

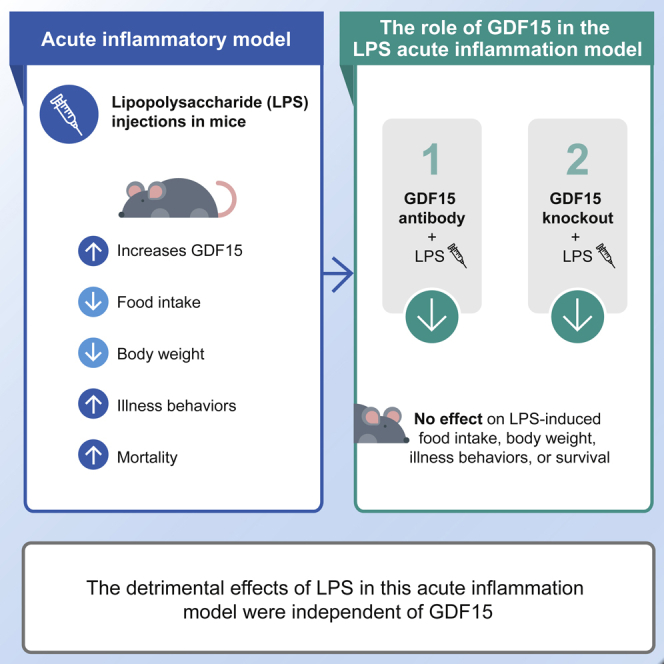
Summary
Growth differentiation factor 15 (GDF15) causes anorexia and weight loss in animal models, and higher circulating levels are associated with cachexia and reduced survival in cancer and other chronic diseases such as sepsis. To investigate the role of sepsis-induced GDF15, we examined whether GDF15 neutralization via a validated and highly potent monoclonal antibody, mAB2, modulates lipopolysaccharide (LPS)-induced anorexia, weight loss, and mortality in rodents. LPS injection transiently increased circulating GDF15 in wild-type mice, decreased food intake and body weight, and increased illness behavior and mortality at a high dose. GDF15 neutralization with mAB2 did not prevent or exacerbate any of the effects of LPS. Similarly, in GDF15 knockout mice, the LPS effect on appetite and survival was comparable with that observed in wild-type controls. Therefore, effective inhibition of circulating active GDF15 via an antibody or via gene knockout demonstrated that survival in the LPS acute inflammation model was independent of GDF15.
Subject area: Cell Biology, Physiology
Graphical abstract
Highlights
-
•
A novel highly potent anti-GDF15 antibody, mAB2, was characterized
-
•
LPS transiently increased GDF15 production in mice
-
•
mAB2 did not prevent or exacerbate the effects of LPS on anorexia or survival
-
•
GDF15 knockout mice showed comparable effects of LPS with wild-type controls
Cell biology; Physiology;
Introduction
Growth differentiation factor 15 (GDF15) is a cytokine that causes anorexia and weight loss in preclinical models including mice, rats, and nonhuman primates (Emmerson et al., 2017; Hsu et al., 2017; Johnen et al., 2007; Mullican et al., 2017; Tsai et al., 2013; Yang et al., 2017) and is associated with weight loss in patients with cancer (Lerner et al., 2015, 2016a). Furthermore, there are numerous reports highlighting elevated GDF15 as a biomarker of poor survival in both acute and chronic illnesses including sepsis, cancer, heart failure, and chronic obstructive pulmonary disease (Kempf et al., 2007; Anand et al., 2010; Lerner et al., 2015; Li et al., 2017; Husebo et al., 2017; Santos et al., 2020). GDF15 regulation of energy balance requires activation of the glial-cell-derived neurotrophic factor receptor alpha-like (GFRAL), which is very narrowly expressed in only the area postrema (AP) and nucleus of the solitary tract of the hindbrain (Emmerson et al., 2017; Hsu et al., 2017; Mullican et al., 2017; Yang et al., 2017; Tsai et al., 2019). In mouse tumor models, circulating GDF15 is chronically elevated, and GDF15 inhibition has been demonstrated to reverse anorexia and weight loss (Lerner et al., 2016b; Hsu et al., 2017).
Circulating GDF15 is also elevated in acute inflammatory models of infection and sepsis including lipopolysaccharide (LPS) and polyinosinic:polycytidylic acid injection and cecal ligation and puncture (CLP) (Luan et al., 2019; Santos et al., 2020). Different from the tumor-bearing mice, the pharmacokinetics of circulating GDF15 is variable and transient in these acute models (Luan et al., 2019; Santos et al., 2020). Whether GDF15 is a key mediator of energy balance and survival in infection and sepsis mouse models is controversial, based on published reports (Li et al., 2018; Luan et al., 2019; Santos et al., 2020; Pereiro et al., 2020). Mice administered recombinant GDF15 had greater survival in the LPS (Li et al., 2018; Luan et al., 2019) and CLP models (Luan et al., 2019), and GDF15 overexpression prevented LPS-induced mortality in zebra fish (Pereiro et al., 2020). Consistent with a protective effect of GDF15, treatment with an anti-GDF15 antibody increased mortality without impacting food intake in mice; however, it is not clear whether the antihuman GDF15 antibody used in the study was effective in neutralizing active mouse GDF15 given the lack of reported efficacy to block anorexia or weight loss (Luan et al., 2019). Furthermore, in contrast to the anti-GDF15 antibody experiments, GDF15 knockout (KO) mice had increased survival in the CLP mouse sepsis model (Santos et al., 2020). Taken together, the role of GDF15 in the acute inflammatory setting is unclear. Therefore, in this study, we carefully characterized a selective and potent anti-GDF15 antibody, mAB2, and examined whether GDF15 neutralization prevents LPS-induced anorexia, weight loss, and mortality in mice.
Results
Characterization of GDF15 neutralizing antibody (mAB2)
A GDF15-specific antibody (mAB2) was generated at Pfizer. Surface plasmon resonance (SPR) analysis was performed to measure the binding of mAB2 to mouse and human GDF15. mAB2 was able to bind both mouse and human GDF15 with high affinity as demonstrated by the apparent KD of 11.8 pM and <10 pM, respectively, and similar ka and kd profiles (Table 1). The analysis used a CM4 sensor chip with immobilized Fc-GDF15 at 38-75 resonance unit Rmax range and full-length immunoglobulin G (IgG) as the analyte. This experimental condition measuring dimer-dimer interaction was likely impacted by avidity. To determine the true KD against mouse GDF15 for in vivo purpose, further SPR study was carried out using optimized experimental conditions with the C1 sensor chip and low immobilized Rmax minimizing avidity and was fitted to a Langmuir 1:1 interaction model with high confidence (Figure 1A). mAB2 was observed to be a fast on/slow off antibody with ka of 5.53 × 106 1/MS and kd of 1.15 × 10−3 1/s and to have strong binding affinity to mouse GDF15 with a true KD of 225.2 pM (Table 2). To test the inhibitory activity of mAB2, we established Chinese hamster ovary-K1 (CHOK1) cells with stable expression of both human GFRAL and human rearranged during transfection (RET). These cells are responsive to GDF15 stimulation as visualized by extracellular signal-regulated kinase (ERK) activation in a dose-dependent manner with an EC50 of 23 pM (Figure 1B). Inhibitory activity of mAB2 was then tested against 150 pM GDF15 (EC80). mAB2 was able to fully inhibit GDF15-mediated ERK phosphorylation with an IC50 of 0.4 nM (Figure 1C). The inhibition activity was specific to GDF15 as mAB2 had no effect on ERK phosphorylation in absence of GDF15 treatment. To evaluate mAB2 in vivo, we generated an adeno-associated virus (AAV) encoding mouse GDF15. Mice dosed with AAV8-mGDF15 typically presented with a 13-fold increase in plasma GDF15 levels 15 days after dosing, going from 133 pg/mL to 1.84 ng/mL (Figure 1D). The elevation of GDF15 was associated with 10% body weight loss achieved 11 days afer AAV dosing. At that time, treating mice with mAB2 (10 mg/kg Q3D) could rapidly reverse the weight loss induced by GDF15 as opposed to mice receiving the control IgG (Figure 1E). To further validate inhibition of GDF15 bioactivity by mAB2, we performed c-fos staining in the AP of the hindbrain where GFRAL-expressing neurons are located (Figure 1F). Mice treated with AAV8-mGDF15 + control IgG had significantly elevated mean c-fos + cell count in the AP region of the hindbrain in comparison with AAV8 control animals. Mice treated with AAV8-mGDF15 + mAB2 had mean c-fos + cell count levels decreased to similar levels as AAV8 control animals, which was significantly less than the AAV8-mGDF15 + control IgG-treated arm (Figure 1G). Our data demonstrate that mAB2 is a potent GDF15-neutralizing antibody.
Table 1.
Surface plasmon resonance analysis using CM4 sensor chip of mAB2 against mouse and human GDF15
| Source | Sensor chip | Analyte | Ligand | ka (1/Ms) | kd (1/s) | Rmax (RU) | Apparent KD (M) | n |
|---|---|---|---|---|---|---|---|---|
| Pfizer | CM4 | mAB2 | mFc-rmGDF15 | 3.58 × 106 | 4.14 × 10−5 | 50.93 | 1.18 × 10−11 | 4 |
| Pfizer | CM4 | mAB2 | mFc-rhGDF15 | 4.72 × 106 | <5.0 × 10−5 | 50.67 | <1.0 × 10−11 | 3 |
Figure 1.

Characterization of GDF15-neutralizing antibody mAB2
(A) Representative Biacore SPR sensorgram plot showing the kinetic binding interaction between mAB2 and mouse GDF15. The colored lines are the double referenced sensorgram data for each mAB2 injection with the overlay of black fit lines generated using a Langmuir 1:1 model and Biacore Insight Evaluation software.
(B) Activation curve for phospho-ERK using dose response of recombinant GDF15 in GFRAL/RET stable cell line.
(C) Inhibition curve for phospho-ERK using dose response of mAB2 against 150 pM of GDF15 (EC80).
(D) Elevation of GDF15 plasma levels in mice after treatment with AAV8-encoding mouse GDF15. ∗∗∗ p value < 0.001 vs. AAV8-control. Data were analyzed using Welch's heteroscedastic F test.
(E) Reversal of GDF15 induced body weight loss with mAB2. ∗∗∗∗ p value < 0.0001 vs. AAV8-control; ^^^^ p value < 0.0001 vs. AAV8-mGDF15 + mAB2 at days 15-18. Data were analyzed using longitudinal mixed effects ANOVA.
(F) Representative images of neuronal activity visualized by c-fos staining in the AP after treatment with AAV8-GDF15 and neutralization with mAB2.
(G) Quantification of c-fos-positive neurons in the AP after AAV8-GDF15 treatment and 24 h after neutralization with mAB2. N = 10 per group. ∗∗∗∗ p value < 0.0001 vs. AAV8-control; ˆˆˆˆ p value < 0.0001 vs. AAV8-mGD15 + mAB2. Data were analyzed using a mixed effects analysis of covariance. Data represented as least squares mean ± SE. Data in (B–E) represented as mean ± SEM.
AAV, adeno-associated virus; ANOVA, analysis of variance; AP, area postrema; ERK, extracellular signal-regulated kinase; GDF15, growth differentiation factor 15; GFRAL, glial cell-derived neurotrophic factor receptor alpha-like; HTRF, homogeneous time-resolved fluorescence; IgG, immunoglobulin G; RET, rearranged during transfection; RU, resonance units; s, seconds; SE, standard error; SEM, standard error of the mean.
Table 2.
Surface plasmon resonance analysis using C1 sensor chip of mAB2 against mouse GDF15
| Source | Sensor chip | Analyte | Ligand | ka (1/Ms) | kd (1/s) | Rmax (RU) | KD (M) | n |
|---|---|---|---|---|---|---|---|---|
| Pfizer | C1 | mAB2 | hFc-rmGDF15 | 5.53 × 106 | 1.15 × 10−3 | 4.87 | 2.25 × 10-10 | 5 |
GDF15-neutralizing antibody mAB2 acutely reverses weight loss and anorexia induced by recombinant GDF15 in mice
To test the efficacy of mAB2 in an acute setting, we first validated a recombinant Fc-hGDF15 protein. In vitro, Fc-hGDF15 was able to activate GFRAL/RET signaling (Figure 2A). In vivo, a single subcutaneous injection of 0.1 mg/kg Fc-hGDF15 resulted in a robust decrease in body weight and food intake over 24 h (Figures 2B and 2C). To test mAB2 in an acute setting, fed and fasted mice were treated with Fc-hGDF15 and simultaneously injected with either mAB2 or IgG control, and food was subsequently reintroduced to assess food intake and physiological postprandial biomarkers (Figure 2D). Food consumption in Fc-GDF15-dosed mice was reduced as soon as 2 h after dosing compared with the vehicle-treated group, and mAB2 treatment was able to fully reverse the anorectic effect of Fc-GDF15 (Figure 2E). Four hours after prandial, insulin and triglycerides were induced and nonesterified fatty acids suppressed by food intake in control mice. In agreement with the food intake data, postprandial responses were strongly blunted by GDF15 in control IgG-treated mice but restored in mAB2-treated mice (Figures 2F–2H). Interestingly, GDF15 was not able to directly modulate plasma insulin, triglycerides, or fatty acids as demonstrated by the absence of response to GDF15 treatment or neutralization when food was not reintroduced. Targeted lipidomic analysis of triglycerides further confirmed that no triglyceride species were regulated by GDF15 in absence of food intake (Figure 2I). These data demonstrate that mAB2 is a potent tool to acutely inhibit GDF15 in vivo and therefore can be used to understand the contribution of GDF15 in the LPS-induced sepsis.
Figure 2.

Validation of mAB2 for acute inhibition of GDF15 in vivo
(A) Activation curve for phospho-ERK using dose response of Fc-GDF15 in GFRAL/RET stable cell line.
(B–C) 24-h body weight (B) and food intake (C) in mice injected with Fc-GDF15 (0.1 mg/kg). ∗∗∗∗ p value < 0.0001 vs. vehicle. Data were analyzed using an unpaired two-tailed t-test.
(D) Schematic representation of the two study designs to test acute inhibition of Fc-hGDF15 by mAB2.
(E) Inhibition of food intake after subcutaneous injection of Fc-GDF15 (0.1 mg/kg) and reversal by cotreatment with mAB2 (10 mg/kg). ∗ p value < 0.004 (2 h), ∗∗∗ p value < 0.0009 (4 h) vs. vehicle + IgG; ˆˆ p value < 0.003 (4 h) vs. Fc-GDF15 + mAB2. Data were analyzed using a longitudinal mixed-effects ANOVA.
(F) Plasma insulin. ∗∗∗∗ p value < 0.0002 vs. vehicle + IgG; ˆ p value < 0.05 vs. Fc-GDF15 + mAB2. (G) NEFA, ∗∗∗ p value < 0.0009 vs. vehicle + IgG; ˆˆ p value < 0.007 vs. Fc-GDF15 + mAB2. (H) Triglycerides, ∗∗∗∗ p value < 0.0001 vs. vehicle + IgG; ˆˆ p value < 0.003 vs. Fc-GDF15 + mAB2, (F–H) Data were analyzed using ANOVA. (I) Heatmap representing fold change in plasma lipid levels when compared with baseline. Data represented as mean ± SEM. N = 10 per group.
ANOVA, analysis of variance; BW, body weight; Concn, concentration; ERK, extracellular signal-regulated kinase; FW, food weight; GDF15, growth differentiation factor 15; GFRAL, glial-cell-derived neurotrophic factor receptor alpha-like; IgG, immunoglobulin G; mpk, mg/kg; NEFA, nonesterified fatty acids; RET, rearranged during transfection; SD, standard deviation; SEM, standard error of the mean; TAG, triglycerides.
Neutralization of GDF15 does not modulate weight loss or anorexia induced with a low dose of LPS in mice
We first assessed if GDF15 was modulated in moderate sepsis induced with a low dose of LPS. Using a 0.1 mg/kg dose of LPS in ad libitum fed conditions, we observed a rapid and transient elevation of GDF15, peaking at 600 pg/mL within 2 h before gradually returning to normal levels within 24 h (Figure 3A), similar to the inflammatory marker KCGRO (Figure 3B). As expected, mice treated with LPS exhibited a 12% reduction in body weight and a 70% reduction in cumulative food intake (4.9 g vs. 1.5 g in vehicle vs. LPS-treated mice, respectively) over 24 h. We then decided to assess the contribution of GDF15 in moderate sepsis induced by a low dose of LPS with or without mAB2 treatment (Figure 3C). Treatment with mAB2 had no significant impact on LPS-induced body weight loss or anorexia (Figures 3D and 3E). We then performed a similar experiment in fasted conditions to increase the potential effect size of LPS on body weight and food intake. As expected under fasted conditions, LPS induced a more dramatic reduction of body weight (20%) and a similar reduction in cumulative food intake (68%) (9.9 g vs. 3.2 g in vehicle vs. LPS-treated mice, respectively) over 24 h. However, treatment with mAB2 resulted in only a marginal increase in body weight and food intake (Figures 3F and 3G). Taken together, these data suggest that GDF15 does not play a significant role in LPS-induced weight loss and anorexia in mice.
Figure 3.

Neutralization of GDF15 does not modulate weight loss and anorexia induced by a low dose of LPS
(A) GDF15 and (B) KCGRO plasma levels in mice after intraperitoneal injection of a 0.1 mg/kg LPS dose. (C) Schematic representation of the 2 study designs. Changes in (D) body weight and (E) cumulative food intake in fed mice after a single dose of LPS (0.1 mg/kg) and treatment with either GDF15-neutralizing mAB2 or control IgG. Changes in (F) body weight and (G) cumulative food intake in fasted mice after single dose of LPS (0.1 mg/kg) and treatment with either GDF15 neutralizing mAB2 or control IgG. ∗∗∗∗ p value < 0.0001 vs. LPS + IgG or LPS + mAB2; ^ p value < 0.05 vs. LPS + IgG. Food intake (fasted mice) in the LPS + mAB2 group tended to be higher than the LPS + IgG (p = 0.052). Data in (D) were analyzed using Welch's heteroscedastic F test. Data in (E, F, G) were analyzed using ANOVA with Tukey’s multiple comparison adjustment. Data represented as mean ± SEM. N = 10 per group.
BW, body weight; FW, food weight; GDF15, growth differentiation factor 15; IgG, immunoglobulin G; LPS, lipopolysaccharide; SEM, standard error of the mean.
Neutralization of GDF15 has a marginal effect on weight loss but not anorexia induced with a low dose of LPS in rats
To understand if these results were specific to mice, we decided to perform a similar experiment in rats. To validate the bioactivity of mAB2 against rat GDF15, we first performed an efficacy study using the Yoshida sarcoma model. Tumor-bearing rats typically presented a 16% body weight loss (Figure 4A) with anorexia (Figure 4B) associated with a 10-fold increase in plasma GDF15 levels within 17 days after tumor cell injection, going from 100 pg/mL to 1.06 ng/mL (Figure 4C). In a remission experiment, when tumor-bearing rats achieved 10% body weight loss, they were treated with either control IgG or mAB2 at 10 mg/kg every 3 days. As shown in Figure 4D, neutralization of GDF15 with mAB2 was able to reverse body weight loss as opposed to control IgG, demonstrating that mAB2 was efficacious against rat GDF15. We then tested the consequence of neutralization of GDF15 with mAB2 in LPS-treated rats. A single dose of 0.25 mg/kg of LPS led to comparable transient rise in plasma GDF15 over 24 h, reaching a maximum of 2 ng/mL 2 h postdose concomitant to an increase in KCGRO (Figures 4E and 4F). We then assessed body weight and food intake over 24 h after 0.25 mg/kg LPS injection in rats that were treated with or without mAB2. The LPS + IgG and LPS + mAB2 groups lost 9% and 7% of body weight, respectively, over 24 h when compared with controls. Although marginal, the improvement of body weight loss in the mAB2-treated rats when compared with the IgG-treated rats was significant (Figure 4G). Similar to our findings in mice, control IgG-treated rats also had a rapid reduction in food intake in response to LPS which was not impacted by mAB2 treatment (Figure 4H). Overall, these results indicated that GDF15 does not play a crucial role in anorexia during moderate sepsis in mice or rats.
Figure 4.

Neutralization of GDF15 marginally modulates weight loss but not anorexia induced by a low dose of LPS in rats
(A) Body weight, (B) food intake, and (C) plasma GDF15 of nontumor-bearing rats and rats injected with Yoshida tumor cells. ∗∗∗∗ p value < 0.0001 vs. NTB. Data were analyzed using Welch's heteroscedastic F test. (D) Body weights of Yoshida-tumor-bearing rats injected with mAB2 and control IgG. ∗∗∗ p value < 0.001 vs. NTB; ^^^ p value < 0.001 vs. Yoshida + mAB2. Data were analyzed using longitudinal mixed effects ANOVA. (E) Plasma GDF15 levels in rats IP injected with 0.25 mg/kg LPS. (F) Plasma KCGRO levels in rats dosed with 0.25 mg/kg LPS at 3 h after dose. ∗∗∗∗ p value < 0.0001 vs. Vehicle; data were analyzed using an unpaired two-tailed t-test. (G) Body weight and cumulative food intake post LPS dose of 0.25 mg/kg. ∗∗∗∗ p value < 0.0001 vs. Vehicle; ˆˆ p value < 0.004 vs. LPS + mAB2; data were analyzed using ANOVA. (H) Cumulative food intake for rats was measured at 1 h, 2 h, 4 h, and 24 h. ∗ p value < 0.05 vs. LPS + IgG or LPS + mAB2. Data were analyzed using Welch's heteroscedastic F test at 24h timepoint. Data represented as mean ± SEM. N = 12 per group.
GDF15, growth differentiation factor 15; IgG, immunoglobulin G; IP, intraperitoneal; LPS, lipopolysaccharide; NTB, nontumor-bearing; SEM, standard error of the mean.
Neutralization of GDF15 does not modulate weight loss, anorexia, or mortality induced by sepsis with a sublethal dose of LPS in mice
A recent report suggesting a role for GDF15 in survival in response to a sublethal dose of LPS prompted us to test if our antibody would show an effect under these more severe sepsis conditions (Luan et al., 2019). At this dose, LPS induction of GDF15 was more robust and durable than with the previous low-dose LPS treatment, reaching a higher maximum concentration (6.34 ng/mL within 90 min) and remained significantly elevated above normal at 0.95 ng/mL after 48 h (Figure 5A). To test the effect of GDF15 inhibition in severe sepsis, mice were pretreated with mAB2 or IgG control antibody 16 h before LPS injection (Figure 5B). Forty-eight hours after LPS injection, there was an average body weight loss of 14% and 16.5% in mice treated with IgG control or mAB2, respectively. This difference in body weight reduction within LPS groups was not statistically significant. By the end of the study, both IgG control and mAB2-treated groups recovered their baseline body weights (Figure 5C). Food intake was completely suppressed for 24 h in the LPS-treated groups compared with vehicle control and then increased over time consistent with body weight. Food intake was similar between IgG control and mAB2-treated groups, although there was a moderate difference in food intake at the 72 h and 96 h time points where the LPS + IgG group recovered faster than the LPS + mAB2 group (Figure 5D). Control animals maintained a normal health condition throughout the study, whereas in the LPS-treated groups, the worst health scores were observed at 6 h aftrer LPS injection, and mice were fully recovered by the 72-h time point with no differences between the IgG control and mAB2 groups (Figure 5E). As expected, LPS treatment resulted in a modest decrease in survival compared with the vehicle control group, and the effect of LPS was similar in magnitude to that observed in the study by Luan et al., 2019 (Figure 5F). Consistent with body weight and health condition, no differences in mortality were observed between the LPS + IgG and LPS + mAB2 treatment groups (Figure 5F). These results suggest that GDF15 does not play a key role in the regulation of body weight, food intake, and health condition during severe sepsis induced with sublethal LPS dose, and GDF15 neutralization did not increase mortality rate under these conditions.
Figure 5.

Neutralization of GDF15 does not modulate weight loss, anorexia, or mortality induced with a sublethal dose of LPS
(A) Plasma GDF15 levels in mice after 5 mg/kg LPS sublethal dose (n = 5 per time point).
(B) Study schematic.
(C) Body weight after LPS and IgG control or mAB2 injection. Body weight is represented as % of the day 0 body weight.
(D) Cumulative food intake after after LPS and IgG control or mAB2 injection.
(E) Health score representing overall condition of animals by their appearance, natural behavior, provoked behavior, and body condition scores after LPS and IgG control or mAB2 injection.
(F) Kaplan-Meier survival curve indicating % survival after LPS injection with IgG control or mAB2 treatments. log rank test p = 0.6. ∗ p value < 0.05, ∗∗∗ < 0.001 vs. LPS + IgG; ^ p value < 0.05, ^^ p value < 0.01, ^^^ p value < 0.001 vs. LPS + mAB2. Data in C and D were analyzed using a longitudinal mixed-effects ANOVA. Failure time data were compared between treatment groups with Kaplan-Meier analysis using the log rank test. Data represented as mean ± SEM. N = 13 per group.
AU, arbitrary unit; BW, body weight; FI, food intake; GDF15, growth differentiation factor 15; HS, health score; IgG, immunoglobulin G; LPS, lipopolysaccharide; PBS, phosphate-buffered saline; SEM, standard error of the mean.
Transgenic mice deficient for GDF15 are not protected against weight loss, anorexia, or mortality induced by sepsis with a sublethal dose of LPS
To understand if long-term deficiency in GDF15 was necessary to influence the progression of detrimental effects including mortality induced by a sublethal dose of LPS, we administered the same sublethal dose of LPS (5 mg/kg) to GDF15 KO mice. In response to LPS, both wild-type (WT) and GDF15 KO mice presented a maximal reduction in body weight 48 h after dosing when compared with saline controls (Figure 6A). There was no difference of LPS-induced body weight loss between WT and GDF15 KO mice at any time point. Similarly, the LPS challenge reduced food intake within 24 h for up to 72 h when compared with saline-injected mice, with comparable food intake between WT and GDF15 KO mice (Figure 6B). Within 6 h after LPS administration, LPS-treated mice exhibited a reduction of health score and fully recovered by 96 h (Figure 6C). Finally, WT and GDF15 KO mice followed a similar survival trajectory with most deaths occurring at 72 and 96 h after LPS administration. Fifty-eight percent and 75% of WT and KO mice survived by the 72-h time point, respectively, with a total of 50% and 77% by 96 h, respectively (Figure 6D). These data demonstrate that chronic inhibition of GDF15 does not affect the impact of severe sepsis on body weight, food intake, and health condition induced by a sublethal dose of LPS and does not increase mortality rate.
Figure 6.

GDF15 deficiency does not modulate weight loss, anorexia, or mortality induced with a sublethal dose of LPS
(A) Body weight and (B) cumulative food intake after 5 mg/kg LPS injection in WT and GDF15 KO mice. ∗ p value < 0.05, ∗∗ p value < 0.01, ∗∗∗ p value < 0.001 vs. WT saline; ^ p value < 0.05, ^^ p value < 0.01, ^^^ p value < 0.001 vs. KO saline; # p value < 0.05 vs. KO LPS. Data were analyzed using a longitudinal mixed-effects ANOVA. (C) Health score representing overall condition of WT and GDF15 KO mice after 5 mg/kg LPS dose based on their appearance, natural behavior, provoked behavior, and body condition scores. (D) Kaplan-Meier survival curve indicating % survival in WT and GDF15 KO mice after LPS injection. ∗ p value < 0.05 vs. WT saline. Survival times are statistically significantly different between treatment groups p < 0.014. Failure time data were compared between treatment groups with Kaplan-Meier analysis using the log rank test. Data represented as mean ± SEM. N = 6–12 per group (n = 12 WT saline, n = 12 WT LPS, n = 6 KO saline, n = 12 KO LPS).
GDF15, growth differentiation factor 15; KO, knockout; LPS, lipopolysaccharide; SEM, standard error of the mean; WT, wild-type.
Discussion
In this paper, we report for the first time the characterization of a highly potent GDF15-neutralizing antibody. This antibody was validated for target engagement in both in vitro and in vivo conditions. Using stringent SPR conditions to determine true affinity and a GFRAL/RET bioactivity assay, our analysis of binding affinities, kinetic parameters, and inhibitory activity demonstrated that mAB2 is a strong binder and potent inhibitor of human and mouse GDF15. In multiple preclinical species, mAB2 could reverse the anorectic actions of GDF15 induced by the use of recombinant protein, AAV-mediated expression, or tumor models. Finally, we could demonstrate that mAB2 reversed the GDF15 mediated activation of neurons in the AP where GFRAL, the only known GDF15 receptor, is expressed.
This thorough characterization and validation of our antibody gave us high confidence in the quality and potency of mAB2 to inhibit GDF15 and allowed us to assess the role of GDF15 in mediating anorexia and survival in LPS-induced sepsis. Despite GDF15 being clearly increased in a dose-dependent manner by LPS injection in both mice and rats fed a standard chow diet, inhibition of GDF15, both acutely with our neutralizing antibody or chronically through genetic ablation, neither improved nor aggravated weight loss and anorexia or increased mortality. These data do not support a role for GDF15 in the regulation of sepsis tolerance.
This study is one of several testing the hypothesis that GDF15 may be an important mediator of the effects of LPS and/or sepsis. Similar to our data, they all demonstrated that GDF15 is rapidly induced during sepsis (Luan et al., 2019; Santos et al., 2020; Pereiro et al., 2020). Also in agreement with the current data, they did not observe differences in the anorectic response to similar doses of LPS used here when mice were treated with a GDF15-neutralizing antibody (Luan et al., 2019). Collectively these data make a strong case that elevated GDF15 is not necessary for the anorectic response to these challenges. Both LPS and CLP induce a wide range of cytokine responses. Many of these cytokines have been reported to be anorectic at least under some conditions. We conclude that the overall milieu that accompanies LPS and CLP likely mediates the potent effects on appetite and that any one factor, including GDF15, may not be sufficient.
On the key end point of survival, treatment with a reported GDF15-neutralizing antibody led to increased death in response to LPS and CLP (Luan et al., 2019). These results imply a beneficial effect of GDF15 to mitigate deleterious effects of infection. However, in both the GDF15 KO model and the administration of a well-validated neutralizing antibody in our study, there was no effect of neutralizing GDF15 to alter survival or other measures of deleterious effects of LPS. The lack of effect is unlikely owing to different mortality rates in the control groups across studies; we intentionally matched our LPS-induced mortality rate (approximately 10%–20%) with that reported by Luan et al. (2019) to allow a sufficient window to test the hypothesis of increased mortality with mAB2 treatment and to provide a valid comparison with the study by Luan et al., 2019.
One potential explanation for the differential outcomes observed between both GDF15-neutralizing antibody studies could be differences in their binding kinetics. Comparing our SPR data with the published SPR data of the antibody used in Luan et al., 2019 (Patent ID: WO2014100689A1, table 27), even in the presence of likely avidity effect for both measurements, it appears that the greatest difference is not on the apparent binding affinities (KD), but in the on-rates and off-rates against recombinant mouse GDF15. The expected accumulation of circulating GDF15 on dosing of a neutralizing antibody, in part owing to the delay in clearance of the antibody-bound GDF15 complex, could become problematic if the antibody used does not offer prolonged target coverage. Collectively, these data suggest that mAB2 used in the present study would have greater target occupancy against mouse GDF15 and therefore could explain the differences in treatment outcomes.
Finally, GDF15 has been proposed to be a regulator of systemic energy homeostasis. As such, GDF15 has been shown to promote lipolysis in humans (Laurens et al., 2020) or to regulate liver triglycerides secretion upon adrenergic simulation (Luan et al., 2019). Our data bring interesting observations in that regard. First, it appears that GDF15 is not a direct regulator of lipolysis as GDF15 did not modulate plasma nonesterified fatty acid levels in fasted mice but was able to prevent suppression of lipolysis through its anorectic action. Similarly, elevation of GDF15 had no direct effects on plasma triglycerides because not a single triglyceride species measured in a targeted lipidomic analysis was found to be modulated by GDF15 in mice without access to food. However, in ab libitum fed mice, elevation of GDF15 did have an inhibitory action on plasma triglycerides consistent with the reduction of food intake.
In conclusion, systemic inflammatory stimuli cause anorexia and weight loss by disrupting the physiological regulation of energy balance. This is achieved by induction of a cytokine storm that comprised multiple redundant signals including GDF15. Several groups have reported the involvement of circulating and brain cytokines in the LPS model (Amiot et al., 1997; Burfeind et al., 2018; Sparkman et al., 2006; Suzuki et al., 2002), any of which could be key drivers mediating the effects of LPS. Our data establish that elevation of circulating GDF15 levels is not necessary to drive the multiple detrimental effects associated with the LPS acute sepsis model.
Limitations of the study
Although we did not specifically measure the pharmacokinetics of mAB2, which could be considered a limitation, we were able to confidently select a dose of mAB2 based on the pharmacokinetic profile of a similar antibody targeting GDF15 (see STAR Methods). Furthermore, we leveraged in vivo data as an indication of its pharmacokinetic profile. For example, in WT mice, we induced weight loss with Fc-GDF15 and then administered a single dose of mAB2 (10 mg/kg) and observed an increase in body weight that was sustained for 6 days, indicating a single dose of mAB2 can cover the entire duration of the LPS experiments reported here. In addition, we measured plasma GDF15 in each rodent study to ensure that the level did not exceed the maximum concentration that this dose of mAB2 was estimated to neutralize.
In the rat study, neutralization of GDF15 with mAB2 was associated with a small, but significant, attenuation of weight loss induced by LPS. A corresponding significant change in food intake was not observed, suggesting a possible effect of mAB2 on energy expenditure. Unfortunately, we do not have a full body composition assessment to understand if the body weight change is attributed to fat or fat-free mass or total body water. Furthermore, although food intake was not statistically different between the LPS + IgG and LPS + mAB2 groups, it is possible that a small change below our limit of detection, coupled with expected increases in water consumption, could be driving the body weight change. We believe, given the small differences and short-term 24-h period of evaluation, that this could offer an alternative explanation to changes in energy expenditure. Further evaluation of this effect would require a longer study duration.
Finally, conclusions concerning the effects of GDF15 on survival are limited by the acute nature of the model (mice are resistant to multiple LPS injections) and the relatively low mortality rates (10–20%) after sublethal injection of LPS. This was intentional to allow optimal testing of the hypothesis that mAB2 or GDF15 KO led to increased mortality. Whether the neutralization of GDF15 can in contrast enhance survival, by using sepsis models with higher mortality rates or in a chronic setting, was not evaluated.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GDF15 mAB (mAB2) | Pfizer (reagent will not be shared due to commercialization consideration) | N/A |
| IgG mAB control | Pfizer | N/A |
| Rabbit anti-c-Fos IgG (FD Neurotechnologies) | Santa Cruz Biotechnology | Cat. # sc-52 |
| Chemicals, peptides, and recombinant proteins | ||
| AAV8-mouse control | Lake Pharma | N/A |
| AAV8-mouse GDF15 | Lake Pharma | N/A |
| Lipopolysaccharide – Sepsis mice studies | Sigma-Aldrich | Cat# L2880, Lot # 059M4031V |
| Lyophilized lipopolysaccharide – GDF15-/- studies | Sigma-Aldrich | Cat# L4391-1MG lot: 088M4067V |
| Lipopolysaccharide – Rat studies | Sigma-Aldrich | Cat# L4391 Lot 067M4036V |
| Methylene chloride, Optima | Fisher Scientific | D151-4 |
| Isopropanol, Optima LC-MS grade | Fisher Scientific | A461-4 |
| Methanol, Optima LC-MS grade | Fisher Scientific | A456-4 |
| Acetonitrile, Optima LC-MS grade | Fisher Scientific | A955-4 |
| Water, Optima LC-MS grade | Fisher Scientific | W64 |
| Formic Acid, Optima LC-MS grade | Fisher Scientific | A117-50 |
| Glyceryl triheptadecanoate | Sigma-Aldrich | T2151 |
| 1,2-Dinonadecanoin | Nu-Chek Prep | D-166 |
| Cholesteryl heptadecanoate | Nu-Chek Prep | CH-816 |
| 1,2-Dilauroyl-sn-glycero-3-phosphocholine | Avanti Polar Lipids | 850335P |
| 1-Heptadecanoyl-2-hydroxy-sn-glycero-3-phosphocholine | Avanti Polar Lipids | 855676P |
| N-Heptadecanoyl-D-erythro-sphingosine | Avanti Polar Lipids | 860517P |
| Ammonium formate, Optima LC-MS grade | Fisher Scientific | A11550 |
| Critical commercial assays | ||
| Mouse /Rat GDF15 Quantikine ELISA kit | R&D Systems | Cat #MCD150 |
| Mouse CXCL1/KC Quantikine ELISA kit | R&D Systems | Cat #MKC00B |
| Mouse Insulin ELISA | Alpco | Cat #80-INSMS-E01 |
| Proinflammatory Panel 2 (rat) kit | MSD | Cat #K15059D |
| Experimental models: cell lines | ||
| Yoshida cell line | Cell Line Services | #RY (ham) Yoshida Sarcoma |
| Experimental models: organisms/strains | ||
| C57BL/6J mice | Jackson Laboratories | #664 |
| GDF-15 knockout mice | University of Michigan | N/A |
| CD© (Sprague Dawley) IGS Rat Crl:CD(SD) | Charles River Laboratories | #001 |
| RNU Nude Rat - Crl:NIH-Foxn1rnu | Charles River Laboratories | #316 |
| Recombinant DNA | ||
| Mouse Fc-tagged mouse recombinant GDF15 (Fc-GDF15) | Pfizer Inc | N/A |
| Human Fc-tagged mouse recombinant GDF15 (Fc-GDF15) | Pfizer Inc | N/A |
| Software and algorithms | ||
| R software | The R foundation | R version 3.6.1 |
| Visiopharm | Visiopharm | Version 2018.4.3.4480 |
| GraphPad Prism | GraphPad Software, Inc | Versions 8.0.2 and 8.4.2 |
| Analyst Software, version 1.7 | Sciex | N/A |
| Multiquant Software, version 3.0 | Sciex | N/A |
| Biacore Insight Evaluation Software | Cytiva | Version 2.0.15 |
| Biacore T200 Evaluation Software | Cytiva | Version 3.2 |
| Other | ||
| Chow diet (mouse studies) | Purina | Cat # 5061 |
| Chow diet (rat studies) | Purina, LabDiet PicoLab | Cat # 5053 |
| Chow diet (GDF15 KO mouse studies) | PicoLab | 5L0D |
| Amine Coupling Kit | Cytiva | BR100050 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Danna Breen (danna.breen@pfizer.com).
Materials availability
Anti-GDF15 mAB (mAB2) will not be shared owing to commercialization consideration.
Data and code availability
No new data sets or codes were generated in this article.
Experimental model and subject details
Mice
C57BL/6J (Jackson Laboratories, Stock# 664) male mice aged 8–10 weeks were acclimated to handling upon arrival for 1 week and were single-housed under a standard 12-h light:dark cycle in a temperature- and humidity-controlled environment (22 ± 1°C). Mice were given ad libitum access to tap water and standard chow (Innovive mouse metal feeder; Purina rodent diet 5061; Purina Mills, St. Louis, MO) except when fasting was required (as detailed in Methods). All procedures were approved by the Pfizer Cambridge Animal Care and Use Committee (Mouse – Animal Use Protocol KSQ-01127). Male GDF15-/- and WT mice were generated using CRISPR/Cas-9 as previously described in the study by Frikke-Schmidt et al., 2019. Thirty-week-old male mice were bred in-house and single-housed with ad libitum access to a chow diet (PicoLab, 5L0D; 2.91 kcal/gm) and access to two forms of enrichment. GDF15-/- and WT mice were kept on a standard 12-h light:dark cycle in a humidity- and temperature-controlled environment (22 ± 1°C). All experiments were approved by the University of Michigan Institutional Animal Care and Use Committee (Animal Use Protocol # PRO00007908).
Rats
Sprague-Dawley male rats (Charles River, Strain #001) aged 9–10 weeks were acclimated upon arrival for handling for 1 week and housed individually to enable accurate food intake measurements. Immune-compromised male rats, RNU #316 - Crl:NIH-Foxn1, aged 10–14 weeks were acclimated upon arrival for handling for 1 week and housed individually under immunocompromised housing conditions to enable accurate food intake measurements. Rats were housed under a standard 12-h light:dark cycle in a temperature- and humidity-controlled environment (22 ± 1°C). Rats were given ad libitum access to water and standard chow (Innovive cages; Purina rodent diet 5053) except when fasting was required. All procedures were approved by the Pfizer Cambridge Animal Care and Use Committee (Rat – Animal Use Protocol KSQ-01197).
Method details
mAB2 in vivo efficacy studies
Mouse GDF15 cDNA was introduced in pAAV-CBA-W plasmid (Lake Pharma). AAV8 control and mGDF15 virus were produced at the MGH Vector Core Facility. C57BL/6J (Jackson Laboratories) male mice aged 8–10 weeks were dosed with either AAV8 control or AAV8-mGDF15 at 2e+10 genome copies per mouse through retro-orbital injections of 100 uL. When AAV8-mGDF15-treated mice achieved 10% weight compared with baseline, mAB2, or IgG control antibodies were administered subcutaneously at 10 mg/kg every 3 days (Q3D). For acute experiments, a mouse Fc-tagged human recombinant GDF15 (Fc-GDF15) was administered subcutaneously at 0.1 mg/kg and simultaneously with either mAB2 or IgG control antibody at 10 mg/kg as a single dose. Fc-GDF15, mAB2, and IgG control were produced at Pfizer Inc. Mice were fasted for 16 h. Blood was collected via the tail snip method. In the refeed study, food was reintroduced 30 min after injection of Fc-GDF15 and mAB2 or IgG control. Food consumption was measured after 1, 2, and 4 h. To be consistent with the timeline, the mice in the fasted study were dosed with Fc-GDF15 and mAB2 for 4.5 h.
Anti-GDF15 antibody mAB2
Anti-GDF15 antibodies were derived from mouse hybridoma where recombinant GDF15 was used as immunogens. Hybridoma clones were selected based on their ability to neutralize mouse and human GDF15 binding to its receptor GFRAL. The selected top clone was affinity matured using phage display. mAB2 is engineered to be a mouse anti-mouse GDF15 antibody that cross-reacts with human GDF15 with similar affinity. mAB2 comprises a mouse IgG1 effector function null D265A (i.e., aspartic acid to alanine at amino acid 265) mutation in the Fc.
GDF15 is a divergent member of the transforming growth factor-β superfamily that shares overall low-sequence homology with other members of this superfamily (<30% sequence identity with its closest member BMP7). Based on sequence alignment, the mAB2-binding epitope on GDF15 is not conserved in BMP7.
mAB2 was diluted in sterile phosphate-buffered saline (PBS) for in vivo studies and dosed subcutaneously. The dose of mAB2 (10 mg/kg) was selected to neutralize greater than 10 ng/mL GDF15 for up to 5 days; this was based on mouse exposures of a similar antibody targeting GDF15. The C-average of the mAB2 dose is approximately 1,000x molar access of 10 ng/mL GDF15. Across rodent studies reported here, circulating plasma GDF15 ranged from 0.8 to 6 ng/mL, all lower than the maximum concentration that this dose of mAB2 was selected to neutralize.
Low-LPS-dose-induced sepsis
Sepsis was induced in C57BL/6J male mice by a single intraperitoneal injection of 0.1 mg/kg lipopolysaccharide (LPS) (Sigma-Aldrich, Catalog # L2880, Lot # 059M4031V). LPS was diluted in sterile water and further diluted in saline + 0.5% bovine serum albumin (BSA). The control group was injected with saline + 0.5% BSA. mAB2 or IgG control antibodies were dosed at 10 mg/kg simultaneously with LPS or vehicle. In the fasted group, mice were fasted 16 h overnight and food was reintroduced along with the treatments. Food consumption and body weight were measured at 1, 2, 4, and 24 h. Blood was collected into ethylenediaminetetraacetic-acid (EDTA)-coated tubes using cardiac puncture at the end of 24 h. Plasma was separated after blood centrifugation for 10 min at 2,000 g and stored at –80°C.
Sprague-Dawley male rats (Charles River) were dosed subcutaneously with mAB2 or IgG control antibodies at 10 mg/kg 1 h before LPS, which was dosed intraperitoneally at 0.25 mg/kg. Food was reintroduced 1 h after LPS dose.
High-LPS-dose-induced sepsis
C57BL/6J (Jackson Laboratories, Stock# 664) male mice aged 8–10 weeks were subcutaneously pretreated with mAB2 at 10 mg/kg dose or with IgG control antibody. Sixteen hours after antibody treatments, sepsis was induced by a single intraperitoneal injection of LPS (Sigma-Aldrich, Catalog # L2880, Lot # 059M4031V) diluted in sterile PBS at sublethal dose of 5 mg/kg. The control group was injected with PBS. Animals were monitored for body weight, food intake, survival, and health condition at 6, 24, 48, 72, and 96 h. Mortality was defined by animal found dead and animal found moribund and sequential euthanization for humane end point. Health condition observation was based on a health assessment scoring system (Table S1).
High-LPS-dose-induced sepsis in GDF15-/-mice
Male GDF15-/- and WT mice with body weights of 29.85 g +/- 0.39 g (mean +/- SEM) were generated using CRISPR/Cas-9 as previously described in the study by Frikke-Schmidt et al., 2019. All mice bred for the experimental cohort were included, as all mice were healthy and naive to prior experimentation. Mice were handled for at least 1 week before experiment by a consistent handler to reduce stress. Lyophilized LPS (Sigma-Aldrich L4391-1MG lot: 088M4067V) was diluted in saline to a stock of 1 mg/mL. On the day of the experiment, 1 mg/mL LPS stock was further diluted in saline to appropriate concentrations. Mice were randomized to determine treatment. Mice were injected with either 5 mg/kg LPS or saline intraperitoneally 2 h after lights on and food was returned to cages. Food intake, body weight, behavior, and appearance scores were measured both at baseline and again at 6, 24, 48, 72, and 96 h after LPS administration. All data were included in the statistical analysis.
Rat Yoshida sarcoma studies
Yoshida cell line (obtained from Cell Line Services: #RY [ham] Yoshida sarcoma) were continuously subcultured in preparation for implantation into animals. The cells were plated on T-500 flasks and split twice each week at a ratio of 1:5 when reaching 90% confluency. The cells were expanded to achieve the desired number to be used in vivo. Immune-compromised rats, RNU #316 – Crl:NIH-Foxn1, aged 9–10 weeks were used for the study. On the day of in vivo implantation, cells were harvested, counted, and suspended into 1:1 sterile Matrigel/PBS and maintained on ice throughout the implantation procedure. After acclimation and baseline food weight/body weights were established, rats were inoculated with 1x106 Yoshida cells/rat (one injection, subcutaneous into the left flank region, in 1:1 sterile Matrigel/PBS; 250-μL volume, flank region). Rats were then monitored for tumor take rate daily until the average tumor size reached a volume in which accurate measurements can be made (∼500 mm3) using digital calipers. When the tumor-bearing rats lost 10% of their tumor-free body weight, 10 mg/kg of mAB2 or control IgG were injected subcutaneously into the rats. At the point when the tumor rats reached approximately 20% weight loss, animals were euthanized, and end points were collected.
Assays
GDF15 and mouse KCGRO were measured using kits from R&D systems (#MGD150 and #MKC00B), and insulin was measured using the Alpco kit (#80-INSMS-E01). Rat KCGRO was measured using the MSD Kit (#K15059D). The protocols provided with the kits were followed.
Lipid profiling
Lipids were extracted from 10 μL of plasma with 900 μL dichloromethane:isopropanol:methanol (25:10:65, v/v/v) containing the following internal standards at a concentration of 200 nM: glyceryl triheptadecanoate, 1,2-dinonadecanoin, cholesteryl heptadecanoate, 1,2-dilauroyl-sn-glycero-3-phosphocholine, 1-heptadecanoyl-2-hydroxy-sn-glycero-3-phosphocholine, and N-heptadecanoyl-D-erythro-sphingosine. Lipid extracts were then dried under N2 and resuspended in isopropanol:acetonitrile:water (2:1:1, v/v/v). Targeted lipidomics analyses were performed on a UPLC-MS/MS using a Waters Acquity UPLC coupled to a Sciex QTrap 5500 mass spectrometer. Lipid classes were separated by reversed-phase chromatography on a Waters Acquity UPLC CSH C18 column, 1.7 micron, 2.1 x 50 mm. The column temperature was set to 55°C and a 10-min gradient was run with the following mobile phase solvents: (i) mobile phase A = 10 mM ammonium formate in water:acetonitrile:formic acid (40:60:0.1, v/v/v) and (ii) mobile phase B = 10 mM ammonium formate in acetonitrile:isopropanol:formic acid (10:90:0.1, v/v/v). Lipid species were then analyzed on a mass spectrometer using positive ion electrospray ionization in the multiple reaction monitoring mode. LC chromatogram peak integration was performed with Sciex MultiQuant software. All data reduction was performed with in-house software.
c-fos quantification in the area postrema by immunohistochemistry
For imaging studies, mice were first perfused with PBS for 3 min via cardiac perfusion to remove blood, followed by 10% formalin for 7 min. Brains were dissected, being very careful to keep the hindbrain and perhaps extend into the spinal cord. The whole brain was then fixed in fresh formalin. Perfusion-fixed mouse brains were shipped based on vendor recommendations to be processed for histological coronal sectioning and staining. Ten animals from each cohort group were used for the colorimetric immunostaining with primary antibody (provided by FD NeuroTechnologies as rabbit anti-c-Fos IgG [Cat.no. sc-52, Santa Cruz Biotechnology, Santa Cruz, CA]) on free-floating, 30-μm coronal cryostat sections of the mouse hindbrain region. Chromogenic IHC using 3,3′-diaminobenzidine (DAB) staining method followed by counterstaining of the c-fos-immunostained sections with cresyl violet solution along with all histological sectioning, immunolabeling, staining, and coverslipping was performed by FD NeuroTechnologies (Columbia, MD). The AP region was coronally sectioned from Bregma –7.20 mm to –7.76 mm; the interval between two stained sections of 120 μm.
Prepared histological brain slices (typically three brain slices per slide) were imaged with a Hitachi HV-F202SCL camera on the Axioscan Z.1 (Carl Zeiss Microscopy, LLC). Transmitted bright-field illumination imaging of whole tissue sections using a Plan-Apochromat 20x/NA 0.8 air objective was performed with a 10% overlap between adjacent image tiles and stitched. Z-stack sections of three optical slices, followed by an extended depth of field processing, was performed during the acquisition to generate a single, 2D image file for each histological slice. All tissue sections were imaged under the same illumination conditions. All raw “.czi” imaging data of scanned histology sections covering the AP were subsequently imported into Visiopharm (version 2018.4.3.4480) for regional annotation and a custom APP within Visiopharm was designed for c-fos segmentation based upon the DAB stain and counted, normalized to the annotated AP area. The Mouse Brain in Stereotaxic Coordinates by Paxinos and Franklin (compact 2nd edition, 2004) and the Allen Brain Atlas (https://mouse.brain-map.org/static/atlas) was used to identify the AP in the histological sections per animal.
mAB2 vs. human and mouse GDF15 SPR methods
SPR was used to characterize the kinetic binding of the murinized anti-GDF15 antibody mAB2 (mAB2) to human GDF15 and mouse GDF15, each fused to mouse Fc (mFc-rhGDF15, mFc-rmGDF15). A CM4 sensor chip (Cytiva, BR100534) was prepared using a Biacore T200 instrument by amine coupling mFc-rhGDF15 and mFc-rmGDF15 to the chip surface. The immobilization level of mFc-rhGDF15 on flow cell 2 was about 9 resonance units (RUs), whereas immobilization levels for mFc-rmGDF15 on flow cells 3 and 4 were 170 RUs and 103 RUs, respectively. Flow cell 1 was used as a reference. The sample and running buffer was HBS-EP+ (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% Tween 20) and the SPR experiment was run at 37°C using a data collection rate of 10 Hz. mAB2 was either 2-fold serially diluted with concentrations ranging from 12.5 nM to 3.13 nM or diluted 3 times from 6 nM to 0.67 nM. The mAB2 dilutions were injected for 60 s at a flow rate of 100 μL/min with dissociation data collected for 600 s, 1,800 s, or 2,000 s. The sensor chip surface was regenerated by removing mAB2 with two injections of 10 mM Glycine pH 1.5 for 60 s at a flow rate of 30 μL/min. Rate constants and apparent affinities of mAB2 were determined by double referencing the resulting sensorgram data (Myszka, 1999) and fit using T200 Evaluation software, version 3.2 (Cytiva).
GDF15/GFRAL/RET signaling cell-based assay
Human GFRAL and RET cDNA were introduced to CHOK1 cells and selected with antibiotics to establish the coexpression stable cell line. Briefly, cells were seeded in 384-well plate the night before assaying. Various doses of mAB2 (in PBS) were added directly into the cells for 30 min at room temperature. Cells were then stimulated with GDF15 for 10 min at room temperature. One hundred fifty picomoles of GDF15 was used to generate IC50 curves. Cells are lyzed after 10 min of treatment and subjected to phospho-ERK assay using Phospho-ERK1/2 (Thr202/Tyr 204) HTRF kit (CISBIO 64ERKPEH). Raw data are captured by Perkin Elmer EnVision Plate Reader.
Statistical analysis
Figure 1. (D) Welch’s heteroscedastic F test was used to compare GDF15 levels due to significant differences in variance (Levene’s test p<0.05) between treated and control animals. (E) Longitudinal mixed effects analysis of variance (ANOVA) models with an AR1 covariance structure and treatment group and days as fixed effects and animals as a random effect were used to estimate body weight over time. The model was used to compare treatment group least squares means averaged across 15 to 18 days in the longitudinal model. (G) Two c-fos imaging studies combined for analysis with a mixed effects analysis of covariance model with treatment group as a fixed effect, animal as a random effect, and study as a covariate to compare treatment group mean-square-root-transformed c-fos+ cell counts using 3–4 slices from the AP region of the hindbrain of each mouse. Figure 2. (B-C) Percent change from baseline body weight at 24 h and cumulative food intake at 24 h were compared between treatment groups using unpaired two-tailed t-test. (E) Cumulative food intake over time was compared between treatment groups using longitudinal mixed effects ANOVA models with an AR(1) covariance structure, treatment group, and h as fixed effects, and animals as a random effect for the study where the animals were refed. (F–H) Two-factor ANOVA (feed and treatment) was used to compare triglycerides and insulin with feed∗treatment interaction pairwise comparisons (significant model interactions for all response variables). No animals were removed from analysis in the LPS experiments. Figure 3. (D) Welch’s heteroscedastic F test was used to compare percent change body weight between groups due to heterogenous variance (Levene’s test p<0.05). (E, F, G) ANOVA with Tukey’s multiple comparison adjustment was used to compare group means. Figure 4. (C) Welch’s heteroscedastic F test was used to compare GDF15 levels due to significant differences in variance (Levene’s test p<0.05) between treated and control animals. (D) Longitudinal mixed effects analysis of variance (ANOVA) models with an AR1 covariance structure and treatment group and days as fixed effects and animals as a random effect were used to estimate body weight over time. The model was used to compare treatment group least squares means averaged across 15 to 16 days in the longitudinal model. (F) Plasma KCGRO was compared between vehicle and LPS dosed group using unpaired two-tailed t-test. (G, H) Welch’s heteroscedastic F test was used for comparison due to significant differences in variance (Levene’s test p<0.05) between treated and control animals. Figure 5. (C, D) Percent change from baseline body weight and cumulative food intake were compared between treatment groups using longitudinal mixed effects ANOVA models with an AR(1) covariance structure, treatment group, and hours as fixed effects, and animals as a random effect. (F) Failure time data were compared between treatment groups with Kaplan-Meier analysis using the log rank test. We used a conservative hazard ratio of 5 to estimate the sample size for the sublethal 5 mg/kg LPS experiment with a sample size of N = 13/treatment arm providing more than 88% power with alpha = 0.05. Figure 6. (A, B) Percent change from baseline body weight and cumulative food intake were compared between treatment groups using longitudinal mixed-effects ANOVA models with an AR(1) covariance structure, treatment group, and h as fixed effects, and animals as a random effect. (D) Failure time data were compared between treatment groups with Kaplan-Meier analysis using the log rank test. The Tukey adjustment for multiple comparisons was used for all models. Histograms, QQ plots, and Shapiro-Wilk test were used to check the normality assumption for linear models. All analyses were performed in R, version 3.6.1.
Acknowledgments
Studies were sponsored by Pfizer. Editorial support was provided by Diane Hoffman, PhD, of Engage Scientific Solutions, and was funded by Pfizer.
Author contributions
Conceptualization, O.B., D.M.B., and R.J.S.; Methodology, N.B.V and K.H.; Formal analysis, D.B.; Investigation, S.J., A.P., C.Z., L.K., E.Z.Q., T.H., M.L., and K.K.; Data curation, S.J. and C.Z.; Writing – Original Draft, O.B. and D.M.B.; Contributed to data or manuscript writing and editing, and approved final version for submission, all authors; Visualization, S.R. and A.P.; Supervision, O.B., D.M.B., and R.J.S.
Declaration of interests
R.J.S. is a member of the advisory boards of Novo Nordisk, Scohia, Kintai Therapeutics, and Ionis. D.M.B., S.J., C.Z., S.J., X.L., L.K., J.P., K.H., E.Z.Q., D.B., M.L., K.K., Z.W., B.B.Z., and L.L. are employees and shareholders of Pfizer. N.B.V., T.H., and O.B. were employees of Pfizer during the conduct of the study and are current employees of Dicerna Pharmaceuticals, Inc., JOINN Biologics, and Cellarity, respectively. A.P. declares no conflict of interest.
Inclusion and diversity
The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: June 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102554.
Supplemental information
References
- Amiot F., Fitting C., Tracey K.J., Cavaillon J.M., Dautry F. Lipopolysaccharide-induced cytokine cascade and lethality in LT alpha/TNF alpha-deficient mice. Mol. Med. 1997;3:864–875. [PMC free article] [PubMed] [Google Scholar]
- Anand I.S., Kempf T., Rector T.S., Tapken H., Allhoff T., Jantzen F., Kuskowski M., Cohn J.N., Drexler H., Wollert K.C. Serial measurement of growth-differentiation factor-15 in heart failure: relation to disease severity and prognosis in the Valsartan Heart Failure Trial. Circulation. 2010;122:1387–1395. doi: 10.1161/CIRCULATIONAHA.109.928846. [DOI] [PubMed] [Google Scholar]
- Burfeind K.G., Zhu X., Levasseur P.R., Michaelis K.A., Norgard M.A., Marks D.L. TRIF is a key inflammatory mediator of acute sickness behavior and cancer cachexia. Brain Behav. Immun. 2018;73:364–374. doi: 10.1016/j.bbi.2018.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerson P.J., Wang F., Du Y., Liu Q., Pickard R.T., Gonciarz M.D., Coskun T., Hamang M.J., Sindelar D.K., Ballman K.K. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017;23:1215–1219. doi: 10.1038/nm.4393. [DOI] [PubMed] [Google Scholar]
- Frikke-Schmidt H., Hultman K., Galaske J.W., Jørgensen S.B., Myers M.G., Jr., Seeley R.J. GDF15 acts synergistically with liraglutide but is not necessary for the weight loss induced by bariatric surgery in mice. Mol. Metab. 2019;21:13–21. doi: 10.1016/j.molmet.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J.Y., Crawley S., Chen M., Ayupova D.A., Lindhout D.A., Higbee J., Kutach A., Joo W., Gao Z., Fu D. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature. 2017;550:255–259. doi: 10.1038/nature24042. [DOI] [PubMed] [Google Scholar]
- Husebo G.R., Gronseth R., Lerner L., Gyuris J., Hardie J.A., Bakke P.S., Eagan T.M. Growth differentiation factor-15 is a predictor of important disease outcomes in patients with COPD. Eur. Respir. J. 2017;49 doi: 10.1183/13993003.01298-2016. [DOI] [PubMed] [Google Scholar]
- Johnen H., Lin S., Kuffner T., Brown D.A., Tsai V.W., Bauskin A.R., Wu L., Pankhurst G., Jiang L., Junankar S. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat. Med. 2007;13:1333–1340. doi: 10.1038/nm1677. [DOI] [PubMed] [Google Scholar]
- Kempf T., von Haehling S., Peter T., Allhoff T., Cicoira M., Doehner W., Ponikowski P., Filippatos G.S., Rozentryt P., Drexler H. Prognostic utility of growth differentiation factor-15 in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007;50:1054–1060. doi: 10.1016/j.jacc.2007.04.091. [DOI] [PubMed] [Google Scholar]
- Laurens C., Parmar A., Murphy E., Carper D., Lair B., Maes P., Vion J., Boulet N., Fontaine C., Marques M. Growth and differentiation factor 15 is secreted by skeletal muscle during exercise and promotes lipolysis in humans. JCI Insight. 2020;5 doi: 10.1172/jci.insight.131870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner L., Gyuris J., Nicoletti R., Gifford J., Krieger B., Jatoi A. Growth differentiating factor-15 (GDF-15): a potential biomarker and therapeutic target for cancer-associated weight loss. Oncol. Lett. 2016;12:4219–4223. doi: 10.3892/ol.2016.5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner L., Hayes T.G., Tao N., Krieger B., Feng B., Wu Z., Nicoletti R., Chiu M.I., Gyuris J., Garcia J.M. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J. Cachexia Sarcopenia Muscle. 2015;6:317–324. doi: 10.1002/jcsm.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner L., Tao J., Liu Q., Nicoletti R., Feng B., Krieger B., Mazsa E., Siddiquee Z., Wang R., Huang L. MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J. Cachexia Sarcopenia Muscle. 2016;7:467–482. doi: 10.1002/jcsm.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Li Y., Tan X.Q., Jia P., Zhao J., Liu D., Wang T., Liu B. Plasma growth differentiation factor-15 is a potential biomarker for pediatric pulmonary arterial hypertension associated with congenital heart disease. Pediatr. Cardiol. 2017;38:1620–1626. doi: 10.1007/s00246-017-1705-7. [DOI] [PubMed] [Google Scholar]
- Li M., Song K., Huang X., Fu S., Zeng Q. GDF15 prevents LPS and Dgalactosamineinduced in fl ammation and acute liver injury in mice. Int. J. Mol. Med. 2018;42:1756–1764. doi: 10.3892/ijmm.2018.3747. [DOI] [PubMed] [Google Scholar]
- Luan H.H., Wang A., Hilliard B.K., Carvalho F., Rosen C.E., Ahasic A.M., Herzog E.L., Kang I., Pisani M.A., Yu S. GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell. 2019;178:1231–1244.e11. doi: 10.1016/j.cell.2019.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullican S.E., Lin-Schmidt X., Chin C.N., Chavez J.A., Furman J.L., Armstrong A.A., Beck S.C., South V.J., Dinh T.Q., Cash-Mason T.D. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 2017;23:1150–1157. doi: 10.1038/nm.4392. [DOI] [PubMed] [Google Scholar]
- Myszka D.G. Improving biosensor analysis. J. Mol. Recognit. 1999;12:279–284. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Pereiro P., Libran-Perez M., Figueras A., Novoa B. Conserved function of zebrafish (Danio rerio) Gdf15 as a sepsis tolerance mediator. Dev. Comp. Immunol. 2020;109:103698. doi: 10.1016/j.dci.2020.103698. [DOI] [PubMed] [Google Scholar]
- Santos I., Colaco H.G., Neves-Costa A., Seixas E., Velho T.R., Pedroso D., Barros A., Martins R., Carvalho N., Payen D. CXCL5-mediated recruitment of neutrophils into the peritoneal cavity of Gdf15-deficient mice protects against abdominal sepsis. Proc. Natl. Acad. Sci. U. S. A. 2020;117:12281–12287. doi: 10.1073/pnas.1918508117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkman N.L., Buchanan J.B., Heyen J.R., Chen J., Beverly J.L., Johnson R.W. Interleukin-6 facilitates lipopolysaccharide-induced disruption in working memory and expression of other proinflammatory cytokines in hippocampal neuronal cell layers. J. Neurosci. 2006;26:10709–10716. doi: 10.1523/jneurosci.3376-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N., Suzuki S., Duncan G.S., Millar D.G., Wada T., Mirtsos C., Takada H., Wakeham A., Itie A., Li S. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002;416:750–756. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- Tsai V.W., Macia L., Johnen H., Kuffner T., Manadhar R., Jorgensen S.B., Lee-Ng K.K., Zhang H.P., Wu L., Marquis C.P. TGF-b superfamily cytokine MIC-1/GDF15 is a physiological appetite and body weight regulator. PLoS One. 2013;8:e55174. doi: 10.1371/journal.pone.0055174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai V.W., Zhang H.P., Manandhar R., Schofield P., Christ D., Lee-Ng K.K.M., Lebhar H., Marquis C.P., Husaini Y., Brown D.A., Breit S.N. GDF15 mediates adiposity resistance through actions on GFRAL neurons in the hindbrain AP/NTS. Int. J. Obes. (Lond.) 2019;43:2370–2380. doi: 10.1038/s41366-019-0365-5. [DOI] [PubMed] [Google Scholar]
- Yang L., Chang C.C., Sun Z., Madsen D., Zhu H., Padkjaer S.B., Wu X., Huang T., Hultman K., Paulsen S.J. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 2017;23:1158–1166. doi: 10.1038/nm.4394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new data sets or codes were generated in this article.