-yl)phenylboronic acid (25a) (134 mg, 0.611 mmol). XPhos Pd G3 (21.54 mg, 0.025 mmol) was used as the catalyst. The reaction mixture was adsorbed onto Celite, and the solvents were removed under reduced pressure. The crude material was purified by silica gel chromatography (0–15% MeOH in EtOAc). Freeze-drying for 1 day afforded white powder (183 mg, 83% yield). 1H NMR (500 MHz, DMSO): δ 8.37 (s, 1H), 8.34 (s, 1H), 7.89 (br s, 1H), 7.58 (br s, 1H), 7.30 (d, J = 8.7 Hz, 2H), 7.05 (d, J = 8.8 Hz, 2H), 6.98–6.94 (m, 2H), 3.85 (s, 3H), 3.23–3.19 (m, 4H), 2.48–2.45 (m, 4H), 2.23 (s, 3H), 2.19 (s, 3H). 19F NMR (471 MHz, DMSO): δ −116.74. HRMS (ESI) for C25H27FN4O2 [M + H]+m/z: calcd, 435.2191; found, 435.2191.
2-Fluoro-4-(5-(4-(4-isopropylpiperazin-1-yl)phenyl)-4-methylpyridin-3-yl)-6-methoxybenzamide (26b)
The title compound was synthesized according to the procedure described for 26a from 24a (150 mg, 0.509 mmol) and 4-(4-isopropylpiperazinyl)phenylboronic acid, pinacol ester (25b) (202 mg, 0.611 mmol). The final compound was white powder (142 mg, 60% yield). 1H NMR (500 MHz, DMSO): δ 8.38 (s, 1H), 8.34 (s, 1H), 7.89 (br s, 1H), 7.58 (br s, 1H), 7.30 (d, J = 8.7 Hz, 2H), 7.04 (d, J = 8.8 Hz, 2H), 6.98–6.95 (m, 2H), 3.85 (s, 3H), 3.23–3.17 (m, 4H), 2.72–2.66 (m, 1H), 2.63–2.57 (m, 4H), 2.19 (s, 3H), 1.02 (d, J = 6.5 Hz, 6H). 19F NMR (471 MHz, DMSO): δ −116.74. HRMS (ESI) for C27H31FN4O2 [M + H]+m/z: calcd, 463.2504; found, 463.2506.
2-Fluoro-6-methoxy-4-(4-methyl-5-(4-((3R,5S)-3,4,5-trimethylpiperazin-1-yl)phenyl)pyridin-3-yl)benzamide (26c)
The title compound was synthesized according to the procedure described for 26a from 24a (156 mg, 0.531 mmol) and (2R,6S)-1,2,6-trimethyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)piperazine (25c) (58.5 mg, 0.177 mmol). The final compound was light yellow powder (36 mg, 43% yield). 1H NMR (500 MHz, CDCl3): δ 8.43 (s, 1H), 8.30 (s, 1H), 7.24 (d, J = 8.6 Hz, 2H), 6.99 (d, J = 8.6 Hz, 2H), 6.77 (dd, J = 9.7, 0.5 Hz, 1H), 6.71 (s, 1H), 6.32 (br s, 1H), 6.09 (br s, 1H), 3.91 (s, 3H), 3.57 (d, J = 11.5 Hz, 2H), 2.86–2.67 (m, 2H), 2.60–2.45 (m, 2H), 2.40 (s, 3H), 2.16 (s, 3H), 1.26 (d, J = 5.8 Hz, 6H). 19F NMR (471 MHz, MeOD): δ −117.13. HRMS (ESI) for C27H31FN4O2 [M + H]+m/z: calcd, 463.2504; found, 463.2510.
2,6-Dimethoxy-4-(4-methyl-5-(4-(4-methylpiperazin-1-yl)phenyl)pyridin-3-yl)benzamide (26d)
The title compound was synthesized according to the procedure described for 26a from 24b (150 mg, 0.489 mmol) and 25a (129 mg, 0.587 mmol). The final compound was white powder (189 mg, 87% yield). 1H NMR (500 MHz, DMSO): δ 8.35 (s, 1H), 8.35 (s, 1H), 7.57 (br s, 1H), 7.30 (d, J = 8.6 Hz, 2H), 7.23 (br s, 1H), 7.05 (d, J = 8.7 Hz, 2H), 6.72 (s, 2H), 3.78 (s, 6H), 3.23–3.19 (m, 4H), 2.48–2.46 (m, 4H), 2.24 (s, 3H), 2.20 (s, 3H). HRMS (ESI) for C26H30N4O3 [M + H]+m/z: calcd, 477.2391; found, 447.2394.
4-(5-(4-(4-Isopropylpiperazin-1-yl)phenyl)-4-methylpyridin-3-yl)-2,6-dimethoxybenzamide (26e)
The title compound was synthesized according to the procedure described for 26a from 24b (150 mg, 0.489 mmol) and 25b (194 mg, 0.587 mmol). The final compound was an off-white powder (179 mg, 75% yield). 1H NMR (500 MHz, MeOD): δ 8.31 (s, 1H), 8.30 (s, 1H), 7.33 (d, J = 8.6 Hz, 2H), 7.13 (d, J = 8.7 Hz, 2H), 6.69 (s, 2H), 3.86 (s, 6H), 3.47–3.36 (m, 4H), 3.15–3.01 (m, 5H), 2.22 (s, 3H), 1.27 (d, J = 6.6 Hz, 6H). HRMS (ESI) for C28H34N4O3 [M + H]+m/z: calcd, 475.2704; found, 475.2705.
2,6-Dimethoxy-4-(4-methyl-5-(4-((3R,5S)-3,4,5-trimethylpiperazin-1-yl)phenyl)pyridin-3-yl)benzamide (26f)
The title compound was synthesized according to the procedure described for 26a from 24b (109 mg, 0.354 mmol) and 25c (58 mg, 0.177 mmol). The final compound was white powder (22 mg, 26% yield). 1H NMR (500 MHz, MeOD): δ 8.31 (s, 1H), 8.29 (s, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.5 Hz, 2H), 6.69 (s, 2H), 3.86 (s, 6H), 3.65 (d, J = 11.8 Hz, 2H), 2.62–2.55 (m, 2H), 2.51–2.44 (m, 2H), 2.37 (s, 3H), 2.23 (s, 3H), 2.16 (s, 2H), 1.23 (d, J = 6.2 Hz, 6H). HRMS (ESI) for C28H34N4O3 [M + H]+m/z: calcd, 475.2704; found, 475.2699.
Kinase Assay
The biochemical potencies of all compounds were measured by Reaction Biology Corporation (RBC) (Malvern, Pennsylvania, United States). Compounds were tested against ALK2/ACVR1 and ALK5/TGFβ-R1 in a 10-dose IC50 mode with a 2-fold serial dilution starting at 1 or 5 μM. Reactions were conducted at an ATP concentration of 10 μM and Casein concentration of 1 mg/mL. LDN-193189 was tested as a control in a 10-dose IC50 mode with a threefold serial dilution starting at 10 μM. Reductions in enzymatic activity were determined relative to DMSO controls.
Cell Culture and Transfection
HEK-293 cells were maintained in Dulbecco’s modified Eagle medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher) and penicillin/streptomycin (Thermo Fisher). HEK-293 cells were transfected with the protein expression or reporter constructs using FuGENE HD (Promega) according to the manufacturer’s instructions. Briefly, DNA was diluted into phenol red-free Opti-MEM (Gibco) at a concentration of 10 μg/mL. Without coming in contact with the sides of the container, 3 μL of FuGENE HD was added for each μg of DNA used. After thorough mixing by inversion, FuGENE HD/DNA complexes were allowed to form by incubation at room temperature for 20 min. Transfection mixture (1 part) was added to 20 parts of HEK-293 cell suspension with a density of 200,000 cells per mL (volume/volume). HEK-293 cells were incubated in a humidified, 37 °C incubator with 5% carbon dioxide for 24 h before they are used in the NanoBRET target engagement assay or dual luciferase reporter assay.
NanoBRET Target Engagement Assay
ALK2-C-terminal nanoluciferase fusion with the GSSG linker was encoded by the pFC32K vector (Promega). ALK2-nanoluciferase fusion construct (1 part) was mixed with 9 parts of Transfection Carrier DNA (mass/mass) (Promega). Transfected cells were trypsinized and resuspended in Opti-MEM at a density of 200,000 cells per mL. Cells (17 μL) were dispensed into each well of 384-well flat-bottom polypropylene plate (Greiner). Working solution (20×) of target engagement tracer PBI-6908 (Promega) was prepared by diluting DMSO stock in tracer dilution buffer (12.5 mM HEPES pH 7.5, 31.25% PEG-400). Stocks (1000×) of test compounds in DMSO (Cell Signaling Technology) were diluted further in Opti-MEM for 10× working solutions. After the addition of 1 μL of 20× target engagement tracer and 2 μL of 10× working solutions, contents of the wells were thoroughly mixed by agitating the plate at 500 rpm for 1 min. Cells were incubated in a humidified, 37 °C incubator with 5% carbon dioxide for 2 h prior to BRET measurement. For bioluminiscence resonance energy transfer (BRET) measurement, the NanoBRET NanoGlo Substrate and Extracellular NanoLuc Inhibitor (Promega) were diluted 166× and 500×, respectively, in Opti-MEM to produce 3× working stock. A PHERAstar FSX microplate reader (BMG Labtech) with the LUM 610-LP 460-80 optical module was used to measure the intensity of dual emission. A measurement interval of 1 s and gain settings of 3600 and 1879 for 610 and 460 nm, respectively, were used. Milli-BRET units (mBU) were calculated by dividing the signal measured at 610 nm with the signal measured at 460 nm and multiplying by 1000. The apparent EC50 values of test compounds were estimated using the [Inhibitor] versus response (three parameters) nonlinear regression curve fitting function of GraphPad Prism 7.
Dual Luciferase Reporter Assay
CAGA-Luc and Renilla-luciferase constructs (a gift of Dr Petra Knaus, Free University of Berlin) were used as the reporter for ALK5 signaling and loading control, respectively. CAGA-Luc construct (4 parts) was mixed with 1 part of Renilla-luciferase construct (mass/mass). Ten thousand transfected cells were seeded into each well of 96-well plate (Corning). Twenty four hours after transfection, the cells were incubated with 10 ng/mL TGF-β1 (Peprotech, 100-21-10) and test compounds simultaneously at the concentrations indicated in a humidified, 37 °C incubator with 5% carbon dioxide. Twenty-four hours later, the cells were harvested, lysed, and processed for the measurement of luciferase activity using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Briefly, the culture medium was aspirated completely, and cells were lysed in 50 μL of 1× PLB with 300 rpm agitation for 30 min. Cell lysate (10 μL) was dispensed into each well of a 384-well flat-bottom polypropylene plate (Greiner). The luminescent signal of firefly and Renilla luciferase activity were measured sequentially using a PHERAstar FS microplate reader (BMG Labtech) after the addition of 25 μL of LARII and Stop & Glo, respectively. A measurement interval of 2 s and gain setting of 3600 were used. The firefly luciferase signal was normalized to the cell number by division with Renilla luciferase signal. The relative luciferase unit (RLU) was obtained by further division with the signal from cells without TGF-β stimulation. The apparent EC50 values of test compounds were estimated using the [Inhibitor] versus response (three parameters) nonlinear regression curve fitting function of GraphPad Prism 7.
Caco-2 Permeability Assay
Caco-2 cells (C2BBe1) were purchased from American Type Culture Collection, ATCC. Caco-2 cell cultures were routinely maintained in T-75 tissue culture flasks in DMEM containing 20% FBS, 0.1 mg/mL normocin, and 0.05 mg/mL gentamicin. These cells were seeded at a density of 40,000 cells/well on the 24-well polyethylene terephthalate (PET) membrane (1.0 μm pore size, 0.31 cm2 surface area) insert plates. Cell monolayers were grown for 21 or 22 days at 37 °C with 5% CO2 in a humidified incubator. The cell culture medium was replaced twice weekly during the cell growth period. Prior to beginning the permeability assay, cell monolayers were rinsed with Hank’s balanced salt solution (HBSS) twice to remove the residual cell culture medium.
The assay buffer comprised HBSS containing 10 mM HEPES and 15 mM glucose at a pH of 7.4. The dosing buffer contained 5 μM metoprolol (positive control), 5 μM atenolol (negative control), and 100 μM Lucifer yellow in the assay buffer. The receiving buffer contained 1% bovine serum albumin (BSA) in the assay buffer. The concentration of the test compound was 5 μM in the dosing buffer (final DMSO concentration was 0.1%). Digoxin at 10 μM was utilized as a Pgp substrate control.
For apical to basolateral (A to B) permeability experiment, 0.25 mL of the dosing buffer was added to the apical chambers, and 1.0 mL of the receiving buffer was added to the basolateral chambers of the assay plate. For the basolateral to apical (B to A) permeability experiment, 0.25 mL of the receiving buffer was added to the apical chambers, and 1.0 mL of dosing buffer was added to the basolateral chambers of the assay plates. The assay plates were then incubated at 37 °C for 120 min on an orbital shaker at 65 rpm. Sample solutions were taken from the donor chambers (10 μL) and receiver chambers (100 μL) after the incubation period. For each sample, there were two technical replicates. The sample solutions from donor chambers were diluted ten times with the receiving buffer. In order to extract test compounds and precipitate BSA from sample solutions, three volumes of acetonitrile (containing 0.5% formic acid and an internal standard) were added, and the plate was vigorously mixed. Sample solutions were then centrifuged at 4000 rpm for 10 min to remove debris and precipitated BSA. Approximately, 150 μL of the supernatant was subsequently transferred to a new 96-well microplate for LC/MS analysis. Narrow-window mass extraction LC/MS analysis was performed for all samples from this study using a Waters Xevo quadrupole time-of-flight (QTof) mass spectrometer to determine relative peak areas of parent compounds. The co-dosed positive and negative controls were also measured for each well to monitor integrity of cell monolayers and well-to-well variability. The apparent permeability coefficient (Papp) and post-assay recovery are calculated using the following equations
where dC/dt is the slope of cumulative concentration in the receiver compartment versus time, Vr is the volume of the receiver compartment, Vd is the volume of the donor compartment, A is the membrane surface area, C0 is the compound initial concentration in the donor chamber, Crfinal is the cumulative receiver concentration at the end of the incubation period, and Cd is the concentration of the donor at the end of the incubation period.
Efflux ratio (ER) is defined as Papp (B-to-A)/Papp (A-to-B).
Liver Microsomal Metabolic Stability Assay
For this assay, stock solutions of test compounds in DMSO (1 mM) were initially diluted to a concentration of 40.0 μM using 0.1 M potassium phosphate buffer (pH 7.4). Test compounds were then added to reaction wells at a final concentration of 1 μM which was assumed to be well below Km values to ensure linear reaction conditions (i.e., avoid saturation). The final DMSO concentration was kept constant at 0.1%. Each compound was tested in duplicate for both time points (0 and 60 min). CD-1 mouse (male) or pooled human liver microsomes (Corning Gentest) were added to the reaction wells at a final concentration of 0.5 mg/mL (protein). The final volume for each reaction was 100 μL, which included the NADPH-regeneration solution (NRS) mix (Corning Gentest). This NRS mix comprised glucose 6-phosphate dehydrogenase, NADP+, MgCl2, and glucose 6-phosphate. Reactions were carried out at 37 °C in an orbital shaker at 175 rpm. Upon completion of the 60 min time point, reactions were terminated by the addition of two volumes (200 μL) of ice-cold acetonitrile containing 0.5% formic acid and an internal standard. Samples were then centrifuged at 4000 rpm for 10 min to remove debris and precipitated proteins. Approximately, 150 μL of supernatant was subsequently transferred to a new 96-well microplate for LC/MS analysis.
Narrow-window mass extraction LC/MS analysis was performed for all samples in this study using a Waters Xevo quadrupole time-of-flight (QTof) mass spectrometer to determine relative peak areas of test compounds. The percentage remaining values were calculated using the following equations
where A is area response after incubation, A0 is area response at initial time point.
hERG Inhibition Assay
hERG IC50 values were generated by Charles River Laboratories (Cleveland, Ohio, United States). Their protocol is described below:
Compounds were tested against cloned hERG potassium channels expressed in HEK293 cells. Chemicals used in solution preparation were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted and were of ACS reagent-grade purity or higher. Stock solutions of test articles and the positive control were prepared in dimethyl sulfoxide (DMSO) and stored frozen. Reference compound concentrations were prepared fresh daily by diluting stock solutions into a Charles River proprietary HEPES-buffered physiological saline (HB-PS) solution which was prepared weekly and refrigerated until use. Because previous results have shown that ≤0.3% DMSO did not affect channels currents, all test and control solutions contained 0.3% DMSO. Each test article formulation was sonicated (model 2510/5510, Branson Ultrasonics, Danbury, CT) at ambient room temperature for 20 min to facilitate dissolution. Cells were cultured in DMEM/nutrient mixture F-12 (D-MEM/F-12) supplemented with 10% FBS, 100 U/mL penicillin G sodium, 100 μg/mL streptomycin sulfate, and 500 μg/mL G418. Before testing, cells in culture dishes were washed twice with HBSS and detached with accutase. Immediately before use in the IonWorks Barracuda system, the cells were washed twice in HB-PS to remove the accutase and resuspended in 5 mL of HB-PS. The test article effects were evaluated using IonWorks Barracuda systems (Molecular Devices Corporation, Union City, CA). HEPES-buffered intracellular solution (Charles River proprietary) for whole cell recordings was loaded into the intracellular compartment of the Population Patch Clamp (PPC) planar electrode. Extracellular buffer (HB-PS) was loaded into PPC planar electrode plate wells (11 μL per well). The cell suspension was pipetted into the wells of the PPC planar electrode (9 μL per well). After establishment of a whole-cell configuration (the perforated patch), membrane currents were recorded using a patch clamp amplifier in the IonWorks Barracuda system. The current recordings were performed one (1) time before test article application to the cells (baseline) and one (1) time after application of the test article. Test article concentrations were applied to naïve cells (n = 4, where n = replicates/concentration). Each application consisted of addition of 20 μL of 2× concentrated test article solution to the total 40 μL of final volume of the extracellular well of the PPC plate. The duration of exposure to each compound concentration was five (5) minutes. The hERG current was measured using a pulse pattern with fixed amplitudes (conditioning pre-pulse: −80 mV for 25 ms; test pulse: +40 mV for 80 ms) from a holding potential of 0 mV (“zero holding” procedure). The hERG current was measured as a difference between the peak current at 1 ms after the test step to +40 mV and the steady-state current at the end of the step to +40 mV.
CYP Inhibition Assay
CYP IC50 values were generated by Pharmaron. Their protocol is described below:
Multiple concentrations (1 μL) of the test compound or positive control compound (CYP1A2: furafylline, CYP2B6: ketoconazole, CYP2C8: quercetin, CYP2C9: sulfaphenazole, CYP2C19: N-3-benzylnirvanol, CYP2D6: quinidine, and CYP3A4: ketoconazole) were transferred to the “Compound Plate”. The concentrations of test compounds and positive control compounds were 0, 0.2, 1, 2, 10, 50, 200, 2000, and 10,000 μM. The master solution was prepared with MgCl2 solution (20 μL of 50 mM solution), phosphate buffer (100 μL of 200 mM solution), ultrapure water (56 μL), human liver microsomes [2 μL of 20 mg/mL stock concentration (Corning UltraPool HLM 150, Mixed Gender Cat. no.: 452117)], and 1 μL of substrate [CYP1A2: phenacetin (8 mM stock concentration), CYP2B6: bupropion (10 mM stock concentration), CYP2C8: paclitaxel (1 mM stock concentration), CYP2C9: tolbutamide (40 mM stock concentration), CYP2C19: mephenytoin (10 mM stock concentration), CYP2D6: dextromethorphan (2 mM stock concentration) and CYP3A4: midazolam (1 mM stock concentration), and testosterone (10 mM stock concentration)]. The master solution was prewarmed in a water bath at 37 °C for 5 min. The incubated master solution (179 μL) was transferred to the compound plate. In the mixed system, the final concentrations of the test compounds and positive control compounds were 0, 0.001, 0.005, 0.01, 0.05, 0.25, 1, 10, and 50 μM. All experiments were performed in duplicate. The reaction was started with the addition of 20 μL of 10 mM NADPH solution at the final concentration of 1 mM. The reaction was stopped by the addition of 1.5 volumes of methanol with IS (100 nM alprazolam, 200 nM imipramine, 200 nM labetalol, and 2 μM ketoprofen) to the “Incubation Plate” at the designated time points (20 min for CYP1A2, 2B6, 2C9, 2C19, and 2D6, 5 min for midazolam-mediated 3A4, and 10 min for testosterone-mediated 3A4). The “Incubation Plate” was centrifuged at 3220 g for 40 min to precipitate the protein. An aliquot of 100 μL of the supernatant was diluted using 100 μL ultrapure H2O, and the mixture was used for LC/MS/MS analysis. The formation of metabolites was analyzed using LC/MS/MS. A decrease in the formation of the metabolites in the peak area to vehicle control was used to calculate an IC50 value (test compound concentration which produces 50% inhibition) using Excel XLfit.
In Vivo Pharmacokinetic Studies
The pharmacokinetic profiles of 8b, 26a, and 26b were assessed by Pharmaron. Their protocol is described below:
Test compounds were dissolved first in DMSO, then mixed with 47.5% PEG400 and 47.5% DI water with 10% Tween80. The solutions were thoroughly vortexed after each step and stored at room temperature. Solutions were freshly prepared on the day of dosing. Female CB17 SCID mice (n = 3) (6–8 weeks old, 17–20 g weight) were orally administered a 10 mg/kg dose (10 mL/kg dose volume, 1 mg/mL concentration) of the test compound. Blood samples were taken via the dorsal metatarsal vein at 0.25, 0.5, 1, 2, 4, 8, and 24 h post-dosage. Blood samples were transferred into plastic microcentrifuge tubes containing the anticoagulant Heparin-Na and centrifuged at 4000 g for 5 min at 4 °C to obtain plasma. The samples were stored in a freezer at −75 ± 15 °C prior to analysis.
To determine brain concentrations, female CB17 SCID mice (n = 3) (6–8 weeks old, 17–20 g weight) were orally administered a 100 mg/kg dose (10 mL/kg dose volume, 10 mg/mL concentration) of the test compound. 4 h post-dose, the animals were terminally anaesthetized by an increasing concentration of CO2. Their chest cavities were opened to expose the heart and an incision at the right auricle using surgical scissors was done. A syringe full of gentle saline was pushed into the heart slowly via the left ventricle (saline volume: ∼10 mL). The animal was placed head down at a 45° angle to facilitate blood removal. Brain samples were collected and kept frozen at −75 ± 15 °C. All brain samples were weighed and homogenized with phosphate buffered saline by brain weight (g) to buffer volume (mL) ratio 1:3 before analysis. The actual concentrations were the detected value multiplied using the dilution factor.
Concentrations of the test compound in the plasma samples were analyzed using a LC/MS/MS method. WinNonlin (Phoenix, version 8.0) or other similar software was used for pharmacokinetic calculations. The following pharmacokinetic parameters were calculated, whenever possible from the plasma concentration versus time data:
PO administration: T1/2, Cmax, Tmax, AUClast, AUCinf, and F.
All animal procedures were in accordance with the regulations of Institutional Animal Care and Use Committee (IACUC) at Pharmaron Inc.
Cocrystallization of ALK2 with M4K2149
Protein Expression and Purification
Constructs were prepared by ligation-independent cloning. The kinase domain of ALK2 (residues 201–499; Uniprot ID, Q04771) was cloned into pFB-LIC-Bse for the baculoviral expression. The construct was verified by sequencing. ALK2 was expressed in Sf9 insect cells grown at 27 °C. Some 72 h postinfection, cells were harvested and lysed using ultrasonication. ALK2 was initially purified by nickel affinity chromatography before subsequent purification by size exclusion chromatography (Superdex 200 16/600). The eluted protein was stored in 50 mM HEPES, pH 7.5, 300 mM NaCl, 10 mM DTT. The hexahistidine tag of ALK2 was cleaved using tobacco etch virus protease after initial nickel purification.
Crystallization
Crystallization was achieved at 4 °C using the sitting-drop vapor diffusion method. ALK2 was preincubated with 1 mM M4K2149 at a protein concentration of 11 mg/mL and crystallized using a precipitant containing 0.1 M citrate pH 4.9, 1 M ammonium sulfate, and 0.2 M sodium/potassium tartrate. Viable crystals were obtained when the protein solution was mixed with the reservoir solution at 2:1 volume ratio. Crystals were cryoprotected with mother liquor plus 25% ethylene glycol, prior to vitrification in liquid nitrogen.
Data Collection
Diffraction data were collected at the Diamond Light Source, station I03 using monochromatic radiation at a wavelength 0.9763 Å.
Phasing, Model Building, Refinement, and Validation
Data were processed with Xia2 and subsequently scaled using the program AIMLESS from the CCP4 suite.40,41 Initial phases were obtained by molecular replacement using the program PHASER and the structure of ALK2 (Protein Data Bank code 6SRH) as a search model.42 The resulting structure solution was refined using Phenix Refine and manually rebuilt with COOT.43,44 The complete structure was verified for geometric correctness with MolProbity.45 Data collection and refinement statistics can be found in Supporting Information Table 1.
Cocrystal images in the article were processed using Molsoft MolBrowser 3.8.
Acknowledgments
This work was funded by the Cancer Therapeutics Innovation Pipeline program at the Ontario Institute for Cancer Research (OICR). The OICR is funded by the Government of Ontario. Funding from The Brain Tumour Charity was used to support the cellular and crystallographic research conducted at the University of Oxford. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute [OGI-055], Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA, Darmstadt, Germany, MSD, Novartis Pharma AG, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. We thank Reaction Biology Corporation for their pro bono contributions in testing the biological activity of our compounds. D.P. is grateful to NSERC for the Collaborative Research and Training Experience 432008-2013 grant. We would like to thank Dr. Gennady Poda for generating the Oracle database where the compound data were stored and for providing his expertise on molecular modeling. We would also like to thank Mehakpreet Saini for contributing to the writing and editing of ADME protocols.
Glossary
Abbreviations
- ALK2
activin receptor-like kinase-2
- ALK5
activin receptor-like kinase-5
- ATP
adenosine triphosphate
- AUCinf
area under the curve (extrapolated to infinity)
- BBB
blood–brain barrier
- BMP
bone morphogenetic protein
- B/P
total brain-to-plasma ratio
- Cb,u
unbound brain concentration
- cLogP
calculated lipophilicity
- Cmax
maximum concentration
- CNS
central nervous system
- DIPG
diffuse intrinsic pontine glioma
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- FOP
fibrodysplasia ossificans progressiva
- GS
glycine–serine-rich domain
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- HBD
hydrogen bond donor
- HDAC
histone deacetylase
- hERG
human ether a-go-go related gene
- HLM
human liver microsome
- MLM
mouse liver microsome
- PK
pharmacokinetic
- P-gp
P-glycoprotein
- SAR
structure–activity relationship
- STKR
serine/threonine kinase receptor
- TFA
trifluoroacetic acid
- TGFβ-R1
transforming growth factor beta receptor 1
- tPSA
topological polar surface area
- WT
wild-type.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00395.
Accession Codes
PBD ID codes: ALK2-M4K2149, 6T6D; ALK2-LDN-213844, 4BGG.
Author Present Address
∇ Dalriada Therapeutics Inc., 3252 Respond Road, Mississauga, Ontario L5M 7X4, Canada.
Author Contributions
Analogues were synthesized by D.E., D.S., and D.P. The compounds were designed and their syntheses devised by D.S., M.B.I., C.A.Z.-V., and D.E. All NanoBRET data was generated by J.F.W. and A.H. Crystallography experiments were performed by E.P.W. and R.A. Caco-2 and microsomal stability studies were conducted by T.K. HRMS were also generated by T.K. A.N.B., A.A., J.A.O., M.B.I., and R.A.-a. were involved in the experimental design, the interpretation of data, and they monitored project progress. O.G.R. and A.M.E. initiated the project and edited the paper. The manuscript was written by D.E. and revised by D.S., C.A.Z.-V., M.B.I., and R.A.-a.
The authors declare no competing financial interest.
Notes
Authors will release the atomic coordinates and experimental data upon article publication.
Supplementary Material
References
- Heffron T. P. Small molecule kinase inhibitors for the treatment of brain cancer. J. Med. Chem. 2016, 59, 10030–10066. 10.1021/acs.jmedchem.6b00618. [DOI] [PubMed] [Google Scholar]
- Di L.; Rong H.; Feng B. Demystifying brain penetration in central nervous system drug discovery. J. Med. Chem. 2013, 56, 2–12. 10.1021/jm301297f. [DOI] [PubMed] [Google Scholar]
- Pacifici M.; Shore E. M. Common mutations in ALK2/ACVR1, a multi-faceted receptor, have roles in distinct pediatric musculoskeletal and neural orphan disorders. Cytokine Growth Factor Rev. 2016, 27, 93–104. 10.1016/j.cytogfr.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmierer B.; Hill C. S. TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- Massagué J.; Blain S. W.; Lo R. S. TGFβ signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Taylor K. R.; Vinci M.; Bullock A. N.; Jones C. ACVR1 mutations in DIPG: lessons learned from FOP. Cancer Res. 2014, 74, 4565–4570. 10.1158/0008-5472.can-14-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K. R.; Mackay A.; Truffaux N.; Butterfield Y. S.; Morozova O.; Philippe C.; Castel D.; Grasso C. S.; Vinci M.; Carvalho D.; Carcaboso A. M.; de Torres C.; Cruz O.; Mora J.; Entz-Werle N.; Ingram W. J.; Monje M.; Hargrave D.; Bullock A. N.; Puget S.; Yip S.; Jones C.; Grill J. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R. K.; Rutka J. T. Diffuse intrinsic pontine glioma: clinical features, molecular genetics, and novel targeted therapeutics. J. Korean Neurosurg. Soc. 2018, 61, 343–351. 10.3340/jkns.2018.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren K. E. Diffuse intrinsic pontine glioma: poised for progress. Front. Oncol. 2012, 2, 205. 10.3389/fonc.2012.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.; Diaz A. K.; Paugh B. S.; Rankin S. L.; Ju B.; Li Y.; Zhu X.; Qu C.; Chen X.; Zhang J.; Easton J.; Edmonson M.; Ma X.; Lu C.; Nagahawatte P.; Hedlund E.; Rusch M.; Pounds S.; Lin T.; Onar-Thomas A.; Huether R.; Kriwacki R.; Parker M.; Gupta P.; Becksfort J.; Wei L.; Mulder H. L.; Boggs K.; Vadodaria B.; Yergeau D.; Russell J. C.; Ochoa K.; Fulton R. S.; Fulton L. L.; Jones C.; Boop F. A.; Broniscer A.; Wetmore C.; Gajjar A.; Ding L.; Mardis E. R.; Wilson R. K.; Taylor M. R.; Downing J. R.; Ellison D. W.; Zhang J.; Baker S. J. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho D.; Taylor K. R.; Olaciregui N. G.; Molinari V.; Clarke M.; Mackay A.; Ruddle R.; Henley A.; Valenti M.; Hayes A.; Brandon A. D. H.; Eccles S. A.; Raynaud F.; Boudhar A.; Monje M.; Popov S.; Moore A. S.; Mora J.; Cruz O.; Vinci M.; Brennan P. E.; Bullock A. N.; Carcaboso A. M.; Jones C. ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Commun. Biol. 2019, 2, 156. 10.1038/s42003-019-0420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastas J. N.; Zee B. M.; Kalin J. H.; Kim M.; Guo R.; Alexandrescu S.; Blanco M. A.; Giera S.; Gillespie S. M.; Das J.; Wu M.; Nocco S.; Bonal D. M.; Nguyen Q.-D.; Suva M. L.; Bernstein B. E.; Alani R.; Golub T. R.; Cole P. A.; Filbin M. G.; Shi Y. Re-programing chromatin with a bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG. Cancer Cell 2019, 36, 528–544. 10.1016/j.ccell.2019.09.005. [DOI] [PubMed] [Google Scholar]
- Hopkins C. R. Inhibitors of the bone morphogenetic protein (BMP) signaling pathway: a patent review (2008-2015). Expert Opin. Ther. Pat. 2016, 26, 1115–1128. 10.1080/13543776.2016.1217330. [DOI] [PubMed] [Google Scholar]
- Cuny G. D.; Yu P. B.; Laha J. K.; Xing X.; Liu J.-F.; Lai C. S.; Deng D. Y.; Sachidanandan C.; Bloch K. D.; Peterson R. T. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4388–4392. 10.1016/j.bmcl.2008.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P. B.; Cuny G. D.; Mohedas A. H.; Bloch K. D.; Peterson R. T.. BMP Inhibitors and Methods of Use Thereof. WO2014138088A1, 2014.
- Sanvitale C. E.; Kerr G.; Chaikuad A.; Ramel M.-C.; Mohedas A. H.; Reichert S.; Wang Y.; Triffitt J. T.; Cuny G. D.; Yu P. B.; Hill C. S.; Bullock A. N. A new class of small molecule inhibitor of BMP signaling. PLoS One 2013, 8, e62721 10.1371/journal.pone.0062721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohedas A. H.; Wang Y.; Sanvitale C. E.; Canning P.; Choi S.; Xing X.; Bullock A. N.; Cuny G. D.; Yu P. B. Structure-activity relationship of 3,5-diaryl-2-aminopyridine ALK2 inhibitors reveals unaltered binding affinity for fibrodysplasia ossificans progressiva causing mutants. J. Med. Chem. 2014, 57, 7900–7915. 10.1021/jm501177w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P. B.; Cuny G. D.; Mohedas A. H.. Compositions and Methods for Inhibiting BMP. WO2015148654A1, 2015.
- Reinecke M.; Ruprecht B.; Poser S.; Wiechmann S.; Wilhelm M.; Heinzlmeir S.; Kuster B.; Médard G. Chemoproteomic selectivity profiling of PIKK and PI3K kinase inhibitors. ACS Chem. Biol. 2019, 14, 655–664. 10.1021/acschembio.8b01020. [DOI] [PubMed] [Google Scholar]
- Lahn M.; Herbertz S.; Sawyer J. S.; Stauber A. J.; Gueorguieva I.; Driscoll K. E.; Estrem S. T.; Cleverly A. L.; Desaiah D.; Guba S. C.; Benhadji K. A.; Slapak C. A. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des., Dev. Ther. 2015, 9, 4479–4499. 10.2147/dddt.s86621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unpublished results. Data can be viewed in presentations archived at https://m4kpharma.com/blog/ (accessed Apr. 1, 2020).
- Morgan M. R.; Roberts O. G.; Edwards A. M. Ideation and implementation of an open science drug discovery business model - M4K Pharma. Wellcome Open Res. 2018, 3, 154. 10.12688/wellcomeopenres.14947.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A.; Alfano I.; Kerr G.; Sanvitale C. E.; Boergermann J. H.; Triffitt J. T.; von Delft F.; Knapp S.; Knaus P.; Bullock A. N. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem. 2012, 287, 36990–36998. 10.1074/jbc.m112.365932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic Z. CNS drug design: balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- tPSA, pKa and cLogP values were calculated using ChemDraw Professional 19.0.
- Johnson T. W.; Richardson P. F.; Bailey S.; Brooun A.; Burke B. J.; Collins M. R.; Cui J. J.; Deal J. G.; Deng Y.-L.; Dinh D.; Engstrom L. D.; He M.; Hoffman J.; Hoffman R. L.; Huang Q.; Kania R. S.; Kath J. C.; Lam H.; Lam J. L.; Le P. T.; Lingardo L.; Liu W.; McTigue M.; Palmer C. L.; Sach N. W.; Smeal T.; Smith G. L.; Stewart A. E.; Timofeevski S.; Zhu H.; Zhu J.; Zou H. Y.; Edwards M. P. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 2014, 57, 4720–4744. 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Mohr P.; Stahl M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601–2611. 10.1021/jm100087s. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Johnson T. W.; Gallego R. A.; Edwards M. P. Lipophilic efficiency as an important metric in drug design. J. Med. Chem. 2018, 61, 6401–6420. 10.1021/acs.jmedchem.8b00077. [DOI] [PubMed] [Google Scholar]
- Sekimata K.; Sato T.; Sakai N.; Watanabe H.; Mishima-Tsumagari C.; Taguri T.; Matsumoto T.; Fujii Y.; Handa N.; Honma T.; Tanaka A.; Shirouzu M.; Yokoyama S.; Miyazono K.; Hashizume Y.; Koyama H. Bis-heteroaryl pyrazoles: identification of orally bioavailable inhibitors of activin receptor-like kinase-2 (R206H). Chem. Pharm. Bull. 2019, 67, 224–235. 10.1248/cpb.c18-00598. [DOI] [PubMed] [Google Scholar]
- Khalili F.; Henni A.; East A. L. L. pKa values of some piperazines at (298, 303, 313, and 323) K. J. Chem. Eng. Data 2009, 54, 2914–2917. 10.1021/je900005c. [DOI] [Google Scholar]
- Jiang J.-k.; Huang X.; Shamim K.; Patel P. R.; Lee A.; Wang A. Q.; Nguyen K.; Tawa G.; Cuny G. D.; Yu P. B.; Zheng W.; Xu X.; Sanderson P.; Huang W. Discovery of 3-(4-sulfamoylnaphthyl)pyrazolo[1,5-a]pyrimidines as potent and selective ALK2 inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 3356–3362. 10.1016/j.bmcl.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Ding C. Z.; Dragovich P.; Fauber B.; Gao Z.; Labadie S.; Lai K. W.; Purkey H. E.; Robarge K.; Wei B.; Zhou A.. Piperidine-dione Derivatives. WO2015140133A1, 2015.
- Mascitti V.; McClure K. F.; Munchhof M. J.; Robinson R. P. Jr.. 4-(5-Cyano-pyrazol-1-yl)-piperidine Derivatives as GPR 119 Modulators. WO2012069948A1, 2012.
- Zhan W.; Ji L.; Ge Z.-m.; Wang X.; Li R.-t. A continuous-flow synthesis of primary amides from hydrolysis of nitriles using hydrogen peroxide as oxidant. Tetrahedron 2018, 74, 1527–1532. 10.1016/j.tet.2018.02.017. [DOI] [Google Scholar]
- Whitehead A.; Ornoski O.; Raghavan S.; Berger R.; Garfunkle J.; Yang Z.; Ji G.; Jiang F.; Fu J.. Fused Pyrazine Derivatives Useful as Soluble Guanylate Cyclase Stimulators. WO2017200825A1, 2017.
- Balkovec J. M.; Bensen D. C.; Borchardt A.; Brady T. P.; Chen Z.; Lam T.; Tari L. W.. Small Molecule Inhibitors of Dihydrofolate Reductase. WO2016201219A1, 2016.
- Pouzet P.; Hoenke C.; Nickolaus P.; Goeggel R.; Fox T.; Fiegen D.; Klinder K.. Novel Piperazino-dihydrothienopyrimidine Derivatives. WO2009050236A1, 2009.
- Guerin D. J.; Bair K. W.; Caravella J. A.; Ioannidis S.; Lancia D. R. Jr; Li H.; Mischke S.; Ng P. Y.; Richard D.; Schiller S. E. R.; Shelekhin T.; Wang Z.. Thienopyridine Carboxamides as Ubiquitin-specific Protease Inhibitors. WO2017139778A1, 2017.
- Winter G. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010, 43, 186–190. 10.1107/s0021889809045701. [DOI] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/s0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/s0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B. III; Snoeyink J.; Richardson J. S.; Richardson D. C. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.